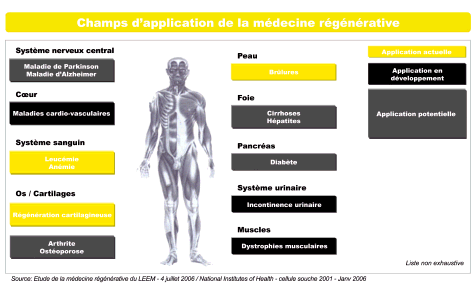
N° 3602 _______ ASSEMBLÉE NATIONALE CONSTITUTION DU 4 OCTOBRE 1958 DOUZIÈME LÉGISLATURE Enregistré à la Présidence de l'Assemblée nationale le 23 janvier 2007 RAPPORT D'INFORMATION DÉPOSÉ PAR LA DÉLÉGATION DE L'ASSEMBLÉE NATIONALE POUR L'UNION EUROPÉENNE (1), sur les médicaments de thérapie innovante ET PRÉSENTÉ par Mme Anne-marie Comparini, Députée. ________________________________________________________________ (1) La composition de cette Délégation figure au verso de la présente page. La Délégation de l'Assemblée nationale pour l'Union européenne est composée de : M. Pierre Lequiller, président ; MM. Jean-Pierre Abelin, Mme Elisabeth Guigou, M. Christian Philip, vice-présidents ; MM. François Guillaume, Jean-Claude Lefort, secrétaires ; MM. Alfred Almont, François Calvet, Mme Anne-Marie Comparini, MM. Bernard Deflesselles, Michel Delebarre, Bernard Derosier, Nicolas Dupont-Aignan, Jacques Floch, Pierre Forgues, Mme Arlette Franco, MM. Daniel Garrigue, Michel Herbillon, Marc Laffineur, Jérôme Lambert, Robert Lecou, Pierre Lellouche, Guy Lengagne, Louis-Joseph Manscour, Thierry Mariani, Philippe-Armand Martin, Jacques Myard, Christian Paul, Axel Poniatowski, Didier Quentin, André Schneider, Jean-Marie Sermier, Mme Irène Tharin, MM. René-Paul Victoria, Gérard Voisin. SOMMAIRE _____ Pages INTRODUCTION 7 I. UNE ARCHITECTURE ADAPTÉE AUX TROIS OBJECTIFS POURSUIVIS : LA QUALITÉ DES SOINS ; L'UNITÉ DU MARCHÉ INTÉRIEUR ; LA PLACE DES FIRMES EUROPÉENNES DANS LES MÉDICAMENTS DU FUTUR 13 A. Le diagnostic sur les médicaments de thérapie innovante : vide juridique et fragmentation du marché intérieur 13 1) Le droit communautaire présente une lacune sur les médicaments de thérapie innovante 13 a) Les dispositions applicables sont réparties entre plusieurs textes 13 b) Une lacune a été identifiée en ce qui concene l'ingénierie tissulaire 14 2) L'absence d'harmonisation s'accompagne d'une situation hétérogène, certains Etats membres, dont la France, ayant prévu leurs propres règles 15 3) La Commission propose d'établir un cadre harmonisé pour rendre plus homogène le niveau des soins, sécuriser les projets des chercheurs comme des entreprises et assurer ainsi l'avance de l'Europe dans un domaine essentiel et prometteur 18 a) L'élévation du niveau des soins 18 b) La recherche et l'industrie pharmaceutiques, notamment les PME, ont d'ores et déjà une stratégie sur les médicaments de thérapie innovante 18 c) Un cadre juridique est nécessaire pour leur indiquer comment seront évalués et éventuellement mis sur le marché puis suivis les résultats de leurs recherches 20 d) Plus tôt les normes européennes seront établies, plus leur autorité sera forte au plan international 20 B. Les options partagées retenues par la Commission : un régime d'autorisation de mise sur le marché plus strict que pour les médicaments de droit commun ; des incitations en faveur des entreprises concernées 21 1) Une procédure centralisée, unique et plus exigeante de délivrance des AMM par l'Agence européenne du médicament, avec notamment la création d'un comité d'experts spécifique, une obligation de traçabilité et un système renforcé de suivi après autorisation 22 2) Des aménagements d'ordre administratif et financier 25 a) Un dispositif d'évaluation et de certification de la qualité des données « de développement initial » pour faciliter la valorisation des travaux des petites et moyennes entreprises innovantes 26 b) La réduction des honoraires perçus pour la fourniture de conseils scientifiques par l'Agence européenne du médicament 27 3) L'absence de débat majeur sur l'essentiel des dispositions nouvelles 27 II. DES AMÉNAGEMENTS A PREVOIR SUR TROIS ELEMENTS : LES HÔPITAUX ; LES PRODUITS COMBINÉS ASSOCIANT UN DISPOSITIF MÉDICAL ET UN MÉDICAMENT DE THERAPIE INNOVANTE ; LES PRÉCAUTIONS D'ORDRE ÉTHIQUE 29 A. Rectifier la portée de l'exception hospitalière pour garantir la sécurité sanitaire et permettre un développement dynamique et harmonieux de ce secteur 29 1) Un débat essentiel qui divise les Etats membres 29 2) La nécessité de faire prévaloir le principe de sécurité sanitaire et d'offrir en outre au secteur hospitalier l'opportunité de tirer parti de toutes ses potentialités 31 B. Préciser le champ respectif des dispositifs médicaux et du médicament pour les produits combinés 32 C. Permettre à l'Agence européenne du médicament de faire appel, en tant que besoin, à une instance consultative en matière d'éthique 34 ANNEXE : Liste des personnes entendues par la rapporteure 41 Mesdames, Messieurs, Le rythme sans précédent des découvertes et innovations qui a marqué au cours des six dernières décennies le secteur des médicaments à usage humain, a permis des progrès spectaculaires. Administrés en masse après la mise au point de la fabrication de la pénicilline, laquelle a été produite dès le début des années 1940 dans les pays anglo-saxons et diffusée en France à la Libération, les antibiotiques ont notamment été à l'origine d'une véritable révolution thérapeutique tant par le champ des pathologies traitées que par l'importance du nombre des patients concernés, chacun d'entre nous. Sans être exhaustif, d'autres substances d'usage moins courant mais non moins significatives ont permis l'amélioration de la qualité des soins, qu'il s'agisse notamment de la découverte des neuroleptiques en France, ou de celles des bêta-bloquants, des antiviraux et, naturellement, des traitements du cancer par chimiothérapie. Au fil du temps cependant, certains de ces progrès interviennent dans des conditions de plus en plus complexes. Les plus récentes et les plus prometteuses de celles qui commencent à poindre relèvent de la biomédecine. Trois approches sont plus particulièrement concernées : la thérapie génique, qui repose sur le principe du transfert de gène ; la thérapie cellulaire somatique, qui utilise des cellules dont les caractéristiques biologiques ont été sensiblement modifiées pour obtenir un effet thérapeutique, diagnostique ou préventif ; l'ingénierie tissulaire, enfin, qui vise à régénérer, réparer ou remplacer des tissus humains existants. Le dernier d'entre eux, le domaine de la médecine régénérative, est en lui-même très vaste, ne serait-ce que par l'ampleur des procédés et techniques qu'il met en jeu : les cellules peuvent provenir du patient lui-même, selon une démarche autologue, soit d'un donneur selon une approche allogénique, soit encore d'un animal, dans un cadre xénogénique. Ces nouvelles approches médicales entraînent des changements profonds dont l'ampleur et la portée ne peuvent être en l'état que difficilement mesurées. En premier lieu, le champ de leurs applications est pour l'instant assez réduit. Pour ce qui est de la médecine régénérative, l'usage courant de ses procédés ne concerne pour l'instant que le sang, les cartilages et la peau. Des développements sont en cours pour le cœur et le système circulatoire, les muscles et le système urinaire. Pour le futur, à échéance encore indéterminée, les chercheurs et les médecins espèrent être en mesure de combattre les maladies d'Alzheimer et de Parkinson, l'arthrite et l'ostéoporose ou encore le diabète et les pathologies du foie, cirrhoses et hépatites. Le schéma suivant récapitule ces éléments :
Pour ce qui est de la France, seule la thérapie cellulaire hématopoïétique, qui repose sur des greffes de cellules de la moelle osseuse ou du sang contre certaines pathologies du sang ou certaines immunothérapies anti-tumorales, est entrée en phase d'application thérapeutique étendue. La plupart des produits envisagés sont sinon encore dans le champ de la recherche clinique voire fondamentale. Selon les informations communiquées, l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) a ainsi autorisé une cinquantaine d'essais cliniques dans le domaine de la thérapie génique. Il convient donc de rester prudent quant à ces perspectives et d'éviter de nourrir de fols espoirs qui risqueraient d'être déçus. Claude Huriet, président de l'Institut Curie, rappelait lors d'un colloque de l'Académie des Sciences en septembre 2006 que « face aux espoirs soulevés par cette médecine régénératrice, il faut savoir garder et ne pas donner de faux espoirs au public »(1). L'éthique de la responsabilité interdit, en effet, de faire rêver les actuels patients et de leur présenter comme réalisables des perspectives qui ne sont, en l'état, au mieux qu'éventuelles. Elle s'impose d'autant plus que comme l'a estimé le professeur Axel Kahn(2), la thérapie cellulaire notamment représente collectivement le « rêve multi-centenaire de la médecine » : réparer et remplacer les pièces de la machine humaine. En deuxième lieu, ces nouveaux procédés thérapeutiques modifient la notion même de médicament, à plusieurs titres : - un remède n'est plus nécessairement un produit, un principe actif, chimique ou biologique, administré à l'homme. La réparation du tissu humain détérioré repose davantage sur un procédé appliqué à une partie du corps humain ou à des éléments vivants ; - acquièrent ou peuvent acquérir, par ailleurs, une valeur thérapeutique des tissus, cellules et organes auparavant considérés comme des « déchets », notamment hospitaliers. Tel est entre autres le cas du cordon ombilical, pour s'en tenir à l'exemple le plus emblématique. Il y a quelque jours encore, un article publié dans le numéro de janvier 2007 du mensuel Pediatrics a relaté comment le sang du cordon ombilical, congelé au moment de sa naissance de l'enfant, avait permis de guérir d'une forme de leucémie un enfant alors âgé de 3 ans. Celui-ci ne présente plus, depuis maintenant 3 années, de symptôme. Face à de telles évolutions, la Commission européenne s'est légitimement interrogée sur les aménagements à apporter au régime juridique actuel du médicament, lequel est fixé au niveau communautaire tant pour des raisons d'harmonisation des politiques de santé publique que de bon fonctionnement du marché intérieur. A l'issue d'un examen attentif dont elle retrace les éléments essentiels dans une étude d'impact qui n'a malheureusement pas été traduite en français (document SEC (2005) 1444), sa réponse a été très claire. Tant des impératifs de santé publique que de marché intérieur, ainsi que de compétitivité des entreprises européennes, selon les prescriptions de la stratégie de Lisbonne, exigent une évolution. En conséquence, elle a présenté, le 25 novembre 2005, la présente proposition de règlement, soumise à l'examen de la Délégation en application de l'article 88-4 de la Constitution. Après plusieurs mois d'un fonctionnement sans heurt, la procédure de codécision, dont relève cette dernière, a connu en septembre des développements inhabituels, au Parlement européen. Le rapport du rapporteur, M. Miroslav Mikolá_ik (PPE-DE, Slovaquie), a été rejeté, le 13 septembre, lors du vote final de la commission de l'environnement, de la santé publique et de la sécurité alimentaire, sur des points liés à l'éthique. La procédure parlementaire a donc dû repartir de son point de départ. Depuis, le même rapporteur a diffusé le 8 novembre dernier un nouveau projet de rapport. Pour la commission des affaires juridiques est de nouveau désignée, comme rapporteure pour avis, Mme Hiltrud Breyer (Verts-ALE, Allemagne). Les circonstances conduisent donc la Délégation pour l'Union européenne à se prononcer très en amont de la procédure, puisque le Parlement européen ne devrait le faire qu'au printemps et que la Présidence allemande a inscrit ce texte, dans la perspective d'un accord politique entre les Etats membres, à l'ordre du jour du Conseil Emploi Politique sociale, Santé et Consommateurs du 30 mai prochain. Pour sa part, la Délégation est dès maintenant en mesure de conclure que la proposition de la Commission étant dans l'ensemble adaptée à ses objectifs sanitaires et économiques, elle n'appelle à ce stade d'aménagement que sur trois points essentiels dont les deux premiers concernent son champ d'application et le dernier un renforcement des précautions d'ordre éthique. I. UNE ARCHITECTURE ADAPTÉE AUX TROIS OBJECTIFS POURSUIVIS : LA QUALITÉ DES SOINS ; L'UNITÉ DU MARCHÉ INTÉRIEUR ; LA PLACE DES FIRMES EUROPÉENNES DANS LES MÉDICAMENTS DU FUTUR A. Le diagnostic sur les médicaments de thérapie innovante : vide juridique et fragmentation du marché intérieur 1) Le droit communautaire présente une lacune sur les médicaments de thérapie innovante a) Les dispositions applicables sont réparties entre plusieurs textes Les dispositions communautaires relatives aux différents produits et instruments thérapeutiques relèvent de plusieurs textes, selon une construction qui résulte de la « révision pharmaceutique » achevée en mars 2004 : - la directive 2001/83/CE du 6 novembre 2001 instituant un code communautaire relatif au médicament, dont le dispositif initial a été ultérieurement modifié tant par la directive 2003/63/CE de la Commission dans son annexe, que par la directive 2004/24/CE du 31 mars 2004 sur les médicaments à base de plantes et la directive 2004/27/CE du même jour pour les autres médicaments à usage humain ; - la directive 2003/94/CE du 8 octobre 2003 établissant les principes et lignes directrice de bonnes pratiques de fabrication concernant les médicaments à usage humain et les médicaments expérimentaux à usage humain ; - le règlement (CE) n° 726/2004 du 31 mars 2004, qui fixe la procédure d'autorisation de mise sur le marché centralisée par l'Agence européenne du médicament, basée à Londres ; - la directive 2004/23/CE du 31 mars 2004, dite « tissus et cellules », relative à l'établissement de normes de qualité et de sécurité pour le don, l'obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissus et cellules humains. Pour l'essentiel, le dispositif juridique ainsi établi pour les médicaments repose sur : - l'exigence d'une autorisation de mise sur le marché (AMM) avant toute commercialisation d'un médicament produit industriellement, ce qui laisse de côté les préparations spécifiques ; - la délivrance de cette AMM soit par l'Agence européenne du médicament, qui dispose même d'un monopole dans certains domaines (tel est notamment le cas pour les médicaments issus des biotechnologies), soit par une autorité nationale compétente (l'Agence française de sécurité sanitaire des produits de santé - des règles harmonisées d'étiquetage, de notice, de distribution en gros et de publicité ; - un dispositif de pharmacovigilance, de suivi des effets indésirables des médicaments sur l'homme et des cas de mésusages comme d'abus graves ; - des normes communes de qualité et de sécurité pour les tissus et cellules humains, s'appliquant d'un bout à l'autre de la filière. b) Une lacune a été identifiée en ce qui concene l'ingénierie tissulaire Sur le plan juridique, aucune initiative n'est nécessaire pour préciser le régime dont relèvent, d'une part, les médicaments de thérapie génique et, d'autre part, ceux de thérapie cellulaire somatique. La dernière révision pharmaceutique communautaire les a en effet explicitement inclus dans le cadre de la directive précitée 2001/83/CE. Tel n'est cependant pas le cas pour l'ingénierie tissulaire, qui ne relève actuellement d'aucun texte. La directive précitée 2004/23/CE a, en effet, pour seul objet de fixer des normes de qualité et de sécurité, et d'encadrer l'ensemble du processus et des étapes, de la collecte des tissus ou cellules jusqu'à leur cession. Elle précise ainsi les règles relatives au don, à l'obtention, au contrôle, à la transformation, à la conservation, au stockage et à la distribution de tissus et cellules humains, quel que soit leur usage. Elle oblige de manière concrète les États membres à superviser les activités d'obtention et de contrôles de tissus et cellules - agrément, inspection, traçabilité et registre. Elle fixe également les règles en matière de qualité et de sécurité des tissus et cellules. Elle ne prévoit pas de règles propres aux thérapies cellulaires non somatiques ou à la médecine régénérative. Comme la Commission, on constate par conséquent un vide juridique entre, d'une part, les règles posées par la directive 93/42/CEE qui fixe les règles de certification CE des dispositifs médicaux, et par la directive 90/385/CEE relative aux dispositifs médicaux implantables actifs et, d'autre part, l'ensemble des dispositions sur le médicament à usage humain. 2) L'absence d'harmonisation s'accompagne d'une situation hétérogène, certains Etats membres, dont la France, ayant prévu leurs propres règles Comme l'a relevé la Commission dans son étude d'impact précitée annexée à la proposition de règlement, l'absence de cadre juridique communautaire sur la médecine régénérative, ingénierie tissulaire et thérapie cellulaire non somatique, a créé des situations très diverses selon les Etats membres. L'absence de cadre harmonisé ne fait, en effet, obstacle ni aux initiatives de certains Etats membres, ni à l'inaction. Certains pays ont prévu des cadres spécifiques. Tel est notamment le cas de la France et du Royaume-Uni. D'autres se sont appuyés sur les règles existantes qu'elles concernent les dispositifs médicaux ou les médicaments à usage humain, ou bien en créant de nouveau dispositifs pour les seuls médicaments de thérapie cellulaire. Le tableau suivant récapitule la situation telle que l'a constatée la Commission. Réglementation sur les produits d'ingénierie tissulaire humains, autologues ou allogéniques dans les Etats membres
Les symboles indiquent les réglementations concernant les produits d'ingénierie tissulaire humains : Source : Commission européenne, étude de mars 2004. Du point de vue du patient, ces divergences entre les pays provoquent deux types d'inégalités : - d'une part, sur le principe de l'accès ou non aux nouveaux médicaments ; - d'autre part, sur la qualité des soins qu'il peut recevoir, puisque le niveau des exigences n'est pas le même. En ce qui concerne les laboratoires pharmaceutiques, le régime juridique d'un produit varie selon l'Etat membre considéré. Le marché intérieur est en pratique fragmenté. La France dispose depuis la loi « Huriet » n°94-654 du 29 juillet 1994 relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal, dont le contenu a été modifié par la loi Les produits cellulaires relèvent tous d'une catégorie précise. Le statut de médicament est conféré à ceux d'origine animale et aux produits de thérapie génique. Un cadre a été créé pour les produits cellulaires à finalité thérapeutique qui ne comportent que des cellules d'origine humaine. L'encadrement juridique des activités de prélèvement et d'administration est adapté au contexte, en fonction de la gravité des pathologies et du caractère plus ou moins invasif, c'est-à-dire susceptible d'affecter l'organisme, des prélèvements. Les prélèvements légers sont ainsi autorisés en cabinet libéral ou en établissement de santé ne bénéficiant pas d'une autorisation spécifique. En revanche, l'activité de préparation des cellules ne peut être réalisée que par des établissements ou organismes dûment autorisés par l'AFSSAPS. Les activités doivent être conformes à des règles de bonne pratique établies sur avis de l'Agence de biomédecine. Au Royaume-Uni, le cadre actuel est plus flexible que celui en vigueur en France, selon éléments communiqués à la rapporteure. 3) La Commission propose d'établir un cadre harmonisé pour rendre plus homogène le niveau des soins, sécuriser les projets des chercheurs comme des entreprises et assurer ainsi l'avance de l'Europe dans un domaine essentiel et prometteur a) L'élévation du niveau des soins Sur le plan sanitaire, l'objectif de la Commission, qui est d'avoir en Europe, dans un cadre communautaire applicable à tous les Etats membres, et de la manière la plus homogène possible, des soins de qualité grâce à un niveau élevé d'exigences, est incontestable. Il n'appelle pas d'observation particulière. b) La recherche et l'industrie pharmaceutiques, notamment les PME, ont d'ores et déjà une stratégie sur les médicaments de thérapie innovante En matière de nouveaux médicaments, deux types d'opérateurs interviennent : d'une part, la recherche publique, notamment la recherche hospitalière, qui détient en France, en la matière, le premier rôle ; d'autre part, les entreprises pharmaceutiques. Ces dernières ont dans l'ensemble commencé à définir leur approche sur les médicaments de thérapie innovante, à plusieurs niveaux. Il s'agit de répondre aux besoins d'un domaine en plein développement. Le marché biopharmaceutique au sens large, qui comprend aussi les médicaments traditionnels issus des biotechnologies, devrait représenter 14 % du marché mondial du médicament en 2011. Sur un plan général, dans le cadre d'un partenariat public-privé conclu par la Fédération européenne d'associations et d'industries pharmaceutiques (FEAIP) et auquel participent la Commission, des chercheurs, des organisations représentant les patients et des régulateurs nationaux, le secteur a publié, le 18 septembre dernier, son agenda stratégique sur l'Initiative médicaments innovants (IMI). L'objectif est de créer les conditions d'un développement en Europe des médicaments leaders en améliorant, sur le plan méthodologique, dans un cadre coordonné, le procédé de découverte et de développement de nouveaux médicaments, grâce à des protocoles plus sûrs et plus efficaces pour les patients. L'IMI est candidate aux initiatives conjointes (ITC) proposées dans le cadre du 7e PCRD, les médicaments innovants figurant parmi les six domaines éligibles aux ITC. Le secteur a chiffré l'enveloppe nécessaire au projet à 3 milliards d'euros pour la période allant de 2007 à 2013, soit 460 millions d'euros par an. Il estime que la moitié de cette somme devrait provenir du 7e PCRD, qui verserait les fonds correspondants aux organismes publics concernés. Sur le plan national, l'industrie pharmaceutique française dispose notamment d'une structure de coordination, le comité biotechnologies, au sein du LEEM (les entreprises du médicament). Dans ce cadre, des études prévisionnelles, telle que « Etat des lieux, les biomédicaments en France en 2004 » sont notamment réalisées. Le comité a également pour objet d'analyser les contraintes du secteur et comme a pu le constater la rapporteure au cours d'une réunion à laquelle il lui a été permis d'assister, ses sessions donnent également lieu à l'examen des perspectives d'aboutissement de projets de thérapies nouvelles, notamment en matière d'ingénierie tissulaire. En ce qui concerne plus spécifiquement les entreprises pharmaceutiques à titre individuel, certaines se sont d'ores et déjà lancées dans les nouveaux médicaments. Il s'agit cependant, en France comme dans les autres pays, plutôt d'entreprises de taille moyenne ou de PME ayant des stratégies spécialisées et employant pour l'essentiel du personnel très hautement qualifié. Selon les statistiques d'IMS Health, société américaine qui fournit des services d'information à l'industrie pharmaceutique, ces stratégies spécialisées s'avèrent en l'état les plus adaptées aux marchés et expliquent que les entreprises moyennes du secteur ont connu 2005 une progression de leur activité supérieure à celle des plus grandes entreprises. c) Un cadre juridique est nécessaire pour leur indiquer comment seront évalués et éventuellement mis sur le marché puis suivis les résultats de leurs recherches Pour que la recherche pharmaceutique privée intervienne dans les nouvelles solutions thérapeutiques en Europe, et que le secteur conserve ainsi selon les engagements de l'Agenda de Lisbonne une avance technique offrant des perspectives d'emplois de haut niveau, il lui faut disposer d'un cadre juridique clair. D'une manière générale, on estime que les charges qui doivent être engagées pour produire un médicament s'élèvent à quelque 650 millions d'euros (800 millions de dollars). Comme l'ont indiqué à la rapporteure les services de la Commission, de tels investissements ne sont consentis que dans un cadre juridique clarifié, notamment quant aux conditions d'évaluation et de validation des projets de nouveaux médicaments qui seront proposés. Naturellement, ce cadre peut et doit évoluer, à la lumière des rapports d'évaluation régulièrement présentés par la Commission sur les règles communautaires applicables. d) Plus tôt les normes européennes seront établies, plus leur autorité sera forte au plan international En raison du niveau scientifique qu'ils exigent, du coût des recherches et de l'étroitesse du marché de chacun d'entre eux, les médicaments de thérapie innovante seront d'abord commercialisés dans l'un des grands Etats ou blocs économiques sur lesquels opèrent les principales entreprises pharmaceutiques et où les assurances sociales permettent l'accès de la totalité ou de la majeure partie des patients aux soins que nécessite leur état de santé : Europe, Etats-Unis et Japon, ainsi qu'Australie et Nouvelle-Zélande. Par conséquent, pour les opérateurs européens, l'accès aux marchés américains et japonais dépendra de deux facteurs : - d'une part, leur avance sur les firmes locales ; - d'autre part, les conditions exigées dans l'Etat concerné, pour accorder une autorisation de mise sur le marché. Ces exigences techniques et règlementaires font l'objet du processus d'harmonisation dit ICH (International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use), cadre dans lequel des lignes directrices communes sont notamment définies. Les entreprises européennes seront donc d'autant plus familiarisées avec les procédures harmonisées que l'Europe sera en avance, aura pu par son expérience éclairer la démarche de ses principaux partenaires américains et japonais, et faire ainsi bénéficier de son expertise le processus qui sera entrepris dans le cadre de l'ICH. B. Les options partagées retenues par la Commission : un régime d'autorisation de mise sur le marché plus strict que pour les médicaments de droit commun ; des incitations en faveur des entreprises concernées Pour clarifier le statut des médicaments de thérapie innovante, la Commission avait le choix entre modifier, par des insertions et des ajouts, le règlement (CE) 726/2004 et la directive 2001/83/CE qui fixent le cadre juridique harmonisé du médicament au niveau européen, ou bien prévoir un nouveau règlement. Son option pour un nouveau règlement, qui s'applique non seulement à l'ingénierie tissulaire, laquelle faisait l'objet d'une lacune juridique, mais également aux médicaments de thérapie génique et de thérapie cellulaire somatique, présente, outre l'avantage de la lisibilité, celui de la cohérence. La complexité de la matière, la primauté et la spécificité du procédé par rapport au produit, l'importance des stratégies d'évaluation des risques et de la pharmacovigilance justifient un cadre spécifique et par voie de conséquence commun à ces trois nouvelles branches du médicament. 1) Une procédure centralisée, unique et plus exigeante de délivrance des AMM par l'Agence européenne du médicament, avec notamment la création d'un comité d'experts spécifique, une obligation de traçabilité et un système renforcé de suivi après autorisation La proposition de la Commission repose sur un aménagement de la procédure d'autorisation centralisée de mise sur le marché (AMM centralisée) par l'Agence européenne du médicament, actuellement fixée par le règlement précité n° 726-2004. Le principe d'une AMM centralisée, a rapidement été considéré acquis par la Commission, puis par la plupart des Etats membres comme par les commissions compétentes du Parlement européen, en dépit de l'intérêt de certains pays, parmi lesquels la France et l'Allemagne, pour une éventuelle double compétence, communautaire et nationale. Il est vrai que la Commission a pu s'appuyer sur le précédent de l'obligation d'une AMM européenne pour les médicaments issus de la biotechnologie, notamment pour les médicaments de thérapie génique et de thérapie cellulaire somatique. Par rapport à des systèmes purement nationaux, l'AMM centralisée présente deux avantages : d'une part, elle donne à son bénéficiaire directement accès aux marchés de l'ensemble des Etats membres, alors que l'AMM nationale n'a le même effet qu'après mise en jeu de la procédure de reconnaissance mutuelle ; d'autre part, elle offre au patient des garanties supérieures quant à l'homogénéité du niveau des soins sur l'ensemble du territoire de l'Union. Afin de rendre la délivrance de l'AMM encore plus rigoureuse pour les médicaments de thérapie innovante, les ajustements prévus concernent tous les stades de la procédure. S'agissant en premier lieu de l'instruction de la demande d'AMM, la Commission propose de faire appel aux compétences spécifiques qu'exigent les médicaments de thérapie innovante en créant aux côtés du Comité du médicament à usage humain (CMUH), un Comité des thérapies innovantes (CTI, ou CAT pour committee on advanced therapy en anglais), dont la mission sera de rendre des avis sur l'ensemble des questions liées aux médicaments de thérapie innovante, notamment en ce qui leur évaluation scientifique avant la délivrance de l'AMM. De manière à regrouper l'ensemble des compétences nécessaires pour couvrir un domaine aussi large et complexe, la Commission propose la composition suivante : cinq membres et cinq suppléants issus du CMUH, un membre et un suppléant désigné par chaque Etat membre qui ne sera pas déjà représenté, ainsi que quatre membres nommés par la Commission sur la base d'un appel public à manifestation d'intérêt, dont deux médecins et deux représentants d'associations de patients. La rareté de ces compétences en Europe est d'ailleurs l'un des éléments invoqués par la Commission à l'appui de l'option pour une procédure unique d'AMM centralisée, de manière à éviter de poser problème aux Etats ayant une faible population comme à ceux dont la médecine serait orientée vers d'autres spécialisations. La responsabilité du CAT en ce qui concerne la sécurité sanitaire sera essentielle. Le deuxième aménagement proposé par rapport aux dispositions de droit commun a trait aux exigences techniques que doivent satisfaire les demandes d'AMM. Afin de prendre en compte la spécificité de l'ingénierie tissulaire, ou des éléments spécifiques résultant de la viabilité des cellules et de la nécessité d'éviter tout risque de prolifération, la Commission propose de préciser le type, la quantité et la qualité des données précliniques et cliniques exigibles pour démontrer la qualité, la sécurité et l'efficacité thérapeutique d'un nouveau produit, en modifiant les annexes de la directive 2003/81/CE précitée et en complétant ces nouvelles dispositions par des lignes directrices établies en concertation avec les partie intéressées. Le troisième élément sur lequel un ajustement de la procédure de droit commun de délivrance de l'AMM centralisée, est suggéré par la Commission, est le suivi, après leur administration, des médicaments de thérapie innovante. Cette exigence est motivée non seulement par la complexité des médicaments de thérapie innovante, mais également par l'ampleur de la période au cours de laquelle ils sont destinés demeurer dans le corps humain. Ce point est particulièrement important pour la thérapie cellulaire. Les cellules utilisées pour le traitement d'un patient ont un métabolisme qui leur est propre et peuvent avoir une certaine mobilité à l'intérieur du corps humain. Elles peuvent également présenter le risque de véhiculer des maladies infectieuses. Il est également nécessaire d'éviter tout effet cancérogène. Deux éléments sont prévus : - d'une part, un suivi post-AMM renforcé avec, d'une part, l'obligation pour le demandeur de préciser les mesures de suivi de l'efficacité du médicament proposé, au-delà des règles de pharmaco-vigilance de droit commun et, d'autre part, la faculté pour la Commission, sur avis de l'Agence européenne du médicament, d'exiger la mise en place d'un dispositif particulier de gestion des risques ainsi que, le cas échéant, des rapports spécifiques et des rapports additionnels sur l'efficacité des systèmes de gestion des risques ; - d'autre part, la mise en place d'une traçabilité complète du médicament, ainsi que de l'ensemble de ses matériaux et matières constitutifs. Cette exigence est conçue d'une manière large, afin de couvrir toutes les matières et substances en contact avec les éventuels tissus ou cellules, depuis la source jusqu'à l'établissement où le produit est utilisé, en passant par l'ensemble des étapes de fabrication, de conditionnement, de transport et de livraison. La durée prévue pour la conservation des données correspondantes est en outre particulièrement longue, à raison de 30 ans au moins : il est proposé que la Commission puisse faire d'une durée supérieure l'une des conditions de délivrance de l'AMM centralisée. 2) Des aménagements d'ordre administratif et financier Comme le rappelle la Commission dans l'exposé sommaire de la proposition de règlement, les médicaments bénéficient déjà sur le plan communautaire de règles spécifiques destinées à favoriser leur compétitivité : - l'accès au marché des 27 Etats membres, sauf recours exceptionnel aux facultés d'interdiction nationale en raison notamment de normes éthiques ou des dispositions propres aux tissus et cellules ; - une harmonisation de la période de protection des données issues des essais cliniques qui interdit de déposer une demande d'AMM leur faisant référence pendant la période couverte. Cette dernière est de huit ans avec des facultés de prorogation dans certains cas ; - la possibilité d'être qualifié de médicament orphelin et de bénéficier des avantages correspondants, notamment du droit exclusif de commercialisation de 10 ans ; - la possibilité d'une procédure d'évaluation accélérée ; - la délivrance, le cas échéant, d'une autorisation de mise sur le marché conditionnelle ou de mise sur le marché dans des circonstances exceptionnelles ; - des mesures d'incitation financière spécifique et une aide administrative et financière en faveur de PME. La Commission suggère dans la proposition de règlement deux mesures supplémentaire pour les seuls MTI : l'une d'ordre administratif ; l'autre d'ordre financier. a) Un dispositif d'évaluation et de certification de la qualité des données « de développement initial » pour faciliter la valorisation des travaux des petites et moyennes entreprises innovantes Afin de favoriser l'innovation, notamment de la part des entreprises qui n'atteindraient pas la dimension suffisante pour assurer jusqu'au bout le développement d'un médicament de thérapie innovante, la Commission propose que les petites et moyennes entreprises qui mettent au point un tel médicament puissent demander à l'Agence d'évaluer d'un point de vue scientifique et de certifier les données sur la qualité et les données non cliniques requises pour les AMM, c'est-à-dire les données de développement initial. Cette procédure serait indépendante de celle de mise sur le marché. Elle apparaît pertinente et doit être retenue. En cas de certification, les PME pourront ainsi faire commercialiser les résultats de leurs travaux, ce qui favorisera un partage des tâches, assez courant dans l'industrie pharmaceutique entre, d'une part, l'innovation et la conception, qui incombent aux PME et, d'autre part, dans le cadre d'une licence d'exploitation de la technologie, les essais cliniques et le développement ultérieur des produits, pour lesquels les plus grandes entreprises sont mieux adaptées. Elles seules peuvent d'ailleurs faire de telles opérations dans certains cas. En complément, il est prévu que toute personne ou bien toute entreprise ayant mis au point un produit à base de cellules ou de tissus puisse demander à l'Agence de déterminer, dans le cadre d'une recommandation, si ce produit répond bien aux critères du médicament de thérapie innovante. La teneur de cette recommandation est prévue pour être accessible au public, dans le respect des règles de la confidentialité commerciale. b) La réduction des honoraires perçus pour la fourniture de conseils scientifiques par l'Agence européenne du médicament La deuxième incitation prévue par la proposition de règlement concerne la réduction, à concurrence de 90 % de la somme initiale, des honoraires perçus par l'Agence européenne du médicament au titre des conseils scientifiques et qu'elle fournit soit, selon les prescriptions de l'article 57 du règlement précité n° 726/2004, sur la conduite des essais et études nécessaires pour démontrer la qualité, la sécurité et l'efficacité des médicaments, soit sur la conception et la réalisation de la pharmacovigilance et du système de gestion des risques après délivrance de l'AMM. Selon les estimations de la Commission, l'économie réalisée est de l'ordre de 63 000 euros pour son bénéficiaire. Dans son projet de rapport du 8 novembre dernier, le rapporteur du Parlement européen a souhaité modulé cet avantage selon la taille de l'entreprise, le taux de réduction de 90 % étant réservé aux seules PME et les autres demandeurs bénéficiant d'un taux de réduction moins favorable, à raison de 65 %. 3) L'absence de débat majeur sur l'essentiel des dispositions nouvelles Dans sa note du 20 novembre dernier au Conseil « Emploi, Politique sociale, santé et consommateur », le groupe « produit pharmaceutique et dispositifs médicaux » a constaté que la plupart des éléments de la proposition ont fait l'objet d'un large accord entre les Etats membres. Elle n'appelle pas, en effet, d'ajustement substantiel. II. DES AMÉNAGEMENTS A PREVOIR SUR TROIS ELEMENTS : LES HÔPITAUX ; LES PRODUITS COMBINÉS ASSOCIANT UN DISPOSITIF MÉDICAL ET UN MÉDICAMENT DE THERAPIE INNOVANTE ; LES PRÉCAUTIONS D'ORDRE ÉTHIQUE Les débats entre Etats membres dans la perspective du Conseil ont montré que les difficultés se focalisaient sur deux éléments du champ d'application du futur règlement : d'une part, les règles applicables au secteur hospitalier ; d'autre part, le régime des médicaments combinés, composés de tissus ou de cellules, d'origine humaine ou animale, et d'un dispositif médical. En outre, l'expérience du débat du Parlement européen, où le rapporteur a présenté des amendements d'ordre éthique sur des questions hors champ, montre qu'il faut apporter un complément aux garanties et précautions déjà prévues, pour éviter les craintes infondées. Tels sont en définitive les trois points importants sur lesquels il revient à la Délégation de prendre position. A. Rectifier la portée de l'exception hospitalière pour garantir la sécurité sanitaire et permettre un développement dynamique et harmonieux de ce secteur 1) Un débat essentiel qui divise les Etats membres D'une manière générale, la directive 2001/83/CE ne prévoit, sur le plan communautaire, d'autorisation de mise sur le marché que pour les seuls médicaments « produits industriellement », ce qui laisse hors champ les préparations spécifiques hospitalières notamment. Il en est naturellement de même de l'activité de recherche. Dans sa proposition, la Commission suggère de compléter cette exclusion automatique et implicite des hôpitaux, et également de la recherche, par une disposition supplémentaire, à l'article 28, excluant explicitement du champ des dispositions sur l'AMM de la directive 2001/83/CE, les « médicaments de thérapie innovante (...) à la fois préparés entièrement et utilisés dans un hôpital, selon une prescription médicale destinée à un malade déterminé ». Cette dernière exclusion, qui ne concernerait donc que les seuls médicaments fabriqués industriellement, partage profondément les Etats membres, à raison, pour l'essentiel, de 5 qui lui sont favorables, de 6 qui souhaitent une extension et de 7, dont la France, qui sont réservés. Ces divergences s'expliquent pour l'essentiel par trois éléments. D'une part, la question se pose dans des termes très différents d'un Etat membre à l'autre. Les règles sanitaires n'y sont pas du même niveau, comme c'est actuellement le cas pour l'ingénierie tissulaire. Les services de santé ne sont pas organisés de la même manière. Les parts relatives du secteur public et du secteur privé, les modalités de fonctionnement des hôpitaux, les relations qu'ils entretiennent entre eux constituent autant d'éléments qui influent sur la perception du problème par chaque pays. D'autre part, la controverse ne porte pas sur le principe d'un encadrement des conditions dans lesquelles les prescriptions hospitalières doivent intervenir, mais sur le niveau auquel celles-ci doivent intervenir : cadre national ou cadre européen. Enfin, si l'exclusion était décidée dans les termes proposés par la Commission ou des termes voisins, les hôpitaux pourraient néanmoins, volontairement, solliciter une AMM communautaire, si tel était leur intérêt. 2) La nécessité de faire prévaloir le principe de sécurité sanitaire et d'offrir en outre au secteur hospitalier l'opportunité de tirer parti de toutes ses potentialités Les contraintes d'une AMM communautaire, délivrée par une agence basée à Londres, sont pour les hôpitaux et les organismes qui pourraient leur être assimilés, uniquement d'ordre administratif et financier : la procédure est jugée lourde et coûteuse. Du point de vue sanitaire en revanche, lequel doit prévaloir conformément au principe de précaution, l'application du droit commun à toutes les préparations fabriquées industriellement, même si elles le sont dans les hôpitaux et les établissements assimilés, présente d'indéniables avantages. Il permet notamment : - de garantir au patient, sur l'ensemble du territoire de l'Union, une harmonisation vers le haut tant de la qualité des produits comme des soins qui lui sont prodigués, que du niveau global des systèmes de santé. Si les pays les plus avancés comme la France ont moins à gagner, tel n'est pas le cas de plusieurs autres Etats membres. Il n'apparaît pas admissible de ne pas faire partager à leurs ressortissants les bénéfices des progrès futurs de la médecine. Il n'y a, en effet, pas plusieurs catégories de patients au sein de l'Union ; - d'appliquer avec la rigueur nécessaire le principe de précaution. Les risques liés à la préparation en nombre d'un médicament sont d'ordre sériel, c'est-à-dire liés à la fabrication en série. Les exigences sanitaires qui les entourent visent à réduire le plus possible ces risques et sont par conséquent indépendantes du statut de l'entité qui les prépare. Les règles qui en découlent doivent donc être les mêmes ; - d'éviter les complexités inutiles. La distinction entre ce qui est industriel et ce qui ne l'est pas permet déjà d'exclure du champ de l'AMM des activités telles que celles de l'Etablissement français du sang, selon les informations communiquées à la rapporteure. Par conséquent, il n'apparaît pas justifié de maintenir dans le texte de la proposition de règlement l'exclusion explicite des hôpitaux, telle qu'elle est prévue, qui se surajoute à celle de droit commun prévue par la directive 2001/83/CE. En outre, de leur propre point de vue, l'application du droit commun des médicaments de thérapie innovante, pour les produits fabriqués industriellement, aux hôpitaux concernés, ne peut être que favorable, à terme. Elle leur permet d'envisager de valoriser directement, au niveau européen, les résultats de leurs travaux, recherches et investissements, dès lors qu'ils auront été en mesure de mettre en place les procédures et structures leur permettant de tirer parti de ces immenses potentialités, qui sont, souvent, encore inexploitées. Le deuxième grand débat entre les Etats membres est un problème de frontière. Les innovations thérapeutiques de l'ingénierie tissulaire se situent, en effet, aux confins entre le domaine du médicament et celui du dispositif médical. Quelles règles convient-il d'appliquer aux produits combinés associant un ou plusieurs dispositifs médicaux à une partie cellulaire ou tissulaire ? La catégorie des dispositifs médicaux est extrêmement large avec quelque 10.000 articles très divers, allant des lunettes et pansements jusqu'aux stimulateurs cardiaques. Elle est régie par la directive 93/42/CEE du Conseil du 14 juin 1993, actuellement en cours de révision. L'enjeu est double : d'une part, il convient d'appliquer les règles sanitaires non seulement les plus élevées, mais également les plus adaptées ; d'autre part, il est indispensable d'éviter toute lacune juridique, situation où aucun des deux cadres ne s'appliquerait. Il est important : les médicaments sont soumis à la procédure exigeante, complexe et lourde de l'AMM ; les dispositifs médicaux font l'objet d'une procédure différente, d'évaluation et de certification par un organisme dit notifié, opération qui se traduit en pratique par l'accès au marquage « CE ». Le contrôle opéré n'est pas similaire, même si les normes dont le respect est exigé pour les dispositifs médicaux seront bientôt renforcées, à l'issue de la révision de la directive précitée qui les régit. Lors des débats, deux critères sont apparus pour opérer la distinction : - d'une part, celui de la viabilité : seraient classés comme médicament de thérapie innovante les produits combinés contenant des cellules viables. Cette position a été soutenue par plusieurs Etats membres, notamment les deux présidences successives de l'année 2006, la présidence autrichienne et la présidence finlandaise. Elle se fonde sur le principe suivant lequel la viabilité confère aux tissus et cellules concernés des propriétés métaboliques, immunologiques ou pharmacologiques qui impliquent d'appliquer le régime du médicament ; - d'autre part, celui de l'accessoire et du principal. Le partage se ferait ainsi selon le caractère principal ou accessoire de la partie dispositif médical et de la partie cellulaire ou tissulaire du produit combiné. La controverse est d'autant plus délicate qu'implicitement, la question de savoir si ce qui est viable peut être accessoire est aussi posée... Pour sa part, la rapporteure estime que, dans ce cas également, le principe de précaution doit jouer et que le niveau d'exigence doit être le même pour les dispositifs médicaux comprenant une partie tissus et de cellules ou, plus généralement, issus de la matière vivante, que pour les médicaments de thérapie innovante. Cette position est d'ailleurs cohérente avec la procédure prévue dans le cadre de la proposition de règlement par la Commission pour les produits combinés, avec : - d'une part, l'évaluation par l'Agence européenne du médicament de l'ensemble du produit concerné, y compris la partie dispositif médical. Lorsque l'évaluation de cette dernière par un organisme notifié est déjà intervenue, l'Agence tient compte des résultats obtenus pour sa propre évaluation du médicament concerné ; - d'autre part, il est également prévu que, dans sa composition, le comité des thérapies innovantes dispose en son sein de compétences en matière de dispositifs médicaux. La rédaction proposée prévoit qu'elle « assure une couverture appropriée » notamment sur les dispositifs médicaux. En complément, il convient naturellement que, par coordination, les mesures prévues dans la proposition de directive modifiant celle de 1993 sur les dispositifs médicaux, fassent l'objet d'un ajustement similaire. C. Permettre à l'Agence européenne du médicament de faire appel, en tant que besoin, à une instance consultative en matière d'éthique En ce qui concerne les questions de bioéthique, toujours présentes, au moins à l'arrière-plan, dès lors qu'il s'agit d'intervenir sur le vivant, la proposition de règlement ne prévoit aucune disposition communautaire nouvelle. Elle respecte également le principe de subsidiarité tel qu'il est appliqué en la matière, en n'affectant pas les règles nationales existantes. Les équilibres nationaux, qui varient d'un Etat membre à l'autre, sont donc harmonieusement préservés. C'est à juste titre que la proposition s'abstient de prévoir quelque nouvelle règle que ce soit, puisque les problèmes que posent les thérapies innovantes concernent l'application des résultats de la recherche. Ils ne sont pas de même nature et donc de même ampleur que ceux que pose la recherche fondamentale. Les questions aussi essentielles et controversées des cellules souches et des embryons surnuméraires, ou l'utilisation des cellules toti-, pluri- ou multipotentes, qui mettent en jeu les convictions, les craintes ou les réflexes les plus intimes des uns et des autres, sont donc hors champ. Cependant, comme il convient d'éviter toute ambiguïté - la Charte des droits fondamentaux de l'Union européenne ; - la Convention sur la protection des droits de l'homme et de la dignité de l'être humain, dite convention d'Oviedo, signée mais non encore ratifiée par la France ; - la directive précitée 2004/23/CE, sur les exigences sur les normes de qualité et de sécurité concernant les tissus et les cellules humains, qui recommande notamment les principes du don volontaire et non rémunéré, de l'anonymat du donneur et du receveur, de l'altruisme du donneur et de la solidarité entre donneur et receveur ; - la directive 2001/20/CE sur le rapprochement des bonnes pratiques en ce qui concerne les essais cliniques dans le domaine du médicament à usage humain : il est cependant demandé à la Commission d'établir des lignes directrices détaillées en ce qui concerne les médicaments de thérapie innovante ; - et enfin, les dispositions nationales applicables en matière de bioéthique, dans le strict respect du principe de subsidiarité. Ce dernier point fait, comme les deux qui le précèdent, pour plus de clarté, l'objet d'une disposition explicite, suivant laquelle n'est pas affectée « l'application des législations nationales interdisant ou limitant l'utilisation de tel ou tel type de cellules humaines ou animales, ou la vente, la fourniture ou l'utilisation de médicaments contenant de telles cellules, consistant dans de telles cellules ou issus de celles-ci. Les Etats membres sont tenus de communiquer à la Commission les législations nationales concernées. » Toutefois, deux questions ne sont pas abordées par la proposition de la Commission et appellent des développements. La première a trait à compatibilité de la commercialisation de produits issus du corps humain, avec les principes de la gratuité et de l'altruisme qui président au don. Il y a, en effet, a priori une contradiction entre ces deux éléments. Tel n'est cependant plus le cas dès lors qu'entre en considération l'importance du procédé auquel sont soumis les tissus et cellules initiaux, procédé qui ne peut intervenir que s'il y a valorisation. Le deuxième point concerne le traitement des questions d'ordre éthique qui pourraient éventuellement se poser, au sein de l'Agence européenne du médicament, pour la mise en œuvre du futur règlement sur les médicaments de thérapie innovante. Il n'est pas certain que cette hypothèse se réalise et, si tel est le cas, les questions qui seront le cas échéant soulevées n'auront pas la même ampleur que celles évoquées plus haut. Néanmoins, il apparaît préférable de faire preuve de sens de l'anticipation et de prévoir les modalités permettant d'établir une solution dans l'harmonie avec les Etats membres. Dans sa proposition, la Commission suggère de régler la question par la composition du comité des thérapies innovantes : lors de la désignation de ses membres, le directeur exécutif de l'Agence et les Etats membres coopèreraient pour s'assurer d'une « couverture appropriée et équilibrée des domaines scientifiques, y compris l'éthique ». Compte tenu de l'ampleur des compétences que devra par ailleurs réunir le comité des thérapies innovantes, les équilibres seront vraisemblablement très complexes et la solution que propose la Commission apparaît donc comme a minima. L'exemple du Groupe européen d'éthique des sciences et des nouvelles technologies, instance neutre, indépendante, pluraliste et pluridisciplinaire, composée de quinze experts nommés par la Commission européenne pour leur expertise et leurs qualités individuelles et ayant pour mission d'examiner les questions éthiques liées aux sciences et aux nouvelles technologies, ainsi que de soumettre des avis à la Commission européenne, représente, en revanche, un précédent intéressant. Celui-ci devrait être transposé auprès de l'Agence européenne du médicament, soit dans le cadre d'une extension de ces compétences, soit par la création d'une structure consultative similaire ad hoc. La Délégation s'est réunie le mardi 23 janvier 2007, sous la présidence de M. Christian Philip, Vice-président, pour examiner le présent rapport d'information. L'exposé du rapporteur a été suivi d'un débat. M. François Guillaume, tout en considérant que la délivrance de l'AMM par l'Agence européenne représentait un avantage incontestable dont tous les Etats membres pourraient tirer profit, s'est interrogé sur les conditions de fonctionnement concrètes de cette procédure, notamment de son coût. Il a souhaité savoir si l'Agence ferait appel au concours des agences nationales, comme c'est le cas de l'Agence européenne de sécurité alimentaire, ce qui, à ses yeux, pourrait constituer un risque d'encombrement et de chevauchement des compétences. La rapporteure a rappelé que l'Agence européenne du médicament avait commencé ses activités en 1995 et qu'elle seule pouvait déjà délivrer les AMM pour les médicaments issus des biotechnologies. Elle a estimé que, pour disposer d'un marché plus large, il était intéressant pour les PME d'avoir accès à la procédure d'AMM communautaire, plutôt que nationale. M. Jérôme Lambert a demandé si la fixation du prix du médicament interviendrait de manière coordonnée avec la délivrance de l'AMM. Telle peut être la pratique en France. Le prix d'un MTI sera-t-il le même dans tous les Etats membres ? La rapporteure a précisé que l'AMM était délivrée au regard de l'efficacité thérapeutique du médicament. En revanche, il appartient aux Etats membres de fixer les prix selon les procédures qu'ils ont prévues. En réponse à M. Christian Philip, Président, la rapporteure a indiqué que l'adoption de la proposition de règlement pourrait intervenir au cours de la Présidence allemande. Puis la Délégation a adopté les conclusions dont le texte figure ci-après. CONCLUSIONS ADOPTEES PAR LA DELEGATION La Délégation, ANNEXE : I. A PARIS - Le Pr. Claude Huriet, président de l'Institut Curie, ancien sénateur, vice-président du comité international de bioéthique de l'Unesco ; - Son Exc. M. Gabriel Keller, ambassadeur chargé des questions de bioéthique ; - Mme Marine Jeantet, conseillère technique pour les produits de santé au cabinet du ministre de la santé et des solidarités, M. Xavier Bertrand ; - M. Xavier Luquet, conseiller diplomatique au cabinet du ministre de la santé et des solidarités, M. Xavier Bertrand ; - Le Pr. Didier Houssin, directeur général de la santé ; - Mme Catherine Dessein, directrice générale de l'Etablissement français du sang ; - M. Jean Marimbert, directeur général de l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) ; - Le Dr Bernard Loty, directeur médical et scientifique de l'Agence de la biomédecine ; - Mme Aline Bessis-Marais, directeur des affaires publiques du LEEM, ainsi que M. Pierre-Yves Arnoux ; - M. Gérard Vincent, délégué général de la Fédération hospitalière de France (FHF). ¬ Au titre du Comité consultatif national d'éthique pour les sciences de la vie et de la santé (CCNE) - Le Pr. Sadek BÉloucif, professeur à l'université Picardie Jules Verne ; - Le Père Olivier de Dinechin, membre de la Compagnie de Jésus, membre du département d'éthique biomédicale du centre Sèvres (facultés jésuites de Paris) ; - M. le Rabbin Haïm Korsia, aumônier général israélite de l'Armée de l'air ; - Mme Marie-Hélène mouneyrat, secrétaire générale. ¬ Lors d'une réunion du Comité biotechnologies du LEEM - M. Marc de Garidel, vice-président Europe du Sud-Ouest d'Amgen ; - M. Didier Hoch, président de Sanofi Pasteur MSD ; - M. Laurent Arthaud, Pharmavent ; - M. Alain Clergeot, président du Laboratoire Chugai Pharma France ; - M. Manuel Gea, président-directeur général de Bio-Modeling Systems ; - M. Alain Huriez, président-directeur général de Neovacs SA ; - Mme Annick Schwebig, président-directeur général du Laboratoire Actelion Pharmaceuticals France ; - Mme Catherine Lassale, directeur des affaires scientifiques, pharmaceutiques et médicales du LEEM ; - M. Luc Aguilar, conseiller de la présidence des Laboratoires Pierre Fabre Dermo-cosmétique ; - Mme Sophie Chappuis, Club Alfa ; - M. Patrick d'Humieres, directeur de la communication du LEEM ; - Mme Isabelle Delattre, responsable éditoriale du LEEM ; - M. Pinhas Melul, directeur général du Laboratoire Chiron France ; - Mme France Normand-Plessier, vice-présidente du Comité Adebiotech ; - M. Didier Veron, directeur des affaires publiques et communication du Groupe Ipsen. II. A LYON - Mme Odile Damour, responsable de la banque des tissus et cellules des Hospices civils de Lyon (Hôpital Edouard Herriot) ; - Dr Laurence Barnouin, directrice de TBF-Génie tissulaire. III. A BRUXELLES - Mme Françoise Grossetête, député européen ; - M. Miroslav Mikolá_ik, député européen, rapporteur de la proposition de règlement relative aux médicaments de thérapie innovante ; - M. Philippe Brunet, directeur-adjoint de cabinet de M. Markos Kyprianou, commissaire à la santé et à la protection des consommateurs ; - Le Dr Jean-Baptiste Brunet, conseiller santé, Représentation permanente de la France auprès de l'Union européenne ; - M. Nicolas Rossignol, Direction générale Entreprises, Commission européenne. 1 () Résumé du colloque de l'Académie des Sciences, organisé les 8 et 9 septembre 2006 sur le thème de la thérapie cellulaire régénérative. 2 () Revue Médecine Sciences, avril 2002. |