Rapport d'information
de la mission d'information commune préparatoire
au projet de loi de révision des lois « bioéthiques » de juillet 1994
TOME II
Auditions - volume 4
Les auditions sont présentées dans l'ordre chronologique des séances tenues par la mission d'information
(la date de l'audition figure ci-dessous entre parenthèses)
- MM. Éric MOLINIÉ, directeur général de l'Association française contre les myopathies, Philippe CHAUMET-RIFFAUD, directeur délégué au développement des thérapeutiques, Mmes Anne-Laure MORIN, juriste, et Jeanne-Hélène di DONATO, banque des tissus pour la recherche de l'Association française contre les myopathies (mercredi 13 décembre 2000)
- M. Salomon KESSOUS, médecin généraliste (mercredi 13 décembre 2000)
- M. le Professeur Didier HOUSSIN, directeur général de l'Établissement français des greffes (mercredi 10 janvier 2001)
- M. le Professeur Dominique DURAND, chef du service de néphrologie-hypertension artérielle-dialyse-transplantation du centre hospitalier de Toulouse Rangueil
(mercredi 10 janvier 2001)
- MM. Dominique BECQUART, président du Lions Club de Mouvaux le Bosquiel et
Pierre DUCHATELLE (mercredi 10 janvier 2001)
- M. Philippe KOURILSKY, professeur au Collège de France et Mme Geneviève VINEY, professeur à l'Université de Paris I (mercredi 17 janvier 2001)
- Mmes Christiane AIZENFISZ, membre du conseil fédéral et Françoise MOATTI, membre de la commission consultative bioéthique, de la Grande Loge Féminine de France
(mercredi 17 janvier 2001)
- MM. Jacques BRUNSCHWIG, médecin, Émile PAPIERNIK, médecin-chef de la maternité de Port-Royal, Pierre-Charles RANOUIL, avocat, et Mme Isabelle SCHOONWATER, magistrat (mercredi 24 janvier 2001)
- MM. Thierry SUEUR, président du comité de la propriété intellectuelle, et François CHRÉTIEN, président du groupe de travail biotechnologies, du Mouvement des Entreprises de France (mercredi 24 janvier 2001)
- MM. Gilles AULAGNER, président du Syndicat national des pharmaciens praticiens hospitaliers et praticiens hospitaliers universitaires, Bernard CERTAIN, pharmacien hospitalier, et Mme Dominique GOERY, directrice scientifique du centre d'étude et de formation hospitalières (mercredi 24 janvier 2001)
- Mme Noëlle LENOIR (mercredi 31 janvier 2001)
- M. Jacques DUMONT, président, Mmes Odile CORBIN, directeur général, CRESPON, directeur des affaires techniques, du syndicat national de l'industrie des technologies médicales, MM. Philippe ROUARD, le Professeur Luc TÉOT et Mme Françoise ROCA, membres du syndicat de l'industrie des dispositifs de soins médicaux (mercredi 31 janvier 2001)
- M. Charles GURY, secrétaire général du syndicat des pharmaciens des établissements publics de santé et de Mme Hélène BARRETEAU (mercredi 31 janvier 2001)
Sommaire
des auditions
Audition de M. Éric MOLINIÉ, directeur général de l'A.F.M.
(Association française contre les Myopathies),
M. Philippe CHAUMET-RIFFAUD,
directeur délégué au développement des thérapeutiques,
Mme Anne-Laure MORIN, juriste,
et de Mme Jeanne-Hélène di DONATO,
banque des tissus pour la recherche,
de l'Association française contre les myopathies
(Extrait du procès-verbal de la séance du 13 décembre 2000)
Présidence de M. Alain Claeys, rapporteur.
M. Alain Claeys, rapporteur. - Chers collègues, je suis heureux d'accueillir au nom de la mission les représentants de l'Association française contre les myopathies. Je vous remercie, mesdames et messieurs, d'avoir accepté de répondre à notre invitation. Nous nous étions déjà vus, pour certains d'entre nous, le 6 septembre dernier à l'occasion de la visite de Génopole, mais nous étions convenus de nous rencontrer à l'occasion d'une audition de notre mission.
Il n'est plus besoin de présenter votre association, qui a encore beaucoup fait parler d'elle le week-end dernier. En effet, le Téléthon est devenu un rendez-vous annuel, quasi institutionnel. Cette année, les dons ont été à la hauteur des espérances puisqu'il semblerait que vous dépassiez l'objectif des 500 millions de francs que vous vous étiez fixé.
Votre association est aujourd'hui un acteur majeur de la recherche médicale en France. Grâce à votre action, le laboratoire Généthon a été créé ; les équipes françaises ont été les premières à proposer des cartes du génome humain entier au début des années 90. En 1998, l'AFM s'est engagée, pour cinq ans, dans la grande tentative de mise au point de techniques de thérapies géniques. Votre volonté de lutter contre la fatalité vous a donné un rôle majeur dans la recherche en génétique.
Enfin, grâce entre autres à l'AFM, plusieurs enfants soignés à l'hôpital Necker par le Professeur Alain Fisher et Mme Marina Cavazzana-Calvo, que nous avons reçue l'été dernier, ont pu quitter leur bulle.
Je tiens également à souligner un autre aspect de votre action, qui n'est pas le moindre et sera un sujet central dans les années qui viennent, celui du handicap. Celui-ci est souvent évoqué dans nos débats, sans être jamais nommé, en considérant les progrès de la médecine qui nous permettent d'espérer qu'il sera éradiqué. En effet, les techniques de dépistage de maladies, toujours plus efficaces, assorties du diagnostic prénatal, permettent d'éviter la naissance d'enfants atteints, ce dont tout le monde se félicite. Mais, comme s'interroge le docteur Delcey, rapporteur d'un groupe de réflexion issu de l'Association des Paralysés de France, un enfant né avec une déficience ne risque-t-il pas de devenir une erreur médicale ? Quel sera le regard de la société sur lui et ses parents devenus, en quelque sorte, suspects de l'avoir laissé venir au monde ?
Votre association organise régulièrement, en partenariat avec l'Association des paralysés de France, des journées d'information comme celle du 18 mai 1999 dont le mot d'ordre était : « Les personnes handicapées - arrêtons d'en faire des gens à part ! »
L'un de vos objectifs est la reconquête de la citoyenneté des personnes handicapées. Or, nous sentons bien qu'une des conséquences de toutes les récentes découvertes et techniques et du diagnostic prénatal est que la tolérance au handicap devient de plus en plus faible dans notre société et que l'on s'achemine vers l'élimination du f_tus au moindre doute. Tout récemment, un arrêt de la Cour de cassation a provoqué une grande émotion à cet égard.
Nous aimerions, en introduction, entendre ce que vous avez à dire sur ce sujet. Ensuite, nous nous proposons, si vous l'acceptez, de vous poser des questions sur les principaux aspects de la loi bioéthique, telle qu'elle a été votée en 1994, mais aussi après les annonces faites par M. le Premier ministre sur la recherche sur l'embryon et l'utilisation des cellules souches. Nous aborderons ainsi le problème du respect de l'individu et de sa dignité face à la recherche médicale.
Le président de votre association s'était ému en 1994 de ce que la loi Huriet « avait été rédigée dans un but de sauvegarde des malades au détriment du secteur de la recherche ». Je souhaiterais que vous puissiez revenir sur ce sujet car ce sera certainement un élément important dans nos travaux de révision des lois bioéthiques.
M. Eric Molinié. - Il est vrai que si l'Association française contre les myopathies est connue, c'est grâce ou à cause du Téléthon. La proximité de cet événement rend les choses d'actualité et les Français, c'est exact, ont bien été, cette année encore, au rendez-vous de cette action de solidarité et de générosité. L'AFM est un peu moins connue et il me revient de dire rapidement qui nous sommes.
Nous sommes une association de malades et de parents de malades, exclusivement, qui, face à l'impuissance de la médecine et l'ignorance de la science, se sont regroupés autour de maladies rares et de maladies génétiques, que sont les maladies neuromusculaires, avec deux objectifs clairement affichés dès l'article premier de nos statuts : d'une part, guérir les maladies et les personnes atteintes de ces maladies neuromusculaires ; d'autre part, aider la personne en attendant la guérison - ce qu'on appelle, dans notre jargon médico-social, réduire le handicap - et aller, comme vous l'avez dit, vers plus de citoyenneté.
Lorsqu'on dit aider et guérir, et parce que nos maladies sont d'origine génétique, nous avons vite compris que pour bien aider la personne et pour aller sur le chemin des thérapeutiques, il fallait comprendre. Et ce comprendre passe par la génomique, par la compréhension des mécanismes génétiques des pathologies et, comme l'a rappelé le sociologue Michel Callon dans un ouvrage qu'il nous a consacré, intitulé Le pouvoir des malades, pour l'AFM, la science n'est qu'un détour, un détour de l'homme malade à l'homme guéri.
Toute l'activité et l'action de l'AFM se veulent comme la réponse aux familles qui sont nos adhérents - plus de dix-huit mille adhérents et sympathisants en France -, marquées par un seul but : la guérison de ces maladies qui présentent cette double caractéristique d'être génétiques et rares.
Parce que nous avons aussi compris, dès le début, que nous ne pourrions pas guérir seuls ces maladies neuromusculaires et qu'il fallait pour les guérir accroître la masse de savoirs en matière de connaissance du génome et de thérapeutiques issues de celle-ci, nous avons très vite, et grâce aux moyens du Téléthon, ouvert et créé des outils d'intérêt collectif, qui ont été nos laboratoires successifs : Généthon 1, Généthon 2 et Généthon 3.
Généthon 1 traite de la génomique fondamentale et a permis de publier, dès 1992, grâce à l'introduction de méthodes industrielles dans la génétique, les premières cartes du génome qui furent offertes à la communauté scientifique, à l'UNESCO, dès novembre 1992.
Généthon 2 s'intéressait au décryptage du génome, avec ses cartes permettant l'identification et la localisation de plus de six cents maladies génétiques à ce jour.
Aujourd'hui, nous abordons un virage très net vers les thérapeutiques, avec la mutation complète de notre laboratoire vers le Généthon 3 qui sera consacré exclusivement à la vectorologie et, donc, à la thérapie génique, la position de l'AFM étant que nous en savons assez, aujourd'hui, sur quelques maladies modèles, pour essayer de faire la preuve de cette thérapie génique. C'est la grande tentative lancée en 1998.
Deux ans après le lancement de cette grande tentative, nous avons pu annoncer, lors du Téléthon, trois premières mondiales d'importance.
Ce sont tout d'abord les enfants bulles d'Alain Fisher, qui n'avaient pas de défenses immunitaires et qui sont maintenant cliniquement guéris. Le professeur Fischer, prudent, n'emploie pas ce mot de guérison parce qu'il ne sait pas combien de temps le traitement durera. Mais aujourd'hui, ces enfants sont chez eux, ne suivent plus aucun traitement et les prélèvements de sang, effectués tous les trois mois, montrent que leurs défenses immunitaires sont pérennes.
C'est ensuite la greffe de myoblastes sur une personne de soixante-douze ans à qui on a prélevé des cellules musculaires de la cuisse pour les injecter dans un c_ur infarcié. Ces cellules ont colonisé le c_ur et réduit l'infarctus. Aujourd'hui, cette personne a retrouvé une activité électrique de son c_ur beaucoup plus forte, pas encore normale, mais lui permettant de vivre à peu près normalement.
Enfin, troisième point, qui touche aux questions éthiques qui font l'objet de notre présence ici, c'est la greffe de cellules f_tales sur le cerveau de cinq personnes atteintes de la maladie de Huntington qui, aujourd'hui, grâce à cette greffe, ont retrouvé une vie non pas normale mais avec des séquelles bien moindres que celles qu'elles auraient eues sans ce traitement. Quelqu'un qui ne pouvait plus se tenir assis et qui avait des mouvements complètement désordonnés s'est retrouvé à pouvoir rejouer de la guitare et refaire du vélo.
J'ai terminé ma présentation rapide de l'AFM qui, à la différence de l'APF, n'a pas jugé bon de créer de comité d'éthique en son sein. Nous estimons être là pour défendre les malades et répondre aux souffrances des familles et, par-là même, ne pas avoir à édicter de positions globales pour l'ensemble de nos adhérents. Nous écoutons leurs questions, nous sommes sensibles à leurs souffrances et nous sommes là pour essayer d'y répondre du mieux que nous pouvons, sachant que la réponse ultime est la guérison de ces maladies génétiques et maladies rares.
M. Pierre Hellier. - Utilisation des embryons surnuméraires et création d'embryons pour la recherche. Quelle est votre position sur ces deux sujets ?
M. Alain Claeys, rapporteur. - Lors de notre visite au Génopole, nous avions parlé de son label éthique. Je pense que, pour terminer votre présentation, il pourrait être utile que vous nous en disiez quelques mots.
M. Eric Molinié. - Nous faisons partie du Génopole. Nous avons été à l'origine de la création de ce Génopole d'Évry qui, de la collecte du sang des malades et de la banque ADN du Généthon à la thérapeutique, passe par le séquençage par des laboratoires publics. L'ensemble de ce site emploie aujourd'hui plus de deux mille personnes à Évry. Pierre Tambourin, qui en est le directeur général, prône une génétique citoyenne et, par l'intermédiaire d'un site Internet très puissant, de cafés du gène et de débats citoyens autour de la génétique, s'attache à faire en sorte que les citoyens s'approprient la génétique et que ne se reproduisent pas les mêmes erreurs que sur le nucléaire. Il faut que les débats aient lieu en amont, au moment où nous élaborons une politique, sachant que la science évolue à toute vitesse, plutôt que de nous retrouver devant le fait accompli d'avoir des centrales nucléaires et une population qui n'a pas suivi le mouvement.
Nous allons créer une école de l'ADN, que nous appellerons École de l'ADN et des Génothérapies, parce que nous ne pouvons pas dissocier pour nos maladies, la connaissance de l'ADN de la thérapeutique. À la différence de l'école de Nîmes, dont nous aurons une licence d'exploitation, nous tenons à associer tout de suite le mot de thérapeutique.
Pour répondre à la question de M. Hellier, notre rôle, en tant qu'association de patients, n'est pas de prendre position au nom de malades et, sous prétexte que j'ai des convictions ou que le président en a, de nous mettre à les imposer à tout le monde. Nous avons cependant demandé aux scientifiques que nous finançons de nous dire quelles étaient, à leur avis, les pistes les plus prometteuses en matière de thérapeutique pour demain. Ils ont relevé l'importance des cellules dites souches.
Ces cellules souches qui existent chez l'embryon, mais dont on sait depuis peu qu'elles existent aussi chez l'adulte et dans le cordon ombilical, sont capables de se différencier selon l'instruction qu'on peut leur donner et de se transformer en tissu musculaire et autres. Nous savons donc l'intérêt très fort que présentent ces cellules souches, que l'on ne trouve pas uniquement dans l'embryon, et nous privilégions le travail intrinsèque sur ces cellules.
Mme Yvette Benayoun-Nakache. - Je souhaiterais vous poser une question sur les maladies rares qu'on appelle aussi orphelines. J'aimerais en connaître la liste - je sais qu'elle est assez longue - parce que, dans nos circonscriptions, nous rencontrons nombre d'associations qui revendiquent l'avancée en matière de recherches sur ces maladies orphelines. Quelle est la relation entre votre association nationale et ces associations ? Existe-t-il un échange permanent entre vous ? Elles sont très nombreuses. Certaines sont bien identifiées, d'autres moins.
M. Philippe Chaumet-Riffaud. - Les notions de maladies rares et orphelines sont souvent associées, mais comportent pourtant une petite différence.
La notion de maladie rare est basée sur la prévalence. Ce sont des affections qui sont définies dans le règlement européen, adopté en décembre 1999, comme présentant une prévalence qui doit être inférieure à cinq pour dix mille au niveau de l'Europe.
La notion de maladie orpheline est distincte. Ce terme est employé pour dire que ce sont des maladies qui n'ont pas de traitement, qu'il s'agisse de traitement médicamenteux ou chirurgical. Or il existe malgré tout quelques maladies rares qui ont des traitements. C'est en cela que les deux termes ne sont pas synonymes. Rare, c'est la prévalence ; orpheline, c'est l'absence de traitement, soit que celui-ci n'existe pas, soit que personne, aucun industriel, ne veuille s'occuper de fabriquer les médicaments. L'AFM consent des efforts très importants pour, dans les cas que nous avons identifiés, essayer d'aider au développement de ces produits.
M. Eric Molinié. - L'action de l'AFM par rapport aux maladies rares est multiforme. En effet, nous nous sommes vite rendu compte que nous ne pourrions nous en sortir seuls. Aussi, et c'est le résultat d'une stratégie, nous avons décidé de chercher à travailler avec d'autres associations parce que leur maladie était modèle par les hasards de la médecine et de la science. Je citerai la mucoviscidose, les maladies lysosomales du foie, les rétinites pigmentaires qui rendent aveugle peu à peu, tout un ensemble de pathologies sur lesquelles nous avons des programmes thérapeutiques ciblés. Ce fut un premier élément de réponse.
Deuxième élément, nous avons aidé à la création d'Eurordis, European organisation for rare disorders, basée à Bruxelles, et qui, en termes de lobbying, est intervenue fortement pour l'adoption du règlement européen sur les médicaments orphelins. Ce fameux règlement permet à des biotech et à des entreprises pharmaceutiques d'obtenir des dégrèvements fiscaux, une liberté d'exploitation plus longue et d'autres avantages sur lesquels je passe.
Cependant, cela ne suffit pas parce que nous sommes dans des thérapeutiques nouvelles, révolutionnaires, qui sont sans commune mesure avec les révolutions médicales précédentes que peuvent avoir été les vaccins, la pénicilline, etc. Nous sommes vraiment dans l'infiniment petit, la correction du vivant. Il faut agir avec beaucoup de délicatesse et de prudence, mais c'est un enjeu formidable que de pouvoir soigner toutes ces maladies.
Il est clair qu'en amont de tout cela, il faut que la recherche sur les maladies génétiques et les maladies rares soit promue. On s'aperçoit très vite que pour qu'une biotech s'intéresse à ces maladies, il faut que l'État, des associations ou des tiers aient avancé suffisamment loin la recherche, dans le stade du développement, parce que cela coûte cher.
Il faut donc aller plus loin. C'est pour cela qu'a été créée l'Alliance maladies rares, qui regroupe aujourd'hui plus de soixante-dix associations de maladies rares et se veut le porte-parole d'un collectif de personnes atteintes de maladies rares, à la fois pour favoriser la reconnaissance d'une vie plus citoyenne et pour pratiquer un lobbying en faveur d'une véritable recherche en ce domaine.
M. Jean-Paul Bacquet. - Cela concerne les malades mais j'aurais aimé connaître votre réflexion quant à un enfant à naître ?
M. Eric Molinié. - Je redirai ce que j'ai déjà dit, monsieur. On ne peut et on n'a pas à édicter de position au nom d'une association. C'est à la famille, éclairée et renseignée sur la pathologie à risque qu'elle porte dans ses gènes, de faire son choix. Nous n'avons pas de position dogmatique en la matière.
M. Jean-Paul Bacquet. - Je formule à nouveau ma question. Vous êtes totalement libre de répondre ou de ne pas répondre. Je voulais connaître votre réflexion personnelle.
M. Eric Molinié. - J'estime que je n'ai pas à la donner.
Mme Christine Boutin. - Notre rapporteur vous a demandé quelle était la position de votre association, si vous en avez une, sur l'arrêt Perruche. Vous n'avez pas répondu.
M. Eric Molinié. - Nous n'avons pas encore défini notre position. Nous sommes en train d'étudier l'arrêt en essayant de connaître, au-delà du brouillage médiatique, la réalité des faits en question, les tenants et les aboutissants. Il serait prématuré de vous répondre. Autant nous sommes prêts aujourd'hui à vous parler de la brevetabilité du génome - et je serais heureux de pouvoir le faire - et de la révision de la loi Huriet, sujets sur lesquels nous avons travaillé et sur lesquels nous avons des éléments à vous apporter, autant en ce qui concerne l'affaire Perruche, l'événement est trop récent, trop grave aussi, pour que nous puissions arrêter une position de but en blanc. Mais nous y travaillons bien évidemment.
M. Alain Claeys, rapporteur. - Votre président propose une procédure simplifiée pour le prélèvement de l'ADN afin d'établir une banque qui permettrait, d'après lui, de progresser en matière de connaissance des maladies rares. Pouvez-vous donner des précisions sur ce sujet ? Si tel était le cas, faudrait-il revenir sur les modalités du consentement ?
Mme Anne-Laure Morin. - En matière de constitution et d'utilisation des collections d'échantillons biologiques humains à des fins de recherche, nous avons des propositions et des réflexions à vous soumettre parce que nous considérons qu'il est important que le législateur prenne en compte ces questions qui doivent aboutir à une utilisation optimale des collections dans le respect des personnes prélevées et, ce, en l'absence d'une législation unifiée ou du moins applicable, puisque l'on attend toujours le décret d'application concernant les collections à visée de recherche.
Effectivement, l'activité de la banque d'ADN et de cellules de Généthon et les campagnes de collectes qui ont été financées par l'AFM nous ont conduit à nous interroger sur la possibilité d'utilisation et de réutilisation des collections déposées. Généthon a le projet de mettre à la disposition de la communauté scientifique le catalogue des collections existantes via Internet. C'est un projet pour lequel nous interrogeons actuellement le CCNE.
Le régime actuel de constitution des collections, qui dépend partiellement de la loi Huriet du 20 décembre 1988, ne nous paraît pas du tout adapté aux recherches génétiques, si bien qu'il est même contourné en pratique puisqu'il arrive que des prélèvements d'ADN soient faits autrement que sous le régime de la loi Huriet, alors que l'on est clairement dans une hypothèse de recherche biomédicale. C'est, par exemple, le cas lors de prélèvements à des fins de diagnostic car, en la matière, la frontière entre diagnostic et recherche est assez poreuse. Dans la pratique, ce régime est également mal utilisé. En ce sens, il n'est pas de nature à respecter et à protéger l'intérêt des patients.
Il conviendrait donc de trouver un nouveau compromis entre, d'une part, les règles qui régissent le statut juridique du prélèvement biologique humain et, d'autre part, les principes éthiques internationaux ou nationaux qui régissent ce type de prélèvement. Ce nouveau compromis aboutirait, à notre sens, à l'institution d'un nouveau régime qui s'appuierait sur quatre grands piliers.
Premier pilier, il s'agirait de mettre en place un régime allégé issu de la loi Huriet, ou un régime différencié, dont M. Philippe Chaumet-Riffaud pourra vous parler plus explicitement.
Cela passerait également, deuxième pilier, par l'élaboration d'une nouvelle forme de consentement qui permettrait aux patients de distinguer clairement clinique et recherche, et permettrait également de stipuler la durée de la conservation de l'ADN, d'indiquer clairement la possibilité de mise en banque et d'échanges éventuels avec d'autres équipes, puisque ce sont des pratiques très courantes, et d'autoriser les utilisations dans un même axe de recherche - par exemple, dans un champ de pathologies donné.
Ce nouveau consentement devrait permettre d'établir un nouveau contact avec le patient prélevé en cas d'utilisation dans un autre axe de recherche et offrirait ainsi la possibilité à celui-ci de se retirer de la recherche - ce dernier point est déjà prévu par la loi Huriet mais n'est pas appliqué dans la pratique actuelle -, en obligeant la banque à détruire l'échantillon si le patient le désire. Il ouvrirait, enfin, la possibilité de l'exploitation par le biais de la propriété industrielle. Telle pourrait être l'élaboration du nouveau consentement.
Le troisième pilier de ce nouveau régime serait le maintien du lien avec le patient. L'étude génétique d'un prélèvement peut mettre en évidence un statut génétique qui peut se révéler utile pour le patient, comme pour sa famille. En cela, nous considérons qu'il doit être possible de maintenir le lien avec le patient, ce qui, par ailleurs, peut permettre de répondre à des objectifs de respect de la traçabilité.
Le quatrième pilier serait l'utilisation des collections dites « en dormance », terme relevé par le Conseil d'État dans son étude. C'est un point qui est très important pour nous, surtout dans le cas de maladies rares.
M. Alain Claeys, rapporteur. - Pourriez-vous préciser ce principe pour notre mission ?
Mme Anne-Laure Morin. - Le principe d'utilisation des collections en dormance émane de la Déclaration universelle sur le génome humain qui édicte un devoir de solidarité envers les familles et les groupes vulnérables, particulièrement un devoir d'encourager les recherches au profit des maladies rares. Ce principe a également été évoqué par le CCNE et par le Conseil d'État dans leurs travaux, quand ils parlent de « droit implicite » ou « d'attente légitime » pour les familles de voir avancer la recherche sur leurs maladies.
L'utilisation de ces collections aboutirait à l'instauration, dans le droit, d'une priorité accordée aux chercheurs, aux termes de laquelle il serait possible de faire utiliser la collection par d'autres équipes de recherche, sous réserve évidemment du consentement des personnes prélevées. Ainsi, l'utilisation des collections, qui sont riches sur le plan scientifique, ne serait plus hypothéquée à l'avenir, comme c'est le cas actuellement, par l'inadéquation d'un consentement, la disparition du lien avec le patient, ou par l'inaction ou le non-aboutissement d'une équipe de recherche.
Mme Christine Boutin. - Je ne suis pas scientifique. Je n'ai pas compris tout ce que vous nous avez dit mais j'essaie malgré tout de comprendre et, très sincèrement, mais j'aimerais que vous me redisiez clairement l'objectif que vous poursuivez, parce qu'il me semble avoir perçu une contradiction entre vos propos initiaux et les moyens que vous proposez pour éviter ce que vous dénoncez aujourd'hui, qui justifierait le nouveau consentement.
Il me semble, en effet, que ce que vous proposez est excessivement compliqué et que ce dont vous avez besoin - je vous pose la question très directement - c'est en fait l'autorisation de pouvoir faire des recherches supplémentaires.
M. Philippe Chaumet-Riffaud. - Je vais essayer de prendre la question selon l'angle, pratique, des recherches biomédicales.
Nous parlons des études de génétique, c'est-à-dire d'études réalisées sur des patients et des familles, dont l'objet est essentiellement d'identifier des gènes ou des facteurs prédictifs. Du point de vue législatif, ces études sont toutes soumises à quatre régimes. Ce sont les lois de 1994, la loi CNIL amendée en 1994 et la loi Huriet, dont certains articles posent entre eux des problèmes d'articulation extrêmement délicats, qui n'ont jamais été résolus. Je me rappelle de discussions durant lesquelles les juristes de la direction générale de la santé ou des hôpitaux et d'autres juristes consultés avaient des avis divergents sur l'interprétation de ces lois.
Concrètement, cela signifie qu'il faut que, dans le cadre de ces recherches, des cliniciens puissent examiner des patients et réaliser des prélèvements de sang. Ces recherches qui ont pour but d'identifier des gènes sont qualifiées, selon la loi Huriet, de « recherche sans bénéfice individuel direct ». Cette notion est éminemment discutable, très difficile à définir. On ne peut pas, à l'heure actuelle, quand on dit qu'on va rechercher un gène promettre un bénéfice individuel car, d'une part, on ne sait pas si on va trouver ce gène et, d'autre part, même si on le trouve, le bénéfice pour les gens ne se fera malheureusement pas à court terme.
Ces recherches étant sans bénéfice individuel direct, elles doivent être réalisées dans des lieux qui ont des autorisations de recherche sans bénéfice. En conséquence, pour des recherches qui nécessitent des prélèvements de sang en quantité extrêmement limitée, on doit demander des régimes d'autorisation relativement lourds, alors que si vous classez avec bénéfice des études thérapeutiques, chirurgicales ou mettant en _uvre des médicaments relativement dangereux, il ne vous est demandé aucune autorisation.
En fin de compte, au lieu de protéger comme c'est l'objectif de la loi, la complexité des demandes d'autorisation pour les lieux de recherche produit exactement l'inverse. Nous sommes amenés à penser que toutes les recherches en génétique ne sont pas obligatoirement menées comme on le souhaiterait.
L'idée est donc d'adapter le niveau de protection à la gravité du geste. Ces études de génétique sont des prises de sang ; des prises de sang qui, pour certaines pathologies, ne peuvent pas être menées dans des centres hospitaliers. C'est le médecin qui se déplace, qui va dans la famille pour les réaliser. Il n'y a pas d'autorisation.
Je l'ai vécu à l'Assistance publique des Hôpitaux de Paris : vous devez passer par un système dit de dérogations préfectorales, qui vous oblige à demander des autorisations auprès de je ne sais combien de structures. L'idée, nous avions vu des projets dans ce sens, serait de garder le régime de qualification sans bénéfice individuel direct mais de pouvoir avoir un assouplissement selon lequel les CCPPRB pourraient juger que certains types de recherche, pour lesquels les gestes sont vraiment des gestes relativement sans risque, puissent, tout en restant qualifiés sans bénéfice individuel direct, être réalisés en dehors des centres pourvus de matériel plus adapté à de la réanimation qu'à autre chose.
Ces commentaires sont tirés de mon expérience en tant que responsable pendant sept ans des essais cliniques à l'Assistance publique-Hôpitaux de Paris. Un grand nombre d'essais ou d'études était soumis chaque année à l'AP-MP. Une des difficultés redoutables était que le niveau d'exigence et d'obligations requis pour certaines études simples était identique à celui demandé pour des études potentiellement beaucoup plus dangereuses. D'où les réactions de certains chercheurs qui ont vraisemblablement préféré passer à côté plutôt que suivre le droit chemin.
Le but n'est donc pas de faire plus d'études génétiques, mais de rendre la réalisation de ces études possible et de faire en sorte que les gens respectent la loi. La loi Huriet est très bien conçue dans son ensemble, mais certaines dispositions ont été beaucoup plus conçues pour des essais de médicaments et de dispositifs que pour des prises de sang. Il n'est pas possible dans le cas de certaines pathologies, d'envisager d'autoriser un petit hôpital ou le local d'un médecin.
Mme Christine Boutin. - Je vous remercie. J'ai mieux compris votre propos. Cela dit, nous sommes là exactement, monsieur le rapporteur, dans l'application de la question que nous nous étions déjà posée au sein de cette mission, mais avec des religieux ou des philosophes - avec M. Alain Bauer, me semble-t-il - de savoir quelle est l'application de la loi et les limites de cette application. Votre programme s'inscrit typiquement dans cette réflexion. Vous dites qu'aujourd'hui, les contraintes sont trop lourdes, et qu'en conséquence, vous constatez des dérives et qu'il faut adapter la loi en raison d'effets pervers que vous dénoncez.
Vous essayez donc de proposer un système qui permettra d'y remédier, sans doute. Je ne vais pas mettre en doute votre proposition même si elle mérite d'être affinée. En tout cas, je ne mets pas en cause votre bonne foi. Mais vous proposez d'amender ce qui a été fait et je ne suis pas du tout persuadée que la loi ne sera pas à nouveau détournée, dans d'autres domaines d'application et que, lorsque nous réviserons les lois bioéthiques dans cinq ans, nous ne constations pas à nouveau que ce consentement n'est plus adapté et qu'il faut accorder d'autres facilités.
C'est un problème que nous avons à régler et votre intervention était très intéressante à cet égard.
M. Alain Claeys, rapporteur. - Pensez-vous que la Haute autorité que M. le Premier ministre envisage d'introduire dans la loi puisse arbitrer ce genre de questions ?
M. Philippe Chaumet-Riffaud. - Les amendements que nous suggérons sont du domaine réglementaire. Tout le système des autorisations des lieux de recherche SBID relève du domaine réglementaire par des circulaires.
Au début des années 1990, M. Huriet avait indiqué à plusieurs reprises qu'il n'envisageait qu'environ 80 à 100 lieux de recherche SBID sur la France. Dans le cadre de mes anciennes fonctions à l'AP-HP, nous avions reçu plus de 700 demandes distinctes d'autorisation. Il faut en effet noter qu'il y a quatre systèmes distincts d'autorisation : médicaments, dispositifs médicaux, études de physiologie et utilisation de produits radioactifs.
L'idée n'est pas d'aller contre l'esprit de la loi, mais de protéger en tenant compte tout de même de caractéristiques spécifiques. On ne peut pas avoir les mêmes exigences pour une étude de phase 1 de tolérance à un nouveau médicament sur des sujets volontaires sains, et pour un simple prélèvement.
Il faut cependant conserver des garde-fous. Il faut qu'une commission indépendante puisse dire que telle recherche, qui ne demande qu'un simple prélèvement, même si elle garde sa qualification sans bénéfice individuel direct qui a l'avantage de donner le meilleur régime en termes d'assurance pour les patients ou pour les volontaires qui participent à l'essai, est autorisée.
M. Alain Claeys, rapporteur. - Quelle est votre position sur le thème du consentement ?
M. Philippe Chaumet-Riffaud. - Le problème des consentements pour ces études tient à la finalité de ces échantillons : à quoi vont-ils servir ?
C'est aussi une discussion qui intéresse énormément les chercheurs qui essaient de faire de leur mieux. Les études génétiques sont habituellement réalisées dans des services spécialisés.
La question de fond du consentement est l'étendue des recherches autorisées. Quand une équipe a pu obtenir des prélèvements pour son travail, par exemple l'identification de gènes de prédisposition pour la maladie d'Alzheimer, de Nuntintgton ou de Parkinson, et que deux ans plus tard cette même équipe de recherche dispose de nouveaux marqueurs pour travailler sur une autre affection neurologique peut-elle effectuer cette nouvelle recherche et dans quelles conditions ? Que doit-elle demander aux patients et aux membres de la famille ?
Les chercheurs s'interrogent sur la formulation des consentements. Que faut-il faire ? Faut-il demander un consentement global ? Peut-on envisager d'avoir des consentements plus larges comme, par exemple, un consentement pour des travaux dans le cadre des maladies neurodégénératives ? Ou faut-il s'en tenir à un consentement extrêmement restrictif ? Nous ne savons pas.
M. Alain Claeys, rapporteur. - La question n'est pas mineure.
M. Philippe Chaumet-Riffaud. - Il ne faut pas oublier le coût humain. Un des principes de fond de tout projet de recherche est d'éviter au maximum d'inclure des gens dans des recherches pour rien. Le problème est de savoir s'il faut automatiquement refaire une cohorte alors qu'éventuellement, le matériel est déjà disponible. Si on décide d'utiliser ce matériel, faut-il reprendre contact avec toutes les personnes concernées pour avoir leur consentement ?
En fait, nous vous livrons plus des éléments de réflexion, que nous souhaitions vous faire partager, que des solutions.
Mme Christine Boutin. - Petite question technique : comment sont mises en application les sanctions ?
M. Philippe Chaumet-Riffaud. - À ma connaissance, aucune sanction relative à une infraction portant sur la loi Huriet n'a été rapportée à ce jour. Vous savez que l'IGAS avait conduit à une certaine époque une enquête sur les essais en cancérologie et qu'un certain nombre de constatations avaient été faites.
Sauf erreur de ma part, aucune sanction n'a été appliquée. Cette situation pourrait témoigner du fait que globalement la loi Huriet est efficacement mise en application.
Mme Jeanne-Hélène di Donato. - J'ajouterai à propos des consentements, qu'ils existent aussi pour tous les autres prélèvements qui peuvent être collectés pour la recherche dans un autre cadre que celui de la loi Huriet. Je pense notamment aux prélèvements faits lors des autopsies f_tales où le consentement est largement préconisé par le Comité consultatif national d'éthique. En règle générale, il est dit à la mère pour quel axe de recherche sera prélevé ce tissu.
C'est vrai aussi pour les exérèses opératoires qui sont actuellement utilisées sans un consentement formel. Il est préconisé qu'un consentement soit posé pour toutes les utilisations d'exérèses opératoires.
Dès lors qu'un consentement serait demandé, ne serait-il pas souhaitable que le patient puisse avoir le droit de dire s'il accepte que ses tissus, qui sont éliminés lors d'une chirurgie ou prélevés lors d'une autopsie, puissent être utilisés dans tel domaine de recherches pathologiques, pour telle visée thérapeutique, pour être mis en banque ou utilisés dans un domaine beaucoup plus lucratif ?
À mon avis, il faut donner au patient le droit de dire ce qu'il a envie que l'on fasse de ses propres tissus ou de ses propres cellules.
Mme Christine Boutin. - Vous me poseriez la question, je serais incapable de répondre.
Mme Jeanne-Hélène di Donato. - Le problème se pose parce que si je prends l'exemple des tissus prélevés lors d'autopsies sur des majeurs, la loi impose actuellement d'avoir un consentement exprimé, soit directement, soit par l'intermédiaire de la famille. Ce consentement est très difficile à mettre en place. Il n'existe aucune instance publique pour récolter ces prélèvements, qui puisse organiser ces autopsies quand cela se fait spécifiquement pour la recherche.
Nous avons des patients qui, d'eux-mêmes, écrivent des lettres dans lesquelles ils expriment leur consentement à ce que tel ou tel organe puisse être prélevé pour être étudié dans leur pathologie. Quand nous les contactons à nouveau en leur disant qu'il faut plutôt joindre un service d'anatomopathologie, parce qu'ils envoient généralement leurs consentements aux associations ne sachant pas où les adresser puisqu'il n'existe pas d'instance publique prenant cela en charge, et que nous leur demandons s'ils accepteraient aussi que ces prélèvements puissent servir à d'autres recherches fondamentales, je puis vous assurer que la grande majorité accepte.
Il faut au moins leur poser la question et leur donner le droit d'accepter ou de refuser que les tissus soient utilisés.
M. Jean-Paul Bacquet. - Je suis surpris d'entendre ce qui vient d'être dit. Si le patient peut choisir à qui il attribue la potentialité d'utiliser ces prélèvements, c'est l'ouverture à toutes formes de lobbying. Ce n'est pas spécifique à ce domaine, cela s'est déjà vu.
Quant aux sanctions, votre réponse ne peut nous satisfaire. Je rappelle le fonctionnement des CCPPRB, dans lesquels la méthodologie est tout à fait exceptionnelle mais, malheureusement, quand on doit juger de la protection des personnes, on peut donner un jugement favorable parce qu'il n'y a pas de risque pour les personnes à propos de quelque chose que l'on sait n'être qu'une étude totalement « bidon », ou pour des motifs de pure commercialisation. Cela n'a strictement aucun intérêt médical. Or, le comité n'a qu'un pouvoir de protection des personnes et ne peut pas « lever le lièvre » de ce qui est totalement hors sujet.
M. Philippe Chaumet-Riffaud. - Concernant les CCPPRB, je pense que l'aspect scientifique et médical est bien validé. Néanmoins, il serait sans doute souhaitable que les associations de patients y soient représentées.
À l'heure actuelle, il y a une représentation par quatre personnes de la société civile qui sont choisies en fonction de compétences juridiques et psychologiques - ce sont les termes employés, me semble-t-il. Si nous voulons atteindre un meilleur équilibre, il serait souhaitable que les associations de patients soient présentes.
Par ailleurs, afin que les CCPPRB soient plus représentatifs, il faut trouver des moyens qui permettent aux membres qui ont d'autres activités, pour être très clairs aux membres issus de professions libérales, de ne pas avoir des difficultés à assister aux réunions. Si vous analysez les comptes rendus des réunions, vous vous rendrez compte clairement qu'au sein des CCPPRB, les représentants du corps médical sont sur représentés.
M. Jean-Paul Bacquet. - Les médecins libéraux ?
M. Philippe Chaumet-Riffaud. - Pas les libéraux, les hospitaliers.
M. Jean-Paul Bacquet. - J'y ai participé à ces comités en tant que membre d'une profession libérale. Cela ne règle pas le problème. Le CCPPRB peut dire qu'il n'y a pas de danger pour les personnes et répondre à une question qui n'a aucun intérêt, parce que nous savons que scientifiquement le médicament - hélas, nous ne pouvons rien y faire - ne présente strictement aucun intérêt ou seulement un intérêt lucratif !
M. Philippe Chaumet-Riffaud. - C'est dommage parce que l'un des articles princeps de la loi Huriet est que les recherches doivent être utiles pour l'augmentation des connaissances. C'est le problème de l'évaluation des CCPPRB. Les taux sont, à l'heure actuelle, de 99,98 % d'avis favorables accordés.
M. Alain Claeys, rapporteur. - Sans abuser de votre temps, d'une façon générale, quelle appréciation portez-vous sur les tests de dépistage génétiques ? Comment, selon vous, doivent-ils être encadrés et autorisés ? Redoutez-vous l'usage qui pourrait en être fait par les assurances ?
Mme Anne-Laure Morin. - Nous avons réfléchi sur le sujet, relativement à la discrimination. Nous avions réfléchi, dans le cadre du groupe qui s'est réuni pour l'examen des lois de bioéthique, à l'utilisation par les assureurs des tests génétiques. Cela dit, nous avons constaté que dans le projet de loi sur la modernisation du système de santé, nos revendications étaient déjà prises en compte.
Nous sommes donc en plein accord avec les termes de ce projet, qui consistent à faire de la discrimination en raison du patrimoine génétique un nouveau cas de discrimination. Et, par ailleurs, nous sommes d'accord pour, dans l'article pénal, ne pas dispenser en matière d'assurance de personnes, les assureurs de cette infraction quand il s'agit de tests asymptomatiques ou prédictifs. Nous espérons que la discussion de ce projet de loi va maintenant avancer et que cette infraction y sera incluse.
Cela ne nous dispense pas de nous interroger sur le débat de fond que nous devons avoir, plus généralement, avec les assureurs en matière de risque de santé.
M. Philippe Chaumet-Riffaud. - Il est un dernier sujet que nous souhaitions aborder avec vous, ce sont les conséquences de l'article L. 672-5 du code de la santé publique qui interdit, à l'heure actuelle, le prélèvement de tissus ou de cellules sur les personnes vivantes mineures, à la seule exception de la greffe de moelle.
Or, comme nous vous l'avons dit, un essai récent a montré qu'à partir de la cuisse d'un adulte, on pouvait arriver à cultiver des cellules, à les réimplanter dans une paroi myocardiale infarciée et récupérer ainsi une partie de la fonction mécanique.
D'autres arguments nous permettent de penser que la transplantation de myoblastes, c'est-à-dire de cellules qui sont précurseurs des fibres musculaires, pourrait être une piste extrêmement intéressante pour l'approche des myopathies.
À l'heure actuelle, tel qu'est formulé cet article, nous allons être considérablement gênés puisque les myoblastes sont surtout présents chez les enfants jeunes. Si cette piste se révélait efficace, nous souhaiterions que l'article soit modifié. Nous avons envisagé, par exemple, qu'il prévoie « la possibilité d'effectuer des prélèvements sur une personne vivante mineure au bénéfice de ses frères et s_urs ou à son propre bénéfice », puisqu'on pourrait prendre ses myoblastes, les modifier par thérapie génique et les lui réinjecter, « dans le cadre de maladies d'une particulière gravité, suivant la procédure déjà édictée pour le prélèvement de moelle osseuse ».
Le prélèvement de moelle osseuse fait en effet l'objet d'une étude par une commission particulière, qui statue au cas par cas. Ce n'est pas une liberté totale de prélever, ce n'est pas l'ouverture à n'importe quoi. C'est une possibilité légale qui est prévue et qui fait l'objet d'une étude, au cas par cas, devant une commission.
Mme Christine Boutin. - Seuls les enfants sont concernés ?
M. Philippe Chaumet-Riffaud. - Non. C'est que les myoblastes, qui sont des cellules jeunes, ont un potentiel beaucoup plus important.
En outre, les myopathies étant souvent très rapidement évolutives, ce sont des jeunes que nous allons traiter. Les essais en cours concernent avant tout des enfants de moins de quinze ans. Une fois atteint l'âge de vingt ou vingt-cinq ans, pour certains types de myopathies, il est trop tard. Cela se ferait dans le cadre de ce qu'on appelle des autogreffes, c'est-à-dire en prenant les propres cellules du sujet, soit par des allogreffes, en prenant des cellules d'un donneur de la fratrie.
M. Alain Claeys, rapporteur. - Nous vous remercions de nous avoir consa-cré tout ce temps.
Audition de M. Salomon KESSOUS,
médecin généraliste
(Extrait du procès-verbal de la séance du 13 décembre 2000)
Présidence de M. Bernard Charles, président.
M. Alain Claeys, rapporteur. - Vous êtes médecin généraliste à Paris. En vous recevant, nous aimerions aborder, au plus près des patients, de leurs préoccupations et des difficultés d'être à leur écoute, un sujet sur lequel nous avons déjà reçu le témoignage de spécialistes.
Nous avons d'abord envie de vous interroger sur l'attente des patients, leur demande, les détresses auxquelles vous devez répondre en tant que médecin. Comment les patients viennent-ils vous voir ? Comment appréhendent-ils les différentes techniques de procréation ? Quel est leur niveau d'information ? Jusqu'où des couples sont-ils prêts à aller, car le « droit à l'enfant » est un sujet qui nous préoccupe ?
Ensuite, nous aimerions savoir comment vous vivez cela, en tant que généraliste : comment abordez-vous ces questions avec vos patients ? Comment pouvez-vous suivre l'évolution des techniques en la matière ? Comment s'organisent les relations entre le médecin généraliste et les structures spécialisées dans l'assistance à la procréation ?
Enfin, nous aimerions connaître votre avis sur une technique comme l'ICSI, sur l'anonymat du don de gamètes, etc.
M. Bernard Charles, président. - En fait, nous souhaitons connaître l'appréciation d'un généraliste au c_ur du dispositif et sa perception de la situation.
M. Salomon Kessous. - Tout d'abord, il faut savoir que, de nos jours, nous sommes rarement sollicités en première intention pour traiter ce problème. Je pratique depuis vingt ans. En vingt ans, j'ai pu constater que le nombre de demandes directes à un généraliste a eu tendance à diminuer. Ces demandes n'ont jamais été débordantes : de trois à quatre par an au maximum ; et encore est-ce un nombre qu'il convient de relativiser car, lorsque j'ai commencé ma pratique, j'étais en campagne et il faut faire la différence entre les grandes villes, les villes de province et la campagne.
Au fil des années, ce nombre a diminué et il serait malhonnête de dire qu'aujourd'hui, nous avons encore des demandes. La seule exception, cependant, mais qui est peut-être due à la particularité de l'endroit où j'exerce, le XVIIIème arrondissement, arrondissement à forte population immigrée, est justement celle de la population immigrée. Ce sont les seuls cas où nous sommes sollicités en première intention. Et encore, même ces cas s'amenuisent au fil du temps, peut-être parce qu'il s'agit justement d'une grande ville comme Paris.
Quelle est l'attente des patients et des couples ? C'est tout, tout de suite, de la manière la plus efficace : si c'était possible, un enfant dans les deux mois.
Quelle est l'attitude d'un généraliste face à un couple demandeur d'enfant ? Au sortir de la faculté, nous avions appris qu'après des tentatives ininterrompues pendant deux ans, on entrait dans un processus d'infertilité chez les femmes en période « d'activité génitale ». La pratique a montré que face à la demande pressante et à l'hyper-médiatisation, il fallait réduire ce délai.
Après avoir interrogé les malades sur leurs relations sexuelles, car ces questions basiques ont leur intérêt avant de s'engager dans un parcours d'assistance médicale à la procréation, je réalise des examens complémentaires que je qualifierai aussi de basiques : une échographie chez la femme, avec un dosage hormonal, et un spermogramme chez l'homme. Si une anomalie est détectée, je les adresse immédiatement à un centre spécialisé.
M. Bernard Charles, président. - Quelles sont vos relations avec le centre spécialisé ?
M. Salomon Kessous. - Nous n'avons pas des relations très entretenues. Une fois que nous avons adressé nos patients à un centre spécialisé, nous recevons généralement une lettre de réponse signalant la prise en charge du couple, puis, quasiment plus rien, sauf lorsqu'il y a une maladie intercurrente pendant le traitement, n'importe laquelle, ou des phénomènes secondaires liés aux injections hormonales.
Cela nous pose problème parce que le retour d'information en provenance de ces centres est source de formation continue pour nous. Ils peuvent nous informer en deux phrases : tel produit peut donner ceci ou cela, des choses qui ne sont pas forcément dans le Vidal !
Par ailleurs, quand, pour une maladie intercurrente, nous sommes amenés à donner un médicament à une patiente, il nous faut nous assurer que nous n'allons pas gâcher tout le processus et le protocole dans lequel elle est engagée. Or, pouvoir joindre le correspondant dans les centres spécialisés, à un instant donné, n'est pas forcément facile.
Enfin, nous voyons ces couples, et surtout les femmes, dans une autre circonstance : ils ou elles reviennent nous voir après l'échec. C'est alors tout le problème de la prise en charge du couple sur le plan psychologique, avec les problématiques que cela peut engendrer, problématiques de couple ou problématiques propres à chacun de ses membres.
Telles sont les relations que nous avons avec ces centres.
Si le bilan basique que j'ai demandé est normal, alors, je m'accorde le délai des deux ans. Si au terme de ce délai, il n'y a pas eu de résultats, je les adresse à un centre spécialisé. C'est ma démarche, qui est celle de plusieurs de mes confrères.
M. Alain Claeys, rapporteur. - Qu'en est-il du degré d'information de vos patients ?
M. Bernard Charles, président. - Quel accueil ont-ils reçu dans ces centres ? En colloque singulier avec vous, ils sont sans doute plus à même de dire comment ils le ressentent ? Vous avez des connaissances médicales, vous pouvez le savoir. Sentez-vous qu'il y a une prise en compte globale ? L'environnement médico-social ou social vous paraît-il suffisant ? Voyez-vous des pistes de réflexion à suggérer ?
M. Salomon Kessous. - Globalement, lors d'une consultation pour infertilité, et encore plus dans le cadre d'une AMP, la demande d'explication est malgré tout fonction du niveau socioculturel. Mais, très souvent, le climat psychologique est tel qu'ils ont tout entendu et rien entendu. Ils ont tout compris et rien compris.
M. Bernard Charles, président. - Sont-ils fixés sur un objectif ?
M. Salomon Kessous. - Absolument. C'est une obsession, une véritable obsession et plus rien n'existe à côté, tant et si bien, comme d'ailleurs pour d'autres spécialités, que l'on revient vers nous pour nous demander des explications. Or, chaque cas étant un cas individuel, la technique ne va pas être identique pour tous. Même si elle est globalement standardisée, elle est unique et peut ne pas être la même pour un couple, qui a tel problème, que pour un autre.
C'est là où nous sommes en difficulté, en tout cas, où je suis en difficulté. N'ayant pas de retour de la part de ces centres, je peux difficilement leur répondre. En plus, il faut être honnête, je suis une formation continue, j'en suis même plusieurs, mais l'AMP est un domaine quasiment de « sur-spécialité ». Sincèrement, ce n'est pas notre première préoccupation en matière de formation.
Je suis curieux par nature, j'essaie de m'informer à travers la presse médicale mais, très souvent, les couples arrivent devant nous avec des informations médiatiques sur des techniques dont nous-mêmes n'avons pas connaissance et il est difficile de leur apporter une réponse. Nous sommes donc obligés de nous informer et d'aller chercher des notions toute théoriques en ne sachant pas quelle méthode leur sera appliquée mais en essayant de leur délivrer tout de même une information. Certains s'engagent dans le processus sans savoir qu'il faut un spermogramme, qu'il faut une émission de sperme. C'est parfois aussi simple que cela. Il faut expliquer tout le basique. Certaines femmes ne savent pas que le prélèvement d'ovocytes se fait par une technique bien particulière.
Ils se disent qu'on va prendre l'ovule, le spermatozoïde, leur fabriquer un enfant. Mais ce n'est pas aussi simple que cela. C'est un vrai parcours.
En fait, nous sommes plus sollicités sur le plan psychologique. Nous faisons plus de la psychothérapie de soutien que du médical pur.
Mme Yvette Roudy. - Je suis totalement profane, mais plus j'avance en information, plus je trouve que l'on ne sait pas où l'on va quand on démarre un parcours d'assistance médicale à la procréation. J'entends dire, par exemple, qu'il faut un traitement de stimulation ovarienne, mais cela demande, malgré tout, plusieurs injections, n'est-ce pas ?
M. Salomon Kessous. - Oui.
Mme Yvette Roudy. - Des piqûres. Après cela, il y aura sans doute une anesthésie pour vérifier si les résultats sont bons. Puis, si tout va bien, il faut que la femme revienne et subisse une nouvelle anesthésie pour l'implantation. Si cela n'a pas marché, on est prêt à recommencer plusieurs fois. Je me dis que ce n'est pas sans risque pour l'équilibre psychologique du couple, probablement, mais aussi pour l'équilibre physiologique et biologique de la femme, qui peut être instrumentalisée dans cette affaire.
Je me demande donc s'il ne serait pas bon que figure dans le texte une obligation d'organiser des entretiens très sérieux, au cours desquels serait donnée une explication très concrète et détaillée de ce qui peut les attendre, où leur serait aussi indiqué que l'on n'est pas sûr que cela réussisse, et précisé que l'on ne peut pas aller au-delà d'un certain nombre d'essais. Je me demande si, durant ces entretiens, il ne serait pas bon d'essayer également de leur suggérer d'adopter un enfant. Leur parle-t-on de l'adoption avant de les encourager à se lancer dans ce parcours ?
En outre, j'ai appris récemment que certaines femmes, certains couples qui sont impatients d'avoir des enfants, même sans être stériles, peuvent avoir recours à une stimulation ovarienne. Ne faut-il pas vraiment y réfléchir ?
Je pense que se pose aussi la question de l'éducation du médecin, dont vous avez l'air de dire qu'elle n'existe pas. Cet aspect me semble insuffisamment traité.
Nous nous laissons « embarquer » dans une sorte d'exploitation par une équipe médicale, et l'on comprend fort bien qu'elle puisse se passionner pour l'enjeu. C'est très exaltant. Mais ne faut-il pas proposer une pause et poser certains freins dans la mesure où l'on ne contrôle pas tout ? De plus, chaque fois, on crée des embryons surnuméraires, ce qui pose tout de même un problème.
M. Salomon Kessous. - Vous avez tout à fait raison. S'engager dans une démarche d'AMP, j'appelle cela « faire le parcours du combattant ». C'est un parcours long, psychologiquement difficile, très difficile.
Sur la formation, pour vous donner un ordre de grandeur, quand nous étions étudiants, les stérilités faisaient l'objet d'une heure de cours, la contraception de deux heures. Depuis, cela a évolué. Je suis d'une ancienne promotion.
En ce qui concerne les hyperstimulations ovariennes, je rejoindrai vos propos parce que je suis un observateur assez privilégié. Je travaille dans un petit groupe comprenant deux médecins généralistes, un dentiste et une infirmière. Il m'arrive souvent de voir mes patientes attendre dans la salle d'attente. Je vois qu'elles n'ont pas rendez-vous et qu'elles vont chez l'infirmière : « Que vous arrive-t-il ? C'est pour des piqûres ». Et je constate que ce sont des piqûres de stimulation ovarienne.
M. Bernard Charles, président. - Et vous n'êtes pas au courant de ces piqûres alors que vous êtes le médecin traitant ?
M. Salomon Kessous. - Je ne suis absolument pas au courant.
M. Bernard Charles, président. - Ce que vous dites là est important. Vous êtes leur médecin ?
M. Salomon Kessous. - Je suis le médecin traitant.
Mme Yvette Roudy. - Qui les a prescrites ?
M. Bernard Charles, président. - Le centre.
M. Salomon Kessous. - Quand c'est fait dans le centre, à la limite, il y a une certaine logique. Dans la mesure où l'on va vers une fécondation in vitro, il est logique de faire une hyperstimulation. S'il est révélé que la femme a une insuffisance ovarienne, le traitement est légitime car il convient de remédier à cette insuffisance. Après tout, quand la thyroïde ne fonctionne pas correctement, on prescrit bien des hormones thyroïdiennes.
Là où je rejoindrai Mme Roudy, c'est quand ces femmes ne consultent pas un centre spécialisé mais voient des gynécologues. Sans vouloir jeter l'opprobre sur mes confrères, car ils subissent une réelle pression de la part de ces couples, je dois dire qu'ils finissent par prescrire des stimulations ovariennes alors que la femme est peut-être infertile pour une raison x, ou que c'est l'homme qui l'est, ou, allez savoir, qu'ils n'ont peut-être pas de sexualité. C'est, je l'ai dit, une question importante à poser dès le début. Ces femmes, sans peut-être avoir de véritables insuffisances ovariennes, subissent des hyper-stimulations ovariennes qui finiront par poser problème tant pour elle que pour l'enfant à naître.
Toute la problématique est là, à mon sens : ne faut-il pas réserver le traitement de l'infertilité, dans sa globalité, FIV ou pas FIV, à des centres spécialisés ou à des gynécologues ayant reçu une formation et ayant une pratique spécifique ?
La question est tellement complexe que cela nécessite une approche multidisciplinaire. J'assimilerai cela à la cancérologie. En cancérologie, même quand tout s'est très bien passé, les patients rencontrent presque systématiquement un psychologue. De la même manière, il serait bon qu'ils en voient un en cas d'AMP, peut-être même la consultation du psychologue devrait-elle précéder celle du gynécologue.
C'est un point de vue. Mais cela me semble nécessaire pour avoir des garanties de qualité dans la prise en charge de ce couple et particulièrement de la femme. Avec l'ICSI, le problème se pose à un degré moindre. L'homme ne subit pas un parcours du combattant comme la femme...
M. Bernard Charles, président. - C'est d'ailleurs une raison du succès.
M. Salomon Kessous. - Il faut être honnête et le reconnaître. En revanche, la femme suit un véritable parcours du combattant. Elle prend un traitement, son organisme supporte une dose d'hormones dont on ne connaît pas l'effet à long terme. Elle a son enfant, c'est très bien, mais qu'adviendra-t-il d'elle à cinquante ans et lorsqu'elle sera confrontée à des problèmes majeurs ?
M. Bernard Charles, président. - J'en viens à un thème que nous avons déjà eu l'occasion de soulever. En effet, j'avais déjà, l'année dernière, lors de la discussion sur la loi de financement de la sécurité sociale, déposé un amendement qui avait été rejeté. Mais je ne désespère pas de le faire adopter dans la loi de modernisation sociale, puisqu'il vient d'être accepté en commission et que nous avons l'accord des médecins, ce qui n'aurait sans doute pas été possible il y a encore quinze ans de la part du Conseil de l'Ordre. Il s'agit d'un amendement qui vise à ce que certaines pratiques, qui présentent des risques, soient réservées à un personnel médical ayant subi une formation spécialisée.
Tous les organismes professionnels l'ont admis. On constate donc une évolution qui répond à votre préoccupation car on voit très bien qu'en la matière, il existe trois niveaux : le généraliste, le gynécologue non spécialisé et le centre. Il est évident que la spécialisation pour ce type de traitement ne peut être admise sans une mise en pratique quasi quotidienne, tant sur le plan technique et scientifique que sur le plan psychologique.
À cet égard, ce que vous venez de dire à propos de l'orientation psychologique nous intéresse beaucoup.
M. Salomon Kessous. - J'apporterai toutefois une nuance à ce que je vous disais tout à l'heure. Autant dans les années 1988-1992, je constatais qu'un assez grand nombre de mes patientes faisaient l'objet d'hyper-stimulations ovariennes en les rencontrant dans la salle d'attente, autant c'est un phénomène qui a bien diminué. Un « holà » a sans doute été mis au sein de la communauté médicale. Je crois que celle-ci souhaitait aussi cette correction.
M. Jean-Michel Dubernard. - Les médecins ne peuvent pas être formés à tout. Ce n'est pas possible. C'est à l'intérieur d'une discipline et de façon continue que l'on doit se former. Actuellement encore, un médecin peut tout faire.
M. Bernard Charles, président. - C'est pour cela que je signalais cet amendement fondamental qui tend à ce que tout le monde accepte que certains actes médicaux, vu les techniques et les connaissances nécessaires pour les mettre en _uvre, soient réservés à des spécialistes. C'est un amendement important.
M. Jean-Michel Dubernard. - Plus on avance, plus on bâtit de belles théories pour organiser un système idéal qui correspond à une médecine dite de luxe, alors qu'au quotidien tout ce que l'on voit dans la pratique a tendance à s'effondrer. C'est ce que je vis depuis quelques années.
Voici ma question, que j'oserai à peine poser devant Mme Roudy : ne pensez-vous pas que, dans cette affaire, l'homme est négligé ? On parle du parcours de combattant de la femme. Très bien. C'est vrai. Mais l'homme ?
J'ai une formation d'urologue et je vois des patients arriver, en couple ou seuls, effondrés parce qu'on leur a trouvé une azoospermie ou une oligospermie, ils viennent en cachette demander un avis complémentaire en se disant que leur généraliste ne doit rien y connaître puisqu'il y a quand même deux millions de spermatozoïdes, c'est donc qu'ils en ont. Et il nous faut leur expliquer que ce n'est pas suffisant.
De plus, la prise en charge qui est faite est une prise en charge du couple et, au sein du couple, on a tendance à considérer le « mâle » comme un sous-facteur du système. C'est du moins la vision que j'en ai.
M. Bernard Charles, président. - Oui, mais parfois aussi le « mâle » fuit ses responsabilités dès qu'il peut.
Mme Yvette Roudy. - Je comprends ce que vous dites. Je le dis très souvent : je pense qu'en effet, on peut casser le couple.
M. Salomon Kessous . - Cela le casse souvent.
Mme Yvette Roudy. - Il paraît qu'on leur interdit d'avoir des relations sexuelles à certains moments pour ne pas contrarier le processus.
M. Jean-Michel Dubernard. - Tout à fait.
Mme Yvette Roudy. - J'insiste plus sur la femme parce que le traitement qu'elle reçoit est lourd : piqûres, anesthésies, et ce à plusieurs reprises. Mais il est vrai qu'il faut prévenir le couple d'un point de vue psychologique, parce qu'ils sont tous deux un peu trop souvent traités comme du matériel humain : « Vous voulez un enfant, donnez-nous vos gamètes et nous, nous nous occuperons du reste ! »
M. Salomon Kessous. - Vous avez raison lorsque vous parlez de l'homme comme élément négligé mais peut-être, historiquement, a-t-on considéré que la stérilité était plus un problème de femmes que d'hommes et donc s'intéressait-on plus aux femmes. Puis, on a fini par comprendre que c'était un problème de couple et qu'il fallait explorer le cas des deux membres du couple de la même manière. Je dois avouer - c'est ma vision quotidienne - que les femmes s'y soumettent plus volontiers, avec presque plus de docilité, que les hommes.
Mme Yvette Roudy. - Ce n'est pas étonnant, on les y a habituées !
M Bernard Charles, président. - Cela ne figurera pas au compte rendu.
Mme Yvette Roudy. - Mais j'y tiens. Leur éducation les habitue à être dociles.
M. Salomon Kessous. - Il y a assimilation pour l'homme entre l'infertilité et la virilité. L'infertilité est ressentie comme une atteinte à la virilité. Il est vrai que cela évolue.
M. Bernard Charles, président. - Vous notez une évolution ?
M. Salomon Kessous. - Incontestablement.
Mme Yvette Roudy. - À quoi est-elle due ?
M. Salomon Kessous. - Je pense à la médiatisation, à l'information. Ils sont de mieux en mieux informés.
M. Bernard Charles, président. - Vous notez un changement ?
M. Salomon Kessous. - En effet.
M. Jean-Michel Dubernard. - Être « clair », comme on disait puisque c'était le mot utilisé...
Mme Yvette Roudy. - Cela signifiait être stérile ?...
M. Jean-Michel Dubernard. - Oui. C'était très mal vécu. Demandez aux médecins de campagne... Cela reste toujours très mal vécu. Il est vrai que cela a un retentissement psychologique. Il ne faut pas négliger l'homme dans le couple.
M. Bernard Charles, président. - Chaque fois que l'on peut avoir le couple globalement impliqué dans la démarche, c'est beaucoup mieux, afin que l'un ne se sente pas en première ligne et l'autre rejeté.
M. Jean-Michel Dubernard. - Quand vous avez une consultation et douze malades qui attendent dans la salle d'attente, vous ne pouvez pas organiser un entretien ou vous le faites mal.
Mme Yvette Roudy. - C'est impossible. Mais, après tout, il existe des entretiens pour les demandes d'IVG ; on peut en faire pour l'assistance médicale à la procréation.
M. Salomon Kessous. - Le problème se pose de manière aiguë avec les populations immigrées. Nous avons affaire à des cultures différentes et cela prend une tournure de drame. Il m'est arrivé plus d'une fois de voir des Maghrébins ou des Africains repartir avec leur femme au pays, la répudier et revenir avec une autre. C'est dramatique. Ces personnes n'ont pas forcément accès à une information, ne serait-ce que par la télévision. Autant on peut critiquer la médiatisation de la médecine telle qu'elle est faite parce qu'elle pousse à croire que tout est possible et pour tout le monde...
M. Bernard Charles, président. - Oui, c'est la porte ouverte à tous les fantasmes.
M. Salomon Kessous. - ...alors que l'on sait bien que chaque cas est un cas individuel, autant ces personnes n'ont même pas accès à cette information. La consultation tourne court. Elle est très compliquée, très difficile, vraiment plus difficile parce que, même si vous les envoyez dans des centres spécialisés, vous savez qu'il y n'aura pas forcément le personnel pour favoriser le dialogue et la communication. Et encore, je m'en tiens au seul dialogue et à la seule communication, je ne parle même pas de techniques.
Mme Yvette Benayoun-Nakache. - Vous venez de répondre à l'une des deux questions que je voulais poser.
Voici ma seconde question : quand, lors des consultations, vous interrogez vos patients, que le dialogue s'établit, que le parcours du combattant est amorcé, ou pas d'ailleurs, que le couple et la femme prennent conscience au fur et à mesure de son déroulement que ce parcours est difficile, à un moment donné, la notion d'adoption n'apparaît-elle pas ? Les couples, le couple lui-même ou l'un de ses membres, ne pensent-ils pas finalement à adopter ?
Quand on voit la misère de l'adoption en France et ce qu'on fait subir à ces couples et à ces femmes, on ne peut pas ne pas se poser cette question. Ne vaudrait-il pas mieux parfois les engager dans cette voie ? Cette question est-elle même évoquée ?
M. Salomon Kessous. - J'aimerais pouvoir l'évoquer, mais je dois avouer que je l'évoque rarement, ou timidement, parce qu'en fait, deux cas se présentent à moi.
Quand est-ce que je rencontre ces patients ?
Il y a trois tentatives de fécondation in vitro remboursées par la sécurité sociale. Je les vois donc entre la première et la seconde tentative, entre la deuxième et la troisième et après la dernière. Évidemment, à chaque fois, j'ai affaire à des gens qui sont au plus bas. C'est l'échec et le retentissement de cet échec sur le couple est considérable ; c'est à se demander s'il faut refaire des tentatives et si ce couple est encore en situation d'avoir un enfant. Ce sont des questions qu'il faut légitimement se poser.
Comment voulez-vous faire ? Je suis face à des gens qui sont dans un psychose obsessionnelle : l'enfant, l'enfant, l'enfant ! Entre la première et la deuxième tentative, je ne vais pas leur parler d'adoption. Encore moins entre la deuxième et la troisième.
Nous nous retrouvons après la dernière. Deux cas de figure s'offrent alors à moi.
Il y a ceux qui sont dans la douleur, qui doivent « digérer » leur échec. Il faut d'abord les aider à franchir ce cap difficile pour envisager ensuite de leur parler, timidement, d'adoption. Très souvent, c'est un processus qu'ils engagent, et pour certains, spontanément même.
Puis, il y a les autres. Vous m'avez demandé jusqu'où ils étaient prêts à aller. Je puis vous garantir qu'ils sont prêts à tout. À tout !
M. Bernard Charles, président. - Cela nous a été dit à plusieurs reprises.
M. Salomon Kessous. - À la limite...
M. Bernard Charles, président. - Partir à l'étranger, dépenser de l'argent...
M. Salomon Kessous. - Vous m'avez enlevé les mots de la bouche. Ils sont prêts à partir, à dépenser n'importe quelle somme dans des circuits, dont je me demande s'ils sont sérieux ou douteux, à des prix ahurissants. Parfois, je me demande s'ils ne subissent pas une forme de mutilation médicamenteuse et psychologique.
Mme Yvette Roudy. - Cet aspect est très important et l'on n'en tient pas suffisamment compte dans les débats et dans les textes parce que l'exaltation est telle, au regard du merveilleux de ces techniques, que l'on ne parle pas suffisamment du prix humain à payer, de la dégradation des couples, du prix psychologique.
Il faut vraiment réfléchir à ce que nous pouvons préconiser. Peut-être un suivi psychologique et des entretiens réguliers du début à la fin ? Des formes de thérapies, parce que parfois, l'échec est pire qu'un deuil. Le travail de deuil est parfois encore plus difficile parce que l'on est atteint dans son intégrité psychologique et qu'on a le sentiment d'avoir été utilisé.
Cet aspect, nous n'en tenons pas suffisamment compte dans nos textes, nos débats et nos appréciations. Il faudrait en parler : dégâts sociaux, psychologiques, dégradation des couples, etc.
Mme Yvette Benayoun-Nakache. - Il faudrait aborder d'emblée cette discussion. Cela permettrait au couple d'avoir cet élément en tête et peut-être d'y trouver un recours s'il n'arrive pas à avoir un enfant par cette méthode.
M. Bernard Charles, président. - Mais s'ils partent directement dans les centres ?
M. Salomon Kessous. - Comme le disait M. le président, très souvent, nous ne sommes pas au courant du processus...
M. Bernard Charles, président. - Alors que vous êtes le médecin de famille.
M. Salomon Kessous. - Mais cela est valable aussi pour d'autres pathologies, même bénignes. On peut poser le problème du nomadisme médical intrahexagonal et extrahexagonal...
M. Bernard Charles, président. - Surtout en milieu urbain.
M. Salomon Kessous. - Cela vaut pour toutes les spécialités. C'est le premier point. Le second, c'est qu'il faut avoir des psychologues qui travaillent dans ces centres, qui ne fassent que cela. J'ai utilisé tout à l'heure la comparaison avec la cancérologie. Il y a une psychologie bien particulière de ces patients, qui relève du psychologue spécialisé.
La pratique nous montre que la prise en charge psychologique d'un patient cancéreux atténue un peu sa douleur. Pourquoi ne pas faire la transposition à l'AMP en disant que la prise en charge psychologique doit se faire sur tout le parcours ? « Un échec : rendez-vous immédiat avec le psychologue ». Cela atténuerait un peu leur douleur. Ils ont une telle attente. Il faut aussi leur dire quel est le pourcentage d'échec. Il faut les y préparer d'emblée. Mais, peut-être leur a-t-on dit et ils n'ont rien entendu.
M. Bernard Charles, président. - C'est une pensée obsessionnelle.
Mme Yvette Roudy. - Il n'y a pas que cela dans la vie. Mais comment le leur dire ? On exalte tellement ces nouvelles techniques.
M. Salomon Kessous. - Il y a peut-être aussi un besoin de communication au sens large du terme, pas seulement dans la communauté médicale mais également tel que les médias peuvent le rapporter. Que voulez-vous ? Un couple qui a des problèmes d'infertilité et qui regarde la dernière technique réalisée à Johannesburg ou ailleurs adhère aussitôt à l'espoir qui lui est donné. De la manière dont cela lui a été présenté, il ne va pas réaliser un instant que cela ne s'applique absolument pas à lui.
Professeur Dubernard, vous devez être confronté au même problème ?
M. Jean-Michel Dubernard. - Sans arrêt.
M. Bernard Charles, président. - Les gens viennent au miracle.
M. Jean-Michel Dubernard. - Il n'y a rien à faire pour que les journalistes donnent une information complète, objective dans ce domaine. Il existe bien quelques émissions spécialisées sur des chaînes câblées confidentielles qui sont très bien faites, mais pour le reste...
M. Bernard Charles, président. - C'est flagrant lors de ces talk shows qui reçoivent des personnes qui viennent témoigner cachées derrière des lunettes noires. Ces thèmes de fertilité, de transsexualité, etc. sont les thèmes préférés de ce genre d'émissions.
Nous vous remercions beaucoup d'être venu et d'avoir apporté ce témoignage qui nous sera fort utile.
Audition de M. le Professeur Didier HOUSSIN,
directeur général de l'Établissement français des greffes
(Extrait du procès-verbal de la séance du 10 janvier 2001)
Présidence de M. Jean-Michel Dubernard, vice-président,
puis de M. Bernard Charles, président.
M. Jean-Michel Dubernard, vice-président. - Nous avons le plaisir de recevoir le professeur Didier Houssin, directeur général de l'Établissement français des Greffes et, par ailleurs, chef de service de Chirurgie de l'hôpital Cochin.
Monsieur le directeur général, je vous remercie d'autant plus d'avoir accepté de répondre à l'invitation de notre mission que la date initiale de votre audition a été reportée à la suite d'impératifs liés au calendrier parlementaire.
Vous le savez, notre mission marque le commencement de la phase parlementaire de la révision des lois bioéthiques de 1994 avec deux ans de retard.
Nos travaux de ce jour sont consacrés au prélèvement d'organes. Le contexte dans lequel s'était déroulée la discussion de la loi de 1994 était assez difficile, en raison d'une certaine forme d'inquiétude qui régnait alors dans l'opinion publique. Vous nous direz si, d'après vous, la loi de 1994, qui a créé l'Établissement français des Greffes et le registre des refus, a réussi à apaiser cette inquiétude.
Vous nous direz également en quoi les évolutions scientifiques et médicales qui sont intervenues depuis 1994, ou celles qui sont annoncées, rendent nécessaires des adaptations de la loi de 1994.
M. Alain Claeys, rapporteur. - M. le directeur général, je renouvelle nos excuses pour avoir dû décaler ce rendez-vous. Les auditions de ce jour sont pratiquement les dernières dans le cadre de cette mission d'information.
Professeur, pour introduire nos débats, il serait intéressant que vous puissiez nous dresser le bilan de l'Établissement français des Greffes. En vous demandant cela, je pense aux structures qui pourraient être proposées dans le futur projet de loi, et qui concerneraient d'autres aspects des lois bioéthiques. L'éclairage sur l'expérience de l'Établissement français des Greffes nous sera utile.
La loi de 1994 a créé cet établissement, qui succédait à France Transplant. Que pensez-vous de la composition et du fonctionnement de son conseil d'administration, des missions et du rôle de son directeur général, en particulier, des relations entre son directeur général et le conseil médical et scientifique ?
Notre deuxième préoccupation est le bilan de l'activité des greffes d'organes en France.
L'Établissement français des Greffes est notamment chargé de gérer la liste nationale des patients, la répartition et l'attribution des greffons. Une première évaluation des résultats des greffes en France a été publiée en 1999. Pouvez-vous nous en rappeler les principales conclusions ?
Le deuxième aspect de ce bilan est le constat de la pénurie d'organes en France. On en parle beaucoup et l'on considère qu'elle provient, pour l'essentiel, du manque de donneurs. Or, une étude plus fine semble faire ressortir que des blocages demeurent au sein même des équipes de praticiens. J'aimerais connaître votre sentiment sur cet aspect de la question.
Enfin, toujours à propos de ce bilan, quel est votre sentiment sur la recommandation du Conseil d'État qui demande expressément que soit inscrit dans la loi que tout prélèvement est une activité médicale. Nous ne sommes pas, pour la plupart d'entre nous, médecins et voudrions comprendre en quoi cette précision pourrait améliorer l'activité de prélèvement.
Dernier aspect, le consentement et, donc, le registre des refus. Si je me réfère aux conclusions du Conseil d'État - puisque nous avons pris le périmètre d'étude de ce dernier pour organiser notre réflexion - celui-ci préconise, concernant le prélèvement sur donneur décédé, de substituer au témoignage de la famille celui des proches.
Mon interrogation à ce propos sera double. D'une part, les équipes médicales ont-elles souhaité cette modification, et pourquoi ? D'autre part, comment celle-ci pourrait-elle se traduire en pratique ? Par exemple, comment s'assurer que nous avons bien affaire à un proche ?
Par ailleurs, quelles observations vous inspire la pratique du registre des refus, même si ce dernier a mis un certain temps à se mettre en place puisque les décrets ne l'ont instauré que très tardivement ? A-t-elle répondu à la volonté de clarification souhaitée en matière de consentement ? Des critiques auraient été émises sur le long délai pour obtenir une réponse. Qu'en est-il dans la pratique ?
Deux autres points pour terminer : l'information en faveur du don d'organes est réalisée sous la responsabilité du ministre chargé de la santé. Le Conseil d'État a suggéré d'y associer le ministère de l'éducation nationale. Qu'en pensez-vous ?
Enfin, concernant le prélèvement d'organes à des fins scientifiques, l'autopsie est définie comme un prélèvement à des fins scientifiques ayant pour but de rechercher les causes directes ou indirectes du décès. Quelles modifications des règles de consentement vous apparaissent comme nécessaires en ce domaine ?
M. Didier Houssin. - M. le président, M. le rapporteur, je vous remercie de votre invitation et m'efforcerai de suivre dans mes réponses le plan que vous avez indiqué.
Premièrement, il s'agissait d'« apaiser l'inquiétude » selon le propos de M. Jean-Michel Dubernard. Je reprendrai ensuite les différents points évoqués par M. Alain Claeys et, plus brièvement, j'évoquerai ensuite les évolutions scientifiques, ce qui amènera à ouvrir quelques perspectives.
Apaiser l'inquiétude.
De fait, en 1994, lorsque la loi de bioéthique a été promulguée et lorsque l'Établissement français de Greffes a été créé, le climat était un climat de crise. Les questions soulevées étaient celles de grandes inquiétudes en matière de sécurité sanitaire sur les tissus, de suspicion de trafic de priorité, d'un pourcentage très élevé de patients non résidents sur certaines listes d'attente, d'un dysfonctionnement notable en matière de consentement à l'occasion de l'affaire dite « d'Amiens ».
Bref, le monde des greffes était plongé en état de crise, lequel contrastait avec l'efficacité thérapeutique de la greffe qui s'annonçait et se confirmait pour certains types de greffes, d'autant que, jusqu'au début des années 1990, on avait assisté à une augmentation apparemment sans limite du nombre des greffes réalisées dans les hôpitaux.
En 1992-93, c'est donc la crise marquée par une chute des prélèvements de cornées de près de 30 % et par celle des prélèvements d'organes de près de 20 %. Lorsqu'en 1994, la loi a été promulguée et l'Établissement français des greffes créé, nous étions donc dans cette situation que l'on pouvait certes qualifier de critique.
L'inquiétude s'est-elle apaisée aujourd'hui ? La situation est-elle redevenue normale ? J'aurais tendance à répondre positivement, du moins partiellement.
J'aborderai, si vous en êtes d'accord, un aspect d'abord purement quantitatif. L'activité de prélèvements et de greffes de cornées a retrouvé son niveau de départ, et l'a même largement dépassé aujourd'hui. L'activité de prélèvement d'organes a remonté une partie de la chute : nous sommes à 1 016 prélèvements d'organes effectués sur des personnes décédées pour l'année 2000 ; au pic, en 1991, il y en avait eu 1 090.
Pour certains types de greffes, le nombre des greffes réalisées chaque année a déjà largement dépassé le pic antérieur puisque, l'an dernier, 806 greffes de foie ont été effectuées alors que le pic était de 697 en 1991. Pour d'autres, comme celle du rein, par exemple, nous ne sommes pas totalement parvenus à récupérer la chute.
Pour d'autres types de greffes, le contexte médical est différent. Cette activité a considérablement évolué, c'est le cas de la greffe cardiaque, qui est réalisée beaucoup moins souvent que dans ces années-là.
Voilà pour l'évolution sur le plan quantitatif.
Sur le plan médiatique, la situation est plus sereine. L'analyse très régulière et le suivi très précis que nous faisons des productions médiatiques, écrites ou audiovisuelles, montrent que le climat est transformé. Les présentations sont beaucoup plus positives. Dans certains cas, nous voyons même se développer une forme de participation des médias au soutien de cette activité.
Cela dit, c'est une situation qui reste fragile. Le moindre dysfonctionnement en termes de sécurité sanitaire, en termes d'équité dans la répartition des greffons ou dans le recueil du consentement pourrait avoir des conséquences dévastatrices. C'est un aspect qui mérite d'être souligné.
La deuxième interrogation de M. Jean-Michel Dubernard portait sur le bilan de la loi de bioéthique. Celui-ci se caractérise en terme de suivi, et vous l'avez souligné, par une certaine lenteur dans la production réglementaire, mais il faut reconnaître que celle-ci est largement dosée : seize décrets devaient être pris à la suite de la loi bioéthique. Il s'agit d'un travail sur un sujet assez technique. Il convient d'avoir conscience qu'il n'était pas possible, pour les administrations centrales chargées de rédiger ces décrets, de les publier rapidement et simultanément. Cela explique un certain retard dans la mise en place du registre national des refus.
Il existe aussi le cas de décrets qui ne sont pas parus pour des raisons autres. Par exemple, celui sur la vigilance n'est pas paru en raison de la mise en place récente et survenue entre-temps de l'agence de sécurité sanitaire.
S'agissant de l'analyse que l'on peut faire de la loi de bioéthique dans ses principes fondamentaux.
À mon sens, le principe de gratuité est parfaitement respecté. Il n'y a pas eu, à ma connaissance, en France, de suspicion, et encore moins de réalité, de trafic et de commerce d'organes. À ma connaissance, au cours des six ans passés, deux patients ont été greffés à l'étranger après avoir acheté le greffon à l'étranger. Sur le territoire national, aucun indice n'a été rapporté de ce genre de pratique.
Quant au consentement, la procédure mise en place pour recueillir le consentement sous la forme d'un régime de consentement présumé, avec création d'un registre national des refus et recueil du témoignage de la famille sur la position du défunt, est un dispositif nuancé, subtil, de mon point de vue, de très bonne qualité, mais difficile à expliquer et sans doute assez mal compris par la population. Il n'en reste pas moins qu'aucun dysfonctionnement sévère n'a été noté dans cette procédure de recueil du consentement.
À l'analyse, le registre national des refus joue son rôle. Il répond à une attente. Il serait dommage de le remettre en cause. Il rassemble aujourd'hui près de 50 000 personnes. Celles-ci étaient suffisamment opposées pour aller se faire inscrire sur un registre informatique, ce qui est une initiative assez lourde. De plus, ces personnes ont la possibilité de changer d'avis. Je peux dire que ce registre rassemble à peu près autant d'hommes que de femmes, que la répartition géographique est homogène, même si l'on ne produit pas d'étude sociologique détaillée de ce genre de registre.
Il est aujourd'hui consulté sans difficulté à partir des hôpitaux, avec un délai de réponse de l'ordre de quelques minutes. En l'an 2000, il a été interrogé 7 426 fois pour un prélèvement à fin thérapeutique, 1 032 fois pour un prélèvement à visée d'autopsie et 366 fois pour un prélèvement dans le cadre d'une recherche à caractère scientifique. Le nombre d'inscrits était hier de 46 852.
C'est un registre qui existe dans d'autres pays, comme le Portugal, la Pologne et l'Autriche qui ont aussi adopté le principe du consentement présumé. Il rend service et me paraît tout à fait congruent avec l'adoption d'un principe de consentement présumé.
Pour dresser le bilan de l'Établissement français des Greffes, je ne suis certainement pas le meilleur juge. Je peux cependant vous signaler que j'avais demandé, en 1997, un audit de l'Inspection générale des affaires sociales qui a porté un premier jugement sur l'Établissement français des Greffes. Elle avait d'ailleurs fait à cette occasion des suggestions qui ont été suivies. Par ailleurs, nous avons actuellement un contrôle régulier de la Cour des comptes, qui peut certainement vous apporter des informations.
Pour ma part, je pense que l'Établissement français des Greffes est une agence qui renforce l'État dans un domaine où, pour des raisons sanitaires mais surtout de justice, de solidarité et d'information du public, l'État a besoin d'être renforcé.
La modalité de constitution d'une agence à large compétence, traitant des tissus, des organes et des cellules, m'a paru un choix original et, finalement, intéressant. L'Établissement français des Greffes a des contreparties analogues dans certains pays européens. Il me semble important que cette agence n'apparaisse pas comme une agence de sécurité sanitaire. L'Établissement français des Greffes n'est pas que cela. Il s'agit d'un organisme qui doit tenir la crête entre des intérêts contradictoires : ceux des malades - et il a un rôle important de promotion à jouer pour obtenir des greffons, sur lequel je reviendrai - et ceux de la société, qui a besoin d'être informée et rassurée puisqu'elle est invitée à participer jusque dans sa chair, dirai-je, à cette activité. Tenir la crête, cela signifie qu'il faut répondre à ces deux exigences : agir en priorité pour les malades mais tout en tenant compte de l'intérêt et de la position de la société sur cette question.
Le conseil d'administration est un conseil que je considère comme classique, où les tutelles et l'État sont majoritaires. Il ne me semble pas poser de difficultés particulières, hormis celle concernant les praticiens qualifiés. En effet, ce conseil d'administration compte cinq praticiens qualifiés qui ont eu tendance au début, moins maintenant, à voir « passer les trains scientifiques » en ayant le sentiment de ne pas être bien à leur place.
De ce point de vue, la suggestion qui a été faite, et qui pourrait être retenue, est que, comme dans les hôpitaux, ces praticiens qualifiés puissent être des représentants de l'instance scientifique qu'est le conseil médical et scientifique, ou délégués par elle, et ne soient pas des praticiens qualifiés indépendants, si je puis dire, de cette instance scientifique. Dans les hôpitaux, les médecins présents au conseil d'administration sont des représentants de la CME. Cela me paraît une analogie intéressante. Nous irions volontiers vers cette proposition, que nous avions suggérée à l'IGAS qui ne l'a cependant pas retenue.
Quant au directeur général, son rôle est de faire fonctionner l'établissement. L'Établissement français des Greffes est la première agence sanitaire à avoir élaboré un contrat d'objectifs et de moyens avec ses tutelles. Ce contrat a été signé l'année dernière. En la matière, le rôle du directeur général a bien sûr été d'orienter la réflexion et la définition des objectifs pour l'Établissement en liaison avec les tutelles. Il remplit un rôle essentiel d'harmonisation de la relation avec les tutelles, d'harmonisation avec les interlocuteurs, au premier rang desquels les hôpitaux, les agences régionales d'hospitalisation et, évidemment, les équipes médicales.
Le directeur général saisit le conseil médical et scientifique. Un des reproches faits par l'Inspection générale des affaires sociales était que le conseil médical et scientifique menait la politique de l'Établissement. Cette analyse ne me paraît pas tout à fait juste car, en fait, ce conseil est interrogé par le directeur général. Mais il est cependant exact que lorsqu'il émet un avis, et que celui-ci est pris à l'unanimité, il est difficile au directeur général de ne pas en tenir compte.
C'est donc un conseil qui me paraît adapté à la mission de l'Établissement et je ne vois pas aujourd'hui de grands dysfonctionnements qui tiendraient à la composition ou aux modalités de travail du conseil médical et scientifique. Les présidents du conseil d'administration ont été un scientifique et un médecin. Ils auraient pu avoir d'autres compétences.
Pour prolonger la réflexion dans le sens que vous évoquiez, la suggestion avait été faite, en son temps, aux tutelles et au Conseil d'État que les missions de l'Établissement pourraient se voir élargies jusqu'à ce qu'il devienne une véritable agence ayant compétence sur tout le champ de la bioéthique. Il y avait des arguments pour, d'autres contre.
Sans passion extrême, j'avais souligné les arguments favorables. Il semble que l'on s'oriente vers une agence autonome, soit ! Mais il pouvait y avoir certains avantages à profiter de l'expérience et de l'acquis d'une certaine « vitesse de croisière » dans le fonctionnement de ce type d'établissement. De plus, cela permettait de croiser des compétences et des champs d'action. Or l'interdisciplinarité me paraît essentielle : mettre en présence des spécialistes des greffes et des spécialistes des cellules embryonnaires et de leur perspective thérapeutique à venir me paraissait assez positif. Les questions relatives au consentement et à la sécurité sanitaire ne sont pas non plus si éloignées.
En revanche, il est vrai qu'il y avait des arguments contre, en particulier, l'utilité éventuelle d'avoir une structure bien individualisée, dans un champ aussi sensible que celui qui touche aux questions liées à la procréation. Je m'en tiendrai à ces brefs commentaires. Le Gouvernement semble avoir défini sa position et cela m'a paru une option tout à fait bonne.
En ce qui concerne l'évaluation des résultats des greffes, l'Établissement français des Greffes a conduit une action originale - en tout cas, une première en France _ d'évaluation d'une thérapeutique équipe par équipe, avec publication en clair des résultats. Le processus qui a été conduit est à mettre, à mon avis, au crédit des équipes de greffes qui ont accepté de se prêter à ce type d'évaluation.
Celle-ci a permis de faire ressortir des éléments rassurants. Premièrement, les résultats des équipes de greffes d'organes en France sont finalement assez voisins d'un centre à l'autre et ne justifient pas, en tout cas, que les malades se précipitent à l'autre bout de la France au prétexte d'un meilleur résultat. Ce résultat rassurant peut s'expliquer par le fait qu'il s'agit d'une thérapeutique assez spécifique, dans un monde hospitalier assez bien ciblé. En tout cas, cela a permis de montrer que l'on peut, en France, mener des comparaisons par équipe et en rendre publics les résultats, voire les commenter dans la presse.
Le travail avait été fait pour les greffes d'organes. Cette année, il l'a été pour les greffes de moelle osseuse. De même, ont été présentées, pour la première fois cette année, les différences de durée d'attente, en particulier en matière de greffe de rein. C'est un point crucial.
Cela m'amène à la question du manque de greffons. C'est la question la plus difficile. Elle hante un peu l'esprit de tous ceux qui s'occupent de prélèvements et de greffes.
En matière de tissus, c'est une question d'organisation. On peut facilement parvenir, dans les deux ans qui viennent, à résoudre totalement le manque de greffons cornéens. C'est en revanche beaucoup plus difficile pour les organes, pour une raison très simple et méconnue : la rareté des greffons tient avant tout à la rareté de l'état de mort encéphalique.
Cette notion est méconnue. C'est un état tout à fait exceptionnel qui n'est observé que dans 2 000 à 3 000 décès hospitaliers. En France, sur 550 000 décès chaque année, 350 000 sont des décès en milieu hospitalier et, sur ces derniers, on ne peut guère constater beaucoup plus que 2 à 3 000 décès en état de mort encéphalique. Pourquoi ? Parce que les circonstances qui conduisent à l'état de mort encéphalique sont exceptionnelles.
En fait, la grande raison du manque de greffons, et ceci est vrai dans tous les pays, tient au fait que cette rareté est presque structurelle, pourrait-on dire, inhérente à la pathologie qui conduit à cette atteinte cérébrale.
La difficulté supplémentaire, c'est qu'à cette rareté de base s'ajoutent des facteurs de rareté complémentaires.
Ces facteurs sont, tout d'abord, l'incapacité de certains hôpitaux à dépister ou à identifier l'état de mort cérébrale. C'est là peut-être que se pose la question du blocage que vous évoquiez. Il est vrai qu'en France, jusqu'à ces derniers temps, certains établissements de santé ne s'investissaient pas de manière très active dans le dépistage des états de mort encéphalique. Ensuite, il y a la possibilité du refus témoigné par la famille ou exprimé par le défunt. Enfin, des obstacles médicaux de toute nature peuvent faire que le prélèvement n'est pas raisonnable ou impossible.
Je reviens sur les deux premiers points pour dire que c'est la raison pour laquelle le prélèvement doit être considéré comme une activité médicale à part entière. Cela restaurerait l'image que l'on en a dans l'hôpital. Il ne serait plus considéré comme une activité de second rang, que l'on pratique à la nuit tombante, mais comme une activité noble qui aboutit au traitement de malades. Positionner l'activité de prélèvement comme une activité médicale à part entière me semble un élément absolument capital.
Le second aspect est, bien sûr, la mobilisation du tissu hospitalier français. A ce propos, l'initiative du Plan Greffes, prise par Mmes les ministres Martine Aubry et Dominique Gillot, au mois de juin 2000, est un élément capital, car il donne justement aux établissements de santé français les moyens de réaliser, dans de bonnes conditions, ce dépistage et cette identification de la mort encéphalique. Il devrait permettre à la France de se situer, dans les trois ans, au niveau des moyens mis en place en Espagne en termes de ratio de personnel médical et non médical dans les établissements de santé se consacrant à l'activité de prélèvement. Je suis, à cet égard, relativement optimiste. On peut difficilement faire plus pour améliorer la faisabilité du prélèvement dans les hôpitaux.
Cela ne signifie pas qu'il en résultera une disponibilité totale des greffons et une disparition des listes d'attente. Le penser serait irréaliste. Mais cela peut permettre de mieux répondre aux demandes, en particulier, dans le cas de greffes de reins ou d'organes thoraciques.
Le contrat d'objectifs et de moyens de l'Établissement français des Greffes, signé en 2000, comporte un objectif prioritaire, qui est de passer de quinze à vingt prélèvements par million d'habitants. En 1994, nous en étions à quatorze ; aujourd'hui, nous en sommes à dix-sept, et j'ai bon espoir qu'en 2002-2003, nous en soyons à vingt.
J'en viens maintenant à l'un des derniers points que vous avez évoqués, celui de l'information du public. Évidemment, le refus est l'un des obstacles au prélèvement. Ce refus est, bien sûr, la résultante de l'opposition du défunt, qui peut d'ailleurs s'être inscrit sur le registre national des refus, mais il peut aussi être la conséquence des circonstances dans lesquelles la famille est invitée à dialoguer, circonstances suffisamment dramatiques pour qu'elle oppose parfois un refus catégorique lié au contexte dans lequel elle vit ce drame. C'est ici que l'information du public hors tout contexte émotionnel prend toute son importance et que l'Établissement français des Greffes joue un rôle en la matière, bien sûr, avec l'aide de son ministère de tutelle.
L'idée de confier au ministère de l'Éducation nationale une tâche dans ce domaine serait, je pense, extrêmement utile. Initiative heureuse, en liaison avec les hospices civils de Lyon, l'année dernière, le rectorat de Lyon a organisé un concours de philosophie sur ce thème, qui a montré l'appétit des jeunes et des enseignants pour ce type de thème. Je crois que des projets sont en cours d'élaboration avec les professeurs de biologie mais il pourrait être effectivement très utile qu'un texte de haut niveau accorde une certaine responsabilité à l'Éducation nationale dans ce domaine.
Enfin, pour finir de répondre aux points que vous avez évoqués et avant de conclure sur les aspects touchant à l'évolution scientifique, je dirai que la perspective d'une harmonisation du recueil du consentement, c'est-à-dire d'un alignement du consentement pour un prélèvement à visée d'autopsie, destiné à connaître les raisons de la mort, sur le consentement pour le prélèvement à visée thérapeutique, nous serait précieuse. Aujourd'hui, nous nous heurtons à des difficultés liées à ce décalage. Si en 1994, on pouvait comprendre qu'existe une sorte de hiérarchie, aujourd'hui, celle-ci gagnerait à être gommée.
J'ajouterai un mot sur le donneur vivant, qui sera sans doute l'objet de débats. En fait, l'Établissement français des Greffes a été peu sollicité sur cette question, peu interrogé. Nous avons effectivement reçu deux ou trois courriers de personnes disant qu'étant mariées depuis trente ans et aimant leur conjoint, elles ne comprenaient pas pour quelles raisons on leur interdisait de lui donner un rein, pourquoi on rendait le don si difficile. Mais cela reste un nombre de cas très limité. La réflexion et le débat qui auront lieu sur cette question seront certainement intéressants. Nous pouvons imaginer un élargissement mais à la condition d'instaurer un dispositif de contrôle, car il serait dangereux d'ouvrir de manière incontrôlée la possibilité de greffes avec donneur vivant à des personnes non apparentées.
Je conclus mon propos en abordant les aspects scientifiques. Celui qui revêt le caractère le plus important, en dehors de greffes multi-tissus - et peut-être pourrons-nous en discuter si vous le souhaitez - est, à mon avis, la perspective thérapeutique de l'utilisation de cellules souches. Cellules souches adultes ou embryonnaires, je crois que la perspective est lointaine. En tout cas, en ce qui concerne les greffes d'organes, elle est excessivement lointaine.
Cet éloignement justifie-t-il de conserver une certaine réserve ? C'est un point de vue que l'on peut défendre. Quoi qu'il en soit, l'Établissement français des Greffes sera intéressé à suivre cette évolution thérapeutique. Mon point de vue est que l'utilisation de cellules souches embryonnaires est une perspective thérapeutique très lointaine. En conséquence, toute la question est de savoir s'il faut modifier des dispositions sensibles pour de telles perspectives. Elle est ouverte. J'avoue que je me contenterai assez volontiers de la solution qui sera choisie.
M. Bernard Charles, président. - Je vous remercie. Mes collègues vont maintenant pouvoir vous poser toutes les questions qu'ils souhaitent mais, auparavant, je voudrais vous interroger sur une question que nous avions évoquée lors de nos précédentes rencontres portant sur l'idée de valorisation du don. À propos du Plan Greffes, vous m'aviez indiqué qu'il vous semblerait bon d'inscrire dans la loi une mention valorisant le don. Comment la verriez-vous car ce sera certainement une question à l'ordre du jour lorsque nous en viendrons à l'examen du texte ?
M. Didier Houssin. - C'est un point très important pour la raison suivante : le législateur, dès le début, a choisi de placer ce qui, objectivement, est un prélèvement sur le corps d'une personne décédée, dans l'esprit du don - manière assez belle de l'habiller. Cet « habillage » a toute sa légitimité et, personnellement, je le défends. Mais la logique doit être poursuivie jusqu'à son terme.
Le don est le geste qui consiste à donner, à recevoir... et à rendre. Vous connaissez la réflexion conduite par Marcel Mauss sur la question. Je pense qu'il est souhaitable de mener cette logique à son terme et de faire apparaître dans la loi que le « rendre » existe aussi. Cette reconnaissance exprimée par la Nation et la société à ceux qui ont donné, même si elle s'exprime de manière symbolique, serait un complément très utile et ouvrirait la porte à des manifestations justement de caractère symbolique. Il en existe déjà, mais un texte les renforcerait. À Lille, à Clermont-Ferrand, à Caen, à Flers, à l'hospice de Bicêtre, un arbre a été planté dans l'hôpital pour manifester la reconnaissance aux donneurs et à leurs familles.
L'accompagnement des familles qu'assurent les coordinations hospitalières est aussi une manière de rendre et d'être à l'écoute. Si cela figurait dans la loi, sans être pompeux naturellement, ce serait précieux.
M. Jean-Michel Dubernard. - J'aurais, bien sûr, de nombreuses questions à vous poser mais, pour éclairer mes collègues, je n'aborderai, monsieur le directeur général, que quatre points.
Premièrement, concernant l'Établissement français des Greffes, pourriez-vous nous indiquer son budget, le nombre de personnes qu'il emploie et celui des personnes employées indirectement, par l'intermédiaire des hôpitaux en Province et à Paris, par les coordinations locales ?
Deuxièmement, concernant les greffes à partir de sujets en état de mort encéphalique, vous avez cité le chiffre de 1 016 donneurs en 2000. Cela représente théoriquement 2 032 reins. Combien de greffes de reins ont-elles été effectuées ? Par quel mécanisme, le différentiel, qui se produit obligatoirement, peut-il s'expliquer ?
Sur le même sujet, la valorisation du don est très nécessaire, mais ne faudrait-il pas avoir le courage d'aborder la valorisation de l'acte ? Considérer l'activité de prélèvement comme un acte médical à part entière, cela ne signifie-t-il pas que les gens qui passent des nuits à s'occuper de sujets en état de mort cérébrale ou à prélever des organes doivent être rémunérés de façon significative ? C'est, à mon sens, ce qui explique que les résultats espagnols soient largement supérieurs aux nôtres dans ce domaine.
Le seul point qui posera véritablement problème dans la révision de la loi bioéthique est, me semble-t-il, celui de l'élargissement éventuel des donneurs vivants. Ce sujet sera le c_ur du débat. Je rappelle qu'actuellement, la loi autorise les prélèvements intra-familiaux et, en cas d'urgence, ceux à partir du conjoint. Mais le Premier ministre s'est déjà engagé, il y a quelques jours, en disant que ce cadre sera élargi et il a même parlé des amis de longue date.
À ce sujet, j'ai plusieurs questions à vous poser.
Avez-vous le pourcentage de donneurs, hommes et femmes, quand il s'agit de conjoints ?
Pouvez-vous nous fournir les résultats actuels concernant les greffes de reins à partir de donneurs vivants par rapport aux prélèvements sur personnes décédées ? Comment peut-on expliquer les différences que l'on constate entre ce qui se passe actuellement et ce qui se passait il y a vingt ans ?
Quel est le risque médical pour le donneur ? Nous venons de vivre à Lyon un drame : la perte d'un donneur vivant de foie. Nous avions vécu, il y a trente ans, un drame similaire pour un donneur de rein. Quel est ce risque, qui me semble encore accru avec la coelioscopie et le développement de la laparoscopie ?
M. Bernard Charles, président. - Qu'est-ce que la laparoscopie ? Pourriez-vous l'expliquer aux non-médecins que nous sommes.
M. Jean-Michel Dubernard. - C'est une technique de chirurgie par laquelle on opère sans ouvrir la paroi, mais en faisant une simple incision par laquelle on sortira le rein. Cela concerne les prélèvements de reins. C'est essentiel à connaître pour pouvoir fournir une réponse qui ait du sens à propos de cet élargissement.
Le développement actuel des greffes par donneur vivant apparenté ne s'explique-t-il pas, ou - c'est une nuance qui a du sens - ne se justifie-t-il pas uniquement par la pénurie d'organes ? Nos décisions doivent prendre en compte le risque de développement d'un commerce d'organes. Quand on parle de commerce d'organes, cela ne veut pas forcément dire paiement, ce peuvent être aussi des pressions psychologiques directes ou indirectes. On se retrouve alors dans des situations d'échange qui ne sont pas dramatiques en soi, mais qui renvoient aux sources de la bioéthique, monsieur le président, parce qu'elles mettent en cause la dignité de l'homme, celle du corps qui peut être considéré comme le véhicule de la dignité humaine. Nous retombons alors sur l'éthique et la morale.
Dernier point, vous avez fait une allusion au fait que la distinction entre cellule, tissu, organe a de moins en moins de sens actuellement. Il faut, bien entendu, introduire dans la loi les termes de « greffe de tissu composite », pour qu'on ne se trouve pas face à un vide juridique quand se développeront, dans les années à venir, les greffes associant plusieurs tissus.
M. Pierre Hellier. - Apparemment, vous êtes satisfait, monsieur le directeur général, du fonctionnement de votre agence, puisque les moyens dont vous disposez sont suffisants. Mais que pourrait-on imaginer, non pas pour améliorer son fonctionnement, mais celui du système des greffes en France ?
M. Didier Houssin. - L'Établissement français des greffes possède un budget de l'ordre de 80 millions de francs. Il emploie cent vingt personnes. Il est important d'ailleurs de bien distinguer les personnels de l'Établissement français des Greffes, qui est un organisme national disposant de petites structures dans certaines régions, des personnels hospitaliers, tels que ceux des coordinations hospitalières des prélèvements. Il est capital que ces derniers apparaissent bien comme des personnes qui répondent à une mission de l'hôpital.
Il faut bien distinguer les moyens consacrés au prélèvement dans les hôpitaux
- c'est en cette matière que notre pays souffre de faiblesses - des moyens de l'Établissement français des Greffes, qui est une agence qui dispose aujourd'hui des moyens lui permettant de fonctionner. Le contrat d'objectifs et de moyens que celui-ci a passé avec ses tutelles lui permettra surtout de renforcer son système d'information, qui est un aspect crucial. Il s'agit, non pas du système d'information de l'Établissement, mais de celui concernant l'ensemble des prélèvements et des greffes en France. Je crois qu'il a les moyens de mener son action.
Concernant les donneurs décédés, sous réserve de vérification des données, l'année dernière, 1 922 greffes de reins ont été faites et 1 016 prélèvements pratiqués. Chaque donneur ayant a priori deux reins, on aurait pu s'attendre à un peu plus de greffes de reins, mais, dans certains cas, soit les reins n'apparaissent pas de bonne qualité et ne sont pas susceptibles d'être greffés ; soit, malheureusement, le prélèvement est interrompu avant que l'on ait pu prélever les deux reins. Il y a donc des circonstances qui font que le « rendement » d'un prélèvement d'organe n'est pas de quatre - ce ne sont pas deux reins, un c_ur et un foie qui sont prélevés, mais de 3,4 organes prélevés en moyenne.
M. Jean-Michel Dubernard. - Ce chiffre a-t-il tendance à baisser ?
M. Didier Houssin. - Effectivement, car malheureusement la qualité des donneurs a tendance à s'altérer du fait que l'on identifie des donneurs de plus en plus âgés. Fort heureusement, les accidents de la voie publique ont diminué, et nous espérons que cela continuera, et les greffons seront de plus en plus souvent prélevés sur des sujets en état de mort encéphalique liée à des accidents vasculaires cérébraux et non plus à des accidents de la voie publique et à des traumas crâniens.
Je n'avais pas répondu à la question concernant les proches et les familles. Dans certains cas, et de manière croissante, les coordinations hospitalières des prélèvements sont confrontées au fait que les personnes qui entourent le défunt sont des amis, et pas forcément membre de sa famille. Ces amis sont parfois investis d'un véritable témoignage ; ce sont eux qui ont entendu le défunt dire qu'il était pour ou contre un prélèvement d'organes. J'imagine que c'est la raison pour laquelle le mot « proches » remplacerait celui de famille. Ces situations restent cependant minoritaires et je ne pense pas que ce soit un élément absolument déterminant pour la faisabilité ou la non-faisabilité du prélèvement.
Parlant de la valorisation du don, vous avez mis le doigt sur un point tout à fait important, celui de la rémunération de l'acte de prélèvement. On peut comprendre les hésitations qu'il y aurait à payer celui-ci à l'acte car, effectivement, il y aurait une sorte d'intérêt à agir. D'aucuns considèrent, peut-être en partie à raison d'ailleurs, que si les Espagnols ont une telle efficacité, c'est que leurs médecins ont un intérêt financier puissant à réaliser les prélèvements. L'analyse que l'on a pu faire de la situation espagnole est la suivante : en 1990, l'Espagne était en forte démographie médicale, confrontée à de nombreux jeunes médecins sans possibilité d'insertion, mal payés et il est exact que certains d'entre eux se sont lancés dans l'activité de prélèvement. Avec un salaire très modeste et des gardes ou des astreintes, le fait d'être rémunérés lorsqu'ils pratiquaient un prélèvement leur apportait un complément de salaire non négligeable. Cela a sûrement été, pour l'Espagne, un dispositif incitatif.
En France, la situation n'est pas tout à fait identique. Les médecins ont un équilibre entre salaire et rémunération des astreintes ou des gardes différent. Il faut rester prudent sur la rémunération du prélèvement à l'acte. Tous les dispositifs incitatifs doivent être étudiés mais, en optant pour celui-là, on risquerait de se voir opposer la critique qu'il y a un intérêt financier direct à agir, ce qui ne manquerait pas de produire un effet néfaste dans l'esprit de la population.
En ce qui concerne le donneur vivant, l'Établissement français des Greffes a été peu sollicité. Je mentionnerai simplement qu'en Inde, où on pratique beaucoup de greffes avec donneur vivant, les donneurs sont souvent des femmes. C'est la déviation que l'on peut toujours craindre. En ce qui concerne ces greffes, il est vrai que les résultats se sont améliorés parce que les immunosuppresseurs permettent aujourd'hui de pallier les difficultés liées à l'absence de lien en termes de génétique, c'est-à-dire de compatibilité tissulaire entre donneur et receveur. Comme, parallèlement, il est possible que la qualité des greffons prélevés sur des personnes décédées se soit un peu altérée, les courbes statistiques font ressortir une légère supériorité des résultats des greffes avec donneur vivant par rapport à celles avec donneur décédé.
Je partage tout à fait votre avis sur le risque médical pour le donneur. La hantise de tout chirurgien ou de tout anesthésiste qui réalise ou participe à une activité de prélèvement sur donneur vivant est le décès du donneur. C'est une catastrophe totale dont le risque ne pourra jamais être totalement annulé. Il est important de garder cela présent à l'esprit. Cela explique, à mon avis, la grande prudence dont il faut faire preuve. Si la loi française élargit le système de la greffe avec donneur vivant, dans la pratique, en résultera-t-il nécessairement des modifications importantes en termes de niveau d'activité sur le plan quantitatif ? Je ne suis pas sûr, parce que justement la crainte est que le donneur ne meure et que cette crainte est extrêmement présente.
En ce qui concerne les moyens de l'Établissement français des Greffes, le contrat d'objectifs et de moyens qui a été signé lui donne les moyens de fonctionner pour les trois ans à venir. Si un effort doit être fait, et il a été décidé, c'est dans les hôpitaux qu'il doit porter. Si le Plan Greffes, qui démarre bien dès 2001, avec des budgets affectés aux agences régionales d'hospitalisation, se poursuit, ce que j'espère, nous devrions être en mesure d'atteindre l'objectif.
Cependant, il y a des éléments incontrôlables, en particulier, les facteurs épidémiologiques. Si demain, par exemple, le nombre des accidents de la voie publique est divisé par deux ou si les médecins inventent un système permettant d'éviter l'_dème cérébral dans les accidents vasculaires cérébraux, il peut ne plus y avoir beaucoup de greffons à prélever.
Quant aux greffes de tissus composites, il est important que la loi prévoie la possibilité de les faire, car c'est une thérapeutique prometteuse, même si elle n'en est qu'à ses débuts. Il est important que le texte ne crée pas d'entraves et rende cette thérapeutique possible, les difficultés tenant essentiellement, et sans spécificité, au problème du consentement du défunt et de la position de sa famille.
Mme Yvette Benayoun-Nakache. - Je reviens, M. le directeur général, sur le registre du refus. En France, les dons d'organes restent encore un sujet tabou parmi d'autres et il est vrai que ce n'est franchement pas un thème que nous abordons facilement avec nos concitoyennes et nos concitoyens. J'ai noté votre avis de ne pas supprimer le registre du refus et vous disiez qu'il compte 46 852 inscriptions. Ce chiffre me semble relativement peu élevé.
Vous avez aussi suggéré d'élargir ce débat, comme commence à le faire l'Éducation nationale, aux enfants et aux adolescents qui sont peut-être plus à même d'être sensibles à cette valorisation du don d'organes. Pourriez-vous préciser cela ?
J'aimerais également connaître votre avis sur le phénomène grandissant de l'incinération en France, notamment chez ces jeunes. N'y a-t-il pas là quelque chose « qui nous échappe » par rapport aux possibilités de dons d'organes ?
Si M. le Président me le permet, je formulerai également la question que souhaitait poser M. Jean-Paul Baquet. Elle concerne la situation très difficile de 1994. Quelle est l'évolution réelle du nombre des greffes en fonction, premièrement, de la sélection des sites agréés, deuxièmement, de la notification en intervention de la greffe et, troisièmement, de l'évolution des greffes et des besoins supplémentaires ?
Mme Catherine Génisson. - Ma question concerne un sujet qui est au c_ur de notre problématique : le donneur vivant. Comment envisagez-vous la définition des proches qui pourraient être donneurs d'organe par rapport au receveur ? Quelles limites doit-on poser pour autoriser le prélèvement dans ces conditions, sachant tous les risques que cela représente, risques immédiats comme sur le moyen terme ?
M. Didier Houssin. - Il est vrai que le don d'organes reste un sujet tabou. Il n'est pas facile d'en parler et l'une des plus grandes difficultés est sans doute d'amener la population d'un pays à envisager sa propre mort, à la regarder en face et à en discuter en famille le soir au dîner. C'est là une tâche extrêmement délicate.
Nous avons la chance, si j'ose dire, d'avoir adopté le principe du consentement présumé qui permet d'éviter d'avoir à mettre en _uvre des politiques excessivement incitatives dans ce domaine. Aux États-Unis, on a vu des présidents successifs s'adresser à la population, à la télévision, en demandant de donner ses organes. Nous ne sommes pas, en France, contraints de le faire. Cela nous permet de mettre en place une information un peu plus neutre. Il me semble que c'est une bonne chose.
C'est la raison pour laquelle je n'ai pas de souhait concernant le registre national des refus. Je constate en effet que le nombre de gens inscrits, s'il est faible en pourcentage, n'est pas nul. En valeur absolue, 50 000 personnes, cela représente tout de même une bonne ville de France. Il est essentiel de pouvoir mettre à disposition, de manière régulière, des documents d'information : 11 millions l'ont été. Cet effort doit se poursuivre, ne serait-ce que par le biais des médecins et des pharmaciens. Peut-être également faut-il envisager d'autres solutions.
Je ne pense pas qu'il y ait de contradiction entre la tendance - mais après tout pourquoi pas ? - au développement de l'incinération et la possibilité de réaliser des prélèvements en vue de greffes. Dans le don d'organes, le corps n'est pas abandonné par la famille, ce n'est pas comme dans le don du corps à la science. Le corps est prêté transitoirement dans un hôpital pour que soit pratiquée une intervention chirurgicale, puis est rendu à la famille qui se chargera des obsèques selon les voeux du défunt, inhumation ou incinération. Ce sont deux choses assez indépendantes et je ne pense pas que l'extension éventuelle de l'incinération ait un impact sur la pratique du don d'organes, sauf à imaginer des liens plus fondamentaux, anthropologiques, sur l'idée qu'on se fait du corps, de sa destination, etc.
Concernant la question de M. Jean-Paul Baquet, j'espère l'avoir suffisamment comprise et je vais essayer d'y répondre. En France, 230 établissements de santé ont l'autorisation de pratiquer des prélèvements. Ce chiffre ne représente pas la totalité des établissements qui pourraient les pratiquer mais notre effort, qui, me semble-t-il, commence à porter ses fruits, a été d'amener peu à peu les établissements de santé à s'inscrire dans cette démarche.
Nous rencontrons d'ailleurs des difficultés dans certaines régions. Les choses évoluent mais il y a peu de temps encore de grands hôpitaux ne participaient pas à l'activité de prélèvement. Il en reste encore aujourd'hui qui n'y participent pas, alors qu'ils en ont théoriquement les moyens. Je ne les citerai pas pour ne pas commettre d'impair et de dommages irréparables ! Mais c'est un point tout à fait important.
La notification, c'est-à-dire la capacité à recenser dans un établissement de santé les personnes décédées en état de mort encéphalique, relève, quant à elle, du rôle de la coordination hospitalière des prélèvements. C'est un des indicateurs importants de la qualité de cette activité que de voir augmenter le recensement. Pour vous donner une idée, en 1997, on a recensé 1 664 sujets en état de mort encéphalique et, en 2000, on en a recensé 1 990. C'est une nette amélioration et il me semble que c'est un indicateur intéressant à suivre et à voir progresser.
Mme Yvette Benayoun-Nakache. - M. Jean-Paul Bacquet s'interrogeait aussi sur l'évaluation des greffes.
M. Didier Houssin. - L'évaluation des résultats des greffes ? Ce qui est sûr c'est que plus se dessinera la tendance de recenser des personnes plus âgées - et aujourd'hui, un prélèvement d'organe peut se faire après soixante ans - plus on s'exposera à ce que les résultats des greffes, liées à des greffons de moins bonne qualité, se dégradent. Mais c'est un mal que l'on peut difficilement éviter.
À titre personnel, je partage le point de vue de M. Jean-Michel Dubernard sur la question du donneur vivant. J'ai un certain nombre de craintes concernant le prélèvement sur donneur vivant car j'ai peur de l'accident pour le donneur, plus que des questions de dérives, même si celles-ci sont aussi à considérer sérieusement. De plus, je constate que la demande exprimée par les malades et leur famille n'est pas si considérable.
Par ailleurs, la demande exprimée par les professionnels de santé en France n'est pas non plus si forte que cela. Il ne me semble pas que vous ayez été submergés de courriers de médecins disant qu'on les empêche de prélever, qu'il faudrait pour sauver les malades qu'on les autorise à le faire plus largement. Certes, des médecins estiment, et disent même, que l'on n'en fait pas assez en France en comparaison d'autres pays, mais la France est la France. J'aurais, pour ma part, tendance à souhaiter une petite ouverture, mais prudente.
M. Bernard Charles, président. - Monsieur le directeur général, je vous remercie d'être venu, de vous être exprimé et d'avoir répondu à nos questions.
Audition de M. le professeur Dominique DURAND,
chef du service de néphrologie-hypertension artérielle-dialyse-transplantation
du centre hospitalier de Toulouse Rangueil
(Extrait du procès-verbal de la séance du 10 janvier 2001)
Présidence de M. Bernard Charles, président.
M. Bernard Charles, président. - Nous avons maintenant le plaisir d'accueillir M. Dominique Durand, chef du service de néphrologie-hypertension artérielle-dialyse-transplantation du CHU de Toulouse-Rangueil.
La mission d'information sur la révision des lois bioéthiques organise des rencontres thématiques. Avant vous, nous recevions le directeur général de l'Établissement français des Greffes, car nous souhaitons compléter notre information sur le thème des prélèvements et transplantations d'organes. Nous attendons de vous un éclairage sur les problèmes concrets rencontrés par les praticiens et les chercheurs.
Outre vos fonctions hospitalières, vos activités de recherche, notamment sur les questions d'immunologie, vous donnent une expérience qui peut être importante pour notre réflexion.
De plus, vous avez présidé, de 1995 à 1998, le conseil médical et scientifique de l'Établissement français des greffes. Nous aimerions connaître votre appréciation sur le fonctionnement de cet établissement ainsi que sur les pistes que nous avons déjà évoquées avec M. le professeur Didier Houssin, qui nous permettraient d'améliorer ce secteur mais aussi les perspectives offertes par les thérapies cellulaires et les conséquences des progrès en matière de transplantation d'organes.
M. Alain Claeys, rapporteur. - Monsieur le professeur, j'aurais quatre séries de questions à vous poser pour introduire le débat.
Pourriez-vous nous faire part de vos remarques sur le fonctionnement de l'Établissement français des Greffes dont vous avez été président du conseil médical et scientifique ?
Deuxièmement, à propos de l'activité de greffes d'organes en France, nous aimerions que vous nous fassiez part de votre expérience de praticien. Quels obstacles rencontrez-vous dans l'exercice de votre activité ? On parle de pénurie d'organes en France, liée essentiellement au manque de donneurs. En tant que praticien, quelle analyse faites-vous des causes de cette situation ?
Ma troisième série de questions concerne le consentement et le registre des refus. Que pensez-vous de la suggestion sur Conseil d'État concernant le prélèvement sur donneur décédé, de substituer au témoignage de la famille celui des proches ? Ce sera un des sujets importants de la révision des lois bioéthiques.
Quelles remarques vous inspire la pratique du registre des refus ?
Par ailleurs, c'est important pour les membres de cette mission qui ne sont pas médecins, pouvez-vous nous rappeler précisément les caractéristiques du prélèvement à des fins thérapeutiques et du prélèvement à des fins scientifiques ? Dans quelle catégorie, classer les prélèvements en vue de rechercher les causes de la mort ? Comment définir l'autopsie ?
Quatrièmement, le Conseil d'État a préconisé d'élargir la possibilité de prélèvement sur une personne vivante. Qu'en pensez-vous ?
Enfin, pouvez-vous nous exposer brièvement les effets attendus de la recherche sur les cellules souches par la communauté médicale ?
M. Dominique Durand. - Comme vous le savez, l'Établissement français des Greffes a été installé dans une période difficile pour la transplantation, difficile parce qu'il y avait incontestablement, à cette époque, une rupture de confiance entre la société et le monde médical de la transplantation. Vous en connaissez les causes. Le ministre de l'époque a considéré qu'il fallait que l'activité de transplantation soit encadrée par les pouvoirs publics et a créé, en conséquence, cet établissement. Celui-ci s'installe donc dans des conditions difficiles.
Cette agence a tenté d'organiser plusieurs aspects.
Le premier objectif a été d'essayer de rétablir la confiance. Pour cela, elle a proposé des projets de texte visant à encadrer toutes les phases de la transplantation. Cela a commencé par la définition de la mort cérébrale. Cette loi a été un moment important car elle disait que la mort cérébrale était la mort. Toutes les conditions du prélèvement ont été redéfinies.
Le deuxième objectif a été d'encadrer parfaitement les listes d'attente. Aujourd'hui, pour qu'un malade soit inscrit, il faut que cette inscription soit validée par l'Établissement français des greffes.
Un point tout à fait fondamental est la distribution des organes. Dans un contexte de pénurie, il est clair que l'attribution des organes est extrêmement difficile. Il était donc très important que des textes régissent cette attribution. Il convenait, à cet égard, que le texte élaboré, qui a été le fruit d'un compromis extrêmement difficile à trouver, l'ait été en grande collaboration avec les praticiens. Les textes ont été notamment élaborés après des réunions des praticiens de la cellule de transplantation qui avaient auparavant fait des propositions. Ils sont donc le fruit d'une collaboration et d'un compromis entre ce qui peut apparaître comme la nécessité d'une égalité parfaite vis-à-vis de la transplantation et des nécessités médicales de gestion des organes. Le choix, qui n'est pas celui fait dans tous les pays, a été de laisser un certain degré de liberté aux équipes à l'échelon local.
Comme vous le savez, il existe trois échelons : un échelon de priorité nationale, puis, un de priorité régionale, puis, ensuite, la gestion locale des équipes. C'est le choix français, avec une priorité nationale accordée à l'enfant. Les enfants sont en effet hautement prioritaires et, grâce à cela, la plupart d'entre eux échappent à la contrainte de la dialyse.
Un autre objectif de l'Établissement français des greffes a été d'assurer le suivi. Ce suivi présente plusieurs aspects.
Le premier est celui de l'évaluation des activités. À l'heure actuelle, la greffe est évaluée en tant que technique, mais aussi par équipe. Les résultats de ces évaluations sont régulièrement publiés. C'est probablement une des premières activités médicales dont les résultats sont publiés.
Le second objectif a été un accompagnement très important de sécurité sanitaire. Dans le contexte de l'époque, cet aspect était essentiel car la sécurité sanitaire en matière de transplantation d'organe a été organisée à un très haut niveau, selon des critères extrêmement contraignants. À mon sens, le temps est venu d'une réflexion portant sur la nécessité d'une sécurité sanitaire aussi lourde. C'est extrêmement onéreux pour, dans de nombreux cas, un résultat minime. Maintenant que le système est en place, que l'atmosphère autour de la transplantation est apaisée, il serait sans doute bon de revoir si la sécurité sanitaire après transplantation doit être aussi pesante. Le suivi de ces patients ne nécessite peut-être pas autant de surveillance. À mon sens, on peut parfaitement parvenir, avec moins de moyens, à une efficacité aussi grande.
Un des objectifs de l'Établissement français des greffes est, enfin, la promotion du don d'organe. C'est une mission qui fut mise en place de façon progressive.
L'augmentation du nombre d'organes disponibles se fait par deux voies.
La première passe par l'information de la société. C'est une voie difficile où l'on n'avance qu'à petit pas et qui est, finalement, assez peu productive. Il est probable que les résultats seront enregistrés bien plus tard. Il va falloir laisser le temps à la société, devant un progrès technique très rapide, de mûrir sa réflexion.
Il est intéressant de constater que le pourcentage de refus n'a pas diminué depuis de longues années, non seulement en France mais aussi dans des pays qui ont fait des efforts plus importants que le nôtre, comme l'Espagne, par exemple. En Espagne, qui est un modèle pour nous puisque c'est un pays dans lequel on prélève énormément d'organes, le pourcentage de refus est presque aussi important qu'en France, malgré tous les efforts consentis. Il y a peut-être là une image d'un refus très profond de la société vis-à-vis d'une activité difficile.
En revanche, il est très important de rendre toutes les procédures de prélèvement efficientes. Cela veut dire que cette chaîne fragile, qui part du moment où une équipe de réanimation prend en charge un grand blessé sur une route, qu'elle amène dans un centre de réanimation, et qui va jusqu'au prélèvement, doit être renforcée. Si l'on veut davantage d'organes, c'est à ce niveau qu'il faut mettre des moyens. L'État l'a déjà considéré puisque, récemment, des moyens importants ont été débloqués pour que les coordinations de prélèvement, qui sont les personnes qui s'occupent de cette activité, soient renforcées.
C'est là que doit porter l'effort, tout en continuant l'information du public, mais en sachant que cette dernière est un long périple.
À côté des organes, il y a eu l'organisation de la greffe des tissus. Il y a cinq ans, il n'existait rien dans ce domaine en France, strictement rien. Les chirurgiens osseux prenaient des têtes fémorales, lorsqu'ils avaient une prothèse de hanche à faire, ils les mettaient dans un réfrigérateur et, le lendemain, ils greffaient.
Tout cela a été organisé, entouré d'un environnement de sécurité sanitaire extrême. Je tiens à le souligner parce qu'en dépit de l'absence d'organisation, de contrôle, de vigilance, il n'y a pratiquement pas eu, dans le monde, d'accident de maladie transmissible à partir de tissus. Les cas sont très rares. Sur des milliers de transplantations, on a peut-être recensé une dizaine de cas publiés.
Pour reprendre ce que j'ai déjà dit, il existe, en ce domaine aussi, une vigilance interne extraordinairement lourde du donneur, du receveur, au moment du prélèvement, trois mois après, six mois après. C'est peut-être moins utile puisque, dans ce domaine, le rapport qualité-prix, si je puis dire, n'est pas très favorable.
Enfin, l'organisation a aussi porté sur l'activité de greffe de cellules hématopoïétiques, mais cette activité était déjà très organisée car les sociétés savantes d'hématologistes étaient déjà structurées en termes de protocoles et d'organisation.
La transplantation est une activité qui coûte extrêmement cher à la société, pour un tout petit nombre de malades. L'installation de l'Établissement français des greffes, qui est une agence relativement lourde, a été utile, nécessaire. Aujourd'hui, je ne sais quel chemin elle va prendre.
Il me semble cependant que les transplanteurs pensent qu'il faudrait que son rôle s'élargisse et qu'au-delà de ses missions, qui ont été définies par le ministre, il joue un rôle directement utile à la transplantation et aux transplanteurs.
Je m'explique. Dans des établissements, existe un système d'information extrêmement développé, permettant un enregistrement de données qui servent à la vigilance et à l'évaluation. Nous souhaiterions que cet Établissement se voie aussi confier une mission scientifique, car celle-ci est très peu marquée en son sein, disponible pour les transplanteurs, ce serait une source d'épanouissement pour notre communauté.
Tels sont les éléments dont je pouvais vous faire part en introduction. Je suis à votre disposition si vous souhaitez me poser des questions.
M. Jean-Michel Dubernard. - Ma première question portera sur les prélèvements d'organes sur les donneurs en état de mort encéphalique. Nous avons - et Didier Houssin le disait tout à l'heure - la chance de vivre en France sous le régime du consentement présumé, chacun étant considéré comme un donneur potentiel à moins que, de son vivant, il n'ait fait le choix de refuser le don d'organes - un choix qui est respectable, quelle qu'en soit la raison - et d'inscrire son nom sur un registre. Or, la loi de 1994, que nous devons réviser, n'est pas complètement inscrite dans cette logique du consentement présumé. Elle dispose que les organes appartiennent au donneur et à lui seul, puisqu'elle fait intervenir la famille, le préleveur devant informer la famille du prélèvement qu'il va opérer.
Ce système fait dévier le consentement présumé. C'est ce que l'on constate, dans la pratique, en tout cas, dans la région où je vis. En réalité, on demande l'autorisation aux familles. Nous en sommes pas du tout dans la notion de consentement présumé.
M. Bernard Charles, président. - On nous le dit sans arrêt.
M. Jean-Michel Dubernard. - Comment faire, sans être trop schématique et dictatorial, pour que le consentement présumé, qui a beaucoup de sens, soit respecté ?
Ma deuxième question concerne les greffes à partir de donneurs vivants apparentés. La tendance est à une certaine forme d'élargissement. Au cours de l'audition précédente, nous avons évoqué les risques que cela comporte pour le donneur. Que pensez-vous d'un éventuel élargissement ? Faut-il élargir ou conserver le texte actuel qui limite ces dons à la famille « génétique », si je puis dire, et au conjoint ou conjointe, en cas d'urgence ? Faut-il remplacer « urgence » par « à titre exceptionnel » ?
Mme Yvette Benayoun-Nakache. - Juste une précision. Vous parliez de la simplification de la chaîne, qui va de la prise en charge du grand blessé sur une route jusqu'au service de transplantation. Quels moyens supplémentaires voyez-vous pour que cette chaîne soit renforcée, et peut-être plus rapide dans l'acheminement ?
M. Dominique Durand. - Je me suis peut-être mal exprimé. Je ne parlais pas de rapidité mais d'efficience.
L'activité de réanimation d'un patient en état de mort cérébrale et de prélèvement est extrêmement lourde. Cela a été longtemps, et c'est encore, pour beaucoup, considéré comme un échec. Lorsqu'un patient est hospitalisé en service de neurochirurgie, après un hématome intercérébral, qu'on l'opère et que l'évolution est défavorable, pour le neurochirurgien, c'est un échec. Et il faut qu'il continue.
Le premier obstacle est qu'il n'est pas certain que tous les médecins réanimateurs et neurochirurgiens en France considèrent que le prélèvement d'organes est un acte thérapeutique aussi simple, au moins dans sa conception, qu'une intervention.
Le deuxième obstacle, ce sont les établissements publics, les hôpitaux, en particulier les hôpitaux généraux. Ils jouent un rôle très important dans le maillage car il apparaît clairement que, dans une région donnée, le nombre de prélèvements est proportionnel au nombre d'hôpitaux qui prélèvent. La région lyonnaise en est un exemple positif. Notre région de Midi-Pyrénées en est un négatif parce que l'on prélève peu dans nos hôpitaux généraux. Nous avons d'ailleurs lancé une politique active pour renverser cette tendance.
Il faut donc que l'institution, que ces hôpitaux généraux adhèrent au projet. Or, tout est fait pour qu'ils n'adhèrent pas, car c'est une activité extrêmement contraignante : un prélèvement demande douze heures, s'effectue la nuit ; cela désorganise complètement les gardes, les interventions du lendemain, etc. S'il y a de nouveaux moyens à donner, ce sont certainement des moyens de motivation pour les équipes des hôpitaux généraux. Cela me paraît essentiel.
Le troisième obstacle, à mon sens, est que nous avons segmenté les équipes de prélèvement. De notre temps - je me tourne vers Jean-Michel Dubernard - le transplanteur faisait tout. Et avec enthousiasme. Aujourd'hui, les choses sont segmentées, mais elles se sont segmentées d'une façon qui a dépassé l'objectif poursuivi.
La loi indique - et c'est une évidence - que les médecins et chirurgiens responsables des équipes de transplantation ou de prélèvement ne doivent pas participer au diagnostic de mort cérébrale. Ce doivent être des médecins indépendants. Mais dès que la mort cérébrale est déclarée, dès cet instant, il me paraîtrait extrêmement important qu'une coordination des équipes de réanimation et des médecins chirurgiens transplanteurs, lesquels peuvent venir soit comme experts soit pour renforcer les équipes au moment de la réanimation, participe à cette activité. Cela me paraît nécessaire. C'est un des éléments qui peut probablement aider.
M. Jean-Michel Dubernard. - N'est-ce pas l'Établissement français des greffes qui a créé un mur entre les équipes ? Comment faire pour que cela change ?
M. Dominique Durand. - C'est à l'échelon local que cela doit se faire. En d'autres termes, dans les hôpitaux périphériques. L'Établissement français des greffes intervient par ses coordonnateurs et l'équipe de coordination. Il faut qu'en périphérie se mette en place une organisation interne à l'hôpital qui intègre le dynamisme des transplanteurs. Cela, aucun texte ne l'interdit. C'est une piste complémentaire parce que c'est en ajoutant des éléments les uns aux autres que nous gagnerons du terrain.
Mme Yvette Roudy. - S'agissant des prélèvements sur une personne vivante, nous nous interrogions sur la notion de « proches ». Jusqu'à présent, on parlait du père, de la mère, etc. Or, dans ses propositions, le Premier ministre a parlé de « proches ». Faut-il définir ce terme à tout prix ? Faut-il d'ailleurs que ce soit un proche ? Je me dis qu'après tout, il peut se trouver une personne qui, dans un geste de grande générosité, ait envie de faire un don sans préciser pour autant à qui. Même si c'est rare, cela peut se produire. Pourquoi écarter cette possibilité ?
M. Dominique Durand. - C'est un problème fondamental.
Vous parliez du donneur vivant. Il faut savoir qu'en France, à l'heure actuelle, les possibilités offertes par la loi, qui est finalement assez restrictive, ne sont pas pleinement utilisées, parce que les médecins et les chirurgiens montrent toujours une profonde réticence à prendre un organe chez quelqu'un en bonne santé, avec les risques que cela comporte. Nous avons connu des accidents. Et encore récemment. Et en augmentant le nombre de prélèvements, nous aurons des accidents.
Il existe donc une réticence de la part des médecins mais aussi dans la société car, souvent, le don en filiation directe, de parents vers les enfants, est assez fréquent. Mais pour les adultes, c'est rarement possible ; aussi, c'est la fratrie qui est concernée. Or la fratrie, dans notre pays, n'est pas très généreuse.
Mme Yvette Benayoun-Nakache. - Peut-être n'est-elle pas très informée ?
M. Dominique Durand. - D'après notre expérience, la difficulté est de savoir si la volonté de donner est profondément libre. Lorsqu'un frère ou une s_ur est donneur, nous faisons tous les examens. Une fois que nous savons que la transplantation est possible, nous convoquons le donneur potentiel dans l'intimité d'un bureau en lui disant : « Si vraiment vous ne voulez pas, nous dirons que c'est impossible pour des raisons médicales ». Un nombre important de personnes prennent cette brèche.
M. Bernard Charles, président. - C'est un signe.
M. Dominique Durand. - Faut-il élargir ? Chacun le ressent de façon personnelle. Mon avis personnel est que je ne vois pas quelle règle éthique pourrait empêcher quelqu'un de donner à un ami.
M. Jean-Michel Dubernard. - Le principe éthique de base peut l'empêcher. C'est le commerce ! Il ne faut pas exagérer. Je pense au cas où le frère, même motivé et généreux au moment du don, a rencontré ultérieurement des problèmes et, plus tard, s'est retourné vers son frère en disant : « Je t'ai donné mon rein. Maintenant, tu m'entretiens ».
Mme Yvette Benayoun-Nakache. - C'est un cas.
M. Jean-Michel Dubernard. - Non. Ce n'est pas un cas. Quand on parle de gens généreux qui ne connaissent pas le receveur, il y a toujours une motivation psychologique. Cela peut être celui qui a écrasé un gamin et qui a fui, qui se demande comment se racheter.
Mme Yvette Roudy. - Pourquoi pas ? Puisque l'anonymat doit être respecté entre donneur et receveur...
M. Jean-Michel Dubernard. - Pas pour le donneur vivant.
Mme Yvette Roudy. - Ah, oui ?
M. Dominique Durand. - Les pays anglo-saxons, notamment l'Angleterre, ont résolu ce problème en nommant une commission d'experts qui comprend des médecins non experts, qui essaient de valider la réalité d'une volonté de don.
Mme Yvette Roudy. - C'est pragmatique.
M. Dominique Durand. - Mais c'est extrêmement compliqué.
Mme Yvette Roudy. - Oui, mais on manque de donneurs.
M. Jean-Michel Dubernard. - Et cela présente toujours un risque pour le donneur.
M. Dominique Durand. - Vous avez émis une idée que je ne partage pas. Vous avez dit : « On en manque ». Je crois profondément que le manque d'organes ne doit pas être un moteur du don familial. Le moteur doit être une véritable volonté de don qu'il faut identifier. En revanche, le manque d'organes doit nous rendre extrêmement dynamique pour prélever des patients en état de mort cérébrale.
M. Bernard Charles, président. - Vous pensez que la lourdeur pour effectuer les prélèvements dans certains centres hospitaliers généraux, où l'on sait qu'il y a pénurie de personnels, fait que l'on n'a pas réuni, sauf dans des régions que vous avez indiquées, tous les éléments qui permettraient d'avoir suffisamment d'organes provenant de personnes en état de mort cérébrale.
M. Jean-Michel Dubernard. - Ne pensez-vous pas que le développement des greffes de donneur vivant risque de diminuer les greffes de donneur mort ?
M. Dominique Durand. - Je ne le crois pas. Personne ne peut imaginer que ce soit la solution. Enormément d'efforts ont vraiment été faits pour organiser la transplantation à partir du donneur cadavérique. Certes, il n'y a pas encore de résultats suffisants, mais les efforts réalisés pour aller dans ce sens, vont, j'en suis sûr, contribuer à augmenter les dons.
M. Jean-Michel Dubernard. - Moi aussi.
Mme Catherine Génisson. - Nous sommes au c_ur du problème parce que si des réticences s'expriment, c'est parce que l'on souhaite que le prélèvement sur cadavre se fasse dans de meilleures conditions. Vous avez deux grandes difficultés. Il y a encore une réticence du corps médical. Il serait sans doute nécessaire de le mobiliser. La meilleure façon serait sans doute de le faire dès l'enseignement de la médecine. Il serait bon également que le prélèvement d'organe soit un acte thérapeutique.
Je suis d'accord avec le professeur Dubernard quand il dit que le consentement présumé c'est idéal en théorie, mais que cela ne se concrétise pas bien sur le terrain. En fait, non seulement nous informons les familles, mais nous leur demandons même leur autorisation. J'ai l'expérience d'un hôpital général. Cela se passe comme cela partout, parce qu'actuellement, le registre des refus n'est pas « grand public ». On ne peut arguer de la position qu'on va prendre en disant que nous sommes seulement obligés d'informer. Nous devons réfléchir à la façon dont nous pouvons parvenir à une meilleure solution.
M. Dominique Durand. - Nous avions étudié la question. Il est intéressant de noter que dans les pays où il y a un consentement explicite, ceux dans lesquels, par exemple, chacun indique sur sa carte d'identité s'il est donneur ou pas, le pourcentage de refus est le même.
Deuxièmement, la loi est parfaitement cohérente : nous sommes généreux, et nous sommes donneurs ; nous sommes libres, donc, l'expression de cette liberté est de pouvoir dire que l'on refuse d'être donneur. Selon le texte, le médecin doit rechercher auprès de la famille le témoignage de la volonté du défunt. Il reste deux ambiguïtés, bien entendu.
Premièrement, le terme de « famille » doit-il demeurer ? J'ai le sentiment que si on élargit le champ des donneurs vivants, il devrait y avoir une nécessité de cohérence. Il faudra bien retenir, ici aussi, « les proches ». Je ne vois pas d'autre solution.
Deuxièmement, la famille, confrontée à la pire situation qui soit, ne sait pas faire la nuance entre témoigner d'une volonté et avoir une attitude patrimoniale. Elle considère qu'elle est l'héritière de la volonté du défunt. Je ne vois pas de solution dans l'écriture d'un texte pour résoudre ce problème. Peut-être saurez-vous le faire ? À mon avis, cela passe par l'information.
M. Jean-Michel Dubernard. - Il faut que ce soit clair, que l'on dise que nous sommes dans le système du consentement présumé, que l'on informe la famille du décès - c'est l'esprit de la loi Veil - que l'on avertisse que l'on va prélever ses organes dans la mesure où le nom ne figure pas au registre des refus. Parfois, cela créera peut-être des situations difficiles qu'il faudra alors gérer.
M. Bernard Charles, président. - L'orientation serait donc d'être plus directifs.
M. Dominique Durand. - Je connais votre opinion, professeur, nous en avons déjà discuté souvent. Je ne suis pas sûr que notre société soit prête à affronter une telle démarche.
Mme Yvette Roudy. - On peut supposer qu'elle l'est.
M. Dominique Durand. - Je n'en suis pas sûr, madame.
M. Bernard Charles, président. - Il ne nous reste que très peu de temps. Quel est votre point de vue sur les prélèvements à des fins thérapeutiques ?
M. Dominique Durand. - Le fait que les textes réglementaires imposent strictement le consentement direct pour procéder à des prélèvements scientifiques a conduit à ce que l'on ne fasse plus de tels prélèvements. C'est une catastrophe. Il faut absolument modifier ces textes. Il est terrifiant de voir tout simplement que, lorsqu'un transplanté du rein a une artère rénale malade, si l'on veut prélever une autre artère pour faire une recherche, nous n'en avons pas le droit. Cela pose un problème majeur.
Ce problème s'étend. Il y a des zones de non-droit, notamment concernant les résidus opératoires. Il n'y a pas de définition réglementaire. C'est un frein considérable. En ce qui concerne l'autopsie, elle reste possible si l'on veut rechercher la cause de la mort mais, à titre scientifique, en pratique, c'est impossible.
J'ajouterai que nous essayons, vous le savez, d'aller vers un maximum de possibilités de prélèvements. Il y a une situation dans laquelle on peut prélever dans les pays étrangers. C'est en cas de mort cérébrale. Ceux que nous prélevons, ce sont les malades en état de mort cérébrale, à c_ur battant. Mais il y a aussi des situations de prélèvement à c_ur arrêté. Certes, c'est plus difficile, les résultats sont moins bons, mais c'est parfaitement possible. En France, on ne peut pas le faire.
Mme Yvette Benayoun-Nakache. - À c_ur arrêté depuis combien de temps ?
M. Dominique Durand. - Par exemple, un malade arrive en urgence dans le service de réanimation, le c_ur s'arrête. Dans ces cas, immédiatement, on l'amène au bloc opératoire. Instantanément.
Mais, pourquoi ne peut-on pas le faire, à ce moment-là ? Parce que nous n'avons pas le consentement et nous n'avons pas le temps d'attendre la famille. Les prélèvements que nous faisons à c_ur arrêté se font donc sur des malades en mort cérébrale, à c_ur battant lorsque nous avons demandé à la famille, et dont le c_ur s'arrête.
M. Bernard Charles, président. - Nous avons rencontré le même problème dans l'élaboration de la loi de 1988, à propos des expérimentations sur les malades trouvés sur la voie publique qui n'avaient aucune famille. Nous avions eu un débat qui a duré trois ou quatre séances. Les réanimateurs nous disaient qu'ils ne pouvaient pas expérimenter dans ces cas-là, qu'ils ne pourraient jamais car ils n'avaient pas le consentement libre et éclairé. Nous avons trouvé un système, qui n'est d'ailleurs pas très bon, qu'il faudra peut-être revoir.
Mme Yvette Bernayoun-Nakache. - Concernant le registre des refus, qui est le seul élément que nous ayons pour l'instant, à l'époque d'Internet ne serait-il pas possible de faire en sorte que ce registre des refus soit connu ? Il ne l'est pas. Il n'y a pas d'information. Ne serait-il pas possible de trouver une voie, par le biais des moyens de communications actuels ? Dès lors que l'information serait assurée, tous ceux qui ne seraient pas sur le registre du refus pourraient vraiment être présumés consentants.
M. Dominique Durand. - Lorsque ce registre du refus a été mis en place, une des grandes difficultés a été de savoir comment le faire connaître au public. En général, cela s'est fait de façon imparfaite, en mettant les documents dans les pharmacies. Il est temps maintenant de passer à un autre niveau. Peut-être la puissance publique doit-elle considérer qu'elle doit être le moteur pour informer de façon plus officielle ?
Vous m'aviez posé également une question sur les cellules souches. Je ne sais pas répondre. Qu'est-ce que l'embryon ? Quelle est sa définition ? Nous pourrions en parler des heures.
M. Alain Claeys, rapporteur. - Pour poser la question autrement, le législateur doit-il, aujourd'hui, se priver d'un certain nombre de pistes de recherche ?
M. Dominique Durand. - Ma réponse est non. J'ai le sentiment qu'il y a, à l'heure actuelle, des instances, des garde-fous, qui font que je n'ai jamais vu de dérapage dans notre pays. Il existe assez de garde-fous pour pouvoir être dynamiques. Je serais donc plutôt serein de ce point de vue.
M. Bernard Charles, président. - Nous vous remercions.
Audition de M. Dominique BECQUART,
président du Lions club de Mouvaux le Bosquiel
et de M. Pierre DUCHATELLE
(Extrait du procès-verbal de la séance du 10 janvier 2001)
Présidence de M. Alain Claeys, rapporteur
M. Alain Claeys, rapporteur. - Monsieur Becquart, je vous remercie d'être parmi nous. Vous êtes président du Lions club de Mouvaux le Bosquiel, qui a mené, en 1998, une campagne de sensibilisation au don d'organes, sur le thème « J'aime la vie. Je dis oui au don d'organes ».
M. Dominique Becquart. - « S'informer est essentiel ».
M. Alain Claeys, rapporteur. - Nous voudrions vous entendre sur votre conception de l'information à donner au public. Quel public faut-il toucher ? Par quel intermédiaire ? Existe-t-il des moments propices pour sensibiliser au don d'organes ? Pensez-vous que le ministère de l'Éducation nationale ait un rôle spécifique à jouer ?
Nous aimerions que vous nous présentiez le bilan de votre campagne car, avec le recul, vous pouvez certainement en tirer un certain nombre d'enseignements.
Mais, en introduction à notre débat, j'ai aussi envie de vous interroger sur la reconnaissance témoignée aux donneurs. Certains préconisent l'organisation d'une journée annuelle de réflexion sur le thème du don d'organes. Qu'en pensez-vous ? Comment pourrait-elle être organisée ?
Comment le Lions club a-t-il eu l'idée de cette campagne ?
M. Dominique Becquart. - Nous tenons d'abord à vous remercier de nous accueillir. Cela nous honore. C'est un bon point final à cette action.
Je dirai quelques mots sur le Lions club. Vous nous connaissez au moins de nom. Nous sommes un club service, c'est-à-dire une réunion de femmes et d'hommes dont le but est d'_uvrer à des actions sociales et humanitaires. Ce mouvement, créé en 1905 par un avocat américain, compte aujourd'hui 1 200 clubs en France, plus d'un million d'adhérents dans le monde, dont 34 000 dans notre pays.
Nous nous réunissons deux fois par mois en essayant, bien évidemment, d'entretenir des liens d'amitié mais aussi d'honorer notre vocation qui est « We Serve »
- nous servons. Lions signifie d'ailleurs Liberty, Intelligence, Our Nation's Safety - liberté et compréhension sont la sauvegarde de notre nation.
Nous menons des actions qui peuvent être locales, régionales et nationales. Ce sont des actions que nous choisissons.
Notre club de Mouvaux, une petite ville située à proximité de Lille, fait partie du district du Lions club du nord de la France. Notre club a toujours été sensibilisé au thème du don d'organes pour des questions qui nous appartiennent et que je n'évoquerai pas ici pour ne pas alourdir mon propos.
Nous nous y sommes donc toujours intéressés, ce qui nous a amené, en 1993-1994, à demander à nos amis du club du Nord de porter cette campagne d'information sur le plan régional.
Nous avons alors été choisis pour la conduire et, pendant deux ans, nous nous sommes formés grâce, essentiellement, à la collaboration d'un médecin : Marie-Dominique Besse qui était à l'époque la coordinatrice de France Transplants pour le Nord. Nous comptons bien entendu, au sein de nos cercles Lions, des médecins qui ont également pu nous délivrer une information. Pendant ces deux ans, nous nous sommes « rodés », si je puis dire, à diffuser ce message d'information. Je dirai ensuite auprès de qui nous l'avons diffusé.
À la fin de ce délai, l'affaire nous plaisait. Nous trouvions que c'était une très bonne action Lions. Nous l'avons portée au suffrage d'un congrès national, à Tours, en 1995, devant nos 34 000 membres des 1 200 clubs, et nous avons eu l'honneur d'être élus.
Fin 1995, nous avons été investis de la mission de diffuser ce message : « J'aime la vie, je dis oui au don d'organe. S'informer est essentiel ».
Notre première action a immédiatement été de nous rapprocher de l'Établissement français des greffes, dirigé par Didier Houssin, qui venait d'être créé. Puisque cet établissement est chargé de l'information sur le don d'organes, il était normal que nous nous adressions à lui, ne serait-ce que pour valider le document dont nous disposions.
Nous avons reçu un accueil très favorable de la part de Didier Houssin, qui fut très content de pouvoir compter sur le « maillage » - le mot est de lui - des Lions clubs. Il est vrai que ceux-ci sont disséminés dans toute la France. Nous possédons ainsi une influence non négligeable. Généralement, le Lions club recrute dans une couche sociale de cadres moyens-supérieurs, des personnes qui ont une certaine influence. Didier Houssin nous a donc encouragés et beaucoup aidés. Nous lui avons présenté l'ensemble de nos documents.
Et nous avons démarré, en ayant quelques idées bien claires à l'esprit.
La première était que ce message sur le don d'organes était un message qui ne devait pas être diffusé par des médecins. Mon ami Pierre Duchatelle, ici présent, est médecin, mais il se range tout à fait à mon avis. Nous pensions que ce message devait être diffusé par nous, en dehors du corps médical.
À l'époque, en 1995-1996, le don d'organes ne se portait pas tellement bien. L'histoire du sang contaminé et beaucoup d'autres de ce genre amenaient les gens à penser, à tort, que l'image du corps médical était atteinte par tous ces problèmes. En présentant nous-mêmes ce message, et sans relation avec le corps médical, nous pouvions avoir plus d'impact. Nous avons toujours tenu ce discours et nous avons toujours pensé qu'il nous appartenait, à nous, « civils » - par « civils », j'entends que nous n'appartenions pas au corps médical - de le délivrer à des jeunes.
N'étant qu'une trentaine de membres dans notre club, notre première tâche a été d'informer les 34 000 Lions, par l'intermédiaire desquels nous espérions toucher les 60 millions de Français. C'était une tâche assez importante.
En conséquence, il nous fallait, premièrement, former nos membres. Nous avons visité les clubs, nous les avons informés, nous leur avons donné des documents et nous avons organisé des conférences.
Puisque nous étions trente-trois dans le club, nous nous sommes divisés en binômes, chacun d'entre nous ayant la responsabilité d'un secteur géographique. Nous avons « inondé » tous ces secteurs de visites, de conférences que nous faisions nous-mêmes qui, souvent, étions des « civils ».
Ensuite, nous leur avons indiqué que maintenant qu'ils étaient formés, leur tâche était d'informer à leur tour. D'informer essentiellement les jeunes et, si possible, en cercle restreint. Nous leur avons conseillé de ne pas faire de grandes conférences de cinq cents personnes, car n'y viendraient que les personnes convaincues de la nécessité du don d'organes, mais de s'adresser plutôt à de petits comités, à l'occasion de petites manifestations Lions (tournois de bridge, de golf, bourses des collectionneurs) ou bien dans des établissements scolaires, à des classes de terminale.
Il s'agissait de parler du don d'organes devant une soixantaine de personnes, en respectant les valeurs du lionisme, auxquelles nous tenons, notamment en s'appuyant sur un message de tolérance. Il ne s'agissait pas de demander aux auditeurs de faire le choix de donner leurs organes mais d'aller les voir en leur expliquant qu'un problème de société se pose actuellement et qu'il existe une méthode pour aider des gens, que 5 à 6 000 personnes attendent cette thérapie et que, malheureusement, le public est mal informé. Aussi venions-nous les voir pour les informer de l'existence de cette possibilité. Chacun est libre de donner ou de ne pas donner ses organes, mais au moins nous les invitions à s'informer afin de pouvoir prendre une décision en toute connaissance de cause et adhérer ou non au projet.
Nous tenons beaucoup à cette notion de tolérance. C'est, pour nous, un aspect essentiel. Du reste, toute notre documentation est basée sur elle et nous rejoignons en ce sens l'esprit de Didier Houssin et de l'Établissement français des greffes.
Vous m'avez posé la question de l'évaluation. Vaste sujet !
Nous nous honorons très modestement d'avoir un peu contribué, sans pouvoir le chiffrer, à l'augmentation des dons d'organes constatée en 1998. Cette année-là marque une petite remontée. Nous voulons bien croire que nous y sommes pour quelque chose.
L'évaluer est malheureusement impossible. Comment pourrions-nous le faire ? Les Lions clubs ont distribué 20 000 bandes dessinées, 600 000 tracts, 10 000 affiches, 500 cassettes. Puis-je dire que, grâce à cela, il y a eu une progression ? Il est certain que nous étions « dans le train » et que nous avons diffusé un message qui, je l'ajoute, continue encore.
Cette action nationale est maintenant terminée puisque nous votons ce type d'action pour deux ans, mais celle-ci a tellement plu au club, parce qu'elle rejoignait, je le répète, les valeurs du lionisme - solidarité, tolérance, humanisme - que beaucoup de clubs continuent encore à nous interroger.
Nous sommes responsables, l'un et l'autre, d'une cellule de veille que nous avons mise en place au sein du club. Nous maintenons notre relation avec l'Établissement français des greffes. Nous continuons à former les clubs qui le désirent.
Que puis-je donner comme recommandations ?
Très modestement, nous pensons qu'il serait intéressant qu'il y ait davantage d'informations dans les lycées, en direction des seize-dix-huit ans, des classes de terminale. Pas dans les collèges. Cette information pourrait se faire dans le cadre d'une instruction civique, en s'appuyant sur les thèmes de solidarité et de fraternité, ou dans celui des sciences de la vie, en insistant très fortement sur la notion de mort encéphalique, qui est un peu technique mais qui n'est pas évidente pour qui n'est pas médecin.
Une deuxième action qui me paraît intéressante serait l'édition d'un timbre. En 1996, j'avais écrit à l'organisme qui s'en occupe et j'avais reçu une réponse, très correcte, m'indiquant que d'autres actions passeraient avant notre suggestion. On voit La Poste éditer aujourd'hui des timbres « Bon anniversaire ! » ou « C'est un garçon » ou « C'est une fille ». Ce ne serait pas mal si on pouvait diffuser un message équivalent pour le don.
Mme Catherine Génisson. - Participez-vous au Téléthon ?
M. Dominique Becquart. - Le Téléthon est une action du Lions club, effectivement. Du moins, lorsque vous téléphonez au Téléthon, vous vous entretenez avec un membre du Lions club ou l'un de ses proches.
Mme Catherine Génisson. - Comment présentez-vous le registre des refus alors que vous faites une campagne positive ?
M. Dominique Becquart. - Une campagne est forcément positive. Quand on parle information, cela le sous-entend.
Mme Catherine Génisson. - Le thème de votre campagne était positif.
M. Dominique Becquart. - Oui : « J'aime la vie, je dis oui au don d'organes, s'informer est essentiel ».
Mme Catherine Génisson. - S'informer est essentiel. Néanmoins, cela doit déboucher sur quelque chose de très concret, c'est-à-dire que la personne, qui a totale liberté de choix, doit connaître la possibilité d'exprimer son refus du prélèvement, donc, d'avoir accès au registre des refus. Votre information va-t-elle jusqu'à la présentation du registre des refus ?
M. Pierre Duchatelle. - Je n'ai pas les dates en mémoire mais notre campagne pour informer sur le don d'organes était, me semble-t-il, juste antérieure à la création du registre des refus. Nous l'avons donc présenté comme une éventualité qui allait surgir sans que nous ayons à en discuter car il n'était pas encore officialisé par l'Établissement français des greffes. Actuellement, quand nous en parlons, nous indiquons qu'il existe un registre des refus à Marseille.
Aujourd'hui, nous donnons donc l'information sur ce registre mais nous n'avions pas eu à le faire lors de notre campagne car ce registre n'était pas encore officiel.
M. Dominique Becquart. - Le message que nous transmettons est exactement celui-là. J'ai encore fait récemment une petite conférence dans un établissement scolaire. Nous concluons en leur conseillant, puisque ce sont des jeunes, d'en parler d'abord en famille et, ensuite, s'ils désirent donner leurs organes dans le cas où, malheureusement, il leur arrivait quelque chose, de l'indiquer à leurs parents et, s'ils le souhaitent, de porter une carte attestant de cette volonté.
Dans le cas contraire, nous leur conseillons pareillement de l'indiquer, de porter un papier sur eux l'indiquant et, s'ils le souhaitent, de s'inscrire au registre des refus à Marseille. C'est très clair.
M. Jean-Michel Dubernard. - À ce sujet, je voulais poser la même question que Catherine Génisson, en soulignant toutefois que toutes ces actions positives ne doivent pas créer de confusion. Elles doivent informer sur le registre des refus.
Les cartes de donneurs, par exemple, posent un énorme problème parce que les gens croient que si on possède la carte, on peut être prélevé et que si on ne l'a pas, on ne peut pas être prélevé. Les familles pensent que nous sommes sous le régime du consentement explicite.
J'ai eu à ce sujet de nombreuses discussions avec les membres de l'ADOT. Les cartes de donneur sèment une confusion. Cependant, si vous informez sur le registre des refus, c'est parfait.
Mme Catherine Génisson. - Merci de cette clarification.
M. Dominique Becquart. - Il est vrai que cette information doit être extrêmement claire. « Vous dites oui, vous agissez de cette manière, vous dites non et vous devez faire ainsi ». Comme cela, les gens sont pleinement responsables.
Mme Catherine Génisson. - En réalité, il est difficile de faire comprendre le concept de consentement présumé. Les gens sont, pour la plupart d'entre eux, dans une logique de consentement explicite. Or nous sommes dans un régime de consentement présumé et cette notion est difficile à expliquer au grand public. Elle passe par une connaissance, par tous, de la mise en place du registre du refus mais c'est une campagne qui est plus difficile à réaliser que ce ne serait le cas pour la logique de consentement explicite.
M. Dominique Becquart. - À notre humble avis, c'est pour cela qu'il est plus facile de transmettre ce message - et donc d'avoir un débat - au sein d'une salle d'une cinquantaine d'adolescents, plutôt que de le faire sur un plateau de télévision. Certaines émissions sur le sujet n'ont pas été très brillantes. Il faut être vigilant à cet égard.
Nous pensons que les lycéens sont la cible la plus appropriée pour une telle campagne. À dix-sept ans, ils sont ouverts et, en règle générale, cela se passe admirablement bien.
Mme Yvette Benayoun-Nakache. - Je souhaitais poser une question concernant l'information que vous donnez. Nous essayons d'avoir des données de terrain très précises. Vous avez parlé de l'information des lycéens. Je pense que nous pourrions même viser de plus jeunes gens car ils sont très réceptifs à tout ce qui les entoure.
Vous avez évoqué deux disciplines au titre desquelles l'Éducation nationale pourrait donner cette information : l'instruction civique et les sciences de la vie. Je pencherais plutôt pour cet aspect scientifique car l'instruction civique enseigne déjà sur les élus, la citoyenneté, etc. Qu'en pensez-vous ?
M. Dominique Becquart. - Personnellement, je préfère agir dans le cadre de la citoyenneté, sur les notions de fraternité plutôt que sur des notions scientifiques où on va faire appel à l'immunologie et à la compatibilité tissulaire, qui sont relativement secondaires.
Dans le message du don d'organes, ce qui est beau, c'est « Liberté, Égalité, Fraternité ». Ce sont ces notions qui sont belles ainsi que celle de la tolérance. Devant un grand problème, on est libre de choisir, on est libre de répondre. Il y a une notion d'égalité, car anonymat est synonyme d'égalité, et de fraternité.
C'est un très beau message.
Mme Catherine Génisson. - Avez-vous eu, durant votre campagne, des contacts avec le ministère de l'Éducation nationale ?
M. Pierre Duchatelle. - Avec le rectorat de Lille, mais pas sur le plan national.
Mme Yvette Benayoun-Nakache. - Pour prolonger cette réflexion, je serais aussi assez opposée au fait de divulguer un tel message par le bais des grands médias. Je préfère ces initiatives de citoyenneté dans les collèges, les lycées, voire même dans les écoles primaires, en classe de CM1-CM2, où les enfants sont aussi très réceptifs, plutôt que d'organiser une grande campagne nationale par le biais des médias, qui me semblerait compromettre le but que l'on s'est fixé de « faire connaître ». Qu'en pensez-vous ?
M. Dominique Becquart. - Attention, les enfants de CM1 sont mineurs.
M. Alain Claeys, rapporteur. - La question est de savoir s'il faut des campagnes de proximité ou destinées au grand public ?
M. Dominique Becquart. - Il faut des campagnes de proximité.
M. Jean-Michel Dubernard. - À mon avis, les deux sont nécessaires. Les campagnes de proximité sont indispensables, les campagnes faites par les médias, si elles sont bien pensées et que le choix du média et des journalistes qui s'occupent de l'émission est judicieux, sont aussi une bonne chose. Mais il ne faut jamais mettre de médecins. Il n'y a rien de pire. (Sourires.) C'est vrai ! Surtout pas de médecins ! En revanche, la présence de patients est très bénéfique. Rien n'est aussi fort que le témoignage d'un enfant qui a reçu une greffe. Là, cela prend beaucoup de sens.
M. Dominique Becquart. - Lors des petites conférences que nous organisons, si nous avons la chance de pouvoir apporter le témoignage de quelqu'un qui a reçu, c'est fantastique. Ou aussi le témoignage de la famille qui a accepté le prélèvement.
M. Jean-Michel Dubernard. - La famille qui a accepté le prélèvement, c'est assez ambigu.
M. Dominique Becquart. - Justement, j'en profite pour revenir sur le fameux article L. 671-7 du code de la santé publique : « si le médecin n'a pas directement connaissance de la volonté du défunt, il doit s'efforcer d'obtenir le témoignage de sa famille ».
Je ne suis pas juriste et je m'interroge sur ce qu'est une « famille » du point de vue du droit. Où s'arrête-t-elle et où commence-t-elle ? À deux heures, dans le couloir d'un hôpital, ce n'est pas forcément évident.
Le médecin doit « s'efforcer », s'il n'a pas « directement » connaissance, qu'est-ce que cela veut dire ? Le texte est totalement ambigu.
Mme Catherine Génisson. - Le texte est totalement ambigu et pose beaucoup de problèmes pour pratiquer une greffe. Actuellement, nous sommes dans la situation où, non seulement, nous demandons son témoignage à la famille, mais où, de fait, nous sommes quasiment obligés de demander son accord à la famille.
Je voulais réagir au fait que vous demandiez à des familles qui ont accepté le prélèvement de témoigner.
M. Dominique Becquart. - Nous ne leur demandons pas, mais certaines viennent spontanément.
Mme Catherine Génisson. - Faut-il favoriser cette situation ou même la laisser se produire spontanément dans la mesure où, me semble-t-il, c'est un acte très délicat ?
M. Dominique Becquart. - Il faut laisser de tels témoignages émerger spontanément. Nous ne pouvons faire pression.
Mme Catherine Génisson. - Nous sommes d'accord. Il ne faut pas entacher ce message de voyeurisme ni laisser s'exprimer des témoignages qui sont trop douloureux à obtenir. Il faut rester pudique en la matière.
M. Dominique Becquart. - Tout à fait.
Pour revenir sur les recommandations, l'Établissement français des greffes multiplie les centres de prélèvement. Nous ne pouvons qu'être d'accord car un meilleur recensement des morts encéphaliques ne peut qu'améliorer le système.
Nous proposons également que la loi puisse, si elle le peut, davantage reconnaître la famille de ceux qui donnent. Par l'intermédiaire d'un témoignage de reconnaissance très difficile à concrétiser. Nous avons nous-mêmes longtemps cherché et nous avons pensé à la plantation d'un arbre de la reconnaissance. Nous en avons planté un dans l'enceinte du CHU de Lille.
Cette initiative a été reprise ailleurs puisque les Lions clubs en ont planté sept dans sept autres villes de France, en accord, bien entendu, avec l'Établissement français des greffes. C'est une manifestation hautement symbolique, merveilleuse, qu'il faudrait développer.
Planter un ginkgo biloba, cet arbre qui a résisté à la bombe atomique, est un symbole fort.
Mme Yvette Benayoun-Nakache. - L'information de nos concitoyennes et nos concitoyens est vraiment ma préoccupation. Toutes les personnes que nous auditionnons et nous-mêmes sommes, pour la plupart, initiés et sensibilisés mais, de tous les textes de loi que nous aurons votés, celui-ce me paraît particulièrement important. Nous ne pouvons pas passer à côté de l'information, à côté de ce débat citoyen que nous appelons de nos v_ux car nous allons fixer certaines normes éthiques qui devront être respectées et qui engagent, je n'ai pas peur de le dire, l'humanité, notamment en France.
De grandes associations comme la vôtre aident à cette information. Tout à l'heure, M. Dubernard disait qu'on pouvait obtenir que de nombreuses personnes témoignent. Pas seulement les médecins, mais aussi les médecins, sans oublier les chercheurs, les juristes, les assureurs... Il y a vraiment tout un panel de personnes, nous l'avons bien vu dans nos auditions.
D'après vous, sans tomber dans le grand monde associatif, quelles associations ou organisations comme la vôtre seraient susceptibles de distiller la bonne information, sans que cela devienne un reality show où l'on dit tout et n'importe quoi ? Comment parvenir à toucher réellement toute la population de façon informative et citoyenne ?
Mon souci est celui-là. Comment faire en sorte que cette information ne soit pas déformée...
M. Dominique Becquart. - Se fasse de façon continue...
Mme Yvette Benayoun-Nakache. - ...et que les personnes n'aient pas que le mot clonage en tête ? En effet, en ce moment, pour les gens, la bioéthique est synonyme de clonage.
Comment apporter cette information ? Vous qui êtes sur le terrain, et surtout, sur la France entière, comment ressentez-vous ce souci d'information et la façon de la délivrer ?
M. Dominique Becquart. - Vous nous demandez un peu de juger les associations qui s'en occupent ?
Mme Yvette Benayoun-Nakache. - Non, pas de juger...
M. Dominique Becquart. - Mais de donner un avis. Il n'est pas facile de répondre. Sans vouloir nous prendre au sérieux, je pense, en toute objectivité, que nous avons fourni un bon travail.
Il paraît difficile de ne pas évoquer l'ADOT. J'en étais administrateur et j'ai démissionné il y a trois ans. C'est gênant.
M. Pierre Duchatelle. - L'ADOT avait sans doute une information un peu trop positive. Elle était plutôt à la recherche d'adhérents à l'association que convaincue de la nécessité de diffuser le message. Les personnes concernées par un problème de don d'organes devaient faire partie de l'association pour avoir accès à l'information.
Pour notre part, nous sommes libérés de cet aspect car les associations comme le Lions club, ou le Rotary, peu importe, ne demandent pas d'adhésion aux gens pour divulguer l'information. Elle l'est de manière totalement désintéressée.
À propos de la divulgation de l'information, je reviens, en tant que médecin, sur un point important. J'ai tout de suite dit que le don d'organes n'était pas un problème de médecins, qu'il fallait échapper à la médecine et diffuser un message de générosité et d'humanisme, qu'il ne fallait surtout pas se laisser prendre au piège du message sur la transplantation car, souvent, les gens dévient rapidement sur cette activité et demandent quels en sont les résultats.
Ce message comporte deux aspects sur lesquels il est très difficile d'informer. Ce sont les deux points qui achoppent, moins avec les jeunes qu'avec les gens qui font partie de la société en général.
La première difficulté touche au problème de la mort encéphalique parce que le doute reste toujours présent à l'esprit : « Est-elle bien repérée ? Êtes-vous sûr que les gens que vous allez prélever sont bien morts ? »
La deuxième difficulté est apparue lorsque nous sommes passés au prélèvement de plusieurs organes parce qu'entendre dire que l'on prélève sur un sujet un c_ur, un foie, deux reins, choque beaucoup. Quand on disait que l'on prélevait un rein, c'était mieux ressenti et plus facilement accepté.
Je voudrais également insister sur ce qui se passe en aval de la greffe. En dehors du problème de l'information, il me semble que l'on pourrait progresser en augmentant, au sein des hôpitaux, les structures d'accueil de façon à ce que les familles qui acceptent le prélèvement ne soient pas « laissées pour compte » - c'est plus rare aujourd'hui parce que des infirmières ont été formées à cela -, que ces familles aient la possibilité ou de rencontrer facilement l'infirmière de l'Établissement français des greffes ou une personne qui puisse les accompagner dans leur démarche, éventuellement un psychologue.
Actuellement, en cas d'ennui grave, de catastrophe naturelle ou d'accident important, des psychologues prennent en charge les familles. Nous devrions pouvoir offrir, pareillement, à la famille, un appui psychologique car elle traverse une période difficile, la perte d'un proche. On va accepter un prélèvement et, le lendemain, on se retrouve seul.
Dans cette optique, le dernier point qui me paraît intéressant serait, toujours dans l'anonymat le plus complet, de donner aux familles une information sur le devenir des organes, ce à quoi ils ont servi. La famille se dit qu'elle a donné des organes mais elle ne sait pas ce qu'il en est advenu. Ils ne savent pas s'ils ont été vraiment greffés, si la greffe s'est bien passée pour celui qui l'avait reçue. Je pense qu'une information pourrait être donnée, naturellement dans l'anonymat le plus strict.
Mme Catherine Génisson. - Je trouve que cette proposition est très séduisante mais, en même temps, elle me paraît dangereuse.
M. Dominique Becquart. - Il ne faut pas d'énumération.
Mme Catherine Génisson. - À partir du moment où on commence à donner un premier renseignement, on sait que la famille va se raccrocher au fait d'avoir des nouvelles et, ensuite, elle va vouloir, pas forcément connaître l'identité du receveur, mais connaître son devenir. Cela risque peut-être de l'empêcher de faire son deuil. Je pense que cette proposition peut être intéressante pour certaines familles mais pas pour toutes.
M. Pierre Duchatelle. - Il faut la laisser à la demande de la famille qui a accepté le prélèvement...
Mme Catherine Génisson. - Elles le demandent toutes.
M. Pierre Duchatelle. - Toutes ne le demandent pas mais elles essaient de savoir si les prélèvements ont été utiles et les organes distribués. On le comprend.
M. Alain Claeys, rapporteur. - Je vous remercie de nous avoir consacré un peu de votre temps.
Audition de M. Philippe KOURILSKY,
professeur au Collège de France,
et Mme Geneviève VINEY,
professeur à l'Université de Paris I
(Extrait du procès-verbal de la séance du 17 janvier 2001)
Présidence de M. Alain Claeys, rapporteur.
M. Alain Claeys, rapporteur. - Nous avons décidé d'aborder le principe de précaution, même si celui-ci n'est pas un élément spécifique à la révision des lois bioéthiques.
Pour ouvrir le débat et votre intervention préalable, nous pourrions partir du travail que vous avait confié, en mars 1999, M. le Premier ministre qui avait souhaité une réflexion approfondie sur le principe de précaution.
Vous lui avez remis ce rapport en novembre 1999. La conclusion de votre étude était sous forme de mise en garde : « Le législateur, l'autorité réglementaire et le juge peuvent faire du principe de précaution le meilleur ou le pire des usages. Le meilleur, s'ils adoptent des mesures qui améliorent véritablement la sécurité des citoyens, tout en évitant l'abstention systématique devant les risques ; le pire s'ils en font un carcan dépourvu de souplesse et un frein à l'innovation et au progrès. » C'est par cette voie étroite que nous abordons, nous aussi, la révision des lois bioéthiques. Nous qui ne sommes pas habitués à ce concept, même si nous l'utilisons tous, souhaiterions que vous nous apportiez des précisions sur sa définition et sa portée.
Selon vous, ce principe a-t-il une portée morale, politique ou juridique ? Quelle est son origine et quelle définition peut-on, aujourd'hui, en donner ? Quelle distinction peut-on faire entre la prudence, la prévention et la précaution ? Si ce principe de précaution a une portée normative, quelle sanction permet, à ce jour, d'en assurer le respect ?
À partir de cette définition et de la portée de ce principe, je souhaiterais revenir à la situation actuelle. La multiplication des poursuites pénales à l'encontre des responsables publics ne pourrait-elle pas conduire à une interprétation très large du principe de précaution, au risque d'induire une inhibition des acteurs publics face aux choix auxquels ils sont confrontés ? C'est un sujet qui nous touche tous en tant que responsables politiques, mais plus largement tous les responsables publics.
L'autre conséquence de ce principe est la procédure qu'il impose de suivre, c'est-à-dire le caractère public du débat. C'est également un élément important. La transparence exigée dans les pratiques scientifiques et les décisions politiques impose de nouveaux modes d'organisation du débat public pour contrôler l'activité scientifique. Sur ce thème, vos remarques nous intéressent également.
Ma dernière question concerne la mise en _uvre de ce principe : quelles modifications ou adaptations jugez-vous nécessaires d'apporter à notre système institutionnel et administratif ? Je pense notamment aux agences administratives indépendantes, type d'agence envisagé dans la révision des lois bioéthiques en ce qui concerne l'assistance médicale à la procréation ou, plus largement, la recherche sur l'embryon.
Mme Geneviève Viney. - Votre première question sur le caractère moral, politique ou juridique de ce principe est souvent posée. À mon avis, c'est d'abord un principe de programmation pour l'action du législateur et de l'autorité réglementaire.
C'est également un principe qui a acquis une portée juridique, car il est retenu par le juge, notamment le juge administratif, pour fonder certaines décisions et apprécier, dans certains cas, la responsabilité des pouvoirs publics. C'est ainsi que le tribunal administratif de Marseille a retenu, au mois de mai 2000, la responsabilité de l'État pour ne pas avoir pris les mesures nécessaires, au moment voulu, à propos de l'amiante. Le Conseil d'État est saisi aujourd'hui de ce problème, mais c'est l'exemple d'une décision qui a reposé sur un raisonnement s'appuyant explicitement sur le principe de précaution.
Le Conseil d'État s'est, d'ailleurs, également appuyé sur ce principe, le 22 novembre 2000, lorsqu'il a eu à statuer définitivement sur la question de l'autorisation d'inscription des variétés de maïs transgénique. Ce faisant, il a admis que le principe de précaution avait une incidence, bien qu'en l'espèce, il ait reconnu que ce principe avait été respecté et qu'il n'y avait pas lieu de reconnaître d'erreur manifeste de l'administration.
M. Alain Claeys, rapporteur. - C'est donc un principe juridique.
Mme Geneviève Viney. - C'est un principe juridique. En même temps, c'est un principe qui inspire l'action du législateur et qui est, par conséquent, un principe politique et moral. Il ne me semble pas qu'il y ait d'opposition entre l'un ou l'autre de ces caractères.
M. Alain Claeys, rapporteur. - Si l'on devait donner une définition de ce principe, quelle serait-elle ?
Mme Geneviève Viney. - Nous avons proposé une définition. Il y en existe une dans la loi Barnier, mais il est difficile de le faire en deux mots...
M. Alain Claeys, rapporteur. - Quand est-il apparu pour la première fois ?
Mme Geneviève Viney. - Bien que cela soit discuté, c'est au cours des années 1970, à la Conférence de la Mer du Nord, qu'il a été, me semble-t-il, évoqué l'une des premières fois. L'origine serait allemande, mais c'est aussi un point discuté.
La définition est délicate. Celle de la loi Barnier est assez controversée, car elle est propre à l'environnement, fait état de risques graves et irréversibles - cette dernière notion étant également fortement contestée - et elle demande que les mesures soient effectives et proportionnées. C'est une définition contestée par certains car elle est considérée comme trop étroite. En outre, elle n'est probablement pas adaptée aux domaines autres que l'environnement. Cette notion d'irréversibilité notamment n'est pas très probante dans d'autres domaines - celui de la santé, par exemple.
M. Philippe Kourilsky. - Une définition importante du principe de précaution est celle fixée par son contraire, si j'ose dire. L'un des problèmes est bien de nature sémantique et touche véritablement à la compréhension de ce qu'est la précaution, sachant que le contresens de la précaution est, bien sûr, de ne rien faire en attendant que.... Or, le principe de précaution, de notre point de vue, est tout le contraire de l'immobilisme. C'est, à l'inverse, une obligation d'agir au mieux en situation d'incertitude et, donc, d'utiliser des procédures particulières puisque l'incertitude impose d'employer des méthodes et des modes d'action différents.
L'un de ces modes d'action est l'obligation de recherche : puisque l'on ne sait pas, il faut chercher à savoir. Doivent donc être régulièrement inscrits dans des procédures qui s'appuient sur le principe de précaution, les moyens permettant de lever l'incertitude au plus vite, ce qui est de l'intérêt public, sachant que la proportionnalité des mesures que l'on prend est évidemment associée à un principe de réversibilité qui n'est pas toujours facile à mettre en _uvre ni toujours bien compris.
L'une des bases sur laquelle nous avons travaillé est, au fond, de penser que le principe de précaution répond à une attente sociologique, celle de la population, que l'on peut analyser sous différents angles, et correspond également à une démarche intellectuelle qui se traduit par une sorte de glissement épistémologique. Autrement dit, ce glissement serait qu'en situation d'incertitude, on remplace la certitude ou la connaissance de l'exacte vérité par une certitude quant aux procédures suivies. En d'autres termes, on passe d'une incertitude sur le contenu d'une situation à une certitude sur les procédures que l'on emploie, qui garantissent que l'on fait au mieux en fonction des connaissances de l'instant.
M. Pierre Hellier. - C'est une obligation de moyens et pas de résultats.
M. Philippe Kourilsky. - Essentiellement, me semble-t-il.
Mme Geneviève Viney. - On ne peut pas systématiser.
M. Philippe Kourilsky. - En effet. Je pense avoir dit l'essentiel. Effectivement, un contresens sur les fondements peut conduire à des conséquences relativement désastreuses.
J'ajouterai, pour répondre à ce que vous demandiez sur la portée politique, morale ou juridique de ce principe, que la transcription en termes d'attitude fait que le principe de précaution est l'un des plus grands exercices du Politique. Le risque, en effet, pour le Politique est de gérer l'image du risque plutôt que le risque lui-même, sachant que l'image du risque se propage dans les populations, parce que cela répond à des attentes, que le système médiatique le permet, le favorise ou le développe, je ne sais, et qu'il est absolument essentiel, de ce point de vue, d'avoir une transparence compréhensible.
Cela m'amène à répondre à une autre de vos questions. Dans l'attitude résolument procédurale qui nous paraît importante, une procédure, qui s'appuie sur ce que l'on appelle des démarches qualité ou d'assurance qualité, est par essence transparente et traçable. En d'autres termes, le simple fait d'avoir une bonne procédure offre la garantie de transparence et de traçabilité.
Vous me direz que cette transparence, il faut la communiquer. Mais, si l'on communique une chose qui n'est pas claire et compréhensible, on n'est pas transparent. La transparence est bien le produit de deux attitudes : la première est d'avoir un dispositif lisible et compréhensible ; la seconde est le fait de le communiquer largement et au grand public.
M. Alain Claeys, rapporteur. - Le dernier point que j'avais évoqué concernait le système institutionnel et les agences. Prenons un cas d'école qui s'est posé récemment à nous, politiques, à propos des tests systématiques. Quel doit être, dans le dispositif de décision d'un responsable politique quel qu'il soit, le rôle des agences ou des organismes indépendants que l'on met en place ? Est-ce un préalable à la décision ou cela vient-il éclairer une décision ? Comment situez-vous, dans ce dispositif institutionnel, ces structures qui se multiplient en marge de l'administration ou à côté de l'administration ?
Mme Geneviève Viney. - Il me semble très important qu'existent ainsi des organismes qui soient en permanence chargés de préparer les décisions ou de réunir les informations qui permettront une meilleure prise de décision. Le système des agences est, dans l'ensemble, à recommander, mais sous certaines conditions. Il faut notamment, me semble-t-il, préserver l'indépendance de l'agence vis-à-vis du pouvoir politique ainsi que la liberté de la décision qui relève du Politique. Le statut de ces agences n'est pas facile à déterminer. Celui qui convient le mieux est sans doute celui d'une autorité administrative indépendante.
M. Alain Claeys, rapporteur. - Cela fait-il partie de la gestion de l'image du risque ?
Mme Geneviève Viney. - Je le pense.
M. Philippe Kourilsky. - L'exemple du nucléaire a suffisamment montré à quel point il était important d'avoir, au moins, des systèmes qui font les mesures, qui soient indépendants des pouvoirs. À l'égard du nucléaire, le soupçon public s'est jeté, à la fois, sur les institutions qui avaient des intérêts économiques, mais également sur le pouvoir politique, tous deux soupçonnés d'avoir des intérêts dans l'opération. Avoir des systèmes de mesures indépendants et, de ce fait, vérifiables et crédibles, est extrêmement important. Mutatis mutandi, je dirais que, dans le domaine de la santé, la création de ces agences possédant un certain degré d'autonomie est un progrès.
M. Claude Evin. - Dans le prolongement des interrogations de notre rapporteur, je crois que l'on touche là à une réelle difficulté, et, sans esprit de polémique entre nous, majorité et opposition, je voudrais prolonger la réflexion à ce sujet en prenant un exemple assez récent qui nous oblige à relativiser cette question d'autonomie.
Je suis convaincu pour différentes raisons, y compris d'expérience personnelle, de la nécessité de disposer d'outils techniques indépendants. Nous avons vu très précisément, avec l'interdiction des farines animales, combien il pouvait y avoir de décalage entre cette autonomie juridique de la technique et la décision politique, puisque nous nous sommes trouvés dans une situation d'entendre certains responsables politiques répondre, dans un premier temps, à la question qui leur était posée d'interdire ou non les farines animales, qu'ils avaient saisi les experts - en l'occurrence, l'Agence de sécurité sanitaire des aliments - et qu'ils attendaient la réponse. Puis, un emballement du calendrier politique, pour diverses raisons, a fait que le Gouvernement a dû prendre une décision sans attendre, indépendamment du travail technique des agences.
Mon propos n'est pas une question en soi, mais un élément de réflexion montrant que si, juridiquement, l'autonomie et la déconnexion des responsabilités sont tout à fait saines et fondées, il peut parfois arriver que ce mode d'organisation ne cadre pas avec le processus de la décision politique. Je tenais à partager cette interrogation avec vous. Peut-être avez-vous une réaction à ce sujet ?
M. Philippe Kourilsky. - J'ai, en effet, une réaction rapide : le principe de précaution étant destiné à éviter les crises, pour l'instant, on l'invoque essentiellement en situation de crise - cela signifie incidemment que tout ne va pas si mal dans le pire des mondes, puisque l'on ne parle absolument pas de tout ce qui s'est bien passé et de toutes les crises qui ont été évitées.
Il faut bien voir, et vous avez raison de le souligner, que les périodes de crises sont liées à des problèmes de calendrier. Il est intéressant, y compris dans le cas des farines animales, de voir que lorsque l'on analyse l'historique du processus, il est sûr qu'il y a eu crise, parce que l'on n'avait pas géré de façon suffisamment précautionneuse en amont. Cela renvoie malgré tout, je le conçois, de façon un peu circulaire, à la problématique que nous abordons aujourd'hui.
M. Pierre Hellier. - Pardonnez-moi mais, ce n'est pas parce qu'il n'y a pas autonomie qu'il y a eu décalage dans les positions des politiques et celles des experts. Ces derniers n'ont pas pu donner de réponse en temps utile, les politiques ont donc été obligés de prendre une décision avant. Que ces experts aient été liés, d'une manière ou d'une autre, au pouvoir politique, n'aurait rien changé. Ils n'auraient pas pu donner leur réponse ou alors ils auraient rendu un avis contraint et forcé par le pouvoir politique. Cette illustration ne me semble pas remettre en cause l'autonomie.
M. Claude Evin. - Je ne mettais pas en cause l'autonomie parce que je pense qu'elle est importante. Je mettais seulement l'accent sur les limites de ce genre de fonctionnement.
M. Pierre Hellier. - Il n'y a pas de remise en cause puisqu'en fait, ce n'est pas parce que l'agence aurait été liée de façon plus étroite au Politique qu'il aurait dû rendre une décision plus rapide.
M. Jean-Michel Dubernard. - Dans le même ordre d'idées, j'ai entendu aujourd'hui, aux questions d'actualité, une réponse du ministre de l'Agriculture qui, grosso modo, voulait dire qu'il fallait bien écouler les stocks de plats cuisinés préparés avant l'application du décret interdisant les viandes de bovins de plus de trente mois. On voit bien les limites d'application du système.
Beaucoup de personnes sont aussi très hostiles à ce principe de précaution en disant qu'il est inhérent à l'espèce humaine et, plus généralement, à toutes les espèces animales qui ont réussi à survivre sur cette terre et que, transféré sur le plan politique, la gestion du risque doit être faite par les politiques disposant de services, qui ne soient pas forcément ces agences dont on sait qu'elles sont, au bout d'un certain temps, toutes plus ou moins influencées par les lobbies. Il suffit pour s'en convaincre de se référer aux États-Unis pour toutes les agences équivalentes. Vous connaissez cela sans doute ; l'indépendance des agences me paraît très discutable.
Ma question se rapporte à ce que vous disiez au début de votre propos. Je me demande si appliquer ce principe à l'extrême ne revient pas à le transformer en principe d'immobilisme car il peut aussi être le meilleur moyen de tuer toute innovation et tout progrès dans la recherche. Il n'existe pas de recherche sans risque et il y a un grand danger à voir se saisir de cette innovation des personnes, souvent la justice, qui s'appuient sur des règles pouvant empêcher de progresser.
M. Philippe Kourilsky. - Je suis en désaccord avec vous quant au fait de pousser à l'extrême le principe de précaution. J'entends par là que c'est sa définition extrême qui doit être réfutée avec la plus grande vigueur. Il faut donc l'adopter, la publier et la corriger chaque fois que nécessaire afin que la définition active du principe de précaution prévale sur celle d'immobilisme.
Ce que j'essaie de dire à ce sujet, c'est qu'à côté des principes de droit, il y a une méthodologie de la précaution. Le droit est une belle chose, mais l'exercice de la précaution requiert une méthodologie à l'intérieur de laquelle s'inscrivent des processus assez simples - d'hygiène intellectuelle, si je puis dire - comme, par exemple, ce fameux principe de comparer la décision de faire à celle de ne pas faire.
Ce n'est pas du tout évident parce que, dans les faits, il est beaucoup plus facile d'essayer de raisonner sur le processus en tant que tel sans s'obliger aux comparaisons systématiques, qui pourtant devraient être menées à chaque fois. Cela double, bien sûr, le travail intellectuel et celui d'expertise. C'est donc un processus lourd.
Mais le principe de précaution bien compris et bien appliqué nous paraît positif.
Mme Geneviève Viney. - De toute façon, on ne peut pas aller contre parce qu'il est aujourd'hui intégré dans la conscience collective. Il répond à une exigence des citoyens à l'égard du Politique. On pourra dire tout ce que l'on voudra, ce principe existe. Il faut donc s'attacher à le cadrer et à lui donner sa portée réelle sans en faire un mécanisme systématique et utilisé de façon erronée. Il faut le définir justement.
M. Pierre Hellier. - Je suis évidemment favorable à ce principe de précaution. On ne peut pas être contre, mais il faut malgré tout s'en méfier, car aucun industriel pharmaceutique n'essaierait de mettre aujourd'hui l'aspirine sur le marché. Personne ne prendrait le risque de présenter le dossier de mise sur le marché de ce produit, il n'arriverait pas à franchir toutes les étapes de l'autorisation de mise sur le marché tellement le principe de précaution est énorme en la matière. Pourtant, l'emploi raisonnable de l'aspirine n'est contesté par personne.
M. Alain Claeys, rapporteur. - Pour prolonger la remarque de notre collègue, nous sommes tous d'accord avec la définition que vous donnez de ce principe de précaution. C'est un principe d'action, mais les poursuites pénales engagées ou abouties le sont sur une définition beaucoup plus inhibante que celle que vous donnez. Cela a des conséquences, de fait, sur le mode d'action des acteurs publics qui, naturellement, s'appliquent à eux-mêmes un principe de précaution beaucoup plus restrictif. Cela rejoint les remarques que nous avons entendues.
Mme Geneviève Viney. - Sur le plan pénal, aucune condamnation n'est prononcée en se fondant sur le principe de précaution. Le droit pénal est fondé sur le principe de légalité, c'est-à-dire que les incriminations sont nécessairement définies par le législateur. Or il n'existe pas d'incrimination du défaut de précaution.
M. Jean-Michel Dubernard. - Et les transfusions pour les hémophiles ?
Mme Geneviève Viney. - En ce qui concerne le problème des hémophiles, ce n'est pas sur le fondement du principe de précaution que la question a été posée.
M. Jean-Michel Dubernard. - Mais il y a deux manières d'interpréter ce qui s'est passé dans l'affaire Garretta.
Mme Geneviève Viney. - Le droit pénal n'est pas adapté à la sanction du principe de précaution et, en aucun cas, il n'a été invoqué comme fondement des poursuites ni des condamnations.
M. Alain Claeys, rapporteur. - N'est pas adapté ?
Mme Geneviève Viney. - En soi, le principe de précaution est un principe qui présente une forte imprécision dans son maniement. Or, le principe fondamental de droit pénal est le principe de légalité et l'interdiction de tout ce qui peut être une extension de l'incrimination pénale. C'est une interprétation stricte.
M. Jean-Michel Dubernard. - Il y a une limite qui est très claire à propos du sang contaminé, est-ce qu'aujourd'hui on ne peut pas interdire les transfusions en raisonnant logiquement et en imaginant qu'elles transfèrent des agents qu'on ne connaît pas ? Vous devriez me répondre « non » puisque l'on ne les connaît pas et qu'aucune agence scientifique, technique, n'a apporté d'informations en ce sens, sauf que vous allez interdire au moment où l'agent nouveau sera découvert. Et l'on se trouve dans la situation où le risque devient absolument ingérable par les politiques !
M. Claude Evin. - J'irai tout à fait dans le sens de Mme Viney. Je n'ai pas de statistiques sur le sujet et peux, peut-être, être pris en défaut sur tel ou tel point, mais en l'occurrence, il n'y a pas eu d'incrimination pénale sur la base du principe de précaution.
En revanche, il est vrai que la problématique du principe de précaution peut conduire un décideur à modifier ses décisions devant la crainte d'être soumis à une procédure de justice pour ne pas avoir pris telle ou telle décision. C'est là une mauvaise lecture du principe de précaution. Cependant, admettons-le, il y a un mouvement d'opinion qui consiste à dire que telle chose n'ayant pas été faite, il y a donc eu manquement dans la prise de décision.
Cela ne signifie pas que cela conduira à une condamnation. Il serait néanmoins intéressant que vous reveniez sur la question de savoir si l'on ne peut pas rapprocher la notion de principe de précaution de celle de principe de proportionnalité dont il faut rappeler qu'il est un des principes fondamentaux du code de déontologie médicale où il est inscrit.
C'est, en pratique médicale, l'un des principes fondamentaux de tout acte thérapeutique, me semble-t-il. N'étant pas moi-même médecin, je me retourne vers mes collègues médecins. Comme on est constamment obligé de prendre des risques, la question étant que, quand on veut soigner quelqu'un, que ce soit par le biais de la recherche ou dans la pratique concrète thérapeutique, on est en permanence obligé de se poser la question de savoir ce qui arriverait de pire au patient si l'on ne faisait pas ce choix. Cette notion de proportionnalité est une notion permanente en médecine.
Pour ma part, j'ai le sentiment qu'elle renvoie au principe de précaution. C'est, d'une certaine manière, l'une des définitions que l'on pourrait donner du principe de précaution.
Mme Geneviève Viney. - La loi Barnier prévoit d'ailleurs la propor-tionnalité.
M. Philippe Kourilsky. - Il est intéressant de constater que le principe de précaution sert maintenant à réinterpréter des situations antérieures, que l'on revoit à l'aune de ce nouveau principe - parfois d'ailleurs avec les ambiguïtés sémantiques qui y sont associées.
Je me permets de relever que ce qui a été dit au sujet de l'aspirine n'est pas exact. Si nous voulions mettre l'aspirine sur le marché aujourd'hui, nous n'y arriverions pas en effet. Cependant, ce ne serait pas en raison du principe de précaution, mais en raison du fait que les effets secondaires seraient jugés inacceptables dans ce que nous considérons comme la normalité du jour.
Cela me permet de dire, devant cette mission qui s'occupe de bioéthique, qu'un véritable problème d'éthique se cache derrière cela. Nous savons très bien que les normes qui sont aujourd'hui acceptées comme éthiques, notamment pour certains essais cliniques, freinent la recherche et empêchent de mettre au point un certain nombre de choses. C'est une responsabilité qui est prise, mais qui est préexistante à toute notion de précaution.
Personnellement, je m'interroge de temps en temps en me demandant s'il n'y a pas là une certaine dérive de l'éthique et si une autocritique de l'éthique ne devrait pas être inscrite dans le processus même de la réflexion éthique, notamment lorsque des délais administratifs, inacceptables à mes yeux, diffèrent d'un à deux ans des essais cliniques parfaitement légitimes qui, finalement, sont acceptés.
Je reviendrai sur la question du sang contaminé, qui est extrêmement intéressante à « revisiter » aujourd'hui. L'application du principe de précaution dans le drame du sang contaminé a été effective. Elle s'est traduite par la circulaire de 1983. Le problème, c'est que cette circulaire n'a pas été appliquée dans les faits. Le principe de précaution a donc bien été employé : c'était la bonne décision, au bon moment. Le problème est bien plus de s'interroger, comme des sociologues l'ont fait, Michel Setbon et d'autres, sur la question de savoir pourquoi cette circulaire issue des autorités de santé n'a pas été appliquée dans les faits. Si elle l'avait été, le drame du sang contaminé n'aurait jamais pris, dans notre pays, les proportions qu'il a prises. Il faut remettre la vérité, me semble-t-il, à ce niveau.
Cette affaire nous montre aussi l'importance d'avoir une histoire de l'expertise. C'est absolument capital, parce qu'il a été extrêmement difficile de reconstituer l'histoire douze ou quatorze ans après. C'est ce qui s'est fait en prétoire devant la Cour de justice de la République où, soudain, une histoire a été écrite qui est devenue suffisamment consensuelle pour qu'on puisse juger en fonction d'elle. C'est du moins l'interprétation du citoyen que je suis.
J'en déduis qu'il y aurait grand intérêt à ce que les expertises soient datées afin de permettre, ultérieurement, de repérer ce qu'était le « vrai accepté » du moment. Sinon, des voix dissidentes s'élèveront toujours pour dire qu'elles avaient bien prévu ceci ou cela.
Je reviendrai sur ces problèmes d'expertise qui sont très importants, mais ajouterai encore un mot au sujet de la transfusion sanguine. Vous avez raison de dire qu'elle est dangereuse. Un nouveau virus a encore été découvert, il y a une quinzaine de mois, dont on ne sait pas trop ce qu'il fait. Il est présent dans 10 à 15 % des dons de sang.
Mais que voulez-vous faire ? Car, dans ce cas, ce qui s'applique, c'est bien entendu la question de l'analyse par symétrie ou par réciprocité. On sait très bien qu'en arrêtant toute transfusion, on aura des centaines de milliers de morts, qui n'auront pas pu être soignés. Donc, on transfuse, avec un risque.
M. Roger Meï. - Dans le cas précis de l'ICSI, où est le principe de précaution ? L'ICSI a été immédiatement appliquée et plusieurs centaines d'enfants sont nés. Ils ont aujourd'hui cinq ou six ans. Que se passera-t-il, si l'on se rend compte, par la suite, que l'ICSI engendre des troubles graves ? Incriminera-t-on la législation d'avoir laissé faire car, en l'occurrence, le principe de précaution, sciemment, n'a pas été appliqué.
Mme Geneviève Viney. - Quelle serait l'incrimination ? Il n'y a pas de texte. En droit pénal, il faut un texte et, de toute façon, la loi du 10 juillet 2000 tendant à préciser la définition des délits non intentionnels a considérablement réduit les risques de poursuites en la matière, sur le plan pénal.
M. Alain Claeys, rapporteur. - Pour le responsable public, le risque commence lorsqu'il s'occupe du problème, lorsqu'il engage la décision.
M. Claude Evin. - On peut parfois avoir ce sentiment.
Mme Geneviève Viney. - Il faut distinguer entre la mise en examen ou la poursuite - l'exercice de l'action - et la condamnation.
M. Alain Claeys, rapporteur. - Mais pour suivre le raisonnement et reprendre l'exemple cité par M. Philippe Kourilsky, si la circulaire n'avait pas existé, il n'y aurait pas ce support. N'est-ce pas un risque ?
M. Claude Evin. - On peut aussi être responsable du fait de ne pas avoir agi.
M. Alain Claeys, rapporteur. - Tout à fait.
Mme Geneviève Viney. - Sur le fondement d'une incrimination qui serait alors l'incrimination d'homicide ou de blessure involontaires ?
M. Claude Evin. - D'homicide involontaire ou de non-assistance à personne en danger.
Mme Geneviève Viney. - Depuis la loi de juillet 2000, ce risque est infime, puisqu'il faut une violation délibérée d'une règle de prudence ou de sécurité mettant en danger la vie. La définition est extrêmement restrictive pour les décideurs publics, et pour les décideurs privés aussi, d'ailleurs.
M. Claude Evin. - On parle beaucoup de faits qui sont antérieurs à cette loi.
M. Philippe Kourilsky. - La notion d'expertise est l'objet d'un certain nombre de malentendus. Comme l'a très bien écrit Philippe Roqueplot dans des ouvrages très intéressants et très compréhensibles, en situation d'incertitude, l'expert ne sait pas et ce qu'il a de mieux à offrir à la collectivité, ce sont les limites de son incertitude. Nous vivons, et en tant que scientifique j'y suis très sensible, dans un malentendu permanent : la recherche opère dans une sphère de doute perpétuel et lorsque la recherche est terminée et que les résultats sont vraiment acquis et vérifiés, ils sont entrés dans les bases de données, et ce n'est plus de la recherche.
L'expertise faite par les chercheurs dans cette sphère d'incertitude est une expertise particulière. Ce n'est pas du tout l'expertise qui s'applique à un produit fini, en sachant exactement ce que l'on doit trouver et pour laquelle la réponse est oui ou non. Il y a donc des dispositifs particuliers sur le plan de la compréhension même de l'expertise et de son cahier des charges, c'est un premier point.
Le second est que je suis convaincu que, dans ce pays, nous manquons de systèmes de mesure. De ce point de vue, je me réjouis du développement d'agences ou plus exactement des moyens accordé à ces agences. Ils pourraient être localisés ailleurs, mais je suis persuadé qu'en matière de santé publique, notre pays n'est pas suffisamment, et de loin, équipé à de nombreux niveaux, y compris celui de la recherche.
Ceci étant dit, je voudrais attirer votre attention sur un point sur lequel nous avions travaillé et qui figure dans notre rapport. À notre avis, il reste un vrai travail, nécessaire et utile, à faire sur le statut de l'expert. En milieu scientifique, la plupart des expertises se font très souvent de façon non contractuelle : on appelle un expert pour lui demander son avis sur telle ou telle question. Or, il y aurait une grande utilité à définir une sorte de statut ou de charte qui précise les droits et les devoirs des experts, qui éventuellement le protège car, dans certains cas, l'expert prend des risques à articuler certains types de conclusions.
Nous avions identifié avec Mme Geneviève Viney que cela serait d'autant plus important dans les sphères internationales. Dans ces sphères, les experts scientifiques sont assez souvent délégués comme représentants de leur gouvernement et non de l'état de la science. Nous assistons donc à des débats d'experts dans des structures internationales - au sein de l'OMC, par exemple - qui, en fait, reflètent très précisément la géographie et les puissances respectives des États et pas du tout une « recherche de la vérité ». Cette notion de statut serait certainement un travail peu onéreux et qui pourrait avoir une grande utilité.
M. Alain Claeys, rapporteur. - Dans les autres pays européens, savez-vous si un travail de réflexion se fait sur ce principe de précaution ? Un travail sur ce thème a-t-il été réalisé au sein de la Communauté européenne ? L'idée d'agence se développe-t-elle de façon systématique ? Quelles sont les tendances chez nos voisins ?
Mme Geneviève Viney. - Les lignes directrices du principe de précaution furent définies, en février 2000, lors d'une communication du Conseil de la Communauté européenne. En 2001, au Sommet de Nice, une résolution de la présidence préconise ce principe. Mais je ne crois pas que cette nouvelle résolution prévoie la création d'une agence européenne.
M. Claude Evin. - Je ne le pense pas parce que les agences sont, en fait, identifiées sur chaque sujet. De plus, la procédure d'agence ne répond pas nécessairement à toute la problématique du principe de précaution. Ce n'est pas parce que l'on va déléguer à une expertise le soin de traiter un problème que l'on a résolu le problème.
M. Jean-Michel Dubernard. - Pour aller dans ce sens, si on prend l'exemple de la Food and Drug Administration, c'est bien une agence mais on peut se demander si elle est réellement indépendante. Théoriquement, c'est le cas, mais en pratique, elle est soumise à tous les lobbies économiques, industriels en particulier. Dans le domaine du médicament ou de l'instrumentation médicale, c'est un véritable scandale, elle fait du protectionnisme à outrance. Cela montre bien les limites de ce genre d'agence.
Mme Geneviève Viney. - Si en droit français, on avait un statut des agences administratives indépendantes, peut-être pourrait-on éviter ces inconvénients. Le Conseil de la concurrence, par exemple, est régi par des règles qui lui confèrent une certaine indépendance par rapport aux intérêts économiques. C'est également le cas de la Commission des opérations de bourse. Ce sont des organismes dont le statut garantit, en principe, l'indépendance. Si cela ne suffit pas, il faut renforcer ces garanties.
M. Claude Evin. - C'est fort intéressant d'un point de vue juridique. Vous avez fait allusion à des autorités administratives indépendantes qui ont effectivement un statut qui garantit leur autonomie. Ce n'est pas tout à fait le statut qui a été retenu pour les agences, notamment sanitaires. Mais d'autres règles, comme l'autonomie du directeur, permettent peut-être de garantir cette autonomie. Sur le plan juridique, pourriez-vous nous éclairer sur le choix d'un cadre susceptible de garantir cette indépendance ?
Mme Geneviève Viney. - Je ne sais pas si le modèle de la COB ou du Conseil de la concurrence est transposable aux agences sanitaires mais je pense qu'il y a des constantes. Mais quand vous parlez de cadre juridique, dans quel sens l'entendez-vous ?
M. Claude Evin. - Cette notion d'autorité administrative indépendante est une notion en droit, qui est identifiable. Lorsque l'on a créé, par exemple, l'Agence sanitaire des produits de santé, nous ne lui avons pas donné le qualificatif d'autorité administrative « indépendante ». C'est un établissement public régit par des règles de fonctionnement, dont certaines, comme le statut du directeur, lui confèrent une certaine indépendance, mais ce n'est pas une autorité administrative indépendante. Elle n'a pas ces fonctions. Puisque se pose la question de la création d'une institution, j'aimerais savoir si l'on peut trouver, dans le droit actuel, un cadre juridique qui donne plus de garanties d'indépendance que d'autres ?
Mme Geneviève Viney. - Je pense effectivement que le Conseil de la concurrence est un modèle du genre. C'est un organisme qui n'est ni vraiment juridictionnel ni vraiment administratif, mais qui possède des traits de l'un et de l'autre. Il faut, à mon avis, s'en inspirer. Les membres qui prennent les décisions ne peuvent pas être révoqués par le pouvoir politique, bénéficiant ainsi d'une certaine inamovibilité qui, sans être comparable à celle des juges, s'en inspire. D'autres garanties demandent réflexion, notamment quant à la possibilité de résister à l'injonction du pouvoir politique et économique.
M. Philippe Kourilsky. - Dans ce cas, il serait logique, et c'est d'ailleurs une tendance qui se dessine, que ces administrations soient soumises à des règles de fonctionnement très précises. Nous avons été très intéressés lorsque nous travaillions sur ce rapport, il y a maintenant plus d'un an, d'apprendre que certains secteurs de l'administration française, notamment la direction générale de la concurrence, de la consommation et de la répression des fraudes (DGCCRF), étaient en train de se mettre aux normes ISO 9001 ou 2 et d'inclure des processus d'assurance qualité dans leurs dispositifs de fonctionnement normaux. C'est extrêmement important.
En d'autres termes, ce que tous, l'État comme les citoyens, peuvent attendre d'agences ou de dispositifs auxquels sont accordés une certaine indépendance, c'est clairement la garantie que « ça tourne ». Or cette garantie passe par des procédures. Ces dernières sont difficiles à mettre en _uvre mais, une fois en place, elles autorisent ce que tout le monde réclame, à savoir la transparence, la lisibilité et la traçabilité - cette fois, non plus sur le plan des produits, mais sur celui des décisions.
Mme Geneviève Viney. - Je pense à un autre point tout à fait différent. Pour que le principe de précaution - ou la sécurité - soit respecté, l'élément essentiel est la remontée de toutes les informations issues de la vigilance. Sur ce point précis, il reste beaucoup de progrès à faire.
Il faudrait que les professionnels en possession d'informations ayant trait à la santé publique ou pouvant influer sur une décision mettant en cause la santé publique ou la sécurité soient obligés de la transmettre.
C'est très délicat car, bien souvent, de telles informations sont couvertes par le secret professionnel ou le secret industriel. Il faudrait donc qu'elles soient transmises à des autorités tenues au secret à l'égard des concurrents, par exemple. Un véritable aménagement de la remontée de l'information paraît indispensable, ainsi d'ailleurs que la « redescente » de celle-ci vers ceux qui ont besoin de la connaître.
M. Roger Meï. - Qu'en est-il du principe de précaution sur une question comme celle de l'effet de serre car il ne s'applique plus sur quelques semaines, voire quelques mois ? Avez-vous réfléchi à cette question de durée ?
Mme Geneviève Viney. - Comme le disait M. Philippe Kourilsky, c'est une question qui se posera au juge. S'il est saisi de poursuites, il aura à dater les décisions et à reconstituer les faits dans leur vérité historique.
M. Philippe Kourilsky. - La gestion d'un phénomène de très long terme, comme celui de l'effet de serre, n'est pas du tout la même que celle d'un phénomène de court terme. La gestion d'un phénomène de long terme relève d'un autre élément du raisonnement qui est aussi très délicat. En théorie, la gestion du phénomène par précaution ne devrait rien entraîner d'irréversible car, si la mesure de précaution est erronée, il faut pouvoir, en théorie, revenir en arrière.
En matière d'effet de serre, en raison des énormes conséquences économiques associées, des personnes travaillent aujourd'hui à essayer de calculer les trajectoires les moins onéreuses et les plus adaptées, de façon à « coller » au mieux, par des mesures réversibles et progressives, à l'évolution anticipée des connaissances. Cela devient un jeu extraordinairement technique et complexe et, à certains égards, tout à fait aléatoire. La précaution poussée à ce niveau devient un art plus qu'une science.
M. Alain Claeys, rapporteur. - Que pensez-vous des débats citoyens, qui se développent actuellement, pour faire le lien entre scientifiques et décision politique ? Est-ce la gestion de l'image du risque vis-à-vis de nos concitoyens ou une pratique qui a son efficacité ?
M. Philippe Kourilsky. - J'ai, à ce sujet, une attitude personnelle très claire. À mon sens, il est du devoir du scientifique de s'exposer au dialogue public. Je sais que beaucoup de mes collègues y sont moins prêts que moi. Je pense pourtant que c'est fondamental. La recherche scientifique est largement financée par le public. Ce public a donc des droits vis-à-vis de la recherche. Je suis de ceux qui pensent que ces devoirs des chercheurs doivent être exprimés dans la pratique.
Cela dit, il faut bien reconnaître que quand on se donne beaucoup de mal pour expliquer ce qui relève de sa compétence et que le message que vous avez cherché à délivrer à grande peine est détruit par trente secondes de télévision, cela vous sape un peu le moral. Il n'empêche, je pense que c'est un devoir.
M. Claude Evin. - Les hommes politiques sont confrontés à ce problème en permanence.
M. Philippe Kourilsky. - Cela nous renvoie au problème de l'image et du système qui engendre et distribue les images, système qui n'est pas uniquement le système médiatique, qui met aussi en cause les pourvoyeurs d'informations et il faut bien dire que dans la sphère scientifique, et dans celle qui fournit des informations, vous avez aussi des manipulateurs d'informations, comme j'imagine qu'il en existe dans d'autres sphères d'activité, peut-être même dans la sphère politique ! (Sourires.)
Il y a un vrai problème de qualification des informations. Évidemment, en matière de précaution et d'incertitude, nous sommes sur un terrain particulièrement propice au glissement de sens et aux argumentations contradictoires. Cela ne fait que renforcer la nécessité de dialoguer et de débattre.
M. Alain Claeys, rapporteur. - Je vous remercie.
Audition de Mme Christiane AIZENFISZ,
membre du conseil fédéral,
et de Mme Françoise MOATTI,
membre de la commission consultative bioéthique,
de la Grande Loge Féminine de France.
(Extrait du procès-verbal de la séance du 17 janvier 2001)
Présidence de M. Alain Claeys, rapporteur.
M. Alain Claeys, rapporteur. - Je vous remercie d'avoir accepté de répondre à notre invitation.
Notre mission a tenu à recevoir chaque famille philosophique ou spirituelle. Chacune d'elles ayant sa position sur la révision des lois bioéthiques, en fonction des choix et de la morale qui lui sont propres, il nous est apparu utile que vous puissiez vous exprimer devant cette mission. C'est donc dans cet esprit total de liberté que nous vous avons invitées et que je vous propose de vous exprimer devant nous aujourd'hui.
J'engagerai le débat par quelques interrogations.
Premier point, quelle appréciation la Grande Loge Féminine de France porte-t-elle sur la place des femmes dans les différents comités d'éthique, y compris les comités de protection des personnes qui se prêtent à des recherches biomédicales ?
Deuxième point, l'assistance médicale à la procréation est un parcours toujours délicat au cours duquel la femme est mise fortement à l'épreuve et est souvent sujette à des pressions. Pour la Grande Loge Féminine de France, comment garantir le libre consentement de la femme ? Qui est le meilleur juge de l'intérêt de l'enfant ? Ce sujet est revenu régulièrement dans nos débats.
Troisième point, plus général, quelle appréciation portez-vous, après cinq ans, sur cette loi bioéthique ? Sans doute avez-vous eu connaissance de l'étude du Conseil d'État. Certaines de ses propositions suscitent-elles des réflexions ou des remarques particulières de votre part ?
Mme Christiane Aizenfisz. - Je vous remercie d'avoir d'emblée préparé des questions qui, bien évidemment, nous interpellent au premier chef en tant que femmes, d'une part, et en tant que franc-maçonnes, d'autre part. Nous essayons de représenter au mieux les 11 000 femmes de la Grande Loge Féminine de France.
Pour répondre à votre première question concernant la place des femmes dans les comités d'éthique, je vous indique que nous avons fait, cette année, candidature auprès du Comité consultatif national d'éthique. Nous sommes heureuses qu'une femme ait été nommée. Il s'agit de Mme Dominique Schnapper, qui ne fait pas partie de notre obédience. Je ne la trahis en aucun cas, même si nous sommes un peu peinées que la candidature à la Grande Loge Féminine de France n'ait pas été acceptée.
Quelle est notre appréciation des lois de bioéthiques après cinq ans ?
Nous avons eu naturellement connaissance de l'étude du Conseil d'État qui constitue un document extrêmement riche et intéressant. Nous ne pouvons que regretter que cette révision n'ait été envisagée qu'avec un certain retard et que de nombreux décrets d'application n'aient pas été pris rapidement à la suite de la promulgation des lois. Certains d'entre eux attendent toujours d'être promulgués.
L'étude nous a semblé extrêmement pertinente, mais nous souhaiterions commencer par vous faire part de notre appréciation du discours de M. le Premier ministre et de l'avant-projet de loi.
M. Alain Claeys, rapporteur. - Avant de vous laisser poursuivre, j'apporterai une précision. Le propos de notre mission n'est pas de discuter de l'avant-projet de loi, que nous aborderons au sein de la commission spéciale qui sera créée à sa suite. En outre, nous n'en avons pas encore discuté entre nous.
En revanche, nous sommes à votre écoute pour connaître vos points d'accord, vos interrogations et, éventuellement, vos points de désaccord avec l'intervention de M. le Premier ministre.
Mme Françoise Moatti. - Le premier document que nous vous avons distribué est la synthèse de notre réflexion sur la révision de cette loi de bioéthique, synthèse que vous avions faite en 1999. Cinq ans après l'adoption de ces lois, et finalement plutôt sept ans - ce retard est d'ailleurs un peu fâcheux - en relisant ce premier document, qui n'est pas obsolète, nous réalisons combien les choses ont évolué.
M. Alain Claeys, rapporteur. - Si je puis me permettre d'apporter une précision, la révision des lois bioéthiques de 1994 a bien commencé cinq ans après, conformément à la loi, par des rapports élaborés par l'Office parlementaire d'évaluation des choix scientifiques et technologiques. La réflexion a donc bien commencé.
Mme Françoise Moatti. - J'atténue donc ma remarque. Mais il est vrai que les choses vont très vite. On se rend compte que certains points ont été sensiblement modifiés. L'essentiel du texte reste le même, mais, sur l'assistance médicale à la procréation - je rejoins votre question - et sur la définition et le statut de l'embryon, notre position reste inchangée : nous évitons de le définir. C'est fâcheux parce qu'il n'est pas très pratique de ne pas avoir de définition, mais, sinon, on entre dans des problèmes ontologiques ou dans des vues si profondément différentes, selon la sensibilité de chacun, que nous sommes presque dans une aporie. Nous continuons donc de penser qu'il ne faut pas définir de statut de l'embryon.
Les conditions d'accès à l'assistance médicale à la procréation restent les mêmes. Cependant, elles vont certainement évoluer par rapport au couple et en fonction de l'évolution de la société. Mais nous sommes, pour l'instant, dans une phase de révision de loi bioéthique et non dans la réflexion sur ce qu'est un couple, la façon dont il se forme, etc. Pour le moment, il nous semble souhaitable qu'elles demeurent les mêmes, tout en sachant que dans cinq ans, voire avant, elles seront probablement modifiées.
Toutefois, un point nous a quelque peu étonnées, concernant le transfert de l'embryon post mortem. La position de l'Académie de médecine et du CCNE sur l'implantation de l'embryon chez la mère, après le décès du géniteur, nous laisse réservées. Nous savons qu'une jurisprudence avait posé problème à Toulouse. Mais on peut tout de même s'interroger sur ce point car, selon les psychologues, se pose un grand problème d'autant que le projet fait état d'un délai allant de trois mois minimum à un an. Je ne vois pas comment l'on peut estimer ainsi la durée d'un travail de deuil et d'un événement provoquant un très grand choc, en le fixant dans une loi.
En revanche, nous savons aussi - il est vrai que la Grande Loge Féminine de France n'est pas univoque sur tout, sauf sur ce qui a trait à la défense des principes d'humanisme -, que certaines, parmi nous, ont beaucoup insisté pour obtenir cette possibilité au nom de la femme et de sa liberté. Le débat reste ouvert mais cette notion de délai nous laisse réservées.
L'anonymat du tiers donneur est un bon exemple de ce qui a évolué dans la société. Nous disions, à l'époque, que nous options pour son maintien, sans exclure une reprise du débat, la position de certains États étant différente de celle de la France. Et nous en parlions comme d'un aspect qui allait poser problème dans les années à venir, et qui devrait être débattu.
En fait, la situation a évolué très vite et, en deux ans, ce grand bouleversement qu'a été la remise en question de l'accouchement sous X remet vraiment l'anonymat en cause non seulement pour l'accouchement sous X mais aussi pour le don de gamètes et, particulièrement, celui d'ovocytes. Il y a lieu d'en tenir compte. Or la loi est ainsi faite que le don d'organes ou autres était régi par l'anonymat, la gratuité et le secret. Il s'agit de notions très imbriquées. Des psychologues et des psychiatres, non des moindres, ont pris des positions extrêmement claires sur le sujet, indiquant que le don d'ovocytes notamment, puisqu'on parle des femmes, devait être un don reconnu, un don en dehors de l'anonymat.
Le débat sur l'anonymat est donc complètement ouvert et mérite d'être approfondi. Il faut, de toute façon, modifier son caractère irréversible.
Concernant le devenir de l'embryon, nous restons favorables à l'autorisation de recherches sur l'embryon. Ce point fut l'objet un grand débat auquel ont participé l'Académie de médecine, la CNMBRDP et le Conseil d'État. De l'avis unanime, il convient de desserrer les contraintes qui pèsent sur cette possibilité d'accès à la recherche sur l'embryon. Naturellement, cela pose des problèmes éthiques puisque tout le monde n'a pas les mêmes opinions sur ce sujet, mais il s'agit de simplement bien poser les conditions de contrôle, dans lesquelles cette recherche interviendra. D'ailleurs, M. le Premier ministre l'a bien vu en proposant une CNMBRDP renovée ou remplacée par une agence.
En ce qui concerne le don d'organes prélevés sur les personnes vivantes, nous proposions un élargissement des liens de parenté exigés préalablement. Je crois que cette évolution va se mettre en place, cependant, nous souhaitons que cela soit soumis à un comité d'experts car les personnes « proches » peuvent se trouver sous dépendance. Une employée peut être une proche. Il faut bien veiller à ce qu'il n'y ait pas de lien de subordination ou de pression morale entre donneur et receveur. Mais cet élargissement est éminemment souhaitable. C'est la raison pour laquelle il est nécessaire qu'un comité d'experts effectue ce contrôle. Il en est un qui est opérationnel et qui pourrait servir de modèle : celui chargé du don de moelle osseuse chez des enfants mineurs. C'est un bon comité dont on pourrait s'inspirer.
Quant aux dons d'organes, en 1999, nous suggérions de prévoir des additifs aux lois concernant le clonage et l'induction d'ovulation.
Concernant le clonage reproductif humain, il faut être ferme. Il y a unanimité, même dans d'autres pays, pour l'interdire mais je ne sais pas si notre loi l'interdit de façon aussi explicite que cela.
M. Alain Claeys, rapporteur. - Il est interdit.
Mme Françoise Moatti. - S'il l'est, c'est très bien.
Reste le problème du clonage thérapeutique qui nous préoccupe. C'est un grand sujet de débat. Le groupe européen d'éthique l'avait déconseillé mais la Grande-Bretagne va s'y engager. Cela pose réellement problème car, ces dernières années, nous avons certainement pâti d'avoir bloqué la recherche. Il est donc difficile de décider législativement de bloquer la recherche sur un sujet qui prendra forcément de l'importance, sauf à créer chez les chercheurs un risque de transgression.
Cependant, l'essentiel est de voir le risque et, à nos yeux de femmes, ce risque est celui de l'instrumentalisation du corps de la femme. Comme vous devez le savoir, le clonage thérapeutique passe par l'ovocyte et s'il se fait à grande échelle, le besoin en ovocytes deviendra forcément très important. Or il n'est pas si facile de les obtenir. Comment les obtiendra-t-on ? On peut imaginer toute sorte de pressions et de dérives.
Il faut en être conscient et c'est ce que nous exprimons dans notre document sous la forme suivante : « La demande d'ovocytes peut se faire pressante, voire conduire à un véritable marché d'ovocytes, d'où la nécessité absolue d'un encadrement très strict pour la loi à venir ».
Nous avions aussi soulevé le problème de l'induction d'ovulation. Cet acte, je vous le rappelle, a pour but de stimuler les ovaires en vue de la production d'ovocytes. Le problème se situe à deux niveaux.
D'une part, n'importe quel médecin peut, aujourd'hui, prescrire des inducteurs d'ovulation. Je suis pharmacienne et je le constate. Or cette prescription devrait être encadrée car c'est une technique qui peut avoir des conséquences sur la femme et qui est un peu abusivement employée. Nous demandions, et nous espérons que cela sera repris, un contrôle des compétences de ceux qui effectuent ces prescriptions ainsi qu'un suivi à long terme des traitements administrés, parce que le nombre de stimulations ovulaires que certaines femmes ont subies est assez spectaculaire. Il serait souhaitable que l'on sache ce qu'il advient des femmes qui les ont subies.
D'autre part, l'inducteur peut être prescrit aussi dans le cadre de l'assistance médicale à la procréation. La stimulation d'ovulation ainsi engendrée conduit à produire beaucoup d'ovules et donc beaucoup d'embryons. Il faut bien alors se poser la question des embryons surnuméraires, le but étant, bien sûr, de réduire leur nombre le plus possible et de limiter les problèmes éthiques posés par l'existence de ces embryons surnuméraires.
Mme Christiane Aizenfisz. - Avant d'aborder le thème de la génétique, je voudrais, en quelques mots, revenir sur notre interprétation du discours de M. le Premier ministre.
Dans les grandes lignes, nous avons relevé quatre grands points. Premièrement, autoriser la recherche sur l'embryon. Nous souhaitons que la prise en compte de valeurs fondamentales encadre, sans la rendre impossible, l'avancée des connaissances scientifiques et leur application dans une double perspective : l'assistance médicale à la procréation (AMP) et le traitement de maladies à partir de cellules souches obtenues soit à partir d'embryons surnuméraires - mais se pose alors la question de ce qui se passera quand les stocks seront épuisés -, soit par transfert de cellules somatiques - est-ce une assimilation au clonage thérapeutique ? -, soit à partir du sang du cordon.
Pourquoi l'alternative des cellules d'adultes n'est-elle pas envisagée ?
M. Alain Claeys, rapporteur. - Elle l'est.
M. Alain Claeys, rapporteur. - D'après les propos de M. le Premier ministre et dans l'avant-projet, aucune piste de recherche n'est abandonnée.
Mme Christiane Aizenfisz. - Bien sûr, mais on aurait pu mettre l'accent sur cette voie.
Le deuxième point est le transfert d'embryon post mortem, si l'existence de la volonté parentale en était affirmée avant le décès et si le délai plus de trois mois et moins d'un an est bien respecté, avec le soutien psychologique.
Troisième point, le don d'organes par des personnes vivantes serait élargi aux concubins et aux conjoints - serait supprimée la condition d'urgence - et il serait, pour ces derniers, soumis à l'examen d'une commission d'experts.
Enfin, le quatrième point concerne la création d'une haute instance de suivi composée de dix-huit membres, dont les avis seront publics.
D'après l'avant-projet, cette loi se composerait de trois grands chapitres venant remplacer, si nous avons bien compris, les lois bioéthiques de 1994.
Dans l'examen des caractéristiques génétiques, il y a modification des articles 16-10 et 16-11 du code civil et modification du code de santé publique avec le consentement de l'intéressé expressément recueilli avant identification et utilisation de ses caractères génétiques.
Trois interrogations ont retenu notre attention concernant la médecine prédictive et les tests génétiques : les limites éthiques du dépistage génétique, l'impact des informations génétiques sur la vie privée des personnes et la façon dont les pouvoirs publics doivent prendre en compte ces évolutions, notamment au regard des banques et des assurances, et des employeurs.
La médecine prédictive ne doit pas aboutir à une discrimination des personnes en raison de leur patrimoine génétique. Connaître ou non son destin biologique est une responsabilité écrasante vis-à-vis de soi-même et des proches. Le droit à ne pas savoir protégerait aussi l'individu d'une information potentiellement traumatisante. Cependant, la médecine prédictive constitue un progrès indéniable dans l'appréciation des risques d'une maladie sur laquelle on peut intervenir par la prévention ou un éventuel traitement approprié.
En ce qui concerne le don et l'utilisation des éléments et produits du corps humain, cet avant-projet de loi modifie de nombreux articles du code de santé publique relatifs aux prélèvements sur vivants et décédés, aux prélèvements et préparations des tissus, cellules et produits du corps humain, et à leurs dérivés, aux dons de gamètes et aux dispositions spécifiques aux cellules souches embryonnaires.
Je dois avouer que nous avons été surprises par la suggestion de témoigner la reconnaissance de la Nation aux donneurs.
Nous avons noté l'élargissement du don d'organes par conjoints et concubins à partir d'une relation étroite et stable après consentement éclairé et libre soumis à l'autorisation d'experts. L'importance accordée à l'Établissement français des greffes nous a semblé de bon aloi. En cas de danger pour la santé publique, le prélèvement sans consentement pour rechercher les causes de la mort pourrait être autorisé.
Concernant la préparation, conservation et utilisation des tissus, cellules et gamètes, cet avant-projet retient les points suivants : le donneur n'est plus obligé de faire partie d'un couple ayant procréé mais doit avoir lui-même procréé et une limitation est posée à dix enfants au lieu de cinq antérieurement. Est prise une disposition spécifique aux cellules souches embryonnaires obtenues avant le stade de différenciation tissulaire, ne pouvant conduire à l'obtention d'un embryon in vitro ; est soulignée l'importance thérapeutique des cellules ES pour des pathologies graves ; enfin, apparaît la notion de produit de thérapie génique et de produit cellulaire d'origine animale à finalité thérapeutique.
Le troisième grand chapitre de cet avant-projet de loi est relatif à la procréation, l'embryologie et la génétique humaine. L'article de 16-4 du code civil impose explicitement une interdiction du clonage reproductif. Douze articles du code de santé publique visent la création de l'Agence de procréation, embryologie et génétique.
L'assistance médicale à la procréation comporte quatre points. La stimulation d'ovulation est une pratique d'AMP reconnue comme telle, préalable à la FIV. Les modalités de prescription et de suivi seront soumises à un arrêté.
Nous avons déjà parlé du transfert d'embryons post mortem avec consentement exprès avant décès. Cependant, un point a suscité une interrogation. Il s'agit de l'« entrée ou de la sortie d'embryons dans le champ territorial du présent code ». Je ne sais pas si cela fait référence à l'import-export d'embryons. Vous nous le direz peut-être. Il s'agit de l'article L. 2141-11 du code de la santé publique.
L'ancien article L.152-8 du code de la santé publique est totalement remanié pour devenir l'article L. 2141-12.
Mes commentaires sur ce troisième chapitre sont les suivants : clonage reproductif humain explicitement interdit ; extension à l'embryon in vitro d'un questionnement éthique ; retour sur l'interdiction des recherches sur l'embryon par rapport à la loi de 1994 en maintenant l'interdiction de la conception in vitro d'embryons à des fins de recherche mais en autorisant la recherche sur les embryons congelés surnuméraires abandonnés de projet parental et ne faisant pas l'objet de don à un autre couple.
On peut conclure que l'on trouve dans ces différents textes un souci constant de la recherche d'un équilibre entre les avancées des sciences de la vie et la nécessité d'assurer les principes de sauvegarde de la dignité de la personne.
Mme Yvette Benayoun-Nakache. - Je n'ai pas de question à poser sur ce que vous venez de dire, avec beaucoup de précision d'ailleurs. Je voudrais seulement préciser que nous sommes une mission d'information préparatoire au projet de loi de révision des lois bioéthiques. Notre réflexion est, maintenant, faussée parce que les auditions que nous faisons se fixent sur l'avant-projet de loi. Notre mission se transformera certes en commission mais, pour l'instant, ce n'est pas encore le cas. Cela brouille la réflexion. Je ne dis pas qu'il faille forcément ne pas en tenir compte, mais il serait souhaitable de continuer à faire ce distinguo.
M. Alain Claeys, rapporteur. - Mme Benayoun-Nakache a raison d'appor-ter cette précision mais nous allons pouvoir faire cette gymnastique intellectuelle...
Mme Christiane Aizenfisz. - Je me permettrai de vous dire, madame, que nous sommes à l'extérieur de votre préoccupation et que nous avons, dans un premier temps, travaillé sur les lois bioéthiques de 1994 en vue de leur révision. Cela correspond au premier texte que nous vous avons soumis.
Malheureusement, nous n'avons pas pu être reçues l'année dernière par M. Claeys, mais nous l'avons été par l'Office parlementaire d'évaluation des choix scientifiques. Il se trouve qu'entre temps, comme vous le savez bien sûr, de très nombreuses évolutions se sont produites, allant bien au-delà des possibilités de compréhension de tout un chacun, c'est-à-dire des profanes mais aussi des personnes spécialisées dans le sujet. Lorsque nous avons eu connaissance du discours de M. Lionel Jospin aux Journées d'éthique, il nous a semblé que pour bien travailler et faire des propositions basées sur une certaine réalité, il était préférable d'avoir la possibilité de consulter l'avant-projet de loi car nous ne pouvions pas donner des points de vue et des additifs à notre position préalable sans savoir ce qui était envisagé dans l'avenir.
Mme Yvette Roudy. - Nous pouvons ne pas être trop formels. Nous discutons très librement dans cette mission. Il est bien évident que, chaque fois qu'un texte devient disponible, que se passe un événement ou que nous lisons un article dans la presse, nous nous en saisissons, nous y réfléchissons et nous en discutons pour l'intégrer à la réflexion générale. Pour ma part, cela ne me gêne pas que nous parlions du discours de M. le Premier ministre, et de l'avant-projet, ou d'autres textes encore. Tout cela entre dans le champ de la réflexion générale.
Je suis impressionnée par l'ampleur de votre réflexion. Vous avez déjà répondu à beaucoup de questions que nous nous posions et que nous sommes encore en train de nous poser. J'ai pourtant quelques interrogations à formuler sur des cas dont nous avons entendu parler par la presse. Je pense notamment au cas, qui se passe aux États-Unis, d'un enfant que l'on a appelé Adam, qui a été procréé en utilisant la méthode du diagnostic préimplantatoire. Sa s_ur était atteinte d'une maladie terrible qui pouvait se soigner grâce à une transfusion d'un enfant très proche, présentant des caractéristiques susceptibles de la soigner - un médicament, en quelque sorte. Les parents ont donc eu recours au dépistage préimplantatoire bien que n'étant pas stériles. Ce fut ensuite le processus et le parcours de la fécondation in vitro. Puis cet enfant, Adam, est né et son premier acte en arrivant au monde a été de soigner sa s_ur.
À ce sujet, nous avons eu des débats, nos positions ont évolué. Je disais que c'était un acte d'amour, d'autres acquiesçaient mais ajoutaient qu'il fallait rester prudent.
Puis, il y a eu Valentin, né dans une famille où les trois premiers enfants sont morts d'une maladie héréditaire. Les parents souhaitaient avoir un enfant sain et ont donc eu recours au dépistage préimplantatoire. On a fait une sélection, on a examiné les embryons et trié ceux qui n'étaient pas atteints de cette maladie. En France, cela s'est fait dans le service de M. René Frydman. Or M. Frydman qui avait dit que la naissance d'Adam ne lui plaisait pas beaucoup, trouvait que celle de Valentin était très bien. Il est vrai que c'est lui qui l'avait permise et pratiquée. (Sourires.)
Je m'interroge : pourquoi serait-ce bien dans un cas et pas dans l'autre ?
Ma dernière question n'a rien à voir avec cela, mais a trait à un sujet que vous n'avez pas abordé. Avez-vous réfléchi à l'ICSI, cette méthode qui permet de prélever un spermatozoïde sur un homme stérile pour, ensuite, l'injecter dans l'ovocyte de sa femme. Cette méthode, très développée en Belgique, est aussi utilisée en France, le Comité consultatif national d'éthique étant très réservé à son égard.
M. Alain Claeys, rapporteur. - Cette pratique de l'ICSI, qui n'était pas connue au moment du vote des lois bioéthiques de 1994, est la principale méthode utilisée pour l'assistance médicale à la procréation.
Mme Françoise Moatti. - Adam et Valentin, c'est la même chose scientifiquement ou techniquement ; c'est un diagnostic préimplantatoire. Il appartient à M. René Frydman d'avoir son opinion. Cependant, il est vrai, les finalités ne sont pas tout à fait les mêmes.
Dans le premier cas, on a l'impression qu'on a fait un enfant pour en sauver un autre. Seuls les psychologues ou les psychiatres pourraient nous dire quelle est la motivation. L'enfant peut-il penser qu'il n'a été fait que pour l'autre, comme, après le décès d'un enfant, le suivant peut se percevoir comme un enfant de remplacement ? C'est cet aspect qui peut être contestable ou poser problème et sur lequel on peut réfléchir.
Le second cas entre complètement dans le cadre de la loi puisque l'AMP n'est pas seulement autorisée pour remédier à la stérilité mais aussi pour des couples qui encourent un risque de transmettre à leur enfant une maladie particulièrement grave. Par ailleurs, permettez-moi de vous faire remarquer que ce diagnostic préimplantatoire de Valentin a été extrêmement médiatisé mais qu'il est « banal ». Le diagnostic préimplantatoire avait été autorisé par la loi, qui était pourtant très restrictive, en 1994. Après d'âpres discussions, il avait été concédé que, dans des cas très exceptionnels, il était praticable. Si bien que les décrets sont parus très tardivement, celui du diagnostic préimplantatoire a été publié très récemment.
C'est la raison pour laquelle nous n'avons pu, en France, pratiquer ce diagnostic préimplantatoire. Il ne s'agit donc pas, en fait, d'une performance scientifique ni même d'une nouveauté puisque les diagnostics préimplantatoires se pratiquent ailleurs. Simplement, il a été très médiatisé.
Cela rejoint un des éléments de notre conclusion : il faut se méfier des grandes informations bioéthiques sensationnelles. Il faut toujours chercher à savoir qui se trouve derrière. Les scientifiques sont des gens que nous aimons et respectons, mais derrière eux, se profilent aussi des laboratoires qu'il faut défendre, des budgets qu'il faut obtenir et des réputations qui sont en jeu.
Vous avez également demandé ce que nous pensions de l'ICSI. Sur ce sujet, nous avons été pris de court car ces découvertes scientifiques ont été très rapides. Vous vous souvenez sans doute de ce célèbre article de Jean-Yves Nau intitulé : Le viol de l'ovule, qui relevait vraiment du sensationnel. L'ICSI a plutôt été de l'ordre du fait accompli. Elle a commencé en Belgique ; elle y a été tolérée et est même utilisée un peu abusivement, me semble-t-il, parce que, si par cette méthode, on est sûr que cela va plus vite, il n'est pas toujours sûr que cela ne concerne que les azoospermies. Je crois que c'est interdit au stade spermatide.
Il me semble que dans le projet de loi, il est prévu que dès qu'apparaîtra une nouvelle découverte, elle sera soumise à expérimentation.
Mme Yvette Roudy. - Je n'ai rien lu de ce type dans le texte.
Mme Christiane Aizenfisz. - Le texte ne dit rien de l'ICSI, Mme Roudy a tout à fait raison. En revanche, l'article L. 2141-5 concernant l'assistance médicale à la procréation, dit qu' « aucune nouvelle technique d'assistance médicale à la procréation ne peut être mise en _uvre avant une évaluation préalable permettant d'assurer son innocuité. ».
Comme le disait Mme Moatti, l'ICSI a été utilisée comme une méthode pratique sans être autorisée législativement et, rétrospectivement, on en tire les enseignements.
M. Pierre Hellier. - Vous nous avez fourni un texte très précis qui est le témoin d'une réflexion forte. Vous nous avez reproché la lenteur de la révision de ces lois bioéthiques. Le délai de cinq ans peut en effet paraître long. Aussi ma question sera la suivante : à qui confier, entre les révisions de ces lois, l'autorisation d'agréer ou pas les avancées scientifiques ? L'ICSI, personnellement, ne me choque pas. C'est un moyen qui me paraît extrêmement astucieux d'assistance à la procréation. Mais l'ICSI pose le problème de sa réglementation lors de son apparition. À cet égard, que proposez-vous entre les révisions de la loi ? Quel genre de structure pourrait donner l'agrément à de nouveaux progrès scientifiques ?
Mme Christiane Aizenfisz. - M. Lionel Jospin et l'avant-projet de loi font état de la création future d'une agence et d'une haute instance de dix-huit membres, dont les avis seront publics. À la différence de ceux émis par le Comité consultatif national d'éthique qui n'étaient que des avis consultatifs, un pas supplémentaire est franchi, si j'ai bien compris.
M. Alain Claeys, rapporteur. - Mais la décision reviendra au Politique ; le Gouvernement acceptera ou non.
Mme Christiane Aizenfisz. - Tout à fait. La décision revenant au Politique mais étant faite...
Mme Yvette Roudy. - Sur proposition de cette agence.
M. Alain Claeys, rapporteur. - Et l'avis sera public
Mme Christiane Aizenfisz. - Cela me semble très positif mais se pose tout de même une question de délai par rapport aux nouveautés. Le seul délai de cinq ans pour la révision de loi, si tant est qu'il soit maintenu, ne permettrait pas de réactualiser la loi au fur et à mesure des avancées scientifiques.
M. Pierre Hellier. - Comme notre collègue Mme Benayoun-Nakache, je suis beaucoup moins à l'aise maintenant, depuis l'intervention du Premier ministre qui a fixé indiscutablement le cadre de cette révision. Nous avons désormais l'impression que nous devons, au sein de cette mission, débattre de la bioéthique dans ce cadre. Je le regrette.
Mme Yvette Roudy. - Nous sommes tout à fait libres d'aller au-delà.
M. Pierre Hellier. - Je l'espère.
M. Alain Claeys, rapporteur. - Notre mission d'information s'achèvera avec la session parlementaire. Lorsque le projet de loi aura été déposé, notre mission d'information deviendra alors une commission spéciale, dont la tâche sera de l'étudier et de l'amender. Je pense que jusqu'à présent, les membres de cette mission ont toujours pu s'exprimer. Cela continuera.
Mme Yvette Roudy. - Je sais que vous avez une structure permanente qui suit de manière régulière et attentive, on peut le constater, ces questions. Il serait intéressant que les relations que nous avons établies aujourd'hui puissent se poursuivre. Je suis d'accord avec vous pour dire qu'il va falloir discuter les propositions du Gouvernement. Il serait souhaitable que la structure qui va se mettre en place soit assez ouverte pour rencontrer, régulièrement, non seulement des savants, des techniciens, des scientifiques, des chercheurs ou des médecins mais aussi des représentants de la société civile qui ont passé du temps à réfléchir au problème et qui en ont tiré des conclusions.
Bien que je sorte complètement de l'épure, je souhaiterais dire ici qu'il faudra encourager ces relations, qui sont très riches et peuvent être très utiles à la réflexion de la future structure.
M. Alain Claeys, rapporteur. - La commission spéciale fixera ses propres règles de fonctionnement. Elle pourra, bien sûr, à tout moment, auditionner des personnalités que nous aurons déjà rencontrées pour éclairer sur tel ou tel aspect du projet de loi. Je crois à l'utilité de garder cette démarche d'ouverture vers l'extérieur.
Mme Françoise Moatti. - Si vous permettez, puisqu'on évoque ces commissions, je voudrais souligner l'une de nos interrogations quant à la CNMBRDP, qui était vraiment scientifique, et au CCNE. Comment tout cela s'organisera-t-il ? Comment les commissions vont-elles s'articuler les unes par rapport aux autres ? En outre, il ressortait du discours du Premier ministre, que pour la représentation de la société civile, il n'y aurait que des associations de malades. M. le Premier ministre a été très précis de ce point de vue. C'est plus qu'un simple cadre. Sans doute faudrait-il peut-être aller au-delà des associations de malades. Et aussi, tenir compte de la parité !
M. Alain Claeys, rapporteur. - Je vous remercie.
Audition de MM. Jacques BRUNSCHWIG, médecin,
Émile PAPIERNIK, médecin-chef de la maternité de Port-Royal,
Pierre-Charles RANOUIL, avocat,
et deMme Isabelle SCHOONWATER, magistrat
(Extrait du procès-verbal de la séance du 24 janvier 2001)
Présidence de M. Alain Claeys, rapporteur,
puis de M. Bernard Charles, président.
M. Alain Claeys, rapporteur. - Le président et la mission ont souhaité rencontré les membres de votre groupe de travail et de réflexion qui travaille sous l'égide du Conseil de l'Europe et étudie l'antinomie entre le droit au secret médical et la non-assistance à personne en danger dans le cas du sida. Vos préoccupations recoupent certaines de nos propres réflexions. Ces interrogations communes concernent des enjeux de santé publique importants, une sensibilité particulière de l'opinion publique, des tensions éthiques qu'il faut concilier entre un objectif de santé publique et la protection des droits de la personne, et des avancées thérapeutiques qui rendent nécessaires d'adapter les dispositions de surveillance épidémiologique.
J'en viens au sujet qui nous permettra d'introduire vos débats et qui concerne les décisions prises en matière de surveillance épidémiologique. On sait que les avancées thérapeutiques sont à la base de ces décisions. En France, cette décision résulte du décret du 6 mai 1999 qui inscrit sur la liste des maladies à déclaration obligatoire l'infection par le virus de l'immunodéficience humaine (VIH), quel qu'en soit le stade. Seule la déclaration du sida était auparavant obligatoire, au terme d'un décret de juin 1986 qui inscrivait également sur cette liste l'infection par le virus de l'hépatite B. Le Conseil national du sida a émis des réserves sur le décret du 6 mai 1999 qui n'a pas pris en compte son opposition à toute déclaration obligatoire de l'infection au VIH, telle qu'elle a été exprimée dans son avis de janvier 1998.
Le 29 octobre 1999, le CCNE a, lui, estimé que l'inscription de l'infection par le VIH et le VHB sur la liste des maladies obligatoires devait être maintenue. Il y a donc désaccord entre ces deux instances.
Quelle appréciation porter sur l'articulation entre un comité d'éthique généraliste et un organisme consultatif spécialisé (en l'espèce, le Conseil national du sida) ? En quoi leurs raisonnements se recoupent-ils, se distinguent-ils ou se contredisent-ils ?
Dans son avis critiquant l'instauration de la déclaration obligatoire, le Conseil national du sida relevait que l'obligation suppose un système illusoire de sanctions en cas de non-respect de la réglementation, voire un aménagement législatif du code de la santé publique d'autant plus inutile qu'il risque de voir la loi non totalement suivie, ce qui est, selon lui, prévisible. Quelle appréciation porter sur l'utilité des sanctions pénales dans ce domaine et plus généralement dans le domaine bioéthique ?
M. Jacques Brunschwig. - Notre thème de réflexion concerne le sida. Lorsqu'un individu est porteur du virus de l'immunodéficience humaine (VIH), comment informer le partenaire sexuel pour éviter une propagation de l'épidémie et sauver des vies humaines ? Dans le contexte actuel, en France, nous proposons une modification du code pénal.
Tous les quatre, nous étions très sensibles et formés aux problèmes posés par la bioéthique et les avancées des sciences dans ce domaine. Depuis deux ans, nous avons créé un groupe de travail pour réfléchir sur l'antinomie entre le droit au secret médical et la non-assistance à personne en danger.
Lorsqu'une personne est infectée par le virus de l'immunodéficience humaine, comment informer son, sa ou ses partenaires ? Ceci pose un problème éthique : il s'agit de sauver des vies humaines. C'est la raison de notre présence devant vous.
Notre groupe de travail est composé de M. Émile Papiernik, médecin-chef de la maternité de Port-Royal qui a une expérience exceptionnelle ; le professeur Pierre-Charles Ranouil, avocat, qui faisait partie du réseau Éthique, médecine et droits de l'homme au Conseil de l'Europe, Mme le juge d'instruction Isabelle Schoonwater, et moi-même, médecin stomatologiste à la retraite après quarante ans d'exercice libéral, membre du Conseil national de l'Ordre des médecins, membre du réseau Éthique médecine et droits de l'Homme du Conseil de l'Europe.
Le virus de l'immunodéficience humaine atteint toutes les couches sociales, hommes et femmes. C'est une maladie gravissime et très contagieuse. La trithérapie permet parfois de gagner quelques années de vie mais elle ne guérit pas. Prendre chaque jour quinze comprimés de trithérapie, plus une vingtaine d'autres comprimés, pour éviter des complications est une véritable épreuve.
N'oublions pas que les rapports sexuels entre un individu infecté et un partenaire non infecté sont le moteur de l'épidémie de VIH. Le VIH a provoqué une épidémie mondiale beaucoup plus importante que celle que l'on avait estimée il y a dix ans. Les chiffres de 1991 ont été multipliés de 50 %.
Je vous indique quelques chiffres provenant des estimations de l'ONUSIDA : en 1999, 34 300 000 personnes vivent avec le VIH sida dans le monde. En 2000, nous passons à 36 100 000 personnes ! Le dernier rapport, publié en décembre 2000, donne une première estimation de l'épidémie de VIH dans le monde. Cette estimation sera rectifiée dans le rapport définitif qui paraît chaque année en juin. En 1999, en France, pour une population de 59 millions d'habitants, l'estimation est de 130 000 personnes ; pour l'an 2000, on passe à 150 000 personnes.
En Espagne, pour 40 millions d'habitants, 120 000 cas. Épidémie plus développée qu'en France. En Italie, pour 57 millions d'habitants, 95 000 cas. En Suède, pour 9 millions d'habitants, 3 000 cas ; c'est-à-dire en proportion de la population française de l'ordre de 18 000, alors que, je le rappelle, la France est à 130 000 cas en 1999 et 150 000 en l'an 2000. Dans l'Europe de l'Est, de 420 000 en 1999, on passe à 700 000 cas en 2000. Il s'agit bien d'une explosion en Russie, due à la drogue.
Certains phénomènes évoluent, mais l'épidémie est toujours grandissante.
D'une part, depuis son apparition, en 1985, cette maladie ne concerne plus uniquement les homosexuels. En France, le dépistage du VIH est si déficient que 50 % des individus découvrent leur séropositivité avec l'apparition de la maladie. Sur le plan mondial, ce chiffre est de 70 %. D'autre part, phénomène nouveau, les femmes sont de plus en plus souvent contaminées : en France, 25 % des cas de maladie touchent des femmes, en Afrique, 55 %.
Dans cette situation, quel est l'avenir des enfants ? Pour Peter Piot, directeur exécutif de l'ONUSIDA : « Éviter à une nouvelle génération de jeunes la maladie et la mort précoce est une responsabilité de la plus haute importance. »
L'individu est responsable de ses actes envers lui-même, mais on oublie qu'il est responsable aussi envers autrui et la société.
M. Emile Papiernik. - L'apparition du sida dans notre société a provoqué, depuis 1985, des réactions de toutes sortes, mais aussi une terrible crainte d'ostracisme, d'exclusion. On a donc fait un effort remarquable pour protéger le privé. On a réussi à obtenir que les gens atteints de VIH soient soignés comme tout le monde. On a réussi une très importante mobilisation des moyens financiers. On a permis à la recherche thérapeutique de découvrir de nouveaux médicaments et de transformer incontestablement le sort des personnes atteintes.
Je fais partie de l'une des équipes qui ont, depuis des années, pris en charge les femmes séropositives et qui ont proposé un accueil respectueux et techniquement irréprochable, qui garantit un sort très différent aux femmes ainsi qu'une diminution formidable de la contamination des enfants pendant l'accouchement. On est à 1 ou 2 % au lieu des 30 % que l'on observe sans médicament.
C'est dans l'exercice de ce métier que j'ai découvert des situations qui m'ont alarmé. J'ai été bouleversé par certaines femmes qui demandaient de ne pas le dire à leur mari parce qu'elle ne le lui avait pas annoncé. Dans la situation actuelle, nous n'avons pas le droit de dire au mari, ou au compagnon, que la personne que l'on soigne est séropositive. Et la femme se dit incapable de le dire elle-même, parce qu'elle a peur de ses réactions, en particulier de la rupture de la relation.
Cela nous rappelle des souvenirs. Ce n'est pas la première fois que les médecins sont confrontés au problème d'informer le partenaire de la maladie sexuelle transmissible parce que, contrairement à ce qui est prescrit dans la loi, le sida, la séropositivité VIH est une maladie sexuelle transmissible. Après avoir été, pendant un certain temps, une maladie transmise par l'usage de la seringue, chez les usagers des drogues, elle est devenue une maladie sexuelle transmissible, quand elle s'est répandue dans la communauté homosexuelle. Mais dans le monde, aujourd'hui, et ce n'est pas négligeable en France, c'est le mode de transmission hétérosexuel qui est le mode de transmission majeur.
Quelle que soit l'information que vous lisiez, vous avez tous la même image de la transmission homosexuelle. Or, même en Russie, c'est la prostitution, et en Asie du sud-est, c'est le formidable commerce du sexe qui sont le vecteur de la transmission de la maladie. Et en Inde, la transmission hétérosexuelle est la première cause de la transmission de la maladie.
M. Jacques Brunschwig a dit que sur 36 millions de personnes atteintes, 43 % sont des femmes ; mais qu'en Afrique, ce chiffre passe à 55 %, parce que dans la relation sexuelle, les femmes sont plus souvent atteintes que les hommes.
En France, pour prendre des chiffres précis dans le bulletin de santé de l'ORS
- il s'agit de données pour 1998, car nous avons eu des difficultés à connaître la situation en raison de la grève des médecins inspecteurs de la santé -, sur 5 000 personnes contaminées par an, dont 2 000 en Ile-de-France, on sait que l'épidémie est aujourd'hui différente de ce qu'elle était à son début et qu'elle est autant observée chez les femmes que chez les hommes par transmission hétérosexuelle. Or, pour des raisons de protection de la personne, on n'a pas voulu mettre en place, pour le sida, les techniques utilisées jusque là pour lutter contre les maladies sexuelles transmissibles et on n'a pas osé aborder la question de la protection du partenaire ou de l'information due au partenaire. Alors que quand on a eu affaire à la syphilis et à la gonococcie, cela paraissait normal et simple. Ces maladies, dans les années 30, étaient aussi redoutables que le VIH aujourd'hui. Si la gonococcie ne tuait pas, la syphilis tuait. Il a fallu attendre la pénicilline, en 1945, pour constater un progrès véritable. Mais on avait décidé de lutter contre la contamination en établissant des règles de déclaration - question que vous avez à résoudre aujourd'hui.
Jusqu'à aujourd'hui, dans les déclarations de grossesse ou de certificats prénuptiaux, le dépistage de la syphilis est obligatoire, alors que celui du VIH est seulement conseillé. Il faut que la personne le demande. Pour des raisons de protection de la personne atteinte et pour contrer la peur de l'ostracisme, on a pris des attitudes collectives très différentes mais ce faisant, peut-être n'a-t-on pas complètement abordé les deux questions suivantes : a-t-on fait tout ce qu'il fallait pour diminuer la diffusion de la maladie et pour que les personnes non atteintes puissent être au moins informées ?
Les règles de confidentialité ont été réellement renforcées. Les pays d'Europe
- il n'existe pas encore de règle communautaire en la matière - ont retenu des règles plus ou moins différentes. Dans certains pays, comme la France, on a renforcé les règles du secret médical, c'est-à-dire que le médecin n'a jamais le droit de dire au partenaire qu'il y a un risque de contamination pour lui. D'autres pays d'Europe ont procédé différemment en libérant, dans certaines circonstances, le médecin du secret médical.
Ce n'est pas ce que nous allons vous proposer. Il faut arriver à trouver un accord entre le respect du secret médical, que nous estimons fondamental, et la possibilité d'informer. Nous faisons des propositions sur la façon d'organiser nos relations sociales, pour que l'on puisse, à la fois, être informé du risque et trouver les moyens d'encourager cette évolution. Nous pensons qu'il faut trouver une solution pour que les partenaires soient protégés contre la contamination, ou, qu'en tout cas, violence ne leur soit pas faite en les exposant à une contamination sans qu'ils aient été informés.
Pour conclure, notre opinion sur le rôle des médecins dans la qualité de l'information donnée au partenaire n'est pas fameuse, les médecins n'aimant pas du tout faire cette information, et une étude méticuleuse...
M. Pierre Hellier. - Ils n'en ont pas le droit.
M. Emile Papiernik. - Je parle des pays où ils en ont le droit. En France, on n'a pas le droit. Les médecins américains ont l'obligation d'informer les partenaires. Une étude américaine précise que 95 % des médecins qui soignent une personne contaminée n'ont pas fait l'information du partenaire. Le médecin est mal placé parce que, d'abord, il ne sait pas qui sont les partenaires et qu'il n'y a aucune raison qu'il le sache, et, ensuite, il n'estime pas qu'il soit en situation de le faire. Ce n'est donc pas du tout l'évolution que nous vous proposons, pensant que le respect du secret médical vaut plus que cela. En revanche, il faut trouver une autre solution.
M. Jacques Brunschwig. - La législation en France comporte deux volets : la déclaration des maladies vénériennes et la déclaration du sida. Les maladies vénériennes sont les maladies sexuellement transmissibles (MST) : la syphilis primaire, la syphilis secondaire, la gonococcie, le chancre mou et la maladie de Nicolas Favre. Dans les maladies vénériennes, il y a obligation de traitement. L'article L. 255 du code de la santé publique fait obligation au malade vénérien de se faire traiter pendant toute la période jugée contagieuse par son médecin. L'article L. 256 du code de la santé publique fait obligation à tout médecin, lorsqu'il diagnostique une maladie vénérienne contagieuse, de prévenir le patient du genre de maladie dont il est atteint, du danger de contamination et des devoirs que lui impose la loi de se traiter jusqu'à la disparition des risques de contagion. Voilà pour l'obligation de traitement pour la maladie vénérienne.
L'article L. 257 du code de la santé publique rend obligatoire la déclaration de maladies vénériennes en période contagieuse. Le médecin qui ne fait pas la déclaration est passible d'une amende. Cette déclaration se fait sous deux formes : déclaration simple avec la seule mention de l'indicatif du malade et du diagnostic (article L. 258 du code de la santé publique), et déclaration nominale avec mention du nom du malade et du diagnostic, obligatoire lorsque le malade ne commence pas le traitement ordonné, l'interrompt en cours de cure ou ne le reprend pas à la date indiquée sur l'avis de contagiosité qui lui a été remis (article L. 259 du code de la santé publique).
On peut aller plus loin, sur le fondement de l'article L. 275 du code de la santé publique, qui prescrit à l'autorité sanitaire de prendre toute disposition pour faire examiner, traiter, et au besoin hospitaliser d'office le malade. L'établissement de la feuille de déclaration est un carnet à souche propre à chaque médecin. Voilà pour les maladies vénériennes.
L'infection par le VIH n'est pas considérée comme une maladie vénérienne. En revanche, pour le stade évolué VIH sida chez l'adulte (à partir de 15 ans), la déclaration du sida est obligatoire aux termes du décret n° 86-770 du 10 juin 1986. Elle doit être faire à la direction départementale de l'action sanitaire et sociale. Cette déclaration est anonyme et comprend sur l'imprimé administratif l'initiale du nom, le prénom, le sexe, la date de naissance, le numéro de département du domicile et la nationalité.
Cette déclaration concerne les patients ayant une pathologie clinique qui répond aux critères du sida. Le 6 mai 1999, paraît le décret dit « décret Kouchner » n° 99-362 : « L'infection par le virus de l'immunodéficience humaine VIH, quel que soit le stade, fait partie de la liste des maladies devant faire l'objet d'une transmission obligatoire des données individuelles à l'autorité sanitaire. »
La proposition du « décret Kouchner » (article D. 11-1 du code de la santé publique) concernant la lutte contre les épidémies met l'infection VIH dans la liste des maladies infectieuses et dangereuses, au même titre que la typhoïde, la rage, la tuberculose, la peste, la maladie de Kreutzfeld-Jakob, le botulisme, le tétanos, etc. Mais cette disposition n'est pas appliquée car elle n'a pas paru respecter les règles de l'anonymat. Il en résulte une difficulté pour une mise à jour correcte de l'épidémiologie en France.
Actuellement, afin de protéger l'anonymat, un nouveau procédé est à l'étude. Les éléments d'identification seront codés et hachés de façon à être indéchiffrables. Après l'accord du Comité de surveillance du VIH, du Conseil d'État et de la Commission nationale de l'informatique et des libertés, ce procédé devrait être mis en place et une première analyse compréhensive d'épidémiologie sera réalisable fin 2002.
M. Pierre-Charles Ranouil. - C'est donc une maladie banale désormais et, en héritiers des Lumières, nous pensons que le législateur doit intervenir parce que la loi, pour nous, n'est pas seulement une simple mise en forme des m_urs dominantes, mais elle est aussi un phare. Dans cette circonstance, il nous a semblé qu'une loi pouvait être un phare pour éclairer cette société et amener chacun devant ses responsabilités.
Nous avions le choix entre deux hypothèses : soit - comme on le fait en pays étrangers - on délie le médecin du secret professionnel et on lui demande d'être un gardien, un détective, de faire ce qu'il n'est pas là pour faire, soit on essaie de trouver un moyen qui mette devant ses responsabilités la personne atteinte de cette maladie contagieuse - ou de toute autre maladie d'ailleurs - entraînant des risques de mort pour l'entourage.
Nous nous sommes longtemps demandés comment faire. Fort opportunément, une réforme du code pénal a récemment créé le concept de mise en danger de la vie d'autrui (article 223-1 du code pénal) qui considère comme un délit le fait de délibérément mettre en danger la vie d'autrui. C'est un texte remarquable : « Le fait d'exposer directement autrui à un risque immédiat de mort ou de blessure de nature à entraîner une mutilation ou une infirmité permanente par la violation manifestement délibérée d'une obligation particulière de sécurité ou de prudence imposée par la loi ou le règlement est puni d'un an d'emprisonnement et de 100 000 francs d'amende. »
Autrement dit, nous tenions là, à notre sens, le moyen d'arriver au résultat que le malade, solennellement, ne devait pas mettre en danger son entourage. Nous avons pensé qu'il serait assez « simple » de procéder de la manière suivante : d'abord ajouter, par voie législative, un alinéa à cet article du code pénal, libellé comme suit : « Sera puni des mêmes peines quiconque, se sachant atteint d'une maladie contagieuse présentant un risque de mort ou d'atteinte grave à la santé, n'informe pas les personnes qu'il expose délibérément à un risque de contamination. »
Se posait ensuite la question de savoir comment cette personne sera consciente de sa situation et de la situation juridique que va lui faire la loi. Nous avons pensé que, par voie réglementaire, il serait possible de mettre en place une procédure par laquelle le médecin qui rencontrerait un tel malade et qui diagnostiquerait cette maladie, lui signalerait qu'il est dans une situation grave et lui dirait ce qu'il lui incombe de faire.
Nous avons imaginé qu'à cette occasion, le malade pourrait signer un texte : « Le médecin qui portera pour l'un de ses patients le diagnostic d'une maladie grave et contagieuse susceptible de mettre en danger la vie de ses proches ou de porter gravement atteinte à leur santé :
« 1° Mettra solennellement au courant ce patient de sa situation et des risques qu'il fait courir à ses proches, ainsi que de l'obligation où il se trouve de les prévenir ;
« 2° Lui fera connaître la sanction pénale qu'il encourt s'il ne prévient pas ses proches susceptibles de contracter par lui la maladie dont il est atteint ;
« 3° Lui fera signer en double exemplaire - dont l'un sera conservé par ce médecin - le procès verbal suivant :
« Le ..., j'ai été avisé par le docteur X de ce que j'étais atteint d'une maladie grave et contagieuse - on ne précise pas la maladie - susceptible de mettre gravement en danger la vie et la santé de mes proches, et qu'en conséquence, il m'appartenait de les prévenir ; que le défaut d'information de mes proches était pénalement sanctionné par l'article 223-1, alinéa 2, du code pénal qui dispose... Signature. »
Pour terminer, nous pensons que ce dispositif devrait être complété par un renforcement du secret médical dans tous les établissements hospitaliers où, actuellement, ce secret, lorsque ce genre de choses vient à être connu, relève plus du secret de Polichinelle.
M. Bernard Charles, président. - En particulier, quand il faut venir prendre les trithérapies.
M. Jean-Paul Baquet. - Où à la caisse d'assurance maladie, quand on exhibe la feuille de l'assuré qui a le sida.
M. Pierre-Charles Ranouil. - Notre idée était donc de tirer parti de ce nouveau concept pénal, en l'appliquant à un cas particulier : « ...celui qui, sachant qu'il est atteint d'une maladie grave et contagieuse ne prévient pas ses proches ». Cela nous a paru tout à fait adapté à notre société qui met en avant la liberté et la dignité de l'individu. Nous n'attentons pas à sa liberté, mais nous le mettons face à sa responsabilité.
M. Jacques Brunschwig. - Cela définit l'alinéa 2 que nous vous proposons en additif à l'article 223-1 du code pénal.
M. Pierre-Charles Ranouil. - C'est une application pratique de cette notion. Vous avez sans doute entendu parler d'une application refusée de cet article 223-1 du code pénal lorsque la Cour de cassation a estimé que conduire sa voiture à tombeau ouvert sur la route n'était pas un délit. En l'occurrence, il s'agirait d'une application plus concrète : ne pas le faire savoir à son entourage susceptible d'être contaminé par une maladie grave et contagieuse alors qu'on a été solennellement prévenu que l'on en est atteint.
Mme Catherine Génisson. - Pourquoi pas ? Mais cela ne suffira pas.
M. Pierre-Charles Ranouil. - C'est un acte de foi dans nos concitoyens, nous les éclairons par cette loi.
Mme Isabelle Schoonwater. - En conclusion, nous avons choisi la responsabilité individuelle, le renvoi à la responsabilité du citoyen. C'est vrai que nous avons choisi le droit pénal parce qu'un article du code pénal a une forte symbolique qui correspond, pour nous, à la gravité de cette épidémie et à la nécessité urgente d'intervenir.
Nous pensons qu'un texte du type de celui que nous proposons, surtout entrant dans ce titre du code pénal sur la mise en danger d'autrui, peut être très fort, peut avoir une valeur utile de pédagogie et d'exemplarité et peut être dissuasif. Au moins, nous pensons que cela mériterait d'être essayé.
Par ailleurs, je crois que le texte que nous vous proposons entre bien dans le champ du principe de précaution que l'on ne peut pas méconnaître, aujourd'hui, devant une épidémie d'une telle ampleur que le VIH.
M. Pierre Hellier. - Au début de vos interventions, j'ai été quelque peu effrayé par vos propos. La notion de secret médical est pour moi essentielle. Si votre proposition avait tenté de le faire disparaître, j'aurais été extrêmement réservé. Il est exact que le traitement, aussi bien de la part des élus que des médecins, de la maladie du sida a été particulier. Le contexte de départ, qui en faisait pratiquement une maladie « divine », qui tombait sur une population à risque, fait que le législateur a pris des dispositions parfois curieuses et frileuses. C'est vrai que le médecin déroge déjà au secret médical puisqu'il fait la déclaration. Lui demander, en plus, de faire de la délation me paraissait totalement impossible et exclu.
M. Bernard Charles, président. - D'ailleurs, il ne l'a pas fait dans les endroits où on l'y a autorisé.
M. Pierre Hellier. - En outre, cela aurait eu une portée extrêmement faible, parce que l'on aurait pu à la rigueur prévenir le partenaire que l'on pouvait rencontrer, mais prévenir d'autres partenaires était une tâche insurmontable que vous auriez demandée au médecin.
Sous réserve d'une réflexion et d'une analyse plus précise, votre proposition me semble raisonnable. Le médecin garde le secret professionnel. Il informe de façon solennelle - c'est une chose que j'accepterais vraisemblablement - son malade de la situation et lui fait prendre les décisions qui s'imposent. À mon avis, on ne peut pas faire beaucoup plus. Dans notre pays cela va marcher. J'étais au Rwanda récemment, je ne vois pas comment le médecin pourra supprimer ou réglementer le vagabondage sexuel qui est la pratique dans ce pays. Mais dans des pays comme le nôtre, cela devrait avoir une certaine action.
M. Jean-Paul Bacquet. - Sur l'ensemble des membres de notre mission, nous sommes aujourd'hui cinq, dont quatre médecins. Cela veut dire quelque chose. Mon propos sera sans doute décousu mais sera à l'image de ma façon d'aborder les choses.
Notre véritable attitude face à cela est le désarroi. Vous avez eu raison d'aborder le problème sous l'aspect du secret médical et de l'information, car il est évident que c'est là que se situe le problème. Votre proposition me semble parfaitement raisonnable, mais je ne suis pas du tout sûr qu'elle soit efficace. Finalement, cette proposition va permettre au médecin d'être couvert pénalement mais en aucun cas de régler le problème du patient. Pour celui qui dit : « Je me suis fait avoir, il n'y a aucune raison que je prenne les précautions pour les autres que l'on n'a pas prises pour moi »
- situation qui est plus fréquente qu'on ne le croit - vous ne réglerez pas le problème.
Cela étant, on a connu d'autres maladies non vénériennes mais contagieuses : le problème de la tuberculose que l'on a réglé, à une époque où l'inefficacité thérapeutique était quasi totale, par l'isolement - rappelez-vous le sidatorium cher à M. Le Pen -, par un dépistage systématique. Sont-ce de bonnes solutions sur le plan médical ? C'est un autre problème, mais on ne peut pas dire que l'on ne peut rien faire. On peut faire quelque chose. Cela demande des responsabilités très lourdes au législateur pour dégager pénalement le médecin qui, lui, aurait à prendre une décision de ce type, alors que le médecin doit se couvrir pénalement par rapport au procès que peut lui faire le patient, voire l'association de défense du patient qui, souvent a aggravé la situation
- certaines associations en ont conscience aujourd'hui - parce qu'elles ont eu un effet négatif, à l'encontre de ce qu'elles recherchaient.
M. Alain Claeys, rapporteur. - Mais que peut-on alors faire pour dégager la responsabilité pénale ?
M. Jean-Paul Bacquet. - Pour prendre l'exemple du malade dangereux en psychiatrie, il existe un cas particulier, dans lequel on procéde à un internement d'office. On a bien trouvé la solution dans ces cas-là. Je ne dis pas qu'il faut faire un traitement d'office. Je veux dire que l'on exclut d'office une partie des patients au prétexte que, sur le plan pénal, le médecin n'a pas les moyens et que c'est un homme libre. Sauf qu'il a une responsabilité vis-à-vis de la santé publique.
Or, nous n'apportons pas de solution. Ce n'est pas un procès que je vous fais. Je me pose les mêmes questions et je n'ai pas trouvé les solutions. En outre, il y a le comportement des hommes. Cette maladie a été vécue, au début, comme une maladie très culpabilisante. Aujourd'hui, lorsque l'on fait un sérodiagnostic, on doit le faire avec le consentement du patient. Or, tous les jours, dans les hôpitaux et cliniques, les personnes entrent avec des prises de sang où on leur fait d'emblée un test VIH sans leur demander l'autorisation. (Signes de dénégation de Mme Catherine Génisson). C'est un bilan systématique.
Peut-être n'est-ce pas le cas à Port-Royal. Je pourrais citer des services dans lesquels c'est systématiquement fait. Quand le patient vous dit que, de toute façon, on le suspecte de cela, mais qu'il n'en veut pas, vous ne le faites pas, même si tous les symptômes sont là, qu'il fait partie des gens infectés. Il a le droit de refuser. On n'apporte pas la solution, je ne fais que le remarquer.
Et puis, il y a aussi la pratique. Le secret ne sera pas respecté, au niveau administratif, que ce soit à la Sécurité sociale ou dans certains services de médecine du travail. Je peux en donner des exemples concrets. Ce n'est pas la loi qui réglera ce problème.
Enfin, quelle que soit la décision que nous aurons à prendre, si nous n'avons pas le courage de l'assumer médiatiquement - c'est pourquoi, je faisais le rapprochement avec la tuberculose - nous aurons toujours un comportement inexplicable. La déclaration des maladies obligatoires n'est pas à la hauteur du nombre de maladies à déclaration obligatoire, et de loin. Or, la médiatisation de cette maladie était très forte parce qu'elle avait un caractère beaucoup plus moral que médical ; elle est devenue médicale, mais elle était beaucoup plus morale au début.
M. Alain Claeys, rapporteur. - Avez-vous eu une discussion avec les représentants du Conseil national du sida ? Quand ils écrivent : « l'obligation suppose un système illusoire de sanction en cas de non-respect de la réglementation. » Ce n'est pas simplement une déclaration d'intention. Une étude critique des lois de 1994 constate qu'il n'y a pas eu, depuis cinq ans, de sanctions pénales prononcées, au titre des lois de 1994.
M. Emile Papiernik. - Nous ne parlons pas du même problème. Mme Isabelle Schoonwater nous a bien dit que l'objectif que nous vous proposons d'atteindre est celui de la prise de conscience. Aujourd'hui, si une personne contaminée fait un procès, face à son contaminateur, elle perd. Si une personne contaminée fait un procès à quelqu'un qui l'a contaminée sans l'informer qu'il était séropositif, elle perd.
Notre proposition est de dire que la responsabilité ne peut pas être uniquement celle du médecin, mais que le patient, celui qui peut contaminer autrui, a aussi un devoir. Il n'a pas seulement des droits à être soigné. Ils ont été respectés - et réellement on a bien fait - mais il a aussi un devoir. Ce n'est plus l'époque de la tuberculose ou de la lèpre où l'on mettait les gens en exclusion. C'était cela la crainte initiale.
M. Alain Claeys, rapporteur. - Comment expliquez-vous qu'en Suède les chiffres soient beaucoup moins élevés.
M. Emile Papiernik. - Ils ont été plus prudents pour les transfusions, ont distribué très rapidement des seringues propres alors que nous avons attendu très longtemps pour le faire. Ils n'ont pas eu de problème avec les transfusions. Ils ont osé faire la déclaration. Ils ont pris des précautions considérables.
Mme Catherine Génisson. - Notre collègue a bien décrit la situation de départ : c'était une maladie « divine » qui s'abattait sur la communauté que l'on connaît, avec une approche moralisatrice de cette maladie. Cela nous a beaucoup pénalisés.
Au-delà de la proposition que vous faites, qui aura probablement des résultats, mais limités par l'analyse qu'en a faite Jean-Paul Bacquet - on connaît des malades sidéins pervers qui se vengent, c'est une réalité - nous en sommes à un point de l'épidémie de cette maladie où, compte tenu de ce que vous avez décrit, de ce que cela devient une « maladie banale », avec une contamination hétérosexuelle telle qu'on la connaît pour beaucoup de maladies contagieuses, il faut faire en sorte - c'est un v_u pieux que de le dire - que les communautés concernées considèrent que cela devient un problème de santé publique. Il ne faut plus cet environnement moralisateur autour de cette maladie. Il faut en faire un problème de santé publique, celui d'une maladie contagieuse, qui doit entrer dans le circuit normal des maladies contagieuses. Il faut s'engager dans cette voie.
M. Pierre Hellier. - Le contexte politique est différent. À l'époque, un certain parti utilisait des arguments pour son usage personnel. Le contexte est complètement différent aujourd'hui. À la réflexion, votre proposition n'apportera peut-être pas une certitude absolue d'efficacité, mais elle aura quand même une efficacité indiscutable.
Cela dit, en cas de procédure, il faudrait que la personne qui va porter plainte contre ce conjoint qui l'aurait contaminée signe aussi un papier disant qu'elle n'a pas été informée. On peut aller très loin car au fond, pourra-t-on prouver qu'elle avait été informée ?
M. Jean-Paul Bacquet. - Il y a un autre débat qu'il convient de ne pas négliger : quand on s'adresse à une population jeune, la notion de la mort et du risque n'est pas du tout la même. La signature d'une attestation de ce type n'engage pas du tout de la même façon une personne plus âgée qui a la notion de la responsabilité, la notion de la vie. Vous parliez d'accidents de voiture, la notion de la vie et de la mort n'est pas du tout la même chez un jeune de 18-20 ans que chez un quadragénaire. Et sa responsabilité individuelle par rapport à un problème de santé publique - une responsabilité collective - n'est pas vécue de la même façon, et encore moins quand on supprime tout ce qui pouvait donner une responsabilité collective à la société. Je pense notamment à la notion de citoyenneté, au service national etc.
M. Jacques Brunschwig. - Le problème des jeunes que vous évoquez est très important. À cela s'ajoute un facteur particulier : du fait de la trithérapie qui induit une rémission de la maladie pour de nombreux malades du sida, la jeune génération ne voit plus les effets dévastateurs de cette maladie. D'où des prises de risques chez les jeunes. Nous avons le devoir de faire des choses très positives à destination de ces jeunes qui n'ont plus conscience de la gravité des choses. Sur les populations à risque, il y a sur Paris notamment des group parties avec rapport multiples et multi partenaires dans la même soirée et sans protection ! C'est effarant.
M. Jean-Paul Bacquet. - Un très bon article rapportait qu'un journaliste avait visité des établissements de ce type où il n'y a même plus de distributeur de préservatifs.
Sur les lois ou les décrets, sur le problème de la seringue que vous avez évoqué, nous avons mis les médecins dans l'obligation de s'abonner à des systèmes de récupération de déchets médicaux etc. Une question orale a été posée hier matin sur les seringues : pour les diabétiques, il n'y a aucune obligation qui soit faite. Tout va à la poubelle. Quand on fait une loi, il faut qu'elle s'applique à tous. On ne peut pas s'arrêter à la pénalisation du corps médical !
M. Emile Papiernik. - On ne fait pas de santé publique sans préciser les responsabilités de chacun. Nous pensons que la seule stratégie possible, même si elle n'est pas totalement efficace, est « d'afficher la couleur » et de situer clairement la responsabilité de chacun. Pas plus, mais pas moins.
M. Bernard Charles, président. - Nous avons eu un débat de qualité. Nous allons l'intégrer à notre réflexion.
Audition de MM. Thierry SUEUR,
président du comité de la propriété intellectuelle,
et François CHRÉTIEN,
président du groupe de travail biotechnologies,
du Mouvement des Entreprises de France
(Extrait du procès-verbal de la séance du 24 janvier 2001)
Présidence de M. Bernard Charles, président.
M. Bernard Charles, président. - Vous avez souhaité être auditionnés par notre mission. Nous procédons à des auditions dans le cadre de la révision des lois de bioéthique de 1994.
Nous avons reçu deux entreprises de biotechnologie : Genset et genOway. Outre les aspects éthiques et scientifiques, nous avons souhaité être mieux éclairés sur les enjeux économiques des évolutions scientifiques. C'est un élément important pour nous. Dans certains secteurs, que je connais assez bien, nous avons vu que le manque d'organisation, à la différence d'autres pays d'Europe, nous avait affaiblis. Je pense en particulier aux problèmes globaux de l'industrie pharmaceutique. Et nous ne souhaiterions pas rencontrer les mêmes problèmes, à terme, dans le domaine des biotechnologies.
M. Alain Claeys, rapporteur. - Madame, messieurs, vous avez souhaité être entendu par notre mission d'information. Comme l'a rappelé le président, nous avons déjà rencontré des entreprises travaillant dans le domaine des biotechnologies.
Trois aspects nous intéressent plus particulièrement. Tout d'abord, de votre point de vue, quelles sont les forces et les faiblesses des industries biotechnologiques dans l'industrie européenne par rapport aux États-Unis, et comment les industries françaises se situent-elles dans ce contexte européen ?
Le deuxième aspect porte sur les brevets, sujet d'actualité. Je souhaiterais que vous soyez le plus précis possible. Qu'est-ce que qui distingue l'approche de la question des brevets en Europe et aux États-Unis ? Des exemples précis peuvent-ils être donnés de ce qui relève de la découverte et de ce qui relève de l'invention pour l'application du droit des brevets ? Notre mission a eu des informations parfois contradictoires. Je pense que, même si ce sujet ne figure pas directement dans l'avant-projet de loi du Gouvernement, il n'est pas impossible que nous devions l'aborder, soit à travers les lois bioéthiques, soit à travers la transcription de la directive européenne.
Deuxième thème : les tests génétiques. Pour vous, quels sont les enjeux économiques des tests génétiques ? Quelle est la place des entreprises françaises dans l'industrie des tests génétiques ? Des craintes ont été exprimées devant les conséquences discriminatoires de l'utilisation des tests génétiques à l'embauche. Selon vos informations, de tels tests ont-ils déjà était pratiqués, comment et sous quelle forme ?
Dernier point, pour que l'on comprenne bien les enjeux scientifiques et économiques des recherches en cours, concrètement, quelles sont les applications possibles du décryptage du génome humain aujourd'hui, de la recherche sur les cellules souches ? Quelles sont les applications possibles dans l'industrie du médicament ? Dans quels pays et dans quelles conditions ces recherches sont-elles déjà conduites et à quel horizon est-il raisonnable de penser que ces recherches trouveront leurs premières applications thérapeutiques ? Quelles en sont aujourd'hui les retombées à court terme ?
M. Thierry Sueur. - L'étendue des questions que vous avez posées étant assez vaste, je ne suis pas sûr d'avoir la compétence pour répondre sur tout. Je propose d'introduire ce débat en précisant que je suis probablement le plus incompétent des membres présents dans la mesure où, si je préside le comité de la propriété industrielle ou intellectuelle du Medef, je travaille à Air Liquide, qui n'a pas d'activité dans ce domaine.
Je propose donc de vous donner quelques éléments sur la propriété industrielle et plus particulièrement sur le brevet. On a pu lire beaucoup de choses dans ce domaine qui montrent que l'outil, en tant que tel, n'est pas parfaitement appréhendé. Je présenterai quelques principes de base qui s'appliquent à la propriété industrielle, et au brevet spécifiquement, quels que soient les domaines d'activité.
Tout d'abord, le brevet est quelque chose d'important pour les entreprises parce que c'est l'outil par excellence qui permet de progresser et d'innover. Il n'y a pas d'innovation durable sans brevet. Je dirai pourquoi tout à l'heure. Mais le brevet n'est pas le début et la fin de toutes choses. Cela a des avantages, des inconvénients et des limites assez strictes.
Et puis, il y a des choses que le brevet n'est pas. Il n'est pas un droit de propriété sur un objet. Quand vous avez un brevet, cela ne donne pas droit à quoi que ce soit, si ce n'est d'interdire quelque chose. Quand vous avez un brevet, en particulier dans le domaine du médicament, cela ne vous donne pas le droit d'avoir une activité. Je peux avoir tous les brevets du monde, si je n'ai pas une autorisation de mise sur le marché (AMM) donnée par un État, mon brevet ne reste qu'un diplôme. Je ne peux rien faire en tant que tel. Cela ne donne pas une propriété sur un objet. Cela donne un droit d'empêcher un tiers que je n'ai pas autorisé à reproduire mon invention. C'est tout. Je peux interdire. Par différence, je peux autoriser, c'est-à-dire que je peux donner une licence. Voilà tout ce que je peux faire avec un brevet, ni plus ni moins, et ce, pendant une durée généralement limitée à vingt ans au maximum.
M. Bernard Charles, président. - Le certificat complémentaire de protection permet à certains secteurs de repousser quelque peu l'échéance.
M. Thierry Sueur. - Si l'on examine les statistiques, la durée des brevets est de six ou sept ans parce que les inventions sont rapidement obsolètes ou entrent dans le domaine public.
Deuxième point : avec un brevet, j'ai dit que je peux interdire à un tiers de faire la même chose que moi. Mais pas toujours ! Par exemple, je ne peux pas interdire l'expérimentation qui est libre. C'est d'ailleurs l'un des intérêts du brevet.
Revenons en arrière : le brevet existe depuis un peu plus de 200 ans. En matière technique, le principe de base existant dans tous les pays est que quand on parle d'une invention, on la décrit, on la publie, on la met dans le domaine public. À partir du moment où j'ai expliqué comment on fabrique tel objet, n'importe qui dans le monde a le droit de le faire. Les États se sont aperçus, à l'aube de la civilisation industrielle, que ce n'était peut-être pas le meilleur moyen d'inciter à innover. Si je dépense de l'argent pour innover et que tout le monde va pouvoir en profiter, je dépense de l'argent pour le roi de Prusse, j'hésite quelque peu. La réaction des gens est donc de cacher leur copie, de cacher ce qu'ils font, de garder le secret, comme au Moyen Âge ou dans le compagnonnage. Le brevet est l'antithèse de cette approche.
La philosophie du brevet est de déposer l'invention et de la faire publier. Sachant que tous les brevets sont publiés au bout de dix-huit mois, tout le monde en aura connaissance. On va donc enrichir le patrimoine technique et la connaissance de l'humanité. Il faut savoir que le brevet est la première source de documentation technique, devant toutes les publications existantes. En contrepartie de cet apport que je fais, chacun des États va me donner un monopole temporaire. C'est une sorte de deal, de contrat : on décrit son invention, on la divulgue, tout le monde pourra en profiter le jour où le brevet tombera dans le domaine public ; en contrepartie, on a le monopole temporaire. Voilà la philosophie du brevet.
J'ai déjà précisé que je peux opposer ce brevet à une activité commerciale, mais je ne peux pas l'opposer à une expérimentation. On voit l'un des premiers intérêts du brevet : il va enrichir la connaissance puisqu'il va être publié, il va permettre à chacun
- laboratoires nationaux, d'entreprises - de faire des tests, des essais, d'expérimenter et donc de perfectionner. Pour donner des exemples de la vie quotidienne, la société pour laquelle je travaille a des concurrents, même si nous sommes le numéro un mondial dans notre domaine. Chaque jour, je regarde les brevets de nos concurrents. Parfois, je vois des brevets où je sens que nous aurons des difficultés. Que faire ? Première solution, on essaie de détruire ce brevet, de l'annuler. Parfois, on y réussit, parfois, on ne le peut pas. Nos inventeurs sont là et cela les stimule, lorsque nous organisons des séances de brainstorming pour trouver une solution alternative. C'est cela qui fait le progrès de tous les jours : faire mieux que l'autre, trouver une solution plus performante, moins chère, d'un meilleur rendement pour un progrès remarquable. Voilà toute la philosophie du brevet.
M. Alain Claeys, rapporteur. - Vous nous décrivez l'aspect positif des brevets. C'est vrai, cela protège l'invention et cela permet de divulguer, d'informer par la publication. Mais nous sommes là dans le domaine du vivant. Pour ceux qui sont opposés à la transcription de la directive européenne, on entend deux types d'arguments : des arguments éthiques et des arguments économiques.
Les arguments éthiques sont de dire que l'on ne peut pas breveter une partie du vivant. Très schématiquement, dans le séquençage du génome, nous découvrons un gène qui a une propriété, une protéine qui pourra éventuellement apporter des solutions thérapeutiques. Que brevète-t-on ? Brevète-t-on le gène ? C'est donc une partie du vivant. En brevetant le gène, on va breveter éventuellement d'autres applications que celle appartenant à celui qui a découvert la protéine donnée. Ceci est l'aspect éthique de l'invention dans le domaine du vivant.
Mais il y a aussi, par rapport aux intérêts bien compris des industries françaises ou européennes, un argument commercial, économique, disant qu'il ne faut peut-être pas généraliser le « brevetage » du vivant. En raison du retard que nous avons aujourd'hui par rapport à des firmes américaines, les entreprises françaises ou européennes ne vont-elles pas, au-delà de l'expérimentation, être totalement dépendantes des périmètres brevetés par l'industrie américaine ?
Ces deux aspects, qui peuvent paraître contradictoires, se retrouvent parmi les signataires de la pétition élaborée par notre collègue Jean-François Mattéi. Des personnes ont signé de bonne foi cette pétition sur des valeurs éthiques disant que l'on ne peut pas breveter le vivant, d'autres ont abordé cela sous l'angle économique.
M. Thierry Sueur. - Je répondrai à la deuxième question concernant la matière économique et je demanderai à François Chrétien de répondre à la première.
D'abord, ce sont des préoccupations économiques et éthiques qui concernent tout le monde, y compris des chefs d'entreprises. C'est un vrai souci et je me réjouis de cette discussion ; j'espère que nous vous apporterons des éléments mais j'attends aussi que l'on m'éclaire un peu. Les choses ne sont pas toujours tranchées et demandent quelques explications.
Pour revenir à l'aspect économique, ce n'est pas la première fois que ce débat a lieu. Le débat sur la question de savoir s'il faut breveter ou non a eu lieu au moins deux fois dans les cinquante dernières années. Il a eu lieu sur la chimie : fallait-il breveter les molécules ? Certains considéraient qu'il n'était pas possible de breveter les molécules chimiques, que l'on risquait ainsi de scléroser complètement le progrès. On parlait des limites entre découvertes : je découvre une molécule existant dans la nature ou je la synthétise. L'histoire a tranché : à l'époque, la plupart des pays - tous aujourd'hui - ont protégé leurs molécules chimiques. L'industrie chimique n'a jamais été aussi florissante pour le bénéfice de tous !
S'agissant du médicament, j'ai également retrouvé un débat similaire remontant aux années 40-50. Le brevet n'était pas acceptable pour la santé publique, les gens risquaient de ne plus pouvoir se soigner, de conserver les médicaments. Il faudra un jour dessiner la carte des premiers pays qui ont accepté la brevetabilité des médicaments et la calquer sur les pays qui ont une industrie pharmaceutique : ce sont les mêmes. Quant aux pays qui ont refusé, à l'époque, de breveter, ils correspondent à ceux qui, aujourd'hui, n'ont pas d'industrie pharmaceutique.
M. Alain Claeys, rapporteur. - Vous considérez que le brevet est pour l'industrie française un moyen de rattraper le retard ?
M. Thierry Sueur. - C'est une bonne question, et l'approche « épidermique » consistant à dire que « les jeux sont déjà faits » est celle que j'ai entendue, depuis vingt ans, de la part des pays en développement. Ce fut le leitmotiv des pays non industrialisés, convaincus que le brevet était mauvais pour eux et qu'ils ne rattraperaient jamais leur retard. Aujourd'hui, des pays comme l'Inde ou la Jordanie affirment qu'ils ont besoin du brevet pour attirer les investissements et susciter l'innovation.
Cela me paraît intéressant de le souligner parce que l'on a un précédent. Ce n'est pas de la théorie. J'ai entendu récemment reposer la même question concernant la protection des logiciels. Les Américains dominent le marché, ne vaut-il mieux pas abandonner ? C'est une attitude défensive, de vaincu, qui au bout du compte tue l'activité économique.
M. François Chrétien. - Le droit harmonisé du brevet en Europe existe depuis la convention de 1973 sur le brevet européen. Bien évidemment, c'est l'époque où sont arrivés les brevets de biotechnologie.
La question s'est posée de savoir si l'on pouvait prendre des brevets sur des inventions qui impliquaient la matière biologique. Les Américains nous avaient quelque peu devancés avec la décision Chakrabarty de 1980 de la Cour suprême sur des micro-organismes mangeurs d'huiles et de pétrole, estimant qu'il n'y avait pas lieu de refuser à l'inventeur le bénéfice du droit classique des brevets.
Le droit européen s'en est lui-même préoccupé, mais la convention de 1973 n'avait pas donné de consignes précises à ce sujet. Cependant, un article 53 A précisait que tout ce qui est contraire à l'éthique est exclu du droit des brevets. On retrouve cette exclusion dans la loi française et dans toutes les lois.
Qu'est-ce qui pouvait tomber sous le coup de cet article ? L'exemple classique est celui de la lettre piégée. Mais pour les armes, ce n'est pas le brevet qui va décider de l'utilisation de l'arme : vous pouvez assassiner votre voisin mais aussi vous défendre...
Avec les biotechnologies, cette question est revenue au devant de la scène. On pouvait se poser les questions éthiques touchant l'ordre public et les bonnes m_urs, au sens de la loi. Et comme le texte de 1973 était imparfait, c'est la jurisprudence qui a actualisé le droit. En 1994, une décision sur le gène de relaxine a repris la problématique en se demandant si, en droit européen, il était possible de prendre des brevets sur la matière biologique et sur ces fameuses séquences de l'ADN. La réponse a été clairement oui. Il y a eu d'autres décisions sur la brevetabilité des plantes et des animaux.
Ensuite est arrivée la directive 98-44, dont la discussion a commencé en 1988 et que j'ai suivie depuis le commencement. Je peux dire que l'on est parti d'un texte essentiellement technique, pour savoir comment adapter le régime général des brevets aux inventions concernant la matière biologique, cette dernière ayant deux caractéristiques particulières : elle est animée, alors que la plupart des inventions son inertes, et elle se reproduit ou on peut la faire reproduire, ce qui était totalement nouveau. Le processus d'adoption a été extrêmement long - dix ans - avec une procédure assez exceptionnelle puisqu'un texte de conciliation a été rejeté en mars 1985 à la surprise quasi générale.
Sur ce problème de protection des ADN d'origine humaine, la Commission, le Parlement et les industriels ont alors constaté qu'il y avait un défaut de communication, un défaut d'explication et qu'il était indispensable de mieux expliquer ce qu'est le brevet et ce que sont ses implications par rapport au vivant.
M. Alain Claeys, rapporteur. - L'article 5 de la directive est contesté aujourd'hui, à la fois par le président de la République et le Premier ministre, qui en souhaitent une relecture. Pourriez-vous nous préciser les choses. Quel est l'enjeu de cette discussion ? Pensez-vous qu'elle est justifiée ?
M. François Chrétien. - Si j'ai bien compris la position de Jean-François Mattei, il est globalement en faveur de la directive parce qu'il pense qu'il faut une protection de l'innovation. Ce qui lui pose problème, c'est la brevetabilité produits des ADN impliqués dans les inventions brevetées.
Nous avons préparé un petit schéma explicatif. Nous sommes dans le domaine génétique. Nous avons affaire principalement à deux types de matière génétique : les gènes et les protéines dans la mesure où certains gènes codent pour des protéines, c'est-à-dire qu'ils commandent la formation et la création de ces protéines.
Première remarque, ces produits sont des molécules chimiques définies. Même s'il y a un caractère informatif particulier, cet aspect est très important parce qu'il ouvre la voie à la question de la protection produits. Or, M. Thierry Sueur vous a expliqué que la protection produits constitue un élément décisif dans la protection par brevet, tout simplement parce que celui qui a trouvé un produit, même s'il n'en a trouvé qu'une première application, dans un premier temps, a bien trouvé, le premier, ce produit. Il est donc bien inventeur de ce produit et il a le droit à un monopole, limité dans le temps, sur cet aspect produit. Voilà pour la règle globale.
Pour entrer dans le détail, ces matières génétiques sont réparties entre deux grands groupes et un ensemble de sous-groupes. Pour les gènes, on est parti du plus général au particulier. Sous la forme la plus globale, c'est ce que l'on appelle le génome, il s'agit de toute l'information génétique contenue dans une cellule humaine. Un sous-ensemble est constitué par les chromosomes, ce sont des ensembles de gènes. Ensuite, on a des éléments beaucoup plus particuliers, c'est-à-dire, l'unité que l'on appelle le gène, qui, lorsqu'il est dans la nature, est capable de coder naturellement pour une protéine. Il y a ensuite l'ADN, dont une séquence nue correspond à ce que l'on appelle la partie codante de la protéine.
Au niveau de la protéine, on retrouve, de façon similaire, des ensembles de protéines quand on parle du génome et des chromosomes ; mais quand on parle d'un gène ou d'une séquence ADN, c'est une protéine particulière qui est concernée.
M. Alain Claeys, rapporteur. - Dans votre tableau où commence l'invention ?
M. François Chrétien. - J'y arrive. La première colonne, pour les gènes, précise clairement que toutes les formes, les sous-types de matière génétique, quand elles sont dans le corps humain, sont non brevetables. C'est une exclusion d'ordre éthique reprise par l'article 5 de la directive.
Pour aller plus loin, lorsqu'on isole cette entité génique du corps humain et qu'on la séquence, on caractérise tous les maillons de cette chaîne et leur nature. Et on s'arrête là. Vous avez fait une découverte absolument sensationnelle mais aucune invention. Autrement dit, à ce stade là, vous ne pouvez pas obtenir de brevet.
Certes, des gens ont essayé de déposer des brevets, mais cela ne suffit pas. Encore faut-il les obtenir. Il faut passer un examen au fond, sur la qualité de brevetabilité de l'invention. Elle est étudiée par des organismes experts comme l'Office européen des brevets, ou les offices américains ou japonais des brevets, dont les examinateurs sont tout à fait aptes à porter un jugement sur ce type de demande.
M. Alain Claeys, rapporteur. - Un gène séquencé est une découverte mais pas une invention, il n'est donc pas brevetable.
M. François Chrétien. - Il est donc exclu de la brevetabilité.
M. Thierry Sueur. - C'est un principe général. La découverte, c'est la constatation de ce qui existe. Ce n'est pas brevetable, et ce, dans tous les domaines d'activités.
M. Bernard Charles, président. - Même isolé. On découvre, mais on ne peut pas le protéger.
M. François Chrétien. - Pour les praticiens des brevets, cela coule de source. Cela fait partie de l'enseignement de base en matière de propriété intellectuelle. Je me souviens des débats au Parlement européen. Nous avons très bien compris que ces débats avaient eu un impact particulier en raison même du sujet et qu'il fallait donc rappeler certaines règles dans l'article 5 afin d'obtenir une meilleure visibilité vis-à-vis du Politique. Mais cela n'a pas changé le droit des brevets. La propriété industrielle était capable de faire son propre tri, indépendamment des questions éthiques, simplement par l'application des règles traditionnelles qui distinguent l'invention de la découverte.
On passe ensuite au sous-type « protéine » : ensemble de protéines ou une protéine particulière. Pour les mêmes raisons que précédemment, si la protéine est dans le corps humain ou si elle est isolée et séquencée, vous ne pouvez pas obtenir un brevet.
Cela change seulement lorsque, après avoir détecté, isolé, séquencé le gène, vous lui avez trouvé une fonction technique de production de la protéine et qu'en outre, la protéine a une application industrielle. En effet, si vous obtenez la protéine et que vous ne savez pas à quoi cela peut servir, vous n'aurez pas de brevet. Il faut aussi une application industrielle.
M. Alain Claeys, rapporteur. - On peut breveter uniquement quand il y a application industrielle.
M. François Chrétien. - C'est une énorme nuance. Pour la partie génome, la partie la plus globale, il n'est pas possible de prendre un brevet. Parce que c'est tellement complexe que vous ne répondrez jamais aux critères de définition d'une invention.
En outre, il faut tenir compte de l'initiative sur le programme du génome humain qui recommande à tous les laboratoires qui font du séquençage de publier les séquences qu'ils auront trouvées. Quelles en sont les conséquences ? Cela répond au souci du professeur Jean-François Mattei. Dans ce cas-là, la molécule, en tant que telle, est connue ; elle n'est donc pas nouvelle. Elle est du domaine public. Celui qui veut prendre un brevet ne peut le faire que sur la partie application.
Pour conclure, il faut relever que si le séquençage offre un degré de précision qui s'améliore, il demeure des « niches », c'est-à-dire des gènes particuliers qui ne sont pas encore trouvés. Dans la mesure où vous avez satisfait aux conditions que j'ai indiquées, qu'en application des règles générales de brevetabilité, le caractère nouveau, inventif, c'est-à-dire non évident par rapport à l'état de la technique, a bien été démontré, qu'en outre, l'application industrielle a été exposée de façon concrète et ne doit pas être seulement une vague possibilité, alors, et alors seulement, vous pouvez avoir une protection de propriété industrielle sur l'invention.
Il y a deux cas : ou bien l'ADN est connu ou il ne l'est pas. Quelle est la différence ? C'est très important. Pour celui qui n'aura pas la protection produit, l'ADN pourra être utilisé pour d'autres sources sans son autorisation.
M. Alain Claeys, rapporteur. - Un gène isolé ou séquencé ne faisant pas partie du corps humain, fournissant des protéines sur une application industrielle est brevetable dans la directive européenne.
M. François Chrétien. - Et en droit français aussi !
M. Alain Clayes, rapporteur. - Le gène qui produit cette protéine, cette partie du vivant, est-il brevetable ?
M. François Chrétien. - Oui. S'il est nouveau, bien sûr !
M. Alain Claeys, rapporteur. - Potentiellement, les autres applications possibles font partie de la sphère du brevet.
M. Bernard Charles, président. - Notre problème est là.
M. François Chrétien. - On tombe dans les problèmes de dépendance signalés par M. Alain Claeys.
M. Bernard Charles, président. - Alain Claeys situe notre débat de législateur. Un brevet répondant aux critères que vous avez explicités (isoler, séquencer, fonction et application industrielle) peut bloquer l'invention d'une nouvelle fonction, à laquelle le premier inventeur n'avait pas pensé. C'est cela qui nous pose problème.
M. Alain Claeys, rapporteur. - Je maintiens que cela peut poser un problème économique pour l'industrie. Pourquoi aux États-Unis mettent-ils tant d'argent ? C'est à la fois pour l'application qu'ils ont trouvée, mais aussi pour les applications potentielles dans le périmètre du brevet.
M. Bernard Charles, président. - Je suis allé aux États-Unis chez Celera Genomics. J'ai vu tout ce qu'ils faisaient. Je suis allé voir les responsables de l'Office des brevets américain. Ils disaient n'avoir aucun contact avec le FDA ni avec le ministère de la santé. Ils « font des brevets ». Le reste - éthique ou autre - n'est pas leur problème, cela relève d'autres personnes.
La question soulevée par notre rapporteur est celle de la puissance financière qui peut bloquer, à l'aide de brevets, des inventions ou des applications industrielles qui n'ont pas été trouvées et qui pourraient encore être trouvées, plus tard, par d'autres que le premier titulaire du brevet.
M. François Chrétien. - Votre question est claire. La première partie de la réponse a été donnée par Thierry Sueur. Quand on se trouve dans une position « dépendante » par exemple, quand on a trouvé une deuxième application au gène...
M. Bernard Charles, président. - Il est arrivé en chimie que l'on trouve une molécule et qu'ensuite, on lui trouve une autre application.
M. François Chrétien. - C'est classique dans le domaine du médicament. Ce qu'a dit Thierry Sueur est très important : le brevet à l'état de demande est publié et vous pouvez travailler dessus, le perfectionner et trouver la deuxième application.
M. Bernard Charles, président. - Si l'on trouve la deuxième application, peut-on la breveter ?
M. François Chrétien. - Bien sûr !
M. Bernard Charles, président. - C'est là tout le débat.
M. François Chrétien. - C'est ce que Thierry Sueur vous disait. La dépendance, au lieu d'être subie, doit stimuler.
M. Alain Claeys, rapporteur. - Nous avons des réponses contradictoires.
M. Thierry Sueur. - C'est un problème général. Cette question s'est posée très longtemps dans le domaine du médicament. La position était que l'on pouvait protéger une application, mais pas les suivantes. La législation a changé et aujourd'hui, on peut protéger d'autres applications pharmaceutiques. J'invente l'aspirine ; c'est une nouvelle molécule qui soigne le mal de tête. J'ai une protection. Si demain on trouve que l'aspirine guérit une autre affection, une demande de brevet peut être déposée qui appartiendra au second inventeur pour protéger l'application nouvelle, inventive.
Des dispositifs dans la loi permettent d'assurer ces liens de dépendance. Il y a des licences de dépendance qui permettent à chacun d'obtenir des licences pour d'autres emplois. Les mécanismes sont, pour nous, classiques, et c'est peut-être pourquoi nous ne les expliquons pas bien. Le tout est de voir si dans le gène cela pose une question différente ou pas, mais la question est légitime.
M. François Chrétien. - J'attire votre attention sur le second graphique qui répond un peu à votre question. Il visualise le brevet dans le développement d'un produit. Pour le brevet, vous avez deux temps : le temps d'acquisition des droits, c'est-à-dire les problèmes de brevetabilité, et le temps de l'exploitation du brevet. Quand on a un problème d'accès commercial, la solution est de ne pas attaquer les critères de brevetabilité en tant que tels parce que l'on a des engagements internationaux communautaires, européens, et mondiaux dans le cadre des traités ADPIC.
On ne peut pas discriminer un domaine en termes de brevetabilité. En revanche, il y a un autre moyen qui est, lui aussi, reconnu au niveau international. Dans des cas particuliers, et notamment quand l'intérêt public est en cause, la possibilité existe de mettre au point une licence obligatoire, soit licence d'office, soit licence dépendante. Si le titulaire du brevet ne veut pas, le juge va l'obliger à donner licence pour pouvoir exploiter l'invention. Même si ces systèmes sont peu utilisés, un cadre juridique adapté existe bien pour faire face à ce type de situation.
Mme Yvette Benayoun-Nakache. - La durée des critères de brevetabilité est de dix-huit mois ?
M. François Chrétien. - Non. « Les critères de brevetabilité » sont la notion qui intervient pour statuer sur la validité du droit que vous obtenez. C'est le temps de l'acquisition qu'il faut considérer. Et ensuite est mentionné le temps de l'exploitation.
Mme Yvette Benayoun-Nakache. - Celui-ci est de dix-huit mois.
M. Thierry Sueur. - La publication est obligatoire dans les 18 mois. La procédure est généralement la suivante : vous déposez une demande de brevet qui est publiée au bout de dix-huit mois. C'est l'Office européen qui s'en charge. Au bout d'un certain temps - deux, trois ou quatre ans - l'Office européen des brevets va examiner votre brevet ; vous allez en discuter avec lui. Il va vous dire si votre invention est brevetable ou pas. Imaginons qu'il accorde votre brevet avec une certaine protection. Cela arrive entre quatre ou cinq ans après le dépôt, parfois plus. Parfois, c'est dix à quinze ans en raison d'oppositions.
M. Bernard Charles, président. - Merci beaucoup messieurs.
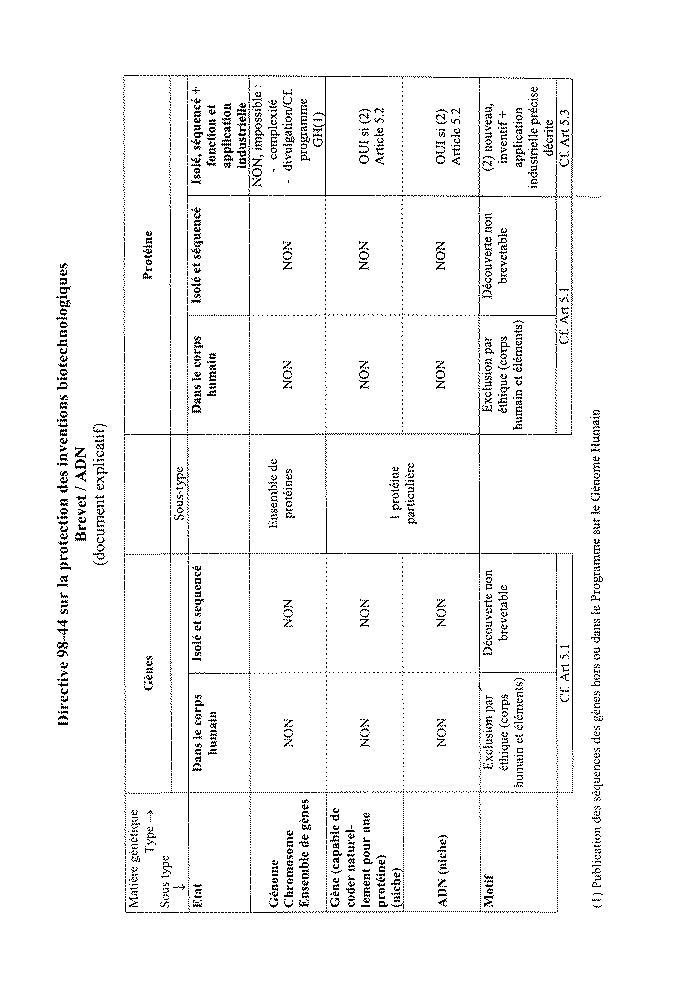
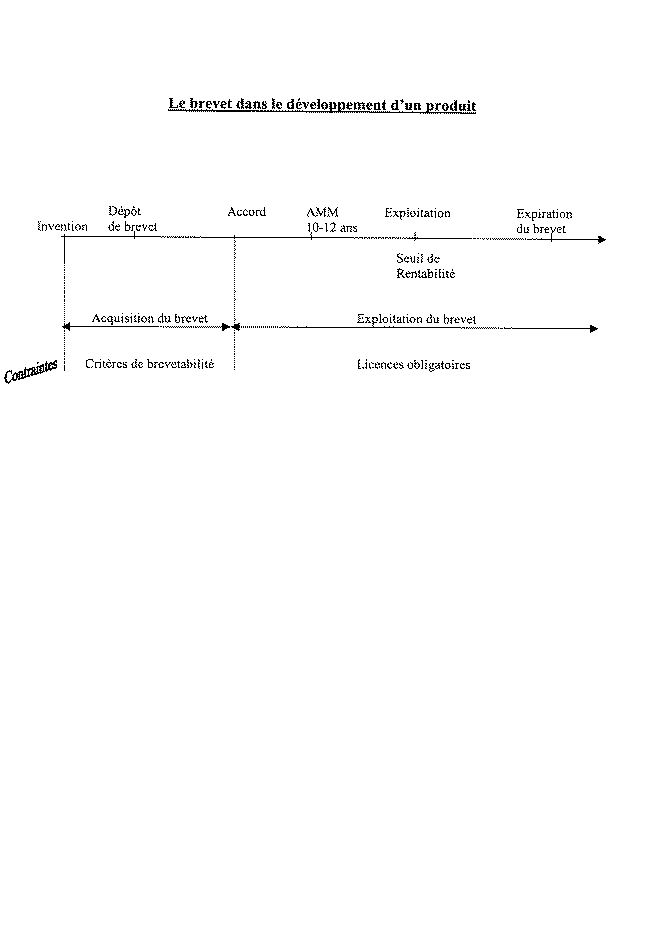
Audition de MM. Gilles AULAGNER,
président du Syndicat national des pharmaciens praticiens hospitaliers
et praticiens hospitaliers universitaires,
Bernard CERTAIN, pharmacien hospitalier,
et Mme Dominique GOERY, directrice scientifique
du centre d'étude et de formation hospitalières
(Extrait du procès-verbal de la séance du 24 janvier 2001)
Présidence de M. Bernard Charles, président.
M. Bernard Charles, président. - Nous avons le plaisir d'accueillir aujourd'hui des praticiens hospitaliers pour évoquer un élément quelque peu parallèle à notre réflexion sur la révision des lois bioéthiques. Même si ce n'est pas inclus dans l'avant-projet de loi du Gouvernement, nous avons souhaité examiner les modifications qu'il serait nécessaire d'apporter à la loi de 1988 relative à la protection des personnes qui se prêtent à la recherche biomédicale. Je connais assez bien cette loi, la première loi bioéthique, appelée communément loi Huriet-Sérusclat, puisque j'en ai été le rapporteur, en 1988, à l'Assemblée nationale. Avec Alain Claeys, nous avons souhaité inclure cette loi dans le champ de notre réflexion. C'est d'ailleurs ce qu'il avait fait lui-même, à l'Office parlementaire d'évaluation des choix scientifiques et technologiques, dans le rapport d'évaluation des lois bioéthiques qu'il a réalisé avec notre collègue sénateur Claude Huriet.
Les pharmaciens hospitaliers jouent un rôle central dans la mise en _uvre de cette loi. Ils ont la responsabilité de la gestion de ce dispositif dans les établissements. J'ai donc le plaisir d'accueillir MM. Gilles Aulagner et Bernard Certain, qui sont président et membre du Syndicat national des pharmaciens praticiens hospitaliers et praticiens hospitaliers universitaires ainsi que le docteur Dominique Goery, pharmacien hospitalier et directeur scientifique du centre d'étude et de formation hospitalières.
Nous souhaiterions que, dans un exposé liminaire, vous nous fassiez part de vos réflexions, en tant que praticiens, sur le fonctionnement de la loi Huriet-Serusclat, sur les modifications législatives qui permettraient d'améliorer cette loi, ainsi que sur ses carences. En outre, vous avez appelé notre attention sur le problème des produits dérivés du génome. Nous verrons quelle est votre position sur ces produits en tant que responsables hospitaliers de la gestion des médicaments.
M. Gilles Aulagner. - Monsieur le président, merci de nous recevoir. Nous voulions évoquer tout d'abord la loi Huriet. Nous souhaitions d'abord souligner que certaines dispositions réglementaires ou législatives, paraissant dans certains cas marginales ou annexes, peuvent, par leur présence ou leur absence, entraîner le succès ou l'échec d'une loi. Un des amendements que le président Bernard Charles avait présenté est devenu l'article L. 5126-11 du code de la santé publique qui a confié aux pharmacies hospitalières le contrôle de la mise en place des essais. C'est l'un des éléments qui a permis le succès de cette loi dans le domaine des médicaments. Notre opinion est plus nuancée sur les autres produits de santé.
Je voudrais revenir sur la gratuité des produits qui ne résulte pas d'une disposition législative, mais réglementaire. À notre sens, cela peut poser un certain nombre de problèmes. Hormis lorsque le promoteur est une société savante ou une grande agence de recherche, la question ne se pose pas pour les médicaments. Cela se passe très bien. Je reviendrai sur le problème des sociétés savantes.
M. Bernard Charles, président. - Nous avons reçu des courriers de certaines sociétés savantes qui disaient leurs difficultés à réaliser certaines expérimentations quand elles voulaient le faire en dehors de l'industrie pharmaceutique. Le problème était de disposer gratuitement des produits pour le site d'expérimentation.
M. Gilles Aulagner. - Sur les dispositifs médicaux, le problème est généralement plus complexe, ce qui amène à observer, sur le terrain, si ce n'est un non-respect, du moins des contournements de la loi. En effet, on a affaire à des produits beaucoup plus complexes, dont le volume financier n'a rien de comparable avec ce qui existe dans le domaine du médicament et où la gratuité pose incontestablement un problème. Cela amène les investigateurs à ne pas déposer de demande de recherche.
M. Bernard Charles, président. - Nous avons des associations de malades qui nous écrivent à ce sujet.
M. Gilles Aulagner. - Nous avons essayé de réfléchir à la façon de régler ce type de difficultés. Pour prendre un exemple caricatural - la prothèse de genou - on sait très bien que le nombre de prothèses posées n'a rien à voir avec le nombre de comprimés utilisés pour tel ou tel produit médicamenteux. D'un autre côté, l'implication d'une PME est beaucoup plus importante. Nous souhaiterions donc que les dispositifs médicaux aient un prix de référence qui soit le prix de référence du produit comparable. Il faudrait qu'au cours de l'essai, l'établissement hospitalier ou la Caisse nationale d'assurance maladie, si c'est un malade ambulatoire, paye le coût du produit de référence et non pas le complément de ce que peut être le caractère innovant du produit.
M. Bernard Charles, président. - Vous ne l'envisagez pas pour tous les produits, mais pour une certaine catégorie de produits. Il y a quand même le cas des produits courants.
M. Gilles Aulagner. - Quand on a affaire à des produits courants, des aiguilles par exemple, la problématique est identique à celle du médicament. Le cas qui fait difficulté est celui des produits qui ont une valeur individuelle très élevée. On peut avoir des dispositifs médicaux qui représentent plusieurs dizaines de milliers d'euros.
M. Bernard Charles, président. - Nous devons auditionner les représentants de l'industrie des dispositifs de soins médicaux afin d'évoquer les problèmes posés aux PME françaises, face aux grandes entreprises, américaines ou autres, qui ont les moyens financiers de donner les produits pour les expérimentations.
M. Bernard Certain. - La loi Huriet elle-même n'a jamais parlé de gratuité des échantillons remis à titre d'essai. C'est uniquement l'administration. Lors d'un colloque au mois de mai dernier, ces problèmes ont été évoqués et les parlementaires interrogés sur ce problème ont répondu que ce n'était pas leur volonté, mais celle du pouvoir réglementaire. C'est pourquoi nous reparlons de ce problème même s'il peut paraître trivial aux législateurs que vous êtes. Sinon, il n'y a pas de raison que l'administration change la position qui est la sienne. Les caisses d'assurance maladie considèrent que c'est très bien ; les services des directions ministérielles considèrent que c'est très bien. La seule chose que nous constatons sur le terrain est qu'il n'y a pas ou pratiquement pas d'essais relatifs aux dispositifs médicaux.
M. Bernard Charles, président. - Nous demanderons à Mme Dominique Goery qui a été longtemps membre d'un comité de protection des personnes ainsi qu'à M. Gilles Aulagner de nous dire comment fonctionnent ces comités chargés de vérifier que les projets d'expérimentation déposés dans les régions respectent la loi. On verra d'ailleurs que les chiffres d'expérimentation pour les dispositifs médicaux sont infiniment plus faibles que ceux des médicaments. Cela montre bien qu'il existe un problème pour les expérimentations de dispositifs médicaux.
M. Gilles Aulagner. - Selon les chiffres de la Conférence nationale des comités de protection, les dispositifs médicaux ne représentent que 10 % du nombre des essais.
M. Bernard Charles, président. - Ce n'est pas la réalité du monde hospitalier que vous connaissez.
M. Gilles Aulagner. - Au niveau hospitalier, la réalité est que le budget médicament est égal au budget des dispositifs médicaux.
M. Bernard Certain. - Il faut quand même parler de la diversité des produits. Un hôpital fonctionne avec 2 200 références. Il y a de 40 000 à 50 000 références dans le domaine du consommable médical.
M. Gilles Aulagner. - En ambulatoire, il y a 9 000 spécialités pharmaceutiques et en dispositif médical, 72 000 références au niveau européen.
M. Bernard Certain. - Votre question était : « Est-ce pour tous les produits ou pas ? » La suggestion que nous formulons est qu'il y ait un putatif. Il y a possibilité d'ouvrir une expérimentation avec des produits achetés. En fait, dans toutes les expérimentations, il y a toujours une convention entre le promoteur, l'expérimentateur et l'établissement où se déroule l'expérimentation. Mais, je dirais qu'aujourd'hui la réglementation impose une situation univoque où tout doit être gratuit.
M. Bernard Charles, président. - On fait des expérimentations cachées avec des produits que l'on paie parce que l'on n'a pas déclaré l'expérimentation.
M. Gilles Aulagner. - Avec un déficit de protection des personnes qui peut être plus ou moins important selon le type de produit ! Lorsque l'on a affaire à une société savante, c'est l'hôpital qui paie le produit soumis à expérimentation. Ce n'est pas un cas particulier.
Par ailleurs, nous souhaitons insister sur la nécessité d'avoir une réflexion sur la capacité des pharmaciens, justifiant d'une expérience appropriée et validée dans le domaine des dispositifs médicaux ou dans le domaine de la pharmacocinétique et de la biodisponibilité, de pouvoir assurer la direction et la surveillance de certaines recherches médicales.
M. Bernard Charles, président. - Aujourd'hui, le problème se pose aussi pour la recherche dans le domaine spatial. Pour les épreuves d'effort, ce ne sont pas nécessairement des médecins qui interviennent.
M. Gilles Aulagner. - Comme dans les recherches psychologiques.
M. Bernard Charles, président. - Dans la loi Huriet, nous avions précisé que le médecin devait être l'investigateur principal. Comme je l'ai signalé à Claude Huriet, les pharmaciens ont le rôle primordial pour certains produits, mais ils ne peuvent pas faire les expériences appropriées. Le problème se pose dans le domaine spatial où les physiciens font des expérimentations et aussi des recherches comportementales en psychologie. On accorde des dérogations à certaines expérimentations pour que l'investigateur principal ne soit pas exclusivement un médecin, sous certaines réserves bien entendu.
M. Gilles Aulagner. - Pour d'autres domaines, il est évident que nous plaidons pour qu'il y ait un dispositif médical des pharmacocinétiques et biodisponibilités.
Un second aspect de nos réflexions sur la loi Huriet concerne les superpositions de réglementations ou de législations que nous pouvons rencontrer. Par exemple, un produit stupéfiant va relever de toutes les réglementations relatives à cette classe de produit ; mais d'un autre côté, il va aussi relever de la réglementation des essais cliniques. Nous vous proposerions que la réglementation des essais cliniques s'applique et que l'on évite de multiplier les références, les interprétations contradictoires et les enregistrements multiples qui sont superfétatoires.
M. Bernard Certain. - Pour donner un exemple, il existe actuellement un essai clinique sur l'utilisation d'un produit chimique issu du cannabis. Par nature, ce produit est un stupéfiant. C'est un problème parce que c'est un stupéfiant et que la conservation de ce produit se fait dans un réfrigérateur. Il n'y a pas encore de coffres-forts réfrigérateurs.
Interrogée, l'Agence du médicament répond que le produit doit aller au réfrigérateur ; les représentants de la DGS, les inspecteurs en pharmacie répondent, eux, que cette substance vénéneuse est à mettre sous clef ! Il faudrait que le législateur corrige cette superposition des réglementations. L'expérimentation clinique fait partie d'un protocole. On peut préciser dans ce protocole certaines conditions du maniement de ce type de produit, par exemple de le mettre sous pli fermé, ou le rendre anonyme etc.
M. Bernard Charles, président. - Le législateur n'avait pas « balayé » le code de la santé publique en fonction de ces aspects.
M. Gilles Aulagner. - On s'en rend compte sur le terrain. À l'époque, nous n'avions pas réfléchi à cet aspect.
Nous souhaitons aborder deux points ayant trait aux CCPPRB. À l'origine de la loi Huriet, les membres des CCPPRB étaient tirés au sort parmi des volontaires. Pour des raisons de simplification, ils sont maintenant nommés par le préfet. On constate une dérive que je trouve choquante, à savoir que l'on nomme des gens « politiquement corrects ». On essaie de nommer ceux « qui ne font pas de vagues » et qui ne soulèvent pas les problèmes. Nous souhaiterions que l'on revienne à un fonctionnement plus démocratique : le système du tirage au sort. Une conférence nationale des comités de protection avait été créée ; j'en avais été l'un des fondateurs. Du jour où l'on est passé aux nominations, aucun des membres du bureau n'a été renouvelé.
M. Bernard Charles, président. - À l'époque, nous avions tenu au tirage au sort pour qu'il n'y ait pas la mise en place d'une cooptation des membres des comités de protection. La DGS avait dit que ce serait très compliqué.
M. Gilles Aulagner. - Il ne faut sans doute pas non plus avoir des membres de CCPPRB professionnels.
M. Bernard Charles, président. - Mme Dominique Goery, vous qui avez été secrétaire d'un comité pendant huit ans, comment avez-vous apprécié la mise en _uvre de la loi ? Quels sont les problèmes posés par les dispositifs médicaux, etc. ?
Mme Dominique Goery. - J'ai été secrétaire du CCPPRB de Toulouse I de 1991 à 1998. Au départ, les nominations résultaient d'un tirage au sort. Même si je pense qu'à un moment donné on peut avoir envie de laisser sa place, il serait dommage que ce soient toujours les mêmes personnes qui soient nommées par cooptation.
Je peux confirmer la plupart des propos de MM. Gilles Aulagner et Bernard Certain. À Toulouse, 10 % des essais concernaient les dispositifs médicaux, soit trois ou quatre sur quarante dossiers suivis, pour la plupart des études de médicaments. Quelques essais concernaient la médecine spatiale, tout ce qui était expérimentation physiologique. Toulouse travaillait beaucoup sur les études comportementales et physiologiques, sur les comportements du corps humain dans l'espace en relation avec d'autres centres européens.
Il y avait ce problème de coordination des essais cliniques par le médecin. Des infirmières, des cadres soignants coordonnaient certaines études comportementales ou même certaines expérimentations sur les soins, sur l'éducation des personnes, sur l'hygiène de vie. On était toujours confronté à cette obligation d'avoir un médecin comme investigateur principal.
La plupart des problèmes rencontrés venaient essentiellement de la différenciation entre essai avec bénéficie individuel direct et essai sans bénéfice individuel direct. Il était délicat, à l'époque, de classer un essai de médicament, dans le cadre d'une maladie grave, avec bénéfice individuel direct quand le médicament était comparé à un placebo. Cela nous est arrivé à plusieurs reprises. On avait du mal en tant que rapporteur à classer l'expérimentation avec bénéfice individuel direct, alors qu'il y avait comparaison, non pas par rapport à un médicament de référence, mais par rapport à un placebo.
Je ne peux que confirmer le contournement de la loi. En tant que rapporteur pendant huit ans et en tant que pharmacien au centre hospitalier de Montauban, je dois souligner que nous avons fait peu d'essais sur les dispositifs médicaux. C'est toujours le cas puisque l'une de mes assistantes, pharmacien hospitalier, qui fait partie du CCPPRB I de Toulouse, m'a confirmé qu'il n'y avait aucune évolution.
M. Bernard Charles, président. - La direction des hôpitaux a soulevé également le problème. Il faut trouver une solution.
M. Gilles Aulagner. - Un problème important se pose en ce qui concerne la notion de « avec ou sans bénéfice individuel direct ». Je parle en tant que pharmacien d'un hôpital pédiatrique où cela crée un problème important. Comme les enfants ont une protection particulière, les essais sans bénéfice individuel direct ne sont pas autorisés. Moyennant quoi - il faut en voir la conséquence en aval - nous n'avons pas les médicaments pour traiter les enfants, ce qui entraîne, en cascade, une multiplication du nombre de préparations faites en pédiatrie.
M. Bernard Charles, président. - Par l'adaptation de produits adultes à la pédiatrie.
M. Gilles Aulagner. - Il y a là probablement une réflexion à mener. Aux États-Unis, la FDA a publié, en 1997, une réglementation applicable pendant quatre ans qui sera reconduite compte tenu de son succès. Elle impose aux titulaires d'une autorisation de mise sur le marché de proposer en même temps la forme pédiatrique. C'est un élément qui me paraît très important dans le domaine de la pédiatrie.
Pendant trois ans, j'ai été président d'un CCPPRB. Il me paraît nécessaire d'institutionnaliser une formation pour les membres « non médicaux » afin d'avoir une réflexion de même niveau. Faute de quoi, les membres « non médicaux » se sentent exclus du débat.
M. Bernard Charles, président. - On a voulu les inclure pour que cela ne se résume pas à un débat de spécialistes. C'est aussi notre problème en ce qui concerne la future agence pour l'assistance médicale à la procréation.
M. Gilles Aulagner. - Il me paraît indispensable de leur donner une formation pour expliquer ce qu'est un essai et comment cela fonctionne. Faute de quoi ils sont très vite marginalisés. Nous en avions ressenti le besoin et, dans mon CCPPRB, j'avais consacré à chaque séance une demi-heure de formation à destination des gens n'appartenant pas au milieu médical.
Pour en venir aux thérapies génique et cellulaire, il est évident qu'aujourd'hui, on se situe plus au stade des essais qu'à celui de la thérapeutique véritable. Mais cette situation va évoluer et il faut savoir l'anticiper. Il nous paraît nécessaire d'avoir une réflexion qui implique les professionnels. Nous avons été choqués de voir qu'il y avait des concertations européennes auxquelles participaient l'AFFSSAPS dans lesquelles les professionnels n'étant pas impliqués.
Quels sont les points importants en matière de thérapies génique et cellulaire ? Je rapprocherai également tout ce qui est dénommé « produits thérapeutiques annexes ». Il s'agit de produits qui servent à conserver organes et cellules. Il nous paraît tout à fait important de dissocier la préparation du prescripteur. Cela a toujours été fait dans le domaine du médicament pour éviter des dérapages possibles...
Le souci de la qualité est garanti par la présence de gens extérieurs qui ne sont pas impliqués. Quand vous êtes impliqué dans l'acte de prescription ou dans l'acte d'administration - on le voit pour un certain nombre de médicaments préparés dans les services -, il peut y avoir des dérapages, assez rapides, en cherchant à valoriser l'acte thérapeutique et le progrès par rapport à la qualité véritable du produit.
Dans ce domaine, il nous paraît fondamental de faire en sorte que celui qui prépare soit différent de celui qui prescrit ou qui administre. Je crois que c'est un élément qui n'apparaît pas suffisamment dans la loi.
D'autre part, la réglementation sur les produits thérapeutiques annexes ne correspond pas toujours aux réalités. Cette réglementation donne l'impression que ces produits sont préparés par l'industrie, alors qu'ils sont préparés majoritairement par les pharmacies hospitalières.
M. Bernard Charles, président. - Le problème est de savoir si on le met dans la loi bioéthique ou dans la loi de modernisation du système de santé.
M. Gilles Aulagner. - J'ai conscience que se pose aussi le problème du sérum neurof_tal utilisé dans les cultures de cellules.
Dernier élément : il nous paraît important de stimuler les formations dans le domaine des thérapies génique et cellulaire, soit sous la forme de DEA soit de DESS. Que ce soit en médecine ou pharmacie, il y a un déficit de formations.
Un domaine encore nous pose question, celui des études pharmaco-économiques.
M. Bernard Charles, président. - Ce n'est pas du tout du ressort de la loi bioéthique.
M. Gilles Aulagner. - On est à la limite de la loi Huriet. Aujourd'hui, on a de plus en plus d'études pharmaco-économiques qui ont des conséquences sur les prescriptions. Il nous paraît absolument nécessaire d'y apporter un peu d'éthique comme l'a fait la loi Huriet avec les CCPPRB.
M. Bernard Charles, président. - On ne pourra pas l'évoquer dans ce cadre.
Merci pour toutes les informations que vous nous avez apportées.
Audition de Mme Noëlle LENOIR
(Extrait du procès-verbal de la séance du mercredi 31 janvier 2001)
Présidence de M. Bernard Charles, Président.
M. Bernard Charles, président. - Mes chers collègues, nous avons le plaisir d'accueillir Mme Noëlle Lenoir, présidente du groupe européen d'éthique des sciences et des nouvelles technologies auprès de la Commission européenne et membre d'honneur du comité international de bioéthique de l'UNESCO. Vous êtes également membre du Conseil constitutionnel.
Madame la présidente, je vous remercie d'avoir accepté de répondre à l'invitation de notre mission. Votre connaissance parfaite des questions que nous évoquons, dans leur dimension à la fois française, européenne et internationale, donne du prix à votre intervention. En 1991, le Premier ministre, M. Michel Rocard, vous avait chargée d'une mission sur le droit de la bioéthique et les sciences de la vie. Vous avez donc suivi l'élaboration des lois de 1994 depuis leur point de départ.
Vous avez également présidé pendant cinq ans le comité international de bioéthique de l'UNESCO. Votre regard sur les approches suivies dans les différents pays nous intéresse donc beaucoup. Enfin, vous avez participé au groupe de travail du Conseil d'État sur l'adaptation des lois de 1994. Pour toutes ces raisons, nous serons très attentifs à votre intervention et à vos réflexions.
M. Alain Claeys, rapporteur. - Madame Noëlle Lenoir, à un moment où notre mission d'information, préparatoire au projet de révision des lois bioéthiques, achève ses auditions, il nous est apparu nécessaire d'aborder avec vous les aspects internationaux des lois bioéthiques. Vous êtes sans aucun doute la mieux placée aujourd'hui pour aborder cet aspect fondamental.
Le groupe européen d'éthique cherche à préserver une voie médiane entre les préoccupations éthiques, le respect du champ de compétence de l'Union et les différentes approches nationales de ces questions. Il est vrai que l'éthique n'est pas une abstraction pure. Elle repose sur des valeurs universelles, mais elle est aussi mise en _uvre concrètement, selon des modalités qui varient en fonction de l'histoire, de la culture de chaque pays. La délégation de notre mission qui a rencontré, à Berlin, nos homologues allemands, a pu constater le poids de la culture sur la législation nationale.
Nous souhaiterions que vous nous présentiez les missions et les travaux du groupe européen d'éthique, ainsi que la manière dont cet organe peut rendre compte de ces diversités nationales. Il serait également intéressant que vous puissiez nous faire part de votre expérience à l'UNESCO et nous donner votre point de vue sur le débat éthique dans la sphère internationale : quelles sont les principales fractures qui apparaissent dans ce domaine, comment les pays du Sud abordent-ils ces problèmes ?
Nous souhaiterions également vous poser un certain nombre de questions sur deux sujets d'actualité, sur lesquels le groupe européen d'éthique a travaillé. Tout d'abord, sur l'avis du 14 novembre 2000 relatif à la recherche sur les cellules souches humaines. Pouvez-vous nous exposer les fondements éthiques et juridiques de cet avis ? L'avis évoque la nécessité de respecter le principe de proportionnalité et une approche de précaution : quel est le sens donné à ces deux notions ?
Conformément à l'article 22 de la Charte des droits fondamentaux, comment concilier des approches nationales très différentes, voire divergentes, par exemple entre le Royaume-Uni et l'Allemagne ? Dans l'avis du 14 novembre dernier, l'accent a également porté sur la nécessité de mettre en place un contrôle public adapté sur les activités de recherche sur les cellules souches embryonnaires. Selon vous, comment ce type de contrôle peut-il s'exercer ? Dans quel cadre institutionnel ?
Enfin, l'avis du groupe européen d'éthique met en avant la responsabilité de l'Union européenne, en qualité de financeur, dans le contrôle des recherches sur les cellules souches. Comment de tels contrôles peuvent-ils s'exercer, selon quelle procédure et avec quels moyens ?
Second sujet d'actualité : la brevetabilité du corps humain. Le groupe européen d'éthique, qui s'appelait alors groupe des conseillers pour l'éthique de la biotechnologie, a rendu un avis, le 25 septembre 1996, sur le projet de directive relatif à la protection juridique des inventions biotechnologiques. Dans cet avis, sont fixés des principes auxquels nous ne pouvons qu'adhérer : la non-commercialisation du corps humain, le libre consentement des personnes sur lesquelles les prélèvements sont réalisés, la subordination des impératifs économiques aux exigences éthiques.
Cet avis affirme la nécessité de distinguer la découverte, non brevetable, de l'invention, sachant que pour qu'un brevet soit délivré pour une invention réalisée à partir de la connaissance d'un gène ou d'une séquence génétique, il est indispensable d'en identifier la fonction et d'en décrire avec précision l'application.
Cet avis a été adopté à l'unanimité, mais l'un des membres a souhaité apporter une nuance. Or cette nuance nous ramène à l'actualité. Il a estimé que le brevet pouvait couvrir l'application technique issue de la connaissance du gène mais en aucun cas le gène lui-même. À partir de cette décision, deux séries de questions me viennent à l'esprit.
Tout d'abord, l'avis rendu par le groupe européen d'éthique n'est pas très clair sur la distinction qui doit être faite entre le gène, la fonction et l'application. Pouvez-vous préciser la manière dont le groupe entend que soit concrètement établie cette distinction, qui est la clé du maintien des principes éthiques énoncés lors de la mise en _uvre du droit des brevets en matière de biotechnologie ?
Ensuite, la difficulté qui apparaît aujourd'hui, dans plusieurs État membres, notamment en France, pour transposer cette directive n'est-elle pas le signe d'une incapacité de l'Union européenne à dégager une voie médiane entre les exigences éthiques et les impératifs économiques, alors que les États-Unis, par exemple, ne semblent pas s'embarrasser de telles contradictions ?
Mme Noëlle Lenoir. - Monsieur le président, monsieur le rapporteur, mesdames, messieurs les députés, la séance a été parfaitement introduite, et je souhaiterais donc revenir sur les grands sujets relatifs à la bioéthique dont la plupart ont été mentionnés par le rapporteur.
Le premier concerne les « matériaux » de la recherche que sont devenus pratiquement tous les éléments et produits du corps humain. Ceux-ci sont destinés en effet à être un jour entièrement recyclés et utilisés comme produits thérapeutiques.
Le deuxième sujet est celui de la brevetabilité du vivant. Celle-ci suscite des interrogations en Europe comme aux Etats-Unis d'ailleurs. Certes, aux États-Unis, éthique et la brevetabilité ne sont pas aussi intimement mêlés qu'Europe. Pour autant, l'application du droit des brevets à la matière vivante (gènes, séquences de gènes, cellules...) donne lieu encore aujourd'hui à controverse tant il est vrai qu'il n'est pas simple de déterminer les conditions de la brevetabilité d'un organisme ou d'un produit vivant existant dans la nature, et qui n'est donc pas fabriqué par l'homme.
Le troisième sujet est relatif aux droits sociaux, et il me paraît devoir retenir l'attention du législateur. Comment accompagner la diffusion des tests génétiques, non plus seulement les diagnostics anténataux, mais les tests pratiqués au cours de l'existence pour éviter de stigmatiser ceux qui ont des antécédents génétiques défavorables ? Ces tests vont arriver sur le marché européen, car il est beaucoup plus facile en effet de mettre au point un test que de trouver un traitement - on l'a vu avec le sida.
Dernier sujet majeur : la recherche sur l'embryon. Les perspectives nouvelles offertes par le clonage « thérapeutique » obligent à repenser la question, bien qu'il faille à mon avis, rester prudent avant de s'engager résolument dans la voie de la production d'embryons par transfert nucléaire, c'est-à-dire par clonage.
Je reviendrai sur ces sujets, mais je voudrais dire auparavant quelques mots à propos du Groupe européen d'Éthique. Née de la vision qu'avait Jacques Delors, en tant que président de la Commission, d'une Europe fondée sur des valeurs communes, cette instance a un rôle consultatif auprès des autorités communautaires. Le Président Delors avait défini des champs sur lesquels il souhaitait approfondir sa réflexion, par exemple, le dialogue social, la prospective et l'éthique. A cette dernière préoccupation a répondu la création du Groupe qui s'appelait à l'époque « Groupe des Conseillers pour l'Éthique des Biotechnologies ». Trois objectifs lui ont d'emblée été assignés : d'abord, aider la Commission européenne à identifier les problèmes de société susceptibles de se poser à terme, compte tenu de l'évolution des biotechnologies ; ensuite, contribuer à améliorer la compréhension de ces problèmes en tenant compte des différentes cultures qui coexistent en Europe, non seulement du reste au niveau des pays, mais aussi au sein des institutions communautaires. La Commission est en effet orientée vers la promotion de la croissance de l'économie et le développement des technologies tandis que le Parlement très diversifié dans sa représentation politique, renvoie davantage à une culture que l'on pourrait dire de « société civile » en lien avec les aspirations des citoyens européens. L'idée était donc que le Groupe puisse apporter sa contribution à une meilleure synergie européenne, en préparant des débats sur certaines questions délicates et en aidant ainsi à la prise de décision communautaire.
C'est ainsi que le Groupe a pris l'habitude d'organiser des tables rondes ouvertes à une large audience, au cours desquelles il procède à des auditions publiques. Ces tables rondes, très suivies, sont devenues son image de marque. Le Groupe actuel bien que renforcé dans son statut est demeuré dans ses missions très proche de l'intention initiale. Il est néanmoins bien mieux armé du point de vue de l'expertise technique, pouvant par exemple faire appel à des experts, européens ou internationaux. En effet, on ne peut plus parler de bioéthique en termes généraux dès lors que les avancées de la génétique rendent les questions de plus en plus techniques et complexes. On ne peut plus se borner à évoquer, par exemple, les tests génétiques ou la question du clonage en général ; il faut vraiment connaître la réalité des pratiques scientifiques. Prenons l'exemple du clonage dit « thérapeutique ». Il s'agit de clonage, en ce sens que des embryons sont produits par la technique du clonage - celle qui a permis la naissance de Dolly - pour permettre de prélever des cellules souches au patrimoine génétique identique à celui dont le noyau cellulaire a servi au dit clonage. Mais on ne va pas jusqu'à la naissance d'enfants par clonage et, en outre, pour l'instant, les cellules souches susceptibles d'être prélevées sur ces clones embryonnaires n'ont aucune vertu thérapeutique. La notion de « thérapeutique » indique que ce type de recherche sur l'embryon n'est pas dirigé vers l'amélioration de la reproduction humaine. Le terme de « thérapeutique » est aussi utilisé pour promouvoir cette recherche et lui permettre d'avoir un soutien, tant financier que moral. Ce n'est pas illégitime en soi, mais néanmoins il faut savoir de quoi l'on parle avant de pouvoir apprécier une pratique d'un point de vue éthique.
D'une manière générale, la recherche en biologie et en génétique est de plus en plus liée au marché. Ce n'est plus une activité uniquement destinée à la quête des connaissances. La valorisation économique de ses résultats est un enjeu essentiel, dans un contexte de compétition acharnée entre laboratoires privés, ainsi qu'entre chercheurs du secteur public et du secteur privé. La biotechnologie a à cet égard beaucoup fait évoluer les conditions socio-économiques de la recherche, ses structures et ses modes de financement.
Je répondrai maintenant à la question de M. le rapporteur sur l'état des législations en matière de bioéthique à travers le monde. Ces législations sont très limitées, voire inexistantes, dans les pays en développement. Tout ce que je puis dire à propos de ces pays, c'est que les ressources en génétique, qu'il s'agisse des ressources humaines ou non humaines (animales et végétales), sont très également réparties à travers le monde. Le vrai défi pour les pays en développement est d'arriver à avoir une certaine maîtrise de l'exploitation de leurs ressources. Les multinationales elles-mêmes adaptent leurs stratégies en fonction des besoins du Tiers-monde ; par exemple, en donnant des facilités pour cultiver du « golden rice » (qui contient de la vitamine A), en abandonnant - sous la pression de la communauté internationale - certains droits sur des brevets pour favoriser la vente de certains médicaments à plus bas prix, ou encore en aidant à la mise en place de services sanitaires. Depuis que la compagnie Monsanto a été confrontée aux difficultés que l'on connaît en Europe du fait du refus des pays et des consommateurs européens d'accepter les OGM produits par la firme, les industries diversifient leur approche selon les continents, menant parfois même des actions dans des pays en développement parallèlement, pourrait-on dire, à l'OMS, la FAO, ou les Nations-Unies. Je ne suis pas certaine que sur ces thèmes de l'agriculture et du développement durables, on ait au demeurant toujours clairement conscience de ce que cela peut représenter en termes de présence, à la fois culturelle et économique.
A côté des législations nationales, rares comme je l'ai dit dans les pays du Sud, il existe maintenant des textes internationaux sur la bioéthique, en premier lieu la Déclaration sur le génome humain et les droits de l'homme de l'Unesco. Celle-ci a été approuvée à l'unanimité par l'Assemblée générale des Nations-Unies en 1998. Ceci est d'autant plus remarquable, que les États-Unis, loin de s'être opposés au texte, l'ont au contraire appuyé entraînant d'autres pays derrière eux. La France, quant à elle, a vraiment joué un rôle de leader. Certains pays du sud, dont notamment ceux possédant des centres de recherche en génétique, ont plus ou moins activement participé au processus d'élaboration et d'adoption de la Déclaration. Je songe notamment à l'Inde, qui est très en avance dans le domaine de la génétique. Des pays d'Amérique latine, tels que l'Argentine et le Brésil, qui mènent des recherches très pointues, ont aussi soutenu la démarche avec d'autres pays comme Cuba, la Colombie et l'Uruguay. Quant à la Chine, qui a des lois eugéniques, elle s'est tenue dans une position de neutralité au contraire du Japon, qui a intégré maintenant la bioéthique dans sa législation et sa réflexion, et est intervenue très positivement dans les discussions sur la Déclaration sur le génome humain.
La bioéthique donne par ailleurs un point de vue intéressant de l'évolution de l'Union européenne. De prime abord, l'Union se caractérise par la diversité des législations nationales, en particulier en matière de recherche sur l'embryon. Ainsi, alors que les Suédois et surtout les Britanniques ont une législation libérale, l'Allemagne, l'Autriche et l'Irlande interdisent formellement cette recherche. La recherche sur l'embryon est même impossible pour des raisons d'ordre constitutionnel en Irlande. En Allemagne et en Autriche, le recours à la fécondation in vitro est en outre pratiquement exclu car l'on n'autorise pas les embryons surnuméraires. En Espagne, les deux lois de 1988 sur la recherche sur l'embryon et sur l'assistance médicale à la procréation sont en revanche extrêmement libérales. En Belgique, aux Pays-Bas, et surtout en Italie, où il n'y a pas de loi, les pratiques sont aussi très libérales. En revanche, au Portugal, en l'absence de loi, la recherche sur l'embryon paraît être hors de question.
Mais la diversité des législations en Europe tend à s'effacer peu à peu. Depuis quelques années, en raison des perspectives médicales offertes par ces recherches en effet, les mentalités et le positionnement des responsables politiques évoluent. Un changement s'est surtout opéré à la suite de l'annonce fin 1998 de découvertes très prometteuses sur les cellules souches humaines. Les pays européens qui n'avaient pas de loi, vont maintenant se doter d'une législation sur la recherche sur l'embryon : aux Pays-Bas, en Italie, et peut-être en Belgique (où un projet de loi est calqué sur le modèle anglais a été rendu public). Même l'Allemagne semble s'ouvrir à des possibilités de recherche sur l'embryon. Ce qui ressort des déclarations du chancelier Gerhard Schröder. Le Gouvernement allemand a, il est vrai, investi des crédits considérables dans le secteur des biotechnologies, avec l'idée d'être en pointe dans ce domaine en Europe. Ce qui est effectivement le cas aujourd'hui. Il me semble donc que les différences observées dans les pays européens, selon leurs cultures et l'influence des religions, sont en train de s'estomper pour des raisons essentiellement économiques. Par ailleurs, l'annonce au public de possibles progrès médicaux liés à la recherche sur l'embryon, a un effet naturellement très attractif sur l'opinion qui attend à juste titre beaucoup, à terme, de ces progrès pour soulager la souffrance humaine.
Autre facteur d'harmonisation des pratiques biomédicales en Europe : la régulation. En dépit de l'absence de compétence législative communautaire dans les domaines de la médecine et de la recherche, les sciences de la vie sont de plus en plus encadrées au niveau communautaire, et c'est ce dont j'aimerais parler maintenant.
Regardons les principales avancées des sciences de la vie :
D'abord, la médecine de « réparation ». Le vivant humain (les gènes, les protéines, les cellules, les tissus, les organes, voire des éléments transgéniques tels que les greffons d'animaux « humanisés ») sera de plus en plus à la base de thérapeutiques de « réparation » : greffes, thérapie génique ou cellulaire. Nous n'en sommes qu'au commencement ; mais déjà, l'on sait que ces produits vont largement circuler sur le marché européen, ce qui justifiera des réglementations communautaires.
Deuxième grande avancée : la maîtrise de la reproduction. En Europe, le clonage reproductif est interdit, de même qu'en principe aujourd'hui la thérapie germinale. Aux États-Unis, on se dirige au contraire vers cette thérapie et peut-être même vers des techniques d'amélioration des traits génétiques. Si la question de la thérapie germinale est à l'ordre du jour aux États-Unis, il est évident qu'elle se posera chez nous. Et comme elle est d'une certaine manière abordée au niveau communautaire, par exemple dans la directive de 1998 sur les brevets, l'Union Européenne aura à s'en mêler.
La dernière grande avancée est liée à la mise au point de tests génétiques dits « prédictifs ». Encore parfois approximatifs dans leurs résultats, et limités dans leurs applications, ces tests vont se développer dans les prochaines années et être commercialisés sur le marché européen.
Quel est donc le point de vue communautaire sur ces sujets ? Tout d'abord, l'utilisation des substances et produits humains fait l'objet d'une approche de santé publique, dès lors qu'il existe des risques de transmission de maladies virales ou infectieuses. L'article 152 du traité d'Amsterdam donne des compétences à la Communauté européenne en matière de santé publique, essentiellement pour vérifier la sécurité sanitaire des produits humains - tissus, organes, cellules, gènes, etc. Toutefois beaucoup reste à faire pour donner véritablement corps à une politique européenne de santé publique. Par ailleurs, il serait utile de clarifier le statut juridique des éléments et produits humains, au niveau communautaire. Curieusement, lorsqu'ils ne sont pas transformés génétiquement, ceux-ci sont assimilés à des dispositifs médicaux ; ce qui veut dire qu'ils ont à peu près le même statut qu'un scanner ou un doppler. Prenons l'exemple des cellules souches humaines. Utilisées telles qu'elles, elles sont considérées comme un dispositif médical, alors que génétiquement modifiées, elles deviennent des OGM. Dans les deux cas - directive sur les dispositifs médicaux et directive OGM - c'est la logique de marché qui est retenue. Elle me paraît insuffisante et il faudrait réglementer l'usage des tissus humains d'un point de vue strictement de santé publique.
En dehors de la santé publique, les préoccupations européennes touchent à la protection des droits fondamentaux. La protection de la vie privée, est l'une d'elle, notamment en ce qui concerne les collections d'échantillons biologiques (qui circulent beaucoup, y compris entre l'Europe et les États-Unis) ainsi que la mise en place de « biobanques ». Aux États-Unis, le secret médical n'existe pas véritablement. Notamment lorsque l'employeur est assureur, il a accès au dossier médical de ses salariés. Le Gouvernement américain a dû se résoudre à faire un pas dans le sens de la protection du secret médical, du fait de la directive européenne de 1995 relative à la protection des données. En vertu de ce texte, en effet, les pays européens peuvent s'opposer au transfert de données vers des pays qui n'assurent pas un niveau de protection équivalent à celui assuré par la directive.
Un troisième type de préoccupation a trait à la propriété intellectuelle. La directive de 1998 a été très discutée. Elle a mis dix ans avant d'être adoptée et il faut l'appliquer. Le sujet est, il est vrai, extrêmement compliqué. Ce sera, à mon avis, l'un des grands sujets portés devant l'Organisation mondiale du commerce, l'OMC, dont les accords cadres dits Trips demeurent controversés par les ONG qui font des biotechnologies le symbole d'une mondialisation dont elle dénonce les effets. La brevetabilité du vivant devrait faire l'objet d'un accord international, car la recherche est mondiale.
Enfin, et ceci mérite d'être souligné, la recherche sur l'embryon n'est pas absente de la réglementation communautaire. Le Groupe européen d'Éthique a même été invité par Mme Édith Cresson, commissaire à la recherche, à rendre un avis sur le sujet en 1998. Cette saisine faisait suite à un amendement d'un député allemand adopté au Parlement, visant à interdire tout financement de la recherche sur l'embryon au titre du programme cadre de recherche communautaire. L'idée de l'auteur de cet amendement était la suivante : dès lors qu'une activité est contraire à l'ordre public national (en l'occurrence en Allemagne), il n'est pas conforme à l'esprit de l'Europe que les ressortissants de ce pays aient à financer, à travers la participation de leur pays au budget de la Communauté européenne, des activités non éthiques au regard de leur propre législation. Le Groupe européen d'Éthique a rappelé que de même qu'en matière d'IVG, l'Europe n'était pas compétente pour décider si l'on devait autoriser ou non la recherche sur l'embryon. Il en a déduit qu'il n'y avait pas de raison d'interdire le financement communautaire de la recherche sur l'embryon dans les pays où elle est permise. En décider autrement serait entamer l'idée même d'une Union européenne. Pour autant, le Groupe a souligné la nécessité de procéder à une évaluation éthique de la recherche sur l'embryon au niveau européen, de façon à pouvoir rendre compte au public de la façon dont se déroulait cette recherche de caractère très sensible. D'où la seconde proposition du Groupe de soumettre en tout état de cause ces recherches à un strict contrôle public. Il n'en est pas ainsi aux États-Unis. En effet, le Congrès y vote, chaque année, dans le cadre du budget, une disposition interdisant tout financement public de la recherche sur l'embryon tout en laissant totalement dérégulée la recherche dans le privé. Aucun contrôle n'étant prévu dans le secteur privé, tout y est donc possible.
Les techniques de reproduction n'entrent pas plus que la recherche sur l'embryon dans les compétences communautaires. Pourtant, la question du clonage humain est directement abordée dans plusieurs textes. Le clonage reproductif est interdit dans la Charte des droits fondamentaux. De même, le programme cadre de recherche cite-t-il le clonage humain parmi les pratiques excluant tous financements de l'Union. Les procédés tendant au clonage humain sont par ailleurs mentionnés comme contraires à l'ordre public et aux bonnes m_urs par la directive de 1998 sur la protection juridique des inventions biotechnologiques et exclus, en tant que tels, de la brevetabilité.
La situation est similaire s'agissant de la thérapie germinale. Exclue de la brevetabilité par la directive susvisée de 1998, la modification de la lignée germinale humaine se voit aussi exclue des financements du cinquième programme cadre de recherche tel qu'arrêté par une décision du Parlement et du Conseil de 1998.
Dernier sujet : les tests génétiques commencent à être à l'ordre du jour au niveau européen. Voici quelques semaines, le Parlement européen a créé une commission temporaire sur la génétique humaine présidée par l'un de ses députés, M. Robert Goebbels. J'ai évoqué devant elle la question des tests génétiques non pas seulement sous l'angle médical, mais en fonction de leur possible utilisation par les employeurs et les assureurs. Les droits des travailleurs migrants et les assurances sont en effet deux domaines très largement communautaires. Comme l'avait notamment proposé le GEE, des parlementaires européens, en particulier Mme Pervenche Béres, ont ainsi fait introduire dans la Charte des droits fondamentaux, une disposition suivant laquelle « nul ne peut faire l'objet de discrimination en fonction de ses caractéristiques génétiques ». On remarquera que le critère « génétique » n'est pas mentionné dans l'article 13 dit « clause générale de non-discrimination » du traité sur l'Union, et qu'il s'agit donc d'une novation. Reste maintenant à en interpréter la portée qui doit être nuancée.
La bioéthique renvoie à des questions à la fois juridiques, politiques et économiques. Je voudrais simplement rappeler quelques-unes d'entre elles en conclusion.
Premièrement, quelle est en la matière la place respective de la réglementation au sens large, qu'elle soit communautaire ou nationale, et de la jurisprudence ? Que faut-il réglementer par des textes ? Les progrès sont si rapides qu'il est difficile de tout encadrer.
Deuxièmement, dès lors que nombre de législations prévoient une forme d'autorité administrative indépendante pour contrôler les évolutions et pratiques biomédicales (greffes, recherche sur l'embryon, reproduction humaine, etc...) quels pouvoirs confier à une telle autorité ? Un pouvoir réglementaire ? Un pouvoir d'octroi d'autorisations individuelles ? Un pouvoir seulement consultatif ? Donner à ce type d'instance un pouvoir uniquement consultatif n'est pas la meilleure solution car l'on se rend compte, en général, que l'administration qui a moins de moyens, suit, dans 99 % des cas, les avis émis par une autorité de ce type ; ce qui allonge d'autant les délais de la prise de décision. En matière de reproduction et de recherche sur l'embryon, la formule de la Haute Autorité britannique sur la Fertilisation Humaine et l'Embryologie est pour moi la meilleure.
Enfin, comment concilier les impératifs du marché et les exigences éthiques ? Suivant la jurisprudence, les pratiques biomédicales sont maintenant considérées comme des prestations de services au sens communautaire du terme. La Cour de justice de Luxembourg a par exemple considéré que l'interruption volontaire de grossesse était une prestation de service. De même la High Court de Londres a-t-elle considéré qu'une femme, s'étant vu refuser une insémination post-mortem en Grande-Bretagne, pouvait se rendre en Belgique pour bénéficier de cette intervention. L'existence d'un marché unique a une grande influence sur l'exercice des droits et des libertés individuels. Et c'est pourquoi, il faut croire en l'avenir d'une bioéthique européenne.
M. Alain Claeys, rapporteur. - Ce point est certainement le plus important
- mais la mission n'y a pas tellement travaillé - car il peut rendre caduque toute une série de dispositifs dans le droit national.
Mme Noëlle Lenoir. - Au plan civil, oui ; au plan pénal, c'est moins sûr, car la compétence du juge pénal national reste fondée sur le principe de la territorialité ; encore que cela puisse évoluer dans le cadre de la communautarisation du pilier « Affaires Intérieures-Justice ».
De toutes les façons, en matière de bioéthique, il faudra déterminer ce qui doit être du ressort national et ce qui devrait faire l'objet d'une régulation européenne ou internationale. Pour ma part, je pense que la réglementation de la recherche sur l'embryon ne peut être, du moins en l'état actuel des choses, que nationale. En revanche, la régulation de l'emploi des tests génétiques dans le monde du travail et dans celui des assurances relève largement du droit communautaire. Enfin, le problème des brevets est, à mon avis, un sujet mondial qui touche au commerce international.
M. Pierre Hellier. - Madame Lenoir, nous avons recueilli, lors de nos auditions, un certain nombre d'avis discordants sur le danger de breveter ou de ne pas breveter ; certains nous ont affirmé qu'il n'y avait aucun risque, aucun problème, que tout ne sera pas breveté et que l'on pourra toujours travailler même s'il y a des brevets.
Second point : je suis choqué de l'achat du capital génétique de certaines peuplades ! Et en vous écoutant, on peut se demander si l'on a véritablement les moyens de résister. Enfin, nous allons passer inévitablement de la thérapeutique à la prévention des pathologies, puis à l'eugénisme. Tout cela est inquiétant.
Mme Noëlle Lenoir. - Je partage votre souci quant à la surestimation du déterminisme génétique, monsieur le député. S'agissant en revanche des brevets, il faut être conscient du fait que la brevetabilité du vivant est admise depuis vingt ans. En effet, un arrêt de la Cour suprême des États-Unis de 1980 a reconnu le droit de breveter une substance qui existe dans la nature, mais dont on a pu repérer certaines fonctions utilisables dans l'industrie. Avant 1980, on ne pouvait pas breveter en tant que telle une substance naturelle. Aujourd'hui, il est tout à fait possible de breveter une hormone et même un animal transgénique (la fameuse souris oncogène de « Harvard »). Breveter le « produit » vivant lui-même est beaucoup plus protecteur que de se limiter en effet à breveter le procédé ayant permis d'obtenir ce produit. C'est la raison pour laquelle la Cour suprême des États-Unis a décidé d'accorder cette protection aux inventions biotechnologiques dans une optique à l'évidence favorable à la recherche et à l'industrie, ce qui a contribué à l'avance prise par les États-Unis dans ce secteur. Même si certains pays en Europe sont en train de rattraper leur retard, les États-Unis restent en pointe. Il est donc difficile, et à mon avis non souhaitable, de revenir sur la brevetabilité du vivant. En revanche, il importe de mettre de l'ordre dans les pratiques des Offices de Brevet. En fonction de l'état des connaissances il y a quelques années, les Offices aux États-Unis et en Europe, notamment, ont accordé des brevets d'une portée beaucoup trop large (cela s'était passé de la même manière dans le domaine de la chimie). La découverte du gène de telle ou telle maladie paraissait extraordinaire, alors qu'on se rend compte aujourd'hui qu'il existe souvent des centaines de mutations génétiques à l'origine d'une même maladie, et qu'un gène n'a pas une seule, mais de multiples fonctions.
M. Alain Claeys, rapporteur. - Dans l'article 5 de la directive européenne, le premier alinéa est extrêmement clair, alors que le deuxième alinéa donne l'impression de revenir sur ce qui est affirmé ; quelle est votre interprétation ? N'y a-t-il pas une ambiguïté ?
Mme Noëlle Lenoir. - L'ambiguïté existe effectivement. À mon avis, cependant, les deux alinéas de l'article 5 de la directive doivent être interprétés ensemble. Le premier indique que l'on ne peut pas breveter quelque chose dont l'applicabilité industrielle serait trop approximative ; le deuxième, quant lui, précise que l'on maintient la jurisprudence selon laquelle on peut breveter un produit naturel si l'on peut décrire ses propriétés physico-chimiques. La directive valide donc la jurisprudence américaine que j'ai évoquée il y a un instant et qui est d'ailleurs appliquée en Europe depuis des années. Prenons l'exemple de la relaxine, hormone qui détend les muscles. Identifiée, isolée et reconstituée par recombinaison génétique, elle a fait l'objet d'un brevet délivré par l'Office Européen des Brevets de Munich. Tout fabricant d'un médicament qui utilise les propriétés de la relaxine doit donc payer des royalties à celui qui en a découvert les propriétés. L'article 5 de la directive aurait pu s'appliquer à ce brevet, me semble-t-il.
M. Jean-Pierre Foucher. - L'ambiguïté existe quand c'est pour une autre application que son application naturelle.
Mme Noëlle Lenoir. - Vous avez raison. Du fait de la délivrance de brevets d'une portée très large et étant donné que les Américains ont été les premiers à s'investir dans ce domaine, les États-Unis ont acquis une position dominante. Toutefois, eux aussi se sont aperçus des inconvénients d'une conception trop extensive de la brevetabilité. Ainsi, sous la pression du Gouvernement Clinton, l'Office américain des Brevets a-t-il été contraint de resserrer ses critères de brevetabilité pour mieux circonscrire la portée de la protection de chaque brevet.
Le brevet permet à la recherche d'être une activité rentable en amont de la fabrication du produit industriel qui sera mis plus tard, souvent beaucoup plus tard, sur le marché. Notamment le brevet assure des ressources aux start-up dont les financeurs et, quand elles sont cotées en bourse, les actionnaires, veulent être rémunérés. Le brevet, avant même d'ailleurs qu'il soit exploité, rentre en ligne de compte dans l'estimation de la valorisation d'une compagnie de biotechnologie. Les recherches en génétique exigent, il faut en être conscient, des investissements souvent importants et à long terme. Or le brevet permet un retour d'investissement nécessaire. Pour autant, il me parait indispensable de clarifier, au niveau européen, les critères de la brevetabilité du vivant : inventivité, caractère nouveau, applicabilité industrielle. Ces trois critères doivent être remplis effectivement sinon la délivrance d'un brevet ne se justifie pas. La question de la brevetabilité ou non des séquences partielles de gènes « non signifiantes » (97 % des gènes humains seraient non signifiants) est particulièrement complexe. En effet, si ces séquences génétiques ne remplissent pas les critères de brevetabilité, leur identification a néanmoins coûté beaucoup d'argent. Ainsi, le 15 février prochain, une version quasi intégrale d'un génome humain entier va être pour la première fois publiée. Le Consortium public anglo-américain (les Anglais y participent à hauteur d'un tiers, les Américains de deux tiers et les Français pour environ 2 %) ayant procédé au séquençage de ce génome publiera ses résultats dans la revue scientifique Nature, et rendra ses données accessibles gratuitement à la communauté scientifique internationale. La société privée créée par le biologiste Craig Venter, Celera Genomics, doit elle rémunérer ses actionnaires. C'est pourquoi, tout en faisant paraître ses données dans la revue Science, elle ne permettra pas de les copier et continuera à demander aux industries qui veulent les utiliser de payer un abonnement de plusieurs millions de dollars par an. MM. Clinton et Blair se sont émus des pratiques commerciales de Craig Venter. Mais comment et au nom de quels principes obliger une compagnie privée à livrer gratuitement le produit de son savoir-faire, alors que la compagnie en question a investi des sommes très importantes dans le séquençage du génome ? C'est toute la question.
L'âpreté de la concurrence entre laboratoires est une dimension majeure des enjeux, à la fois éthiques et économiques, d'une recherche véritablement sans précédent dans l'histoire des sciences.
M. Bernard Charles, président. - Madame Lenoir, je vous remercie infiniment.
Audition de M. Jacques DUMONT, président,
Mme Odile CORBIN, directeur général
et Mme CRESPON, directeur des affaires techniques,
du syndicat national de l'industrie des technologies médicales,
de M. Philippe ROUARD, M. le professeur Luc TÉOT
et de Mme Françoise ROCA,
membres du syndicat de l'industrie des dispositifs de soins médicaux
(Extrait du procès-verbal de la séance du 31 janvier 2001)
Présidence de M. Bernard Charles, président.
M. Bernard Charles, président. - Nous avons le plaisir d'accueillir aujourd'hui des représentants du Syndicat national de l'industrie des technologies médicales, le SNITEM, et du Syndicat de l'industrie des dispositifs de soins médicaux, l'APAMED. Le SNITEM est représenté par M. Jacques Dumont qui en est le président. Il est accompagné de Mme Odile Corbin et de Mme Crespon. Nous avons également le plaisir d'accueillir M. Philippe Rouard, Mme Roca et M. Luc Téot, qui représentent l'APAMED.
Je vous remercie d'avoir accepté de répondre à notre invitation. Comme vous savez, nous avons commencé d'auditionner, la semaine dernière, sur le bilan de la loi Huriet relative à la protection des personnes qui se prêtent à des recherches biomédicales.
À plusieurs occasions, M. Huriet et moi-même, ainsi que d'autres de nos collègues, avons senti qu'il y avait une différence dans les recherches cliniques entre les médicaments et les dispositifs médicaux et que la loi Huriet témoignait de quelques faiblesses dans le domaine des dispositifs médicaux. Des membres de comités de protection des personnes nous l'ont d'ailleurs confirmé, montrant la portion assez faible, par rapport au chiffre d'affaires hospitalier, des dispositifs médicaux qui passent dans les CCPPRB comparés au médicament.
J'ai donc souhaité l'audition de représentants des industriels pour nous permettre de voir comment, à l'occasion de la révision des lois bioéthiques, nous pourrions améliorer certains aspects de cette loi de 1988.
Sans doute pourriez-vous, dans un premier temps, nous préciser brièvement la notion de dispositif médical, car elle est souvent assimilée, à tort, à celle de médicament.
Vous pourriez également nous présenter les difficultés de mise en _uvre de la loi Huriet et nous présenter les aménagements qui pourraient y être apportés.
M. Jacques Dumont. - Ce qui est au centre du débat d'aujourd'hui est cette assimilation, trop souvent faite, entre l'industrie pharmaceutique et notre propre industrie. Le détail des différences qui nous séparent est important dans la mesure où la loi Huriet, du moins son interprétation, a fait une assimilation entre les deux qui nous a posé des problèmes très importants.
Pour définir en quelques mots qui nous sommes, je dirai que le SNITEM est le Syndicat national des industries de technologies médicales. Il regroupe environ deux cents entreprises, de taille extrêmement variable. Il faut préciser que notre industrie est très disparate, tant dans ses produits que dans ses technologies. Nous comptons une quarantaine de grosses sociétés, multinationales, qui opèrent sur un plan mondial et, à l'opposé, quelque cent vingt sociétés comptant entre cinquante et cent employés, travaillant sur des niches de produits et des technologies très spécifiques. Toute cette variété est régie par les mêmes réglementations, ce qui pose parfois certaines difficultés.
La multitude de nos produits est extrêmement importante par rapport à celle de la pharmacie. Il y a 3 500 molécules pharmaceutiques. Nous comptons, dans notre domaine, près de 8 000 lignes de nomenclature et 400 000 produits, une complexité extrême donc. Nous allons de l'imagerie au fauteuil roulant, en passant par des produits consommables comme les implants orthopédiques, les implants actifs tels les pacemaker, etc. C'est donc un éventail extrêmement large et disparate, même si tous ces produits ont été confrontés au même problème dans le cadre de la recherche.
M. Philippe Rouard. - L'APAMED est l'autre syndicat de l'industrie des dispositifs de soins médicaux. Il n'est pas concurrent mais plutôt complémentaire du SNITEM puisqu'il opère dans un domaine différent, celui des pansements, des contentions, des textiles de santé concernant des pathologies du domaine du traitement de la plaie, de l'insuffisance veineuse chronique et de l'orthopédie.
Nous avons une cinquantaine d'adhérents. Le chiffre d'affaires global de la profession est d'environ quatre milliards de francs, principalement réalisé en ville, c'est-à-dire dans les officines, car la plupart de nos produits sont inscrits au TIPS. De ce fait, nous sommes concernés par les questions liées à la prise en charge et par la future réglementation, qui tarde à sortir, sur les évaluations cliniques pour les nouveaux produits.
APAMED est un petit syndicat mais nous essayons d'être présents dans le domaine de la santé car nos produits sont de première nécessité et, bien souvent, utilisés par les patients eux-mêmes.
Mme Odile Corbin. - Je commencerai par une remarque liminaire générale et rappellerai qu'il nous est très vite apparu que le dispositif de la loi Huriet avait été conçu dans un contexte extrêmement marqué par la culture du médicament plutôt que par celle du dispositif médical.
À l'origine, il était même question de faire une loi qui assurerait la protection des volontaires sains. Puis, il est apparu qu'il convenait également de protéger toutes les personnes susceptibles d'entrer dans ce type de processus, et la loi Huriet a été élaborée dans ce sens. Je ne nie pas que c'est un dispositif qui a dû poser des problèmes à l'industrie pharmaceutique, il n'empêche que les difficultés rencontrées par les fabricants de dispositif médical sont d'une nature et d'une dimension tout à fait différentes.
Pour illustrer mon propos, je prendrai simplement un exemple et je rappellerai que l'article L. 209-9 du code la santé publique, qui traite du consentement de la personne entrant dans une étude, indique que cette personne dispose, bien entendu, de la possibilité de retirer son consentement à tout moment. C'est une notion dont tout citoyen peut se féliciter, mais il n'empêche que si cela représente un inconvénient pour l'industrie du médicament, il est clair que pour l'industrie du dispositif médical et, notamment pour les fabricants de dispositifs implantables, comme les prothèses ou les pacemaker, le problème se pose dans des termes différents, puisque l'on n'« explante » pas et si les personnes se retirent de l'étude, elles gardent le dispositif dont elles ont été bénéficiaires.
M. Bernard Charles, président. - Nous aurons également à étudier cette notion de « rétractabilité » car, dans les études génétiques, qui est un des thèmes de notre réflexion, nous rencontrerons le même problème même si ce n'est pas tout à fait comparable au cas d'une prothèse. Nous aurons à étudier le problème spécifique posé, à la fois pour les dispositifs médicaux et pour la génétique, par cette notion de rétractabilité que nous avons souhaitée inscrire dans la loi de 1988.
Mme Odile Corbin. - Je vais essayer de vous décrire rapidement les spécificités de ces produits qui ne sont pas des médicaments, qui ont des caractéristiques propres, et pour lesquels le dispositif de la loi Huriet a constitué un ensemble de difficultés plus ou moins mal surmontées, donnant lieu, sur un point au moins qui touche à la gratuité des produits, à des « bidouillages », c'est-à-dire à la recherche de solutions pour que chacun « s'y retrouve », mais qui ne sont pas satisfaisantes parce que ce sont des solutions non formalisées et qui sont dérogatoires par rapport à la règle commune.
Je vais vous donner quelques indications sur ce que sont nos produits ou, tout au moins, vous dire quelles sont les spécificités qui s'y attachent.
Par rapport au médicament, une des premières caractéristiques des dispositifs médicaux réside dans la différence de durée de vie. Pendant une dizaine d'années, j'ai fait partie d'un laboratoire français. Nous étions dans la fin des années 80, certains produits dataient des années 75. Dans le monde des dispositifs médicaux, nous ne connaissons pas ce type de situation. Des modifications incrémentales interviennent très rapidement et font qu'un dispositif médical a une durée de vie moyenne de l'ordre de deux ans. Il est clair qu'après ce délai, le dispositif qui arrive sur le marché n'est pas radicalement différent, c'est un dispositif modifié, mais ce n'est plus le même que le précédent.
Une autre spécificité qui s'attache au dispositif médical est la grande difficulté, voire l'impossibilité, lorsque l'on développe des études, de trouver des placebos ou des comparateurs. Le problème des placebos marque la différence avérée entre dispositif et médicament et pose un véritable problème puisque nous sommes dans une culture du médicament et que les textes et les procédures qu'ils prévoient s'inscrivent dans cette culture. Nous avons dû beaucoup discuter avec les autorités de tutelle pour faire admettre qu'il n'est pas toujours possible de trouver un placebo qui puisse entrer dans l'étude en même temps que le dispositif qui fait l'objet de cette étude.
Une autre caractéristique que l'on ne retrouve pas forcément dans d'autres produits, est celle des gammes. Dans le domaine des produits fabriqués par les adhérents de l'APAMED qui tournent tous autour des produits textiles - pansements et autres -, les effets de gamme sont extrêmement clairs. Un produit donné est décliné en une gamme. Par exemple, tel pansement sera décliné pour le coude, pour le genou, etc. On se demande s'il convient de faire entrer leur étude dans un dispositif Huriet. Mais cela rejoindra la problématique des difficultés rencontrées et de ce qu'il faudrait faire pour améliorer la situation.
Autre problème, les dispositifs médicaux qui coûtent très cher et ceux qui coûtent peu cher sont indifféremment soumis au même dispositif de la loi Huriet. Il y a là, de notre point de vue, une absence de proportion entre le coût de ces dispositifs et les coûts induits par le fait d'entrer dans un dispositif d'étude de type loi Huriet.
M. Bernard Charles, président. - Vous voulez parler de la fourniture gratuite des produits soumis à étude ?
Mme Odile Corbin. - Absolument, ainsi que des coûts associés. Ce problème sera développé par MM. Rouard et Téot.
On remarque également une très grande différence entre les deux secteurs car, globalement, même s'il se présente sous diverses formes, un médicament est une molécule alors que les dispositifs médicaux n'ont pas grand chose de commun entre eux. Entre une seringue, un pansement, un fauteuil roulant et un défibrillateur, il existe un extraordinaire éclatement des différents types de produits.
À cette fragmentation du marché, s'ajoute une différence marquée entre le secteur de la pharmacie et celui du dispositif médical, liée à la nature du tissu industriel. Dans notre secteur, nous comptons aussi de grandes entreprises, filiales de groupes internationaux, elles ne sont cependant pas aussi importantes que dans le domaine du médicament. Nous avons surtout tout un tissu de petites entreprises dont certaines sont proches de l'artisanat, puisque le plus petit de nos adhérents est constitué de trois personnes. C'est l'exemple extrême mais de nombreuses entreprises membres de notre syndicat fonctionnent avec quinze personnes, ce qui pose un problème d'échelle lorsqu'il faut se lancer dans l'essai de certains types de dispositif.
Le recrutement des patients est également un point de différenciation avec le secteur du médicament car nous nous heurtons, pour certains dispositifs, à énormément de difficultés, voire à une impossibilité, de réunir des cohortes comparables à celles qui existent pour le médicament. Il n'est pas question, dans notre secteur, de recruter des milliers de patients. Sur certaines études, nous sommes déjà très contents de compter une dizaine de patients. De ce point de vue aussi, se pose un problème d'échelle et de proportion.
Il nous semble enfin que nous avons parfois du mal à trouver des investigateurs en nombre suffisant qui soient à la fois compétents, au courant des contraintes de notre secteur et mobilisables sur les études que nous avons à mener. Parallèlement à ce manque d'investigateurs, nous constatons que peu de CCPPRB ont une réelle compétence en ce qui concerne les produits pour lesquels leur avis est requis.
M. Philippe Rouard. - Je parlerai en binôme avec le professeur Luc Téot, qui est président de la Société française et francophone de plaie et cicatrisation. Nous avons souhaité avoir un représentant médecin et praticien pour qu'il nous apporte sa vision du problème.
Le réel problème que pose le dispositif Huriet est celui de la gratuité des produits. C'est la véritable pierre d'achoppement. On se heurte aux coûts unitaires d'un certain nombre de produits, liés soit au coût unitaire élevé dans le cas d'un défibrillateur ou d'un scanner, soit à un coût de traitement qui, dans la durée, peut aboutir, en ce qui concerne par exemple le pansement, à des budgets extrêmement conséquents. Ce problème de la gratuité est essentiel et nous souhaitons vraiment que puisse être envisagée une prise en charge totale ou partielle de ces dispositifs médicaux, sous une forme qui reste à déterminer. En tout cas, il faudrait que puisse exister une compensation vis-à-vis du fabricant qui réalise l'étude, qui, bien évidemment, en tire un bénéfice, mais les patients et les personnes qui utilisent ces produits en tirent aussi bénéfice.
Le deuxième point est le montant des coûts associés, nous l'avons déjà évoqué. Lorsque l'on additionne les frais de dossiers, d'assurance et tout ce qui entoure la mise en place d'une étude clinique, on constate qu'ils constituent également un frein important parce que peu d'entreprises peuvent les supporter.
L'autre point important, sur lequel le professeur Téot va pouvoir nous apporter quelques éclairages, est la difficulté d'obtenir le consentement éclairé dans un certain nombre de cas. Professeur, vous pourriez sans doute nous livrer deux ou trois exemples dans le domaine du traitement de la plaie.
M. Luc Téot. - Le traitement de la plaie intéresse essentiellement une frange de la population âgée. En termes médicaux, la situation a beaucoup évolué depuis plusieurs années lorsqu'est apparu un principe très simple, celui du respect de la cicatrisation en milieu humide, c'est-à-dire lorsqu'est apparue une nouvelle classe de pansements qui ont pour effet essentiel d'absorber. Ces pansements ont transformé le pronostic cicatriciel et permettent à des personnes qui mouraient de leurs plaies, essentiellement les escarres, importantes chez les personnes âgées, d'être maintenues en vie, voire de guérir le plus souvent.
Une des difficultés que nous rencontrons dans ce cas précis est que notre population cible, pour ces pansements, est âgée. La plupart du temps, elle a perdu le contact avec la réalité, temporairement bien souvent, mais il est cependant difficile d'obtenir leur accord pour les engager dans une démarche même si celle-ci nous paraît louable. Nous tournons donc autour du problème, en essayant de faire venir la famille pour qu'elle valide, mais nous faisons aussi parfois appel à une tierce personne, la surveillante du service. En fait, nous ne sommes pas face à une situation très claire au regard de l'application de la loi Huriet.
M. Philippe Rouard. - C'est un point important. On constate aussi une disparité en fonction des coûts des investigations. C'est un problème sur lequel il va falloir se pencher.
Par ailleurs, faire trop d'études cliniques, lorsque ce n'est pas nécessaire, n'est pas forcément éthique parce que l'on expose beaucoup de gens. Il faut donc savoir précisément dans quel cadre l'étude doit se réaliser et s'il est nécessaire qu'elle se fasse toujours dans un cadre aussi contraignant que celui de la loi Huriet.
En conclusion, les industriels ont une volonté affichée de développer des études, mais toutes les difficultés que nous venons d'évoquer les incitent parfois à délocaliser les études cliniques. Ils ne les font plus en France mais dans des pays étrangers où les contraintes sont moins pesantes et où ils espèrent rencontrer de plus grandes facilités, techniques et financières, pour pouvoir les mener. C'est un aspect que vous souhaitiez développer, me semble-t-il, professeur Téot ?
M. Luc Téot. - En effet, il est au c_ur du débat. On voit apparaître des technologies nouvelles qui seraient extrêmement porteuses mais qui ne peuvent pas être mises en valeur du fait de structures trop petites ou insuffisamment financées, la moindre étude contre placebo coûtant à ces entreprises une somme considérable.
Le deuxième point est la valorisation nécessaire des pansements, par exemple, pour répondre à cette logique validante, cette logique d'evidence based medicine, cette médecine par les preuves, qui, depuis quinze ans, est la clé de notre validation scientifique. On bute sur un obstacle parce que le coût en France est élevé. Plusieurs firmes françaises n'hésitent plus à faire leurs études en multicentrique européen, dans les pays où la législation est légèrement différente, soit même à l'extérieur de l'Europe, dans des pays où la législation est moins stricte. Il est fort regrettable que l'on ne puisse pas valoriser nos propres forces.
M. Philippe Rouard. - Un autre élément, que nous avons déjà évoqué, est le fait que la perception, par les CCPPRB, de la spécificité des dispositifs médicaux est tout à fait insuffisante. Il serait souhaitable qu'ils en tiennent compte dans l'exercice de leurs compétences et dans les décisions qu'ils prennent et qu'ils ne soient pas totalement « imprégnés » de la culture médicament pour juger de la validité d'un protocole.
En résumé, nos deux souhaits principaux sont, d'une part, la prise en compte des spécificités des dispositifs médicaux et, d'autre part, ce qui semble le plus important, le principe de la gratuité sur lequel il convient de réfléchir ainsi que les modalités à mettre en _uvre pour que cette gratuité soit, en partie au moins, compensée par quelque chose qui reviendrait à l'industriel.
M. Jean-Luc Préel. - Vous venez d'évoquer des questions importantes. Je voudrais revenir sur celle des études contre placebo. Il me semble, en effet, que celles-ci se justifient moins que celles contre dispositif de référence déjà existant. Je voudrais connaître votre avis sur ce problème.
Dans ce cadre, on pourrait envisager que la gratuité ne soit plus obligatoire, en se disant que l'on met un nouveau pansement par rapport à un pansement déjà existant.
De même, pour le matériel implantable, prothèses biliaires ou cardiaques, c'est effectivement un produit par rapport à un autre, la question étant de savoir si le nouveau dispositif va apporter un service supplémentaire ou non. Et ce qui devrait permettre de le valider.
M. Philippe Rouard. - J'adhère totalement à ce que vous dites. Je pense que le « produit de référence » fait partie du traitement standard habituel et n'a plus lieu d'être fourni gratuitement.
La seule difficulté est la standardisation du protocole sur laquelle nous travaillons déjà depuis cinq ans. Nous sommes assez nombreux à nous y intéresser puisqu'il y a quinze jours, nous étions 3 400 réunis par cette question au Palais des Congrès. Mais l'harmonisation des pratiques, dirai-je pour rester neutre, ne se fait pas en un tour de main et il faut bien comprendre que 40 % encore du territoire ne comprend pas un traître mot à ce qu'est réellement un protocole de référence, tel que nous l'entendons ce soir. Se pose un problème d'information de l'ensemble des professionnels, information que nous nous efforçons de mener à bien mais qui demandera encore quelques années.
Mme Yvette Benayoun-Nakache. - Je souhaiterais poser une question technique parce que je n'ai pas une très grande connaissance en matière de pansement. Vous avez parlé de pansements absorbants pour les escarres. Avez-vous enregistré des avancées notoires sur les pansements pour grands brûlés ?
M. Luc Téot. - Nous entrons là dans la technique.
En quelques mots, ce qui différencie une plaie due à des escarres et une plaie due à une brûlure est l'absence d'infection locale dans le cas de l'escarre. Habituellement, l'escarre n'est pas infectée alors que la brûlure ne demande qu'à s'infecter très rapidement dès qu'elle dépasse le deuxième degré profond. Pour être clair et schématique, ces pansements absorbants sont utilisés en cas de brûlures tant que celles-ci ne dépassent pas une certaine profondeur. Au-delà, nous sommes obligés d'utiliser des topiques anti-infectieux, c'est-à-dire des crèmes qui contiennent des antibiotiques locaux.
M. Jean-Luc Préel. - Les dispositifs médicaux sont tellement variés et d'un coût si important parfois, ne pensez-vous pas qu'il serait judicieux de les séparer en plusieurs catégories ? J'imagine que les poches de stomisés, les pansements, etc. représentent une catégorie bien identifiable, pour laquelle on peut avoir des études en série relativement faciles. En revanche, des matériels implantables de type défibrillateur, dont les coûts sont complètement différents, relèvent plus d'une logique vraiment industrielle. Avez-vous des propositions pour distinguer diverses catégories sur lesquelles l'on pourrait avoir des études différentes ?
M. Jacques Dumont. - Ce ne sera pas simple. Nous avons 8 000 lignes de produits dans la nomenclature. Il y a effectivement des produits de grande consommation comme les poches de stomie qui se vendent par millions, le pansement également, mais il existe aussi un très grand nombre de produits, notamment en ce qui concerne les implants, qui représentent une variété extraordinaire de types de matériels. De là à les classifier et à avoir des règles de classification... En fait, il est extrêmement difficile de définir les bases sur lesquelles fonder une classification. Il faudrait trouver une articulation.
M. Philippe Rouard. - Une première articulation pourrait être invasif-non invasif ou implantable-non implantable. Cette césure, très grossière, serait déjà significative en elle-même.
Mme Odile Corbin. - Pour répondre à M. Préel sur la gratuité, je dirai qu'en fait des systèmes ont déjà été mis en place mais qu'ils varient énormément d'un établissement hospitalier à l'autre. Il existe, par exemple, des systèmes de prise en charge partagée entre industriel et établissement, qui ne sont toutefois pas satisfaisants parce qu'ils ne sont pas formalisés et que, de facto, ils se trouvent totalement dérogatoires. Il est certain qu'il y a des idées à approfondir de ce point de vue.
M. Bernard Charles, président. - Il est vrai que la gratuité est une base de réflexion dans la loi Huriet puisque celle loi a été essentiellement basée sur le médicament. Les évolutions ont fait, et c'est l'un de vos soucis, que l'Agence du médicament est devenue l'Agence des produits de santé et que le Comité économique des médicaments est devenu le Comité économique des produits de santé. On sent très bien une évolution qui tend à faire entrer sous le vocable « produits de santé » différentes catégories : médicaments, dispositifs médicaux, et peut-être bientôt d'autres, avec la génétique.
À l'occasion de la révision des lois bioéthiques de 1994, la loi Huriet ne sera pas revue sur le fond, mais nous souhaitons profiter de cette mission pour en mieux connaître les problèmes spécifiques d'application.
Mme Odile Corbin. - Que ce soit pour les médicaments ou pour les dispositifs médicaux, les études soumises à la loi Huriet concernent toutes les investigations, qu'elles soient pré ou post marquage CE. Or, pour un médicament, une fois que la molécule a passé le cap de l'AMM, c'est une molécule qui est fixée et figée, qui ne va plus se transformer. En revanche, dans le domaine du dispositif médical, c'est un phénomène qui, lui, est très spécifique, des études sont réalisées qui ne sont pas des études cliniques, qui sont, dans le fond, des études d'amélioration souvent conçues en collaboration avec des professionnels.
J'employais tout à l'heure le mot d'« incrémental », parce que je ne trouvais pas d'autre terme. Ce sont ces petits pas qui sont faits dans la vie des dispositifs, qui aboutissent parfois à des modifications conséquentes. Pour les études prémarquage CE, la loi Huriet peut s'appliquer mais on peut se poser la question de savoir si elle doit l'être pour les études postérieures à la mise sur le marché.
M. Bernard Charles, président. - Notre difficulté est de ne pas ouvrir de petites fenêtres par lesquelles tous risquent de tenter de passer. C'est tout le problème du législateur. Nous avons eu un débat soutenu, M. Claude Evin s'en souvient, concernant les phases IV du médicament. On nous avait dit que si nous encadrions ces phases IV, elles iraient toutes se faire à l'étranger.
Finalement, l'encadrement s'est fait et nous nous sommes rendu compte que cela ne marchait pas trop mal. Je comprends votre propos, surtout pour des PME car vous avez un tissu d'entreprises qui est totalement différent de celui de l'industrie du médicament. Mais notre rôle est de trouver l'équilibre. Si on ouvre un peu, on s'aperçoit que le système dérive rapidement.
M. Jacques Dumont. - J'irai dans votre sens, d'autant plus que, pour l'instant, les pansements sont relativement simples sur le plan technologique, mais que, dans peu de temps, nous verrons apparaître le biologique et l'interactif.
M. Bernard Charles, président. - Je vous remercie.
Audition de M. Charles GURY,
secrétaire général du syndicat des pharmaciens
des établissements publics de santé
et de Mme Hélène BARRETEAU
(Extrait du procès-verbal de la séance du 31 janvier 2001)
Présidence de M. Bernard Charles, président.
M. Bernard Charles, président. - Notre objectif principal est la révision de la loi de bioéthique de 1994. Mais, comme l'avait fait le Conseil d'État dans son analyse des lois bioéthiques, nous avons souhaité étudier une autre loi bioéthique, la loi Huriet-Sérusclat sur la protection des personnes qui se prêtent à des recherches biomédicales. Cette loi, qui avait fait l'objet d'un consensus à l'Assemblée nationale en 1988 - j'en étais d'ailleurs le rapporteur - est une loi qui a aujourd'hui plus de douze ans. C'est la raison pour laquelle nous souhaitions vous entendre à son sujet.
M. Charles Gury, vous êtes secrétaire général du Syndicat national des pharmaciens des établissements publics de santé, et vous êtes accompagné de Mme Hélène Barreteau, membre de ce syndicat.
Étant donné que les pharmaciens sont présents dans les CCPPRB et qu'en tant que gestionnaire des hôpitaux, vous avez la responsabilité de la gestion de tout ce qui est produit pour les expérimentations, nous voulions connaître votre avis sur les problèmes posés par cette loi.
Nous avons reçu, avant vous, les représentants de l'industrie des dispositifs médicaux qui nous ont fait part de leurs préoccupations spécifiques. Nous souhaiterions que vous nous apportiez votre témoignage afin que nous puissions voir les points qu'il vous semble nécessaire d'améliorer après douze ans de fonctionnement.
Nous en avons longuement parlé avec Claude Huriet, il y a quelques mois. Il est tout à fait favorable à l'idée de saisir l'opportunité de cette révision des lois bioéthiques de 1994 pour tenter d'améliorer certains aspects de celle de 1988, étant entendu que nous ne reviendrons pas sur le fond de son dispositif.
M. Charles Gury. - Je vous remercie de nous avoir conviés à cette réflexion. Tout d'abord, il faut, de notre point de vue, faire un bilan positif concernant l'aspect gestion et délivrance des unités thérapeutiques dans le cadre de la loi Huriet. Celle-ci nous permet de fournir des prestations qui vont dans le sens d'une sécurité accrue pour le patient, ce qui est le fondement et la philosophie de cette loi. Les textes répondent donc bien à cet objectif.
Je ne sais si je dois entrer dans le détail de ce que la gestion pharmaceutique des unités thérapeutiques peut apporter. Je pense que c'est connu. Je ne vous éclairerai pas sur ce sujet. Pour nous, le bilan est tout à fait positif.
Néanmoins, quelques problèmes se posent, qui tiennent principalement à l'inadéquation entre le statut des pharmacies à usage interne et celui d'établissements pharmaceutiques. Je pense à un point en particulier, concernant les autorisations d'importation des unités thérapeutiques. Ce problème se pose principalement lorsque, le promoteur - privé, la plupart du temps - est étranger et que l'essai est organisé par la maison mère ; donc, à l'étranger. Généralement, sa filiale française est mise complètement à l'écart de cet essai.
Se pose donc le problème de l'importation des unités thérapeutiques. Nous nous retrouvons dans la situation de voir de plus en plus de laboratoires nous demander, en tant que pharmaciens hospitaliers, de signer une demande d'importation. Or les textes actuels nous l'interdisent totalement puisque nous ne sommes pas un établissement pharmaceutique. Les textes sont très clairs à cet égard. Lorsque nous acceptons, demeure le problème de la responsabilité du contrôle pharmaceutique parce qu'à partir du moment où nous signons cette autorisation d'importation, nous en assumons la responsabilité pleine et entière.
M. Bernard Charles, président. - Recevez-vous de plus en plus de ces demandes ?
M. Charles Gury. - Aux dires de nos collègues, ces requêtes se produisent de plus en plus fréquemment dans le cadre d'essais multicentriques d'une maison mère. Naturellement, deux cas de figures s'offrent à nous. Le premier est celui d'essais provenant d'une maison mère au sein de la Communauté européenne, pour lesquels il y a une harmonisation des modèles des exigences de contrôle. Cela induit que nous pouvons nous engager sans crainte.
M. Bernard Charles, président. - Si un contrôle est accepté dans un État de la Communauté, vous pouvez considérer, en tant que signataire, que le contrôle est accepté et assumé. Il n'y a pas à réaliser de double contrôle.
M. Jean-Luc Préel. - Mais les laboratoires pharmaceutiques ont tous, ou quasiment, une filiale française. Pourquoi ne passent-ils pas par celle-ci ?
M. Charles Gury. - C'est une façon très simple de solutionner le problème.
M. Bernard Charles, président. - Il semblerait qu'il existe des blocages.
M. Charles Gury. - À mon avis, c'est un blocage interne des laboratoires.
M. Jean-Luc Préel. - Ce n'est pas un problème lié à la législation ?
Mme Hélène Barreteau. - Il n'y a pas que cela. Bien souvent, il est fait appel à des sociétés de services qui n'ont pas de pharmaciens, mais seulement des médecins.
M. Bernard Charles, président. - Il s'est en effet mis en place des sociétés de services pour les essais cliniques, souvent parallèles aux structures des purs laboratoires. C'est un phénomène dont je n'avais pas conscience qu'il fût aussi répandu. Vous avez bien fait de soulever cette question.
Sur le plan factuel, confirmez-vous que le pourcentage des dispositifs médicaux qui passent en CCPPRB est très inférieur à ce qu'il devrait être ? On nous parle de 10 % de l'activité. Cela tendrait à prouver qu'il y a des blocages. Sans doute nous faudra-t-il réfléchir à des évolutions qui permettent aux dispositifs médicaux de mieux s'intégrer au système ? Nous avons déjà eu, lors de l'audition précédente, quelques explications sur ces blocages.
M. Charles Gury. - Dans le cas des dispositifs médicaux, l'un des blocages principaux tient à l'un des aspects de la loi Huriet, qui rend obligatoire la gratuité des unités thérapeutiques en essai. Autant cette disposition a été acceptée par les industriels du médicament, autant elle pose problème pour les industriels des dispositifs médicaux, essentiellement d'ailleurs, pour des dispositifs médicaux implantables, l'argument étant que si, dans le cadre d'un essai, vous implantez ce dispositif, vous ne l'implantez pas pour la durée de l'inclusion mais, par définition, à vie.
M. Bernard Charles, président. - On aurait dû en implanter un de toute façon. C'est une substitution par un essai.
M. Charles Gury. - Les industriels disent qu'ils sont d'accord pour faire l'implantation mais que le patient va finalement en bénéficier toute sa vie. La question qu'ils posent est de savoir s'il est normal qu'ils payent au-delà de la période officielle d'inclusion définie pour l'essai ? C'est un des arguments des industriels.
Mme Hélène Barreteau. - De plus, si vous prenez l'exemple des prothèses auditives faites sur mesure, il paraît très difficile d'envisager de produire des séries sur des prothèses sur mesure. C'est là un autre problème.
M. Bernard Charles, président. - D'après vous, la spécificité de certains dispositifs médicaux doit conduire à une révision de la loi. Le problème est qu'il est très difficile de toucher à cet aspect de la loi, étant donné la très grande hétérogénéité de ces produits. Une loi ne serait jamais assez précise pour cela. Nous avons d'ailleurs demandé, lors de l'audition précédente, quels pourraient être les critères d'une classification. Les industriels eux-mêmes avaient du mal à les définir. Implantable/non implantable, disaient-ils. Mais nous ne pouvons pas légiférer en ouvrant ainsi de petites fenêtres.
Nous comprenons bien, et vous nous le confirmez, que la gratuité soit un facteur de blocage et qu'il nous faut aussi prendre en compte les problèmes économiques spécifiques aux PME dans ce domaine car le blocage peut dépasser le seul problème scientifique et celui des malades, pour finir par ne plus être qu'économique. Mais notre rôle est de trouver des solutions simples et générales qui permettent de contourner ce problème. Peut-être pouvez-vous réfléchir à des propositions ?
Nous disposerons de celles des industriels que nous auditionnerons dans le cadre de la commission spéciale qui suivra le dépôt du projet de loi sur le bureau de l'Assemblée nationale. Il nous faudra alors avoir des éléments concrets en main. Nous sentons bien que la loi Huriet, fondée sur la culture du médicament, nécessite une adaptation, après le fort développement que connaissent les dispositifs médicaux, mais il nous faut trouver le moyen de ne pas trop d'ouvrir de fenêtres.
M. Charles Gury. - Je partage votre avis. Autant je comprends que l'on puisse envisager la non-gratuité pour des dispositifs médicaux implantables, autant pour un dispositif médical situé à la frontière du médicament, comme certains produits servant à emboliser, pour les anévrismes, par exemple, qui ne sont utilisés que ponctuellement, il est beaucoup plus discutable qu'un industriel, promoteur privé, demande à faire payer le dispositif en question. Il y a sans doute une séparation à faire.
M. Bernard Charles, président. - La notion d'implantable/non implantable vous paraît donc, en tant que praticien, être une piste de réflexion raisonnable ?
M. Jean-Luc Préel. - Pourquoi en effet ne pas tracer une ligne entre dispositif implantables et non implantables. D'un côté, les premiers sont installés de manière définitive et on ne va pas aller les retirer. Pourtant, il me paraît difficile d'envisager que le défibrillateur implantable qui a permis la survie soit gratuit, vu le coût du matériel qui est, très souvent, élevé. C'est une limite possible.
De l'autre côté, pour les dispositifs médicaux tels que les pansements ou autres, dès lors qu'il ne s'agit pas d'études contre placebo mais d'études contre du matériel déjà existant, le but étant d'apporter un service supplémentaire, on pourrait envisager de le rémunérer au prix du référent. Cela ne me choquerait pas.
M. Bernard Charles, président. - Vous rejoignez là ce que disait le professeur Jaillon que nous avons auditionné, et qui évoquait justement cette possibilité.
M. Jean-Luc Préel. - Je suis d'accord mais, ensuite, les industriels du médicament demanderont la même chose. Il n'y aura pas de raison pour qu'on ne leur rembourse pas au prix du médicament référent.
M. Charles Gury. - Mais cette réglementation est acceptée dans le domaine du médicament.
M. Bernard Charles, président. - Le syndicat des pharmaciens le confirme ; il n'y a pas de discussion à ce sujet. Le problème ne se pose que pour les dispositifs médicaux.
Mme Yvette Benayoun-Nakache. - Une classification s'impose, de fait.
M. Bernard Charles, président. - Il faut trouver un moyen législatif, au moins généraliste, pas trop précis, mais qui permette de bien dégager les pistes qui s'ouvrent à nous.
M. Charles Gury. - « Implantable ». Il faut être prudent quand on emploie ce terme car il existe aussi des dispositifs médicaux implantables momentanément. Peut-être une piste pourrait-elle être la notion de dispositif implantable plus de trente jours, puisqu'il me semble que le texte sur la traçabilité s'imposerait théoriquement sur les dispositifs médicaux implantables pour une durée de plus de trente jours.
Mme Yvette Benayoun-Nakache. - Il y a aussi les implantables avec rejet qui peuvent survenir. Il faut une classification assez précise. Prenez l'exemple des ballonnets que l'on utilise lors des ruptures d'anévrisme.
M. Bernard Charles, président. - Ils sont à la limite du médicament. C'est un acte réalisé dans une action thérapeutique.
Mme Yvette Benayoun-Nakache. - Il y a les actions thérapeutiques et celles que l'on fait à titre expérimental. Si nous revoyons cette loi, il faut essayer de trouver une classification plus précise.
M. Bernard Charles, président. - Concernant la protection des malades et une meilleure traçabilité des produits, vous nous confirmez, en tant que gestionnaire, que la loi Huriet fonctionne correctement. Vous avez une maîtrise des circuits, de la traçabilité. Vous pouvez dire que tel produit en expérimentation est allé avec tel investigateur. C'est très important pour nous. Étudier cet aspect relève de notre rôle de législateur.
Vous confirmez que les dispositifs médicaux posent problème et que la justification de la non-gratuité doit s'étudier sur ceux que l'on implante, en particulier pour une durée supérieure à trente jours. C'est bien dans cette direction que vous nous suggérez de conduire notre réflexion ?
M. Charles Gury. - Tout à fait.
M. Bernard Charles, président. - Je souhaiterais que vous y réfléchissiez entre vous, en tant que praticiens et que vous puissiez nous faire ultérieurement quelques propositions. Il serait intéressant que vous nous remettiez alors des notes assez précises et qu'éventuellement nous vous auditionnons une nouvelle fois.
M. Charles Gury. - C'est entendu.
M. Bernard Charles, président. - Je vous remercie d'être venus.
| 
