

N° 451
______
ASSEMBLÉE NATIONALE
CONSTITUTION DU 4 OCTOBRE 1958
TREIZIÈME LÉGISLATURE
Enregistré à la Présidence de l'Assemblée nationale le 5 décembre 2007.
RAPPORT
FAIT
AU NOM DE LA COMMISSION DES AFFAIRES CULTURELLES, FAMILIALES ET SOCIALES SUR LE PROJET DE LOI, ADOPTÉ PAR LE SÉNAT, ratifiant l’ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament,
PAR Mme CÉcile Gallez,
Députée.
——
Voir les numéros :
Sénat : 340, 460 et T.A. 8 (2007-2008)
Assemblée nationale : 301
INTRODUCTION 5
I.- LE PROJET DE LOI PERMET DE RATIFIER L’ORDONNANCE PRÉVUE PAR LA LOI DU 26 FÉVRIER 2007 SELON UNE PROCÉDURE CONFORME AUX EXIGENCES CONSTITUTIONNELLES 7
A. LA VOIE DE L’ORDONNANCE EST APPARUE COMME LA PLUS ADAPTÉE POUR PERMETTRE À LA FRANCE D’HONORER RAPIDEMENT SES ENGAGEMENTS EUROPÉEN 7
1. La France accusait un retard important dans la transposition de plusieurs directives relatives aux produits de santé 7
2. L’urgence, la technicité et le caractère assez largement contraint de la transposition ont plaidé en faveur du recours à l’ordonnance 9
B. LE PROJET DE LOI SATISFAIT AUX CONDITIONS POSÉES PAR LA LOI DU 26 FÉVRIER 2007 12
1. L’ordonnance du 26 avril 2007 n’excède pas le champ de l’habilitation donnée par le Parlement 12
2. Le projet de loi portant ratification de cette ordonnance permettra de garantir la sécurité juridique de ses dispositions 13
II.- LE PROJET DE LOI PARTICIPE AU RENFORCEMENT DE LA QUALITÉ ET DE LA SÉCURITÉ DES PRODUITS DE SANTÉ 15
A. LES PRINCIPALES AVANCÉES CONTENUES DANS L’ORDONNANCE DU 26 AVRIL 2007 15
1. Les produits d’origine humaine 15
2. Les aliments diététiques, les produits cosmétiques et les médicaments à usage humain 16
3. Les médicaments vétérinaires 17
B. LES NOUVELLES DISPOSITIONS INTRODUITES PAR LE SÉNAT 18
1. Une nouvelle habilitation à légiférer par ordonnances est demandée par le gouvernement 18
2. L’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) deviendrait l’unique autorité compétente en matière de recherches biomédicales 19
3. Il apparaît problématique de reporter l'entrée en vigueur de l'interdiction de redistribution des médicaments non utilisés 19
Article 1er : Ratification de l’ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament 25
Article 1er bis : Habilitation à prendre par ordonnances les dispositions pour transposer la directive du 31 mars 2004 relative aux tissus et cellules humains et aménager les sanctions applicables dans le domaine des produits de santé 37
Article 2 : Contrôle douanier des importations et exportations d’échantillons biologiques, de sang et de ses composants et produits dérivés 39
Article 3 : Correction d’une erreur matérielle 40
Article 4 : Clarification rédactionnelle concernant le régime juridique des insecticides et acaricides à usage humain 42
Article 5 : Compétences de l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) en matière de recherches biomédicales 44
Article 6 : Conditions de collecte, de destruction et de redistribution à des fins humanitaire des médicaments non utilisés 48
ANNEXES 55
ANNEXE 1 : LISTE DES PERSONNES AUDITIONNÉES 55
ANNEXE 2 : ARTICLE 38 DE LA CONSTITUTION 57
ANNEXE 3 : ARTICLE 39 DE LA LOI N° 2007-248 DU 26 FÉVRIER 2007 PORTANT DIVERSES DISPOSITIONS D'ADAPTATION AU DROIT COMMUNAUTAIRE DANS LE DOMAINE DU MÉDICAMENT 59
TABLEAU COMPARATIF 61
ANNEXE : ORDONNANCE N° 2007-613 DU 26 AVRIL 2007 PORTANT DIVERSES DISPOSITIONS D’ADAPTATION AU DROIT COMMUNAUTAIRE DANS LE DOMAINE DU MÉDICAMENT 71
Afin de permettre la réalisation du marché intérieur dans le secteur pharmaceutique, tout en garantissant un niveau élevé de protection de la santé publique, un corpus de règles a été progressivement défini au niveau communautaire, depuis 1965, et a fait l’objet d’une réforme importante, engagée par la Commission européenne en juillet 2001. L’achèvement du marché unique dans ce secteur est en effet essentiel pour garantir l’accès rapide aux médicaments, la protection de la santé des patients ainsi que la promotion de l'innovation thérapeutique et de la compétitivité des entreprises.
Ce projet de loi a pour objet de ratifier l’ordonnance n° 2007-613 du 26 avril 2007, qui a été prise sur le fondement de l’habilitation donnée au gouvernement par la loi n° 2007-248 du 26 février 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament, afin de permettre à la France de transposer, non sans retard, un ensemble de directives adoptées pour la plupart en mars 2004, voire même depuis janvier et février 2003 pour deux d’entre elles.
Concernant des domaines très divers, l’ordonnance du 26 avril 2007 comporte plusieurs dispositions qui vont dans le sens d’un renforcement de la qualité et de la sécurité des produits de santé mis sur le marché. Compte tenu de l’encombrement de l’ordre du jour parlementaire, de l’urgence des mesures nationales de transposition et du caractère très technique d’un certain nombre d’entre elles, sans doute peut-on penser en effet que « les ordonnances ne sont pas les adversaires de la loi mais son nécessaire complément (1) ».
Il n’est cependant pas interdit d’espérer qu’à l’avenir, la transposition de textes communautaires sera préparée plus en amont afin d’éviter que la France ne se distingue pas par le retard pris pour honorer ses engagements européens et que la procédure de l’ordonnance apparaisse de ce fait comme le seul moyen d’y remédier rapidement, de la même façon qu’il serait incontestablement plus respectueux des droits du Parlement que les lois qu’il adopte puissent rapidement entrer en vigueur et, pour cela, que les textes nécessaires d’application soient également préparés plus en amont. Plus généralement, on peut s’interroger également sur les raisons de l’engorgement du calendrier législatif, qui conduit à différer, voire à empêcher, l'inscription à l’ordre du jour de textes prêts à l'examen.
Lors de l’examen de ce projet de loi en première lecture, le mercredi 17 octobre 2007, le Sénat a adopté quatre amendements, dont trois d’initiative gouvernementale. Ainsi, alors qu’il comportait initialement trois articles, le présent texte en comporte désormais sept, les quatre nouveaux articles introduits par le Sénat visant notamment à aménager le régime d’autorisation des recherches biomédicales ainsi que les conditions de collecte et de redistribution des médicaments non utilisés, qui sont rapportés dans les officines dans le cadre du dispositif « Cyclamed ».
I.- LE PROJET DE LOI PERMET DE RATIFIER L’ORDONNANCE PRÉVUE PAR LA LOI DU 26 FÉVRIER 2007 SELON UNE PROCÉDURE CONFORME AUX EXIGENCES CONSTITUTIONNELLES
La possibilité pour le gouvernement de légiférer par ordonnance est enserrée et conditionnée par une « loi du départ », selon la formule du professeur Jean Gicquel, en l’occurrence la loi n° 2007-248 du 26 février 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament, et par une « loi du retour » devant le Parlement, que constitue le présent projet de loi portant ratification de l’ordonnance prise sur son fondement.
A. LA VOIE DE L’ORDONNANCE EST APPARUE COMME LA PLUS ADAPTÉE POUR PERMETTRE À LA FRANCE D’HONORER RAPIDEMENT SES ENGAGEMENTS EUROPÉEN
1. La France accusait un retard important dans la transposition de plusieurs directives relatives aux produits de santé
Afin notamment de prendre en compte l’émergence de nouvelles thérapies, l’expérience acquise dans la mise en œuvre des procédures d’autorisation de mise sur le marché (AMM) et la nécessité de garantir l’accès aux produits de santé, les codes communautaires relatifs aux médicaments à usage humain et vétérinaire ont été profondément modifiés par une série de directives du Parlement européen et du Conseil, adoptées pour la plupart en mars 2004. En particulier, le règlement n° 726/2004 du 31 mars 2004, immédiatement applicable dans les États membres, a permis d’améliorer les conditions d’autorisation, de surveillance et de pharmacovigilance des médicaments à usage humain relevant de la procédure communautaire dite centralisée.
S’agissant en revanche des dispositions communautaires nécessitant des mesures nationales d’exécution, si la loi précitée du 26 février 2007 a permis d’adapter notre droit interne aux prescriptions de la directive n° 2004/27/CE du 31 mars 2004 relative aux médicaments à usage humain, plusieurs autres textes communautaires n’avaient toujours pas été transposés, du moins dans leur intégralité. Ces directives, dont les échéances fixées pour leur transposition ont expiré depuis plusieurs années, comme l’indique le tableau présenté ci-après, concernent des domaines très divers :
– la directive n° 2002/98/CE du 27 janvier 2003 fixe des normes de qualité et de sécurité pour la collecte, le contrôle, la transformation, la conservation et la distribution du sang humain et des composants sanguins ;
– la directive n° 2003/15/CE du 27 février 2003 vise à harmoniser les législations des États membres relatives aux produits cosmétiques ;
– la directive n° 2004/23/CE du 31 mars 2004 établit des normes de qualité et de sécurité pour le don, l'obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissus et cellules humains ;
– la directive n° 2004/24/CE du 31 mars 2004 introduit de nouvelles dispositions concernant les médicaments traditionnels à base de plantes ;
– la directive n° 2004/28/CE du 31 mars 2004 modifie le code communautaire relatif aux médicaments vétérinaires.
Dates limites de transposition par les États membres des directives du Parlement européen et du Conseil dans le domaine des produits de santé
Directives |
Date limite de transposition |
Directive n° 2002/98/CE du 27 janvier 2003 établissant des normes de qualité et de sécurité pour la collecte, le contrôle, la transformation, la conservation et la distribution du sang humain et des composants sanguins |
8 février 2005 |
Directive n° 2003/15/CE du 27 février 2003 modifiant la directive n° 76/768/CEE du Conseil concernant le rapprochement des législations des États membres relatives aux produits cosmétiques |
11 septembre 2004 |
Directive n° 2004/23/CE du 31 mars 2004 relative à l’établissement de normes de qualité et de sécurité pour le don, l'obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissus et cellules humains |
7 avril 2006 |
Directive n° 2004/24/CE du 31 mars 2004 modifiant, en ce qui concerne les médicaments traditionnels à base de plantes, la directive n° 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain |
30 octobre 2005 |
Directive n° 2004/28/CE du 31 mars 2004 modifiant la directive n° 2001/82/CE instituant un code communautaire relatif aux médicaments vétérinaires |
30 octobre 2005 |
Alors que la France a déjà été condamnée par la Cour de justice des communautés européennes (CJCE) pour manquement à ses obligations de transposition en matière d'importations personnelles de médicaments, de nouvelles procédures en manquement risquaient dès lors d’être engagées à son encontre, l’exposant de ce fait à de lourdes pénalités financières. En effet, selon les informations communiquées par le gouvernement :
– une mise en demeure a été adressée à la France pour défaut de transposition dans les délais impartis de la directive n° 2004/23 relative aux tissus et cellules, à laquelle les autorités françaises ont répondu le 12 juin 2007 ;
– la Commission a engagé un recours en manquement devant la CJCE, le 9 février 2007, pour défaut de transposition de la directive n° 2004/24 relative aux médicaments à base de plantes ;
– une autre procédure en manquement a été engagée concernant la directive n° 2004/28 instituant un code communautaire relatif aux médicaments vétérinaires, la CJCE ayant constaté dans un arrêt très récent (2) que la France avait effectivement manqué aux obligations qui lui incombent en application de l’article 3 de cette directive.
2. L’urgence, la technicité et le caractère assez largement contraint de la transposition ont plaidé en faveur du recours à l’ordonnance
L’article 38 de la Constitution permet au gouvernement, pendant un délai limité, de prendre par ordonnances des mesures qui relèvent matériellement du domaine de la loi. Parce qu’elle déroge à l’article 34 de la Constitution, aux termes duquel « la loi est votée par le Parlement », cette procédure est très encadrée et traditionnellement conçue comme exceptionnelle, ce qu’elle fut en effet jusqu’à ces dernières années.
Au fur et à mesure cependant, et de manière clairement plus marquée depuis 2000 comme l’illustre le graphique présenté ci-après, le recours aux ordonnances est devenu beaucoup plus fréquent, au point même qu’en 2005, le nombre d’ordonnances a représenté plus de la moitié des textes intervenus dans le domaine de la loi (3). Ce procédé de législation déléguée permet en effet au gouvernement de contourner l’encombrement du calendrier parlementaire, d’accélérer la mise en œuvre de son programme dans certaines circonstances ou d’alléger l’ordre du jour des assemblées des textes les plus techniques, tels que ceux prévoyant des mesures de codification à droit constant ou encore la transposition de certaines directives. C’est tout particulièrement le cas dans le domaine sanitaire et social, comme en témoignent plusieurs exemples récents concernant l’organisation des professions de santé ou le fonctionnement des établissements de santé (4).
Si le recours à cette procédure pour prendre des mesures d’adaptation au droit communautaire ne saurait être exclu par principe, celui-ci devrait cependant conserver un caractère exceptionnel, justifié par des considérations d’urgence et par l’impossibilité d’assurer, dans des conditions satisfaisantes, par d’autres moyens la transposition de mesures ne soulevant pas d’enjeux politiques majeurs.
Concernant les mesures nationales d’exécutions rendues nécessaires par la réforme d’ensemble de la législation pharmaceutique intervenue au printemps 2004, la plupart de ces conditions semblent en l’occurrence avoir été réunies. En effet, dès lors que les délais prescrits pour la transposition de plusieurs directives avaient expiré depuis plusieurs années, il devenait urgent de combler ce retard, le Conseil constitutionnel a reconnu que l'urgence est au nombre des justifications que le gouvernement peut invoquer pour recourir à l'article 38 de la Constitution.
Par ailleurs, selon les informations communiquées à la rapporteure lors de la préparation de l’examen du projet de loi portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament, la version initiale de celui-ci comportait plus de 90 articles visant à transposer plusieurs directives en date du 31 mars 2004. La nature technique de la plupart des dispositions proposées accentuant encore cette lourdeur, le projet de loi a alors été jugé beaucoup trop important pour qu’il soit examiné dans de bonnes conditions par le Parlement. Il a donc été décidé d’alléger le texte tout en préservant sa cohérence, en limitant son champ à la transposition de la directive n° 2004/27 précitée relative aux médicaments à usage humain.
À titre d’exemple, concernant le caractère très technique d’un certain nombre de prescriptions communautaires, les conditions particulières de délivrance des AMM des médicaments « destinés uniquement à être utilisés pour les poissons d’aquarium, oiseaux d’appartements, pigeons voyageurs, petits rongeurs, furets et lapins de compagnie » ou encore de commercialisation des génériques « destinés aux poissons et aux abeilles ou à d’autres espèces considérées comme mineures » (cf. l’article 32 de l’ordonnance n° 2006-613 du 26 avril 2007 en annexe), pour juridiquement nécessaire qu’il soit de les inscrire dans un texte de loi, ne semblent pas soulever d’enjeux politiques d’une importance telle qu’elle aurait pu valablement justifier de s’opposer au recours à l’ordonnance afin d’y procéder. Au surplus, le degré de précision atteint par un certain nombre de directives, sans doute nécessaire pour parvenir à l’achèvement du marché intérieur dans le secteur pharmaceutique, rend d’autant plus contrainte la définition des mesures nationales d’exécution.
Il est vrai cependant que toutes les mesures communautaires restant à transposer ne présentent pas la même importance et qu’en particulier, on aurait pu imaginer que les mesures d’adaptation au droit communautaire dans le domaine des produits d’origine humaine fassent l’objet d’un débat plus large, s’agissant notamment des questions relatives au don de gamètes et à l’assistance médicale à la procréation. L’engorgement de l’ordre du jour parlementaire puis la suspension des travaux de l’Assemblée nationale à l’occasion des élections présidentielle et législatives du printemps dernier auraient toutefois conduit à reporter de plusieurs mois, et sans doute davantage, l’entrée en vigueur de ses dispositions, alors qu’une mise en demeure a déjà été adressée à la France pour défaut de transposition de la directive n° 2004/23 relative aux tissus et cellules.
Pour l’ensemble de ces raisons, l’article 39 de la loi n° 2007-248 du 26 février 2007 a ainsi permis au gouvernement de prendre par ordonnances les mesures nécessaires à la poursuite de l’adaptation de notre réglementation au droit communautaire dans le domaine du médicament.
Évolution du nombre d’ordonnances publiées et expressément ratifiées par le Parlement depuis 1984
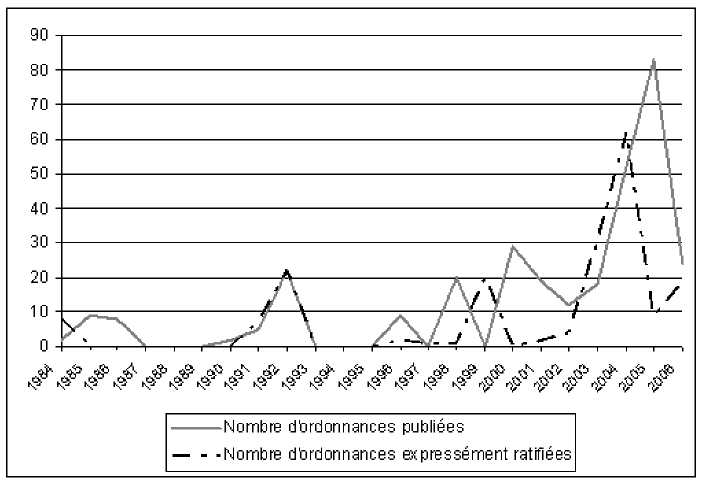
Source : graphique réalisé d’après les données présentées dans l’étude réalisée par le service des études juridiques du Sénat, « Les ordonnances, bilan au 31 décembre 2006 » (janvier 2007).
B. LE PROJET DE LOI SATISFAIT AUX CONDITIONS POSÉES PAR LA LOI DU 26 FÉVRIER 2007
1. L’ordonnance du 26 avril 2007 n’excède pas le champ de l’habilitation donnée par le Parlement
Aux termes de l’article 38 de la Constitution, l’habilitation à légiférer par ordonnance est donnée au gouvernement « pour l’exécution de son programme ». Selon la jurisprudence du Conseil constitutionnel (5), ce texte doit être entendu comme faisant obligation au gouvernement d’indiquer avec précision au Parlement, lors du dépôt d’un projet de loi d’habilitation et pour la justification de la demande présentée par lui, quelle est la finalité des mesures qu’il se propose de prendre ainsi que leur domaine d’intervention.
À cet égard, il ressort clairement des termes de l’article 39 de la loi n° 2007-248 du 26 février 2007 précitée que le champ et la finalité de l'autorisation délivrée au gouvernement ont été définis avec une précision suffisante pour satisfaire à ces exigences constitutionnelles. Conformément à l’habilitation donnée par le Parlement, l’ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament comporte ainsi un ensemble de mesures visant à :
– assurer la transposition des cinq directives susmentionnées relatives aux produits sanguins, aux médicaments vétérinaires, aux médicaments traditionnels à base de plantes, aux produits cosmétiques et aux tissus et cellules ;
– adapter au droit communautaire les dispositions du code de la santé publique relatives aux autorisations d’importation des médicaments à usage humain, aux insecticides et acaricides destinés à l’homme et au régime juridique des aliments diététiques destinés à des fins médicales spéciales ;
– habiliter les agents de la direction générale de la concurrence, de la consommation et de la répression des fraudes (DGCCRF) à transmettre à l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) les éléments recueillis dans l’exercice des pouvoirs d'enquête qui leur sont dévolus concernant certains produits de santé ;
– mettre en cohérence le dispositif existant dans le cadre du code de la santé publique en matière de classification des substances et préparations dangereuses et vénéneuses avec les dispositions issues du droit communautaire.
S’inscrivant dans les limites de l’habilitation accordée par le Parlement, le champ de l’ordonnance du 26 avril 2007 est même légèrement en deçà, puisque deux mesures prévues par l’article 39 de la loi du 26 février 2007 n’ont pu être prises par ordonnance dans les délais prescrits concernant, d’une part, le régime des sanctions pénales et administratives dans le domaine des produits de santé et, d’autre part, certaines dispositions nécessaires à la transposition de la directive n° 2004/23/CE relative aux tissus et cellules humains (cf. infra).
2. La ratification de l’ordonnance par le Parlement permettra de donner valeur législative à ses dispositions
Dans son troisième et dernier paragraphe, l’article 39 de la loi du 26 février 2007 a fixé deux échéances visant à limiter dans le temps la possibilité offerte au gouvernement de légiférer par ordonnances, conformément aux dispositions prévues par l’article 38 de la Constitution.
Il apparaît en premier lieu que l’ordonnance du 26 avril 2007 a été prise avant l’expiration du délai fixé par la loi d’habilitation, à savoir huit mois suivant la publication de celle-ci pour une grande partie des mesures prévues par l’article 39 précité, et notamment l’ensemble de celles visant à transposer une directive, et de trois mois à compter de cette même date dans les autres cas. En second lieu, le présent projet de loi, qui a été déposé au Sénat le 20 juin 2007, satisfait à la deuxième exigence posée par le Parlement, qui prévoyait que pour chaque ordonnance, un projet de loi de ratification est déposé « au plus tard le dernier jour du deuxième mois à compter de la publication de l’ordonnance », sous peine de caducité des ordonnances prises.
Ces deux conditions étant réunies, l’ordonnance est donc devenue applicable depuis sa publication en avril 2007. Il convient toutefois de rappeler que ce texte demeure un acte de nature réglementaire, qui peut dès lors être contesté devant le juge administratif, tant que sa ratification n’est pas intervenue, soit par le vote d’une loi de ratification ou d’une disposition législative ayant explicitement cet objet, soit en conséquence d’une modification apportée par le législateur aux dispositions de l’ordonnance qui peut, dans certains cas, être assimilée à une ratification implicite.
Par ailleurs, l’inscription à l’ordre du jour parlementaire du présent projet de loi mérite d’être souligné au regard du nombre d’ordonnances ayant été expressément ratifiées par le Parlement, qui est resté longtemps relativement faible, celui-ci représentant en effet moins d’une trentaine entre 1960 et 1990, sur les 158 ordonnances prises sur le fondement de l’article 38 de la Constitution.
En lui conférant une valeur législative, le projet de loi ratifiant l’ordonnance n° 2007-613 du 26 avril 2007 permettra ainsi d’assurer la sécurité juridique des mesures d’harmonisation, de simplification et d’amélioration de la qualité des produits de santé qu’elle comporte.
II.- LE PROJET DE LOI PARTICIPE AU RENFORCEMENT DE LA QUALITÉ ET DE LA SÉCURITÉ DES PRODUITS DE SANTÉ
Le projet de loi a pour principal objet de ratifier l'ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d'adaptation au droit communautaire dans le domaine du médicament. Sans détailler l’intégralité de ses dispositions, d’inégale importance et parfois très techniques, plusieurs d’entre elles méritent plus particulièrement d’être soulignées, en raison des avancées qu’elles comportent en matière de sécurité sanitaire des produits pharmaceutiques et humains.
Les nouvelles dispositions introduites par le Sénat, lors de l’examen de ce texte en première lecture, le 17 octobre 2007, contribuent par ailleurs à en élargir sensiblement la portée puisqu’il comprend désormais, notamment, des mesures relatives aux recherches biomédicales ou encore, ce qui semble autrement plus problématique, aux conditions de redistribution des médicaments non utilisés.
Les 53 articles que comporte cette ordonnance, dont l’article 1er du projet de loi porte ratification, sont regroupés dans huit chapitres consacrés respectivement aux médicaments traditionnels à base de plantes, aux produits d’origine humaine, aux produits cosmétiques, aux aliments diététiques destinés à des fins médicales spéciales, aux médicaments vétérinaires, à la classification des substances et préparations chimiques dangereuses et enfin, à d’autres dispositions.
L’utilisation croissante de produits d'origine humaine à des fins thérapeutiques, tels que des tissus oculaires, cardiovasculaires, des cellules reproductrices ou de la peau, ainsi que le développement des échanges intracommunautaires de ces substances ont rendu nécessaire la définition d’un cadre réglementaire minimal au niveau communautaire. Pour limiter au maximum les risques d’infection par transplantation, la directive n° 2004/23 du 31 mars 2004 a ainsi prévu des normes de qualité et de sécurité pour l’utilisation de ces produits.
Transposant ses dispositions, du moins une grande partie d’entre elles, le chapitre II de l’ordonnance du 26 avril 2007 permet de renforcer le contrôle des établissements qui transforment, conservent, stockent et distribuent des tissus et cellules humains, en prévoyant notamment que chaque établissement désigne une personne responsable de la sécurité et de la qualité de ces substances et des préparations de thérapie cellulaire.
Ce chapitre comporte également plusieurs dispositions reprenant les principes énoncés par la directive n° 2002/98 du 27 janvier 2003, qui prévoit la mise en place d’un ensemble de règles contraignantes tout au long de la filière transfusionnelle, c’est-à-dire lors de toutes les étapes qui vont de la collecte à l’emploi du sang humain et de ses composants. L’importance de l'utilisation thérapeutique des produits sanguins requiert en effet de veiller à ce qu’ils présentent toutes les garanties nécessaires en termes de qualité et de sécurité, en vue notamment de prévenir la transmission des maladies.
Dans cet objectif, les compétences respectives des établissements de transfusion sanguine et des établissements de santé sont précisées en matière de distribution et de délivrance des produits sanguins. En outre, le régime des sanctions prévues en cas de non-respect de la réglementation applicable à l'activité transfusionnelle est aménagé et renforcé. Ces dispositions viennent ainsi compléter celles prévues par le décret n° 2006-99 du 1er février 2006 relatif à l’Établissement français du sang (EFG) et à l’hémovigilance, qui avait permis d’engager la transposition de cette directive.
Enfin, la liste des produits susceptibles de faire l'objet d'un contrôle douanier est complétée pour y ajouter les dérivés de tissus et, comme le propose le nouvel article 2 du présent texte, les échantillons biologiques ainsi que le sang et ses produits dérivés destinés à des fins scientifiques, permettant ainsi de renforcer la surveillance sanitaire des échanges intracommunautaires de ces produits.
S’agissant, en premier lieu, des médicaments à usage humain, le chapitre Ier de l’ordonnance permet d’introduire dans le code de la santé publique la définition des médicaments à base de plantes et institue une procédure d'enregistrement spécifique pour les médicaments traditionnels à base de plantes, qui répondent à certains critères, liés notamment à l’ancienneté d’usage. Certaines dispositions concernant en particulier les autorisations d'importation ainsi que la publicité de médicaments à usage humain leur sont également rendues applicables.
Le chapitre V de l’ordonnance supprime par ailleurs les dispositions du code de la santé publique spécifiques aux insecticides et acaricides, puisque ces produits sont considérés comme des médicaments à usage humain au niveau communautaire, l’article 4 du projet de loi permettant d’apporter une clarification rédactionnelle opportune sur ce point. Enfin, le régime des autorisations d’importation des médicaments à usage humain est sensiblement modifié par le chapitre VIII de l’ordonnance, conformément aux dispositions prévues par l’article 39 de la loi du 26 février 2007.
S’agissant, en second lieu, des autres produits à usage humain, le chapitre III de l’ordonnance permet tout d’abord d’achever la transposition en droit interne de la directive n° 2003/15 du 27 février 2003, qui comportait plusieurs avancées significatives en matière d’étiquetage et d’information du consommateur sur la composition et les effets indésirables des produits cosmétiques. Ainsi, plus de quatre ans après l’adoption de cette directive, l’ordonnance a posé le principe de l'interdiction de réaliser des expérimentations animales ou de mettre sur le marché des produits cosmétiques dont la formulation finale ou les ingrédients ont fait l’objet de telles expérimentations. Cette interdiction doit entrer en vigueur au fur et à mesure de la validation au niveau communautaire de « méthodes alternatives » à ces expérimentations.
La définition et le régime juridique des aliments diététiques destinés à des fins médicales spéciales ont, d’autre part, été modifiés par le chapitre IV de l’ordonnance, afin notamment de reprendre la définition posée par la réglementation communautaire, en précisant qu’ils doivent être utilisés sous contrôle médical, de mieux définir la sous-catégorie d’aliments diététiques soumis à une prescription médicale obligatoire mais aussi de prévoir la définition de principes de bonnes pratiques en matière de fourniture et la délivrance.
Concernant, en troisième lieu, les mesures de contrôle et de gestion des risques, les agents des directions générale de la concurrence, de la consommation et de la répression des fraudes (DGCCRF), des douanes et des impôts ont désormais la possibilité de transmettre directement à l’AFSSAPS les éléments recueillis dans l'exercice de leurs pouvoirs d'enquête et de contrôle pour certains produits de santé, qui permettra d’en accroître sensiblement l’efficacité (chapitre VIII de l’ordonnance).
Enfin, le chapitre VII de ce texte vise à améliorer le dispositif de classification des substances et préparations chimiques dangereuses, en prenant mieux en compte les risques allergisants et toxiques pour la reproduction de certains de ses produits.
Le chapitre IV de l’ordonnance apporte plusieurs modifications au régime juridique des médicaments vétérinaires, concernant principalement les conditions de délivrance de l’autorisation de mise sur le marché (AMM) par l’Agence française de sécurité sanitaire des aliments (AFSSA), l’introduction de la définition des médicaments génériques, biologiques et biologiques similaires, l’aménagement de la procédure d’enregistrement des médicaments homéopathiques ainsi que la détermination de principes de bonnes pratiques par l’AFSSA, dans le domaine notamment de la fabrication et de la pharmacovigilance.
Il convient à cet égard de souligner que ces nouvelles mesures sont en grande partie similaires à celles prévues par la loi n° 2007-248 du 26 février 2007 concernant les médicaments à usage humain. L’article 3 du projet de loi permet par ailleurs de rectifier une erreur matérielle de rédaction figurant dans ce chapitre de l’ordonnance.
Lors de l’examen du présent projet de loi par le Sénat, le gouvernement a sollicité une nouvelle habilitation, pour une durée de quatre mois à compter de sa publication, afin de conduire jusqu’à leur terme deux projets d’ordonnance, qui avaient été engagés sur le fondement de l’article 39 de la loi du 26 février 2007. Ces textes n’ont pu en effet être publiés dans les délais prescrits en raison notamment du nombre des concertations requises et du caractère très technique de certaines de leurs dispositions.
L’article 1er bis du projet de loi prévoit ainsi d’autoriser le gouvernement à prendre les mesures nécessaires pour harmoniser et de compléter les dispositions pénales relatives aux médicaments vétérinaires et aux produits soumis au contrôle de l’AFSSAPS, ainsi que pour instaurer, en tant que de besoin, des sanctions administratives dans les domaines qui n’en disposent pas. Selon les informations recueillies par la rapporteure, le projet d’ordonnance en cours de préparation viserait à établir un dispositif pénal cohérent de sanctions pour l’ensemble des produits de santé. Il permettrait également d’actualiser et d’harmoniser entre les différents produits de santé les montants des amendes encourues et, le cas échéant, les durées d’emprisonnement, par catégorie d’infraction. De nouvelles infractions pénales seraient également créées, par exemple en cas de non-respect des obligations incombant aux industriels en matière de cosmétovigilance.
Le projet de loi prévoit, d’autre part, de poursuivre par ordonnance la transposition de la directive n° 2004/23 du 31 mars 2004 relative aux tissus et cellules, s’agissant de ses dispositions relatives au don de gamètes et à l’assistance médicale à la procréation (AMP). Le projet d'ordonnance, préparé en collaboration étroite avec l’Agence de la biomédecine et en cours de concertation avec les professionnels de santé, aurait notamment pour objet de :
– simplifier et d’adapter les dispositions législatives relatives aux conditions d’autorisation des activités d’AMP et subordonner l’autorisation de ces activités à l’avis de l’Agence de la biomédecine concernant les procédés mis en œuvre pour leur réalisation ;
– mettre en place un régime d’autorisation spécifique pour les activités d’importation ou d’exportation de gamètes et de tissus germinaux, assorti d’un dispositif de sanctions administratives et pénales ; sur le fondement de l’article 4 de la directive, il serait notamment prévu que seuls les gamètes obtenus dans le respect des principes 16 à 16-8 du code civil, relatifs au caractère volontaire, anonyme et gratuit des dons, puissent faire l’objet d’une importation ;
– prévoir la désignation d’une personne responsable de la qualité et de la sécurité des gamètes dans chacune des structures autorisées à pratiquer des activités d’AMP, tels que les établissement de santé et les laboratoires d’analyses, cette personne devant satisfaire à des exigences de qualification et d’expérience dans ce domaine ;
– délivrer au donneur de gamètes certaines informations sur les principes éthiques et les risques liés au don.
Dans l’ensemble, ces dispositions auraient donc essentiellement pour objet, conformément à la réglementation communautaire, de définir des normes de qualité et de sécurité dans le domaine de l’AMP, sans remettre en cause les principes posés par le Parlement dans la loi n° 2004-800 du 6 août 2004 relative à la bioéthique. Il convient par ailleurs de souligner que le champ de l’habilitation demandée par le gouvernement, tel que défini par l’article 1er bis du présent projet de loi, reprend les mêmes termes que les dispositions, déjà adoptées par le Parlement, figurant à l’article 39 de la loi n° 2007-248 du 26 février 2007 précitée.
2. L’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) deviendrait l’unique autorité compétente en matière de recherches biomédicales
La loi n° 2004-806 du 9 août 2004 relative à la politique de santé publique a procédé à une réforme importante du régime des recherches biomédicales afin de transposer les dispositions de la directive n° 2001/20/CE du Parlement européen et du Conseil du 4 avril 2001 relative aux essais cliniques de médicaments.
Comportant plusieurs dispositions ayant permis de renforcer significativement l’information et la protection des personnes y participant, cette loi a notamment prévu de soumettre la réalisation des recherches biomédicales à l’avis favorable du comité de protection des personnes et à une autorisation préalable de l’autorité compétente, c’est-à-dire l’AFSSAPS pour les médicaments et autres produits mentionnés à l’article L. 5311-1 du code de la santé publique ou le ministre chargé de la santé dans les autres cas.
Sur proposition du gouvernement, l’article 5 du présent projet de loi prévoit de transférer à l’AFSSAPS l’ensemble des missions qui incombent aujourd’hui au ministère de la santé en tant qu’autorité compétente pour certaines recherches. Selon le gouvernement, il s’agirait ainsi d’utiliser au mieux les moyens d’expertise de l’AFSSAPS mais aussi de simplifier le dispositif actuel par la création d’un guichet unique pour les promoteurs. Compte tenu notamment des moyens limités de la direction générale de la santé (DGS) pour la gestion des autorisations de recherche, cette mesure viserait également à renforcer la surveillance sanitaire de ces activités.
3. Il apparaît problématique de reporter l’entrée en vigueur de l’interdiction de redistribution des médicaments non utilisés
L’association « Cyclamed » a été créée en 1993 par l’industrie pharmaceutique afin de satisfaire à l’obligation posée par le décret n° 92-377 du 1er avril 1992, en vertu duquel tout producteur de déchets est tenu de contribuer ou de pourvoir à l’élimination de l’ensemble des déchets d’emballage. Au-delà de ces objectifs environnementaux, l’institution d’un dispositif de collecte des médicaments non utilisés (MNU) par les pharmacies devait permettre de limiter les risques sanitaires liés à ce type particulier de déchets et de redistribuer certains de ces médicaments à des fins humanitaires dans les pays en voie de développement.
Malgré ces objectifs séduisants, le dévouement des bénévoles et le travail exemplaire de plusieurs associations, ce dispositif s’est toutefois avéré décevant et source de nombreux problèmes, dénoncés par un rapport récent de l’Inspection générale des affaires sociales (6).
En premier lieu, le nombre de MNU utilisables est en baisse continue depuis dix ans comme l’indique le tableau ci-après, ce qui s’explique en grande partie par la nécessité qui s’est imposée de mieux sélectionner les médicaments pouvant être redistribués. En effet, les MNU sont souvent inadaptés aux besoins locaux, puisqu’ils comptent un nombre important de psychotropes et de médicaments destinés au traitement de maladies associées au vieillissement mais insuffisamment d’anti-infectieux ou de génériques par exemple. De surcroît, ces médicaments ne sont pas toujours envoyés sous une forme leur permettant d’être utilisés, en raison notamment de problèmes de langue ou de quantités insuffisantes pour initier un traitement.
Évolution de la réutilisation humanitaire des médicaments collectés via Cyclamed
(en tonnes)
1995 |
1999 |
2000 |
2001 |
2002 |
2003 | |
Total des médicaments collectés |
- |
3 355 |
3 016 |
3 054 |
3 446 |
3 211 |
Total des MNU expédiés (1) |
1 515 |
831 |
713 |
646 |
629 |
210 |
Déchets permettant une récupération énergétique |
5 379 |
10 385 |
10 994 |
12 151 |
13 444 |
14 207 |
Valorisation totale (2) |
6 894 |
11 216 |
11 707 |
12 797 |
14 073 |
14 717 |
Taux de valorisation humanitaire (1/2) |
0,22 |
0,07 |
0,06 |
0,05 |
0,04 |
0,03 |
Source : rapport de l’Inspection générale des affaires sociales (IGAS) sur le dispositif de recyclage des médicaments « Cyclamed » (janvier 2005)
En second lieu, les MNU ne présentent clairement pas toutes les garanties requises en termes de traçabilité et de qualité (conservation dans des conditions inadéquates, dates de péremptions dépassées, envoi de produits retirés du marché, etc.). Par ailleurs, l’existence d’un circuit parallèle de distribution des médicaments entraîne des risques de trafics et de fraudes, y compris en France. Enfin, l’envoi de ces médicaments conduit à fragiliser les politiques pharmaceutiques locales, fondées sur la structuration des achats de médicaments et des réseaux de distribution, du fait notamment de la revente à la sauvette de ces médicaments par des personnes non qualifiées, ce qui, outre d’évidents risques sanitaires, a pour effet d’affaiblir les pharmaciens et grossistes locaux, qui peuvent difficilement lutter contre cette concurrence.
Plusieurs organisations internationales, au premier rang desquelles l’Organisation mondiale de la santé (OMS), la Banque mondiale et le Haut comité pour les réfugiés, ont dès lors recommandé que les MNU ne fassent plus l’objet de dons, sentiment partagé par un nombre croissant d’associations humanitaires. Conformément à ces préconisations, la loi n° 2007-248 du 26 février 2007 précitée a ainsi prévu d’interdire la redistribution des médicaments collectés dans les officines en prévoyant toutefois, avant son entrée en vigueur, une période transitoire de dix-huit mois suivant la publication de la loi, soit jusqu’en août 2008.
Cette période devait notamment permettre aux associations humanitaires de s’adapter et de trouver de nouvelles sources d’approvisionnements en médicaments neufs. Depuis lors, un groupe de travail a été constitué à ce sujet par le ministère de la santé et les associations ont entrepris de définir précisément leurs besoins. Toutefois, outre le fait que le décret d’application prévu par l’article 32 de loi du 26 février 2007 n’a toujours pas été publié, des interrogations subsistent sur les modalités d’organisation et de financement de ce nouveau dispositif, les entreprises du médicament (LEEM) ayant en effet accepté le principe d’une contribution par des dons de médicaments à des actions humanitaires, mais pas de prendre en charge la totalité des besoins recensés, dont le coût serait au minimum de 20 millions d’euros par an.
Par ailleurs, en raison notamment des délais requis pour l’agrément des associations et l’obtention d’une autorisation d’ouverture d’un établissement pharmaceutique, nécessaire pour permettre aux associations d’assurer la distribution de médicaments, certaines d’entre elles se sont inquiétées des difficultés à poursuivre leurs activités après août 2008. C’est pourquoi le Sénat a adopté un amendement visant à prolonger la durée de cette période transitoire pendant dix-huit mois suivant la publication du présent projet de loi (article 6).
Il apparaît cependant problématique de différer ainsi au second semestre de l’année 2009 au plus tôt l’entrée en vigueur d’une mesure importante en termes de santé publique, votée par le Parlement il y a à peine quelques mois, et ce d’autant plus qu’un consensus a émergé sur la nécessité de mettre un terme à la redistribution des MNU. À l’initiative de la rapporteure, la commission a adopté un amendement prévoyant de maintenir l’échéance fixée par loi du 26 février 2007 pour l’entrée en vigueur de l’interdiction de redistribution des MNU, soit en août 2008. En tout état de cause, il apparaît nécessaire que les textes réglementaires d’application de la loi du 26 février 2007 soient rapidement publiés mais aussi qu’une réflexion soit engagée sur le conditionnement des médicaments et les actions de nature à promouvoir leur bon usage. Si les associations doivent disposer des moyens nécessaires pour poursuivre leurs activités, il convient également que le ministère de la santé et l’AFSSAPS examinent dans les meilleurs délais les demandes d’agrément et d’autorisation d’ouverture d’établissement pharmaceutique qui seraient déposées par les associations. Enfin, cette question doit être replacée dans le cadre plus général de la politique de coopération française, afin d’apporter une aide réellement adaptée aux besoins des pays en voie de développement.
L’article 6 du projet de loi vise, d’autre part, à permettre aux centres et structures disposant d’équipes mobiles de soins aux personnes en situation de précarité ou d’exclusion, qui sont gérés par des organismes à but non lucratif, de délivrer gratuitement les médicaments nécessaires à leurs soins. Afin de mieux encadrer ce dispositif dérogatoire et garantir la sécurité et la qualité des soins dispensés à ces personnes en situation de précarité, la commission a adopté deux amendements prévoyant l’agrément par le ministre chargé de la santé de ces organismes ainsi que la délivrance des médicaments sous la responsabilité d’un médecin ou d’un pharmacien.
La commission a examiné le présent projet de loi, sur le rapport de Mme Cécile Gallez, au cours de sa séance du mercredi 5 décembre 2007.
Un débat a suivi l’exposé de la rapporteure.
M. Christian Kert, président, a félicité la rapporteure pour la qualité de son exposé.
Mme Catherine Lemorton s’est déclarée globalement en accord avec les dispositions prévues par le projet de loi, en regrettant cependant le retard avec lequel le gouvernement a entrepris la transposition de ces directives par rapport aux autres pays européens. Concernant les médicaments non utilisés, les problèmes liés à leur redistribution ont été soulevés, dès la fin des années 1990, par des organisations non gouvernementales (ONG), telles que Pharmaciens sans frontières et Médecins sans frontières, ainsi que par l’Inspection générale des affaires sociales (IGAS), dans un rapport de janvier 2005, et l’Organisation mondiale de la santé (OMS). Comme l’a souligné la rapporteure, il n’apparaît donc pas opportun de reporter l’entrée en vigueur des dispositions prévoyant l’interdiction de la redistribution humanitaire des médicaments non utilisés au-delà du délai de dix-huit mois. De plus, il n’est pas acceptable que l’industrie pharmaceutique puisse empêcher le développement des médicaments génériques dans certains pays en voie de développement ou émergents, comme cela s’est produit récemment en Inde, où un procès a été engagé par un laboratoire contre l’État indien afin de contrer la commercialisation de certaines classes de médicaments génériques. À l’initiative du groupe socialiste, le Parlement européen est d’ailleurs intervenu en faveur du retrait de la plainte déposée par cette entreprise pharmaceutique.
En application de la loi du 26 février 2007, les associations humanitaires, qui s’impliquent dans la délivrance de médicaments, en particuliers dans des situations d’urgence, disposent d’un délai de dix-huit mois pour obtenir le statut d’établissement pharmaceutique, ce qui est une bonne chose. Toutefois, l’association Tulipe, fondée par les entreprises du médicament (LEEM), ne doit pas continuer à fonctionner dans une certaine opacité, au risque d’empêcher la mise en place d’une véritable politique du médicament dans les pays en voie de développement. Le ministère des affaires étrangères devrait sans doute intervenir pour clarifier cette situation. Il convient également de souligner qu’aucun laboratoire commercialisant des génériques n’est adhérent de Tulipe.
Compte tenu de l’importance de ces questions, il serait par ailleurs souhaitable que la ministre de la santé, de la jeunesse et des sports soit présente lors de l’examen de ce projet de loi en séance publique, plutôt que la secrétaire d’État chargée de la solidarité comme c’était le cas au Sénat, et ce d’autant plus que la politique du médicament n’a pas aujourd’hui la place qu’elle devrait avoir dans notre pays. Il est donc très positif que des travaux soient actuellement engagés sur cette question par la Mission d’évaluation et de contrôle des lois de financement de la sécurité sociale (MECSS), sous la présidence de M. Pierre Morange et M. Jean Mallot.
Il convient par ailleurs de se féliciter que le présent projet de loi ne comporte aucune mesure concernant les programmes d’observance, alors qu’il avait été initialement envisagé par le gouvernement, en février dernier, de prendre de telles dispositions par ordonnance. Le rapport récent de l’Inspection générale des affaires sociales (IGAS) sur la visite médicale et l’information des médecins souligne d’ailleurs clairement l’importance de la place de l’industrie pharmaceutique dans notre pays. Enfin, si les dispositions du projet de loi peuvent sembler sécurisantes, il est essentiel que les principes posés par la loi du 6 août 2004 relative à la bioéthique, dont la révision est prévue pour 2009, ne soient pas remis en cause. Il convient donc de suivre attentivement cette question.
En réponse, la rapporteure a apporté les précisions suivantes :
– Il est vrai que la France a engagé avec retard la transposition des directives européennes, comme cela a déjà été dit.
– Concernant les médicaments non utilisés, le report de la réforme du dispositif « Cyclamed » conduirait en effet à de nombreux problèmes, en particulier pour les pharmaciens qui ne savent plus aujourd’hui précisément ce qu’ils doivent faire, mais aussi car il pourrait risquer de favoriser la récupération de ces médicaments par des personnes qui ne seraient pas habilitées à le faire. La situation ne peut donc pas perdurer. C’est pourquoi il faut maintenir la date d’août 2008 pour l’entrée en vigueur des dispositions relatives à l’interdiction de redistribution des médicaments non utilisés.
Mme Catherine Lemorton a souhaité que les pouvoirs publics, d’une part, rendent public le rapport de l’IGAS sur l’observance des traitements et, d’autre part, engagent une campagne d’information pour sensibiliser nos concitoyens à l’importance de la collecte des médicaments non utilisés dans les pharmacies. En effet, du fait de la suppression du dispositif de redistribution humanitaire des médicaments non utilisés, il est à craindre que les patients ne jettent les médicaments, en pensant que ce n’est plus la peine de les rapporter dans les officines, sans en mesurer toutes les conséquences, notamment environnementales.
La rapporteure a déclaré partager cette préoccupation.
La commission a examiné les articles du présent projet de loi au cours de sa séance du mercredi 5 décembre 2007.
Article 1er
Ratification de l’ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament
Cet article a pour objet de ratifier l’ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament, prise sur le fondement de l’habilitation accordée au gouvernement par l’article 39 de la loi n° 2007-248 du 26 février 2007, dont le texte est reproduit dans l’annexe n° 3 du présent rapport.
Cette ordonnance comporte huit chapitres, consacrés respectivement aux médicaments traditionnels à base de plantes, aux produits d’origine humaine, aux produits cosmétiques, aux aliments diététiques destinés à des fins médicales spéciales, aux médicaments vétérinaires, à la classification des substances et préparations chimiques dangereuses et, enfin, à d’autres dispositions.
1. Les médicaments traditionnels à base de plantes
Le chapitre Ier de l’ordonnance comporte six articles visant à transposer la directive n° 2004/24/CE du 31 mars 2004 relative aux médicaments traditionnels à base de plantes (7).
L’article L. 5121-1 du code de la santé publique, qui énumère les différentes catégories de médicaments à usage humain, est tout d’abord complété pour introduire la définition des médicaments à base de plantes, soit « tout médicament dont les substances actives sont exclusivement une ou plusieurs substances végétales ou préparations à base de plantes ou une association de plusieurs substances végétales ou préparations à base de plantes » (article 1er de l’ordonnance).
Une procédure d’enregistrement spécifique auprès de l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) est par ailleurs instituée pour les médicaments traditionnels à base de plantes répondant à certains critères :
– ils sont conçus pour être utilisés sans l’intervention d’un médecin, à des fins de diagnostic, de prescription ou de suivi des traitements ;
– ils sont exclusivement destinés à être administrés selon un dosage et une posologie spécifiés, administrés par voie orale, externe ou par inhalation ;
– la durée d’usage traditionnel est écoulée et les données sur cet usage sont suffisantes.
Selon les précisions apportées par le ministère de la santé, les médicaments traditionnels à base de plantes relèvent du monopole pharmaceutique et ne peuvent être délivrés que par un pharmacien, dès lors qu’ils répondent à la définition du médicament posée par l’article L. 5121-1 précité. La spécificité du régime de ces médicaments réside dans le fait qu'ils sont soumis à un enregistrement simplifié et non à une autorisation de mise sur le marché (AMM) bien qu'ils soient définis comme des médicaments. En effet, la directive n° 2004/24/CE prévoit que contrairement à la procédure d’AMM, fondée sur le critère d’efficacité prouvée du médicament, l’enregistrement des médicaments traditionnels à base de plantes repose principalement sur une efficacité présumée, du fait notamment de l’ancienneté d’usage.
Si l’AFSSAPS considère que les conditions susmentionnées ne sont pas réunies, la commercialisation des médicaments traditionnels à base de plantes est alors subordonnée à la délivrance d’une autorisation de mise sur le marché (AMM) ou de l’enregistrement prévu pour les médicaments homéopathiques.
Certaines dispositions sont également harmonisées avec celles relatives aux conditions de délivrance de l'AMM des médicaments à usage humain telles qu’issues de la loi du 26 février 2007 précitée. Ainsi, il est notamment prévu que l’enregistrement est effectué pour cinq ans et peut être renouvelé, le cas échéant, pour une durée illimitée (article 2).
Les modalités d’application de cette procédure, concernant notamment le contenu du dossier présenté à l’appui d’une demande d’enregistrement ainsi que les conditions de renouvellement, de modification, voire de suspension de celui-ci, doivent être précisées par un décret en Conseil d’État (article 4).
L’ordonnance comporte par ailleurs d’autres mesures qui sont liées à la transposition de la directive :
– Il est tout d’abord institué, au profit de l’AFSSAPS, une taxe à laquelle sont assujettis les demandeurs de l’enregistrement pour des médicaments traditionnels à base de plantes, dont le montant doit être fixé par décret dans la limite de 10 110 euros, de la même façon que les articles L. 5121-15 et L. 5121-16 du même code, tels que modifiés par la loi précitée du 26 février 2007, prévoient le versement d’un tel droit progressif pour les demandes d’AMM ou d’enregistrement présentées pour d’autres médicaments à usage humain (article 3) ;
– L’entreprise exploitant un médicament traditionnel à base de plantes est par ailleurs tenue de communiquer à l’AFSSAPS la date de commercialisation de chaque présentation de ce médicament (article 5), ainsi qu’il prévu pour les autres médicaments disposant d’une AMM ou d’un enregistrement homéopathique, conformément à l’article L. 5124-5 du même code tel qu’issu de la loi du 26 février 2007 précitée ;
– Enfin, l’article L. 5123-2 du même code, concernant la liste des médicaments pris en charge et utilisés par les collectivités publiques, est complété par la référence aux médicaments traditionnels à base de plantes qui relèvent de la procédure d’enregistrement (article 6).
2. Les produits d’origine humaine
Le chapitre II de l’ordonnance comporte quinze articles, qui sont rassemblés dans deux sections respectivement consacrées aux tissus et cellules et aux produits sanguins.
Les neuf articles de la section 1 de ce chapitre ont pour principal objet de transposer plusieurs dispositions de la directive n° 2004/23/CE du 31 mars 2004, qui définit des normes de qualité et de sécurité concernant les tissus et cellules humains utilisés pour des usages thérapeutiques (8).
Les articles L. 1211-8, L. 1242-1 et L. 1243-6 du code de la santé publique sont tout d’abord modifiés afin d’exclure du champ d’application de son livre II, relatif au don et à l’utilisation des produits du corps humain, ceux de ces produits qui sont prélevés et utilisés à des fins thérapeutiques autologues (c’est-à-dire sur une même personne), dans le cadre d’une seule et même intervention, sans être préparés ou conservés à aucun moment dans un établissement ou un organisme habilité à le faire (article 7).
Le rapport au Président de la République sur la présente ordonnance (9) précise à cet égard que « l’utilisation péri-opératoire de ces produits relève de l'évaluation et de l'encadrement de droit commun des actes médicaux effectués par la Haute autorité de santé et de la déclaration obligatoire à l’autorité compétente des évènements indésirables graves liés à des soins et de la vigilance relative aux actes de soins prévue à l’article L. 1413-14 ».
Cette section prévoit, d’autre part, que les établissements qui transforment, conservent, stockent et distribuent des tissus et cellules humains ne peuvent, sans avoir obtenu au préalable une autorisation des autorités compétentes, apporter de modification substantielle à leurs activités. Ces dispositions permettent ainsi de soumettre à une autorisation les modifications apportées non seulement aux éléments de l’AMM initiale, mais également à l’environnement médical et technique, concernant par exemple les locaux ou les équipements, qui peuvent avoir des conséquences sur la qualité des produits. La liste des modifications substantielles sera définie par un décret en Conseil d’État, les autres modifications devant être déclarées auprès du directeur général de l'AFSSAPS (article 8). Une modification de cohérence est en conséquence apportée à l’article L. 1243-9 du même code (article 11).
L’article 8 de l’ordonnance comporte d’autres dispositions visant notamment à :
– permettre aux établissements qui prélèvent des cellules souches hématopoïétiques (10) issues de la moelle osseuse de les distribuer en vue d’une greffe immédiate, en aménageant une exception à la règle selon laquelle l’autorisation de distribuer des cellules est délivrée aux établissements qui sont également autorisés à les préparer et à les conserver ;
– donner une base légale aux dispositions prévoyant l’obligation de désignation, dans chaque établissement concerné, d’une personne responsable de la qualité et de la sécurité des tissus et des préparations de thérapie cellulaire, selon des modalités déterminées par décret en Conseil d’État. Concernant l’Établissement français du sang, qui regroupe plusieurs établissements dont certains ont des activités de préparation et de conservation des tissus et des préparations de thérapie cellulaire, il est prévu la désignation en son sein d’une personne responsable unique, qui aura autorité sur les directeurs d'établissements de transfusion pour l'exercice de sa mission.
L’article L. 1223-1 du même code, qui définit les missions des établissements de transfusion sanguine, est également modifié afin de faire référence aux établissements qui sont autorisés par l’AFSSAPS à exercer des activités de préparation et de conservation des tissus et cellules (article 9). Sous l’autorité de la personne responsable précédemment mentionnée (cf. article L. 1243-2-1, tel qu’inséré par l’article 8 de l’ordonnance), les directeurs des établissements de transfusion sanguine sont par ailleurs chargés d’assurer la mise en oeuvre des dispositions législatives et réglementaires relatives à la qualité et à la sécurité des tissus et de leurs dérivés et des préparations de thérapie cellulaire (article 10).
En outre, conformément à la directive n° 2004/23 du 31 mars 2004 précitée, plusieurs modifications sont apportées aux conditions d’importation et d’exportation des tissus et cellules issus du corps humain (article 12).
Outre l’harmonisation des dispositions du code de la santé publique applicables en la matière avec la terminologie communautaire, concernant notamment la définition des dispositifs médicaux de diagnostic in vitro, il est notamment prévu que les activités d’importation et d’exportation à des fins thérapeutiques de tissus, cellules ou de préparations de thérapie cellulaire soient subordonnés à la délivrance d’une autorisation par l’AFSSAPS après avis de l’Agence de biomédecine. Par coordination, une modification est de ce fait apportée à l’article L. 1243-5 du même code (article 13).
À titre dérogatoire, l’article 12 de l’ordonnance prévoit également que des établissements et des organismes, par exemple des banques de tissus et de cellules, qui ne disposeraient pas de cette autorisation puissent cependant être habilités par l’AFSSAPS à importer ou à exporter des tissus, cellules et préparations de thérapie cellulaire dans des situations d’urgence et pour un patient donné.
L’article L. 1245-6 du même code est par ailleurs complété afin de confier à l’AFSSAPS l’élaboration de règles de bonnes pratiques pour la distribution des tissus, cellules, préparations de thérapie cellulaire et produits du corps humain utilisés à des fins thérapeutiques, après avis de l’Agence de biomédecine (article 14).
Enfin, à l’article 38 du code des douanes, la liste des produits dits sensibles, qui peuvent faire l'objet d'un contrôle douanier, est complétée afin d’y ajouter les dérivés de tissus. Les services douaniers pourront ainsi s’assurer que les établissements ou organismes qui importent et exportent ces produits sont bien autorisés à exercer cette activité (article 15). Comme le souligne le rapporteur au nom de la commission des affaires sociales du Sénat, M. Gilbert Barbier (11), il s’agit ainsi « de permettre aux agents des douanes de procéder à des contrôles inopinés de ces produits et de s'assurer (…) qu'ils sont présentés dans un bureau des douanes ouvert aux opérations commerciales. »
● Les produits sanguins
La section 2 de ce chapitre comporte six articles ayant pour objet de transposer la directive n° 2002/98/CE du 27 janvier 2003 relative au sang humain et à ses composants (12).
Elle permet en premier lieu d’introduire une distinction entre la « distribution » des produits sanguins labiles, c’est-à-dire leur fourniture en gros conformément aux dispositions prévues par la directive, et leur « délivrance », qui implique une attribution nominative de ces produits. Dans les deux cas, ces actes ne peuvent être réalisés sans qu’il n’ait été procédé à des d’analyses biologiques et à des tests de dépistage de maladies transmissibles (article 16).
Les compétences respectives des établissements de transfusion sanguine et des établissements de santé, concernant la délivrance et la distribution des produits sanguins labiles, sont également précisées. Ainsi, la délivrance de ces produits peut être effectuée par des établissements de transfusion sanguine ainsi que par des établissements de santé, mais ces derniers ne sont pas habilités à procéder à leur distribution (article 17).
Les articles 18 à 21 de l’ordonnance permettent par ailleurs de transposer les dispositions prévues par l’article 27 de la directive n° 2002/98/CE précitée concernant le régime des sanctions applicables en cas d’infraction aux dispositions nationales prises pour son application.
– Ainsi, pour harmoniser la procédure d'agrément des établissements de transfusion sanguine et celle de l’autorisation des dépôts de sang dans les établissements de santé, le nouvel article L. 1221-10-2 du code de la santé publique précise les conditions de suspension ou de retrait de cette autorisation, dans le cas notamment où les prescriptions relatives à la conservation des produits sanguins en vue de leur délivrance n’auraient pas été respectées (article 18).
– De nouvelles dispositions sont également introduites afin de sanctionner la poursuite d’activités ayant déjà fait l’objet d’une décision de retrait ou de suspension d’agrément ou d’autorisation en application des articles L. 1223-5 et L. 1221-10-2 du même code (article 19).
– Enfin, les articles L. 1271-4 et L. 1271-8 du même code, relatifs aux infractions à la réglementation applicable aux activités transfusionnelles, sont modifiés afin d’harmoniser leurs dispositions avec la terminologie communautaire mais également d’en améliorer la rédaction concernant l’énoncé des sanctions pénales encourues (articles 20 et 21).
De nombreuses dispositions de la directive n° 2003/15/CE du 27 février 2003 relative aux produits cosmétiques (13) ont déjà été transposées en droit interne par l’ordonnance n° 2004-1148 du 28 octobre 2004, le décret n° 2004-1219 et un arrêté en date du 17 novembre 2004 ainsi que par le décret n° 2006-62 du 18 janvier 2006. Ces mesures concernent notamment l’étiquetage des produits cosmétiques ainsi que la mise à disposition du public d’informations sur leur composition et leurs effets indésirables.
Toutefois, la France n’avait toujours pas transposé les dispositions prévues par le point 2 de l’article 1er de cette directive, qui prévoit l’interdiction de commercialiser des produits contenant des produits testés sur des animaux et de réaliser des expérimentations animales portant sur des produits cosmétiques finis ou des ingrédients entrant dans leur composition. Il convient à cet égard de rappeler que la France avait déposé, le 3 juin 2003, un recours en annulation de ces dispositions devant la Cour de justice des communautés européennes (CJCE) mais celui-ci a été rejeté par un arrêt en date du 24 mai 2005.
C’est pourquoi le chapitre III de la présente ordonnance comporte deux articles visant à achever la transposition de la directive du 27 février 2003 précitée. Il permet ainsi de poser le principe de l’interdiction de réaliser des expérimentations animales et de commercialiser des produits dont la formulation finale ou les ingrédients ont fait l’objet d’une telle expérimentation. Conformément aux prescriptions communautaires, il est par ailleurs prévu que cette interdiction entre en vigueur au fur et à mesure de la validation au niveau communautaire des « méthodes alternatives », constatée par arrêté du ministre chargé de la santé, pris sur proposition de l’AFSSAPS, et au plus tard le 11 mars 2009 (article 22).
Des sanctions pénales sont par ailleurs instituées en cas de non-respect de cette interdiction (article 23).
4. Les aliments diététiques destinés à des fins médicales spéciales
Le chapitre IV de l’ordonnance vise à aménager le régime juridique des aliments diététiques destinés à des fins médicales spéciales (ADDFMS), comme par exemple les nutriments particuliers destinés à des patients atteints de certaines maladies du métabolisme.
La définition de ces aliments diététiques, posée par l’article L. 5137-1 du code de la santé publique, est tout d’abord modifiée pour reprendre les dispositions prévues par la directive n° 1999/21/CE du 7 juin 1999 (14), en précisant notamment que ces aliments ne peuvent être utilisés que sous contrôle médical (article 24), c’est-à-dire que les personnes auxquelles ils sont destinés sont suivies et conseillées par un médecin.
Les autres dispositions prévues par l’article L. 5137-1, dans sa rédaction antérieure à l’entrée en vigueur de la présente ordonnance, ont également été modifiées et transférées au nouvel article L. 5137-2 du même code, afin de définir plus précisément la catégorie des ADDFMS soumis à une prescription médicale obligatoire et pour lesquels des modalités particulières de délivrance au détail sont prévues. En effet, comme le souligne le rapport au Président de la République précité sur la présente ordonnance, « les professionnels identifiaient avec difficulté (…) la catégorie des aliments diététiques qui, du fait de leur composition, sont susceptibles de présenter un risque pour les personnes auxquelles ils ne sont pas destinés ». C’est pourquoi il lui a été substituée la référence aux ADDFMS qui répondent aux besoins nutritionnels particuliers de personnes atteintes d’une des maladies nécessitant ce type d’apport et figurant sur une liste fixée par arrêté du ministre chargé de la santé, d’une part, et à des caractéristiques déterminées par arrêté, d’autre part (1° et 2° de l’article L. 5137-2 du même code).
De plus, il est désormais prévu que la fourniture et la délivrance de cette catégorie d’aliments doivent être effectuées conformément aux bonnes pratiques définies par arrêté du ministre chargé de la santé. Aussi les pharmaciens et les personnes morales habilitées à les délivrer au détail pourront-ils disposer d'éléments permettant de garantir que le produit est conforme à sa composition.
Enfin, les nouvelles dispositions introduites au même article L. 5137-2, concernant la définition par voie réglementaire des règles relatives à la composition et à la présentation des ADDFMS, permettent d’améliorer l’articulation entre les dispositions du code de la santé publique et celles du code de la consommation concernant ces produits (article 25).
5. Les insecticides, acaricides et antiparasitaires à usage humain
Les trois articles du chapitre V de l’ordonnance suppriment les dispositions du code de la santé publique spécifiques aux insecticides et acaricides, dans la mesure où ces produits sont considérés comme des médicaments à usage humain au niveau communautaire (15).
À cet égard, l’article 4 du présent projet de loi permet de lever une ambiguïté résultant de la modification ainsi apportée à l’article L. 5311-1 du même code par l’article 27 de l’ordonnance, concernant le régime juridique applicable aux insecticides et acaricides à usage humain.
6. Les médicaments vétérinaires
Le chapitre VI de l’ordonnance comporte vingt articles, regroupés dans sept sections, visant à transposer la directive n° 2004/28/CE du 31 mars 2004 relative aux médicaments vétérinaires (16). De nombreuses dispositions de ce texte étant similaires à celles de la directive du 31 mars 2004 relative aux médicaments à usage humain (17), leur transposition suit, logiquement, les mêmes principes que ceux de la loi n° 2007-248 du 26 février 2007 précitée, tout en prenant en compte certaines spécificités présentées par les médicaments vétérinaires.
● Définitions
La section 1 de ce chapitre complète l’article L. 5141-2 du code de la santé publique afin notamment d’introduire les définitions des médicaments génériques vétérinaires, biologiques vétérinaires et biologiques vétérinaires similaires, selon des termes proches de ceux prévus par la loi précitée du 26 février 2007 pour les médicaments à usage humain (article 29). Il est par ailleurs précisé que les additifs et les prémélanges d’additifs, qui sont autorisés conformément au règlement (CE) nº 1831/2003 du Parlement européen et du Conseil du 22 septembre 2003 relatif aux additifs destinés à l’alimentation des animaux, ne sont pas considérés comme des médicaments vétérinaires (article 30).
● Autorisations de mise sur le marché (AMM) et enregistrements
Les règles de délivrance de l’AMM des médicaments vétérinaires par l’Agence française de sécurité sanitaire des aliments (AFSSA) sont précisées et complétées par la section 2 de ce chapitre. Ainsi, comme pour les médicaments à usage humain, l’article L. 5141-5 du même code tel que modifié par l’ordonnance prévoit notamment que l’AMM des médicaments vétérinaires, délivrée pour cinq ans, peut être renouvelée, le cas échéant, pour une durée illimitée et que toute modification des éléments de l’AMM doit être préalablement autorisée par l’AFSSA. Des dispositions relatives à la transparence des travaux de l’agence sont par ailleurs introduites dans le même article (article 31).
Outre les précisions apportées aux conditions de commercialisation des médicaments génériques, cette section introduit également plusieurs mesures plus spécifiques aux médicaments vétérinaires, concernant notamment les conditions particulières de délivrance de l’AMM pour les médicaments destinés à certains animaux de compagnie ou à des animaux dont la chair ou les produits sont destinés à la consommation humaine (article 32).
À l’article L. 5141-6 du même code, les conditions de refus, de modification, de suspension ou de retrait de l'AMM sont précisées et complétées, à travers notamment l’introduction de la notion d’évaluation du rapport « bénéfice-risque » du médicament (article 33). La procédure d'enregistrement des médicaments homéopathiques vétérinaires est également modifiée afin notamment de l’étendre, sous certaines conditions, à des médicaments conçus pour être administrés à des animaux dont la chair et les produits sont destinés à la consommation humaine (article 34).
Un nouvel article L. 5141-10-1 est par ailleurs inséré dans le même code afin de prévoir que dans le cas où un animal doit faire l’objet d’une importation ou d’une exportation depuis ou vers un État non membre de la Communauté européenne et qu’il est soumis à des dispositions sanitaires spécifiques obligatoires, l’AFSSA peut, à titre dérogatoire, autoriser un vétérinaire à administrer à cet animal un médicament immunologique vétérinaire ne disposant pas d’une AMM en France mais autorisé dans le pays concerné (article 35).
Des entreprises ou organismes auront par ailleurs la possibilité de préparer des autovaccins à usage vétérinaire, à la condition d’employer une personne qualifiée ayant obtenu à cet effet une autorisation délivrée par l’AFSSA (article 36). Des dispositions analogues sont également prévues pour la préparation d’allergènes pour un seul animal (article 37). Sont enfin précisées les conditions dans lesquelles les vétérinaires qui sont établis dans un autre État membre peuvent utiliser en France des médicaments vétérinaires autres qu'immunologiques ayant obtenu une AMM dans cet État, lorsque ces médicaments ne sont pas autorisés sur le territoire national (article 38).
● Mesures d’application
L’unique article de la section 3 de ce chapitre prévoit la détermination par voie réglementaire des conditions d’application des nouvelles dispositions relatives aux médicaments vétérinaires, concernant notamment le contenu du dossier de demande d’AMM ou d’enregistrement ainsi que les règles applicables en matière de pharmacovigilance (article 39).
● Préparation industrielle et vente en gros
La section 4 institue tout d’abord un régime de déclaration pour les modifications non substantielles des éléments de l’AMM, les autres modifications, dont la nature sera précisée par décret en Conseil d’État, devant faire l’objet d’une autorisation préalable (article 40). L’AFSSA est par ailleurs chargée de définir les principes de bonnes pratiques, qui devront être respectés en matière notamment de fabrication et de distribution des médicaments vétérinaires et des aliments médicamenteux ainsi que dans le domaine de la pharmacovigilance (article 41).
De nouvelles dispositions, similaires à celles prévues pour les médicaments à usage humain, sont introduites à l’article L. 5142-3 du même code. Elles prévoient de soumettre les entreprises exploitant un médicament vétérinaire à l’obligation d’informer l’AFSSA des dates de commercialisation de chaque présentation de ce médicament et, le cas échéant, de l’arrêt envisagé de sa commercialisation, de son retrait du marché ou encore d’un risque de rupture de stock (article 42). Également inséré par cette section de l’ordonnance, l’article L. 5142-5-1 du même code vise à permettre à l'AFSSA d'interdire, dans certains cas, la fabrication, l’importation, la détention ou la vente de médicaments immunologiques vétérinaires (article 43).
● Préparation extemporanée et distribution au détail
Les dispositions relatives à la préparation extemporanée (c’est-à-dire non réalisée à l’avance) des médicaments vétérinaires sont tout d’abord modifiées par la section 5 de ce chapitre, en posant notamment le principe du respect des règles de bonnes pratiques définies par l’AFSSA (article 42).
La nouvelle rédaction de l’article L. 5143-4 du même code permet, d’autre part, d’élargir les conditions de prescription d’un médicament par le vétérinaire lorsqu’il n’existe pas de médicaments appropriés pour l’espèce ou la maladie concernée, selon la procédure dite « de la cascade », des dispositions particulières étant prévues pour les équidés (article 45). Des aménagements sont également apportés à l’article L. 5143-5 du même code, qui fixe la liste des médicaments vétérinaires dont la délivrance est subordonnée à la rédaction d'une ordonnance par le vétérinaire (article 46).
● Substances pouvant entrer dans la fabrication des médicaments vétérinaires
La rédaction de l’article L. 5144-1 est modifiée afin d’actualiser et de mettre en cohérence avec la réglementation communautaire les références à certains produits qui peuvent entrer dans la fabrication des médicaments vétérinaires, s’agissant notamment de ceux susceptibles de demeurer à l’état de résidus toxiques ou dangereux dans les denrées alimentaires d’origine animale (article 47).
● Dispositions transitoires
La section 7 du chapitre VI de l’ordonnance précise enfin les modalités d’application dans le temps des nouvelles dispositions relatives à la délivrance de l’AMM des médicaments vétérinaires (article 48).
7. La classification des substances et préparations chimiques dangereuses
Le chapitre VII de l’ordonnance a pour principal objet de mettre en cohérence le dispositif de classification des substances et préparations chimiques dangereuses avec les prescriptions communautaires, en particulier la directive n° 1999/45/CE du Parlement européen et du Conseil du 31 mai 1999 (18).
L’article L. 5132-2 du code de la santé publique prévoit en effet que les substances et préparations dangereuses sont classées en neuf catégories selon qu’elles sont : très toxiques, toxiques, nocives, corrosives, irritantes, sensibilisantes, cancérogènes, mutagènes ou toxiques pour la reproduction. Il est en effet apparu nécessaire de prendre en compte l’ensemble des risques identifiés au niveau communautaire, en y intégrant notamment ceux liés au caractère sensibilisant ou allergisant de certains produits ainsi que la classification des produits cancérogènes, mutagènes ou reprotoxiques selon que :
– le risque est avéré chez l’homme et scientifiquement démontré (catégorie 1) ;
– il existe une forte présomption que l'exposition de l'homme à de telles substances peut provoquer ou augmenter la fréquence probable d’un cancer, de défauts génétiques héréditaires, d’effets nocifs dans la progéniture ou d’une atteinte aux fonctions reproductives (catégorie 2) ;
– il y a présomption de risque, sans que les informations disponibles soient suffisantes pour classer ces substances et préparations dans la catégorie 2 (catégorie 3).
Ce faisant, les dispositions du code de la santé publique sont également harmonisées avec celles prévues par le code du travail, dans lequel les prescriptions communautaires en matière de classification et d’étiquetage des substances et préparations dangereuses avaient déjà été transposées par le décret n° 2004-725 du 22 juillet 2004. Cette nouvelle rédaction de l’article L. 5132-2 permet par ailleurs de renforcer le fondement légal des mesures de gestion des risques liés à ces substances et préparations (article 49).
Le chapitre VIII de l’ordonnance comporte quatre articles concernant principalement les médicaments à usage humain.
Il est tout d’abord précisé que l’enregistrement d’un médicament traditionnel à base de plantes ainsi que l'autorisation temporaire d’utilisation (ATU) prévue à l’article L. 5121-12 du même code valent autorisation d’importation sur le territoire douanier des médicaments concernés. Dans le même sens, l’autorisation accordée à un promoteur pour réaliser une recherche biomédicale vaut autorisation d'importation pour tout médicament nécessaire à cette recherche (article 50).
Ce chapitre permet, d’autre part, aux agents des directions générales des douanes, des impôts et de la concurrence, de la consommation et de la répression des fraudes (DGCCRF), qui sont habilités à rechercher et à constater les infractions à la réglementation applicable aux activités et à certains produits relevant de la compétence de l’AFSSAPS, de communiquer directement à celle-ci les documents et informations recueillis au cours de leurs investigations. Alors que la réglementation antérieurement applicable subordonnait la communication de ces éléments à la saisine préalable du procureur de la République, ces nouvelles dispositions ne peuvent que contribuer à accroître l’efficacité des contrôles réalisés par la DGCCRF (article 51).
En outre, les dispositions de l’article L. 5122-3 du même code, qui précise les médicaments pouvant faire l’objet d’une publicité, sont rendues applicables aux médicaments traditionnels à base de plantes, aux médicaments qui sont importés selon la procédure prévue par l’article L. 5124-17-1 du même code ainsi qu’aux médicaments autorisés dans un autre État membre mais non en France et dont l’AFSSAPS permet, à titre dérogatoire, la mise sur le marché pour des raisons de santé publique justifiées (article 52).
Enfin, le dernier article prévoit que le Premier ministre, le ministre de l’économie, des finances et de l’industrie, le garde des sceaux, ministre de la justice, le ministre de l’agriculture et de la pêche et le ministre de la santé et des solidarités sont responsables, chacun en ce qui le concerne, de l’application de la présente ordonnance (article 53).
*
La commission a adopté l’article 1er sans modification.
Article 1er bis
Habilitation à prendre par ordonnances les dispositions pour transposer la directive du 31 mars 2004 relative aux tissus et cellules humains et aménager les sanctions applicables dans le domaine des produits de santé
Issu d’un amendement du gouvernement adopté par le Sénat, suivant l’avis favorable du rapporteur de la commission des affaires sociales, cet article vise à autoriser le gouvernement à prendre par ordonnances deux catégories de mesures relevant du domaine de la loi, pendant une durée limitée à quatre mois à compter de la publication du présent texte.
1. Le champ de l’habilitation demandée par le gouvernement
L’article 39 de la loi n° 2007-248 du 26 février 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament, dont le texte est reproduit dans l’annexe n° 3 du présent rapport, a autorisé le gouvernement à prendre par ordonnances plusieurs dispositions relevant du domaine législatif, et notamment celles nécessaires :
– pour transposer la directive n° 2004/23/CE du Parlement européen et du Conseil du 31 mars 2004 relative à l’établissement de normes de qualité et de sécurité pour le don, l’obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissus et cellules humains (c du I de l’article 39) ;
– pour harmoniser et compléter les dispositions pénales relatives aux produits mentionnés aux articles L. 5141-1 (médicaments vétérinaires) et L. 5311-1 du code de la santé publique (principalement les médicaments et dispositifs médicaux mais aussi d’autres produits à finalités sanitaire, cosmétique et d’hygiène corporelle, qui sont énumérés dans l’encadré présenté sous l’article 5 du présent rapport) ainsi que « pour instaurer, en tant que de besoin, des sanctions administratives dans les domaines qui n'en disposent pas et pour harmoniser leur mise en œuvre avec les sanctions pénales » (2° du II de l’article 39).
Il était par ailleurs précisé que les ordonnances prévues par le I et par le 2° du II du même article 39 ne pourraient être prises que pendant un délai de huit mois suivant la publication de la loi du 26 février 2007, conformément aux dispositions prévues par son III.
Toutefois, si l’ordonnance n° 2007-613 du 26 avril 2007, dont l’article 1er du présent projet de loi prévoit la ratification, a permis de prendre la majeure partie des dispositions prévues par l’article 39 de la loi du 26 février 2007, tel n’a cependant pas été le cas pour les deux catégories de mesures susmentionnées concernant, d’une part, les sanctions pénales et administratives dans le domaine des produits de santé et, d’autre part, la transposition de la directive relative aux tissus et cellules humains, s’agissant notamment de ses dispositions relatives aux gamètes utilisés à des fins de reproduction.
Selon l’exposé des motifs de l’amendement dont résulte le présent article, ce retard s’expliquerait notamment par le caractère « particulièrement vaste » du champ de l’habilitation donnée par le Parlement ainsi que par « la priorité donnée à la transposition des directives sur le médicament [qui] a eu pour conséquence de retarder les deux derniers chantiers qui s'avèrent particulièrement lourds et délicats ».
En effet, le projet d’ordonnance relatif aux sanctions pénales et administratives a tout d’abord requis une collaboration étroite entre les ministères chargés de la santé, de l’agriculture et de la justice ainsi que la participation des services de l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) et de l’Agence française de sécurité sanitaire des aliments (AFSSA), en raison notamment de sa dimension technique.
S’agissant de la directive n° 2004/23/CE relative aux tissus et cellules humains, dont l’ordonnance n° 2007-613 du 26 avril 2007 a déjà permis de transposer plusieurs mesures, le retard pris dans l'élaboration du projet d'ordonnance concernant le don de gamètes et l'assistance médicale à la procréation s’expliquerait également par son caractère très technique et le nombre important des consultations requises. Selon le ministère de la santé, de la jeunesse et des sports, ce texte, préparé en coordination avec l’Agence de la biomédecine, a été adressé à l’ensemble des professionnels de l’assistance médicale à la procréation et transmis aux ministères chargés de la justice et du budget, des comptes publics et de la fonction publique.
Afin de « conduire à leur terme ces deux projets d’ordonnance qui sont déjà très avancés », comme l’a précisé la secrétaire d’État chargée de la solidarité, Mme Valérie Létard, lors de l’examen du présent projet de loi par le Sénat, le 17 octobre 2007, le premier alinéa du présent article vise à accorder au gouvernement une nouvelle habilitation à légiférer par ordonnances, dont le champ est défini selon les mêmes termes que ceux du c du I et du 2° du II précités de l’article 39 de la loi du 26 février 2007.
Le gouvernement aurait ainsi la possibilité de prendre les mesures nécessaires pour transposer la directive du 31 mars 2004 relative aux tissus et cellules ainsi que pour harmoniser et compléter les dispositions relatives aux sanctions pénales dans le domaine des produits de santé et, enfin, pour instaurer, en tant que de besoin, des sanctions administratives, dont la mise en œuvre serait harmonisée avec les sanctions pénales.
2. Les délais de publication des ordonnances et de dépôt du projet de loi de ratification devant le Parlement
Conformément aux exigences posées par l’article 38 de la Constitution (cf. annexe n° 2), le présent article prévoit d’assortir l’habilitation donnée au gouvernement de deux conditions visant à encadrer dans le temps la possibilité qui lui est offerte de légiférer par ordonnances.
Dans son premier alinéa, il est tout d’abord précisé que les ordonnances ne pourront être prises que pendant une durée limitée à quatre mois à compter de la date de publication du présent texte. Le second alinéa prévoit par ailleurs que le projet de loi portant ratification de ces ordonnances devra être déposé devant le Parlement au plus tard le dernier jour du deuxième mois suivant leur publication.
*
La commission a adopté l’article 1er bis sans modification.
Article 2
Contrôle douanier des importations et exportations d’échantillons biologiques, de sang et de ses composants et produits dérivés
Si l’article 2 bis du code des douanes prévoit que ses dispositions ne s’appliquent pas aux échanges intracommunautaires, les agents des douanes disposent, à titre dérogatoire, de prérogatives de contrôle sur quelques produits dits sensibles, qui sont limitativement énumérés par le 4 de l’article 38 du même code et soumis à certaines restrictions de circulation, à des règles de qualité, de conditionnement ou à des formalités particulières.
Parmi ces produits figurent notamment les produits sanguins labiles (comprenant le plasma, le sang total et les cellules sanguines), les pâtes plasmatiques (19), les organes et tissus, les préparations de thérapie cellulaire ainsi que les médicaments à usage humain mentionnés à l’article L. 5124-13 du code de la santé publique.
Les services douaniers peuvent ainsi procéder à des contrôles inopinés et vérifier que les organismes qui importent ou exportent ces produits disposent de l’autorisation requise pour effectuer cette activité.
L’article 15 de l’ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament, dont l’article 1er du présent projet de loi porte ratification (cf. supra), complète cette liste de produits sensibles pour y inclure les « dérivés » de tissus.
Le présent article propose d’étendre de nouveau le champ des dérogations prévues par l’article 38 du code des douanes, afin de soumettre au contrôle douanier les importations et exportations :
– d’échantillons biologiques, d’une part (soit les échantillons issus de tout prélèvement biologique à des fins diagnostique, thérapeutique et scientifique) ;
– de sang et de ses composants et produits dérivés, lorsqu'ils sont importés ou exportés à des fins scientifiques, d’autre part. En effet, selon les précisions apportées par le ministère de la santé, de la jeunesse et des sports, les produits sanguins labiles et les pâtes plasmatiques, qui figurent déjà dans la liste des produits susceptibles de faire l’objet de restrictions de circulation, sont des produits à visée thérapeutique, alors que la référence à la notion plus large de sang et de « ses composants et produits dérivés » permet d’étendre l’application de ces dispositions aux produits sanguins destinés à la recherche.
Il s’agit ainsi d’assurer le respect de la réglementation applicable à ces produits en matière d’importation ou d’exportation. Il convient en effet de rappeler qu’en vertu de l’article L. 1221-12 du code de la santé publique, « l’importation ou l’exportation de sang, de ses composants ou de ses produits dérivés à des fins scientifiques est soumise à l'autorisation du ministre chargé de la recherche prévue à l’article L. 1245-5 ». Ce dernier prévoit un régime d’autorisation des activités d’importation et d’exportation à des fins thérapeutiques des tissus et cellules issus du corps humain et précise, s’agissant des échantillons biologiques, que ces activités ne peuvent être effectuées que par des personnes dont l’activité comporte des analyses de biologie médicale, des examens d’anatomo-cytopathologie, des expertises judiciaires ou des contrôles de qualité ou d’évaluation, notamment de dispositifs médicaux de diagnostic in vitro.
*
La commission a adopté l’article 2 sans modification.
Article 3
Correction d’une erreur matérielle
L’article L. 5141-5-2 du code de la santé publique tel qu’inséré par l’article 32 de l’ordonnance n° 2007-613 du 26 avril 2007, dont l’article 1er du présent projet de loi porte ratification, vise à transposer en droit interne certaines dispositions de la directive n° 2004/28/CE du 31 mars 2004 relative aux médicaments vétérinaires (20).
En effet, ce nouvel article prévoit que les médicaments conçus pour être administrés à des animaux destinés à la consommation humaine, pour obtenir une autorisation de mise sur le marché (AMM), ne doivent contenir qu’une ou des substances pharmacologiquement actives figurant dans l’une des annexes I, II ou III du règlement du Conseil (CEE) nº 2377/90 du 26 juin 1990 modifié établissant une procédure communautaire pour la fixation des limites maximales de résidus de médicaments vétérinaires dans les aliments d’origine animale.
À titre dérogatoire, le dernier alinéa du même article L. 5141-5-2 prévoit un régime spécifique pour les médicaments réservés aux équidés qui ne sont pas destinés à l'abattage pour la consommation humaine. La seconde phrase de cet alinéa précise que, dans ce cas, les médicaments vétérinaires ne doivent pas comporter de substances pharmacologiquement actives figurant à l’annexe IV du règlement, ni être destinés à être utilisés pour le traitement des affections spécifiées dans le résumé « autorisé » des caractéristiques du produit, pour lesquelles un médicament vétérinaire est autorisé pour soigner les animaux de la famille des équidés.
Le résumé des caractéristiques du produit (RCP), dont le contenu est défini par l’article R. 5121-23 du même code, reproduit dans l’encadré présenté ci-après, constitue en effet un élément important dans la procédure de mise sur le marché des médicaments. Ainsi, le résumé doit être joint à la demande d’AMM adressée au directeur général de l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) et soumis à son approbation. Toutefois, tant la réglementation communautaire que le code de la santé publique font aujourd’hui référence à la notion de « résumé des caractéristiques du produit ».
Le contenu du résumé des caractéristiques du produit (RCP)
« Le résumé des caractéristiques du produit doit comporter les renseignements suivants :
1º Dénomination de la spécialité ;
2º Forme pharmaceutique ;
3º Composition qualitative et quantitative en principes actifs et en constituants de l’excipient dont la connaissance est nécessaire à une bonne administration du médicament, en utilisant les dénominations communes internationales lorsqu'elles existent ou, à défaut, les dénominations de la pharmacopée européenne ou française ;
4º Nom ou raison sociale et domicile ou siège social du demandeur de l'autorisation de mise sur le marché ;
5º Nature du récipient ;
6º Conditions de délivrance au public ;
7º Durée de stabilité, si nécessaire après reconstitution du produit ou lorsque le récipient est ouvert pour la première fois ;
8º Précautions particulières de conservation ;
9º Incompatibilités majeures chimiques ou physiques ;
10º Propriétés pharmacologiques et, dans la mesure où ces renseignements sont utiles pour l'utilisation thérapeutique, éléments de pharmacocinétique ;
11º Indications thérapeutiques ;
12º Effets indésirables (fréquence et gravité) ;
13º Mises en garde spéciales ;
14º Contre-indications ;
15º Précautions particulières d’emploi, notamment en cas de grossesse et d’allaitement, d’utilisation par des enfants ou des personnes âgées et dans des circonstances pathologiques particulières. S’il y a lieu, les précautions particulières qui doivent être prises par les personnes qui manipulent le médicament et qui l’administrent aux patients ainsi que les précautions qui doivent éventuellement être prises par le patient ;
16º Effets sur la capacité de conduire des véhicules ou d’utiliser des machines ;
17º Interactions médicamenteuses et autres ;
18º Posologie et mode d'administration ;
19º Surdosage : symptômes, conduite d'urgence, antidotes ;
20º Précautions particulières d'élimination des produits non utilisés ou des déchets dérivés de ces produits, s’il y a lieu ;
21º Date d’établissement du résumé des caractéristiques du produit. »
Source : article R. 5121-23 du code de la santé publique
Le présent article procède en conséquence à la rectification de cette erreur matérielle en supprimant, dans la seconde phrase du dernier alinéa de l’article L. 5141-5-2 du code de la santé publique, le terme inapproprié de résumé « autorisé » des caractéristiques du produit.
La commission a adopté l’article 3 sans modification.
Article 4
Clarification rédactionnelle concernant le régime juridique des insecticides et acaricides à usage humain
Résultant de l’adoption par le Sénat d’un amendement du gouvernement ayant recueilli l’avis favorable du rapporteur de sa commission des affaires sociales, M. Gilbert Barbier, cet article a pour objet de corriger une ambiguïté rédactionnelle issue de l’article 27 du chapitre V de l'ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament.
En effet, ce chapitre V a pour objet de supprimer les dispositions du code de la santé publique spécifiques aux insecticides et acaricides appliqués sur l'homme dans la mesure où que ces produits sont considérés comme des médicaments à usage humain au niveau communautaire et doivent donc être soumis aux mêmes prescriptions.
De ce fait, dans l’objectif d’éviter toute confusion sur le régime juridique applicable à ces produits, le a de l’article 27 de l’ordonnance précitée a introduit la mention des insecticides et acaricides parmi les exemples de médicaments mentionnés au troisième alinéa (1°) de l’article L. 5311-1 du code de la santé publique, qui énumère les différentes catégories de produits relevant du contrôle de l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS).
Ainsi, aux termes du 1° de l’article L. 5311-1, l’AFSSAPS est notamment compétente pour les médicaments, y compris les préparations magistrales, hospitalières et officinales, les substances stupéfiantes, psychotropes ou autres substances vénéneuses utilisées en médecine, « les insecticides, acaricides et antiparasitaires à usage humain », les huiles essentielles et plantes médicinales ainsi que les matières premières à usage pharmaceutique.
Toutefois, en insérant la référence aux insecticides entre celles des psychotropes et des huiles essentielles, l’article 27 de l’ordonnance introduit une ambiguïté dès lors que l’on pourrait en conclure que ces produits sont soumis à des dispositions particulières au même titre, par exemple, que les psychotropes, comme le souligne l’exposé des motifs de l’amendement dont résulte le présent article.
Or cette nouvelle rédaction pose problème compte tenu des dispositions par ailleurs prévues par l’article L. 5414-1 du même code, qui donne compétence aux agents de la direction générale de la concurrence, de la consommation et de la répression des fraudes (DGCCRF) pour rechercher et constater les infractions aux lois et règlements relatifs aux activités et aux produits mentionnés à l’article L. 5311-1 « à l’exception des médicaments et des substances stupéfiantes et psychotropes ou vénéneuses mentionnées à son 1° ».
Compte tenu de la confusion entretenue par la rédaction de l’article L. 5311-1 précité quant au régime juridique applicable aux insecticides et acaricides, il en résulte que les agents de la DGCCRF pourraient rester compétents pour rechercher et constater les infractions aux lois et règlements relatifs à ces produits, contrairement au dispositif prévu pour les autres médicaments.
Afin de lever cette ambiguïté et rappeler que ces produits relèvent bien du régime juridique des médicaments à usage humain, le présent article propose de modifier la rédaction du 1° de l’article L. 5311-1, en déplaçant l’insertion de la référence aux insecticides et aux acaricides au début de cet alinéa. Ainsi, il serait désormais clairement précisé à cet article que l’AFSSAPS est notamment compétente pour « les médicaments, y compris les insecticides, acaricides et antiparasitaires à usage humain ».
*
La commission a adopté l’article 4 sans modification.
Article 5
Compétences de l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) en matière de recherches biomédicales
Issu d’un amendement du gouvernement adopté par le Sénat, lors de sa séance du 17 octobre 2007, suivant l’avis favorable de la commission des affaires sociales, cet article prévoit de transférer à l’AFSSAPS les missions incombant actuellement au ministère de la santé au titre d’autorité compétente pour certaines recherches biomédicales.
1. La répartition actuelle des compétences entre l’AFSSAPS et le ministère de la santé en matière de recherches biomédicales
Aux termes de l’article L. 1121-1 du code de la santé publique, les recherches biomédicales recouvrent celles qui sont organisées et pratiquées sur l’être humain en vue du développement des connaissances biologiques ou médicales.
Le régime de ces recherches a été profondément réformé par la loi n° 2004-806 du 9 août 2004 relative à la politique de santé publique, qui a permis de transposer en droit interne les dispositions de la directive n° 2001/20/CE du Parlement européen et du Conseil du 4 avril 2001 relative aux essais cliniques de médicaments à usage humain. En application de l’article L. 1123-8 du même code, la réalisation des recherches biomédicales est subordonnée à la délivrance d’une autorisation par l’ « autorité compétente », selon la terminologie communautaire. Les principales missions qui lui incombent à ce titre, en matière notamment de suivi et, le cas échéant, de suspension ou d’interdiction de la recherche, sont rappelées dans l’encadré ci-dessous.
Les principales dispositions législatives relatives aux responsabilités
de l’autorité compétente en matière de recherches biomédicales
– Nul ne peut mettre en œuvre une recherche biomédicale sans autorisation de l’autorité compétente délivrée dans un délai fixé par voie réglementaire.
– Si, dans les délais prévus par voie réglementaire, l’autorité compétente informe le promoteur par lettre motivée qu’elle a des objections à la mise en œuvre de la recherche, le promoteur peut modifier le contenu de son projet de recherche et adresser cette nouvelle demande à l’autorité compétente. Cette procédure ne peut être appliquée qu’une seule fois à chaque projet de recherche. Si le promoteur ne modifie pas le contenu de sa demande, cette dernière est considérée comme rejetée.
– Après le commencement de la recherche, toute modification substantielle de celle-ci à l’initiative du promoteur doit obtenir, préalablement à sa mise en œuvre, un avis favorable du comité de protection des personnes (CPP) et une autorisation de l’autorité compétente. Dans ce cas, le comité s’assure qu’un nouveau consentement des personnes participant à la recherche est bien recueilli si cela est nécessaire.
– Les événements et les effets indésirables définis pour chaque type de recherche sont notifiés respectivement par l’investigateur au promoteur et par le promoteur à l’autorité compétente ainsi qu’au CPP compétent. Dans ce cas, le comité s’assure, si nécessaire, que les personnes participant à la recherche ont été informées des effets indésirables et qu’elles confirment leur consentement.
– Lorsqu’un fait nouveau intéressant la recherche ou le produit faisant l’objet de la recherche est susceptible de porter atteinte à la sécurité des personnes qui s’y prêtent, le promoteur et l’investigateur prennent les mesures de sécurité urgentes appropriées. Le promoteur informe sans délai l’autorité compétente et le CPP de ces faits nouveaux et, le cas échéant, des mesures prises.
– L’autorité compétente peut, à tout moment, demander au promoteur des informations complémentaires sur la recherche. En cas de risque pour la santé publique ou en cas d’absence de réponse du promoteur ou si l’autorité administrative compétente estime que les conditions dans lesquelles la recherche est mise en œuvre ne correspondent plus aux conditions indiquées dans la demande d’autorisation de la recherche ou ne respectent pas les dispositions du titre II, relatif aux recherches biomédicales, du livre premier du code de la santé publique, elle peut à tout moment demander que des modifications soient apportées aux modalités de réalisation de la recherche, à tout document relatif à la recherche, ainsi que suspendre ou interdire cette recherche. Sauf en cas de risque imminent, une modification du protocole à la demande de l’autorité compétente ou une décision de suspendre ou d’interdiction ne peut intervenir qu’après que le promoteur a été mis à même de présenter ses observations.
– Le promoteur avise l’autorité compétente et le CPP compétent que la recherche biomédicale est terminée et indique les raisons qui motivent l’arrêt de cette recherche quand celui-ci est anticipé.
– L’autorité compétente établit et gère une base de données nationales sur les recherches biomédicales. Elle est également chargée de mettre en place et de diffuser des répertoires de recherches biomédicales, sauf si le promoteur s’y oppose pour des motifs légitimes.
Source : articles L. 1123-8 et suivants du code de la santé publique
L’article L. 1123-12 du même code précise que, selon la nature des recherches, le terme d’autorité compétente désigne :
– l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) pour les recherches portant sur les produits mentionnés à L. 5311-1, principalement les médicaments et les dispositifs médicaux mais aussi d’autres produits de santé, cosmétiques et d’hygiène corporelle, dont la liste est présentée dans l’encadré ci-après ;
– le ministre chargé de la santé dans les autres cas (par exemple pour des recherches engagées dans les domaines de la physiologie, de la physiopathologie, de la génétique, de l’évaluation des stratégies thérapeutiques ou de l’épidémiologie), qui dispose donc d’une compétence par défaut dans ce domaine, le champ de compétence de l’AFSSAPS étant au contraire défini positivement par le code de la santé publique.
Il en résulte ainsi une dualité d’autorité en matière de recherches biomédicales. Les autorisations délivrées par l’AFSSAPS représentent ainsi environ 70 % des nouvelles recherches, contre 30 % pour celles accordées par le ministère de la santé, de la jeunesse et des sports.
Les produits énumérés par l’article L. 5311-1 du code de la santé publique pour lesquels l’AFSSAPS est compétente en matière de recherches biomédicales
« 1º Les médicaments, y compris les préparations magistrales, hospitalières et officinales, les substances stupéfiantes, psychotropes ou autres substances vénéneuses utilisées en médecine, les insecticides, acaricides et antiparasitaires à usage humain, les huiles essentielles et plantes médicinales, les matières premières à usage pharmaceutique ;
2º Les produits contraceptifs et contragestifs ;
3º Les biomatériaux et les dispositifs médicaux ;
4º Les dispositifs médicaux de diagnostic in vitro ;
5º Les produits sanguins labiles ;
6º Les organes, tissus, cellules et produits d'origine humaine ou animale, y compris lorsqu'ils sont prélevés à l'occasion d'une intervention chirurgicale ;
7º Les produits cellulaires à finalité thérapeutique ;
8º Le lait maternel collecté, qualifié, préparé et conservé par les lactariums ;
9º Les produits destinés à l'entretien ou à l'application des lentilles de contact ;
10º (Abrogé)
11º Les procédés et appareils destinés à la désinfection des locaux et des véhicules dans les cas prévus à l'article L. 3114-1 ;
12º Les produits thérapeutiques annexes ;
13º (Abrogé)
14º Les lentilles oculaires non correctrices ;
15º Les produits cosmétiques ;
16º Les micro-organismes et toxines mentionnés à l'article L. 5139-1 ;
17º Les produits de tatouage. »
Source : article L. 5311-1 du code de la santé publique
2. Le dispositif proposé
Comportant deux paragraphes, le présent article vise à créer un guichet unique pour les promoteurs de recherches biomédicales, en transférant à l’AFSSAPS les compétences actuellement exercées par le ministre de la santé au titre d’autorité compétente pour certaines recherches médicales. Plusieurs raisons sont avancées pour justifier ce transfert.
– En premier lieu, selon le ministère chargé de la santé, la délivrance d’autorisations sur des dossiers individuels n’apparaît pas relever du champ des missions d’une direction d'administration centrale, en l’occurrence la direction générale de la santé (DGS), et ce, d’autant moins que, dans la pratique, « ces autorisations, qui concernent à peu près six cents demandes par an, sont traitées par deux ou trois personnes », comme l’a souligné le rapporteur de la commission des affaires sociales du Sénat.
– Ces dispositions doivent par ailleurs permettre d’optimiser la gestion des moyens consacrés à l’encadrement des recherches biomédicales, en utilisant au mieux l’expertise de haut niveau développée au sein de l’AFSSAPS sur ces questions.
– Les deux autorités compétentes disposent aujourd’hui des mêmes prérogatives pour garantir la sécurité et de protection des personnes, s’agissant en particulier de la possibilité de suspendre ou d'interdire la recherche, conformément à l’article L. 1123-11 du code de la santé publique.
– Il s’agirait également d’un élément de simplification pour les promoteurs des recherches biomédicales, en particulier pour les recherches « mixtes », qui peuvent à la fois relever de la compétence de l’AFSSAPS et de celle du ministère de la santé.
– Il convient enfin de rappeler que l’AFSSAPS perçoit déjà la taxe prévue par l’article L. 1123-8 du même code, lorsqu’une demande d'autorisation d’une recherche est présentée à l'autorité compétente, quelle qu’elle soit.
Le 1° du I de cet article propose en conséquence d’élargir les compétences de l’AFSSAPS à l'ensemble des recherches biomédicales.
En effet, en vertu de l’article L. 5313-1 du même code, l’AFSSAPS est aujourd’hui chargée de contrôler l'application des lois et règlements relatifs aux seuls activités et produits mentionnés à l'article L. 5311-1 précité. Il est donc proposé d’introduire dans ce dernier un nouvel alinéa disposant que « l’agence participe à l’application des lois et règlements relatifs aux recherches biomédicales et prend, dans les cas prévus par des dispositions particulières, des décisions relatives aux recherches biomédicales ».
Il s’agit ainsi de permettre à l’AFSSAPS de contrôler l'ensemble des recherches biomédicales, cette mesure étant de nature à « renforcer la surveillance sanitaire réalisée sur ces activités », selon l’exposé des motifs de l’amendement dont résulte le présent article.
Le 2° du I de cet article modifie, pour les mêmes motifs, la rédaction de l’article L. 1123-12 précité, qui définit la notion d’autorité compétente.
Le a du 2° propose tout d’abord de supprimer les dispositions limitant la compétence de l’AFSSAPS aux seules « recherches portant sur les produits mentionnés à l’article L. 5311-1 » ainsi que la référence à la compétence du « ministre chargé de la santé dans les autres cas ». Tel que réécrit par le présent article, le premier alinéa de l’article L. 1123-12 disposerait donc que l’autorité compétente est l’AFSSAPS.
Le b du 2° modifie par coordination la rédaction du second alinéa du même article L. 1123-12, qui prévoit que lorsqu'une collection d'échantillons biologiques humains est constituée pour les seuls besoins d'une recherche biomédicale, elle est déclarée à l’autorité compétente « pour cette recherche ». Il apparaît en effet nécessaire de supprimer ces dispositions, dès lors qu’il est proposé de poser le principe de la compétence unique de l’AFSSAPS en matière de recherches biomédicales.
Le 3° du I tend également à modifier par cohérence la seconde phrase du septième alinéa de l’article L. 1121-3 du même code, qui prévoit actuellement, pour les recherches biomédicales autres que celles portant sur des médicaments, que des recommandations de bonnes pratiques sont fixées « par arrêté du ministre chargé de la santé » et par décision de l’AFSSAPS « pour les produits mentionnés à l’article L. 5311-1 ».
Il s’agit ainsi de transférer au directeur général de l’AFSSAPS les compétences actuelles du ministère chargé de la santé en matière d'élaboration des recommandations de bonnes pratiques pour les recherches ne portant pas sur les produits mentionnés à l’article L. 5311-1 du même code.
Enfin, le II du présent article prévoit que les dispositions du I entrent en vigueur au 1er avril 2008 afin « de permettre à l’AFSSAPS de s’organiser pour assumer cette nouvelle mission », selon l’exposé des motifs de l’amendement du gouvernement évoqué précédemment.
Pour les recherches autres que celles portant sur des produits mentionnés à l’article L. 5311-1 du code de la santé publique, la deuxième phrase de ce paragraphe prévoit par ailleurs que le ministre chargé de la santé se prononcera sur les demandes d’autorisation déposées jusqu’au 31 mars 2008 inclus, au titre de :
– l’article L. 1123-8 du même code, c’est-à-dire pour la délivrance de l’autorisation nécessaire à la mise en œuvre de la recherche biomédicale ;
– l’article L. 1123-9 du même code, soit pour l’autorisation requise après le commencement de la recherche et préalablement à ce qu’une modification substantielle de celle-ci soit apportée à l’initiative du promoteur.
*
La commission a adopté l’article 5 sans modification.
Article 6
Conditions de collecte, de destruction et de redistribution à des fins humanitaire des médicaments non utilisés
Introduit par le Sénat à l’initiative de M. Jean-Pierre Michel et des membres du groupe socialiste, avec l’avis favorable du gouvernement et du rapporteur de la commission des affaires sociales, cet article vise à modifier l’article L. 4211-2 du code de la santé publique relatif aux conditions de collecte, de destruction et de redistribution à des fins humanitaires des médicaments à usage humain non utilisés.
1. Le dispositif prévu par l’article 32 de la loi n° 2007-248 du 26 février 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament
La création par l’industrie pharmaceutique en 1993 d’un dispositif de collecte et de recyclage des médicaments non utilisés (MNU), coordonné par l’association « Cyclamed », répondait à des objectifs à la fois :
– environnementaux, en réduisant les pollutions ;
– sanitaires, en luttant contre les accidents domestiques ;
– humanitaires, en permettant l’envoi dans les pays en voie de développement de certains des médicaments non utilisés (MNU) rapportés par les particuliers dans les pharmacies, après qu’ils aient été triés et mis à la disposition d’associations humanitaires.
Ce dispositif soulève toutefois plusieurs problèmes, comme l’a souligné très clairement l’Inspection générale des affaires sociales (IGAS), dans un rapport de janvier 2005 (21). En effet, ni la qualité, ni la traçabilité de ces médicaments ne peuvent être vraiment garanties. Par ailleurs, les médicaments collectés ne sont pas toujours adaptés aux besoins des populations destinataires. Enfin, l’expédition des MNU dans les pays en voie de développement peut perturber les politiques pharmaceutiques locales et l’organisation du circuit de la distribution.
Pour l’ensemble de ces raisons, l’article 32 de la loi précitée du 26 février 2007, adopté à l’initiative du gouvernement, a posé le principe de l’interdiction de la redistribution des MNU, en ménageant une période de transition d’une durée maximum de dix-huit mois suivant la publication de la loi avant l’entrée en vigueur de cette interdiction.
L’article L. 4211-2 du code de la santé publique prévoit ainsi aujourd’hui le dispositif suivant.
– Aux termes de son premier alinéa, les officines de pharmacie et les pharmacies à usage intérieur des établissements de santé et médico-sociaux sont tenues de collecter gratuitement les médicaments apportés par les particuliers. Indépendamment de la question de la redistribution des MNU, il a en effet été décidé de maintenir le dispositif spécifique de collecte de ces médicaments par les pharmacies, distinct du circuit normal d’élimination des déchets ménagers, en raison des risques particuliers que présentent les déchets issus de médicaments.
– En vertu de son deuxième alinéa, les médicaments ainsi collectés peuvent être mis gratuitement à la disposition de populations démunies par des organismes à but non lucratif, sous la responsabilité d'un pharmacien.
Toutefois, « à compter d’une date fixée par décret et au plus tard dix-huit mois après la date d'entrée en vigueur » de la loi du 26 février 2007, soit en août 2008, ces dispositions seront supprimées et il y sera substitué l’affirmation du principe selon lequel « toute distribution et toute mise à disposition des médicaments ainsi collectés sont interdites ». Ces médicaments devront alors être « détruits dans des conditions sécurisées », a priori par incinération, s’agissant du mode d’élimination de ces déchets aujourd’hui considéré comme le plus sûr.
Cette période de transition visait notamment à accompagner les organisations non gouvernementales (ONG) lors de ce changement. Lors de la séance du Sénat du 24 janvier 2007, M. Xavier Bertrand, alors ministre de la santé et des solidarités, a en effet jugé nécessaire d’« aider ces organisations, pendant cette période de transition, à formaliser leurs besoins et à trouver de nouvelles sources d'approvisionnement : dons, achats de génériques, obtention de subventions pour les associations particulièrement impliquées », en ajoutant que « le LEEM [Les entreprises du médicament] s'est engagé, y compris par écrit, à poursuivre l'approvisionnement de ces associations en une quantité équivalente de médicaments à destination des pays concernés. D’autre part, j'ai indiqué que, si cela s'avérait insuffisant, l’État s’engagerait à fournir au moins les quantités antérieurement données, voire davantage. »
– Les quatre derniers alinéas du même article L. 4211-2 prévoient la détermination par décret en Conseil d’État des conditions de la collecte et de la destruction des MNU, s’agissant notamment des modalités de financement de celle-ci. Il était également prévu qu’un décret précise les conditions de redistribution de ces médicaments à des associations humanitaires, afin d’encadrer davantage ces pratiques pendant la période transitoire de dix-huit mois, ces dispositions devant être supprimées lors de l’entrée en vigueur de l’interdiction prévue au deuxième alinéa. Plus de neuf mois après la publication de la loi du 26 février 2007, ce décret n’a cependant toujours pas été publié.
2. Les dispositions prévues par le présent article
Selon les auteurs de l’amendement dont résulte le présent article, celui-ci vise à « prolonger le délai transitoire [de dix-huit mois] afin de permettre aux associations, de s'adapter dans des conditions satisfaisantes à l'entrée en vigueur de cette interdiction, notamment par la recherche de nouvelles sources d’approvisionnement pérennes, rationalisées, mieux adaptées et sécurisées ». Il convient toutefois d’observer que cet article a également pour effet de compléter et de modifier, sur d’autres points, le dispositif prévu par l’article 32 de la loi du 26 février 2007 concernant les conditions de destruction et de redistribution des médicaments non utilisés.
Le I de cet article procède tout d’abord à une réécriture globale de l’article L. 4211-2 du code de la santé publique (alinéas 1 à 8 du présent article).
Dans une rédaction inchangée par rapport aux dispositions actuellement prévues par le premier alinéa de l’article L. 4211-2, l’alinéa 2 du présent article prévoit que les officines de pharmacie et les pharmacies à usage intérieur sont tenues de collecter les médicaments à usage humain non utilisés apportés par les particuliers qui les détiennent.
L’alinéa 3 du présent article dispose que les médicaments ainsi collectés « sont détruits dans des conditions sécurisées » – ces dispositions étaient déjà prévues par le II de l’article 32 de la loi précitée du 26 février 2007, mais ne devaient entrer en vigueur qu’au terme de la période transitoire évoquée plus haut –ou mis à la disposition d’organismes à but non lucratif, dont il est précisé qu’ils doivent également avoir une « vocation humanitaire ».
En outre, il est désormais prévu que ces organismes soient agréés par les « autorités administratives » – terme dont on peut regretter l’imprécision, en supposant qu’il reviendra au ministère chargé de la santé de procéder à cet agrément – « après avis du conseil central compétent de l’ordre national des pharmaciens ». Selon les informations communiquées par le ministère de la santé, il s’agirait de donner une base légale à la procédure d’agrément, le projet de décret en cours de préparation sur la valorisation des MNU prévoyant de délivrer celui-ci sur la base de critères de sécurité et de qualité des pratiques de dons des MNU et de soumettre les organismes agréés au respect de bonnes pratiques fixées par arrêté du ministre chargé de la santé.
Cette nouvelle rédaction du deuxième alinéa de l’article L. 4211-2 a par ailleurs pour effet de supprimer la référence au fait que les MNU sont mis « gratuitement » à la disposition, non plus des « populations démunies » par des organismes à but non lucratif, mais uniquement de ces derniers. Enfin, il est à noter que les dispositions prévoyant actuellement que cette redistribution des MNU est effectuée « sous la responsabilité d’un pharmacien » sont également supprimées, même s’il semble que le projet de décret en cours de préparation comporte une disposition analogue.
De manière quelque peu redondante, l’aliéna 4 du présent article prévoit que la récupération des MNU en vue de leur redistribution ne peut être effectuée que par des organismes à but non lucratif et à vocation humanitaire ayant obtenu un agrément.
Reprenant une grande partie des dispositions actuellement prévues par les quatre derniers alinéas de l’article L. 4211-2, les alinéas 5 à 8 du présent article prévoient qu’un décret en Conseil d’État précise ses conditions d’application et notamment les conditions de destruction, y compris des modalités de son financement, et de collecte des MNU. Par cohérence avec les nouvelles dispositions introduites à cet article, « les conditions de l’agrément des organismes à but non lucratif et à vocation humanitaire » et de la mise à la disposition de ces organismes des MNU doivent également être définies par voie réglementaire, la référence au fait que ces médicaments soient mis à la disposition des « populations démunies » par ces associations étant également supprimée.
Le II du présent article prévoit qu’« au plus tard dans un délai de dix-huit mois » (selon une formulation d’ailleurs assez curieuse, puisque contrairement au dispositif prévu par la loi du 26 février 2007, il n’est pas prévu qu’un décret puisse fixer une date plus précoce d’entrée en vigueur de ces dispositions ; il ne semble donc pas y avoir de raison que cette application intervienne « au plus tard » dans dix-huit mois) :
– le deuxième alinéa est remplacé par de nouvelles dispositions prévoyant l’interdiction de toute distribution et de toute mise à disposition des MNU, ces médicaments devant être détruits dans des conditions sécurisées ;
– et qu’en conséquence, les troisième et septième alinéas, relatifs aux conditions de redistribution de ces médicaments, sont également supprimés à cette même date, ces deux mesures étant quasiment identiques à celles déjà prévues par les II et IV de l’article 32 de la loi du 26 février 2007.
Concernant les motifs du report ainsi proposé de l’entrée en vigueur de l’interdiction de la redistribution des MNU, le sénateur M. Jean-Pierre Michel a expliqué qu’en dépit de la mise en place d’un groupe de travail au ministère de la santé sur la mise en place d’autres sources d’approvisionnement des associations humanitaires en médicaments, « il faut bien reconnaître que les dispositions pratiques n’ont pas été prises. Donc, si le délai prévu [par la loi du 26 février 2007] devait être maintenu, les associations se trouveraient dans une situation difficile ».
Si l’auteur de l’amendement avait initialement proposé de porter ce délai à trente mois suivant la publication du présent projet de loi, le Sénat a adopté un sous-amendement du rapporteur de la commission des affaires sociales, avec l’avis favorable du gouvernement, qui a permis de réduire à dix-huit mois la durée de la période transitoire précédant l’entrée en vigueur de l’interdiction de redistribution des MNU. Le rapporteur, M. Gilbert Barbier, a en effet estimé que « le délai de trente mois repousserait à 2010 le règlement d’un problème crucial de santé publique ».
Enfin, le III du présent article a pour objet d’insérer dans le titre II (« Autres services de santé ») du livre III (« Aide médicale urgente, permanence des soins, transports sanitaires et autres services de santé ») de la sixième partie du code de la santé publique, un chapitre V intitulé : « Centres et équipes mobiles de soins aux personnes en situation de précarité ou d’exclusion gérés par des organismes à but non lucratif ».
L’unique article, L. 6325-1, de ce chapitre vise à permettre aux centres et structures disposant d’équipes mobiles de soins aux personnes en situation de précarité ou d’exclusion gérés par des organismes à but non lucratif de délivrer gratuitement les médicaments nécessaires à leurs soins, dans des conditions définies par décret.
S’il est fait référence, de manière très générale, aux personnes en situation de précarité ou d’exclusion, il convient tout d’abord de noter que ce dispositif ne devrait s’adresser qu’à un nombre limité de personnes, dont les sans domicile fixe, compte tenu de l’existence de la couverture maladie universelle (CMU) et de l’aide médicale d’État (AME).
Selon les précisions apportées par le ministère chargé de la santé, les centres de soins et équipes mobiles des associations humanitaires, telles que Médecins du monde, sont aujourd’hui amenées à dispenser des soins et à délivrer des médicaments aux personnes démunies en France, le plus souvent par l’intermédiaire de médecins ou, plus rarement, de pharmaciens. Ces associations s’approvisionnent en MNU et complètent leurs besoins par des dons de l’industrie pharmaceutique ou l’achat de médicaments neufs en officine. Les dispositions prévues au III du présent article viseraient ainsi à sécuriser ces pratiques, en permettant la délivrance de médicaments par les centres et équipes mobiles de soins gérés par ces organismes, en l’absence d’un pharmacien, par dérogation au principe du monopole pharmaceutique.
Il apparaît cependant nécessaire d’encadrer davantage ce dispositif – en précisant par exemple que les médicaments ne peuvent être délivrés que sous la responsabilité d’un médecin ou d’un pharmacien et en prévoyant que ces organismes soient agréés par le ministère chargé de la santé – afin de veiller à ce que ces conditions dérogatoires de délivrance des médicaments présentent toutes les garanties nécessaires en termes de sécurité et de qualité des soins pour les personnes en situation de précarité.
*
La commission a adopté six amendements de la rapporteure ayant pour objet de :
– préciser que la mise à disposition d’organismes à but non lucratif et à vocation humanitaire des médicaments non utilisés, qui sont collectés dans les officines, s’effectue sous la responsabilité d’un pharmacien ;
– clarifier la rédaction de cet article, en précisant que l’agrément des organismes à but non lucratif, autorisés à récupérer les médicaments non utilisés, est délivré par le ministre chargé de la santé ;
– maintenir l’échéance fixée par la loi n° 2007-248 du 26 février 2007 pour l’entrée en vigueur de l’interdiction de la redistribution des médicaments non utilisés, soit le 28 août 2008, alors que les dispositions introduites par le Sénat visaient à la reporter de dix-huit mois après l’entrée en vigueur du présent texte, Mme Catherine Lemorton ayant souhaité cosigner l’amendement ;
– subordonner à l’obtention d’un agrément par le ministre chargé de la santé la possibilité pour les organismes à but non lucratif, qui gèrent des centres et structures disposant d’équipes mobiles de soins aux personnes en situation de précarité, de délivrer gratuitement les médicaments nécessaires à leurs soins ;
– préciser que, dans le cadre de ce dispositif dérogatoire, la délivrance, à titre gratuit, de médicaments aux personnes en situation de précarité doit s’effectuer sous la responsabilité d’un médecin ou d’un pharmacien ;
– supprimer les II et IV de l’article 32 de la loi n° 2007-248 du 26 février 2007, par coordination avec les dispositions prévues par le II du présent article.
La commission a adopté l’article 6 ainsi modifié.
Puis la commission a adopté à l’unanimité l’ensemble du projet de loi ainsi modifié.
*
* *
En conséquence et sous réserve des amendements qu’elle propose, la commission des affaires culturelles, familiales et sociales demande à l’Assemblée nationale d’adopter le projet de loi, adopté par le Sénat, ratifiant l’ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament (n° 301).
ANNEXE 1
LISTE DES PERSONNES AUDITIONNÉES
(par ordre chronologique)
Ø Conseil national de l’ordre des pharmaciens – M. Jean Parrot, président
Ø Collectif Europe et médicament – M. Pierre Chirac, membre de la rédaction de la revue Prescrire, et Mme Laure Lechertier, responsable du département de la politique du médicament à la Mutualité française
Ø Cabinet de la ministre de la santé, de la jeunesse et des sports – Mme Muriel Dahan, conseillère technique pour les produits de santé
Ø Les entreprises du médicament (LEEM) – M. Richard Lerat, secrétaire général, M. Jérôme Soletti, responsable des partenariats, et Mme Aline Bessis-Marais, directrice en charge des affaires publiques
Ø Pharmacie humanitaire internationale (PHI) – M. Jean-Marc Merle, président
Ø Ordre de Malte – M. Jean de Pennart, secrétaire général
« Le Gouvernement peut, pour l'exécution de son programme, demander au Parlement l'autorisation de prendre par ordonnances, pendant un délai limité, des mesures qui sont normalement du domaine de la loi.
« Les ordonnances sont prises en Conseil des Ministres après avis du Conseil d’État. Elles entrent en vigueur dès leur publication mais deviennent caduques si le projet de loi de ratification n'est pas déposé devant le Parlement avant la date fixée par la loi d'habilitation.
« À l’expiration du délai mentionné au premier alinéa du présent article, les ordonnances ne peuvent plus être modifiées que par la loi dans les matières qui sont du domaine législatif. »
ARTICLE 39 DE LA LOI N° 2007-248 DU 26 FÉVRIER 2007
PORTANT DIVERSES DISPOSITIONS D'ADAPTATION AU DROIT COMMUNAUTAIRE DANS LE DOMAINE DU MÉDICAMENT
« I.- Dans les conditions prévues à l'article 38 de la Constitution, le gouvernement est autorisé à prendre par ordonnances les dispositions nécessaires à la transposition des directives ou de celles de leurs dispositions qui n'ont pas encore été transposées, ainsi que les mesures d'adaptation de la législation liées à cette transposition :
a) Directive 2002/98/CE du Parlement européen et du Conseil du 27 janvier 2003 établissant des normes de qualité et de sécurité pour la collecte, le contrôle, la transformation, la conservation et la distribution du sang humain et des composants sanguins, et modifiant la directive 2001/83/CE ;
b) Directive 2003/15/CE du Parlement européen et du Conseil du 27 février 2003 modifiant la directive 76/768/CEE du Conseil concernant le rapprochement des législations des Etats membres relatives aux produits cosmétiques ;
c) Directive 2004/23/CE du Parlement européen et du Conseil du 31 mars 2004 relative à l'établissement de normes de qualité et de sécurité pour le don, l'obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissus et cellules humains ;
d) Directive 2004/24/CE du Parlement européen et du Conseil du 31 mars 2004 modifiant, en ce qui concerne les médicaments traditionnels à base de plantes, la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain ;
e) Directive 2004/28/CE du Parlement européen et du Conseil du 31 mars 2004 modifiant la directive 2001/82/CE instituant un code communautaire relatif aux médicaments vétérinaires.
II.- Dans les mêmes conditions, le Gouvernement est autorisé à prendre par ordonnances les mesures requises :
1° Pour adapter au droit communautaire les dispositions du code de la santé publique relatives aux autorisations d'importation des médicaments à usage humain et celles du même code concernant les insecticides et acaricides destinés à l'homme, ainsi que celles définissant le régime juridique des aliments diététiques destinés à des fins médicales spéciales ;
2° Pour harmoniser et compléter les dispositions pénales relatives aux produits mentionnés aux articles L. 5141-1 et L. 5311-1 du code de la santé publique, pour instaurer, en tant que de besoin, des sanctions administratives dans les domaines qui n'en disposent pas et pour harmoniser leur mise en oeuvre avec les sanctions pénales ;
3° Pour permettre aux agents mentionnés au 1° de l'article L. 215-1 du code de la consommation de recourir à l'Agence française de sécurité sanitaire des produits de santé dans l'exercice des pouvoirs d'enquête qui leur sont dévolus en application de l'article L. 5414-1 du code de la santé publique ;
4° Pour permettre la mise en cohérence du dispositif existant dans le cadre du code de la santé publique en matière de classification des substances et préparations dangereuses et vénéneuses avec les dispositions issues du droit communautaire.
III.- Les ordonnances prévues par le I et le 2° du II sont prises dans un délai de huit mois suivant la publication de la présente loi. Celles prévues aux 1°, 3° et 4° du II sont prises dans un délai de trois mois à compter de cette même date.
Le projet de loi de ratification de chacune des ordonnances prévues par le présent article est déposé devant le Parlement au plus tard le dernier jour du deuxième mois à compter de la publication de cette ordonnance. »
___
Dispositions en vigueur ___ |
Texte du projet de loi ___ |
Texte adopté par le Sénat en première lecture ___ |
Propositions de la Commission ___ |
Projet de loi ratifiant l’ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament |
Projet de loi ratifiant l’ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament |
Projet de loi ratifiant l’ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament | |
Article 1er |
Article 1er |
Article 1er | |
L’ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament est ratifiée. |
Sans modification |
Sans modification | |
Article 1er bis (nouveau) |
Article 1er bis | ||
Dans les conditions prévues à l’article 38 de la Constitution, le Gouvernement est autorisé à prendre par ordonnances, dans un délai de quatre mois à compter de la date de publication de la présente loi, d’une part, les dispositions nécessaires à la transposition de la directive 2004/23/CE du Parlement européen et du Conseil du 31 mars 2004 relative à l’établissement de normes de qualité et de sécurité pour le don, l’obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissus et cellules humains et, d’autre part, les mesures requises pour harmoniser et compléter les dispositions pénales relatives aux produits mentionnés aux articles L. 5141-1 et L. 5311-1 du code de la santé publique, pour instaurer, en tant que de besoin, des sanctions administratives dans les domaines qui n’en disposent pas et pour harmoniser leur mise en œuvre avec les sanctions pénales. |
Sans modification | ||
Le projet de loi portant ratification de ces ordonnances est déposé devant le Parlement au plus tard le dernier jour du deuxième mois suivant la publication de ces ordonnances. |
|||
|
Code des douanes |
Article 2 |
Article 2 |
Article 2 |
Art. 38. - 1. Pour l'application du présent code, sont considérées comme prohibées toutes marchandises dont l'importation ou l'exportation est interdite à quelque titre que ce soit, ou soumise à des restrictions, à des règles de qualité ou de conditionnement ou à des formalités particulières. 2. Lorsque l'importation ou l'exportation n'est permise que sur présentation d'une autorisation, licence, certificat, etc., la marchandise est prohibée si elle n'est pas accompagnée d'un titre régulier ou si elle est présentée sous le couvert d'un titre non applicable. 3. Tous titres portant autorisation d'importation ou d'exportation (licences ou autres titres analogues) ne peuvent, en aucun cas, faire l'objet d'un prêt, d'une vente, d'une cession et, d'une manière générale, d'une transaction quelconque de la part des titulaires auxquels ils ont été nominativement accordés. |
Sans modification | ||
4. Au titre des dispositions dérogatoires prévues à l’article 2 bis, les dispositions du présent article sont applicables aux marchandises relevant des articles 2, 3, 4, 5 et 19 de la loi nº 92 1477 du 31 décembre 1992 relative aux produits soumis à certaines restrictions de circulation et à la complémentarité entre les services de police, de gendarmerie et de douane, aux marchandises visées à l’article L. 5132-9 du code de la santé publique, aux médicaments à usage humain visés à l’article L. 5124-13 du code de la santé publique, aux micro-organismes et aux toxines mentionnés à l’article L. 5139-1 du code de la santé publique, aux médicaments à usage vétérinaire mentionnés à l’article L. 5142-7 du code de la santé publique, aux marchandises présentées sous une marque contrefaite, ainsi qu’aux produits sanguins labiles et aux pâtes plasmatiques mentionnés au 1º et au 2º de l’article L. 1221-8 du même code, aux organes, tissus et leurs dérivés, cellules, gamètes issus du corps humain ainsi qu’aux préparations de thérapie cellulaire mentionnés aux articles L. 1235-1, L. 1243-1, L. 1244-8 et L. 1245-5 dudit code, aux tissus ou cellules embryonnaires ou foetaux mentionnés à l’article L. 2151-6 du même code, aux sources artificielles et naturelles de radionucléides définies à l’article L. 1333-1 du code de la santé publique et relevant des articles L. 1333-2 et L. 1333-4 du même code et aux déchets définis au II de l’article L. 541-1 du code de l’environnement dont l’importation, l’exportation ou le transit sont régis soit par les articles L. 541-40 à L. 541-42 du même code et les dispositions réglementaires prises pour leur application, soit par le règlement (CEE) nº 259/93 du Conseil du 1er février 1993 concernant la surveillance et le contrôle des transferts de déchets à l’entrée et à la sortie de la Communauté européenne, ainsi que par les décisions des autorités communautaires prises en application de ce règlement. Les dispositions du présent article s’appliquent également aux objets de toute nature comportant des images ou des représentations d’un mineur à caractère pornographique visées par l’article 227-23 du code pénal. ………………………... |
À la première phrase du quatrième alinéa de l’article 38 du code des douanes, après les mots : « aux produits sanguins labiles et aux pâtes plasmatiques mentionnés au 1° et au 2° de l’article L. 1221-8 du même code » sont ajoutés les mots : « au sang, ses composants et ses produits dérivés à des fins scientifiques mentionnés à l’article L. 1221-12, » et après les mots : « ainsi qu’aux préparations de thérapie cellulaire » sont ajoutés les mots : « et aux échantillons biologiques ». |
Dans la première phrase du 4 de l’article… … biologiques ». |
|
|
Code de la santé publique |
Article 3 |
Article 3 |
Article 3 |
Art. L. 5141-5-2. - Pour obtenir l’autorisation définie à l’article L. 5141-5, les médicaments conçus pour être administrés à des animaux dont la chair ou les produits sont destinés à la consommation humaine ne doivent contenir qu’une ou des substances pharmacologiquement actives figurant dans l’une des annexes I, II ou III du règlement du Conseil (CEE) nº 2377/90 du 26 juin 1990 modifié établissant une procédure communautaire pour la fixation des limites maximales de résidus de médicaments vétérinaires dans les aliments d’origine animale. ……………………….. |
Sans modification | ||
Par dérogation au premier alinéa, un médicament vétérinaire contenant des substances pharmacologi-quement actives ne figurant pas à l’annexe I, II ou III du règlement précité peut être autorisé pour les animaux particuliers appartenant à la famille des équidés qui ont été identifiés conformément à l’article L. 212-9 du code rural et qui ont été déclarés comme n’étant pas destinés à l’abattage pour la consom-mation humaine. Ces médicaments vétérinaires ne contiennent pas de substances pharmacologiquement actives figurant à l’annexe IV du règlement mentionné ci-dessus et ne sont pas destinés à être utilisés pour le traitement d’affections telles que spécifiées dans le résumé autorisé des caractéristiques du produit, pour lesquelles un médicament vétérinaire est autorisé pour soigner les animaux de la famille des équidés. |
Au dernier alinéa de l’article L. 5141-5-2 du code de la santé publique, les mots : « dans le résumé autorisé des caractéristiques du produit » sont remplacés par les mots : « dans le résumé des caractéristiques du produit ». |
Dans la seconde phrase du dernier alinéa de l’article L. 5141-5-2 du code de la santé publique, après le mot : « résumé », le mot : « autorisé » est supprimé. |
|
Article 4 (nouveau) |
Article 4 | ||
Art. L. 5311-1. –L’Agence française de sécurité sanitaire des produits de santé est un établissement public de l'Etat, placé sous la tutelle du ministre chargé de la santé. L'agence participe à l'application des lois et règlements et prend, dans les cas prévus par des dispositions particulières, des décisions relatives à l'évaluation, aux essais, à la fabrication, à la préparation, à l'importation, à l'exportation, à la distribution en gros, au conditionnement, à la conservation, à l'exploitation, à la mise sur le marché, à la publicité, à la mise en service ou à l'utilisation des produits à finalité sanitaire destinés à l'homme et des produits à finalité cosmétique, et notamment |
Le 1° de l’article L. 5311-1 du code de la santé publique est ainsi rédigé : |
Sans modification | |
1º Les médicaments, y compris les préparations magistrales, hospitalières et officinales, les substances stupéfiantes, psychotropes ou autres substances vénéneuses utilisées en médecine, les insecticides, acaricides et antiparasitaires à usage humain, les huiles essentielles et plantes médicinales, les matières premières à usage pharmaceutique ; |
« 1° Les médicaments, y compris les insecticides, acaricides et antiparasitaires à usage humain, les préparations magistrales, hospitalières et officinales, les substances stupéfiantes, psychotropes ou autres substances vénéneuses utilisées en médecine, les huiles essentielles et plantes médicinales, les matières premières à usage pharmaceutique ; ». |
||
……………………….. |
Article 5 (nouveau) |
Article 5 | |
I. – Le code de la santé publique est ainsi modifié : |
Sans modification | ||
17º Les produits de tatouage. |
1° Après le 17° de l’article L. 5311-1, il est inséré un alinéa ainsi rédigé : |
||
|
……………………….. |
« L’agence participe à l’application des lois et règlements relatifs aux recherches biomédicales et prend, dans les cas prévus par des dispositions particulières, des décisions relatives aux recherches biomédicales. » ; |
||
2° L’article L. 1123-12 est ainsi modifié : |
|||
Art. L. 1123-12. – L’au-torité compétente est l'Agence française de sécurité sanitaire des produits de santé pour les recherches portant sur les produits mentionnés à l'article L. 5311-1, et le ministre chargé de la santé dans les autres cas. |
a) Dans le premier alinéa, les mots : « pour les recherches portant sur les produits mentionnés à l’article L. 5311-1, et le ministre chargé de la santé dans les autres cas » sont supprimés ; |
||
Lorsqu'une collection d'échantillons biologiques humains est constituée pour les seuls besoins d'une recherche biomédicale, elle est déclarée à l'autorité compétente pour cette recherche. |
b) Dans le second alinéa, les mots : « pour cette recherche » sont supprimés ; |
||
Art. L. 1121-3 . - … Les recherches biomédicales portant sur des médicaments sont réalisées dans le respect des règles de bonnes pratiques cliniques fixées par décision de l'Agence française de sécurité sanitaire des produits de santé. Pour les autres recherches, des recommandations de bonnes pratiques sont fixées par arrêté du ministre chargé de la santé et par décision de l'Agence française de sécurité sanitaire des produits de santé pour les produits mentionnés à l'article L. 5311-1. ..................................... |
3° Dans la seconde phrase du septième alinéa de l’article L. 1121-3, les mots : « par arrêté du ministre chargé de la santé et » et les mots : « pour les produits mentionnés à l’article L. 5311-1 » sont supprimés. |
||
II. – Les dispositions du I entrent en vigueur au 1er avril 2008. À cet effet, le ministre chargé de la santé se prononce au titre des articles L. 1123-8 et L. 1123-9 du code de la santé publique sur les demandes d’autorisation déposées jusqu’au 31 mars 2008 inclus concernant les recherches biomédicales autres que celles portant sur des produits mentionnés à l’article L. 5311-1 du même code. |
|||
Article 6 (nouveau) |
Article 6 | ||
I. – L’article L. 4211-2 du code de la santé publique est ainsi rédigé : |
I.- Alinéa sans modification | ||
Art. L. 4211-2. – Les officines de pharmacie et les pharmacies à usage intérieur sont tenues de collecter gratuitement les médicaments à usage humain non utilisés apportés par les particuliers qui les détiennent. |
« Art. L. 4211-2. – Les officines de pharmacie et les pharmacies à usage intérieur sont tenues de collecter gratuitement les médicaments à usage humain non utilisés apportés par les particuliers qui les détiennent. |
Art. L. 4211-2. – Alinéa sans modification | |
Les médicaments ainsi collectés peuvent être mis gratuitement à la disposition de populations démunies par des organismes à but non lucratif, sous la responsabilité d'un pharmacien. |
« Les médicaments ainsi collectés sont détruits dans des conditions sécurisées ou mis à la disposition d’organismes à but non lucratif et à vocation humanitaire, agréés par les autorités administratives après avis du conseil central compétent de l’ordre national des pharmaciens. |
Les… ..sécurisées ou, sous la responsabilité d’un pharmacien, mis à la… …par le ministre chargé de la santé après avis… …pharmaciens. Amendements n°1 et 2 | |
« La récupération des médicaments non utilisés en vue de leur redistribution ne peut être effectuée que par des organismes à but non lucratif et à vocation humanitaire ayant obtenu l’agrément mentionné au deuxième alinéa. |
Alinéa sans modification | ||
Un décret en Conseil d'Etat précise : |
« Un décret en Conseil d’État précise les conditions d’application du présent article et notamment : |
Alinéa sans modification | |
- les conditions de la collecte des médicaments inutilisés mentionnée au premier alinéa ; |
« - les conditions de la collecte des médicaments non utilisés mentionnée au premier alinéa ; |
Alinéa sans modification | |
- les conditions de la destruction des médicaments mentionnée au deuxième alinéa, et notamment les conditions de financement de cette destruction ; |
« - les conditions de la destruction des médicaments mentionnée au deuxième alinéa, et notamment les conditions de financement de cette destruction ; |
Alinéa sans modification | |
- les conditions de mise à disposition des médicaments inutilisés aux populations démunies par les organismes à but non lucratif mentionnée au deuxième alinéa. |
« - les conditions de l’agrément des organismes à but non lucratif et à vocation humanitaire mentionné au deuxième alinéa et de la mise à la disposition de ces organismes des médicaments non utilisés. » |
Alinéa sans modification | |
A compter d'une date fixée par décret et au plus tard dix-huit mois après la date d'entrée en vigueur de la présente loi, le deuxième alinéa du même article est ainsi rédigé : |
II. – Au plus tard dans un délai de dix-huit mois après la date d’entrée en vigueur de la présente loi, l’article L. 4211-2 du même code est ainsi modifié : |
II. – À compter du 28 août 2008, l’article L. 4211-2 du même code est ainsi modifié : Amendement n°3 | |
1° Le deuxième alinéa est ainsi rédigé : |
1°Non modifié | ||
Toute distribution et toute mise à disposition des médicaments ainsi collectés sont interdites. Ces médicaments sont détruits dans des conditions sécurisées. |
« Toute distribution et toute mise à disposition des médicaments non utilisés sont interdites. Ces médicaments sont détruits dans des conditions sécurisées. » ; |
||
Le sixième alinéa du même article est supprimé à compter de la date d'entrée en vigueur du deuxième alinéa de l'article L. 4211-2 du code de la santé publique prévue au II du présent article. |
2° Les troisième et septième alinéas sont supprimés. |
2°Non modifié | |
SIXIÈME PARTIE Etablissements et services de santé LIVRE III Aide médicale urgente, permanence des soins, transports sanitaires et autres services de santé TITRE II Autres services de santé |
III. – Le titre II du livre III de la sixième partie du même code est complété par un chapitre V ainsi rédigé : |
III.- Alinéa sans modification | |
« Chapitre V |
|||
« Centres et équipes mobiles de soins aux personnes en situation de précarité ou d’exclusion gérés par des organismes à but non lucratif |
Division et intitulé sans modification | ||
« Art. L. 6325-1. – Les centres et structures disposant d’équipes mobiles de soins aux personnes en situation de précarité ou d’exclusion gérés par des organismes à but non lucratif peuvent délivrer, à titre gratuit, les médicaments nécessaires à leurs soins, dans des conditions définies par décret. » |
Art. L. 6325-1. – Les… …à but non lucratif agréés par le ministre chargé de la santé peuvent délivrer, à titre gratuit et sous la responsabilité d’un médecin ou d’un pharmacien, les médicaments… …par décret. Amendements n°4 et 5 | ||
IV. – Les II et IV de l’article 32 de la loi n° 2007-248 du 26 février 2007 portant diverses dispositions d'adaptation au droit communautaire dans le domaine du médicament sont supprimés. Amendement n°6 |
ANNEXE AU TABLEAU COMPARATIF
Ordonnance n° 2007-613 du 26 avril 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament
Chapitre Ier
Médicaments traditionnels à base de plantes
Article 1er
À l’article L. 5121-1 du code de la santé publique, il est ajouté un 16° ainsi rédigé :
« 16° Médicament à base de plantes, tout médicament dont les substances actives sont exclusivement une ou plusieurs substances végétales ou préparations à base de plantes ou une association de plusieurs substances végétales ou préparations à base de plantes. »
Article 2
I.- Après l’article L. 5121-14 du même code, il est inséré un article L. 5121-14-1 ainsi rédigé :
« Art. L. 5121-14-1. - Ne sont pas soumis à l’autorisation de mise sur le marché les médicaments traditionnels à base de plantes qui remplissent les critères suivants :
« 1° Ils sont conçus pour être utilisés sans l’intervention d’un médecin à des fins de diagnostic, de prescription ou de suivi du traitement ;
« 2° Ils sont exclusivement destinés à être administrés selon un dosage et une posologie spécifiés ;
« 3° Ils sont administrés par voie orale, externe ou par inhalation ;
« 4° La durée d’usage traditionnel est écoulée ;
« 5° Les données sur l’usage traditionnel du médicament sont suffisantes.
« Ces médicaments font l’objet, avant leur mise sur le marché ou leur distribution à titre gratuit, d’un enregistrement auprès de l’Agence française de sécurité sanitaire des produits de santé.
« Toutefois, si l’agence considère qu’un médicament traditionnel à base de plantes relève, compte tenu de ses caractéristiques, du régime de l’autorisation de mise sur le marché ou de celui de l’enregistrement de médicament homéopathique, l’enregistrement prévu à l’alinéa précédent n’est pas applicable.
« Il est procédé à cet enregistrement si les critères mentionnés ci-dessus sont remplis et si le demandeur est établi dans un État membre de la Communauté européenne ou partie à l’accord sur l’Espace économique européen. Il peut être refusé en cas de danger pour la santé publique.
« L’enregistrement est effectué pour une durée de cinq ans. Il peut être renouvelé, le cas échéant sans limitation de durée, dans des conditions fixées par décret en Conseil d’État. Ce décret détermine également les conditions dans lesquelles il peut devenir caduc.
« L’enregistrement peut être modifié par l’Agence française de sécurité sanitaire des produits de santé.
« L’enregistrement peut être suspendu ou retiré par l’agence si les critères et les conditions auxquelles il est subordonné ne sont plus remplis ou en cas de danger pour la santé publique.
« L’accomplissement des formalités prévues au présent article n’a pas pour effet d’exonérer le fabricant et, s’il est distinct, le titulaire de l’enregistrement, de la responsabilité que l’un ou l’autre peut encourir dans les conditions du droit commun en raison de la fabrication ou de la mise sur le marché du médicament. »
II. - Pour les médicaments traditionnels à base de plantes dont la mise sur le marché a eu lieu avant la date de publication de la présente ordonnance, une demande d’enregistrement doit être déposée dans les conditions fixées au présent article.
À titre transitoire, ces produits peuvent continuer à être commercialisés jusqu’à la notification éventuelle d’un refus d’enregistrement par le directeur général de l’Agence française de sécurité sanitaire des produits de santé, et au plus tard jusqu’au 30 avril 2011.
Article 3
L’article L. 5121-15 du même code est modifié comme suit :
1° Après le premier alinéa, il est inséré un alinéa ainsi rédigé :
« Toute demande d’enregistrement mentionnée à l’article L. 5121-14-1 ou toute demande de modification ou de renouvellement de cet enregistrement donne lieu au versement, au profit de l’Agence française de sécurité sanitaire des produits de santé, d’un droit progressif déterminé en fonction de l’objet de la demande et de l’existence, le cas échéant, d’une autorisation de mise sur le marché délivrée par l’agence antérieurement à la demande d’enregistrement et dont le montant est fixé par décret dans la limite de 10 110. »
2° Au deuxième alinéa, les mots : « Ce droit est recouvré » sont remplacés par les mots : « Ces droits sont recouvrés ».
Article 4
L’article L. 5121-20 du même code est modifié comme suit :
1° Au 11°, après les mots : « ou du titulaire de l’enregistrement de médicament homéopathique », sont ajoutés les mots : « ou du titulaire de l’enregistrement de médicament traditionnel à base de plantes ».
2° Il est rétabli un 12° ainsi rédigé :
« 12° Les modalités de présentation des demandes tendant à obtenir l’enregistrement des médicaments traditionnels à base de plantes prévu à l’article L. 5121-14-1, le contenu du dossier présenté à l’appui de ces demandes, ainsi que les conditions dans lesquelles interviennent l’enregistrement et les décisions, refusant, modifiant, renouvelant, suspendant ou retirant celui-ci. »
Article 5
À l’article L. 5124-5 du même code, après la référence : « L. 5121-8 » sont ajoutés les mots : « à l’enregistrement de médicament traditionnel à base de plantes ».
Article 6
Au premier alinéa de l’article L. 5123-2 du même code, les références : « L. 5121-12 et L. 5121-13 » sont remplacées par les références : « L. 5121-12, L. 5121-13 et L. 5121-14-1 ».
Chapitre II
Produits d’origine humaine
Section 1
Tissus et cellules
Article 7
I.- L’article L. 1211-8 du code de la santé publique est remplacé par les dispositions suivantes :
« Art. L. 1211-8. - Ne sont soumis aux dispositions du présent livre ni les produits du corps humain pour lesquels il est d’usage de ne pas appliquer l’ensemble des principes qu’énoncent les articles L. 1211-1 à L. 1211-7, ni les éléments et produits du corps humain prélevés et utilisés à des fins thérapeutiques autologues dans le cadre d’une seule et même intervention médicale, sans être conservés ou préparés à aucun moment au sein d’un organisme ou d’un établissement autorisé en application de l’article L. 1243-2. »
II.– À l’article L. 1242-1 du même code, il est ajouté un alinéa ainsi rédigé :
« Cet article ne s’applique pas aux éléments et produits du corps humain mentionnés à l’article L. 1211-8. »
III.- À l’article L. 1243-6 du même code, il est ajouté un alinéa ainsi rédigé :
« Cet article ne s’applique pas aux greffes et administration d’éléments et produits du corps humain mentionnés à l’article L. 1211-8. »
Article 8
I.- Le troisième alinéa de l’article L. 1243-2 du même code est remplacé par les dispositions suivantes :
« Toute modification substantielle dont la liste est fixée par décret en Conseil d’État affectant une ou plusieurs des activités exercées par l’établissement ou l’organisme autorisé doit faire l’objet d’une nouvelle autorisation. Les autres modifications sont soumises à une déclaration auprès du directeur général de l’Agence française de sécurité sanitaire des produits de santé.
« Par dérogation aux dispositions du premier alinéa, les établissements de santé autorisés à prélever des cellules hématopoïétiques issues de la moelle osseuse peuvent distribuer des cellules hématopoïétiques issues de la moelle osseuse non transformées en vue d’une greffe immédiate. »
II.- Après l’article L. 1243-2, il est inséré un article L. 1243-2-1 ainsi rédigé :
« Art. L. 1243-2-1. - Le respect, dans les établissements autorisés au titre de l’article L. 1243-2, des dispositions législatives et réglementaires relatives à la qualité et la sécurité des tissus et de leurs dérivés et des préparations de thérapie cellulaire, est garanti par une personne responsable désignée dans des conditions prévues par décret en Conseil d’État.
« Au sein de l’Etablissement français du sang, la personne responsable a autorité sur les directeurs des établissements de transfusion sanguine pour l’exercice de cette mission. »
Article 9
À la troisième phrase du deuxième alinéa de l’article L. 1223-1 du même code, après les mots : « d’autres activités de santé, notamment » sont ajoutés les mots : « les activités prévues à l’article L. 1243-2 et ».
Article 10
Le troisième alinéa de l’article L. 1223-4 du même code est complété par les dispositions suivantes :
« Si l’établissement de transfusion sanguine est autorisé à exercer les activités prévues à l’article L. 1243-2, son directeur assure, sous l’autorité de la personne responsable mentionnée au deuxième alinéa de l’article L. 1243-2-1, la mise en oeuvre des dispositions législatives et réglementaires relatives à la qualité et à la sécurité des tissus et de leurs dérivés et des préparations de thérapie cellulaire. »
Article 11
Au 2° de l’article L. 1243-9 du même code, après les mots : « ainsi que les conditions de modification » sont ajoutés les mots : « de ces autorisations par l’autorité administrative compétente, notamment la liste des modifications devant faire l’objet de l’autorisation prévue au troisième alinéa de l’article L. 1243-2, ainsi que les conditions ».
Article 12
L’article L. 1245-5 du même code est modifié ainsi qu’il suit :
1° Le premier alinéa est remplacé par les dispositions suivantes :
« Seuls peuvent exercer l’activité d’importation et d’exportation à des fins thérapeutiques des tissus, de leurs dérivés, des cellules issus du corps humain, quel que soit leur niveau de préparation, et des préparations de thérapie cellulaire, les établissements ou les organismes autorisés par l’Agence française de sécurité sanitaire des produits de santé en application de l’article L. 1243-2 et qui obtiennent pour cette activité une autorisation spécifique. Cette autorisation est délivrée par l’Agence française de sécurité sanitaire des produits de santé après avis de l’Agence de la biomédecine.
« Tout produit mentionné à l’alinéa précédent qui a été préparé et conservé dans un État membre de la Communauté européenne ou partie à l’accord sur l’Espace économique européen et qui n’a pas fait l’objet de l’autorisation prévue à l’article 6-2 de la directive 2004/23/CE du Parlement européen et du Conseil du 31 mars 2004 relative à l’établissement de normes de qualité et de sécurité pour le don, l’obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissus et cellules humains fait l’objet, préalablement à son importation, de l’autorisation prévue à l’article L. 1243-5.
« Tout produit mentionné au premier alinéa qui a été préparé et conservé dans un Etat non membre de la Communauté européenne ni partie à l’accord sur l’Espace économique européen fait l’objet, préalablement à son importation, de l’autorisation prévue à l’article L. 1243-5.
« Lorsque les produits ne bénéficient pas de l’autorisation mentionnée à l’article L. 1243-5, l’établissement ou l’organisme qui envisage d’exporter ces produits communique à l’Agence française de sécurité sanitaire des produits de santé les motifs pour lesquels cette autorisation n’est pas disponible. L’agence communique ces motifs aux autorités de santé compétentes du pays importateur. L’agence peut, pour des raisons liées à l’absence de qualité ou de sécurité, interdire l’exportation des produits pour lesquels elle a refusé l’autorisation mentionnée à l’article L. 1243-5. »
2° Dans l’ensemble de l’article, le mot : « réactifs » est remplacé par les mots : « dispositifs médicaux de diagnostic in vitro ».
3° Il est complété par un alinéa ainsi rédigé :
« Par dérogation aux dispositions des trois premiers alinéas, des établissements ou organismes ne bénéficiant pas de l’autorisation d’exercer les activités d’importation et d’exportation mentionnée à ces alinéas peuvent, dans des situations d’urgence, être autorisés par l’Agence française de sécurité sanitaire des produits de santé à importer ou à exporter à des fins thérapeutiques des tissus, des cellules, quel que soit leur niveau de préparation, et des préparations de thérapie cellulaire, destinés à un patient. L’Agence de la biomédecine est informée des autorisations délivrées en application du présent alinéa. »
Article 13
À la fin du premier alinéa de l’article L. 1243-5 du même code, après les mots : « ainsi que de leurs indications thérapeutiques » sont ajoutés les mots : « et après avis de l’Agence de la biomédecine ».
Article 14
À l’article L. 1245-6 du même code, après le mot : « conservation, » sont ajoutés les mots : « à la distribution, ».
Article 15
À la première phrase du quatrième alinéa de l’article 38 du code des douanes, après les mots : « organes, tissus » sont ajoutés les mots : « et leurs dérivés ».
Section 2
Produits sanguins
Article 16
Au premier alinéa de l’article L. 1221-4 du code de la santé publique, les mots : « ne peuvent être distribués ni utilisés » sont remplacés par les mots : « ne peuvent pas être distribués, délivrés, utilisés ».
Article 17
Le premier alinéa de l’article L. 1223-2 du même code est complété par la phrase suivante : « L’activité de délivrance des produits sanguins labiles est exercée par l’établissement de transfusion sanguine ou par l’établissement de santé sous l’autorité d’un médecin ou d’un pharmacien. »
Article 18
Il est inséré, après l’article L. 1221-10-1 du même code, un article L. 1221-10-2 ainsi rédigé :
« Art. L. 1221-10-2. - Toute violation dans un établissement de santé et du fait de celui-ci des prescriptions législatives et réglementaires relatives à la conservation des produits sanguins labiles en vue de leur délivrance ainsi que des termes de l’autorisation mentionnée au premier alinéa de l’article L. 1221-10 entraîne la suspension ou le retrait par l’autorité administrative de cette autorisation. Cette suspension ou ce retrait ne peut intervenir qu’après mise en demeure adressée au directeur de l’établissement de santé de prendre toutes mesures propres à remédier à la violation ou au manquement constaté, ou de fournir toutes explications nécessaires. Cette mise en demeure est faite par écrit par l’autorité compétente et fixe un délai d’exécution ou de réponse qui ne peut excéder un mois.
« En cas d’urgence tenant à la sécurité des personnes, une suspension de l’autorisation peut être prononcée à titre conservatoire par l’autorité compétente. »
Article 19
I.- À l’article L. 1271-1 du même code, après les mots : « autorisations prévues aux articles », est ajoutée la référence : « L. 1221-10, ».
II.- Il est inséré, après l’article L. 1271-1, un article L. 1271-1-1 ainsi rédigé :
« Art. L. 1271-1-1. - Est puni de deux ans d’emprisonnement et de 75 000 € d’amende le fait de contrevenir à une décision de retrait ou de suspension d’agrément ou d’autorisation prise en application des articles L. 1223-5 et L. 1221-10-2. »
Article 20
L’article L. 1271-4 du même code est remplacé par les dispositions suivantes :
« Art. L. 1271-4. - Est punie de deux ans d’emprisonnement et de 75 000 € d’amende la distribution, la délivrance ou l’utilisation du sang, de ses composants ou de leurs dérivés, sans qu’il ait été procédé aux analyses biologiques et aux tests de dépistage de maladies transmissibles requis en application de l’article L. 1221-4. »
Article 21
Le second alinéa de l’article L. 1271-8 du même code est remplacé par les dispositions suivantes :
« Est puni des mêmes peines prévues aux mêmes articles le fait :
« - de distribuer ou de délivrer à des fins thérapeutiques un produit sanguin labile ne figurant pas sur la liste prévue à l’article L. 1221-8, à moins qu’il ne soit destiné à des recherches biomédicales ;
« - d’utiliser un produit sanguin labile en violation d’une disposition ou d’une décision édictée par l’Agence française de sécurité sanitaire des produits de santé en application de l’article L. 1221-10-1. »
Chapitre III
Produits cosmétiques
Article 22
Après l’article L. 5131-7-1 du code de la santé publique, il est inséré deux articles ainsi rédigés :
« Art. L. 5131-7-2. - Sans préjudice des obligations générales découlant de l’article L. 5131-4, il est interdit de :
« a) Mettre sur le marché des produits cosmétiques dont la formulation finale, afin de satisfaire aux exigences du présent chapitre, a fait l’objet d’une expérimentation animale au moyen d’une méthode autre qu’une méthode alternative. Cette interdiction entre en vigueur au fur et à mesure de la validation et de l’adoption des méthodes alternatives par la Commission européenne, constatées par arrêté du ministre chargé de la santé pris sur proposition de l’Agence française de sécurité sanitaire des produits de santé et, au plus tard, le 11 mars 2009 ;
« b) Mettre sur le marché des produits cosmétiques contenant des ingrédients ou des combinaisons d’ingrédients qui, afin de satisfaire aux exigences du présent chapitre, ont fait l’objet d’une expérimentation animale au moyen d’une méthode autre qu’une méthode alternative. Cette interdiction entre en vigueur au fur et à mesure de la validation et de l’adoption des méthodes alternatives par la Commission européenne, constatées par arrêté du ministre chargé de la santé pris sur proposition de l’Agence française de sécurité sanitaire des produits de santé et, au plus tard, le 11 mars 2009 ;
« c) Réaliser, afin de satisfaire aux exigences du présent chapitre, des expérimentations animales portant sur des produits cosmétiques finis ;
« d) Réaliser, afin de satisfaire aux exigences du présent chapitre, des expérimentations animales portant sur des ingrédients ou des combinaisons d’ingrédients. Cette interdiction entre en vigueur au fur et à mesure du remplacement de telles expérimentations par une ou plusieurs méthodes alternatives validées constaté par arrêté du ministre chargé de la santé et, au plus tard, le 11 mars 2009. Ces méthodes alternatives sont décrites dans un arrêté des ministres chargés de la santé, de la consommation et de l’industrie, pris sur proposition de l’Agence française de sécurité sanitaire des produits de santé.
« L’interdiction mentionnée aux a et b ci-dessus entre en vigueur au plus tard le 11 mars 2013 pour les expérimentations concernant la toxicité des doses répétées, la toxicité pour la reproduction et la toxicocinétique.
« Art. L. 5131-7-3. - Dans des circonstances exceptionnelles, lorsque la sécurité d’un ingrédient existant de produit cosmétique suscite de graves préoccupations, l’Agence française de sécurité sanitaire des produits de santé peut demander à la Commission européenne d’accorder une dérogation aux dispositions de l’article L. 5131-7-2. Cette demande comporte une évaluation de la situation et indique les mesures dérogatoires jugées nécessaires. »
Article 23
À l’article L. 5431-2 du même code, après le 3°, il est ajouté un 4° ainsi rédigé :
« 4° De mettre sur le marché des produits cosmétiques ou de réaliser des expérimentations animales portant sur des produits cosmétiques finis ou sur des ingrédients ou des combinaisons d’ingrédients en méconnaissance des interdictions prévues à l’article L. 5131-7-2. »
Chapitre IV
Aliments diététiques destinés
à des fins médicales spéciales
Article 24
L’article L. 5137-1 du code de la santé publique est remplacé par les dispositions suivantes :
« Art. L. 5137-1. - On entend par aliments diététiques destinés à des fins médicales spéciales les aliments destinés à une alimentation particulière qui sont spécialement traités ou formulés pour répondre aux besoins nutritionnels des patients. Ils sont destinés à constituer l’alimentation exclusive ou partielle des patients dont les capacités d’absorption, de digestion, d’assimilation, de métabolisation ou d’excrétion des aliments ordinaires ou de certains de leurs ingrédients ou métabolites sont diminuées, limitées ou perturbées, ou dont l’état de santé appelle d’autres besoins nutritionnels particuliers qui ne peuvent être satisfaits par une modification du régime alimentaire normal ou par un régime constitué d’aliments destinés à une alimentation particulière ou par une combinaison des deux.
« Ils ne peuvent être utilisés que sous contrôle médical. »
Article 25
Après l’article L. 5137-1 du même code, il est inséré deux articles L. 5137-2 et L. 5137-3 ainsi rédigés :
« Art. L. 5137-2. - Sont soumis à prescription médicale obligatoire les aliments diététiques destinés à des fins médicales spéciales qui répondent :
« 1° Aux besoins nutritionnels particuliers de personnes atteintes d’une des maladies nécessitant ce type d’apport et figurant sur une liste fixée par arrêté du ministre chargé de la santé ;
« 2° A des caractéristiques déterminées par le même arrêté.
« Ils ne peuvent être délivrés au détail que par les pharmacies à usage intérieur des établissements de santé, les officines de pharmacie, ainsi que par des personnes morales agréées par l’autorité administrative. La demande d’agrément est accompagnée d’un dossier dont le contenu est fixé par arrêté du ministre chargé de la santé. En cas de méconnaissance des dispositions législatives ou réglementaires applicables, l’agrément peut être suspendu ou retiré. La fourniture et la délivrance de ces produits doivent être conformes aux bonnes pratiques dont les principes sont définis par arrêté du ministre chargé de la santé.
« Art. L. 5137-3. - Les règles relatives à la composition et à la présentation des produits mentionnés à l’article L. 5137-1 sont fixées par décret, pris en application de l’article L. 214-1 du code de la consommation. »
Chapitre V
Insecticides et acaricides
Article 26
Au 2° de l’article L. 4211-1 du code de la santé publique, les mots : « la préparation des insecticides et acaricides destinés à être appliqués sur l’homme, » sont supprimés.
Article 27
L’article L. 5311-1 du code de la santé publique est modifié comme suit :
a) Au 1°, après les mots : « utilisées en médecine », sont insérés les mots : « les insecticides, acaricides et antiparasitaires à usage humain, » ;
b) Le 10° est abrogé.
Article 28
Sont abrogés :
1° Le 4° de l’article L. 5132-6 du code de la santé publique ;
2° Le chapitre VI du titre III du livre Ier de la cinquième partie du même code (dispositions législatives) ;
3° Le chapitre VI du titre III du livre IV de la cinquième partie du même code (dispositions législatives).
Chapitre VI
Médicaments vétérinaires
Section 1
Définitions
Article 29
L’article L. 5141-2 du code de la santé publique est modifié comme il suit :
1° Le 1° est abrogé ;
2° Au 2°, sont ajoutés les mots : « , ou tout allergène, défini comme tout produit destiné à identifier ou à provoquer une modification spécifique et acquise de la réponse immunologique à un agent allergisant ; » ;
3° Au 7°, les mots : « produits, substances ou composition appelés » sont remplacés par les mots : « substances appelées, » ;
4° L’article est complété par sept alinéas ainsi rédigés :
« 8° Médicament générique vétérinaire, tout médicament vétérinaire qui, sans préjudice des dispositions des articles L. 611-2 et suivants du code de la propriété intellectuelle, a la même composition qualitative et quantitative en principes actifs et la même forme pharmaceutique qu’un médicament vétérinaire dit de référence et dont la bioéquivalence avec le médicament de référence est démontrée par des études de biodisponibilité appropriées. Un médicament vétérinaire ne peut être qualifié de médicament de référence que si son autorisation de mise sur le marché a été délivrée au vu d’un dossier comportant, dans des conditions fixées par voie réglementaire, l’ensemble des données nécessaires et suffisantes à elles seules pour son évaluation. Pour l’application du présent alinéa, les différentes formes pharmaceutiques orales à libération immédiate sont considérées comme une même forme pharmaceutique. De même, les différents sels, esters, éthers, isomères, mélanges d’isomères, complexes ou dérivés d’un principe actif sont regardés comme ayant la même composition qualitative en principe actif, sauf s’ils présentent des propriétés sensiblement différentes au regard de la sécurité ou de l’efficacité. Dans ce cas, des informations supplémentaires fournissant la preuve de la sécurité et de l’efficacité des différents sels, esters ou dérivés d’une substance active autorisée doivent être données par le demandeur de l’autorisation de mise sur le marché ;
« 9° Médicament biologique vétérinaire, tout médicament vétérinaire dont la substance active est produite à partir d’une source biologique ou en est extraite et dont la caractérisation et la détermination de la qualité nécessitent une combinaison d’essais physiques, chimiques et biologiques ainsi que la connaissance de son procédé de fabrication et de contrôle ;
« 10° Médicament biologique vétérinaire similaire, tout médicament biologique vétérinaire qui, sans préjudice des dispositions des articles L. 611-2 et suivants du code de la propriété intellectuelle, a la même composition qualitative et quantitative en substance active et la même forme pharmaceutique qu’un médicament vétérinaire biologique de référence mais qui ne remplit pas les conditions prévues au 8° du présent article pour être regardé comme un médicament générique vétérinaire en raison de différences liées notamment à la variabilité de la matière première ou aux procédés de fabrication et nécessitant que soient produites des données précliniques et cliniques supplémentaires dans des conditions déterminées par voie réglementaire ;
« 11° Préparation extemporanée vétérinaire, tout médicament vétérinaire qui est préparé au moment de son utilisation ;
« 12° Préparation magistrale vétérinaire, toute préparation extemporanée vétérinaire réalisée selon une prescription destinée à un animal ou à des animaux d’une même exploitation ;
« 13° Préparation officinale vétérinaire, tout médicament vétérinaire préparé en pharmacie inscrit à la pharmacopée ou au formulaire national et destiné à être délivré directement à l’utilisateur final ;
« 14° Temps d’attente, la période nécessaire entre la dernière administration du médicament vétérinaire à l’animal dans les conditions normales d’emploi et l’obtention des denrées alimentaires provenant de cet animal, afin de protéger la santé publique, en garantissant que de telles denrées alimentaires ne contiennent pas de résidus en quantités supérieures aux limites maximales de résidus des substances pharmacologiquement actives, telles que fixées en vertu du règlement du Conseil (CEE) n° 2377/90 du 26 juin 1990 modifié établissant une procédure communautaire pour la fixation des limites maximales de résidus de médicaments vétérinaires dans les aliments d’origine animale. »
Article 30
Le 1° de l’article L. 5141-3 du même code est remplacé par les dispositions suivantes :
« 1° Les additifs et les prémélanges d’additifs autorisés conformément au règlement (CE) n° 1831/2003 du Parlement européen et du Conseil du 22 septembre 2003 relatif aux additifs destinés à l’alimentation des animaux. »
Section 2
Autorisations de mise sur le marché et enregistrements
Article 31
L’article L. 5141-5 du code de la santé publique est remplacé par les dispositions suivantes :
« Art. L. 5141-5. - Tout médicament vétérinaire fabriqué industriellement ou selon une méthode dans laquelle intervient un processus industriel qui ne fait pas l’objet d’une autorisation de mise sur le marché délivrée par la Communauté européenne en application du règlement (CE) n° 726/2004 du Parlement européen et du Conseil du 31 mars 2004 doit faire l’objet, avant sa mise sur le marché ou sa distribution à titre gratuit, d’une autorisation préalable de mise sur le marché délivrée par l’Agence française de sécurité sanitaire des aliments. L’autorisation peut être assortie de conditions appropriées.
« Par exception au premier alinéa, ne sont pas soumis à autorisation de mise sur le marché les aliments médicamenteux, les autovaccins à usage vétérinaire, les allergènes pour un seul animal, les médicaments vétérinaires à base d’isotopes radioactifs, les préparations magistrales et officinales vétérinaires, les médicaments vétérinaires soumis aux essais de recherche et de développement, y compris les essais cliniques.
« Une autorisation de mise sur le marché ne peut être délivrée qu’à un demandeur établi dans un Etat membre de la Communauté européenne.
« Le demandeur de l’autorisation peut être dispensé de produire certaines données et études dans des conditions fixées par voie réglementaire.
« L’autorisation de mise sur le marché est initialement délivrée pour une durée de cinq ans. Elle peut être renouvelée, le cas échéant sans limitation de durée, dans des conditions fixées par décret en Conseil d’Etat, sauf si l’agence décide, pour des raisons justifiées ayant trait à la pharmacovigilance, de procéder à un renouvellement supplémentaire, sur la base d’une réévaluation des effets thérapeutiques du médicament vétérinaire au regard des risques tels que définis au 1° de l’article L. 5141-6. Ce décret détermine également les conditions dans lesquelles elle peut devenir caduque.
« Toute modification des éléments d’une autorisation de mise sur le marché délivrée par l’Agence française de sécurité sanitaire des aliments doit être préalablement autorisée.
« L’accomplissement des formalités prévues au présent article n’a pas pour effet d’exonérer le fabricant et, s’il est distinct, le titulaire de l’autorisation de mise sur le marché, de la responsabilité que l’un ou l’autre peut encourir dans les conditions du droit commun en raison de la fabrication ou de la mise sur le marché du médicament vétérinaire.
« L’agence rend publics sans délai un rapport de synthèse de l’évaluation effectuée pour tout nouveau médicament vétérinaire dans des conditions déterminées par voie réglementaire, ainsi que les décisions d’octroi, de suspension et de retrait de l’autorisation de mise sur le marché mentionnées aux articles L. 5141-5 et L. 5141-6. Elle rend également publics sans délai l’ordre du jour et les comptes rendus, assortis des détails et explications des votes, y compris les opinions minoritaires, à l’exclusion de toute information présentant un caractère de confidentialité commerciale, des réunions des commissions siégeant auprès d’elle et consultées en matière de mise sur le marché des médicaments vétérinaires et de pharmacovigilance vétérinaire, son règlement intérieur et celui de ces commissions. »
Article 32
Après l’article L. 5141-5 du code de la santé publique, sont insérés quatre articles L. 5141-5-1 à L. 5141-5-4 ainsi rédigés :
« Art. L. 5141-5-1. - L’autorisation de mise sur le marché peut être accordée dans des conditions, définies par voie réglementaire, faisant exception aux dispositions de l’article L. 5141-5 dans les cas suivants :
« 1° Dans des circonstances exceptionnelles et sous réserve du respect d’obligations spécifiques, relatives notamment à la sécurité du médicament, à la notification à l’Agence française de sécurité sanitaire des aliments de tout incident lié à son utilisation et aux mesures à prendre dans ce cas, lorsque le demandeur démontre qu’il n’est pas en mesure de fournir des renseignements complets sur l’efficacité et la sécurité du médicament dans des conditions normales d’emploi. Le maintien de cette autorisation est décidé par l’agence sur la base d’une réévaluation annuelle de ces obligations et de leur respect par le titulaire ;
« 2° Lorsque, du fait de la rareté des indications ou du fait de l’état d’avancement de la science, la demande n’est pas assortie de l’ensemble des justifications prévues, sous réserve que le médicament soit délivré sur prescription d’un vétérinaire et administré sous sa responsabilité ;
« 3° Pour les médicaments vétérinaires qui sont destinés uniquement à être utilisés pour les poissons d’aquarium, oiseaux d’appartements, pigeons voyageurs, animaux de terrarium, petits rongeurs, furets et lapins de compagnie exclusivement, sous réserve que ces médicaments ne contiennent pas de substances mentionnées aux I et II de l’article L. 234-2 du code rural.
« Art. L. 5141-5-2. - Pour obtenir l’autorisation définie à l’article L. 5141-5, les médicaments conçus pour être administrés à des animaux dont la chair ou les produits sont destinés à la consommation humaine ne doivent contenir qu’une ou des substances pharmacologiquement actives figurant dans l’une des annexes I, II ou III du règlement du Conseil (CEE) n° 2377/90 du 26 juin 1990 modifié établissant une procédure communautaire pour la fixation des limites maximales de résidus de médicaments vétérinaires dans les aliments d’origine animale.
« Les médicaments contenant des substances pharmacologiquement actives figurant à l’annexe III du règlement précité ne sont autorisés que pour la période correspondant à la limite fixée par le règlement ; dans le cas où cette période est prolongée, l’autorisation peut être reconduite pour une durée égale.
« Par dérogation au premier alinéa, un médicament vétérinaire contenant des substances pharmacologiquement actives ne figurant pas à l’annexe I, II ou III du règlement précité peut être autorisé pour les animaux particuliers appartenant à la famille des équidés qui ont été identifiés conformément à l’article L. 212-9 du code rural et qui ont été déclarés comme n’étant pas destinés à l’abattage pour la consommation humaine. Ces médicaments vétérinaires ne contiennent pas de substances pharmacologiquement actives figurant à l’annexe IV du règlement mentionné ci-dessus et ne sont pas destinés à être utilisés pour le traitement d’affections telles que spécifiées dans le résumé autorisé des caractéristiques du produit, pour lesquelles un médicament vétérinaire est autorisé pour soigner les animaux de la famille des équidés.
« Art. L. 5141-5-3. - Pour un médicament générique vétérinaire défini au 8° de l’article L. 5141-2, l’autorisation de mise sur le marché peut être délivrée avant l’expiration des droits de propriété intellectuelle qui s’attachent au médicament de référence. Toutefois, la commercialisation de cette spécialité générique ne peut intervenir qu’après l’expiration des droits de propriété intellectuelle, sauf accord du titulaire de ces droits.
« Préalablement à cette commercialisation, le titulaire de l’autorisation de mise sur le marché du médicament générique vétérinaire informe le directeur général de l’Agence française de sécurité sanitaire des aliments des indications, formes pharmaceutiques et dosages du médicament vétérinaire de référence pour lesquels les droits de propriété intellectuelle n’ont pas expiré.
« Les dispositions du présent article s’appliquent également aux médicaments biologiques vétérinaires similaires définis au 10° de l’article L. 5141-2 et aux médicaments vétérinaires présentant des caractéristiques communes avec un médicament vétérinaire de référence mais ne répondant pas à la définition du médicament générique en raison de différences portant sur un ou plusieurs éléments de cette définition et nécessitant que soient produites des données supplémentaires dans des conditions fixées par voie réglementaire.
« Art. L. 5141-5-4. - Un médicament générique vétérinaire autorisé en application de l’article L. 5141-5-3 ne peut être commercialisé qu’à l’expiration d’une période de dix ans suivant l’autorisation initiale du médicament vétérinaire de référence.
« Toutefois, la période de dix ans prévue au premier alinéa est portée à treize ans pour les médicaments vétérinaires destinés aux poissons et aux abeilles, ou à d’autres espèces considérées comme mineures et figurant sur un arrêté du ministre chargé de l’agriculture.
« Pour les médicaments vétérinaires destinés à une ou plusieurs espèces productrices de denrées alimentaires, et contenant une nouvelle substance active qui, au 30 avril 2004, n’a pas encore été autorisée dans la Communauté européenne, la période de dix ans prévue au premier alinéa est prolongée d’un an pour chaque extension de l’autorisation à une autre espèce animale productrice de denrées alimentaires, si elle est autorisée dans les cinq ans qui suivent la délivrance de l’autorisation de mise sur le marché initiale.
« Cette période ne peut toutefois dépasser treize ans au total, pour une autorisation de mise sur le marché concernant quatre espèces productrices de denrées alimentaires ou plus.
« L’extension de la période de dix ans à onze, douze ou treize ans pour un médicament vétérinaire destiné à une espèce productrice de denrées alimentaires n’est octroyée qu’à la condition que le titulaire de l’autorisation de mise sur le marché ait également été à l’origine de la fixation de limites maximales de résidus pour les espèces couvertes par l’autorisation.
« Les dispositions du présent article sont également applicables aux médicaments biologiques vétérinaires similaires définis au 10° de l’article L. 5141-2 ou aux médicaments présentant des caractéristiques communes par rapport à un médicament de référence mais ne répondant pas à la définition du médicament générique vétérinaire en raison de différences portant sur un ou plusieurs éléments de cette définition nécessitant que soient produites des données supplémentaires dans des conditions déterminées par voie réglementaire. »
Article 33
L’article L. 5141-6 du même code est remplacé par les dispositions suivantes :
« Art. L. 5141-6. - L’autorisation de mise sur le marché d’un médicament vétérinaire est refusée s’il apparaît que sa mise sur le marché est de nature à compromettre gravement la protection de la santé humaine ou de la santé animale.
« L’autorisation est également refusée si, après vérification de la conformité des renseignements et documents qui doivent être présentés à l’appui de la demande, il apparaît :
« 1° Soit que l’évaluation des effets thérapeutiques positifs du médicament au regard des risques pour la santé humaine ou animale ou la protection de l’environnement liés à sa qualité, à sa sécurité ou à son efficacité n’est pas considérée comme favorable dans les conditions d’emploi indiquées dans la demande d’autorisation de mise sur le marché ou, lorsque la demande concerne des médicaments vétérinaires à usage zootechnique, les bénéfices en matière de santé et de bien-être des animaux ainsi que la sécurité du consommateur sont insuffisamment pris en compte ;
« 2° Soit que le médicament vétérinaire n’a pas la composition qualitative ou quantitative déclarée ou que l’effet thérapeutique annoncé fait défaut sur l’espèce animale de destination ou est insuffisamment démontré par le demandeur ;
« 3° Soit que le médicament vétérinaire est présenté pour une utilisation interdite ;
« 4° Soit, pour les médicaments destinés à être administrés à des animaux dont la chair ou les produits sont destinés à la consommation humaine :
« a) Que le temps d’attente indiqué dans le dossier est insuffisant pour que les denrées alimentaires provenant de l’animal traité ne contiennent pas de résidus à des niveaux supérieurs aux limites maximales de résidus fixées par le règlement (CEE) n° 2377/90 du Conseil du 26 juin 1990 modifié établissant une procédure communautaire pour la fixation des limites maximales de résidus de médicaments vétérinaires dans les aliments d’origine animale ou qu’il est insuffisamment justifié ;
« b) Ou que la ou les substances à action pharmacologique présentes dans le médicament ne figurent pas dans l’une des annexes I, II et III du même règlement ;
« 5° Soit que l’étiquetage ou la notice proposé par le demandeur n’est pas conforme aux dispositions du présent code ;
« 6° Soit que l’administration d’un médicament immunologique vétérinaire interfère avec un programme national pour le diagnostic, le contrôle ou l’éradication d’une maladie des animaux ou entraînerait des difficultés à certifier l’absence de contamination des animaux vivants ou des aliments ou d’autres produits obtenus à partir des animaux traités ou que la maladie contre laquelle le médicament est supposé conférer une immunité est très peu répandue sur le territoire national.
« L’autorisation de mise sur le marché peut être modifiée, suspendue ou supprimée pour les mêmes motifs. Elle peut l’être aussi lorsqu’il apparaît :
« 1° Soit que les renseignements figurant dans le dossier d’autorisation de mise sur le marché, la notice ou l’étiquetage sont erronés ;
« 2° Soit que les contrôles pour s’assurer de la conformité de la composition qualitative et quantitative du médicament vétérinaire n’ont pas été effectués ;
« 3° Soit que les renseignements figurant dans le dossier de demande d’autorisation de mise sur le marché n’ont pas été modifiés ;
« 4° Soit que tout élément nouveau concernant ces mêmes parties du dossier n’a pas été transmis aux autorités compétentes. »
Article 34
L’article L. 5141-9 du même code est remplacé par les dispositions suivantes :
« Art. L. 5141-9. - Par exception aux dispositions de l’article L. 5141-5, ne sont pas soumis à l’autorisation de mise sur le marché prévue à cet article mais à un enregistrement auprès de l’Agence française de sécurité sanitaire des aliments les médicaments homéopathiques vétérinaires autres qu’immunologiques satisfaisant à l’ensemble des critères suivants :
« 1° Une voie d’administration décrite par la pharmacopée européenne ou la pharmacopée française ou, à défaut, par les pharmacopées utilisées de façon officielle dans les autres Etats membres de la Communauté européenne ;
« 2° L’absence d’indication thérapeutique particulière sur l’étiquetage ou dans toute information relative au médicament ;
« 3° Un degré de dilution garantissant l’innocuité du médicament, notamment tel qu’il ne contienne pas plus d’une partie par 10 000 de la teinture mère.
« Il est procédé à cet enregistrement si les critères mentionnés ci-dessus sont remplis et, pour les médicaments homéopathiques conçus pour les animaux dont la chair ou les produits sont destinés à la consommation humaine, s’ils ne contiennent que des souches homéopathiques figurant dans l’une des annexes I, II ou III du règlement du Conseil (CEE) 2377/90 du 26 juin 1990 établissant une procédure communautaire pour la fixation des limites maximales de résidus de médicaments vétérinaires dans les aliments d’origine animale. Il peut être refusé en cas de danger pour la santé humaine ou la santé animale.
« L’enregistrement peut s’appliquer à une série de médicaments homéopathiques vétérinaires obtenus à partir de la ou des mêmes souches homéopathiques.
« L’agence enregistre le médicament pour une durée de cinq ans. L’enregistrement peut ensuite être renouvelé, le cas échéant sans limitation de durée, dans des conditions fixées par décret en Conseil d’Etat. Ce décret détermine également les conditions dans lesquelles l’enregistrement devient caduc.
« L’enregistrement peut être modifié, suspendu ou supprimé si les critères et les conditions prévus au présent article ne sont plus remplis ou en cas de danger pour la santé humaine ou la santé animale. »
Article 35
Après l’article L. 5141-10 du code de la santé publique, il est inséré un article L. 5141-10-1 rédigé comme suit :
« Art. L. 5141-10-1. - Par dérogation à l’article L. 5141-5, dans le cas où un animal fait l’objet d’importation ou d’exportation, depuis ou vers un Etat non membre de la Communauté européenne et est soumis à des dispositions sanitaires spécifiques obligatoires, l’Agence française de sécurité sanitaire des aliments peut autoriser un vétérinaire à administrer à cet animal un médicament immunologique vétérinaire ne disposant pas d’une autorisation de mise sur le marché en France mais autorisé en vertu de la législation de l’autre Etat. »
Article 36
À l’article L. 5141-12 du code de la santé publique, après les mots : « personne qualifiée » sont insérés les mots : « ou une entreprise ou un organisme employant une personne qualifiée ».
Article 37
Après l’article L. 5141-12 du code de la santé publique, il est inséré un article L. 5141-12-1 ainsi rédigé :
« Art. L. 5141-12-1. - La préparation des allergènes pour un seul animal doit être effectuée par une personne qualifiée ou une entreprise ou un organisme employant une personne qualifiée ayant obtenu à cet effet une autorisation délivrée par le directeur général de l’Agence française de sécurité sanitaire des aliments. »
Article 38
L’article L. 5141-15 du code de la santé publique est modifié comme suit :
1° Après les mots : « ayant obtenu une autorisation de mise sur le marché dans cet Etat membre, » sont insérés les mots : « lorsque ces médicaments ne sont pas autorisés en France, » ;
2° Il est ajouté un alinéa ainsi rédigé :
« Ces vétérinaires sont soumis aux règles relatives à la prescription et à la délivrance prévues à l’article L. 5143-5. »
Section 3
Mesures d’application
Article 39
L’article L. 5141-16 du code de la santé publique est modifié comme suit :
1° Le 1° est remplacé par les dispositions suivantes :
« 1° Les règles relatives à l’étiquetage, la notice et la dénomination des médicaments vétérinaires mentionnés aux articles L. 5141-1 et L. 5141-2 ; » ;
2° Il est rétabli après le 1° un 2° ainsi rédigé :
« 2° Les conditions dans lesquelles des autorisations de mise sur le marché peuvent être considérées comme faisant partie d’une autorisation de mise sur le marché globale ; » ;
3° Le 3° est remplacé par les dispositions suivantes :
« 3° Les modalités de présentation des demandes tendant à obtenir l’autorisation de mise sur le marché prévue à l’article L. 5141-5, le contenu du dossier présenté à l’appui de ces demandes, ainsi qu’après la délivrance de l’autorisation les modalités de son actualisation, les conditions dans lesquelles le demandeur peut être dispensé de produire certains éléments du dossier et celles dans lesquelles interviennent les décisions accordant, modifiant, soumettant à des obligations spécifiques, renouvelant, suspendant ou supprimant ces autorisations ; ».
4° Le 5° est remplacé par les dispositions suivantes :
« 5° Les modalités de présentation des demandes tendant à obtenir l’enregistrement d’un médicament vétérinaire homéopathique prévu à l’article L. 5141-9, le contenu du dossier présenté à l’appui de ces demandes, y compris les documents permettant de démontrer la qualité des lots de fabrication de ces médicaments homéopathiques vétérinaires, ainsi que les conditions dans lesquelles interviennent les décisions accordant, modifiant, renouvelant, suspendant, ou supprimant cet enregistrement ; ».
5° Les 7° à 14° sont remplacés par les dispositions suivantes :
« 7° Les modalités de présentation des demandes tendant à obtenir les autorisations temporaires d’utilisation d’un médicament vétérinaire prévues à l’article L. 5141-10, le contenu du dossier présenté à l’appui de ces demandes, ainsi que les conditions dans lesquelles interviennent les décisions accordant, modifiant, suspendant ou supprimant ces autorisations ;
« 8° Les règles applicables en cas de changement de titulaire de l’autorisation de mise sur le marché d’un médicament vétérinaire ou de l’enregistrement d’un médicament homéopathique vétérinaire ;
« 9° Les conditions auxquelles est subordonnée la publicité pour les médicaments vétérinaires ;
« 10° Les règles applicables à la pharmacovigilance des médicaments vétérinaires et des médicaments à usage humain utilisés au titre du 3° de l’article L. 5143-4 ;
« 11° Les modalités de présentation des demandes tendant à obtenir les autorisations de préparer les autovaccins prévus à l’article L. 5141-12 ou de préparer les allergènes pour un seul animal prévues à l’article L. 5141-12-1, le contenu du dossier présenté à l’appui de ces demandes, ainsi que les conditions dans lesquelles interviennent les décisions accordant, modifiant, renouvelant, suspendant ou supprimant ces autorisations ;
« 12° Les modalités d’application du présent titre aux départements d’outre-mer ;
« 13° Les conditions dans lesquelles les vétérinaires mentionnés à l’article L. 5141-15 peuvent utiliser les médicaments vétérinaires mentionnés au même article ;
« 14° Les règles de procédure applicables aux recours ouverts contre les décisions visées aux 3°, 5°, 7° et 11° du présent article ; ».
6° Au dernier alinéa, les mots : « A l’exception du cas visé au 11° » sont remplacés par les termes : « Sauf dans le cas mentionné au 12° ».
Section 4
Préparation industrielle et vente en gros
Article 40
Le dernier alinéa de l’article L. 5142-2 du code de la santé publique est remplacé par un alinéa ainsi rédigé :
« Toute modification substantielle des éléments de l’autorisation initiale est subordonnée à une autorisation préalable. Un décret en Conseil d’Etat fixe les cas de modification substantielle de l’autorisation initiale. Les autres modifications font l’objet d’une déclaration. »
Article 41
L’article L. 5142-3 du même code est ainsi rédigé :
« Art. L. 5142-3. - Les activités exercées dans les établissements mentionnés à l’article L. 5142-1 doivent l’être en conformité avec les bonnes pratiques dont les principes sont définis par décision de l’Agence française de sécurité sanitaire des aliments, notamment :
« 1° La fabrication, l’importation et la distribution en gros de médicaments vétérinaires ;
« 2° La fabrication, l’importation, la distribution en gros et la distribution au détail des aliments médicamenteux ;
« 3° La pharmacovigilance vétérinaire. »
Article 42
Après l’article L. 5142-3 du même code, il est créé un article L. 5142-3-1 ainsi rédigé :
« Art. L. 5142-3-1. - Lorsqu’un médicament vétérinaire soumis aux dispositions du chapitre Ier du présent titre est mis sur le marché, l’entreprise qui l’exploite communique, sans délai, à l’Agence française de sécurité sanitaire des aliments, les dates de commercialisation de chaque présentation de ce médicament.
« Elle informe l’agence, au préalable ou en cas d’urgence de manière concomitante, de toute action qu’elle engage pour en suspendre la commercialisation, le retirer du marché ou en retirer un lot déterminé ainsi que de tout risque de rupture de stock sur un médicament vétérinaire sans alternative thérapeutique disponible ou en raison d’un accroissement significatif et imprévisible de la demande. Elle en indique la raison si celle-ci concerne l’efficacité du médicament vétérinaire ou la protection de la santé humaine ou animale ou de l’environnement. »
Article 43
Après l’article L. 5142-5 du code de la santé publique, il est inséré un article L. 5142-5-1 ainsi rédigé :
« Art. L. 5142-5-1. - L’Agence française de sécurité sanitaire des aliments peut interdire la fabrication, l’importation, la détention et la vente de médicaments immunologiques vétérinaires sur tout ou partie du territoire national s’il est établi :
« 1° Soit que l’administration d’un médicament immunologique vétérinaire interfère avec un programme national pour le diagnostic, le contrôle ou l’éradication d’une maladie des animaux ou entraînerait des difficultés à certifier l’absence de contamination des animaux vivants ou des aliments ou d’autres produits obtenus à partir des animaux traités ;
« 2° Soit que la maladie contre laquelle le médicament est supposé conférer une immunité est très peu répandue sur le territoire national. »
Section 5
Préparation extemporanée et distribution au détail
Article 44
L’article L. 5143-1 du code de la santé publique est remplacé par les dispositions suivantes :
« Art. L. 5143-1. - La préparation extemporanée des médicaments vétérinaires par les personnes mentionnées à l’article L. 5143-2 et, pour les aliments médicamenteux, par les personnes intervenant dans les conditions prévues à l’article L. 5143-3 est réalisée en conformité avec des bonnes pratiques de préparation dont les principes sont fixés par décision de l’Agence française de sécurité sanitaire des aliments. »
Article 45
L’article L. 5143-4 du code de la santé publique est modifié comme suit :
I.- Le 3° est remplacé par les dispositions suivantes :
« 3° Si les médicaments mentionnés aux 1° et 2° n’existent pas :
« a) Soit un médicament autorisé pour l’usage humain ;
« b) Soit un médicament vétérinaire autorisé dans un autre Etat membre en vertu de la directive 2001/82/CE du Parlement européen et du Conseil instituant un code communautaire relatif aux médicaments vétérinaires, pour la même espèce ou pour une autre espèce, pour l’affection concernée ou pour une affection différente, sans préjudice de l’autorisation mentionnée à l’article L. 5142-7 ; ».
II.- La dernière phrase du dernier alinéa est remplacée par les dispositions suivantes : « Si le médicament utilisé n’indique aucun temps d’attente pour les espèces concernées, le vétérinaire fixe le temps d’attente applicable qui ne peut être inférieur au minimum fixé pour la denrée animale considérée, par arrêté des ministres chargés de l’agriculture et de la santé, après avis de l’Agence française de sécurité sanitaire des aliments. »
III.- Il est ajouté quatre alinéas ainsi rédigés :
« Le précédent alinéa ne s’applique pas aux équidés identifiés conformément à l’article L. 212-9 du code rural et déclarés comme n’étant pas destinés à l’abattage pour la consommation humaine. En outre, par exception au même alinéa, le vétérinaire peut prescrire et administrer à un équidé identifié conformément à l’article L. 212-9 du code rural et déclaré comme étant destiné à l’abattage pour la consommation humaine un médicament contenant des substances à action pharmacologique ne figurant dans aucune des annexes I, II, III ou IV du règlement CEE n° 2377/90 du Conseil si les conditions suivantes sont respectées :
« a) Les substances à action pharmacologique qu’il contient sont inscrites sur la liste fixée par le règlement (CE) n° 1950/2006 de la Commission du 13 décembre 2006 établissant, conformément à la directive 2001/82/CE du Parlement européen et du Conseil instituant un code communautaire relatif aux médicaments vétérinaires, une liste de substances essentielles pour le traitement des équidés ;
« b) Le vétérinaire prescrit et administre les médicaments contenant ces substances pour les indications prévues par ce règlement et consigne ce traitement dans le document d’identification obligatoire ;
« c) Le vétérinaire fixe un temps d’attente qui ne peut être inférieur à une durée fixée par arrêté des ministres chargés de l’agriculture et de la santé, après avis de l’Agence française de sécurité sanitaire des aliments. »
Article 46
Le premier alinéa de l’article L. 5143-5 du code de la santé publique est remplacé par les dispositions suivantes :
« Est subordonnée à la rédaction par un vétérinaire d’une ordonnance, qui est obligatoirement remise à l’utilisateur, la délivrance au détail, à titre gratuit ou onéreux, des médicaments suivants :
« 1° Les médicaments vétérinaires contenant des substances prévues à l’article L. 5144-1, à l’exception des substances vénéneuses à doses ou concentrations trop faibles pour justifier de la soumission au régime de ces substances ;
« 2° Les aliments médicamenteux ;
« 3° Les médicaments visés à l’article L. 5143-4 ;
« 4° Les nouveaux médicaments vétérinaires contenant une substance active dont l’usage vétérinaire est autorisé depuis moins de cinq ans.
« Cette ordonnance ne peut prescrire que la quantité de médicaments nécessaire au traitement. »
Section 6
Substances pouvant entrer dans la fabrication
des médicaments vétérinaires
Article 47
L’article L. 5144-1 du code de la santé publique est modifié ainsi qu’il suit :
1° Le c est remplacé par les dispositions suivantes :
« c) Substances à activité anabolisante, anticatabolisante ou bêta-agoniste ; » ;
2° Le e est remplacé par les dispositions suivantes :
« e) Substances pharmacologiquement actives susceptibles de demeurer à l’état de résidus toxiques ou dangereux dans les denrées alimentaires d’origine animale et figurant dans l’une des annexes I ou III du règlement du Conseil (CEE) n° 2377/90 du 26 juin 1990 établissant une procédure communautaire pour la fixation des limites maximales de résidus de médicaments vétérinaires dans les aliments d’origine animale. »
Section 7
Dispositions transitoires
Article 48
I. - Les durées, déterminées par voie réglementaire, qui servent de référence pour la mise en oeuvre du quatrième alinéa de l’article L. 5141-5 du code de la santé publique dans sa rédaction issue de l’article 31 de la présente ordonnance sont applicables dès lors que la demande d’autorisation de mise sur le marché du médicament vétérinaire de référence a été déposée à compter du 30 octobre 2005.
II.- Les dispositions de l’article L. 5141-5-4 du code de la santé publique dans sa rédaction issue de l’article 32 de la présente ordonnance ne sont applicables que lorsque l’autorisation initiale de mise sur le marché du médicament vétérinaire de référence a été délivrée au vu d’une demande déposée à compter du 30 octobre 2005.
Chapitre VII
Classification des substances
et préparations chimiques dangereuses
Article 49
L’article L. 5132-2 du code de la santé publique est remplacé par les dispositions suivantes :
« Art. L. 5132-2. - Les substances et préparations dangereuses mentionnées au 1° de l’article L. 5132-1 sont classées dans les catégories suivantes :
« 1° Très toxiques : substances et préparations qui, par inhalation, ingestion ou pénétration cutanée en très petites quantités, entraînent la mort ou nuisent à la santé de manière aiguë ou chronique ;
« 2° Toxiques : substances et préparations qui, par inhalation, ingestion ou pénétration cutanée en petites quantités, entraînent la mort ou nuisent à la santé de manière aiguë ou chronique ;
« 3° Nocives : substances et préparations qui, par inhalation, ingestion ou pénétration cutanée, peuvent entraîner la mort ou nuire à la santé de manière aiguë ou chronique ;
« 4° Corrosives : substances et préparations qui, en contact avec des tissus vivants, peuvent exercer une action destructrice sur ces derniers ;
« 5° Irritantes : substances et préparations non corrosives qui, par contact immédiat, prolongé ou répété avec la peau ou les muqueuses, peuvent provoquer une réaction inflammatoire ;
« 6° Sensibilisantes : substances et préparations qui, par inhalation ou pénétration cutanée, peuvent donner lieu à une réaction d’hypersensibilisation telle qu’une exposition ultérieure à la substance ou à la préparation produit des effets néfastes caractéristiques ;
« 7° Cancérogènes : substances et préparations qui, par inhalation, ingestion ou pénétration cutanée, peuvent provoquer un cancer ou en augmenter la fréquence :
« - cancérogènes de catégorie 1 : substances et préparations que l’on sait être cancérogènes pour l’homme ;
« - cancérogènes de catégorie 2 : substances et préparations pour lesquelles il existe une forte présomption que l’exposition de l’homme à de telles substances et préparations peut provoquer un cancer ou en augmenter la fréquence ;
« - cancérogènes de catégorie 3 : substances et préparations préoccupantes pour l’homme en raison d’effets cancérogènes possibles mais pour lesquelles les informations disponibles sont insuffisantes pour classer ces substances et préparations dans la catégorie 2 ;
« 8° Mutagènes : substances et préparations qui, par inhalation, ingestion ou pénétration cutanée, peuvent produire des défauts génétiques héréditaires ou en augmenter la fréquence :
« - mutagènes de catégorie 1 : substances et préparations que l’on sait être mutagènes pour l’homme ;
« - mutagènes de catégorie 2 : substances et préparations pour lesquelles il existe une forte présomption que l’exposition de l’homme à de telles substances et préparations peut produire des défauts génétiques héréditaires ou en augmenter la fréquence ;
« - mutagènes de catégorie 3 : substances et préparations préoccupantes pour l’homme en raison d’effets mutagènes possibles mais pour lesquelles les informations disponibles sont insuffisantes pour classer ces substances et préparations dans la catégorie 2 ;
« 9° Toxiques pour la reproduction : substances et préparations qui, par inhalation, ingestion ou pénétration cutanée, peuvent produire ou augmenter la fréquence d’effets nocifs non héréditaires dans la progéniture ou porter atteinte aux fonctions ou capacités reproductives :
« - toxiques pour la reproduction de catégorie 1 : substances et préparations que l’on sait être toxiques pour la reproduction de l’homme ;
« - toxiques pour la reproduction de catégorie 2 : substances et préparations pour lesquelles il existe une forte présomption que l’exposition de l’homme à de telles substances et préparations peut produire ou augmenter la fréquence d’effets nocifs non héréditaires dans la progéniture ou porter atteinte aux fonctions ou capacités reproductives ;
« - toxiques pour la reproduction de catégorie 3 : substances et préparations préoccupantes en raison d’effets toxiques possibles pour la reproduction mais pour lesquelles les informations disponibles sont insuffisantes pour classer ces substances et préparations dans la catégorie 2.
« Un décret en Conseil d’Etat fixe les conditions dans lesquelles la mise sur le marché, la publicité et l’emploi des substances ou préparations mentionnées au premier alinéa du présent article peuvent, pour des raisons de santé publique, faire l’objet de mesures d’interdiction, de restriction ou de prescriptions particulières proportionnées à la nature du danger ou du risque qu’elles comportent pour la santé humaine. »
Chapitre VIII
Autres dispositions
Article 50
À l’article L. 5124-13 du code de la santé publique, le deuxième alinéa est remplacé par les dispositions suivantes :
« L’autorisation de mise sur le marché prévue à l’article L. 5121-8, les enregistrements prévus aux articles L. 5121-13 et L. 5121-14-1, l’autorisation temporaire d’utilisation prévue à l’article L. 5121-12 ou l’autorisation prévue au 12° et au 13° de l’article L. 5121-1 valent autorisation au sens de l’alinéa précédent. L’autorisation prévue à l’article L. 1123-8 vaut autorisation d’importation pour tout médicament nécessaire à la réalisation de la recherche biomédicale autorisée. »
Article 51
L’article L. 5414-1 du même code est complété par un alinéa ainsi rédigé :
« Ces agents peuvent communiquer à l’Agence française de sécurité sanitaire des produits de santé les informations et documents recueillis dans les conditions prévues à l’alinéa précédent afin qu’elle procède à toute évaluation et expertise pour les produits mentionnés au même alinéa. »
Article 52
À l’article L. 5122-3 du même code, les mots : « l’enregistrement mentionné à l’article L. 5121-13 » sont remplacés par les mots : « l’autorisation mentionnée à l’article L. 5121-9-1 ou un des enregistrements mentionnés aux articles L. 5121-13 et L. 5121-14-1 ou qui sont importés selon la procédure mentionnée à l’article L. 5124-17-1 ».
Article 53
Le Premier ministre, le ministre de l’économie, des finances et de l’industrie, le garde des sceaux, ministre de la justice, le ministre de l’agriculture et de la pêche et le ministre de la santé et des solidarités sont responsables, chacun en ce qui le concerne, de l’application de la présente ordonnance, qui sera publiée au Journal officiel de la République française.