

______
ASSEMBLÉE NATIONALE
CONSTITUTION DU 4 OCTOBRE 1958
DOUZIÈME LÉGISLATURE
Enregistré à la Présidence de l'Assemblée nationale le 28 juin 2006.
RAPPORT
FAIT
AU NOM DE LA COMMISSION DES AFFAIRES CULTURELLES, FAMILIALES ET SOCIALES SUR LE PROJET DE LOI (n° 3062) portant diverses dispositions d'adaptation au droit communautaire dans le domaine du médicament,
PAR Mme Cécile GALLEZ,
Députée.
--
INTRODUCTION 9
I.- LA RÉVISION DE LA LÉGISLATION PHARMACEUTIQUE COMMUNAUTAIRE DOIT ÊTRE TRANSPOSÉE TRÈS RAPIDEMENT 11
A. L'UNION EUROPÉENNE EST COMPÉTENTE EN MATIÈRE DE LÉGISLATION RELATIVE À LA FABRICATION ET À LA MISE SUR LE MARCHÉ DES MÉDICAMENTS 11
1. La compétence communautaire relative au médicament est bien établie 11
a) La compétence communautaire est fixée par le traité constitutif 11
b) Le champ de la politique européenne en matière de médicament est devenu de plus en plus large 12
2. Le « paquet » communautaire a connu un parcours heurté 12
a) En 2004, deux directives et un règlement réforment profondément le droit communautaire 12
b) L'historique de l'élaboration de la directive est riche d'enseignements 12
B. LA NÉCESSITÉ DE MODIFIER LE DROIT EUROPÉEN EN MATIÈRE DE MÉDICAMENT ÉTAIT ÉTABLIE 13
1. La modification du code communautaire est progressivement devenue une nécessité 13
a) La législation communautaire exigeait une modernisation d'ensemble 13
b) La nécessité de revoir les dispositions du code communautaire relatif aux médicaments à usage humain est devenue évidente 14
2. Les procédures d'autorisation et de suivi sanitaire des médicaments ont connu des failles troublantes 14
a) La « crise de confiance » du médicament est d'ampleur mondiale 15
b) Des particularités françaises soulignent la nécessité de profonds changements 15
C. EXIGENCE CONSTITUTIONNELLE, LA TRANSPOSITION RAPIDE DU « PAQUET MÉDICAMENT » EST AUSSI UN IMPÉRATIF POLITIQUE 16
1. La France ne doit plus être la dernière de la classe en matière de transposition 16
2. Le gouvernement a déjà entamé le processus de transposition de la directive n° 2004/27 du 31 mars 2004 18
3. Un recours en manquement pourrait être engagé contre la France 18
II.- LE PROJET DE LOI COMPORTE PLUSIEURS AVANCÉES MAJEURES DANS LE DOMAINE DU MÉDICAMENT 21
A. UN ÉQUILIBRE EST AMÉNAGÉ ENTRE LA PROTECTION DE L'INNOVATION ET LA NÉCESSITÉ DE PROMOUVOIR DAVANTAGE LES GÉNÉRIQUES 22
1. En progression depuis plusieurs années, le développement des génériques doit être soutenu 22
2. Les procédures d'autorisation de mise sur le marché des génériques et des « biosimilaires » sont simplifiées 24
a) Les conditions d'autorisation des génériques sont assouplies 24
b) Les notions de médicaments biologiques et « biosimilaires » sont introduites 25
3. Plusieurs mesures visent à renforcer parallèlement la protection des médicaments innovants 26
B. LE PROJET DE LOI PERMET D'ACCROÎTRE LA DISPONIBILITÉ ET LA SÉCURITÉ SANITAIRE DES MÉDICAMENTS 26
1. L'accès aux médicaments est facilité dans certaines circonstances 26
2. La procédure de mise sur le marché est réformée afin de renforcer la sécurité sanitaire des médicaments 27
a) Le processus d'autorisation est recentré sur l'évaluation de la balance bénéfice-risque 27
b) Le renforcement de la pharmacovigilance et du suivi des médicaments était une nécessité 29
c) Le renforcement de la réglementation concerne aussi les matières premières à usage pharmaceutique 32
C. LA TRANSPARENCE DES TRAVAUX DE L'AFSSAPS SERA RENFORCÉE, ET L'INFORMATION DES PATIENTS AMÉLIORÉE 33
1. La publicité des dossiers d'autorisation de médicaments est de nature à renforcer la légitimité et la crédibilité des décisions de l'AFSSAPS 34
a) Le présent projet transpose des dispositions de la directive prescrivant une transparence accrue du fonctionnement des agences chargées du médicament 34
b) Le présent projet réaffirme le principe de la publicité des dossiers d'autorisation 34
2. L'étiquetage et la notice des médicaments seront plus informatifs pour le patient 35
D. LA TRANSPOSITION DE LA DIRECTIVE CONSTITUE UN APPORT INDISPENSABLE À LA LIMITATION DE L'INFLUENCE DE L'INDUSTRIE PHARMACEUTIQUE 35
1. La publicité pour les médicaments continue à faire l'objet d'une grande vigilance 36
a) La publicité pour le médicament prend principalement trois formes 36
b) L'introduction éventuelle de la publicité auprès du grand public pour les médicaments sur ordonnance constitue un enjeu majeur pour l'Europe 37
c) Les programmes d'accompagnement des patients doivent faire l'objet d'une surveillance particulière 38
2. Les relations entre les firmes pharmaceutiques et les professions médicales et pharmaceutiques sont mieux réglementées 39
a) Le dispositif « anti-cadeaux » est renforcé 39
b) Le principe de la déclaration d'intérêts est étendu à tous les personnels travaillant pour l'AFSSAPS 40
TRAVAUX DE LA COMMISSION 41
I.- DISCUSSION GÉNÉRALE 41
II.- EXAMEN DES ARTICLES 43
Chapitre Ier Dispositions relatives aux médicaments 43
Article 1er : Conditions d'exonération de la responsabilité des professionnels de santé, des fabricants et des titulaires de l'autorisation d'utilisation ou de mise sur le marché d'un médicament dans le cas d'une menace sanitaire grave 43
Article 2 : Renforcement la réglementation des relations entre les professionnels de santé et les entreprises pharmaceutiques 47
Article 3 : Actualisation de la définition du médicament et principe de l'application de la réglementation pharmaceutique aux produits dits « frontière » 51
Article 4 : Définition des spécialités et groupes génériques et des médicaments homéopathiques, biologiques et biologiques similaires 54
Article 5 : Modification du régime d'autorisation de mise sur le marché (AMM) des médicaments 59
Après l'article 5 66
Article 6 : Critères de refus de l'autorisation de mise sur le marché (AMM) d'un médicament et conditions de délivrance de l'AMM dans des circonstances exceptionnelles 66
Article 7 : Possibilité donnée à l'AFSSAPS de permettre la mise sur le marché d'un médicament autorisé uniquement dans un autre état membre 70
Article 8 : Obligation pour le titulaire de l'AMM d'un générique d'informer l'AFSSAPS sur les indications, formes et dosages de la spécialité de référence encore protégés par le droit des brevets 72
Article 9 : Conditions de commercialisation des médicaments génériques, biologiques similaires et quasi-génériques 75
Article 10 : Exclusion des études et essais requis en vue de l'obtention d'une autorisation de mise sur le marché (AMM) du champ de protection des brevets 78
Article 11 : Application aux médicaments biologiques similaires et « quasi-génériques » d'un régime juridique proche de celui des médicaments génériques 81
Article 12 : Modification du régime des autorisations temporaires d'utilisation (ATU) 83
Article 13 : Clarification de l'assiette du droit progressif perçu par l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) pour l'enregistrement des médicaments homéopathiques 86
Article 14 : Clarification de l'assiette du droit progressif versé à l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) pour l'autorisation de mise sur le marché (AMM) des médicaments 88
Article 15 : Décrets en Conseil d'État 90
Article additionnel après l'article 15 : Critères de certification des logiciels d'aide à la prescription 92
Article 16 : Renvoi à la compétence règlementaire pour la définition des mentions obligatoires devant figurer sur les publicités relatives au médicament 93
Article 17 : Publicité auprès du public pour les médicaments à usage humain 93
Article 18 : Remise gratuite d'échantillons de médicaments et avantages consentis aux professionnels de santé 98
Article 19 : Remise gratuite d'échantillons de médicaments - Mentions obligatoires devant figurer dans les publicités pour les médicaments - Autorisation des publicités de rappel 101
Article 20 : Transmission à l'AFSSAPS de la date de commercialisation du médicament 103
Article 21 : Informations à fournir à l'AFSSAPS en cas de suspension de commercialisation d'un médicament ou de risque de rupture de stock 105
Article 22 : Autorisation d'importation par un particulier de médicament à usage humain 106
Article 23 : Régime juridique des matières premières à usage pharmaceutique 109
Article 24 : Bonnes pratiques de fabrication et de distribution des matières premières à usage pharmaceutique 113
Article 25 : Pouvoirs d'inspection de l'AFSSAPS en ce qui concerne les matières premières à usage pharmaceutique 115
Article 26 : Publicité de la synthèse des dossiers d'autorisation d'un nouveau médicament 117
Après l'article 26 122
Article 27 : Transmission d'échantillons à titre gratuit à l'AFSSAPS 122
Article 28 : Publicité et annualité des déclarations d'intérêts 124
Chapitre II : Habilitation à prendre des ordonnaces 130
Article 29 : Habilitation du gouvernement à prendre par ordonnances des dispositions dans le domaine du médicament 130
Article 30 : Mayotte, Saint-Pierre-et-Miquelon, Terres australes et antarctiques françaises, Wallis, Futuna, Nouvelle-Calédonie et Polynésie française 136
TABLEAU COMPARATIF 139
AMENDEMENTS NON ADOPTÉS PAR LA COMMISSION 163
ANNEXES 169
ANNEXE 1 : LISTE DES PERSONNES AUDITIONNÉES 169
ANNEXE 2 : SCHÉMA DE LA PROCÉDURE DE CODÉCISION SUIVIE POUR LA RÉVISION DE LA DIRECTIVE N° 2001/83/CE INSTAURANT UN CODE COMMUNAUTAIRE RELATIF AUX MÉDICAMENTS À USAGE HUMAIN 171
Dans le domaine du médicament, dont l'étymologie devrait pourtant inciter à la plus grande prudence, puisqu'en grec le mot pharmakon désigne tout à la fois un remède et un poison, il est en France un paradoxe saisissant.
Alors que plusieurs médicaments ont été mis en cause au cours des dernières années - notamment des molécules anticholestérol, telles que la cérivastatine, ou des anti-inflammatoires prescrits contre l'arthrose, comme le Celebrex ou encore le Vioxx brutalement retiré du marché en 2004 - suscitant de profondes inquiétudes, voire une crise de confiance de l'opinion à l'égard des médicaments, leur consommation n'en continue pas moins de croître et surtout de se banaliser.
C'est tout particulièrement le cas des médicaments psychotropes, comme l'a mis en évidence le très récent rapport de Mme Maryvonne Briot, au nom de l'Office parlementaire d'évaluation des politiques de santé (OPEPS) de l'Assemblée nationale.
En la matière, on peut bien sûr continuer de rêver au « risque zéro », mais il apparaît infiniment plus constructif et surtout protecteur de se poser plutôt la question dans les termes suivantes : de quelle façon prévenir, limiter et gérer au mieux les risques nécessairement liés aux médicaments, dès lors qu'ils ont par définition une action sur le corps humain ? Et comment « restaurer la confiance », pour reprendre l'intitulé du rapport récent de la mission d'information de la commission des affaires sociales du Sénat (1) ?
Face à ces interrogations, l'Europe, historiquement guidée par la nécessité d'harmoniser la réglementation des États membres, afin de supprimer les entraves au développement du marché intérieur et à la libre concurrence, a progressivement défini un corpus de règles « avec pour objectif essentiel la sauvegarde de la santé publique (2) ».
C'est ainsi qu'au printemps 2004, le code communautaire relatif aux médicaments à usage humain a été substantiellement modifié par un « paquet » de directives, en particulier la directive n° 2004/27 du 31 mars 2004 (3), que le présent projet de loi vise à transposer en droit interne.
D'une lecture il est vrai aride, et comportant certaines dispositions très complexes et techniques, ce texte pourrait par bien des aspects sembler éloigné des préoccupations de nos concitoyens. Il n'en est pourtant rien, bien au contraire.
En effet, comme l'a souligné avec justesse la revue indépendante Prescrire, qui s'est fortement impliquée dans le suivi de la réforme de la législation pharmaceutique communautaire, avec d'autres associations européennes représentant les consommateurs, les professionnels de santé, les malades et les mutuelles, réunis au sein du Collectif Europe et médicament : « L'Europe, ce n'est pas trop « loin » : c'est même là où se décide nos choix de société. L'Europe, ce n'est ni « inaccessible », ni « incompréhensible » : il suffit de se retrousser les manches et de ne pas laisser le champ libre aux intérêts particuliers. L'Europe, ce n'est pas trop « technique » : derrière chaque dossier se cachent en fait des choix de société (4) ».
Souvent critiquée pour son opacité et son déficit démocratique, la procédure d'examen ayant conduit à l'adoption de ce « paquet médicament » a donné lieu à des débats particulièrement approfondis et animés, la proposition initiale de la Commission ayant été très substantiellement modifiée et enrichie par le Parlement européen et le Conseil.
Le temps est à présent venu pour les membres du Parlement français de se saisir de ce sujet et de permettre la transposition de l'ensemble de ces textes dans les plus brefs délais, en adoptant ce projet de loi portant diverses dispositions d'adaptation au droit communautaire dans le domaine du médicament.
I.- LA RÉVISION DE LA LÉGISLATION PHARMACEUTIQUE COMMUNAUTAIRE DOIT ÊTRE TRANSPOSÉE TRÈS RAPIDEMENT
La transposition d'une directive européenne est d'abord une obligation juridique à laquelle la France, conformément à ses engagements, doit se soumettre. En l'espèce, la directive n° 2004/27 permettra d'introduire dans le droit interne une série d'avancées dans le domaine du médicament. Ces progrès, bien que de nature très disparate, constituent pour notre pays autant de pas vers la modernisation de notre système de soins, modernisation déjà largement engagée par les trois réformes fondamentales réalisées sous cette législature : la loi n° 2004-810 du 13 août 2004 relative à l'assurance maladie, la loi n° 2004-806 du 9 août 2004 relative à la politique de santé publique et la loi n° 2004-800 du 6 août 2004 relative à la bioéthique.
A. L'UNION EUROPÉENNE EST COMPÉTENTE EN MATIÈRE DE LÉGISLATION RELATIVE À LA FABRICATION ET À LA MISE SUR LE MARCHÉ DES MÉDICAMENTS
Les compétences des institutions européennes en matière de médicament, initialement limitées à des enjeux de politiques industrielles et à la garantie du bon fonctionnement du marché intérieur (marché devenu « unique ») n'ont cessé de s'étendre, en particulier dans le domaine de la protection de la santé publique.
La compétence de l'Union européenne en la matière s'appuie, s'agissant de la directive n° 2004/27, sur l'article 95 du traité instituant la Communauté européenne. Cet article prévoit que le Conseil arrête les mesures relatives au rapprochement des dispositions législatives, réglementaires et administratives des États membres qui ont pour objet l'établissement et le fonctionnement du marché intérieur. Conformément à cet article, la Commission, dans ses propositions en matière de santé, de sécurité, de protection de l'environnement et de protection des consommateurs, « prend pour base un niveau de protection élevé en tenant compte notamment de toute nouvelle évolution basée sur des faits scientifiques. Dans le cadre de leurs compétences respectives, le Parlement européen et le Conseil s'efforcent également d'atteindre cet objectif ».
La particularité de l'élaboration de ce droit européen relatif au médicament est qu'elle est marquée par l'influence de la direction générale de la Commission chargée des entreprises et de l'industrie. La direction générale chargée de la santé et de la protection des consommateurs est cependant de plus en plus impliquée.
En matière de médicament, le but de la législation européenne est de promouvoir le bon fonctionnement du marché intérieur, tout en assurant un niveau élevé de protection de la santé humaine grâce notamment à l'amélioration de l'accès de tous les patients européens aux médicaments. Depuis quelques années, la construction du droit communautaire du médicament s'oriente autour de ces deux axes fondamentaux. Pourtant, au fil du temps, d'autres préoccupations sont également apparues : le souci de promouvoir les médicaments génériques, les conditions de déroulement des essais cliniques, le questions de propriété industrielle et l' « impact environnemental »,... L'Union a aussi joué un rôle moteur dans la promotion des médicaments orphelins. La compétence communautaire ne s'étend pas aux conditions de fixation des prix et de remboursement des médicaments, question qui continue à relever de la compétence nationale.
Trois textes ont été adoptés le 31 mars 2004 par le Parlement européen et par le Conseil de l'Union européenne, après plus de deux ans de négociations :
- le règlement n° 726/2004/CE établissant des procédures communautaires pour l'autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant une Agence européenne des médicaments ; cette norme abroge et remplace le règlement n° 2309/93/CE ;
- la directive n° 2004/27 modifiant la directive n° 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain ;
- la directive n° 2004/28/CE modifiant la directive n° 2001/82/CE instituant un code communautaire relatif aux médicaments vétérinaires, qui ne sera pas traité dans le cadre du présent rapport.
Les propositions de la commission ont fait l'objet de discussions et de négociations au sein du groupe « médicaments » du Conseil. L'ensemble de la réforme a été examiné sur la base de la procédure de la codécision (article 251 du Traité instituant la Communauté européenne et conduisant à impliquer le Parlement et le Conseil). Les trois textes ont été adoptés par le Parlement européen en première lecture en séance plénière le 23 octobre 2002. De très nombreux amendements ont été adoptés à la suite du rapport de Mme Françoise Grossetête, représentante française au Parlement européen. Ces amendements allaient plutôt dans le sens du renforcement de l'aspect « santé publique » des textes, en réaction à un texte jugé trop favorable à l'industrie pharmaceutique.
En novembre et décembre 2003 a eu lieu la deuxième lecture au Parlement. La directive relative aux médicaments à usage humain y a encore fait l'objet de débats nourris. Les principaux points de discussion ont concerné la l'extension de la procédure centralisée d'AMM, le renouvellement quinquennal de l'AMM, le régime juridique des médicaments génériques ainsi que la publicité et l'information délivrée aux patients sur les médicaments.
La discussion autour de la directive a été marquée par la naissance du groupe « Collectif Europe et médicament », qui été créé en mars 2002. Il rassemble plus de soixante organismes, originaires de douze États membres de l'Union européenne. Il regroupe des associations de malades, des organisations familiales et de consommateurs, des organismes d'assurance maladie et des organisations de professionnels de la santé. Ce rassemblement d'acteurs aux intérêts rarement convergents, qu'un de ses représentants qualifie d'« inédit dans l'histoire de l'Union européenne », soutient l'idée que « le médicament n'est pas un produit comme un autre ». Le collectif a suivi en particulier la préparation de la directive en se concentrant sur la publicité directe des laboratoires pharmaceutiques auprès des patients, dont il a dénoncé les effets négatifs (cf. infra).
Les propositions de textes communautaires relatifs aux médicaments ont été approuvées par la délégation de l'Assemblée nationale pour l'Union européenne de l'Assemblée nationale au cours de sa réunion du 11 mai 2003.
L'origine de la politique communautaire remonte à la directive n° 65/65/CE du 26 janvier 1965. La création du code communautaire relatif au médicament à usage humain date de la directive n° 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain. Cette directive avait pour objet de codifier et de compléter sept directives aux objets très différents, datant de 1965 à 1992, concernant notamment les autorisations de mise sur le marché (AMM) et les essais cliniques de médicaments, ainsi que les conditions de distribution en gros, de classification, d'étiquetage et de publicité. Elle a permis de poser un certain nombre de principes en matière de législation européenne relative au médicament, principes qui orientent toujours le droit communautaire en la matière. En particulier, la législation communautaire a apporté une contribution importante à la réalisation de l'objectif de libre circulation, en toute sécurité, des médicaments à usage humain et d'élimination des entraves aux échanges de ceux-ci. L'autre texte fondateur est le règlement (CEE) n° 2309/93 du Conseil du 22 juillet 1993 établissant les procédures communautaires et instituant l'Agence européenne du médicament.
b) La nécessité de revoir les dispositions du code communautaire relatif aux médicaments à usage humain est devenue évidente
La nécessité de revoir la directive est rapidement apparue. Le renvoi aux considérants de la directive n° 2004/27, dont la transposition fait l'objet du présent projet, permet de prendre connaissance des motifs ayant guidé la révision du code communautaire : la promotion du fonctionnement du marché intérieur et le niveau élevé de protection de la santé humaine.
En outre, l'article 71 du règlement (CEE) n° 2309/93 prévoyait que, dans un délai de six ans à compter de son entrée en vigueur, la Commission devait publier un rapport général sur l'expérience acquise sur la base du fonctionnement des procédures d'autorisation de mise sur le marché établies par ledit règlement et par d'autres dispositions de la législation communautaire. À la lumière du rapport de la Commission, il s'est avéré nécessaire d'améliorer le fonctionnement des procédures d'autorisation de mise sur le marché.
L'intégration croissante du marché pharmaceutique européen, la mobilité des personnes, les évolutions technologiques et les enseignements de l'application de la réglementation antérieure ont souligné la nécessité de modifier certains points de la législation communautaire, notamment en ce qui concerne la définition du médicament, l'extension de la procédure centralisée et la promotion des médicaments génériques. La perspective de l'élargissement de l'Union européenne a également joué un rôle dans l'accélération de la construction de la nouvelle directive. Dans un contexte où la concurrence avec l'industrie américaine du médicament est toujours plus âpre, la nécessité de définir un ensemble de règles modernisées et plus centralisées au niveau européen a sans doute aussi été un facteur déterminant.
2. Les procédures d'autorisation et de suivi sanitaire des médicaments ont connu des failles troublantes
Compte tenu de la mondialisation des marchés, la crise de confiance qui a frappé les laboratoires pharmaceutiques a connu une ampleur sans précédent. Elle prend un relief particulier en France, dont la situation présente des spécificités. Le « paquet » communautaire prend en compte la dimension de la crise du médicament.
Un fait a particulièrement joué en faveur de la modification de la législation communautaire relative au médicament : le scandale du médicament Vioxx intervenu à l'automne 2004. Ce scandale à la dimension mondiale touchant la firme Merck, conjugué aux doutes récurrents entourant l'indépendance des experts, voire celle de prestigieuses revues médicales, a mis en évidence la nécessité d'améliorer les procédures d'autorisation de mise sur le marché, de renforcer l'indépendance des agences vis-à-vis de l'industrie pharmaceutique et surtout de promouvoir des dispositifs efficaces de pharmacovigilance. Devant la construction d'acteurs mondiaux de la pharmacie, il devenait nécessaire de mettre en place des institutions et des procédures susceptibles de faire le poids.
Les crises relatives aux médicaments n'ont pas commencé en 2004. On peut citer, par exemple, l'affaire de la thalidomide. En effet, par construction, le médicament doit répondre à un délicat arbitrage entre avantages et risques. La tendance à la revendication d'une sécurité accrue, dans la perspective de l'application du principe de précaution, a tendu à compromettre la perception par le grand public de ce délicat équilibre. Le besoin de sécurité est sans doute devenu plus grand.
En outre, la croissance très forte des dépenses de promotion et de marketing des laboratoires a pu être considérée comme problématique, alors même que, depuis une quinzaine d'année, les laboratoires semblent peiner à sortir des molécules réellement innovantes.
La nécessité de transposer les dispositions de la directive est rendue encore plus aiguë en raison des spécificités françaises en matière de médicaments. Ces spécificités sont largement connues. Il s'agit d'abord d'une surconsommation générale de médicaments, et en particulier d'antibiotiques, de tranquillisants et d'hypnotiques. Par rapport à nos voisins européens, les alternatives à la prise de médicaments (par exemple la psychothérapie ou la phytothérapie) sont insuffisamment développées. Cette situation préoccupante, à la tendance difficile à infléchir, a deux aspects négatifs : d'abord pour la santé publique, puis pour les finances des régimes d'assurance maladie. De même, la France se caractérise par le poids de la visite médicale. On compte ainsi en France un visiteur médical pour neuf médecins, contre un visiteur pour vingt-deux médecins en Allemagne.
De plus, la France, par rapport à ses pays voisins de l'Union européenne, l'Allemagne notamment, connaît des taux de pénétration des médicaments génériques relativement bas. Les vagues récentes de déremboursement de médicaments à service médical rendu insuffisante vont mécaniquement augmenter en France l'automédication, qui appelle une réglementation particulière. Enfin, la France a longtemps été marquée par une certaine opacité dans le circuit du médicament. Cette opacité a depuis quelques années fait part à plus de transparence, mais la situation est encore perfectible.
C. EXIGENCE CONSTITUTIONNELLE, LA TRANSPOSITION RAPIDE DU « PAQUET MÉDICAMENT » EST AUSSI UN IMPÉRATIF POLITIQUE
Il convient au préalable de rappeler qu'aux termes de l'article 249 du Traité instituant la communauté européenne, une directive « lie tout État membre destinataire quant au résultat à atteindre, tout en laissant aux instances nationales la compétence quant à la forme et aux moyens ». Si le règlement s'applique directement dans les États membres, la directive doit ainsi faire l'objet d'une transposition, dans un délai déterminé, pour entrer en vigueur.
L'acte de transposition ne doit cependant pas uniquement être entendu comme une simple retranscription des dispositions d'une directive. Il s'agit au contraire, selon la jurisprudence de la Cour de justice des Communautés européennes (CJCE), de prendre « toutes mesures générales ou particulières, propres à assurer l'exécution de cette obligation ». Cela peut nécessiter l'édiction de nouvelles normes, l'organisation de mécanismes de contrôles, voire la mise en œuvre de sanctions, non prévues par la directive, mais nécessaires pour garantir le plein effet de ses objectifs.
« De la qualité de la transposition en droit interne des directives et des décisions-cadres négociées dans le cadre des institutions européennes dépendent à la fois la sécurité des situations juridiques et le crédit de la France auprès de ses partenaires européens », comme l'a rappelé avec force et justesse la circulaire du Premier ministre du 27 septembre 2004 relative à la procédure de transposition en droit interne des directives.
En effet, un manquement aux obligations communautaires n'affecte pas seulement notre crédit au sein de l'Union, il expose aussi la France à des sanctions contentieuses, y compris pécuniaires, et entrave le bon fonctionnement du marché intérieur, portant préjudice in fine à la compétitivité des entreprises et à la protection des consommateurs.
En matière de transposition de directives, dont il faut rappeler qu'elle constitue une exigence constitutionnelle (5), la France a enregistré des résultats significatifs depuis quelques années. Ainsi, le déficit de transposition - soit le pourcentage de directives du marché intérieur qui n'ont pas encore été transposées et dont le délai de mise en œuvre est dépassé - se situait à 1,7 % à la fin du mois de novembre 2005, contre 4,1 % en mai 2004.
Evolution du déficit de transposition de la France depuis 1997
(en %)
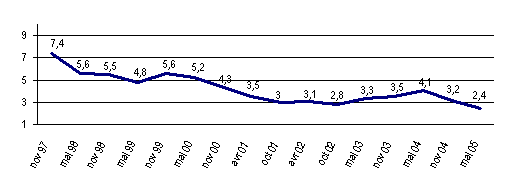
Source : Rapport d'information n° 2447 de M. Christian Philip, au nom de la Délégation pour l'Assemblée nationale pour l'Union européenne, sur la transposition des directives européennes
Encourageants, ces résultats sont toutefois encore insuffisants, dans la mesure où la France se place aujourd'hui au dix-huitième rang des vingt-cinq pays de l'Union européenne, ce dont témoigne également le nombre de procédures en manquements qui ont été engagés à son encontre.
Nombre de procédures en manquements engagées
contre des États membres au 1er octobre 2005
État membre |
Nombre de procédures engagées |
Danemark |
31 |
Finlande |
36 |
Luxembourg |
36 |
Suède |
41 |
Pays-Bas |
44 |
Portugal |
53 |
Royaume-Uni |
66 |
Belgique |
66 |
Allemagne |
101 |
France |
113 |
Espagne |
115 |
Italie |
157 |
Source : Commission européenne (tableau d'affichage du marché unique), février 2006
S'il faut saluer tant la détermination que les mesures annoncées par Mme Catherine Colonna, ministre déléguée aux affaires européenne, dans sa communication sur la transposition des directives présentée au Conseil des ministres le 22 février 2006, des efforts substantiels n'en devront pas moins être déployés pour atteindre l'objectif fixé par le Conseil européen d'un déficit de transposition de 1,5 %.
En particulier, la mise en œuvre de l'ensemble des dispositions prévues par la circulaire du 27 septembre 2004 ainsi qu'une meilleure préparation de la traduction en droit interne d'une directive, en amont de son adoption, pourraient sans doute permettre de rompre avec les délais anormalement longs de transposition qui ont trop longtemps caractérisé la France, en particulier dans le domaine de la santé.
2. Le gouvernement a déjà entamé le processus de transposition de la directive n° 2004/27 du 31 mars 2004
Bien que parcellaire, la transposition en droit du « paquet médicament » a été engagée par le gouvernement dès l'été 2004.
Ainsi, l'article 30 de la loi n° 2004-810 du 13 août 2004 relative à l'assurance maladie a permis d'étendre la définition des génériques aux différents dérivés chimiques d'un même principe actif, tels que les sels, les esters ou les isomères, dès lors qu'ils ont une efficacité et une sécurité équivalente à celle de la spécialité de référence, dite princeps. Transposant l'article 10 de la directive n° 2004/27, ces dispositions visaient également à limiter les pratiques de certains laboratoires consistant à commercialiser des dérivés chimiques de leur princeps dans le seul but de conserver leurs parts de marché au moment de l'expiration des droits attachés au brevet de celui-ci.
Dans un sens très favorable aux génériques, le décret n° 2005-156 du 18 février 2005 a également apporté plusieurs aménagements substantiels au régime d'autorisation de mise sur le marché (AMM) des médicaments, à travers principalement l'introduction de la notion d' « AMM globale » (cf. infra).
Enfin, des efforts significatifs ont été réalisés par l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) concernant la transparence de ses travaux. La modification de son règlement intérieur a ainsi permis de rendre applicables certaines dispositions de la directive n° 2004/27.
S'agissant de la directive n° 2004/27, dont le délai de transposition a été largement dépassé puisqu'il était fixé au 30 octobre 2005, la Commission européenne n'a pas encore engagé de procédure en manquement contre la France, selon les informations communiquées par le ministère de la santé et des solidarités. Lors de la réunion du 2 décembre 2005 du comité pharmaceutique (6), il est en effet apparu que la grande majorité des États membres n'avaient pas encore transposé ce texte, du moins dans sa totalité.
En revanche, des procédures en manquement ont été engagées contre la France pour défaut de transposition des directives n° 2004/24 (7) et n° 2004/28 (8), relatives aux médicaments à base de plantes et aux médicaments vétérinaires, qui doivent être transposées par ordonnance sur le fondement de l'article 29 du projet de loi. Le collège des commissaires européens, lors de sa réunion « infractions » du 28 juin prochain, doit se prononcer sur l'envoi ou non à la France de deux avis motivés concernant ces directives.
Une mise en demeure a par ailleurs été adressée à la France concernant la directive n° 2002/98/CE relative aux composants sanguins (9), à laquelle il a été répondu le 7 décembre 2005. Enfin, une procédure d'astreinte financière a été engagée pour non-exécution d'un arrêt de la Cour de justice des Communautés européennes relatif aux importations personnelles de médicament, la réponse à la mise en demeure ayant été transmise aux autorités communautaires le 9 février dernier. L'article 22 du projet de loi doit permettre de mettre fin à cette procédure.
Il y a donc urgence à transposer rapidement l'ensemble du « paquet médicament », et ce d'autant plus qu'il comporte des avancées majeures pour la santé publique, le renforcement de la compétitivité des industries européennes, l'information et la protection des patients.
A titre liminaire, il convient de préciser que le présent projet de loi n'est pas exclusivement un projet visant à transposer des dispositions du droit communautaire.
En effet, les articles et dispositions suivantes ne relèvent pas de la transposition des textes communautaires stricto sensu, bien qu'ils aient un rapport étroit avec la politique du médicament et qu'ils renforcent les dispositions issues de la transposition des directives :
- la définition du groupe générique, posée par l'article 4, correspond à une notion française ;
- l'article 12 s'inscrit dans le cadre d'une faculté laissée aux États membres par le code communautaire ;
- les articles 13 et 14 constituent pour partie des mesures de clarification de la réglementation nationale ;
- l'article 22 relatif aux importations personnelles de médicaments tire les conséquences d'un arrêt de la CJCE mais ne transpose pas directement la directive ;
- les articles 23 à 25 permettent d'introduire une définition des matières premières à usage pharmaceutique et de renforcer la sécurité sanitaire, mais ne constituent pas une transposition de la directive ;
- l'article 27 (transmission d'échantillons à l'AFSSAPS) n'a pas de lien avec le code communautaire mais est destiné à permettre à l'AFSSAPS de mieux remplir des missions qui relèvent de la mise en œuvre de la directive ;
- s'agissant du II de l'article 29, les 3° (régime juridique des programmes d'observance destinés aux patients) et 4° (faculté donnée aux agents de la direction générale de la concurrence, de la consommation et de la répression des fraudes (DGCCRF), de la direction générale des douanes et de la direction générale des impôts de recourir à l'AFSSAPS) n'ont pas de lien avec le code communautaire.
Les trois grandes orientations proposées par le présent projet consistent à assurer la promotion équilibrée des génériques, la mise en place d'un contrôle accru des médicaments et enfin le renforcement de l'information des patients sur les médicaments et le fonctionnement de l'AFSSAPS.
A. UN ÉQUILIBRE EST AMÉNAGÉ ENTRE LA PROTECTION DE L'INNOVATION ET LA NÉCESSITÉ DE PROMOUVOIR DAVANTAGE LES GÉNÉRIQUES
Selon le rapport de la commission des comptes de la sécurité sociale de juin 2006, les dépenses de médicaments remboursés par le régime général de l'assurance maladie se sont élevées à plus de 16,7 milliards d'euros en 2005. S'agissant d'un secteur caractérisé par une croissance dynamique (+ 5,2 % en 2005), le développement des génériques, dont le prix est inférieur de 30 à 50 % à celui des autres médicaments, représente un enjeu décisif pour la maîtrise des dépenses de santé, à qualité des soins égale. Leur promotion constitue ainsi un axe central de la politique du médicament engagée par le gouvernement, qui a enregistré des résultats très significatifs depuis 2002.
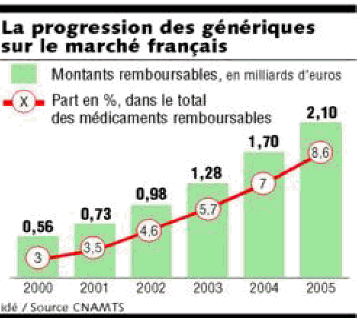
Cette progression est encore plus nette parmi les médicaments pour lesquels il existe une offre générique, c'est-à-dire ceux qui sont inscrits au répertoire des groupes génériques (ces derniers comprenant la spécialité originale de référence, dite princeps, et son ou ses génériques).
En à peine plus de trois ans, le taux de pénétration des génériques au sein du répertoire est en effet passé de 32,5 % à 63 % en décembre 2005. Outre la mobilisation des caisses d'assurances maladie, des professionnels de santé et des assurés, il faut à cet égard saluer la contribution très significative des pharmaciens à cette progression, à travers l'exercice de leur droit de substitution entre les différentes spécialités pharmaceutiques inscrites dans un même groupe du répertoire.
Evolution du taux de pénétration des génériques dans
le répertoire en vigueur au 31 décembre 2005
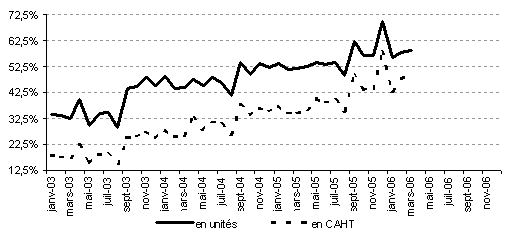
Source : Rapport de la commission des comptes de la sécurité sociale de juin 2006
En dépit de ces progrès tangibles, les génériques conservent toutefois un potentiel de développement significatif : en 2005, ceux-ci ne représentaient en effet que 8,6 % des remboursements de médicaments en valeur et 14,6 % en volume. Sans prétendre à l'exhaustivité, on peut néanmoins identifier plusieurs freins, qui sont autant d'enjeux pour les années à venir et auxquels le présent projet de loi permet partiellement de répondre.
En matière de substitution, l'accord pluriannuel (2006-2008), conclu entre l'Union nationale des caisses d'assurance maladie (UNCAM) et les trois organisations syndicales représentatives des pharmaciens, le 18 janvier dernier, a tout d'abord fixé un objectif ambitieux de 70 % de pénétration des génériques pour la fin 2006.
La promotion de la prescription en dénomination commune internationale (DCI) doit également être renforcée. Lors de sa réunion en date du 31 mai 2006, le collège de la Haute autorité de santé a d'ailleurs décidé que « la certification [des logiciels d'aide à la prescription] devra imposer aux éditeurs la possibilité d'une prescription en DCI ».
L'un des problèmes majeurs demeure toutefois le caractère encore très restreint du répertoire des groupes génériques, puisqu'il représente uniquement 17,5 % du marché total en valeur. Cette situation s'explique notamment par l'importance des médicaments, pour lesquels les droits de propriété intellectuelle attachés au brevet (valable 20 ans) n'ont pas encore expiré, mais aussi par la durée de protection des données cliniques de l'AMM et, dans certains cas, par les stratégies de contournements mises en œuvre par les laboratoires pharmaceutiques, consistant par exemple à accroître les durées de protection ou à diversifier leurs gammes de produits. D'ici fin 2007, le répertoire devrait toutefois être sensiblement élargi, en raison du nombre important de molécules qui vont tomber dans le domaine public et dont le potentiel d'économies s'élève à près de 700 millions d'euros.
Enfin, les délais, ainsi que la lourdeur des procédures d'autorisation de mise sur le marché (AMM) et d'inscription au remboursement et de fixation du prix ont pu contribuer à freiner le développement des génériques. En particulier, le délai de délivrance d'une AMM générique est plus important que pour les autres médicaments, puisqu'il s'élevait à un an en 2005.
2. Les procédures d'autorisation de mise sur le marché des génériques et des « biosimilaires » sont simplifiées
L'article 5 du projet de loi prévoit tout d'abord que seront définies par voie réglementaire les nouvelles modalités de mise en œuvre de la procédure « allégée » d'autorisation, applicable aux génériques, c'est-à-dire les conditions dans lesquelles les demandeurs d'une AMM peuvent être dispensés de produire les résultats de certaines données et études, à la condition notamment que leur bioéquivalence avec la spécialité de référence soit démontrée. Conformément à la directive n° 2004/27, la période de protection des données, qui est actuellement de dix ans, sera ainsi portée à huit ans. Il s'agit là d'une avancée substantielle pour les génériqueurs, puisqu'ils pourront déposer une demande d'AMM dès la fin de la huitième année de protection des données.
La protection des données de l'AMM
« La protection des données consiste à interdire l'utilisation des données de l'évaluation clinique d'un médicament princeps pendant un certain nombre d'années. En France, jusqu'à présent, la durée était de dix ans [ à compter de la délivrance de l'AMM à la spécialité de référence ]. [S'agissant de la directive n° 2004/27], le compromis porte à huit ans la protection des données des essais cliniques, sans possibilité de commercialiser un générique avant dix ans. Cela signifie qu'une firme spécialisée dans les génériques peut constituer un dossier de demande d'AMM en utilisant les résultats des essais cliniques réalisés pour évaluer un médicament princeps dès la fin de la huitième année de protection des données. Il peut ainsi se préparer à commercialiser son générique (...) ».
Source : « Europe et médicament », Revue Prescrire n° 252 de juillet - août 2004
Dans le même sens, l'article 4 du projet de loi permet de consacrer au niveau législatif le principe de l' « AMM globale », qui a déjà été transposée par le décret n° 2005-156 du 18 février 2005 (10). Ainsi, les autorisations obtenues pour pour tout nouveau dosage, forme pharmaceutique ou voie d'administration d'un médicament - c'est-à-dire les « extensions de gamme » - sont désormais considérées comme faisant partie d'une même AMM et ne peuvent plus dès lors donner lieu à des périodes supplémentaires de protection des données. Il est également proposé de modifier la définition des groupes génériques, de façon à inclure ces gammes de médicaments en leur sein et faciliter ainsi la substitution.
Allant de pair la modification de la définition du médicament générique par la loi du 13 août 2004 relative à l'assurance maladie, cette modification du régime des AMM doit permettre de limiter les stratégies de contournement des génériques, qui avaient conduit par le passé à retarder significativement l'arrivée sur le marché de nouveaux génériques.
Parachevant ce dispositif, une clause dite « Bolar » (11) est introduite afin de faciliter la réalisation des études et des essais nécessaires à l'obtention d'une AMM et des actes pratiques qui en résultent, l'article 10 du projet de loi précisant qu'ils ne peuvent être considérés comme contraires au droit des brevets.
Deux décennies après la mise sur le marché communautaire des premiers médicaments produits par des moyens biotechnologiques, plusieurs de ces brevets arrivent maintenant à expiration. De ce fait, les premiers médicaments « biogénériques » ou « biosimilaires » - soit les copies de médicaments biologiques, qui ne peuvent pas toujours être considérés comme des « génériques », en raison notamment des caractéristiques particulières de leurs matières premières ou de leurs procédés de fabrication - sont sur le point d'arriver sur le marché de l'Union européenne. En mars dernier, la Commission a ainsi accordé la première autorisation de commercialisation pour un produit biosimilaire, l'omnitrope, destiné à traiter le manque d'hormone de croissance chez les enfants et les adultes.
Il s'agit là d'une évolution d'autant plus importante que, comme l'a déclaré le vice-président de la Commission européenne, M. Günter Verheugen, « les médicaments biosimilaires présentent de nouvelles opportunités, tant pour l'expansion de notre industrie générique que pour le contrôle des dépenses nationales de santé. Néanmoins, ces produits complexes doivent se conformer aux mêmes normes rigoureuses de qualité, de sécurité et d'efficacité que tout autre médicament, dans l'intérêt des patients européens ».
C'est pourquoi le projet de loi propose tout d'abord de définir les notions de médicaments biologiques et biologiques similaires (article 4) et d'appliquer à ces derniers un régime juridique proche de celui des génériques (articles 5 et 11). Aussi pourront-ils bénéficier d'une procédure allégée d'AMM et obtenir une telle autorisation avant l'expiration du brevet du médicament biologique de référence. En revanche, dans la mesure où il n'y a pas identité totale entre ces deux catégories de médicaments et par application du principe de précaution, la substitution ne sera pas autorisée dans ce cas.
L'article 9 institue tout d'abord une période de protection de marché, d'une durée de dix ans après la délivrance de l'AMM initiale au princeps, pendant laquelle un générique ne pourra être commercialisé, étant précisé que ce dernier aura toutefois la possibilité d'obtenir une AMM ou d'être inscrit au répertoire avant cette échéance.
Par ailleurs, afin de favoriser la recherche sur de nouvelles indications, l'exploitant du princeps pourra bénéficier d'une année supplémentaire de protection, s'il obtient, pendant les huit premières années suivant la délivrance de son AMM, une autorisation pour une ou plusieurs indications thérapeutiques nouvelles de nature à apporter un avantage clinique important par rapport aux thérapies existantes. Ce faisant, cette règle dite des « 8+2+1 » (les huit ans faisant dans ce cas référence à la période au terme de laquelle, un génériqueur peut déposer une demande d'AM) permet ainsi d'introduire une notion de « valeur thérapeutique ajoutée » du médicament.
Afin de mieux veiller au respect effectif des droits conférés par le brevet, le projet de loi soumet le titulaire d'une AMM d'un générique à l'obligation d'informer le directeur général de l'AFSSAPS de toutes les formes, indications et dosages du princeps, qui demeurent protégés par des droits de propriété intellectuelle au moment de la commercialisation du générique (article 11).
La loi n° 2004-806 du 9 août 2004 relative à la politique de santé publique comporte un important volet de mesures destinées à améliorer la capacité de réaction des pouvoirs publics dans le cas d'une menace sanitaire grave, telle qu'une attaque bioterroriste ou un risque de pandémie grippale. L'article 1er du projet de loi permet de renforcer l'efficacité de ce dispositif, en prévoyant l'exonération de la responsabilité des fabricants et des titulaires de l'AMM d'un médicament, dans le cas où son utilisation serait prescrite par le ministre chargé de la santé, en dehors de ses indications ou sans qu'il ne soit autorisé.
Vise également à garantir l'accès de tous aux médicaments, la possibilité donnée à l'AFSSAPS de permettre la mise en marché d'un médicament, autorisé dans un autre État membre mais non en France, si, pour des raisons de santé publique, la situation devait le requérir (article 7).
Avancée majeure pour les personnes atteintes d'une maladie grave, dont le pronostic vital est gravement menacé, en l'état des connaissances scientifiques actuelles, l'article 12 du projet prévoit par ailleurs qu'ils auront désormais la possibilité d'avoir accès à des traitements, qui sont encore en phase d'expérimentation clinique, sous le régime des autorisations temporaires d'utilisation (ATU), délivrées par l'AFSSAPS.
Le présent projet prend également en compte les risques de rupture de stock, éventualité envisagée par l'article 22 de la directive. Ainsi, l'article 21 propose de modifier l'article L. 5124-6 du code de la santé publique et vise à préciser les conditions dans lesquelles les entreprises commercialisant des médicaments doivent alerter l'AFSSAPS en cas de suspension de commercialisation du produit ou de rupture de stock. La rédaction proposée permet de couvrir les entreprises exploitant un médicament depuis un autre État de l'Union européenne. Conformément au projet, l'entreprise exploitant un médicament ou produit devra informer l'AFSSAPS de toute action qu'il a engagée pour en suspendre la commercialisation, le retirer du marché ou en retirer un lot déterminé, ainsi que de tout risque de rupture de stock sur un médicament ou produit sans alternative thérapeutique disponible ou en raison d'un accroissement significatif et imprévisible de la demande.
L'article 22 du présent projet vise également à accroître la disponibilité des médicaments en supprimant les barrières excessives aux importations de médicaments réalisées par les particuliers. A la suite d'une condamnation de la France par la CJCE (cf. supra), il vise à ce que les importations personnelles de médicaments, lorsqu'elles concernent des médicaments ayant fait l'objet d'une AMM dans un État membre ou des médicaments homéopathiques ayant fait l'objet d'un enregistrement dans un État membre, soient autorisées a priori sans qu'une autorisation d'importation auprès des autorités françaises soit nécessaire.
2. La procédure de mise sur le marché est réformée afin de renforcer la sécurité sanitaire des médicaments
a) Le processus d'autorisation est recentré sur l'évaluation de la balance bénéfice-risque
Comme l'affirme très clairement la directive n° 2004/27, les critères de qualité, de sécurité et d'efficacité d'un médicament doivent permettre l'évaluation de son « rapport bénéfice/risque », aussi bien au stade de sa mise sur le marché qu'à tout autre moment que les États membres jugent approprié.
Les notions de nocivité et d'effet thérapeutique d'un médicament ne peuvent en effet être examinées qu'en relation réciproque et n'ont qu'une signification relative, appréciée en fonction de l'état d'avancement de la science et de l'indication du produit concerné. Selon ce principe, les effets secondaires ou les risques associés à un médicament ne peuvent, par exemple, être appréciés de la même manière selon qu'il vise à traiter une grippe ou une leucémie.
Définition communautaire du « rapport bénéfice-risque »
Aux termes du point 28 de l'article 1er de la directive n° 2001/83 consolidée, ce terme désigne : « l'évaluation des effets thérapeutiques positifs du médicament au regard du risque », tel que défini au point 28, premier tiret, soit « tout risque pour la santé du patient ou la santé publique lié à la qualité, à la sécurité ou à l'efficacité du médicament. »
Si le rapport bénéfice-risque est de fait déjà mis en œuvre par l'AFSSAPS, le projet de loi permet de donnant ainsi un fondement légal à l'appréciation plus fine et « sur-mesure », à laquelle doit procéder l'agence avant d'autoriser, de refuser ou de retirer un médicament (article 6).
Ce projet de loi s'inscrit ainsi dans la continuité de la loi du 9 août 2004 relative à la politique de santé publique (12), qui a procédé à une réforme en profondeur du régime des recherches biomédicales. Depuis lors, l'autorisation des protocoles de recherche, pour les essais cliniques de médicaments, doit en effet se fonder sur l'évaluation de leur balance bénéfice-risque.
Dans le sens d'un renforcement de la sécurité sanitaire et d'une meilleure appréciation des risques associés à un produit à usage humain, l'article 3 du projet de loi permet également, en cas de doute, d'imposer l'application de la réglementation pharmaceutique, plus contraignante et protectrice pour les consommateurs, aux produits, par exemple des cosmétiques, qui pourraient répondre à la fois à la définition du médicament et à celles d'autres catégories de produits régies par le droit communautaire ou national.
Par ailleurs, si les AMM des médicaments sont actuellement renouvelables tous les cinq ans, le rapport sur l'expérience acquise dans le fonctionnement des procédures communautaires d'autorisation (13) a permis de mettre en évidence le fait que « le renouvellement des AMM relèvent en pratique de moins en moins souvent d'une réévaluation scientifique, mais davantage d'une simple procédure administrative ».
De fait, compte tenu du nombre de demandes dont elle est saisie, (cf. le tableau présenté ci-après), il est clair que l'AFSSAPS n'a pas actuellement les moyens de procéder une réévaluation scientifique approfondie de l'ensemble des médicaments inscrits à la pharmacopée à l'occasion de chaque renouvellement quinquennal de leur autorisation.
Procédure nationale d'autorisation de mise sur le marché par l'AFSSAPS
1998 |
1999 |
2000 |
2001 |
2002 |
2003 |
2004 | |
Nouvelles demandes d'AMM déposées |
847 |
950 |
982 |
1 005 |
897 |
1 460 |
1 096 |
Décisions sur les AMM |
1 765 |
1 751 |
1 532 |
1 494 |
1 506 |
2 013 |
2 293 |
Octroi |
806 |
782 |
683 |
632 |
559 |
644 |
596 |
Autres notifications |
867 |
690 |
611 |
627 |
772 |
1 099 |
1 395 |
Refus |
72 |
279 |
238 |
235 |
175 |
270 |
302 |
Modifications d'AMM |
6 022 |
8 480 |
8 590 |
8 749 |
10 386 |
12 416 |
15 810 |
Renouvellement |
1 666 |
1 364 |
1 927 |
2 411 |
3 129 |
2 121 |
1 752 |
Notification de retraits |
1 455 |
1 235 |
357 |
417 |
421 |
542 |
241 |
Source : AFSSAPS (rapport annuel pour 2004)
Afin d'alléger ces charges administratives et libérer ainsi davantage de moyens pour l'évaluation des nouveaux médicaments ou ceux présentant des risques particuliers, le projet de loi prévoit qu'après sa délivrance initiale, l'AMM sera valable pour une durée déterminée, qui sera fixée à cinq ans (article 5).
En contrepartie, le dispositif de suivi et de pharmacovigilance des médicaments sera significativement renforcé, comme la délégation pour l'Union européenne de l'Assemblée nationale en avait d'ailleurs formulé le souhait, lors de sa réunion du 22 mai 2003 sur la proposition de directive modifiant la directive n° 2001/83.
Chacun s'accorde aujourd'hui à reconnaître la nécessité et l'urgence qu'il y a à renforcer le système de pharmacovigilance et, plus généralement, la surveillance du marché. C'est d'ailleurs l'un des principaux points mis en exergue par le rapport de la mission d'information sénatoriale précitée, qui avance également plusieurs propositions intéressantes afin d'y remédier.
Dans cet objectif, le projet de loi prévoit tout d'abord que seront définies par décret en Conseil d'État les règles applicables en la matière, s'agissant notamment des obligations de signalement incombant aux membres des professions de santé et aux entreprises pharmaceutiques (article 15).
Ce décret permettra ainsi de transposer les dispositions de la directive n° 2004/27, prévoyant notamment que les « rapports périodiques actualisés relatifs à la sécurité », dans lesquels les titulaires de l'AMM sont tenus d'enregistrer tous les effets indésirables présumés d'un médicament, devront être transmis plus régulièrement aux agences sanitaires. L'article 104 de la directive précise par ailleurs que ces rapports devront désormais être accompagnés d'une « évaluation scientifique du rapport bénéfice-risque du médicament ». De la même manière, pour obtenir une AMM, le demandeur devra produire une description détaillée du système de pharmacovigilance et, le cas échéant, du plan de gestion du risque qui sera mis en place.
Le renforcement de la pharmacovigilance et plus généralement du contrôle du médicament passe aussi par d'autres mesures proposées par le présent projet.
Ainsi, l'article 20 impose aux titulaires d'AMM d'avertir les autorités sanitaires dès que la spécialité est effectivement commercialisée. Cette communication est un élément important du système de pharmacovigilance. Il permet en effet à l'agence de disposer d'un tableau de bord à jour recensant non seulement les spécialités approuvées mais aussi celles effectivement commercialisées.
La disposition de l'article 22 de la directive imposant aux titulaires d'AMM de donner des informations sur les ventes aux agences n'est pas transposée par le présent projet. Selon les informations transmises à la rapporteure, cette disposition devrait faire l'objet d'un décret permettant au directeur général de l'AFSSAPS de demander aux laboratoires de lui communiquer toute information relative au volume des ventes et toute information concernant le volume des prescriptions.
De même, l'article 26 permet à l'AFSSAPS de demander la transmission à titre gratuit d'échantillons de médicaments. Les inspecteurs de l'AFSSAPS ne peuvent prélever des échantillons qu'à l'occasion d'inspections, ce qui limite considérablement le champ des contrôles. L'article propose d'élargir les possibilités, pour l'AFSSAPS, d'obtenir gratuitement des échantillons auprès des entreprises. Si les industriels transmettent sans grande difficulté les médicaments chimiques, il n'en est pas de même pour certains médicaments biologiques, plus onéreux. Cette disposition est de nature à faciliter l'exercice par l'AFSSAPS de ses missions.
L'article 29 du présent projet permet également d'habiliter le gouvernement à rétablir les prérogatives de contrôle des agents des douanes dans les échanges intracommunautaires pour certaines catégories de produits sensibles limitativement énumérés. Le gouvernement pourrait ainsi compléter par ordonnance la liste des produits susceptibles de faire l'objet d'un contrôle douanier.
Ce même article 29 prévoit la promulgation d'ordonnances ayant pour objet de renforcer la sécurité sanitaire. Il permet notamment d'habiliter le gouvernement à transposer par ordonnance la directive 2002/98/CE du Parlement européen et du Conseil du 27 janvier 2003 établissant des normes de qualité et de sécurité pour la collecte, le contrôle, la transformation, la conservation et la distribution du sang humain, et des composants sanguins. Ces dispositions permettraient notamment de mieux délimiter les compétences respectives des établissements de santé et les établissements de transfusion sanguine en matière de distribution et de délivrance. Elles modifieraient les sanctions pénales applicables en cas de non-respect des règles applicables à l'activité transfusionnelle et elles ajouteraient la délivrance de produits sanguins labiles aux actes ne pouvant être réalisés sans que soient réalisés les analyses biologiques et les tests relatifs aux maladies transmissibles.
Une ordonnance transposera également la directive 2004/24/CE du Parlement européen et du Conseil du 31 mars 2004 modifiant, en ce qui concerne les médicaments traditionnels à base de plantes, la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain. L'ordonnance étendrait notamment à ces médicaments des dispositions applicables aux autres médicaments à usage humain et relatives à la publicité, aux autorisations d'importation, à la prise en charge par la sécurité sociale et à certaines sanctions pénales.
L'ensemble de ces mesures permettra ainsi de conforter les actions engagées par l'AFSSAPS dans ce domaine.
Les axes de développement de l'AFSSAPS en matière de pharmacovigilance
1. La création d'un département de surveillance du risque et du bon usage des médicaments.
Ce nouveau département intègre, depuis septembre 2005, les activités d'évaluation susceptibles de contribuer à la surveillance du risque : la pharmacovigilance, la pharmacoépidémiologie, l'évaluation des cas d'abus et de pharmacodépendance et l'information sur le bon usage. Il permet d'organiser une veille continue, coordonnée et transversale de l'usage des médicaments.
2. La mise en place de plans de gestion de risques.
Les plans de gestion de risques sont prévus par la directive du 31 mars 2004 dans les situations justifiant une surveillance renforcée, afin de développer une gestion anticipée du risque. Ils doivent notamment accompagner les demandes d'AMM lorsqu'elles concernent une nouvelle substance active, un médicament générique, un produit princeps à risque, un biosimilaire, un produit ayant subi des changements significatifs ou dont l'utilisation est susceptible d'être large. A la suite des différents retraits très médiatiques ayant ébranlé le monde pharmaceutique, en particulier celui de la cérivastatine, il est en effet apparu souhaitable aux agences nationales de renforcer leurs exigences en matière de pharmacovigilance. A cet effet, un groupe de travail ad hoc a été mis en place à l'automne 2002 pour réfléchir à la problématique de l'« european risk management strategy » (ERMS). Fruit de ce travail, le concept du « risk management plan » apparaît en janvier 2003. Désormais, depuis novembre 2005, l'Agence européenne des médicaments, comme les agences nationales, mettent en œuvre des plans de gestion de risques.
3. Le renforcement du système de pharmacovigilance.
Une actualisation des bonnes pratiques de pharmacovigilance, dont la version initiale date de 1994, a été réalisée pour tenir compte des nouvelles dispositions techniques et réglementaires, notamment communautaires. Elles sont devenues opposables par un arrêté du 28 avril 2005 et diffusées depuis aux professionnels de santé pour les sensibiliser à l'importance de la notification spontanée des effets indésirables. En outre, les compétences des centres régionaux de pharmacovigilance (CRPV) seront désormais sollicitées lors de la mise en place des plans de gestion de risques. Leur réactivité est en effet un facteur essentiel pour assurer une réponse adéquate en situation de crise. Enfin, la nouvelle base informatique de pharmacovigilance devrait améliorer sous peu le traitement et la gestion de l'information recueillie, grâce à la déclaration électronique des effets indésirables.
4. Le développement de la pharmaco-épidémiologie.
La connaissance des conditions réelles de prescription et d'utilisation des médicaments est nécessaire pour évaluer les risques et assurer leur bon usage. La réalisation d'études de pharmaco-épidémiologie s'intègre donc dans les plans de gestion de risques pour compléter les données issues des notifications spontanées. Elles permettent de quantifier les risques, d'en identifier les facteurs mais aussi d'affiner l'évaluation du bénéfice sur la base de données de co-morbidité. Leur réalisation requiert l'utilisation de bases de données complètes. Dans cette optique, l'AFSSAPS a engagé un état des lieux des outils, structures, méthodes et actions disponibles pour assurer ce type d'études. En outre, les CRPV sont invités à renforcer leurs liens avec les équipes universitaires spécialisées en pharmaco-épidémiologie dans le cadre d'études communes, comme celle menée sur les accidents automobiles et les médicaments.
5. Le renforcement des inspections en pharmacovigilance
Les entreprises pharmaceutiques font l'objet d'inspections de leur système de pharmacovigilance, qui s'inscrivent dans un programme systématique d'inspections régulières globales des laboratoires ou dans un programme ciblé d'inspections approfondies.
6. La gestion active de l'iatrogénèse médicamenteuse
Au-delà de la gestion des effets indésirables survenant dans des conditions normales, il s'agit de prendre en compte les accidents liés aux mauvaises pratiques et/ou aux problèmes de conditionnement du médicament. Aussi, un guichet unique a été créé au sein de l'AFSSAPS, dans le cadre du nouveau département de surveillance du risque et d'information sur le bon usage, pour recueillir et coordonner la gestion des signalements et des risques d'erreurs liés à un défaut de présentation du médicament, en particulier d'étiquetage.
7. La promotion du bon usage des médicaments
Cette information de l'AFSSAPS s'adresse aux professionnels de santé et aux patients. Elle peut répondre à une question d'actualité concernant le médicament (mise au point) ou s'inscrire dans une démarche plus générale de stratégie thérapeutique (recommandation de bonne pratique). En décembre 2003 a ainsi été diffusée une mise au point actualisée sur le traitement hormonal substitutif de la ménopause (THS).
Source : Rapport n° 382 de Mme Marie-Thérèse Hermange, au nom de la mission d'information de la commission des affaires sociales du Sénat sur les conditions de mise sur le marché et de suivi des médicaments, « Restaurer la confiance » (juin 2006)
c) Le renforcement de la réglementation concerne aussi les matières premières à usage pharmaceutique
S'agissant de la qualité des médicaments, il est impératif de contrôler tous les composants des médicaments sur toute la chaîne, de la fabrication à la distribution. Ainsi, le considérant 19 de la directive précise qu'il « convient de garantir la qualité des médicaments à usage humain fabriqués ou disponibles dans la Communauté, en exigeant que les substances actives qui entrent dans leur composition soient conformes aux principes relatifs aux bonnes pratiques de fabrication. Il s'avère nécessaire de renforcer les dispositions communautaires relatives aux inspections et de mettre en place un registre communautaire portant sur les résultats de ces inspections ».
Les matières premières à usage pharmaceutique sont une des composantes des médicaments. A ce titre, il convient d'en surveiller la fabrication et toutes les opérations afférentes (vente, cession, distribution ...) afin que les impératifs de sécurité sanitaire soient respectés.
Le régime juridique applicable aux matières premières à usage pharmaceutique fait l'objet des articles 23 à 25 du présent projet. L'article 23 propose que le code de la santé publique définisse les matières premières à usage pharmaceutique et crée une présomption d'usage pharmaceutique pour ces matières. L'article 24 du présent projet définit la composition des matières premières à usage pharmaceutique et propose d'introduire de bonnes pratiques de fabrication et de distribution des matières premières à usage pharmaceutique. Les principes de ces bonnes pratiques seront définis conformément au droit communautaire par une décision de l'AFSSAPS, après avis de l'AFSSA (Agence française de sécurité sanitaire des aliments).
L'article 25 fixe les compétences d'inspection de l'AFSSAPS en ce qui concerne les matières premières à usage pharmaceutique. L'Agence, dans le cadre de ses pouvoirs d'inspection des opérations relatives aux matières premières pharmaceutiques délivrera un « certificat de conformité » lorsqu'elle constate que les opérations respectent les bonnes pratiques.
La directive comporte des mesures très importantes en matière de transparence des travaux des agences sanitaires, qu'il s'agisse de leur fonctionnement interne ou de la publicité de leurs décisions et des motivations de celles-ci. Le fonctionnement des agences chargées de l'autorisation des médicaments et de la pharmacovigilance a fait l'objet de nombreuses critiques. Une critique récurrente porte sur les conditions de la publicité relative aux décisions des agences. Selon certains, l'opacité supposée de ce fonctionnement et notamment l'absence de publicité des motivations des décisions cautionneraient des décisions contestables.
La publicité du processus de décision des agences, dès lors qu'il ne remet pas en cause le secret industriel et la propriété intellectuelle, est un élément renforçant la crédibilité de ces institutions. D'un point de vue démocratique, elle promeut le contrôle des citoyens sur des autorités disposant de compétences très importantes et échappant parfois, en raison de leur statut d'autorité indépendante, à un contrôle politique.
La directive n° 2004/27, de ce point de vue, constitue un progrès dans la voie de la transparence. Le règlement opère des modifications similaires s'agissant de l'agence européenne. En particulier, le deuxième alinéa du paragraphe 126 ter qu'elle introduit dans le code communautaire oblige chaque agence nationale à rendre accessible au public son règlement interne et celui de ses comités, l'ordre du jour de ses réunions, les comptes rendus de ses réunions, assortis des décisions prises, des détails des votes et des explications de vote, y compris les opinions minoritaires.
1. La publicité des dossiers d'autorisation de médicaments est de nature à renforcer la légitimité et la crédibilité des décisions de l'AFSSAPS
L'essentiel des dispositions relatives à l'information des patients et au fonctionnement des agences sanitaires sera transposé par voie règlementaire. En effet, il ne relève pas de la loi de se substituer au règlement intérieur d'une agence sanitaire.
a) Le présent projet transpose des dispositions de la directive prescrivant une transparence accrue du fonctionnement des agences chargées du médicament
L'AFSSAPS a fait des progrès significatifs pour se mettre en conformité avec la directive n° 2004/27. Elle a ainsi publié sur Internet un compte rendu de la commission nationale de pharmacovigilance. Le 25 avril dernier, elle a également mis en ligne pour la première fois un compte rendu de la commission d'autorisation de mise sur le marché (commission d'AMM du 2 mars dernier). Selon la rédaction de la « Revue Prescrire », dans sa livraison du 1er juin dernier, « il ne s'agit pas de véritables « minutes » de la réunion de la commission, mais d'un compte rendu critiquable, publié plus d'un mois après la réunion. » Cependant, on peut considérer qu'il s'agit d'un progrès significatif. De même, il faut souligner que l'AFSSAPS met à disposition du public sur Internet un nombre croissant de résumés de caractéristiques principales (RCP) des spécialités pharmaceutiques. Cependant, l'ordre du jour des réunions des conseils et commissions n'est toujours pas en ligne.
L'article 26 du présent projet propose que l'agence rende publique la synthèse des dossiers d'autorisation des médicaments (il s'agit en fait de la publicité des synthèses des dossiers d'évaluation). Cette disposition consacre en partie la pratique actuelle de l'AFSSAPS, qui publie sur Internet un rapport public d'évaluation pour chaque nouvelle autorisation de mise sur le marché (AMM) ou chaque modification majeure d'AMM. Ce rapport est rédigé conformément à l'article 21 du code communautaire (la loi n° 98-535 du 1er juillet 1998 relative au renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits destinés à l'homme disposait déjà que l'AFSSAPS rend publique une synthèse des dossiers d'autorisation de tout nouveau médicament).
L'agence publie également une fiche de synthèse relative à l'autorisation, fiche qui résume les enjeux scientifiques de l'AMM. Un autre élément indispensable à l'amélioration de la publicité des travaux de l'AFSSAPS a consisté à rendre accessible les déclarations d'intérêts des experts et membres des conseils et commissions de l'Agence (cf. infra).
A l'initiative du Parlement européen, la directive n° 2004/27 comporte plusieurs dispositions visant à améliorer l'étiquetage et la notice des médicaments, dont l'article 15 du projet de loi prévoit la transposition par voie réglementaire.
Pour ne prendre que quelques exemples, il est notamment prévu que lorsque qu'un médicament contient plus de trois substances actives, sa dénomination commune internationale (DCI) doit être portée sur son emballage extérieur et son conditionnement primaire (par exemple un flacon), ce qui pourrait notamment permettre de diminuer les risques de mésusage pour le patient. Le nom du médicament devra également figurer en braille et l'exploitant d'un médicament devra veiller à ce que la notice soit disponible dans des formats appropriés pour les aveugles et les malvoyants.
Dans le même sens, la directive prévoit que les notices doivent refléter les résultats de la consultation de groupes cibles de patients « afin de garantir sa lisibilité, sa clarté et sa facilité d'utilisation », ces résultats devant par ailleurs être communiqués aux autorités compétentes des États membres.
Mieux informé, le patient pourra ainsi se prémunir plus efficacement contre les risques éventuellement présentés par le médicament pour sa santé.
D. LA TRANSPOSITION DE LA DIRECTIVE CONSTITUE UN APPORT INDISPENSABLE À LA LIMITATION DE L'INFLUENCE DE L'INDUSTRIE PHARMACEUTIQUE
Le présent projet permet d'une part de règlementer la publicité pour les médicaments et de limiter l'influence de l'industrie pharmaceutique.
La directive continue d'interdire toute publicité directe auprès du grand public pour les médicaments prescrits sur ordonnance.
On distingue en général trois types de publicité pour les médicaments, selon une typologie qui aurait été élaborée par la Food and Drugs Administration (institution des Etats-Unis chargée notamment de l'autorisation des nouveaux médicaments) :
- la « publicité complète » ou intégrale, qui mentionne la marque et les indications ; ce type de publicité est souvent accompagné de l'obligation légale d'informer sur les risques et les effets secondaires ;
- la « publicité de rappel » (publicité mentionnant exclusivement la marque, la dénomination commune internationale ou le nom du médicament) ;
- la « publicité de demande d'aide » (description d'une situation présentée comme pathologique et suggestion d'aller voir un médecin, sans que soit mentionné le médicament).
Il arrive qu'un produit puisse simultanément faire l'objet d'une campagne de publicité de demande d'aide et d'une campagne de publicité de rappel.
Sur le sujet, on peut se référer utilement à une publication rédigée par le Conseil canadien de la santé (14) ; les Canadiens sont en effet plus sensibilisés à ce problème en raison de la diffusion au Canada sur les grands réseaux de télévision américains de publicités pour les médicaments prescrits sur ordonnance.
Seuls les Etats-Unis et la Nouvelle-Zélande autorisent la publicité auprès du grand public pour les médicaments devant faire l'objet d'une prescription médicale. Les effets positifs allégués d'une telle publicité - si l'on écarte l'augmentation attendue des ventes de médicaments, au bénéfice des laboratoires et de leurs actionnaires - sont principalement l'information procurée aux personnes, aux patients et à leurs proches. La publicité auprès du public pour les médicaments prescrits sur ordonnance, selon ses partisans, permettrait d'accroître le niveau de bien-être des personnes. En effet, elle accroîtrait les possibilités de dépistage et de prévention des pathologies, permettant ainsi de promouvoir les diagnostics précoces. Elle encouragerait l'autonomie du patient et le dialogue avec le médecin. Elle parierait sur la maturité du citoyen consommateur, qui serait capable de recul vis-à-vis des publicités. Dans un contexte où Internet constitue une source de publicité complètement déréglementée, voire dangereuse (notamment au travers des pourriels), la publicité produite et validée par des firmes pharmaceutiques, visant des médicaments approuvés et distribués par les canaux traditionnels de distribution, serait une garantie de sérieux.
b) L'introduction éventuelle de la publicité auprès du grand public pour les médicaments sur ordonnance constitue un enjeu majeur pour l'Europe
La possibilité de l'introduction dans l'Union européenne de la publicité directe aux consommateurs pour les médicaments prescrits sur ordonnance, parfois déguisée en « information des patients » est un enjeu majeur, et cela à de multiples points de vue. Elle pose quatre grands types de questions :
- la santé publique sera-t-elle améliorée ?
- les publicités constituent des masses financières énormes pour les diffuseurs vendant les espaces publicitaires, or ces diffuseurs présentant parfois des programmes d'information relatifs à la santé ; l'indépendance de ces médias sera-t-elle menacée (15) ?
- le prix du médicament sera-t-il augmenté ?
- l'augmentation éventuelle des ventes des laboratoires permettra-t-elle d'alimenter les dépenses de recherche et développement des firmes européennes, leur permettant de rivaliser avec les entreprises américaines ?
Les autorités européennes ont finalement rejeté la possibilité de la publicité directe aux consommateurs pour les médicaments prescrits sur ordonnance. Sur ce point précis de la règlementation de la publicité auprès du grand public pour les médicaments d'ordonnance, l'historique de l'élaboration de la directive est particulièrement intéressant.
En juillet 2001, la Commission européenne a lancé un projet de révision du code communautaire des médicaments à usage humain visant à permettre, pour une période d'essai de cinq ans, la publicité des médicaments d'ordonnance visant trois affections chroniques : le SIDA, le diabète et l'asthme. Ce projet rencontre une vive résistance dans l'opinion publique et parmi les autres institutions de l'Union européenne, le Parlement et le Conseil des ministres. En octobre 2002, une majorité de parlementaires européens vote contre cette proposition. En 2003, le Conseil des ministres se prononce également contre cette initiative.
La version définitive de la directive (article 63) interdit la publicité auprès du public pour les médicaments prescrits sur ordonnance. Elle autorise la publicité pour les médicaments qui, par leur composition et leur objectif, sont destinés à être utilisés sans intervention d'un médecin pour le diagnostic, la prescription ou la surveillance du traitement, au besoin avec le conseil du pharmacien, et conçus dans cette optique. En outre, elle autorise les États à interdire la publicité auprès du public pour les médicaments remboursables. Néanmoins, la rédaction de l'article 63 de la directive indique que la Commission n'a pas totalement renoncé à son projet. L'article prévoit que la Commission présente au Parlement européen et au Conseil un rapport sur les « pratiques actuelles en matière de communication d'information - notamment par Internet - et sur leurs risques et leurs avantages pour les patients ». Il charge la Commission de formuler, s'il y a lieu, des propositions « définissant une stratégie d'information assurant une information de qualité, objective, fiable et non publicitaire sur les médicaments ».
c) Les programmes d'accompagnement des patients doivent faire l'objet d'une surveillance particulière
La rapporteure estime tout à fait opportun que la publicité pour les médicaments prescrits sur ordonnance ne puisse s'adresser au grand public. En effet, la publicité dérèglementée en direction du public peut avoir des effets désastreux sur la santé publique en poussant à la surconsommation de médicaments et en modifiant les rapports entre patients et médecins et pharmaciens. Outre les questions liées à la soutenabilité financière d'un tel processus, les effets iatrogéniques de ces excès soulignent la nécessité de limiter l'influence de la publicité.
Le régime de la publicité auprès du public pour les médicaments est fixé par l'article L. 5122-6 du code de la santé publique, dont le premier alinéa dispose que la publicité auprès du public pour un médicament n'est admise qu'aux conditions suivantes : ce médicament ne doit pas être soumis à prescription médicale, il ne doit pas être remboursable et l'autorisation de mise sur le marché ou l'enregistrement ne comporte pas de restrictions en matière de publicité auprès du public en raison d'un risque possible pour la santé publique. Le dernier alinéa de l'article précise que la publicité auprès du public pour un médicament est nécessairement accompagnée d'un message de prudence et de renvoi à la consultation d'un médecin en cas de persistance des symptômes.
Les conditions de la publicité pour les médicaments auprès du public sont renforcées par le présent projet, qui propose de renvoyer à la compétence règlementaire la définition des mentions obligatoires devant figurer sur les publicités pour les médicaments (mention de l'appartenance à la catégorie des spécialités génériques, conseils de prudence...). De plus, l'enregistrement ou l'autorisation de mise sur le marché pourront non seulement comporter des restrictions en matière de publicité auprès du public, mais aussi comporter une interdiction totale de publicité. En outre, les motifs possibles de cette restriction ou interdiction sont élargis : il est proposé que les restrictions ou l'interdiction du publicité mentionnées par l'autorisation de mise sur le marché ou l'enregistrement soient introduites non seulement « en raison d'un risque possible pour la santé publique » mais aussi « lorsque le médicament n'est pas adapté à une utilisation sans intervention d'un médecin pour le diagnostic, l'initiation, ou la surveillance du traitement ».
En revanche, les publicités dites « de rappel » sont autorisées. L'article 19 du présent projet propose qu'un décret en Conseil d'État autorise les publicités dites « de rappel » ne comprenant que le nom, la dénomination commune internationale ou la marque du médicament.
Enfin, la rapporteure souligne qu'il faudra notamment être vigilant quant aux conditions de conclusion des partenariats privés-publics visant à améliorer l'information fournie aux patients. En effet, le II de l'article 29 du présent projet prévoit d'habiliter le gouvernement à fixer par ordonnance le régime juridique des programmes d'accompagnement menés par les entreprises pharmaceutiques en direction des patients. Compte tenu des dangers potentiels présentés par cette évolution, il faudrait préciser dans l'habilitation législative que les programmes d'observance sont soumis à l'autorisation préalable de l'AFSSAPS.
2. Les relations entre les firmes pharmaceutiques et les professions médicales et pharmaceutiques sont mieux réglementées
Le projet de loi comporte une série de dispositions visant à clarifier les relations entre les firmes pharmaceutiques et les professions de santé.
Son article 2 prévoit ainsi que l'hospitalité, qui peut être offerte à ces derniers, lors de congrès ou de journées d'études par exemple et à la condition qu'elle soit prévue par une convention soumise au conseil de l'ordre compétent, devra désormais être strictement limitée à l'objectif principal de la manifestation. La mise en œuvre du dispositif de formation obligatoire continue, prévue par la loi du 9 août 2004 relative à la politique de santé, permettra par ailleurs de renforcer l'efficacité de ce dispositif.
« Le médecin ne peut aliéner son indépendance professionnelle » :
commentaire de l'article 5 du code de déontologie des médecins
« Aucun avantage personnel ne doit conduire le médecin. Son indépendance vis-à-vis de l'argent doit être claire : indépendance de façon directe (dessous de table) ou indirecte (commissions, ristournes, dichotomies) - cf. articles 22, 23 et 24 du code de déontologie.
La mise en application, confiée à l'Ordre, de l'article L. 4113-6 du code de la santé publique fait apparaître les dangers auxquels est exposée l'indépendance du médecin dans ses relations avec les industriels de la pharmacie, des techniques médicales, aussi bien dans les phases de recherche que dans les périodes d'utilisation des produits.
Pour garantir l'indépendance du médecin, le législateur a souhaité, que sa relation avec l'industrie soit transparente. Pour en permettre le contrôle, la convention passée entre eux doit faire l'objet d'un avis de l'Ordre. Dans cette mission de contrôle, ce qui est pris en compte - quelle que soit la nature de l'avantage consenti, en particulier dans les travaux de recherche - n'est pas le montant de la rémunération en lui-même, mais son adéquation à la charge de travail imposée. Une somme d'argent disproportionnée (ou des prises en charge de frais) ne peut être consentie à un médecin : cela reviendrait à le fidéliser directement ou indirectement, et ainsi à orienter ses prescriptions.
En autorisant les industriels à prendre en charge des frais de formation médicale continue, la loi a, par ailleurs, précisé et même souligné que l'hospitalité (lors des congrès, colloques, journée, etc.) doit être d'un niveau raisonnable et rester accessoire par rapport à l'objectif principal de la réunion (article 24). »
Source : Site Internet du Conseil national de l'ordre des médecins (CNOM)
En outre, l'article 18 du présent projet règlemente la distribution des échantillons de médicaments en posant le principe d'une interdiction de remise « directe » d'échantillons au public à des fins promotionnelles. De plus, il limite les possibilités de « cadeaux » que peuvent remettre les laboratoires aux médecins et pharmaciens, en disposant que les cadeaux fournis doivent avoir un lien avec l'exercice de la médecine ou de la pharmacie.
b) Le principe de la déclaration d'intérêts est étendu à tous les personnels travaillant pour l'AFSSAPS
Un autre enjeu de la promotion de la transparence du fonctionnement des agences est le respect du principe d'indépendance des agents travaillant pour ces autorités. Une des dispositions visant à garantir cette transparence est la déclaration d'intérêts.
Le premier alinéa de l'article 126 ter du code communautaire dispose que les agents de l'agence nationale, les rapporteurs et les experts ne doivent avoir dans l'industrie pharmaceutique aucun intérêt financier ou autre qui pourrait nuire à leur impartialité. Ces personnes doivent faire annuellement une déclaration de leurs intérêts financiers. L'article 126 ter n'indique pas que ces déclarations doivent être rendues publiques. Elles sont donc accessibles en vertu d'une disposition de droit interne.
Le principe de la déclaration d'intérêts est étendu à tous les personnels travaillant pour l'AFSSAPS. En effet, l'article 28 du présent projet propose d'étendre à toutes les catégories de personnels travaillant pour l'AFSSAPS l'obligation de faire une déclaration de leurs intérêts. Cette déclaration porte non seulement sur les intérêts de la personne détenus dans le domaine pharmaceutique, mais aussi dans les sociétés de conseil. L'obligation est étendue aux agents travaillant au sein de l'AFSSAPS et relevant d'un statut de fonctionnaires, ainsi qu'aux agents contractuels de droit public ou privé.
La commission a examiné, sur le rapport de Mme Cécile Gallez, le présent projet de loi au cours de sa séance du 28 juin 2005.
Un débat a suivi l'exposé de la rapporteure.
M. Claude Evin a tout d'abord estimé que si ce texte, assez technique, ne pose pas de problèmes majeurs dans l'ensemble, plusieurs de ses dispositions nécessitent toutefois d'être amendées. En particulier, le recours aux ordonnances, prévu par l'article 29 du projet de loi, est toujours difficilement acceptable pour les parlementaires et il l'est en l'occurrence d'autant moins que le dixième alinéa de cet article ne se limite pas à transposer une directive, puisqu'il concerne également les « programmes d'observance », c'est-à-dire les actions d'accompagnement des patients. Alors que les principes relatifs à la publicité ont été définis par la loi, on ne peut que déplorer que le Parlement soit ainsi privé de la possibilité d'engager un véritable débat sur ce point. L'examen rapide du rapport de Mme Cécile Gallez soulève au surplus des questions d'importance, telles que celle relative au choix entre une procédure déclaratoire ou d'autorisation a priori, choix dont le législateur aurait pu utilement être saisi. Par ailleurs, le projet de loi n'aurait pas été excessivement alourdi s'il avait comporté des dispositions relatives aux conditions de mise en œuvre des programmes d'observance. Le recours aux ordonnances ne peut donc être accepté sur ce point.
Après avoir souligné la clarté de l'intervention de la rapporteure, M. Laurent Wauquiez a souhaité avoir des précisions sur l'articulation entre la procédure d'AMM des génériques et la protection par le brevet des princeps. Il semblerait en effet qu'avant l'expiration des droits de propriété intellectuelle du princeps, un générique puisse obtenir une AMM et être commercialisé, au mépris du droit des brevets. Alors que, dans le cadre d'une politique industrielle, de nombreux pays européens se sont dotés d'outils juridiques destinés à lutter contre ces pratiques, rien n'existe de la sorte en France et les décisions judiciaires seront longues à intervenir. Dès lors, l'examen de projet de loi ne pourrait-il pas permettre de remédier à cette carence, en renforçant le régime de protection des brevets ?
En réponse aux différents intervenants, la rapporteure a apporté les précisions suivantes :
- la technicité de certaines dispositions d'adaptation au droit communautaire dans le domaine du médicament, s'agissant par exemple des cosmétiques, imposait de recourir aux ordonnances ;
- un amendement sera présenté afin de subordonner la mise en œuvre des programmes d'observance à une autorisation préalable de l'AFSSAPS ;
- si un générique peut effectivement obtenir une AMM pendant la période de protection par le brevet du princeps, sa commercialisation ne peut toutefois intervenir qu'à l'expiration des droits de propriété intellectuelle de la spécialité de référence.
M. Laurent Wauquiez a néanmoins souligné que, dans les faits, l'appareil judiciaire français, en raison de ses lenteurs, ne permet pas de protéger efficacement les droits de l'exploitant du princeps en matière d'exclusivité commerciale.
Le président Jean-Michel Dubernard a jugé opportun d'interroger le gouvernement sur ses intentions en matière de protection des brevets, lors de l'examen de ce projet de loi en séance publique.
La commission a examiné les articles du présent projet de loi au cours de sa séance du 28 juin 2005.
Chapitre Ier
Dispositions relatives aux médicaments
Article 1er
Conditions d'exonération de la responsabilité des professionnels de santé, des fabricants et des titulaires de l'autorisation d'utilisation ou de mise sur le marché d'un médicament dans le cas d'une menace sanitaire grave
Cet article a pour objet de transposer les dispositions prévues par l'article 5 de la directive n° 2001/83/CE du 23 octobre 2001 instituant un code communautaire relatif aux médicaments à usage humain (16), telle que modifiée par la directive n° 2004/27 du 31 mars 2004 (17) du Parlement européen et du Conseil.
Il s'agit ainsi de mieux anticiper l'ensemble des mesures qui pourraient s'avérer nécessaires pour faire face à un risque pour la santé publique d'une particulière gravité.
1. Les conditions actuelles d'exonération de la responsabilité des professionnels de santé dans le cas d'une menace sanitaire grave
Inséré par l'article 18 de la loi n° 2004-806 du 9 août 2004 relative à la politique de santé publique, l'article L. 3110-3 du code de la santé publique dispose, dans son unique alinéa, que les professionnels de santé ne peuvent être tenus pour responsables des dommages entraînés par l'utilisation d'un médicament, dès lors qu'elle s'impose en raison d'une menace sanitaire grave.
Ce régime dérogatoire à l'article L. 1142-1 du même code, qui fixe les principes généraux de la responsabilité des professionnels de santé, est toutefois très strictement encadré :
- Il concerne uniquement « les dommages résultant de la prescription ou l'administration d'un médicament », ce qui exclut par exemple les gestes techniques ou les dispositifs médicaux.
- Il n'est applicable qu'à la condition que les « conditions normales d'utilisation prévues par l'autorisation de mise sur le marché » (AMM) du médicament ne soient pas respectées, concernant par exemple sa posologie ou son délai d'administration : les médicaments disposant d'une autorisation temporaire d'utilisation (ATU) ou ceux qui ne sont pas autorisés n'entrent donc pas dans le champ d'application de cet article.
- L'intervention du professionnel de santé doit avoir été rendue nécessaire par « l'existence d'une menace sanitaire grave » : il peut s'agir par exemple d'un risque de pandémie grippale ou encore d'une attaque bioterroriste.
- Enfin, la prescription ou l'administration du produit doit avoir été « recommandée par le ministre chargé de la santé », dans le cadre de ses pouvoirs de police sanitaire, tels que définis par l'article L. 3110-1 du même code. Dans le cas d'une menace sanitaire grave appelant des mesures d'urgence, en particulier une épidémie, le ministre de la santé dispose en effet de la faculté de prescrire toute mesure proportionnée aux risques courus et appropriée aux circonstances de temps et de lieu afin de prévenir et de limiter les conséquences des menaces possibles sur la santé de la population.
Parachevant ce dispositif, l'article L. 3110-4 du même code prévoit enfin que, sans préjudice des actions qui pourraient être exercées dans les conditions de droit commun, la réparation intégrale des accidents médicaux, des affections iatrogènes et des infections nosocomiales, imputables à des activités de prévention, de diagnostic ou de soins, réalisées en application des mesures de police sanitaire prévues par l'article L. 3110-1, incombe à l'Office national d'indemnisation des accidents médicaux, des affections iatrogènes et des infections nosocomiales (ONIAM).
2. Les dispositions de la directive n° 2004/27 modifiant l'article 5 de la directive n° 2001/83
A l'initiative du Parlement européen, l'article 5 de la directive n° 2001/83 a été modifié par l'article 1er de la directive n° 2004/27 afin de renforcer les moyens d'action des pouvoirs publics face à un danger majeur pour la santé publique, quelle que soit son origine (terroriste, accidentelle ou pandémique).
« En réponse à la propagation suspectée ou confirmée d'agents pathogènes, de toxines, d'agents chimiques ou de radiations nucléaires qui sont susceptibles de causer des dommages », l'article 5 modifié permet ainsi aux États membres d'autoriser temporairement, pour des raisons de santé publique justifiées et sous certaines conditions, la distribution d'un médicament non autorisé.
Conformément au paragraphe 3 de cet article, les États membres sont par ailleurs chargés de veiller à ce que la responsabilité civile ou administrative des professionnels de santé, mais aussi des titulaires de l'AMM et des fabricants, ne puisse être engagée, dans ces circonstances particulières, « pour toutes les conséquences résultant de l'utilisation d'un médicament en dehors des indications autorisées ou de l'utilisation d'un médicament non autorisé ».
Il est enfin précisé que ces dispositions s'appliquent indépendamment du fait qu'une autorisation, nationale ou communautaire, ait été accordée ou non.
3. La transposition proposée : l'extension du champ de l'exonération de la responsabilité des professionnels en cas de menace sanitaire grave
Afin de transposer ces nouvelles dispositions en droit interne, le présent article propose de modifier la rédaction de l'article L. 3110-3 précité sur les trois points suivants.
· Le I du présent article élargit tout d'abord les cas dans lesquels la responsabilité des professionnels de santé ne peut être engagée.
Ce régime dérogatoire s'appliquerait ainsi lorsque le médicament a été prescrit ou administré en dehors des conditions normales d'utilisation prévues par son AMM, comme c'est actuellement le cas, mais aussi :
- « en dehors des indications thérapeutiques » prévues par cette même autorisation, par souci de clarification ;
- en dehors des conditions et indications prévues par son autorisation temporaire d'utilisation (ATU) [sur cette notion, cf. infra, le commentaire présenté sous l'article 12 du projet de loi] ;
- enfin, lorsque le médicament ne fait l'objet d'aucune autorisation, qu'il s'agisse d'une AMM ou d'une ATU.
· Le II vise à clarifier la rédaction du même article L. 3110-3 afin de préciser la nature des pouvoirs confiés au ministre chargé de la santé.
Reprenant la terminologie employée par l'article 5 modifié de la directive n° 2001/83, ce paragraphe prévoit en effet que la prescription ou l'administration du médicament doit avoir été recommandée « ou exigée » par le ministre. De fait, cette nouvelle rédaction est plus cohérente avec les dispositions prévues par l'article L. 3110-1, puisque celui-ci prévoit que le ministre peut « prescrire, par arrêté motivé », toute mesure proportionnée aux risques encourus : ses pouvoirs excèdent ainsi ceux d'une simple recommandation.
· Le III complète l'article L. 3110-3 par un nouvel alinéa visant à étendre le champ de l'exonération de la responsabilité des professionnels de santé à deux nouvelles catégories de personnes :
- Est exonéré le fabricant d'un médicament, qui ne peut non plus être tenu pour responsable des dommages résultant de l'utilisation d'un médicament, et ce, sous les mêmes conditions que celles prévues pour les professionnels de santé. La prescription ou l'administration d'un médicament, non autorisé ou lorsque les indications et conditions d'utilisation prévues par son AMM ou son ATU ne sont pas respectées, doit en effet au préalable avoir été recommandée ou exigée par le ministre de la santé, en application de l'article L. 3110-1 précité.
- Sont également exonérés de responsabilité, sous les mêmes réserves, le titulaire de l'AMM (il arrive en effet fréquemment qu'il ne soit pas le fabricant du médicament, ce dernier devant être autorisé en tant qu'établissement pharmaceutique, contrairement au titulaire de l'autorisation), de l'ATU ou de l'autorisation d'importation d'un médicament.
Sur ce dernier point, il convient de rappeler que tout médicament ne disposant pas d'une AMM ou d'une ATU accordée pour des médicaments importés doit faire l'objet, sauf cas particuliers, d'une autorisation délivrée par le directeur général de l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) avant son importation dans le territoire douanier, dans les conditions prévues par les articles R. 5121-109 à R. 5121-114 ou au titre de l'autorisation d'importation parallèle, définie aux articles R. 5121-115 et suivants du même code.
Concernant une question différente mais connexe, on peut d'ailleurs noter que la mission d'information de l'Assemblée nationale sur la grippe aviaire a également soulevé le problème de la responsabilité juridique des fabricants, dans son rapport de janvier dernier (18). Des difficultés risqueraient en effet d'apparaître si un médicament, en l'occurrence un vaccin pandémique, était utilisé dans des circonstances particulières, liées cette fois aux conditions de production en masse et dans l'urgence de celui-ci. A cet égard, le rapporteur, M. Jean-Pierre Door, proposait d'envisager le règlement de cette question au niveau législatif, s'agissant notamment des conditions de prise en charge du risque vaccinal.
La dernière phrase de cet alinéa précise enfin que ces nouvelles dispositions n'exonèrent en aucun cas les fabricants et les titulaires de l'AMM de la responsabilité qu'ils encourent, dans les conditions de droit commun, en raison de la fabrication ou de la mise sur le marché du médicament.
Dans ce sens, il convient en effet de rappeler que, sur proposition du Conseil, le dernier paragraphe de l'article 5 du code communautaire dispose que la responsabilité du fait des produits défectueux, au sens de la directive n° 85/374 du Conseil (19), n'est pas affectée par le paragraphe 3 du même article, relatif aux exonérations de responsabilité.
*
La commission a adopté l'article 1er sans modification.
Article 2
Renforcement la réglementation des relations entre
les professionnels de santé et les entreprises pharmaceutiques
Cet article procède à la transposition des dispositions dites « anti-cadeaux » de la directive n° 2004/27, qui visent à mieux encadrer les relations entre les professionnels de santé et les industries pharmaceutiques.
1. Les dispositions actuellement prévues par l'article L. 4113-6 du code de la santé publique
a) Le principe : l'interdiction pour les professionnels médicaux de recevoir des avantages de la part des entreprises pharmaceutiques
Les personnes habilitées à prescrire des médicaments doivent être à même d'exercer cette responsabilité en toute objectivité, sans être influencées par des incitations financières de nature à aliéner leur indépendance professionnelle.
C'est pourquoi l'article L. 4113-6 du code de la santé publique pose l'interdiction, pour les membres des professions médicales mentionnées au livre Ier du même code, parmi lesquelles les médecins, les sages-femmes et les chirurgiens dentistes, de « recevoir des avantages en nature ou en espèces, sous quelque forme que ce soit, d'une façon directe ou indirecte, procurés par des entreprises assurant des prestations, produisant ou commercialisant des produits pris en charge par les régimes obligatoires de sécurité sociale ».
Cette interdiction s'applique également aux pharmaciens depuis la loi n° 2002-303 du 4 mars 2002 relative aux droits des malades et à la qualité du système de santé.
Il convient par ailleurs de rappeler que les rapports des professionnels de santé avec l'industrie pharmaceutique sont également strictement encadrés par les différentes règles déontologiques qui leur sont applicables.
S'agissant par exemple des médecins, leur code de déontologie dispose ainsi qu'ils ne peuvent aliéner leur indépendance professionnelle sous quelque forme que ce soit (article R. 4127-5 du même code), doivent se garder de toute attitude publicitaire (article R. 4127-13) et ne peuvent solliciter ou accepter un avantage, en nature ou en espèces, pour une prescription ou un acte médical quelconque (R. 4127-24). Ainsi qu'il est dit à l'article R. 4127-19, la médecine ne saurait, en d'autres termes, être exercée comme un commerce.
b) L'exception : l'hospitalité offerte lors de manifestations de promotion ou à caractère exclusivement professionnel et scientifique
L'article L. 4113-6 aménage deux types de dérogations à l'interdiction de principe posée par son premier alinéa. L'une d'entre elles concerne « l'hospitalité offerte, de manière directe ou indirecte, lors de manifestations de promotion ou lors de manifestations, à caractère exclusivement professionnel et scientifique » (par exemple congrès, colloques, journées d'étude ou de formation), sous réserve toutefois que soient réunies les conditions définies par son troisième alinéa :
- l'hospitalité offerte doit tout d'abord être prévue par une convention passée entre l'entreprise et le professionnel, et soumise pour avis au conseil départemental de l'ordre compétent avant sa mise en application ;
- elle ne peut en aucun cas être étendue à des personnes autres que les professionnels directement concernés, par exemple les conjoints ;
- elle doit enfin être « d'un niveau raisonnable » et rester « accessoire par rapport à l'objectif principal de la réunion », ces dispositions reprenant précisément les termes des article 94 et 95 de la directive n° 2001/83, dans leur rédaction antérieure à la publication de la directive n° 2004/27.
Comme le souligne le Conseil national de l'ordre des médecins, le caractère raisonnable et accessoire ne peut cependant « être quantifié ni chiffré » et doit en conséquence faire l'objet d'« une appréciation multifactorielle », qui s'appuie notamment sur les critères définis par l'ordre de la façon suivante.
Critères mis en œuvre par l'ordre national des médecins pour apprécier la conformité des conventions d'hospitalité aux dispositions de l'article L. 4113-6
« L'hospitalité offerte doit être « raisonnable et accessoire » par rapport à l'intérêt médical de la réunion, dont l'objectif principal doit rester médical ; ce caractère raisonnable et accessoire ne peut être ni quantifié ni chiffré. Son appréciation étant multifactorielle, elle est confiée à un conseiller ordinal qui compare l'intérêt médical de la manifestation, la qualification des médecins invités et l'hospitalité proposée. L'avis repose sur l'analyse du programme scientifique détaillé (thème, durée du programme médical par rapport à la durée totale de la manifestation, lieu de son déroulement) et de l'hospitalité offerte (durée de prise en charge, montant, prise en charge totale ou partielle des frais, et évidemment absence de prise en charge de toutes activités de loisirs, sportives ou culturelles...).
- Pour les manifestations départementales ou de proximité, la prise en charge des frais de transport des médecins n'est pas justifiée (exceptions possibles pour intervenants). Il en est de même pour les manifestations régionales ou inter-régionales lorsque le lieu de la manifestation ne répond à aucun impératif médical ni à l'origine géographique des médecins invités (exceptions possibles pour intervenants).
- Pour les manifestations nationales, la prise en charge totale des frais d'inscription, de restauration, d'hébergement et de transport ne semble pas respecter le caractère raisonnable et accessoire de l'hospitalité admise par la loi (exceptions possibles pour intervenants), notamment si le programme médical ménage des plages de temps libre permettant des activités sociales, culturelles ou de loisirs en un lieu attractif ou touristique. Il appartient à l'entreprise organisatrice de fixer le taux de participation financière des médecins invités, par exemple au prorata de la charge de travail et des autres paramètres.
- Pour les manifestations internationales, dont les programmes prévoient assez souvent des plages de repos ou de temps libre, par ailleurs justifiées, il semble normal, pour les raisons déjà exposées ci-dessus, que les entreprises ne prennent pas en charge la totalité des frais d'inscription, de transport, d'hébergement ou de restauration (exceptions possibles pour les intervenants et les médecins ayant une convention de rapporteur du congrès).
Il appartient à l'entreprise de fixer le taux de la participation financière des médecins invités au prorata du temps libre et du principe d'une hospitalité raisonnable et accessoire. Par souci de transparence, le Conseil de l'ordre demande que les frais d'inscription (s'ils sont pris en charge) précisent les droits ouverts. Si des activités socioculturelles sont prévues par les organisateurs avec un montant individualisé dans les frais d'inscription, il appartient au médecin invité de régler directement ces frais, s'il entend participer à ces activités ; si leur coût n'est pas individualisé dans les droits d'inscription (cas fréquent aux Etats-Unis), il appartient aux entreprises de ne pas prendre en charge la totalité des frais d'inscription (...).
Pour toutes les manifestations évoquées ci-dessus, un système d'attestation de présence du médecin à chaque session du programme doit être mis en place, en général sous la forme de liste émargée par les médecins présents ; il s'agit d'une exigence normale qui par ailleurs s'est révélée très utile aux médecins dans le cadre des contrôles de la Direction générale de la concurrence et de la répression des fraudes (DGCCRF) ».
Source : Site Internet du Conseil national de l'ordre national des médecins
Enfin, l'article L. 4163-2 du même code prévoit que toute infraction aux dispositions prévues par l'article L. 4113-6 est punie de deux ans d'emprisonnement et de 75 000 euros d'amende. Des sanctions pénales ont par exemple été prononcées suite à un congrès organisé à Fort-de-France, au cours duquel la prise en charge de l'hébergement des praticiens invités a été étendue à leurs conjoints, ainsi que dans le cas d'une hospitalité offerte à Marbella, qui est apparue sans rapport avec l'objet et la durée de la réunion scientifique.
2. Les modifications proposées par le projet de loi
A l'initiative du Parlement européen, la directive n° 2004/27 a modifié les articles 94 et 95 du code communautaire concernant respectivement les manifestations de promotion des médicaments et celles à caractère exclusivement scientifique et professionnel.
Dans son rapport, au nom de la commission de l'environnement, de la santé publique et de la politique des consommateurs du Parlement européen, sur la proposition de modification de la directive n° 2001/83(20), Mme Françoise Grossetête notait en effet que « les activités de promotion des ventes se sont trouvées sous les feux de l'actualité dans divers États membres car elles ont abouti à un manque d'indépendance des professionnels de la santé », avant de conclure à la nécessité de rendre le dispositif actuel plus contraignant.
Le présent article propose en conséquence de modifier la rédaction du troisième alinéa de l'article L. 4113-6 du code de la santé publique afin de transposer littéralement les dispositions prévues par les articles 94 et 95 du code communautaire, tels que modifiés par la directive n° 2004/27.
Ainsi, l'hospitalité offerte aux professionnels médicaux devra désormais être strictement « limitée » à l'objectif principal de la manifestation, et non plus « accessoire » par rapport à celui-ci et « d'un niveau raisonnable ».
*
La commission a examiné un amendement de suppression de l'article présenté par M. Claude Evin.
M. Claude Evin a contesté le bien-fondé des dispositions prévues par cet article, en jugeant suffisamment protectrices les dispositions actuelles de l'article L. 4113-6 du code de la santé publique, relatives au dispositif dit « anti-cadeaux ».
La rapporteure s'est déclarée défavorable à l'amendement, en rappelant que cet article reprend les mêmes termes que ceux la directive n° 2004/27 du Parlement européen et du Conseil du 31 mars 2004, que la France est tenue de transposer.
La commission a rejeté l'amendement.
Elle a ensuite examiné un amendement de M. Claude Evin précisant que l'hospitalité offerte aux professionnels de santé doit être « d'un niveau raisonnable et limitée à l'objectif scientifique principal de la manifestation ».
Rappelant que cet article prévoit uniquement de limiter l'hospitalité offerte par les entreprises pharmaceutiques à « l'objectif principal de la manifestation », M. Claude Evin a jugé nécessaire de maintenir la condition, actuellement en vigueur, relative au niveau raisonnable de l'hospitalité. En effet, si l'on voulait polémiquer sur ce sujet, on pourrait insinuer qu'en organisant ces manifestations, l'objectif principal poursuivi par les entreprises pharmaceutiques n'est rien d'autre que d'accroître le volume de leurs ventes ! Aussi est-il nécessaire de mettre l'accent sur le caractère nécessairement scientifique de ces manifestations. Si cette proposition ne recueillait pas l'accord de la commission, il serait en tout état de cause nécessaire de compléter cet article par la référence au caractère raisonnable de l'hospitalité offerte, comme le propose l'amendement suivant, de repli, à défaut de quoi, on pourrait en venir à se poser la question du bien-fondé de la manifestation, pourquoi pas jusqu'à Pékin !
La rapporteure s'est opposée à l'amendement, au motif que le terme « scientifique » est trop restrictif au regard non seulement des dispositions de la directive n° 2004/27, mais aussi du fait que ces manifestations peuvent être l'occasion d'évoquer des questions plus générales, d'ordre professionnel. Il serait en revanche opportun de préciser que l'hospitalité doit être d'un niveau raisonnable, comme le propose l'amendement suivant.
M. Claude Evin a convenu que la rédaction de l'amendement est certainement perfectible : la référence à un objectif de santé publique a d'ailleurs été envisagée dans un premier temps, avant d'être finalement abandonnée au profit du terme, plus précis, d'objectif scientifique.
Saluant cette proposition, le président Jean-Michel Dubernard a estimé que la référence à l'objectif scientifique fait sens mais qu'il serait également souhaitable de mentionner l'objectif professionnel. Il est en revanche plus difficile de savoir à quoi renvoie exactement le terme « raisonnable » .
La rapporteure ayant fait observer que l'article L. 4113-6 du code de la santé publique fait référence à des manifestations à caractère professionnel et scientifique, M. Claude Evin a rectifié son amendement afin de préciser que l'hospitalité est « limitée à l'objectif professionnel et scientifique principal de la manifestation ».
Le président Jean-Michel Dubernard a salué cette rectification, tout en considérant que la rédaction en est sans doute encore perfectible.
Mme Jacqueline Fraysse s'est interrogée sur le maintien de dispositions concernant le niveau de la dépense.
La commission a adopté l'amendement ainsi rectifié : en conséquence, l'amendement suivant de M. Claude Evin est devenu sans objet.
La commission a ensuite adopté l'article 2 ainsi modifié.
Article 3
Actualisation de la définition du médicament et principe de l'application
de la réglementation pharmaceutique aux produits dits « frontière »
L'article L. 5111-1 du code de la santé publique distingue actuellement trois formes de médicaments, la dernière étant spécifique à la France :
- le médicament par la présentation : « toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l'égard des maladies humaines » (premier alinéa), ce critère ayant pour objectif de protéger la santé publique en évitant la commercialisation d'un produit dépourvu de tout effet thérapeutique, mais présenté comme un médicament ;
- le médicament par la fonction : « tout produit pouvant être administré à l'homme ou à l'animal, en vue d'établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions organiques » (premier alinéa), cette catégorie ayant été introduite par l'ordonnance n° 67-827 du 23 septembre 1967, transposant en droit interne la directive du 26 janvier 1965, aujourd'hui abrogée et codifiée dans la directive n° 2001/83 ;
- le médicament par la composition : « les produits diététiques qui renferment dans leur composition des substances chimiques ou biologiques ne constituant pas elles-mêmes des aliments, mais dont la présence confère à ces produits, soit des propriétés spéciales recherchées en thérapeutique diététique, soit des propriétés de repas d'épreuve » (deuxième alinéa).
Compte tenu notamment des progrès médicaux, le présent article vise à actualiser la définition du médicament et à préciser la nature des produits qui entrent dans son champ.
1. L'actualisation de la définition du médicament
Comme l'a souligné la Commission européenne dans son rapport général sur l'expérience acquise dans l'application des procédures d'autorisation de mise sur le marché (AMM)(21), la définition du médicament posée par la législation communautaire était « formulée d'une façon qui ne [permettait] pas de couvrir certaines formes de traitement médical, nouvelles ou futures, notamment celles relatives à la thérapie génique et à la thérapie cellulaire ». Pour des raisons de cohérence et d'efficacité, il est par ailleurs apparu préférable d'inclure ces traitements au cadre existant plutôt que d'établir un ensemble de règles distinctes.
C'est pourquoi la directive n° 2004/27 a modifié les dispositions du paragraphe 2 de l'article 1er de la directive n° 2001/83, concernant la définition du médicament par la fonction.
· Le I du présent article propose en conséquence de modifier la rédaction de l'article L. 5111-1 du code de la santé publique. S'il procède à une réécriture globale du premier alinéa de cet article, les modifications apportées par ce paragraphe ne portent en réalité que sur la définition du médicament par la fonction, et ce, sur les trois points suivants.
- La notion de « substance ou composition pouvant être utilisée chez l'homme ou chez l'animal ou pouvant leur être administrée » est tout d'abord substituée à celle, plus restrictive, de « produit pouvant être administré à l'homme ou à l'animal ».
En effet, certains médicaments ne sont pas directement « administrés » au patient, en particulier en matière de thérapie génique et de thérapie cellulaire. Dans ce cas, des cellules sont en effet traitées in vitro puis réadministrées au patient : le médicament est alors l'outil « utilisé » pour modifier les cellules et n'est pas constitué des cellules modifiées elles-mêmes.
Les termes « chez le patient » doivent être entendus comme signifiant « à finalité de traitement du patient » ; il y a bien en effet une utilisation thérapeutique chez l'homme. La question s'était également posée pour certains médicaments radiopharmaceutiques, qui sont définis par le point 6 de l'article 1er de la directive comme « tout médicament qui, lorsqu'il est prêt à l'emploi, contient un ou plusieurs radionucléides (isotopes radioactifs), incorporés à des fins médicales ».
- Ces substances ou compositions doivent, d'autre part, avoir pour finalité d'établir un diagnostic médical, comme c'est actuellement le cas, mais aussi de restaurer, corriger ou modifier leurs fonctions « physiologiques », et non plus organiques, ce terme étant à la fois moins désuet, plus compréhensible et plus général (22).
Ce terme avait d'ailleurs déjà remplacé le mot « organique » dès 2001, lors de la codification des différentes directives relatives aux médicaments en un seul texte, la directive n° 2001/83. En France, l'arrêté du 23 avril 2004 relatif au contenu du dossier d'AMM (23) a également repris cette notion.
- Enfin, le type d'actions que peut exercer le médicament sur les fonctions physiologiques est précisé : il peut ainsi s'agir d'une « action pharmacologique, immunologique ou métabolique ». Le septième considérant de la directive n° 2004/27 a précisé à cet égard que cette énumération d'actions visait notamment à inclure des médicaments tels que les thérapies géniques, les produits radiopharmaceutiques ainsi que certains médicaments à usage local.
En réalité, en tout cas en France, il s'agit moins par ces dispositions d'élargir la définition du médicament que d'expliciter ce qu'elle recouvre actuellement de façon implicite, les actions pharmacologiques, immunologiques et métaboliques étant d'ores et déjà prises en compte pour qualifier un produit de médicament.
2. Le principe de l'application en cas de doute de la réglementation pharmaceutique aux produits dits « frontière »
Soulignant « le caractère dual de certains produits frontière », tels que les cosmétiques, les compléments alimentaires ou les biocides, ainsi que les difficultés subséquentes d'interprétation quant à la législation qui leur est applicable, la Commission européenne a proposé de modifier la rédaction de l'article 2 de la directive n° 2001/83 (paragraphe 2). L'objectif était double : renforcer la sécurité juridique du dispositif actuel mais surtout garantir un niveau élevé de protection des consommateurs, en faisant prévaloir la réglementation pharmaceutique, naturellement plus contraignante que celle applicable à d'autres catégories de produits.
En conséquence, le II du présent article complète l'article L. 5111-1 du code de la santé publique précité par un nouvel alinéa procédant à la transposition quasi littérale des dispositions issues de la directive n° 2004/27.
Cet alinéa prévoit qu'en cas de doute, lorsqu'un produit, eu égard à l'ensemble de ses caractéristiques, est susceptible à la fois de répondre à la définition du médicament et à celles d'autres catégories de produits régies par le droit communautaire ou national, c'est la réglementation pharmaceutique qui doit s'appliquer, ce produit étant alors considéré comme un médicament. Par exemple, si un produit appliqué sur la peau répond à la définition du cosmétique, il sera néanmoins considéré comme un médicament en raison de la substance médicamenteuse qu'il contient.
En revanche, ces dispositions n'ont pas lieu d'être appliquées s'il n'y a pas de doute sur la qualification du produit. Dans son septième considérant, la directive n° 2004/27 précise en effet que « lorsqu'un produit répond de façon évidente à la définition d'autres catégories de produits », notamment les denrées alimentaires, les compléments alimentaires, les biocides ou les produits cosmétiques, il n'entre pas dans le champ d'application de la directive n° 2001/83.
*
La commission a adopté l'article 3 sans modification.
Article 4
Définition des spécialités et groupes génériques et des médicaments homéopathiques, biologiques et biologiques similaires
Composé de trois paragraphes, cet article vise d'une part à actualiser les définitions des médicaments et groupes génériques ainsi que des médicament homéopathiques. Il permet, d'autre part, d'introduire dans le code de la santé publique les notions de médicaments biologiques et biologiques similaires.
1. La modification de la définition des génériques
Le I de cet article procède en premier lieu à la réécriture globale du sixième alinéa (5°) de l'article L. 5121-1 du code de la santé publique, qui définit les spécialités et groupes génériques.
a) Les spécialités génériques
Les spécialités génériques d'une spécialité de référence, dite princeps, désignent actuellement celles qui ont « la même composition qualitative et quantitative en principe actif, la même forme pharmaceutique et dont la bioéquivalence avec la spécialité de référence est démontrée par des études de biodisponibilité appropriées ».
Définitions de la bioéquivalence et de la biodisponibilité
Aux termes de l'article R. 5121-1 du code de la santé publique :
- La biodisponibilité désigne la vitesse et l'intensité de l'absorption dans l'organisme, à partir d'une forme pharmaceutique, du principe actif ou de sa fraction thérapeutique destiné à devenir disponible au niveau des sites d'action ;
- La bioéquivalence se définit par l'équivalence des biodisponibilités.
Il est par ailleurs précisé que les différentes formes pharmaceutiques orales à libération immédiate sont considérées comme une même forme pharmaceutique. Ainsi, une spécialité présentée en comprimés pourra être le générique d'un princeps sous la forme d'un sirop.
Afin de transposer les dispositions introduites à l'article 1er du code communautaire par la directive n° 2004/27, l'article 30 de la loi n° 2004-810 du 13 août 2004 relative à l'assurance maladie a également prévu que « les différents sels, esters, éthers, isomères, mélanges d'isomères, complexes ou dérivés d'un principe actif, sont considérés comme un même principe actif, sauf s'ils présentent des propriétés sensiblement différentes au regard de la sécurité ou de l'efficacité ».
Dans ce cas, des informations supplémentaires fournissant la preuve de la sécurité et de l'efficacité des différents sels, esters ou dérivés d'une substance active doivent cependant être apportées.
Le présent paragraphe permet tout d'abord de clarifier la rédaction du 5° de l'article L. 5121-1 précité, en distinguant plus clairement les définitions des spécialités (a) et des groupes génériques (b).
L'alinéa 3 (a) de cet article reprend tout d'abord les dispositions actuellement applicables aux spécialités génériques, en précisant que :
- les spécialités génériques désignent celles ayant la même composition qualitative et quantitative en « principes actifs » que la spécialité de référence, cette rédaction au pluriel étant plus pertinente et mieux conforme au point 2 (b) de l'article 10 de la directive n° 2001/83 consolidée ;
- par souci de clarification, les différents sels, esters, éthers et isomères sont « regardés comme ayant la même composition qualitative en principe actif », et non plus comme un même principe actif.
Cet alinéa insère, d'autre part, de nouvelles dispositions prévoyant qu'un médicament ne peut être qualifié de spécialité de référence que si son autorisation de mise sur le marché (AMM) a été délivrée sur le fondement d'un dossier comportant « l'ensemble des données nécessaires et suffisantes à elles seules pour son évaluation ».
Il s'agit ainsi, pour les demandes d'AMM des nouveaux médicaments, de donner une base légale à la notion de dossier complet, issue de la directive n° 2004/27 et qui sera précisée par voie réglementaire, par opposition aux procédures dite « allégées », pour lesquels le demandeur d'une AMM, pour un médicament générique par exemple, pourra être dispensé de produire certaines données et études (cf. infra, l'article 5 du projet de loi).
L'alinéa 4 du présent article insère un nouvel alinéa (b) dans le 5° de l'article L. 5121-1 précité, qui reprend tout d'abord les dispositions actuellement en vigueur concernant les groupes génériques, constitués par une spécialité de référence et les spécialités qui en sont génériques.
Il est également précisé, comme c'est aujourd'hui le cas, qu'en l'absence de spécialité de référence, un groupe générique peut être constitué de spécialités ayant la même composition qualitative et quantitative en principes actifs (désormais au pluriel, pour les mêmes raisons que celles évoquées ci-dessus), la même forme pharmaceutique et dont le profil de sécurité et d'efficacité est équivalent. C'est par exemple le cas pour des molécules anciennes comme le paracétamol, pour lesquelles il est difficile de déterminer avec certitude celle qui a été historiquement la spécialité de référence.
Le présent article insère par ailleurs de nouvelles dispositions prévoyant qu'une spécialité remplissant les conditions pour être un princeps, qui présente la même composition qualitative et quantitative en principes actifs et la même forme pharmaceutique qu'un princeps d'un groupe générique déjà existant et dont la bioéquivalence avec cette spécialité est démontrée, peut aussi figurer dans ce groupe générique, à la condition que les deux spécialités relèvent d'une même AMM globale, définie par voie réglementaire.
La notion d'AMM globale, issue de la directive n° 2004/27, a déjà été transposée en droit interne par un décret récent du 18 février 2005 (24), il ne sera donc pas nécessaire à court terme d'édicter un nouveau décret visant à définir ses modalités de mise en œuvre.
L'AMM globale a pour effet de ne pas conférer aux extensions de gamme des médicaments (nouveau dosage ou nouvelle forme pharmaceutique par exemple) des périodes de protection des données de l'AMM supplémentaires, confirmant ainsi la jurisprudence de la Cour de justice des communautés européennes (CJCE). En France, la protection autonome accordée aux données relatives aux extensions de gamme, permise par la jurisprudence du Conseil d'État, avait en effet conduit à retarder l'arrivée sur le marché de nouveaux génériques.
Définition communautaire de l'autorisation de mise sur le marché (AMM) globale
Aux termes de l'article 6 de la directive n° 2001/83, tel que modifié par la directive n° 2004/27 :
« Lorsqu'un médicament a obtenu une première AMM, conformément au premier aliéna, tout dosage, forme pharmaceutique, voie d'administration et présentation supplémentaires, ainsi que toute modification ou extension doivent également obtenir une autorisation ou être inclus dans l'AMM initiale. Toutes ces AMM sont considérées comme faisant partie d'une même autorisation globale, notamment aux fins de l'application de l'article 10, paragraphe 1 », relatif aux conditions d'autorisation et de commercialisation des génériques. »
Les dispositions prévues par le présent article permettront ainsi la constitution de groupes génériques élargis, en vue de favoriser la substitution et promouvoir ainsi la pénétration des génériques sur le marché.
Conformément à l'article L. 5121-23 du même code, les pharmaciens ont en effet la possibilité de délivrer par substitution à la spécialité prescrite une spécialité du même groupe générique, à la condition notamment que le prescripteur n'ait pas exclu cette possibilité, pour des raisons particulières tenant au patient, par une mention expresse portée sur la prescription.
2. L'actualisation de la notion de médicaments homéopathiques
Les médicaments homéopathiques sont actuellement définis par le quatorzième alinéa (11°) du même article L. 5121-1 précité comme tout médicament obtenu « à partir de produits, substances ou compositions appelés souches homéopathiques », selon un procédé de fabrication homéopathique décrit par la pharmacopée européenne ou française ou, à défaut, par les pharmacopées utilisées de façon officielle dans un autre État membre de la Communauté européenne.
Reprenant la terminologie employée par le point 5 de l'article 1er de la directive n° 2001/83, tel que modifié par la directive n° 2004/27, le II de cet article vise à clarifier la rédaction du 11° de l'article L. 5121-1, afin de préciser que les médicaments homéopathiques désignent ceux qui sont obtenus à partir de « substances » appelées souches homéopathiques, ce terme générique étant en effet plus approprié (alinéa 5 du présent article).
Définition communautaire de la notion de « substance »
Aux termes de l'article 1er de la directive n° 2001/83 consolidée, le terme de substance désigne toute matière quelle qu'en soit l'origine, celle-ci pouvant être :
- humaine, telle que : le sang humain et les produits dérivés du sang humain ;
- animale, telle que : les micro-organismes, animaux entiers, parties d'organes, sécrétions animales, toxines, substances obtenues par extraction, produits dérivés du sang ;
- végétale, telle que : les micro-organismes, plantes, parties de plantes, sécrétions végétales, substances obtenues par extraction ;
- chimique, telle que : les éléments, matières chimiques naturelles et les produits chimiques de transformation et de synthèse.
3. L'introduction des notions de médicaments biologiques et biologiques similaires
Le III a pour objet de compléter l'article L. 5121-1 par deux nouveaux alinéas (14° et 15°) relatifs aux médicaments biologiques et biologiques similaires.
a) Les médicaments biologiques
Reprenant les termes de la définition posée par le point 3.2 de l'annexe I du code communautaire, tel que modifié par la directive n° 2004/27, l'alinéa 7 du présent article, définit le médicaments biologique comme tout médicament :
- dont la substance est produite à partir d'une source biologique ou en est extraite ;
- et dont « la caractérisation et la détermination de la qualité nécessitent une combinaison d'essais physiques, chimiques et biologiques, ainsi que la connaissance de son procédé de fabrication et de son contrôle ».
Il peut s'agir par exemple de médicaments dérivés du sang et du plasma humain, de vaccins ou encore de protéines recombinantes.
b) Les médicaments biologiques similaires
L'alinéa 8 et dernier de cet article définit les médicaments biologiques similaires, souvent appelés « biosimilaires », comme :
- tout médicament biologique de même composition qualitative et quantitative en substance active et de même forme pharmaceutique d'un médicament biologique, dit de référence ;
- mais qui ne remplit pas les critères définissant les génériques, tels que définis au 5° du présent article, en raison de différences liées notamment à la variabilité de la matière première ou aux procédés de fabrication ;
- et à la condition que soient produites des données précliniques et cliniques supplémentaires, dans des conditions fixées par voie réglementaire.
De même que pour les médicaments génériques, il est par ailleurs précisé que ces dispositions s'appliquent sans préjudice de l'article L. 611-2 du code de la propriété intellectuelle, relatif aux titres de propriété intellectuelle qui protègent les inventions.
En d'autres termes, les biosimilaires sont des copies de médicaments biologiques de référence, qui ne peuvent pas être juridiquement considérées comme des génériques, dans la mesure où les critères de bioéquivalence mis en œuvre pour les molécules chimiques ne sont pas adaptés aux spécificités présentées par les matières biologiques.
Ces dispositions permettent ainsi de transposer les dispositions prévues par le paragraphe 4 de l'article 10 de la directive n° 2001/83, telle que modifiée par la directive n° 2004/27.
*
La commission a examiné un amendement de M. Claude Evin visant à modifier la rédaction de la dernière phrase de cet article, afin de remplacer le terme de « propriétés » du médicament par celui d' « effets démontrés » au regard de la sécurité ou de l'efficacité.
Suivant l'avis défavorable de la rapporteure, qui a jugé préférable de retenir la terminologie employée par la directive n° 2004/27, la commission a rejeté l'amendement.
La commission a adopté un amendement de clarification rédactionnelle de la rapporteure, précisant que les médicaments biologiques désignent ceux dont la substance « active » est produite à partir d'une source biologique ou en est extraite.
La commission a ensuite adopté l'article 4 ainsi modifié.
Cet article a pour objet de réformer la procédure d'autorisation de mise sur le marché (AMM) des médicaments, concernant tant les conditions de sa délivrance, que sa durée et, le cas échéant, sa caducité.
1. L'amélioration des conditions de délivrance de l'AMM
Le premier alinéa de l'article L. 5121-8 du code de la santé publique subordonne actuellement à la délivrance d'une AMM par l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS), la commercialisation ou la distribution à titre gratuit de « toute spécialité pharmaceutique ou tout autre médicament fabriqué industriellement, ainsi que tout générateur, trousse ou précurseur qui ne fait pas l'objet d'une autorisation de mise sur le marché délivrée par la Communauté européenne en application du règlement (CEE) nº 2309/93 du Conseil du 22 juillet 1993 ».
Définition des générateurs, trousses et précurseurs
- Générateur : tout système contenant un radionucléide parent déterminé servant à la production d'un radionucléide de filiation obtenu par élution ou par toute autre méthode et utilisé dans un médicament radiopharmaceutique.
- Trousse : toute préparation qui doit être reconstituée ou combinée avec des radionucléides dans le produit radiopharmaceutique final.
- Précurseur : tout autre radionucléide produit pour le marquage radioactif d'une autre substance avant administration.
Source : Article L. 5121-1 du code de la santé publique
Ainsi, sont notamment exemptés de l'obligation posée par l'article L. 5121-8 :
- les spécialités pharmaceutiques qui ne sont pas fabriquées industriellement, par exemple les préparations magistrales, préparées extemporanément en pharmacie, selon une prescription destinée à un malade déterminé ;
- les médicaments qui relèvent de la procédure dite « centralisée », c'est-à-dire communautaire. Pour les différentes catégories de médicaments énumérées par le règlement n° 2309/93 du Conseil du 22 juillet 1993, cette procédure, obligatoire ou optionnelle selon les cas, comprend l'évaluation de la spécialité par l'Agence européenne des médicaments, qui délivre une AMM valable sur l'ensemble du territoire communautaire ;
- les préparations de thérapie génique et les préparations de thérapie cellulaire xénogéniques qui font l'objet d'un régime spécifique d'autorisation, en application de la loi n° 2004-800 du 6 août 2004 relative à la bioéthique ;
- certains médicaments homéopathiques, qui doivent cependant être enregistrés auprès de l'AFSSAPS (cf. infra, l'article 13 du projet de loi) ;
- les médicaments qui font l'objet d'une autorisation temporaire d'utilisation (cf. infra, l'article 12 du projet de loi).
b) Les modifications proposées par le présent article
Le 1° du I de cet article a pour objet de substituer aux deux premiers alinéas de l'article L. 5121-8 précité cinq nouveaux alinéas, dont les trois premiers précisent les conditions de délivrance de l'AMM par l'AFSSAPS.
· L'alinéa 3 du présent article apporte les trois modifications suivantes au dispositif actuel.
- Outre les médicaments « fabriqués industriellement », sont désormais également soumis à l'obligation d'une AMM pour être commercialisés, ceux dans la fabrication desquels « intervient un processus industriel ». La référence à un processus industriel permet ainsi de donner aux autorités sanitaires des États-membres la possibilité de distinguer avec plus de précision les produits entrant dans le champ de l'AMM de ceux qui n'y entrent pas.
Toutefois, selon les informations communiquées par le ministère de la santé et des solidarités, le gouvernement souhaite que ce critère soit utilisé de la façon la plus souple possible afin de maintenir dans notre pays le double statut des produits de thérapie cellulaire et génique qui peuvent être soit des médicaments fabriqués industriellement soumis à AMM, soit des préparations non soumises à AMM. Celles-ci sont développées le plus souvent par des promoteurs hospitalo-universitaires ou des établissements de transfusion sanguine dans une double finalité de soins et de recherche clinique. Elles sont dans la plupart des cas préparées en situation autologue pour un nombre limité de patients. Or il pourrait être difficile d'imposer le coût et la lourdeur d'une procédure d'AMM à ces acteurs, qui jouent un rôle important dans la recherche et le développement de produits innovants et complexes.
- Il est désormais fait référence au règlement n° 726/2004 du 31 mars 2004(25), qui a élargi la liste des médicaments relevant de la procédure centralisée, puisque celui-ci a abrogé et remplacé le règlement n° 2309/93 précité.
Catégories de médicaments pour lesquels la procédure centralisée d'AMM est obligatoire
- Médicaments issus de l'un des procédés biotechnologiques suivants : technologie de l'acide désoxyribonucléique recombinant, expression contrôlée de gènes codant pour des protéines biologiquement actives dans des procaryotes et des eucaryotes, y compris des cellules transformées de mammifères, méthodes à base d'hybridomes et d'anticorps monoclonaux.
- Médicaments à usage humain contenant une nouvelle substance active qui, à la date d'entrée en vigueur du présent règlement, n'était pas autorisée dans la Communauté et dont l'indication thérapeutique est le traitement d'une des affections suivantes : syndrome d'immunodéficience acquise (sida), cancer, maladie neurodégénérative, diabète.
- Médicaments désignés comme des médicaments orphelins, conformément au règlement (CE) n° 141/2000 du Parlement européen et du Conseil du 16 décembre 1999.
- Médicaments à usage humain contenant une nouvelle substance active et dont l'indication thérapeutique est le traitement des maladies auto-immunes et d'autres dysfonctionnements immunitaires ainsi que des maladies virales (à compter du 20 mai 2008).
- Médicaments à usage vétérinaire destinés principalement à être utilisés comme améliorateurs de performance pour accélérer la croissance ou pour augmenter la productivité des animaux traités.
Source : Annexe du Règlement n° 726/2004 du 31 mars 2004 précité
- La dernière modification est moins substantielle, puisqu'il s'agit uniquement de préciser que l'AMM peut être assortie de conditions « appropriées » et non plus « adéquates ».
· En application de l'alinéa 4 du présent article, les conditions dans lesquelles le demandeur de l'AMM peut être dispensé de produire certaines données et études seront définies par voie réglementaire.
Il s'agit ainsi de prévoir la transposition des dispositions communautaires relatives aux procédures d'autorisation dites « allégées » ou « abrégées », qui ont été substantiellement modifiées par la directive n° 2004/27.
En particulier, l'article 10 modifié de la directive n° 2001/83 prévoit que, sans préjudice de la législation relative à la protection de la propriété intellectuelle, le demandeur d'une AMM n'est pas tenu de fournir les résultats précliniques et cliniques s'il peut démontrer que le médicament est un générique d'un médicament de référence autorisé depuis au moins huit ans.
Ces dispositions, qui seront transposées par voie réglementaire, constituent une avancée majeure pour les génériques, dans la mesure où la durée de protection des données de l'AMM du princeps sera réduite de dix à huit ans. Les génériqueurs pourront donc déposer plus tôt leurs demandes d'AMM, étant précisé que leur commercialisation ne pourra intervenir qu'à l'expiration d'un délai de dix suivant la délivrance de l'AMM initiale à la spécialité de référence.
· L'alinéa 5 a pour objet de préciser qu'une AMM ne peut être délivrée qu'à un demandeur établi dans un État membre de la Communauté européenne ou partie à l'accord sur l'Espace économique européen.
2. La suppression des renouvellements quinquennaux et l'introduction de la notion de caducité de l'AMM
L'AMM est actuellement délivrée pour une durée de cinq ans et est ensuite renouvelable par période quinquennale, sur demande du titulaire de l'AMM présentée au plus tard trois mois avant la date de son expiration.
Toute modification des éléments d'une AMM délivrée par l'AFSSAPS, quelle que soit son importance, doit être préalablement autorisée. L'AMM peut être modifiée, suspendue ou retirée par l'agence.
b) Les dispositions prévues par la directive n° 2004/27
· Afin notamment d'alléger les charges administratives des autorités compétentes des États membres, l'article 24 de la directive n° 201/83 a été modifié par la directive n° 2001/27 et prévoit désormais que :
- l'AMM est valable pendant cinq ans (paragraphe 1) ;
- elle peut être renouvelée au terme des cinq ans sur la base d'une évaluation du rapport bénéfice-risque effectuée par l'autorité compétente qui délivre l'autorisation. À cette fin, le titulaire de l'AMM doit fournir une version consolidée du dossier en ce qui concerne la qualité, la sécurité et l'efficacité du médicament, y compris toutes les modifications apportées depuis la délivrance de l'AMM, au moins six mois avant que celle-ci n'expire (paragraphe 2) ;
- une fois renouvelée, l'AMM est valable pour une durée illimitée, sauf si l'autorité compétente décide, pour des raisons justifiées ayant trait à la pharmacovigilance, de procéder à un nouveau renouvellement quinquennal (paragraphe 3).
· Compte tenu de ces modifications, la directive n° 2004/27 a par ailleurs introduit deux nouvelles dispositions prévoyant qu'une AMM devient caduque :
- lorsque l'autorisation, dans les trois années qui suivent sa délivrance, n'est pas suivie d'une mise sur le marché effective du médicament dans l'État membre qui l'a délivrée (paragraphe 4) ;
- lorsqu'un médicament autorisé, précédemment mis sur le marché dans l'État membre qui l'a autorisé, n'est plus effectivement sur le marché de cet État pendant trois années consécutives (point 5).
c) La transposition proposée par le présent article
Le présent article propose en conséquence de modifier la rédaction de l'article L. 5121-8 précité sur les deux points suivants :
- L'alinéa 6 prévoit que l'AMM sera délivrée « pour une durée déterminée », qui sera fixée à cinq ans par voie réglementaire, et pourra ensuite être renouvelée « le cas échéant, sans limitation de durée », dans des conditions fixées par décret en Conseil d'État.
- Ce décret déterminera également les conditions dans lesquelles une AMM peut devenir caduque, comme le prévoit la dernière phrase de cet alinéa.
Le 2° du I du présent article (alinéa 7) modifie la rédaction du dernier alinéa du même article L. 5121-8, qui précise actuellement que l'accomplissement des formalités visées par celui-ci, soit l'obtention d'une AMM, n'a pas pour effet d'exonérer le fabricant « ou », s'il est distinct, le titulaire de l'AMM de la responsabilité qu'ils encourent dans les conditions du droit commun en raison de la fabrication ou de la mise sur le marché du médicament.
Dans le cas où le fabricant n'est pas le titulaire de l'AMM, il s'agit ainsi de lever toute ambiguïté sur le fait que il n'y pas qu'une seule personne, l'un ou l'autre d'entre eux, dont la responsabilité peut être engagée, mais éventuellement les deux, l'un au titre de la fabrication du médicament « et » l'autre, en raison de sa mise sur le marché.
Enfin, le II précise que « les durées, déterminées par voie réglementaire qui servent de référence pour la mise en œuvre du deuxième alinéa », relatif aux procédures allégées d'autorisation, s'appliqueront pour les demandes d'AMM, déposées postérieurement au 29 octobre 2005 (alinéa 8). Pour les génériques, l'article 2 de la directive n° 2004/27 prévoit en effet que les dispositions concernant la durée de protection des données, portée à huit ans, s'appliqueront pour les demandes présentées à compter de la date limite de transposition de la directive, soit le 30 octobre 2005.
*
La commission a examiné un amendement de Mme Jacqueline Fraysse précisant que l'autorisation de mise sur le marché (AMM), prévue à l'article L. 5121-8 du code de la santé publique, est délivrée pour une durée de cinq ans.
Suivant l'avis favorable de la rapporteure, qui a précisé qu'il avait été envisagé de transposer ces dispositions par voie réglementaire, la commission a adopté l'amendement à l'unanimité.
La commission a examiné un amendement de Mme Jacqueline Fraysse visant à transposer l'alinéa 3 de l'article 24 de la directive n° 2004/27.
Mme Jacqueline Fraysse a jugé nécessaire de donner à l'AFSSAPS la possibilité de renouveler l'AMM pour une durée limitée, pour des raisons justifiées ayant trait à la pharmacovigilance.
Suivant l'avis favorable de la rapporteure, la commission a adopté l'amendement à l'unanimité.
La commission a ensuite examiné un amendement de Mme Jacqueline Fraysse tendant à créer un lien automatique entre la délivrance de l'AMM d'un médicament et la reconnaissance de sa valeur thérapeutique, d'une part, et son inscription au remboursement, d'autre part.
Suivant l'avis défavorable de la rapporteure, au motif que la délivrance d'une AMM et l'inscription au remboursement d'un médicament relèvent de logiques distinctes, la commission a rejeté l'amendement.
La commission a examiné un amendement de Mme Jacqueline Fraysse tendant à subordonner la délivrance d'une AMM à la présentation d'essais cliniques comparatifs par le demandeur, destinés à établir le caractère réellement novateur de l'apport thérapeutique du médicament. Trop souvent en effet, de nouveaux produits sont mis sur le marché, sans apporter de réelle plus-value d'un point de vue thérapeutique.
La rapporteure s'est opposée à l'amendement, en estimant qu'un tel dispositif serait contraire à la directive n° 2001/83/CE modifiée, qui ne prévoit pas la réalisation de tels essais cliniques comparatifs préalablement à la délivrance d'une AMM : cet amendement constituerait donc une entrave à la libre concurrence et au bon fonctionnement du marché intérieur.
La commission a rejeté l'amendement.
La commission a examiné un amendement de Mme Jacqueline Fraysse prévoyant que les nouveaux dosages, formes pharmaceutiques, voies d'administration ou présentations doivent faire l'objet d'une autorisation ou être inclus dans l'AMM initiale du médicament.
Suivant l'avis défavorable de la rapporteure, qui a rappelé que la notion d'AMM globale a été transposée en droit interne par un décret en date du 18 février 2005, la commission a rejeté l'amendement.
La commission a ensuite adopté l'article 5 ainsi modifié.
La commission a examiné un amendement de Mme Jacqueline Fraysse prévoyant l'étude de l'impact environnemental de toute spécialité pharmaceutique faisant l'objet d'une demande d'AMM, ainsi que des dispositions visant à le limiter.
Mme Jacqueline Fraysse a souligné la nécessité de transposer les dispositions de la directive n° 2004/27, qui comportent une avancée intéressante en matière environnementale.
Suivant l'avis défavorable de la rapporteure, au motif que les dispositions de l'article 8 de la directive n° 2001/83 modifiée, concernant l'évaluation du risque pour l'environnement du médicament, seront transposées par voie réglementaire, de même que l'ensemble de celles relatives au contenu du dossier de demande d'AMM, la commission a rejeté l'amendement.
La commission a ensuite examiné un amendement de Mme Jacqueline Fraysse proposant la création d'un Conseil national du médicament, seul à même d'assurer la nécessaire démocratisation du processus d'élaboration, d'autorisation et de suivi des médicaments. Cet amendement constitue une réponse aux difficultés d'ordre éthique, sanitaire et économique, qui sont au cœur des préoccupations actuelles.
La rapporteure s'est déclarée défavorable à l'amendement, compte tenu notamment des compétences actuelles dans ce domaine du Comité économique des produits de santé (CEPS), de la Haute autorité de santé ou du ministère chargé de la santé.
Après que le président Jean-Michel Dubernard a relevé, dans ce sens, qu'il est certes parfois utile de procéder à un regroupement, mais que tel n'est pas le cas en l'espèce, la commission a rejeté l'amendement.
Suivant l'avis défavorable de la rapporteure, la commission a rejeté un amendement visant à dérembourser et à retirer l'AMM des médicaments dont le service médical rendu (SMR) est insuffisant, Mme Jacqueline Fraysse ayant estimé que seuls les médicaments considérés comme véritablement efficaces doivent être pris en charge par la sécurité sociale.
Article 6
Critères de refus de l'autorisation de mise sur le marché (AMM)
d'un médicament et conditions de délivrance de l'AMM
dans des circonstances exceptionnelles
Cet article vise, d'une part, à modifier les raisons pour lesquelles une autorisation de mise sur le marché (AMM) doit être refusée, et, d'autre part, à mieux encadrer le dispositif de l'AMM dite « exceptionnelle ».
1. Les motifs actuels de refus de délivrance d'une AMM
a) Le principe
Les motifs de refus de l'AMM sont aujourd'hui limitativement énumérés par l'article L. 5121-9 du code de la santé publique, qui a transposé l'article 26 de la directive n° 2001/83, dans sa rédaction en vigueur antérieurement à la publication de la directive n° 2004/27.
Ainsi, une AMM ne peut en aucun cas être délivrée par l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) s'il apparaît :
- soit que le médicament est nocif dans les conditions normales d'emploi ;
- soit qu'il n'a pas la composition qualitative et quantitative déclarée, ou que l'effet thérapeutique annoncé fait défaut ou est insuffisamment justifié par le demandeur ;
- soit que la documentation et les renseignements fournis ne sont pas conformes au dossier qui doit être présenté à l'appui de la demande.
La demande d'autorisation ne peut être rejetée qu'après que le demandeur a été invité à fournir ses justifications. La décision de rejet doit être motivée et mentionner les voies et délais de recours qui lui sont applicables.
Dans ses quatre derniers alinéas, l'article L. 5121-9 prévoit toutefois que, à titre dérogatoire, l'AMM peut être délivrée, sous réserve du respect d'obligations spécifiques, lorsque, pour certaines indications thérapeutiques, le demandeur peut démontrer qu'il n'est pas en mesure de fournir des renseignements complets sur l'efficacité et l'innocuité du médicament dans les conditions normales d'emploi.
Tel sera le cas :
- si les indications prévues se présentent si rarement que le demandeur ne peut raisonnablement être tenu de fournir les renseignements complets ;
- ou si l'état d'avancement de la science ne permet pas de donner les renseignements complets ;
- ou si des principes de déontologie médicale interdisent de recueillir ces renseignements.
Ces dispositions résultent de la transposition de l'article 22 de la directive n° 2001/83, qui prévoyait, antérieurement à l'entrée en vigueur de la directive n° 2004/27, que « dans des circonstances exceptionnelles et après consultation du demandeur, une autorisation peut être soumise à certaines obligations spécifiques, visant à procéder à des études complémentaires après l'obtention de l'autorisation et à notifier les effets indésirables du médicament ».
Il est également précisé que ces autorisations exceptionnelles ne peuvent être accordées que « pour des raisons objectives et vérifiables » et doivent reposer sur l'un des motifs définis en annexe de la directive et repris à l'article L. 5121-9 précité.
2. Les modifications proposées par le présent article
a) L'AMM devra désormais être refusée si la balance bénéfice-risque est considérée comme défavorable
· Reprenant les termes de l'article 26 de la directive n°2001/83, dans sa nouvelle rédaction issue de la directive n° 2004/27, le I propose tout d'abord de modifier la rédaction du premier alinéa de l'article L. 5121-9 précité.
Parmi les motifs justifiant le refus d'une AMM, il s'agit ainsi de remplacer les dispositions qui faisaient référence au caractère « nocif » d'un médicament par celle de « l'évaluation des effets thérapeutiques positifs du médicament ou produit au regard des risques liés à son utilisation [non] considérée comme favorable », c'est-à-dire l'appréciation plus fine de la balance « bénéfice-risque », qui doit notamment tenir compte de l'indication du médicament.
Dans son seizième considérant, la directive n° 2004/27 a en effet souligné la nécessité d'harmoniser au niveau européen et d'adapter les critères de refus, de suspension et de retrait des AMM afin de veiller à ce que le bénéfice lié à l'efficacité d'un médicament l'emporte toujours sur ses risques potentiels.
· Le I du présent article précise, d'autre part, que l'AMM doit être refusée lorsque l'effet thérapeutique annoncé fait défaut ou qu'il est insuffisamment « démontré », et non plus « justifié », par le demandeur, ce terme étant naturellement plus contraignant pour celui-ci, et donc mieux à même de garantir la sécurité sanitaire du médicament.
b) La délivrance de l'AMM exceptionnelle est mieux encadrée
L'article 22 de la directive n° 2001/83 a été modifié par la directive n° 2004/27 afin principalement de renforcer les obligations incombant au titulaire de l'AMM dite exceptionnelle, en matière de sécurité et de pharmacovigilance. Il y est également précisé que cette autorisation doit être octroyée après concertation avec le demandeur.
Le II de cet article propose en conséquence de remplacer les quatre derniers alinéas de l'article L. 5121-9 du code de la santé publique par un alinéa unique, définissant les nouvelles conditions de délivrance d'une AMM « dans des circonstances exceptionnelles ».
Les modifications apportées au dispositif actuel portent sur les trois points suivants :
- Le demandeur doit tout d'abord démontrer qu'il n'est pas en mesure de fournir des renseignements complets sur l'efficacité et « la sécurité » du médicament et non plus son « innocuité », dans des conditions normales d'emploi, ainsi qu'il est dit au point 6 de la partie II de l'annexe de la directive n° 2001/83 modifiée.
- Il est par ailleurs précisé que les « obligations spécifiques », définies par voie réglementaire, au respect desquelles est subordonnée la délivrance d'une AMM exceptionnelle, doivent concerner notamment « la sécurité du médicament » ainsi que la notification à l'AFSSAPS « de tout incident lié à son utilisation et les mesures à prendre ».
Exemples d' « obligations spécifiques » pouvant être prescrites
au titulaire de l'AMM dans des circonstances exceptionnelles
« L'AMM peut dans ce cas être accordée sous réserve d'obligations spécifiques, qui peuvent être les suivantes :
- le demandeur doit mener à son terme un programme d'essais défini dans le délai fixé par l'autorité compétente, dont les résultats serviront à une réévaluation du rapport bénéfice-risque ;
- le médicament en question ne doit pouvoir être délivré que sur prescription médicale et, le cas échéant, son administration peut n'être autorisée que sous contrôle médical strict, éventuellement en milieu hospitalier, et, pour un médicament radiopharmaceutique, par une personne autorisée ;
- la notice et toute information médicale doivent attirer l'attention du médecin sur le fait que, sous certains aspects, nommément désignés, il n'existe pas encore suffisamment de renseignements sur le médicament en question. »
Source : Point 6 de la partie II de l'annexe de la directive n° 2001/83
Sur ce dernier point, il convient de rappeler que la directive n° 2004/27 précise que la liste de ces conditions spécifiques d'autorisation doit être « immédiatement rendue accessible au public, ainsi que les délais et les dates d'exécution », ces dispositions devant être transposées par voie réglementaire.
- Enfin, le maintien de cette autorisation, décidé par l'AFSSAPS, doit se fonder sur une réévaluation annuelle de ces obligations et de leur respect par le titulaire de l'AMM exceptionnelle.
*
La commission a adopté l'article 6 sans modification.
Article 7
Possibilité donnée à l'AFSSAPS de permettre la mise sur le marché d'un médicament autorisé uniquement dans un autre état membre
Comportant deux alinéas, cet article vise à garantir un accès effectif et rapide aux médicaments, en permettant leur distribution, sous certaines conditions, dans le cas où ils ne disposeraient pas d'une autorisation de mise sur le marché (AMM) en France.
1. Les dispositions prévues par le nouvel article 126 bis de la directive n° 2001/83
· Inséré par la directive n° 2004/27, l'article 126 bis du code communautaire donne la possibilité aux États membres d'autoriser la mise sur le marché d'un médicament sur leur territoire, bien qu'il ne dispose pas d'une AMM, ni même qu'une demande en ce sens ait été déposée, mais à la condition qu'il soit autorisé dans un autre État membre et que cette décision soit motivée par « des raisons de santé publique justifiées » (paragraphe 1).
Ces dispositions résultent d'un amendement à la proposition de directive modifiant la directive n° 2001/83 proposé par le Conseil, dans sa position commune du 29 septembre 2003 (26), afin d'« améliorer la disponibilité des médicaments, particulièrement sur des marchés plus petits ». Il s'agissait plus particulièrement de remédier aux problèmes qui pourraient apparaître « dans les nouveaux États membres, dont les marchés, de plus petite taille, n'attirent pas les sociétés pharmaceutiques », comme l'a souligné la Commission européenne (27).
En effet, les entreprises pharmaceutiques ne présentent pas toujours de demande d'AMM dans certains de ces États, car leur intérêt commercial peut être limité par l'étroitesse ou le manque de solvabilité des marchés. En outre, les traités d'adhésion des nouveaux États membres ayant prévu la mise aux normes de la réglementation pharmaceutique communautaire du stock des AMM existantes dans ces États, un certain nombre d'entre elles risquent de disparaître, y compris pour des médicaments largement utilisés dans ces pays.
Les dispositions de l'article 126 bis permettront ainsi aux nouveaux États membres d'accroître en contrepartie leur arsenal thérapeutique et d'avoir accès aux mêmes médicaments que dans le reste de la Communauté.
· Cet article précise également les conditions de mise en œuvre de ce régime dérogatoire d'autorisation des médicaments afin qu'il présente toutes les garanties nécessaires en termes de sécurité sanitaire et de transparence.
- Lorsque l'État membre recourt à cette possibilité, il devra prendre les mesures nécessaires pour garantir le respect des exigences posées par la directive n°2001/83 consolidée, en particulier celles prévues aux titres V, VI, VIII, IX et XI (paragraphe 2).
- L'État membre devra également notifier son intention d'autoriser un médicament en application de cet article au titulaire de l'AMM, dans l'État où le médicament est autorisé, et demander à l'autorité compétente de cet État de lui communiquer une copie du rapport d'évaluation et de l'AMM du médicament, avant qu'une telle autorisation soit délivrée (paragraphe 3).
- La Commission européenne est chargée d'établir un registre accessible au public des médicaments autorisés, actualisé à partir des informations transmises par les États membres et consultable sur son site Internet (paragraphe 4).
- Enfin, l'application de cet article fera l'objet d'un rapport de la Commission, remis au Parlement européen et au Conseil au plus tard le 30 avril 2008, en vue de proposer toute modification nécessaire (paragraphe 5).
2. La transposition proposée par le présent article
Après les articles L. 5121-8 et L. 5121-9 du code de la santé publique, concernant respectivement les conditions de délivrance et de refus de l'AMM (cf. supra, articles 5 et 6 du projet de loi), le présent article propose d'insérer un nouvel article L. 5121-9-1 dans le chapitre Ier (« Dispositions générales ») du titre II (« Médicaments à usage humain ») du livre Ier du même code, afin de transposer les dispositions prévues par l'article 126 bis du code communautaire.
L'article L. 5121-9-1 prévoit ainsi que dans le cas où un médicament ne dispose pas de l'AMM prévue par l'article L. 5121-8 du même code, mais qu'il est autorisé dans un autre État membre de la Communauté européenne ou partie à l'Espace économique européen, l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) peut autoriser sa mise sur le marché pour des raisons de santé publique.
Conformément à la directive n° 2004/27, ces dispositions sont également applicables lorsque aucune demande d'AMM n'est en cours d'instruction pour le médicament concerné.
Afin de transposer les autres dispositions de l'article 126 bis, en particulier celles prévues par ses deuxième et troisième paragraphes, il est enfin précisé que les conditions d'autorisation de la mise sur le marché d'un médicament en vertu de cet article seront précisées par voie réglementaire.
*
La commission a examiné un amendement de Mme Jacqueline Fraysse prévoyant que l'AFSSAPS peut autoriser à titre provisoire la mise sur le marché d'un médicament, autorisé uniquement dans un autre État membre, pour une durée limitée à un an et pour des raisons de santé publique justifiées et rendues publiques.
Mme Jacqueline Fraysse a souligné la nécessité de renforcer la transparence de ce dispositif ainsi que les règles applicables en matière de pharmacovigilance, conformément à la directive n° 2004/27.
La rapporteure a rappelé que l'article 126 bis de la directive prévoit déjà qu'un registre accessible au public des médicaments autorisés en vertu de cet article doit être établi par la Commission européenne et qu'au surplus, une telle limitation à un an de l'autorisation délivrée par l'autorité compétente n'est pas prévue par la directive, en dépit des dénégations de Mme Jacqueline Fraysse.
Suivant l'avis défavorable de la rapporteure, la commission a rejeté l'amendement.
La commission a ensuite adopté l'article 7 sans modification.
Article 8
Obligation pour le titulaire de l'AMM d'un générique d'informer l'AFSSAPS
sur les indications, formes et dosages de la spécialité de référence
encore protégés par le droit des brevets
L'article L. 5121-10 du code de la santé publique précise actuellement les modalités particulières de mise sur le marché et d'inscription au répertoire d'une spécialité générique, ainsi que les conditions dans lesquelles le titulaire de l'AMM de la spécialité de référence, dite princeps, en est tenu informé.
Le présent article a pour objet de modifier la rédaction de l'article L. 5121-10 précité afin principalement de renforcer la protection de l'innovation pharmaceutique.
1. Les dispositions prévues par l'article 11 modifié de la directive n° 2001/83
L'article 8 de la directive n° 2001/83 prévoit qu'en vue de l'octroi d'une autorisation de mise sur le marché (AMM) pour un médicament ne relevant pas de la procédure centralisée (communautaire), un résumé des caractéristiques du produit (RCP) doit être joint à la demande adressée à l'autorité compétente de l'État membre, soit, en France, l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS).
Conformément à l'article 11 de la même directive, le RCP comporte différents renseignements sur le médicament, en particulier : sa composition qualitative et quantitative en substances actives et en composants de l'excipient, sa forme pharmaceutique, ses propriétés pharmacologiques ainsi que des informations cliniques, parmi lesquelles les indications thérapeutiques, la posologie, le mode d'administration et les contre-indications et précautions particulières d'emploi du médicament.
Dans le cas des génériques, les règles définissant le contenu du RCP ont toutefois posé un certain nombre de difficultés et de contentieux. En effet, les laboratoires qui souhaitent faire autoriser leurs spécialités génériques dans plusieurs États membres, dans le cadre de la procédure de reconnaissance mutuelle, doivent déposer un même dossier de demande d'AMM, et donc un même RCP. Surtout, les autorités sanitaires de certains États membres ont posé l'obligation que le RCP du générique soit identique à celui du princeps.
Or, dans le cas par exemple d'un princeps, dont les indications étaient encore brevetées au moment de la commercialisation du générique (28), cette dernière obligation conduisait à faire du génériqueur le contrefacteur du brevet du princeps portant sur la nouvelle application thérapeutique.
La directive n° 2004/27 a permis de remédier à cette difficulté, en complétant l'article 11 de la directive n° 2001/82 par un nouvel alinéa prévoyant que, pour les demandes d'AMM des génériques, ne doivent pas être incluses les parties du RCP d'une spécialité de référence « renvoyant à des indications ou à des formes de dosage qui étaient encore protégées par le droit des brevets au moment où le médicament générique a été mis sur le marché ».
2. Les modifications proposées par le présent article
Comportant quatre alinéas, cet article vise à modifier la rédaction de l'article L. 5121-10 précité sur les deux points suivants.
· Le I de cet article procède tout d'abord à la transposition des dispositions prévues par le dernier alinéa de l'article 11 modifié de la directive n° 2001/83.
Après le troisième alinéa de l'article L. 5121-10, il est proposé d'insérer un nouvel alinéa prévoyant qu'avant la commercialisation d'une spécialité générique, le titulaire de son AMM est tenu d'informer le directeur général de l'AFSSAPS des indications, formes pharmaceutiques et dosages du princeps pour lesquels les droits de propriété intellectuelles n'ont pas expiré.
Selon les informations communiquées par le ministère de la santé et des solidarités, il a été décidé de faire peser cette obligation sur le titulaire de l'AMM d'un générique, dont c'est l'intérêt de se prémunir d'un contentieux, la Commission européenne ayant par ailleurs validé par écrit la mesure de transposition.
Au surplus, l'AFSSAPS n'aurait pas les moyens d'effectuer une telle recherche. Dans ce sens, il convient en effet de rappeler que l'article L. 5121-10 a été modifié par la loi de financement de la sécurité sociale pour 2004 (29) afin précisément de dispenser l'AFSSAPS de la vérification des droits de propriété intellectuelles attachés à un princeps, préalablement à l'inscription d'une spécialité au répertoire des groupes génériques.
· Le II de cet article vise à modifier la rédaction du dernier alinéa de l'article L. 5121-10 précité, qui prévoit actuellement qu'aux seules fins d'en garantir la publicité, le directeur général de l'AFSSAPS doit tenir disponible au public la liste des titres de propriété intellectuelle attachés à une spécialité de référence si le titulaire de l'AMM de celle-ci la lui a communiquée à cet effet, le laboratoire étant seul responsable de l'exactitude des informations fournies.
Il est proposé de supprimer les dispositions de la dernière phrase de cet alinéa, aux termes duquel « les conditions de rémunération du service rendu par l'agence » pour l'établissement de la liste des titres de propriété intellectuelle précitée « sont fixées par une décision de son conseil d'administration ».
Il convient à cet égard de rappeler que, selon la jurisprudence du Conseil d'État (30), présente le caractère d'une redevance pour service rendu « toute redevance demandée à des usagers en vue de couvrir les charges d'un service public déterminé ou les frais d'établissement et d'entretien d'un ouvrage public et qui trouve sa contrepartie directe dans les prestations fournies par le service ou dans l'utilisation de l'ouvrage ».
Selon les informations communiquées par le ministère de la santé et des solidarités, le Conseil d'État a suggéré de déclasser ces dispositions, dans la mesure où elles concernent une rémunération pour service rendu et relèvent en conséquence du domaine réglementaire.
Si l'article 34 de la Constitution réserve à la compétence du législateur la fixation des règles concernant l'assiette, le taux et les modalités de recouvrement des « impositions de toute nature », la rémunération des services rendus par l'État ne peut en effet être instituée que par la voie réglementaire (31), conformément à l'article 4 de la loi organique relative aux lois de finances (32) (LOLF).
*
La commission a adopté l'article 8 sans modification.
Article 9
Conditions de commercialisation des médicaments génériques,
biologiques similaires et quasi-génériques
Actuellement, conformément à l'article L. 5121-10 du code de la santé publique (cf. supra, l'article 8 du projet de loi), une spécialité générique ne peut être commercialisée qu'à l'expiration des droits de propriété intellectuelle de la spécialité de référence, dite princeps, mais peut, avant cette échéance, obtenir une autorisation de mise sur le marché (AMM) et être inscrite au répertoire des groupes génériques.
Après l'article L. 5121-10 précité, le I de cet article a pour objet d'insérer un nouvel article L. 5121-10-1, comportant deux alinéas, dans le chapitre Ier (« Dispositions générales ») du titre II (« Médicaments à usage humain ») du livre premier (« Produits pharmaceutiques ») du code de la santé publique.
Son premier alinéa pose tout d'abord le principe de l'interdiction de commercialiser un générique pendant une période de dix ans suivant la délivrance de l'AMM initiale au princeps. Il convient par ailleurs de préciser que les dispositions de l'article L. 5121-10 demeurent applicables et qu'en conséquence, un générique ne peut non plus être commercialisé avant l'expiration des droits de propriété intellectuelle du princeps.
En France, selon les informations fournies par Les entreprises du médicament (LEEM), la durée d'exclusivité commerciale du princeps, soit le délai entre sa mise sur le marché et la commercialisation du générique correspondant, après l'expiration des droits de propriété intellectuelle, est souvent proche d'une dizaine d'années.
Pour les princeps, le dépôt du brevet, qui correspond à la découverte d'une nouvelle molécule, est en effet bien antérieur à l'obtention d'une AMM, compte tenu des essais cliniques nécessaires pour son obtention ainsi que des délais liés à la procédure administrative de mise sur le marché, comme l'indique le schéma présenté ci-après.
Le cycle de vie du médicament
Recherche exploratoire |
Tests pré-cliniques |
Recherche |
Procédures administratives (AMM, prix, remboursement) |
Phase de commercialisation et pharmacovigilance |
|||||
5 ans |
10 ans |
15 ans |
20 ans |
+ 5 ans maximum | |||||
Expiration du brevet |
|||||||||
Dépôt du brevet |
Si CCP | ||||||||
Source : Les entreprises du médicament (LEEM)
Toutefois, afin de favoriser l'innovation, cet article prévoit également que cette période, dite de protection de marché, peut être portée de dix à onze ans si, pendant les huit premières années suivant la délivrance de l'AMM au princeps, le titulaire de celle-ci obtient une nouvelle autorisation pour une ou plusieurs indications thérapeutiques nouvelles, considérées comme apportant un avantage clinique important par rapport aux thérapies existantes. Ce faisant, il s'agit donc en quelque sorte de prendre en compte la valeur thérapeutique ajoutée du médicament.
Le dernier alinéa du nouvel article L. 5121-10-1 rend ces dispositions applicables :
- aux médicaments biologiques similaires, tels que définis par le 15°de l'article L. 5121-1 du même code, inséré par l'article 4 du projet de loi ;
- aux médicaments dits « quasi-génériques », soit ceux qui présentent des caractéristiques communes avec un médicament de référence, mais qui ne peuvent pas satisfaire à la définition du médicament générique, en raison de différences portant sur un ou plusieurs éléments de cette définition, et qui nécessitent que soient produites des données supplémentaires dans des conditions déterminées par voie réglementaire.
Cette dernière notion permet de transposer les dispositions issues du paragraphe 3 de l'article 10 modifié de la directive n° 2001/83, qui prévoit que « lorsque le médicament ne répond pas à la définition du médicament ou lorsque la bioéquivalence ne peut être démontrée au moyen d'études de biodisponibilité ou en en cas de changements de la ou des substances actives, des indications thérapeutiques, du dosage, de la forme pharmaceutique ou de la voie d'administration par rapport à ceux du médicament du référence, les résultats des essais précliniques ou cliniques appropriés sont fournis ».
2. Les modalités d'application dans le temps de ces dispositions
Conformément à l'article 2 de la directive n° 2004/27, le II précise que les dispositions prévues par l'article L. 5121-10-1 ne s'appliqueront qu'aux spécialités de référence pour lesquels une AMM initiale a été délivrée, suite à une demande déposée à compter du 30 octobre 2005.
*
La commission a examiné un amendement de Mme Jacqueline Fraysse visant à introduire la notion de « valeur thérapeutique ajoutée ».
Mme Jacqueline Fraysse a jugé nécessaire d'encourager la recherche innovante, tout en favorisant le générique et, à cette fin, d'introduire la notion de valeur thérapeutique ajoutée. En effet, le terme d'« avantage clinique important », qui figure dans le projet de loi, n'est pas suffisamment clair et risque de ce fait de limiter l'essor des génériques, en allongeant d'un an la période de protection des spécialités de référence. Afin de préserver l'exclusivité commerciale dont ils bénéficient, les laboratoires pharmaceutiques développent souvent de nouveaux médicaments, dont le principe actif ou la présentation ne sont que très peu modifiés par rapport au princeps. Il faut inverser cette tendance et faire en sorte que seuls les médicaments apportant une réelle valeur thérapeutique ajoutée puissent bénéficier d'une période de protection supplémentaire, les autres médicaments devant plus rapidement tomber dans le domaine public.
La rapporteure s'est déclarée défavorable à l'amendement, en jugeant suffisamment précises les dispositions du projet de loi, qui reprennent celles de la directive n° 2004/27, et en observant que la notion de « valeur thérapeutique ajoutée » n'est pas précisément définie d'un point de vue juridique. En conséquence, la commission a rejeté l'amendement.
La commission a ensuite examiné un amendement de Mme Jacqueline Fraysse prévoyant que l'avantage clinique important des médicaments doit être démontré par des études comparatives indépendantes
Mme Jacqueline Fraysse a précisé que cet amendement poursuit les mêmes objectifs que le précédent, à savoir renforcer le contrôle de l'efficacité thérapeutique d'un prétendu nouveau médicament et permettre aux génériques d'arriver plus rapidement sur le marché, afin de lutter contre les stratégies actuellement mises en œuvre par les laboratoires pharmaceutiques.
La rapporteure s'est déclarée défavorable à l'amendement.
Mme Maryvonne Briot a néanmoins jugée non dépourvue d'intérêt la proposition avancée par Mme Jacqueline Fraysse.
Le président Jean-Michel Dubernard a estimé qu'il convient effectivement de regarder cette question de plus près et d'envisager les éventuelles améliorations qui pourraient être apportées sur ce point, d'ici l'examen du projet de loi en séance publique.
La commission a rejeté l'amendement.
La commission a examiné un amendement de M. Claude Evin visant à supprimer la dernière phrase du troisième alinéa de l'article L. 5121-10 du code la santé publique.
M. Claude Evin a précisé que l'amendement tend à supprimer les dispositions prévoyant que la commercialisation d'un générique ne peut intervenir qu'après l'expiration des droits de propriété intellectuelle de la spécialité de référence, en raison des modifications apportées par cet article, et non pas à abroger les dispositions relatives aux conditions de rémunération du service rendu par l'AFSSAPS, comme il était indiqué dans l'exposé sommaire.
Après que la rapporteure a émis un avis défavorable, en rappelant que le II de l'article 8 du projet de loi prévoit de supprimer les dispositions de l'article L. 5121-10 relatives aux conditions de rémunération du service rendu par l'AFSSAPS, M. Claude Evin a retiré l'amendement.
La commission a ensuite adopté l'article 9 sans modification.
Article 10
Exclusion des études et essais requis en vue de l'obtention d'une autorisation de mise sur le marché (AMM) du champ de protection des brevets
Cet article vise à aménager le régime de protection des brevets, par l'introduction d'une clause dite « Bolar » (33) visant à faciliter la réalisation des études et essais nécessaires en vue de l'obtention d'une autorisation de mise sur le marché (AMM) pour une spécialité générique.
1. Les conditions actuelles de protection de la propriété industrielle dans le domaine du médicament
a) L'objet du brevet
De façon schématique, un brevet donne un droit exclusif d'exploitation d'une innovation à son inventeur, pendant une période limitée, en contrepartie de la diffusion légale de celle-ci. Sont ainsi interdites, à défaut d'accord du propriétaire, la fabrication, l'offre, la mise sur le marché du produit ou du procédé breveté, ou encore du produit obtenu directement par le procédé objet du brevet (article L. 613-3 du code de la propriété intellectuelle).
En application de l'article L. 611-1 du même code, toute invention peut faire l'objet d'un titre de propriété industrielle délivré par le directeur de l'Institut national de la propriété industrielle (INPI). Sont toutefois seules brevetables « les inventions nouvelles impliquant une activité inventive et susceptibles d'application industrielle » (article L. 611-10 du même code).
Les titres de propriété industrielle protégeant les inventions comprennent notamment :
- le brevet d'invention, délivré pour une durée de vingt ans à compter du jour du dépôt de la demande ;
- le certificat complémentaire de protection (CCP) rattaché à un brevet, dont la durée ne peut être supérieure à cinq ans à compter de la date à laquelle il produit effet (article L. 611-3 du même code). Il s'agit en fait d'un système de prolongation de la protection juridique des inventions pharmaceutiques, destiné à tenir compte du fait que, pour les médicaments, la durée du brevet est amputée d'une longue période nécessaire à l'obtention de l'AMM.
b) Les exceptions admises
Les seules exceptions aux droits conférés par le brevet, prévues par l'article L. 613-5 du même code, portent sur :
- les actes accomplis dans un cadre privé et à des fins non commerciales (a) ;
- les actes accomplis à titre expérimental qui portent sur l'objet de l'invention brevetée (b) ;
- la préparation de médicaments faite extemporanément et par unité dans les officines de pharmacie, sur ordonnance médicale, et les actes concernant les médicaments ainsi préparés (c).
c) Le cas particulier des génériques
Les essais de bioéquivalence réalisés par les laboratoires de génériques en vue de l'obtention d'une AMM, avant l'expiration du brevet portant sur la spécialité de référence, ont longtemps été qualifiés de contrefaçon par la jurisprudence de la grande majorité des États membres.
En France, deux jugements du tribunal de grande instance de Paris, en 2001 et 2002 (34), ont cependant autorisé ces essais, sur le fondement des dispositions prévues par l'article L. 613-5 précité, concernant les actes accomplis à titre expérimental qui portent sur l'invention brevetée.
En tout état de cause, la Cour de cassation n'ayant jamais été appelée à se prononcer sur la question des essais de bioéquivalence, une insécurité juridique persistait sur ce point. La question demeurait par ailleurs posée pour l'ensemble des essais et études réalisées par les génériqueurs pour préparer leur demande d'AMM.
2. Les modifications apportées par la directive n° 2004/27
Le rapport général sur l'expérience acquise dans l'application des procédures d'AMM des médicaments (35), publié par la Commission européenne, a souligné que « pour éviter de faire les analyses scientifiques requises pour la préparation d'une demande pour un médicament générique hors Communauté, pour des raisons purement juridiques, il serait approprié d'introduire une disposition permettant ce type d'activité pendant la période de protection par brevet appliquée au produit original ».
C'est notamment pourquoi le dernier paragraphe de l'article 10 du code communautaire, tel que modifié par la directive n° 2004/27, prévoit que « la réalisation des études et des essais nécessaires en vue de l'application des paragraphes 1, 2, 3 et 4 », relatifs à la mise sur le marché des médicaments génériques, « et les exigences pratiques qui en résultent ne sont pas considérés comme contraire aux droits relatifs aux brevets et aux certificats complémentaires de protection pour les médicaments ».
Le demandeur d'une AMM pour un générique pourra ainsi effectuer les essais nécessaires au dépôt du dossier avant la fin de la période d'exclusivité, sans pour autant enfreindre la réglementation relative à la protection industrielle et commerciale.
La référence aux « exigences pratiques qui en résultent » doit notamment permettre d'inclure la présentation d'échantillons, ainsi qu'il a été indiqué par le Conseil de l'Union européenne, dans sa position commune du 29 septembre 2003 (36).
3. La transposition proposée
Le présent article a pour objet de compléter l'article L. 613-5 du code de la propriété intellectuelle précité, par un nouvel alinéa (d), précisant que sont exclus de la protection conférée par le droit des brevets, les études et essais requis en vue de l'obtention d'une AMM pour un médicament, ainsi que les actes nécessaires à leur réalisation et à l'obtention de l'autorisation.
Ces dispositions permettront ainsi de faciliter la réalisation de ces essais et d'éviter qu'une grande partie d'entre eux ne soient effectuées hors de la Communauté européenne.
*
La commission a adopté l'article 10 sans modification.
Article 11
Application aux médicaments biologiques similaires et « quasi-génériques » d'un régime juridique proche de celui des médicaments génériques
L'article 4 du présent projet de loi a permis d'introduire la notion de médicaments biologiques similaires, dits « biosimilaires » (15° de l'article L. 5121-1 du code la santé publique). Bien qu'étant les copies de médicaments biologiques de référence, ils ne peuvent être juridiquement considérés comme des génériques, en raison notamment de la variabilité de leurs matières premières et de leurs procédés particuliers de fabrication.
Les « quasi-génériques » désignent pour leur part des médicaments qui présentent des caractéristiques communes avec un princeps, mais qui ne répondent pas à la définition du générique, en raison de différences portant sur un ou plusieurs éléments de celle-ci (en raison, par exemple, de formes pharmaceutiques différentes), conformément à l'article 9 du projet de loi. Des données supplémentaires doivent dans ce cas être produites afin qu'ils soient considérés comme quasi génériques, ou encore, selon la terminologie européenne, « essentiellement similaires ».
Afin de faciliter l'arrivée sur le marché de ces deux catégories de médicaments, le présent article tend à leur appliquer un régime juridique très proche de celui des génériques.
L'alinéa 1 de cet article propose ainsi d'insérer un nouvel article L. 5121-10-2, comportant six alinéas, dans le chapitre Ier (« Dispositions générales ») du titre II (« Médicaments à usage humain ») du livre premier (« Produits pharmaceutiques ») du code de la santé publique.
Ce nouvel article reprend à l'identique la quasi-totalité des dispositions prévues par l'article L. 5121-10, qui précise les conditions d'autorisation et de commercialisation des génériques, à l'exception toutefois de son actuel troisième alinéa, relatif à l'inscription d'une spécialité au répertoire des groupes génériques, qui permet au pharmacien d'exercer son droit de substitution.
L'alinéa 2 prévoit tout d'abord qu'une autorisation de mise sur le marché (AMM) peut être délivrée pour un biosimilaire avant l'expiration des droits de propriété intellectuelle qui s'attachent au médicament biologique de référence. Le demandeur de l'autorisation doit alors informer le titulaire de ces droits concomitamment au dépôt de sa demande.
D'autre part, lorsque l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) a délivré une AMM pour un biosimilaire, l'alinéa 3 de cet article prévoit qu'elle est tenue d'en informer le titulaire de l'AMM du médicament biologique de référence.
Conformément à l'alinéa 4, la commercialisation du médicament biologique similaire ne peut intervenir qu'après l'expiration des droits de propriété intellectuelle du médicament biologique de référence, sauf accord du titulaire de ces droits.
Avant la commercialisation d'un biosimilaire, l'alinéa 5 pose l'obligation pour le titulaire de son AMM d'informer le directeur général de l'AFSSAPS « des indications, formes pharmaceutiques et dosages du médicament biologique de référence pour lesquels les droits de propriété intellectuelle n'ont pas expiré », ainsi qu'il est prévu par l'article 8 du projet de loi pour les génériques.
Aux seules fins d'en garantir la publicité, l'alinéa 6 de cet article charge le directeur général de l'AFSSAPS de tenir disponible au public la liste des titres de propriété intellectuelle attachés à un médicament biologique de référence, si le titulaire de l'AMM de ce médicament la lui a communiquée à cet effet. Le laboratoire est seul responsable de l'exactitude des informations fournies.
L'alinéa 7 précise enfin que les dispositions du présent article s'appliquent également aux quasi-génériques, dont l'article 9 du projet de loi prévoit qu'ils ne peuvent être commercialisés qu'à l'expiration d'une période de dix ans suivant la délivrance de l'AMM initiale de la spécialité de référence, voire onze ans si le titulaire de l'AMM de cette dernière obtient une nouvelle autorisation pour une indication apportant « un avantage clinique important par rapport aux thérapies existantes ».
*
La commission a adopté l'article 11 sans modification.
Article 12
Modification du régime des autorisations temporaires d'utilisation (ATU)
Cet article tend à modifier les conditions d'autorisations temporaires d'utilisation (ATU), délivrées pour certains médicaments par l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS).
Ces dispositions s'inscrivent dans le cadre de la possibilité ouverte par l'article 5 (paragraphe 1) de la directive n° 2001/83, tel que modifié par la directive n° 2001/47, aux termes duquel :
« Un État membre peut, conformément à la législation en vigueur et en vue de répondre à des besoins spéciaux, exclure des dispositions de la directive les médicaments fournis pour répondre à une commande loyale et non sollicitée, élaborés conformément aux spécifications d'un professionnel de santé agréé et destinés à ses malades particuliers, sous sa responsabilité personnelle directe ».
1. La réglementation actuelle concernant les ATU nominatives
Lorsqu'ils ne disposent pas d'une AMM, les médicaments destinés à soigner des maladies rares ou graves, pour lesquelles il n'existe aucun traitement approprié, peuvent néanmoins faire l'objet d'une autorisation temporaire d'utilisation (ATU) délivrée par l'AFSSAPS, sous certaines conditions limitativement définies, dans le cadre d'un usage compassionnel.
Conformément à l'article L. 5121-12 du code de la santé publique, tel que modifié par la loi n° 2004-806 du 9 août 2004 relative à la politique de santé publique, il existe aujourd'hui deux types d'ATU :
- les ATU dites de cohorte, lorsque l'efficacité et la sécurité de ces médicaments sont fortement présumées, au vu des résultats d'essais thérapeutiques auxquels il a été procédé en vue d'une demande d'AMM, et que cette demande a été déposée ou que le demandeur s'engage à la déposer dans un délai déterminé (a) ;
- les ATU dites nominatives, lorsque « ces médicaments sont prescrits à des malades nommément désignés et, le cas échéant, importés dans ce but, sous la responsabilité de leur médecin traitant, dès lors que leur efficacité et leur sécurité sont présumées en l'état des connaissances scientifiques et qu'ils sont susceptibles de présenter un bénéfice réel » (b).
Dans ce dernier cas, l'AFSSAPS délivre l'ATU, pour une durée limitée, à la demande du médecin traitant des patients concernés. L'autorisation peut par ailleurs être subordonnée à la mise en place d'un protocole d'utilisation thérapeutique et de recueil d'informations.
Quelque soit le type d'ATU, l'autorisation peut être suspendue ou retirée pour des motifs de santé publique ou si les conditions prévues par l'article L. 5121-12 ne sont plus remplies.
2. Les améliorations apportées par le présent article
Le I de cet article procède à une réécriture globale du troisième alinéa de l'article L. 5121-12 précité (b), qui pose les conditions de délivrance des ATU nominatives.
- Il est tout d'abord précisé que les médicaments, le cas échéant importés, doivent être prescrits à « un patient » nommément désigné, ce terme se substituant à celui de « malades », sous la responsabilité « d'un médecin », dont il n'est plus précisé qu'il doit nécessairement être le médecin « traitant ».
- Cet article prévoit, d'autre part, une condition supplémentaire à la délivrance des ATU nominatives : pour en bénéficier, le patient ne doit pas avoir la possibilité de participer à une recherche biomédicale. Pour permettre aux patients d'avoir accès à des traitements nouveaux, qui font encore l'objet de recherches, il est en effet plus logique et surtout plus protecteur que la voie de droit commun soit leur participation à des essais cliniques de médicaments.
Réformé en profondeur par la loi précitée relative à la politique de santé publique, le régime des recherches biomédicales comporte de nombreuses garanties visant à protéger le participant à celles-ci. Le protocole de recherche doit ainsi être autorisé par l'autorité compétente et le comité de protection des personnes, et des modalités particulières d'information et de recueil du consentement de la personne sont prévues, afin de s'assurer du caractère parfaitement « libre et éclairé » de celui-ci.
En outre, contrairement aux ATU, les recherches biomédicales permettent d'évaluer la sécurité, l'efficacité et le rapport bénéfice-risque du médicament, en vue de l'obtention d'une AMM.
- Il est, troisièmement, proposé d'étendre le champ des ATU nominatives aux cas dans lesquels « une issue fatale à court terme pour le patient est, en l'état des thérapeutiques disponibles, inéluctable ». Demeurent par ailleurs applicables les dispositions actuelles prévoyant que les médicaments ne peuvent être prescrits sous ATU que s'ils sont susceptibles de présenter un bénéfice pour le patient.
- Enfin, le médecin qui demande une ATU doit justifier que le patient, son représentant légal ou la personne de confiance qu'il a désignée, en application de l'article L. 1111-6 du même code, tel que modifié par la loi n° 2005-370 du 22 avril 2005 relative aux droits des malades et à la fin de vie, a reçu « une information adaptée à sa situation sur l'absence d'alternative thérapeutique, les risques courus, les contraintes et le bénéfice susceptible d'être apporté par le médicament ».
Définition de la « personne de confiance »
« Toute personne majeure peut désigner une personne de confiance qui peut être un parent, un proche ou le médecin traitant, et qui sera consultée au cas où elle-même serait hors d'état d'exprimer sa volonté et de recevoir l'information nécessaire à cette fin. Cette désignation est faite par écrit. Elle est révocable à tout moment. Si le malade le souhaite, la personne de confiance l'accompagne dans ses démarches et assiste aux entretiens médicaux afin de l'aider dans ses décisions.
« Lors de toute hospitalisation dans un établissement de santé, il est proposé au malade de désigner une personne de confiance dans les conditions prévues à l'alinéa précédent. Cette désignation est valable pour la durée de l'hospitalisation, à moins que le malade n'en dispose autrement.
« Les dispositions du présent article ne s'appliquent pas lorsqu'une mesure de tutelle est ordonnée. Toutefois, le juge des tutelles peut, dans cette hypothèse, soit confirmer la mission de la personne de confiance antérieurement désignée, soit révoquer la désignation de celle-ci. »
Source : Article L. 1111-6 du code de la santé publique
A cet égard, il convient de rappeler que, dans le cas où une recherche biomédicale est envisagée sur une personne majeure hors d'état d'exprimer son consentement et ne faisant pas l'objet d'une mesure de protection juridique, la loi relative à la politique de santé publique a également prévu que l'autorisation est donnée par la personne de confiance, et à défaut de celle-ci, la famille ou une personne entretenant avec l'intéressé des liens étroits et stables. Ces dispositions permettront ainsi d'améliorer l'information du patient et de s'assurer de la réalité de son consentement, au regard des contraintes et des risques encourus.
- La dernière phrase de cet alinéa précise enfin que la procédure suivie doit être inscrite dans le « dossier médical » du patient.
Selon le ministère de la santé et des solidarités, il ne s'agirait cependant pas de reporter ces éléments dans le dossier médical personnel (DMP), mentionné à l'article L. 136-36-1 du code de la sécurité sociale, mais de veiller à qu'ils figurent dans les « informations concernant la santé du patient », mentionnées à l'article L. 1111-7 du code de la santé publique, qui doivent être conservées par les professionnels de santé.
Enfin, le II de cet article modifie la rédaction du quatrième alinéa du même article L. 5121-12 en précisant que la prescription d'un médicament dans le cadre d'une ATU doit être demandée par le médecin « prescripteur », afin que cette possibilité ne soit pas limitée au seul « médecin traitant » du patient, au sens de la loi n° 2004-810 du 13 août 2004 relative à l'assurance maladie.
*
La commission a adopté l'article 12 sans modification.
Article 13
Clarification de l'assiette du droit progressif perçu par l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) pour l'enregistrement des médicaments homéopathiques
Cet article tend à améliorer la rédaction de l'article L. 5121-15 du code de la santé publique relatif au droit d'enregistrement des médicaments homéopathiques.
1. Les conditions d'enregistrement des médicaments homéopathiques
Par dérogation au principe posé par l'article L. 5121-8 du code de la santé publique (cf. supra, article 5 du projet de loi), les médicaments homéopathiques sont exemptés de l'obligation de disposer d'une autorisation de mise sur le marché (AMM) pour être commercialisés, s'ils satisfont à l'ensemble des conditions fixées par l'article L. 5121-13 du même code, soit :
- une administration par voie orale ou externe (1°) ;
- l'absence d'indication thérapeutique particulière sur l'étiquetage ou dans toute information relative au médicament (2°) ;
- le degré de dilution garantissant l'innocuité du médicament ; en particulier, le médicament ne peut contenir ni plus d'une partie par 10 000 de la teinture mère, ni plus d'un centième de la plus petite dose utilisée éventuellement en allopathie, pour les principes actifs dont la présence dans un médicament allopathique entraîne l'obligation de présenter une prescription médicale (3°). En d'autres termes, la concentration en substances actives doit être si faible que ces produits ne peuvent en aucun cas constituer un risque pour la santé.
A contrario, par exemple si l'exploitant souhaite que le médicament homéopathique revendique une indication thérapeutique particulière ou si sa concentration en substances actives est plus élevée que les seuil définis au 3°, celui-ci doit alors faire l'objet d'une AMM, qui est délivrée selon des règles particulières concernant les essais pharmacologiques, toxicologiques et chimiques.
S'ils sont dispensés d'une AMM, les médicaments homéopathiques doivent cependant faire l'objet d'un enregistrement auprès de l'AFSSAPS, avant leur commercialisation ou leur distribution à titre gratuit ou onéreux, en gros ou au détail. Cet enregistrement peut être refusé, suspendu ou supprimé si les conditions prévues au même article L. 5121-13 ne sont pas remplies ou en cas de danger pour la santé publique. De même que pour l'AMM, l'enregistrement est actuellement valable pendant cinq ans et peut être renouvelé par période quinquennale.
L'article L. 1521-14 du même code précise, d'autre part, que l'enregistrement peut couvrir une série de médicaments homéopathiques obtenus à partir de la ou des mêmes souches homéopathiques et que la demande d'enregistrement doit être accompagnée de documents permettant de démontrer la qualité et l'homogénéité des lots de fabrication de ces médicaments.
2. Les modifications proposées par le présent article concernant le droit d'enregistrement des médicaments homéopathiques
En application de l'article L. 5121-15 du code de la santé publique, « toute demande d'enregistrement » mentionnée aux articles L. 5121-13 et L. 5121-14 précités doit donner lieu au versement, au profit de l'AFSSAPS, d'un droit progressif dont le montant est fixé par voie réglementaire, dans la limite de 76 00 euros. Ce droit est recouvré selon les modalités prévues pour le recouvrement des créances des établissements publics administratifs de l'État.
Son montant est actuellement déterminé, selon les différentes catégories de médicaments concernés, par les articles D. 5121-63 et D. 5121-66 du même code, tels qu'issus du décret n° 2000-1194 du 5 décembre 2000.
Ces articles prévoient en particulier que le droit progressif doit être versé non seulement pour toute demande initiale d'enregistrement d'un médicament homéopathique, mais aussi pour toute demande de modification ou de renouvellement quinquennal, bien que cela ne soit pas explicitement prévu par l'article L. 5121-15 précité.
Montant du droit perçu par l'AFSSAPS sur les demandes d'enregistrement des médicaments homéopathiques
CATÉGORIES DE MÉDICAMENTS |
MONTANT |
MÉDICAMENT HOMÉOPATHIQUE UNITAIRE OU SÉRIE DE MÉDICAMENTS OBTENUS À PARTIR : |
|
- DE LA MÊME SOUCHE HOMÉOPATHIQUE |
1 768 € |
- DE DEUX À CINQ SOUCHES HOMÉOPATHIQUES |
2 478 € |
- DE SIX SOUCHES HOMÉOPATHIQUES OU PLUS |
7 600 € |
MÉDICAMENT HOMÉOPATHIQUE UNITAIRE OU SÉRIE DE MÉDICAMENTS AUTORISÉS ET MIS SUR LE MARCHÉ AVANT LE 18 JANVIER 1994 À PARTIR : |
|
- DE LA MÊME SOUCHE HOMÉOPATHIQUE |
760 € |
- DE DEUX À CINQ SOUCHES HOMÉOPATHIQUES |
1 256 € |
- DE SIX SOUCHES HOMÉOPATHIQUES OU PLUS |
3 800 € |
MODIFICATION DU DOSSIER D'ENREGISTREMENT |
496 € |
RENOUVELLEMENT QUINQUENNAL D'ENREGISTREMENT |
380 € |
Source : Articles D. 5121-63 et D. 5121-66 du code de la santé publique
Selon les informations communiquées par le ministère de la santé et des solidarités, le rendement total des taxes versées à l'AFSSAPS au titre de l'enregistrement ou de l'AMM des médicaments homéopathiques s'est élevé en 2005 à 277 596 euros, comme l'indique le tableau présenté ci-après.
Évolution depuis 2001 du rendement des droits versés à l'AFSSAPS
pour les demandes d'AMM et d'enregistrements
des médicaments homéopathiques
2001 |
2002 |
2003 |
2004 |
2005 |
79 121 € |
383 455 € |
300 550 € |
228 466 € |
277 596 € |
Source : Ministère de la santé et des solidarités
Afin de consolider la base législative des articles D. 5121-63 et suivants précités, le présent article tend à clarifier la rédaction du premier alinéa de l'article L. 5121-15 afin de préciser qu'un droit progressif doit être versé à l'AFSSAPS pour toute demande d'enregistrement « ou toute demande de modification ou de renouvellement de cet enregistrement ».
*
La commission a adopté l'article 13 sans modification.
Article 14
Clarification de l'assiette du droit progressif versé à l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) pour l'autorisation de mise sur le marché (AMM) des médicaments
L'article L. 5121-16 du code de la santé publique prévoit que toute demande d'AMM doit être accompagnée du versement d'un droit progressif à l'AFSSAPS, fixé par voie réglementaire dans la limite de 25 400 euros.
Selon les différentes catégories de médicaments concernés, l'article D. 5121-64 du même code, tel que modifié en dernier lieu par le décret n° 2005-309 du 1er avril 2005, détermine le montant de ce droit, dont il est prévu qu'il soit versé non seulement pour les demandes initiales d'AMM, mais également pour les demandes de modification ou de renouvellement quiquennal.
Droits perçus par l'AFSSAPS
sur les demandes d'AMM des médicaments allopathiques
DOSSIER COMPLET D'AMM |
25 400 € |
DOSAGE OU FORME PHARMACEUTIQUE SUPPLÉMENTAIRE PRÉSENTÉ SIMULTANÉMENT À LA PREMIÈRE DEMANDE D'AUTORISATION |
12 700 € |
NOUVELLES INDICATIONS : USAGES THÉRAPEUTIQUES DIFFÉRENTS (DEMANDE OU MODIFICATION D'AMM) |
16 790 € |
NOUVELLES VOIES D'ADMINISTRATION | |
NOUVELLES ASSOCIATIONS | |
RÉFÉRENCE À LA LITTÉRATURE SCIENTIFIQUE | |
DOSAGE OU FORME PHARMACEUTIQUE SUPPLÉMENTAIRE PRÉSENTÉ SIMULTANÉMENT À LA PREMIÈRE DEMANDE D'AUTORISATION ET RELEVANT DES POINTS 3, 4, 5, 6 CI-DESSUS |
8 395 € |
ESSENTIELLEMENT SIMILAIRES |
10 110 € |
NOUVELLES FORMES GALÉNIQUES | |
NOUVEAUX DOSAGES (1 DOSSIER PAR DOSAGE) | |
MÉDICAMENTS À BASE DE PLANTES | |
PRODUITS MENTIONNÉS À L'ARTICLE L 5136-1 DU CODE DE LA SANTÉ PUBLIQUE | |
ALLERGÈNES (PAR FAMILLE DE PRODUITS) | |
PRÉPARATION FIGURANT À LA PHARMACOPÉE |
6 740 € |
FORMULAIRE NATIONAL | |
MODIFICATION |
1 011 € |
RENOUVELLEMENT QUINQUENNAL |
674 € |
Source : Article D. 5121-64 du code de la santé publique
Selon les informations communiquées par le ministère de la santé et des solidarités, le rendement total des taxes perçues par l'AFSSAPS sur les demandes d'AMM des médicaments s'est élevé à près de 30,42 millions d'euros, comme l'indique le tableau présenté ci-après.
Évolution depuis 2000 du rendement des droits perçus par l'AFSSAPS
sur les demandes d'AMM des médicaments allopathiques
2000 |
2001 |
2002 |
2003 |
2004 |
2005 |
18 883 555 € |
21 205 811 € |
20 279 297 € |
26 377 639 € |
26 478 891 € |
30 417 153 € |
Source : Ministère de la santé et des solidarités
Pour les mêmes raisons que celles évoquées à l'article 13 du projet de loi, le présent article propose de modifier la rédaction du premier alinéa de l'article L. 5121-16 de préciser qu'un droit progressif doit être versé à l'AFSSAPS pour toute demande d'AMM mentionnée à l'article L. 5121-8 du même code « ou toute demande de modification ou de renouvellement de cette autorisation ».
*
La commission a adopté l'article 14 sans modification.
Composé de dix paragraphes, cet article vise à modifier l'article L. 5121-20 du code de la santé publique, relatif aux modalités d'application du chapitre premier (« Dispositions générales ») du titre II (« Médicaments à usage humain ») du livre premier du même code.
Il s'agit ce faisant d'améliorer l'architecture et la clarté de cet article, mais aussi de modifier sa rédaction par cohérence avec les dispositions prévues par le présent projet de loi et par la directive n° 2004/27.
Par coordination avec l'article 4 du projet de loi, seront ainsi précisées par décrets en Conseil d'État :
- la définition des critères scientifiques justifiant, le cas échéant, l'exonération des études de biodisponibilité des spécialités génériques, la procédure d'inscription au répertoire, mais aussi, désormais, « les modalités de l'inscription dans un groupe générique existant d'une spécialité remplissant la condition pour être spécialité de référence et de la création de groupes génériques en l'absence de spécialité de référence », en application du I du présent article, qui procède à une réécriture globale du deuxième alinéa (1°) de l'article L. 5121-20 ;
- les conditions dans lesquelles des autorisations de mise sur le marché (AMM) peuvent être considérées comme faisant partie d'une AMM globale, conformément au II du présent article, qui complète le troisième alinéa (2°) de l'article L. 5121-20.
Afin de transposer l'ensemble des dispositions prévues par le titre V (« Etiquetage et notice ») de la directive modifiée n° 2001/83, le III procède à une réécriture du 3° du même article L. 5121-20, en renvoyant à un décret en Conseil d'État la détermination des règles relatives à l'« étiquetage, la notice », et non plus « la présentation », ainsi que la dénomination des médicaments et produits visés par le chapitre premier précité.
Par coordination avec les dispositions prévues par les articles 4 et 5 du projet de loi et pour permettre la transposition par voie réglementaire de celles prévues par la directive n° 2001/83 modifiée, concernant notamment les médicaments homéopathiques, devront également être précisés par décrets en Conseil d'État :
- le contenu du dossier présenté à l'appui d'une demande d'AMM, ainsi que, désormais, « les modalités de présentation des demandes tendant à obtenir l'AMM » et « les conditions dans lesquelles le demandeur peut être dispensé de produire certains éléments du dossier et celles dans lesquelles interviennent les décisions accordant, modifiant, renouvelant, suspendant ou supprimant ces autorisations ainsi qu'après la délivrance de l'autorisation les modalités de son actualisation », conformément au 4° de l'article L. 5121-20, tel qu'il est proposé de le réécrire par le IV du présent article ;
- les modalités de présentation des demandes tendant à obtenir l'enregistrement des médicaments homéopathiques, prévu à l'article L. 5121-13 du même code, le « contenu du dossier » présenté à l'appui de ces demandes ainsi que « les conditions dans lesquelles interviennent les décisions accordant, modifiant, renouvelant, suspendant ou supprimant ces enregistrements », comme le prévoit le V de cet article, modifiant le 6° de l'article L. 5121-20 ;
- les modalités de présentation des demandes tendant à obtenir une autorisation temporaire d'utilisation (ATU), prévue à l'article L. 5121-12 (cf. supra l'article 12 du projet de loi), mais aussi « le contenu du dossier présenté à l'appui de ces demandes ainsi que les conditions dans lesquelles interviennent les décisions accordant, modifiant, renouvelant, suspendant ou supprimant ces autorisations », aux termes du 8° de l'article 5121-20, tel que modifié par le VI du présent article ;
- enfin, conformément au 11° du même article, tel que modifié par le VII du présent article, les règles applicables en cas de changement du titulaire de l'AMM « ou du bénéficiaire de l'enregistrement de médicament homéopathique ».
Le Conseil d'État ayant estimé que la définition des modalités d'application des articles L. 5121-17 et L. 5121-18 du même code, relatifs à la taxe annuelle des médicaments et produits bénéficiant d'une AMM, doit faire l'objet d'un décret simple, le VIII tend à supprimer le 12° de l'article L. 5121-20.
Le titre IX de la directive n° 2001/83 consolidée (articles 101 à 110) comportant d'importantes avancées dans le domaine de la pharmacovigilance, le IX du présent article propose de réécrire le 13° du même article L. 5121-10 afin que les règles applicables en la matière soient définies par décret en Conseil d'État, en particulier « les obligations de signalement incombant aux membres des professions de santé et aux entreprises exploitant un médicament ou un produit soumis aux dispositions du présent titre ».
Enfin, par cohérence avec le V du présent article, le X a pour objet de supprimer le 15° de l'article L. 5121-20, relatif aux « modalités de présentation des demandes tendant à obtenir l'enregistrement des médicaments homéopathiques, la nature du dossier ainsi que les règles relatives à l'étiquetage et à la notice de ces médicaments ».
*
La commission a examiné un amendement de Mme Jacqueline Fraysse visant à transposer une disposition de la directive permettant l'accès de tous, et notamment des personnes aveugles ou malvoyantes, aux informations essentielles concernant le médicament.
Mme Jacqueline Fraysse a précisé que l'amendement prévoit la traduction du nom du médicament en braille sur l'emballage et l'obligation pour le détenteur de l'AMM de veiller à ce que la notice d'information soit disponible, sur demande, dans des formats appropriés pour les aveugles et malvoyants.
La rapporteure s'étant déclarée défavorable à cet amendement, au motif qu'il relève du domaine règlementaire, la commission a rejeté l'amendement.
La commission a ensuite examiné un amendement de Mme Jacqueline Fraysse prévoyant que le dossier présenté pour une demande d'AMM doit comporter une évaluation de l'impact environnemental des spécialités pharmaceutiques.
Suivant l'avis défavorable de la rapporteure, pour les mêmes raisons que celles précédemment évoquées, la commission a rejeté l'amendement.
La commission a examiné un amendement de Mme Jacqueline Fraysse visant à renforcer les principes de pharmacovigilance, suite à l'allègement des procédures d'AMM, conformément aux orientations de la directive n° 2004/27.
La rapporteure ayant émis un avis défavorable à l'amendement, au motif qu'il ne relève pas du domaine de la loi, la commission a rejeté l'amendement.
La commission a ensuite adopté l'article 15 sans modification.
Article additionnel après l'article 15
Critères de certification des logiciels d'aide à la prescription
La commission a examiné un amendement de la rapporteure précisant que les règles de bonne pratique déterminées par la Haute autorité de santé spécifient que les logiciels d'aide à la prescription permettent de prescrire directement en dénomination commune internationale (DCI).
La rapporteure a précisé que la prescription et la dispensation des médicaments en dénomination commune internationale (DCI) offrent deux avantages majeurs. D'abord, la DCI accroît la sécurité des patients : le risque d'absorber deux fois le même médicament ayant deux marques différentes est diminué, la possibilité de trouver un médicament déterminé à l'étranger est garantie. De plus, la prescription en DCI permet d'accroître la substitution par les pharmaciens et donc de favoriser les médicaments génériques. Il convient donc de promouvoir la DCI.
La commission a adopté l'amendement.
Article 16
Renvoi à la compétence règlementaire pour la définition des mentions obligatoires devant figurer sur les publicités relatives au médicament
Les articles 16 et 17 du présent projet de loi comportent des dispositions relatives à la publicité pour les médicaments à usage humain.
L'article L. 5122-1 du code de la santé publique définit de manière particulièrement large la publicité pour les médicaments à usage humain. Il s'agit de « toute forme d'information, y compris le démarchage, de prospection ou d'incitation qui vise à promouvoir la prescription, la délivrance, la vente ou la consommation de ces médicaments, à l'exception de l'information dispensée, dans le cadre de leurs fonctions, par les pharmaciens gérant une pharmacie à usage intérieur».
Le présent article vise à abroger l'article L. 5122-4 du code de la santé publique, qui dispose que « la publicité des spécialités définies au 5º de l'article L. 5121-1 doit mentionner l'appartenance à la catégorie des spécialités générique ». Le renvoi au 5° de l'article L. 5121-1 du code de la santé publique correspond à la définition du médicament générique.
Comme le précise l'exposé des motifs du présent projet, il s'agit de donner compétence au pouvoir réglementaire en matière de définition des mentions obligatoires devant figurer sur les publicités relatives aux médicaments. En effet, la compétence législative en la matière paraît peu établie à la lecture de l'article 34 de la Constitution, sauf à considérer que la fixation des mentions obligatoires devant figurer sur les publicités relatives aux médicaments constitue un « principe fondamental de la sécurité sociale ».
Conformément au II de l'article 19 du présent projet de loi (cf. infra), les conditions dans lesquelles ces mentions sont rendues obligatoires seront fixées par un décret en Conseil d'État, qui déterminera également les dérogations possibles au caractère obligatoire de ces mentions.
*
La commission a adopté l'article 16 sans modification.
Article 17
Publicité auprès du public pour les médicaments à usage humain
Pour des motifs de sécurité sanitaire, la publicité des médicaments à usage humain auprès du public fait l'objet d'une réglementation très stricte.
Le présent article propose de modifier l'article L. 5122-6 du code de la santé publique, qui fixe les conditions de la publicité auprès du public pour les médicaments afin d'élargir les possibilités d'interdiction de publicité pour certains médicaments.
· Le droit en vigueur pose des conditions très contraignantes en matière de publicité pour les médicaments à usage humain auprès du public
La publicité pour les produits pharmaceutiques est étroitement règlementée par la partie législative du code de la santé publique, qui lui consacre un chapitre entier, constitué de seize articles. En particulier, les publicités auprès du public pour les médicaments sont soumises à une procédure d'autorisation préalable de l'AFSSAPS. Dans sa rédaction en vigueur, modifiée par l'article 127 de la loi nº 2004-806 relative à la politique de santé publique du 9 août 2004, l'article L. 5122-6 du code de la santé publique fixe trois conditions cumulatives à l'autorisation de la publicité, aménage une exception et introduit une obligation. En outre, un dispositif spécifique est aménagé pour les médicaments devant faire l'objet d'une radiation de la liste des médicaments remboursables.
Le premier alinéa de l'article L. 5122-6 dresse la liste des conditions autorisant les publicités pour les médicaments auprès du public. Il s'agit de trois conditions négatives excluant un certain nombre de médicaments :
- le médicament ne doit pas être soumis à prescription médicale ;
- il ne doit pas être remboursable par les régimes obligatoires d'assurance maladie ;
- l'autorisation de mise sur le marché ou l'enregistrement (pour les médicaments homéopathiques) ne doivent pas comporter de restrictions en matière de publicité « en raison d'un risque possible pour la santé publique ».
L'exception consiste à autoriser les campagnes publicitaires pour les vaccins ou les produits présentés comme supprimant l'envie de fumer ou réduisant l'accoutumance au tabac (produits visés à l'article L. 5121-2 du code de la santé publique).
La publicité pour un médicament auprès du grand public, lorsqu'elle est possible et autorisée, est enfin obligatoirement accompagnée d'un message de prudence et de renvoi à la consultation d'un médecin en cas de persistance des symptômes.
La loi relative à la politique de santé publique du 9 août 2004 a modifié l'article L. 5122-6 en autorisant qu'un médicament faisant l'objet d'une mesure de radiation de la liste des produits remboursables puisse faire l'objet d'une publicité auprès du public avant l'entrée en vigueur de la mesure de radiation, dans les conditions et sous les réserves suivantes :
- la décision de radiation de la liste des médicaments remboursables doit prévoir la possibilité d'une publicité auprès du public avant l'entrée en vigueur de la radiation ;
- la publicité auprès du public est faite dans des conditions définies par décret ;
- le médicament ne doit pas être soumis à prescription médicale et son autorisation ou son enregistrement ne doivent pas comporter de restrictions en matière de publicité auprès du public en raison d'un risque possible pour la santé publique ;
- le médicament doit être mentionné dans une convention prévue par l'article L. 162-17-4 du code de la sécurité sociale comportant des engagements sur le chiffre d'affaires.
· Les dispositions de la directive en matière de publicité auprès du public interdisent la publicité pour les médicaments délivrés sur prescription médicale
Les articles 62 à 72 de la directive fixent les grandes lignes du régime juridique applicable en matière de publicité pour les médicaments.
L'article 62 réécrit l'article 88-1 du code communautaire relatif aux médicaments à usage humain en posant le principe général de l'interdiction de la publicité des médicaments auprès du public pour deux types de médicaments : les médicaments qui ne peuvent être délivrés que sur prescription médicale et ceux qui contiennent des substances définies comme des psychotropes ou des stupéfiants. Cette interdiction ne s'applique pas aux campagnes de vaccination. L'article 88-3 du code communautaire relatif aux médicaments à usage humain autorise en outre les États membres à interdire la publicité auprès du public pour les médicaments qui sont remboursables ; il ne s'agit donc que d'une faculté pour les États.
L'article 88-2 du code communautaire relatif aux médicaments à usage humain précise que peuvent faire l'objet d'une publicité auprès du grand public les médicaments qui, par leur composition et leur objectif, sont destinés à être utilisés sans l'intervention d'un médecin pour le diagnostic, la prescription ou la surveillance du traitement, au besoin avec le conseil du pharmacien ; ils doivent de plus avoir été conçus à cette fin, cette dernière condition étant ajoutée par la directive aux prescriptions résultant de l'ancienne rédaction de l'article 88-2 du code communautaire.
L'article 64 de la directive modifie l'article 89 de la directive afin d'aménager le régime juridique des publicités dites « de rappel », qui ne comportent que le nom du médicament ou sa dénomination commune internationale, lorsqu'elle existe, ou la marque du médicament.
· Les modifications proposées par le présent article
Il est proposé de renforcer les conditions que doit satisfaire le médicament faisant l'objet d'une publicité auprès du public, alors même que ce médicament remplirait les conditions exigées par le code de la santé publique pour faire l'objet d'une publicité auprès du public (notamment l'aspect non remboursable et l'absence de prescription médicale obligatoire).
Le I de l'article vise à modifier le premier alinéa de l'article L. 5122-6 du code de la santé publique, qui fixe les conditions auxquelles doit satisfaire un médicament dont les promoteurs veulent faire la publicité auprès du public. En particulier, la rédaction en vigueur de cet alinéa prévoit que la publicité pour le médicament auprès du public n'est n'admise que si l'autorisation de mise sur le marché ne comporte pas de restrictions en matière du publicité en raison d'un risque possible pour la santé publique.
Il est proposé (1° du I de l'article) d'ajouter à la notion de « restrictions » celle « d'interdiction ». Dorénavant, l'enregistrement ou l'autorisation de mise sur le marché pourront non seulement comporter des restrictions en matière de publicité, mais aussi une interdiction.
De plus, les motifs possibles de cette restriction ou interdiction sont élargis. En effet, il est proposé (3° du I de l'article) que les restrictions ou l'interdiction de publicité mentionnées par l'autorisation de mise sur le marché ou l'enregistrement soient introduites non seulement « en raison d'un risque possible pour la santé publique » mais aussi « lorsque le médicament n'est pas adapté à une utilisation sans intervention d'un médecin pour le diagnostic, l'initiation ou la surveillance du traitement ».
Il est donc donné possibilité à l'AFSSAPS de délivrer une AMM comportant une restriction ou une interdiction de publicité auprès du public, non seulement en raison d'un risque possible pour la santé publique mais aussi lorsque l'utilisation du médicament n'est pas adaptée sans intervention d'un médecin. Ce dernier cas de figure peut conduire l'AFSSAPS à étendre considérablement les possibilités de restriction ou d'interdiction de publicité.
Cependant, cette extension de compétence de l'AFSSAPS est nuancée car elle concerne l'utilisation de médicaments pour le diagnostic, l'initiation (prescription) ou la surveillance de traitement. Pour les autres cas subsiste comme unique motif de restriction ou d'interdiction de publicité le risque possible pour la santé publique.
Le 2° du I de l'article propose d'élargir la condition relative au caractère non remboursable du médicament en l'étendant à toutes les présentations du médicament. Le médicament pourra faire l'objet d'une publicité auprès du public si aucune de ses différentes présentations ne peut faire l'objet d'un remboursement.
La condition relative au caractère non remboursable est donc considérablement durcie. Dans le cas où le même produit comporte une présentation remboursable et une présentation non remboursable, cette disposition permet d'éviter qu'une publicité faite pour la présentation non remboursable ne soit utilisée pour promouvoir indirectement la présentation remboursable.
Le II du présent article, par parallélisme, propose de modifier les conditions dans lesquelles un médicament faisant l'objet d'une mesure de déremboursement peut faire l'objet d'une publicité auprès du grand public, afin de prévoir les situations où l'autorisation de mise sur le marché ou l'enregistrement comportent une mesure d'interdiction de publicité auprès du public. Dans ce cas, la publicité auprès du public n'est pas permise.
Enfin, le III de l'article propose de supprimer le dernier alinéa de l'article L. 5122-6 du code de la santé publique relatif au message de prudence ou de renvoi à la consultation d'un médecin en cas de persistance des symptômes dans les publicités pour les médicaments s'adressant au public. Conformément à l'article 19 du présent projet de loi (cf. infra), les conditions dans lesquelles ces mentions sont rendues obligatoires - et les éventuelles dérogations à ce principe - seront fixées par un décret en Conseil d'État. Selon les informations transmises à la rapporteure, le décret évoquerait également le recours au pharmacien.
*
La commission a examiné un amendement de M. Claude Evin visant à supprimer le huitième alinéa de cet article.
M. Claude Evin a indiqué que le dernier alinéa de l'article L. 5122-6 du code de la santé publique, dont l'alinéa 8 du présent article propose la suppression, dispose que « la publicité auprès du public pour un médicament est nécessairement accompagnée d'un message de prudence et de renvoi à la consultation d'un médecin en cas de persistance des symptômes ». Supprimer cet alinéa revient à supprimer l'obligation de prudence dans le message publicitaire. Une telle orientation exige d'être retirée sous peine de banaliser la consommation médicamenteuse.
La rapporteure s'est déclarée favorable à cet amendement, ainsi que Mme Jacqueline Fraysse.
La commission a adopté l'amendement à l'unanimité.
La commission a ensuite adopté l'article 17 ainsi modifié.
Article 18
Remise gratuite d'échantillons de médicaments
et avantages consentis aux professionnels de santé
Le présent article vise à règlementer les relations entre les professions de santé et les représentants de l'industrie pharmaceutique, en ce qui concerne la distribution des échantillons de médicaments d'une part et les éventuels « cadeaux » d'autre part.
Compte tenu du poids financier des entreprises fabriquant et commercialisant des médicaments, il est en effet essentiel que la législation garantisse que les professionnels de santé, qui prescrivent et dispensent les médicaments, soient à l'abri de leur influence.
A titre de rappel, l'article 2 du présent projet de loi (cf. supra), proposant de modifier l'article L. 4113-6 du code de la santé publique, transpose un article de la directive n° 2004/27 relatif au dispositif « anti-cadeaux » concernant les relations entre les professionnels de santé et l'industrie. Dans le code de la santé publique, il est inscrit dans le chapitre relatif aux règles communes d'exercice des professions médicales, chapitre figurant dans la quatrième partie du code consacrée aux professions de santé.
Le présent article propose de modifier l'article L. 5122-10 du code de la santé publique, qui se situe dans le chapitre relatif à la publicité pour les médicaments inclus dans la cinquième partie du code de la santé publique relative aux produits de santé. Cet article règlemente la remise à titre gratuit des échantillons et les « cadeaux ».
· La réglementation déjà en vigueur
- Les échantillons gratuits de médicaments
Les cinq alinéas de la rédaction en vigueur de l'article L. 5122-10 du code de la santé publique prévoient déjà des dispositions en la matière. Les quatre premiers alinéas encadrent la remise gratuite d'échantillons :
- les échantillons gratuits ne peuvent être remis que sur demande des personnes habilitées à prescrire ou à dispenser des médicaments dans le cadre des pharmacies à usage intérieur ;
- le contenu des échantillons est limité ; ils ne doivent pas comporter des substances classées comme psychotropes ou stupéfiants ;
- dans les congrès, la remise gratuite d'échantillons gratuits est interdite dans les enceintes ouvertes au public ;
- enfin, les échantillons doivent être identiques aux spécialités pharmaceutiques concernées et porter la mention : « échantillon gratuit ».
Les établissements pharmaceutiques peuvent faire parvenir des échantillons aux professionnels habilités à en recevoir. Jusqu'à récemment, les modalités d'acheminement de ces échantillons ne faisaient l'objet d'aucune disposition législative ou réglementaire. Rien n'interdisait de faire remettre ces échantillons par les visiteurs médicaux, plutôt que de les faire livrer directement. La charte de la visite médicale, conclue le 22 décembre 2004 entre le comité économique des produits de santé (CEPS) et l'industrie pharmaceutique en application de la loi n° 2004-810 du 13 août 2004 relative à l'assurance maladie interdit la remise d'échantillons au médecin par le visiteur médical, sauf dans les départements d'outre-mer. En effet, cette pratique ne donnait pas les garanties nécessaires en termes de sécurité de conservation des produits. Les laboratoires, en revanche, peuvent distribuer des échantillons aux médecins, mais sur leur demande.
- Les avantages consentis aux professionnels de santé
Le cinquième alinéa de l'article L. 5122-10 est relatif aux « cadeaux » ; il dispose que « dans le cadre de la promotion des médicaments auprès des personnes habilitées à les prescrire ou à les délivrer, il est interdit d'octroyer, d'offrir ou de promettre à ces personnes une prime, un avantage pécuniaire ou un avantage en nature, à moins que ceux-ci ne soient de valeur négligeable ». Il s'agit par ce biais de réglementer l'activité des visiteurs médicaux, qui a fait l'objet de la charte de la visite médicale.
· Les dispositions du droit communautaire
S'agissant des échantillons, l'article 96 du code communautaire relatif aux médicaments à usage humain dispose que des échantillons gratuits ne peuvent être remis à titre exceptionnel qu'aux personnes habilitées à prescrire et selon certaines conditions. Le nombre d'échantillons fourni pour chaque médicament par an et par prescripteur doit être limité. Chaque fourniture d'échantillons doit répondre à une demande écrite, datée et signée, émanant du prescripteur. Chaque échantillon doit être identique au plus petit conditionnement commercialisé et doit porter la mention « échantillon médical gratuit ».
La directive modifie la condition relative à la taille de l'échantillon et précise qu'aucun échantillon ne doit être plus grand que le plus petit conditionnement commercialisé. En outre, le 6° de l'article 62 de la directive prescrit que « les États membres interdisent la distribution directe de médicaments au public à des fins promotionnelles par l'industrie ».
S'agissant des cadeaux et avantages, l'article 94 du code communautaire relatif aux médicaments à usage humain précise que dans le cadre de la promotion des médicaments auprès des personnes habilitées à les prescrire ou à les délivrer, il est interdit d'octroyer, d'offrir ou de promettre à ces personnes une prime, un avantage pécuniaire ou un avantage en nature à moins que ceux-ci ne soient de valeur négligeable et n'aient trait à l'exercice de la médecine ou de la pharmacie.
· Les modifications proposées
Le I du présent article concerne la problématique de la remise d'échantillons gratuits. Il propose de modifier les quatre premiers alinéas de l'article L. 5122-10 du code de la santé publique.
Il vise d'abord à préciser que les dispositions du premier alinéa de l'article, prescrivant que les échantillons gratuits sont remis uniquement sur demande, ne visent que les échantillons « de médicaments ». Cette disposition harmonise la rédaction des deux premiers alinéas de l'article L. 5122-10. Il s'agit d'une simple coordination terminologique, car le code de la santé publique utilise plusieurs expressions pour désigner la même chose : « échantillons gratuits », « échantillons », « échantillons de médicaments » et « échantillons gratuits de médicaments ».
L'alinéa visant à ce que les échantillons soient identiques aux spécialités pharmaceutiques concernées et portent la mention « échantillon gratuit », en quatrième position dans la rédaction en vigueur de l'article, est remonté en troisième position.
Il est également proposé de rédiger différemment l'alinéa relatif à l'interdiction de remise d'échantillons dans le cadre des congrès. La rédaction en vigueur, ambiguë, interdit la remise d'échantillons, lors des congrès, dans les enceintes accessibles au public. Cette disposition est modifiée afin de poser une interdiction générale d'interdiction de remise « directe » d'échantillons au public à des fins promotionnelles. Cette modification, qui introduit plus de clarté, est de nature à faciliter l'application de la disposition.
Le II de l'article concerne les « avantages » divers consentis aux personnes habilitées à prescrire (médecins) ou à délivrer des médicaments (pharmaciens) dans le cadre de la promotion des médicaments, particulièrement dans le cadre de la visite médicale. Dans la rédaction en vigueur, la remise de cadeaux, de prime ou d'avantage aux personnes habilitées à prescrire ou à délivrer des médicaments fait l'objet d'une interdiction générale, sauf si leur valeur est négligeable. En revanche, la nature de ces cadeaux n'est pas précisée.
Le II du présent article, qui transpose l'article 94-I du code communautaire relatif aux médicaments à usage humain, propose de n'autoriser ces avantages et cadeaux qu'à deux conditions cumulatives : d'une part, la valeur négligeable de ces avantages et, d'autre part, le lien entre ces cadeaux et l'exercice de la médecine et de la pharmacie. Il s'agit donc de renforcer considérablement la législation nationale en la matière, en interdisant la remise de cadeaux, même de valeur négligeable, n'ayant aucun lien avec la médecine ou la pharmacie. Il est à noter que la charte de la visite médicale interdit au délégué médical de proposer au médecin des cadeaux en nature ou en espèces et de répondre à d'éventuelles sollicitations dans ce domaine émanant du professionnel de santé.
*
La commission a adopté l'article 18 sans modification.
Article 19
Remise gratuite d'échantillons de médicaments -
Mentions obligatoires devant figurer dans les publicités pour les
médicaments -
Autorisation des publicités de rappel
Cet article vise à préciser la compétence réglementaire s'agissant, d'une part, de la remise d'échantillons gratuits et, d'autre part, des mentions obligatoires devant figurer dans les publicités pour les médicaments à usage humain.
L'article L. 5122-16 du code de la santé publique, dont la modification est proposée par le présent article, fixe les dispositions réglementaires devant faire l'objet d'un décret en Conseil d'État.
Le I du présent article est une disposition de coordination relative à la remise d'échantillons gratuits de médicaments. Effectuant une harmonisation avec la rédaction de l'article L. 5122-10 du code de la santé publique telle que modifiée par l'article 18 du présent projet (cf. supra), il n'appelle pas de commentaire particulier.
Le II du présent article propose de créer deux catégories supplémentaires de dispositions devant faire l'objet d'un décret en Conseil d'État : les mentions obligatoires devant figurer dans les publicités pour les médicaments et les dispositions relatives à la publicité de rappel.
· Les mentions obligatoires devant figurer dans les publicités pour les médicaments
Seront définies par voie réglementaire les mentions obligatoires devant figurer dans les publicités pour les médicaments. Pour les publicités visant le public, il s'agit notamment des messages de prudence ou de renvoi à la consultation d'un médecin en cas de persistance des symptômes, messages qui sont visés au dernier alinéa de l'article L. 5122-6 du code de la santé publique et qui feront donc l'objet d'un décret (la suppression de cet alinéa étant proposée par l'article 16 du présent projet, cf. supra). Les mentions obligatoires figurent déjà dans les textes règlementaires suivants : R. 5122-8 pour les publicités destinées aux professionnels et R. 5122-3 pour les publicités visant le grand public.
· La publicité de rappel
Le paragraphe 2 de l'article 89 du code communautaire relatif aux médicaments du médicament autorise la publicité de rappel, en permettant qu'une publicité pour un médicament à usage humain mentionne exclusivement la « dénomination » du médicament. En effet, le b) du I de l'article 89 du code communautaire dispose qu'une publicité pour un médicament à usage humain doit être accompagnée d'un certain nombre de mentions (dénomination du médicament, informations pour un bon usage et invitation expresse à lire les instructions). Le code communautaire autorise (il s'agit d'une faculté pour les États membres) les publicités de rappel auprès du public lorsqu'elles comportent uniquement la dénomination du médicament. L'article 64 de la directive modifie l'article 89 du code communautaire. Il rend obligatoire, dans les publicités, la dénomination et le nom du médicament. Il continue d'autoriser les publicités de rappel auprès du public mais en élargissant les possibilités. En effet, ces publicités peuvent comporter uniquement le nom du médicament, sa dénomination commune internationale (lorsqu'elle existe) ou sa marque. Une publicité de rappel est une publicité qui se contente de faire référence à une publicité antérieure pour un produit ou une marque. Il peut ainsi s'agir d'un très court spot télévisé faisant apparaître un logo sans autre commentaire, sans mention des indications du médicament et sans conseil de prudence.
Le présent article propose donc d'introduire une base légale autorisant par voie réglementaire des dérogations à l'obligation de joindre aux publicités (pour le public et les professionnels) un certain nombre de mentions. Ainsi, aux termes du présent article, un décret en Conseil d'État pourra définir une catégorie particulière de publicité (les publicités de rappel), qui comportera uniquement soit le nom du médicament, soit sa dénomination commune internationale, soit sa marque. Sur ces points, la rédaction du présent article reprend presque littéralement la rédaction de la directive.
L'introduction de la publicité de rappel pour les médicaments représente une nouveauté dans le droit français, en application d'une faculté aménagée par la directive. Selon les informations communiquées à la rapporteure, le décret en Conseil d'État pris pour l'application de cette disposition précisera les mentions que devront comporter ces publicités de rappel en ce qui concerne les médicaments. Pour les publicités auprès du public, les mentions obligatoires pourraient être le nom du médicament, le nom du laboratoire reprenant ses « attributs » (logo) et la mention « ceci est un médicament » ; pour les publicités auprès des professionnels, ces mentions pourraient être le nom du médicament, le nom du laboratoire et le principe actif du médicament (désigné par sa dénomination commune).
La rapporteure remarque que le III de l'article 17 du présent projet (cf. supra) a proposé de confier au gouvernement la compétence de fixer le principe du message de prudence joint aux publicités pour les médicaments s'adressant au public, principe fixé par la loi. De plus, le présent article propose que le règlement fixe conjointement, s'agissant des publicités s'adressant au public et aux professionnels, le principe de ces mentions obligatoires (dont le message de prudence) et les possibilités d'y déroger. Une dérogation explicite est prévue par le présent projet, celle relative aux publicités de rappel. Cependant, la mention de l'adverbe « notamment » dans le dispositif du II du présent article laisse une grande marge d'appréciation pour le gouvernement s'agissant de réglementer les mentions obligatoires devant figurer dans les publicités visant le public.
*
La commission a adopté l'article 19 sans modification.
Article 20
Transmission à l'AFSSAPS de la date de commercialisation du médicament
Cet article vise à préciser les conditions d'application de l'article
L. 5124-5 du code de la santé publique obligeant l'établissement pharmaceutique à communiquer à l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) la date de la commercialisation d'un médicament ou d'un produit soumis à l'autorisation de mise sur le marché.
· Le droit en vigueur
La rédaction en vigueur de l'article L. 5124-5 du code de la santé publique dispose que « lorsqu'un médicament ou produit soumis à l'autorisation de mise sur le marché prévue à l'article L. 5121-8 est commercialisé, l'établissement pharmaceutique qui l'exploite communique, sans délai, la date de cette commercialisation à l'Agence française de sécurité sanitaire des produits de santé. »
La communication de la date de commercialisation à l'agence nationale est un élément important du système de pharmacovigilance. Il permet à l'agence de disposer d'un tableau de bord à jour recensant non seulement les spécialités approuvées mais aussi celles effectivement commercialisées.
· Les dispositions de la directive
Le présent article constitue la transposition de l'article 22 de la directive 2004/27, qui introduit dans le code communautaire relatif aux médicaments à usage humain un article 23 bis nouveau, dont le premier alinéa dispose qu'« après la délivrance d'une autorisation de mise sur le marché, son titulaire informe l'autorité compétente de l'État membre qui l'a délivrée de la date de la mise sur le marché effective du médicament à usage humain dans cet État membre, en tenant compte des différentes présentations autorisées ».
La communication de la date de commercialisation d'une spécialité ayant obtenue une AMM est essentielle dès lors que l'AMM devient caduque si le médicament considéré n'est pas effectivement mis sur le marché. Or, la directive modifie l'article 24 du code communautaire relatif aux médicaments à usage humain en disposant que toute AMM non utilisée pendant trois années consécutives entraîne la caducité de celle-ci.
· Les modifications proposées
Le présent article vise à clarifier la rédaction de l'article L. 5124-5 du code de la santé publique en proposant deux modifications.
D'abord, la mention de l'« établissement pharmaceutique » est remplacée par la mention de l'« entreprise ».
Le régime juridique de l'établissement pharmaceutique est défini par le chapitre IV « Fabrication et distribution en gros » du titre II (« Médicaments à usage humain ») du livre premier (« Produits pharmaceutiques ») de la cinquième partie du code de la santé publique. L'établissement pharmaceutique est une notion juridique large comprenant les officines, les pharmacies à usage intérieur et les entreprises pharmaceutiques (une entreprise peut comporter plusieurs établissements pharmaceutiques). Certaines activités relatives aux médicaments ne peuvent avoir lieu que dans un établissement pharmaceutique ; l'ouverture d'un établissement pharmaceutique est soumise à autorisation.
La mention de « l'entreprise exploitant », par opposition à la mention de « l'établissement pharmaceutique exploitant » permet de couvrir les entreprises exploitant un médicament depuis un autre État de l'Union européenne, lesquelles ne peuvent être soumises au statut d'établissement pharmaceutique prévu par le droit français, puisqu'elles ne sont pas sur notre territoire.
Ensuite, il est proposé que l'établissement transmette à l'AFSSAPS non plus « la date de cette commercialisation » mais, le cas échéant, « les dates de commercialisation de chaque présentation de ce médicament ou produit ». Cette modification permet de prendre en compte les cas où le médicament fait l'objet de présentations différentes. Elle contribue à la mise en place de toutes les conditions nécessaires à l'établissement d'une pharmacovigilance efficace. Le code de la santé publique est plus strict que la directive, puisque la rédaction de l'article L. 5124-5 du code de la santé publique prescrit que la transmission de la date de commercialisation à l'AFSSAPS se fait « sans délai », alors que l'article 23 bis de la directive ne mentionne pas de délai.
*
La commission a adopté l'article 20 sans modification.
Article 21
Informations à fournir à l'AFSSAPS en cas de suspension de commercialisation d'un médicament ou de risque de rupture de stock
Le présent article, proposant de modifier l'article L. 5124-6 du code de la santé publique, vise à préciser les conditions dans lesquelles les entreprises commercialisant des médicaments doivent alerter l'AFSSAPS en cas de suspension de commercialisation du produit ou de rupture de stock.
· Le droit en vigueur
La rédaction en vigueur de l'article L. 5124-6 du code de la santé publique, issue de l'article 23 de la loi nº 2004-806 relative à la politique de santé publique du 9 août 2004, fait d'abord peser sur les entreprises commercialisant des médicaments une obligation d'information de l'AFSSAPS en cas d'action visant à suspendre la commercialisation du produit ou à le retirer du marché. Cette information doit être « immédiate ». L'article prescrit également au fabricant d'indiquer la raison de ces actions si elles sont relatives à l'efficacité du médicament ou à la protection de la santé publique.
L'établissement pharmaceutique doit en outre informer l'AFSSAPS de tout risque de rupture de stock sur deux types de médicament ou produit dont il assure l'exploitation : les médicaments ou produits sans alternative thérapeutique disponible ; les médicaments ou produits connaissant un accroissement brutal et inattendu de la demande.
· La directive
Il est proposé de transposer l'article 22 de la directive, qui introduit dans le code communautaire relatif aux médicaments à usage humain un article 23 bis nouveau ainsi rédigé : « Le titulaire [de l'AMM] prévient également l'autorité compétente si le médicament n'est plus mis sur le marché dans l'État membre concerné, de manière provisoire ou définitive. Cette notification doit avoir lieu, hormis dans des circonstances exceptionnelles, au plus tard deux mois avant l'interruption de la mise sur le marché du médicament ».
Cet article 23 bis prévoit également que les agences peuvent demander aux laboratoires des données sur les quantités de médicament vendues, disposition qui ne fait pas l'objet d'une transposition législative par le présent projet mais qui fera l'objet d'une mesure réglementaire.
· Les modifications proposées
Le I du présent article vise à substituer à la mention « établissement pharmaceutique » la mention de l' « entreprise », substitution faisant l'objet d'une coordination au IV. La motivation de cette substitution est identique à celle de l'article précédent du présent projet : elle permet de couvrir les entreprises exploitant un médicament depuis un autre État de l'Union européenne, lesquelles ne peuvent être soumises au statut d'établissement pharmaceutique prévu par le droit français, puisqu'elles ne sont pas sur notre territoire.
Conformément à la rédaction de la directive, le II supprime la mention du caractère « immédiat » de l'information fournie à l'AFSSAPS par les fabricants en cas d'action visant à suspendre la commercialisation du produit ou à le retirer du marché. Le délai sera fixé par décret (cf. infra, V). L'article 23 bis introduit par la directive dans le code communautaire fixe un délai de deux mois hors circonstances exceptionnelles.
Le III concerne l'information obligatoire à fournir à l'AFSSAPS en cas de risque d'une rupture de stock, obligation visée par la dernière phrase de l'article L. 5124-6 du code de la santé publique dans sa rédaction en vigueur. Le III propose une modification rédactionnelle consistant à déplacer le contenu de cette dernière phrase et à le joindre à la fin de la première phrase de l'article L. 5124-6, dont la rédaction deviendrait : « L'entreprise exploitant un médicament ou produit soumis aux dispositions du chapitre I du présent titre informe l'Agence française de sécurité sanitaire des produits de santé de toute action qu'il a engagée pour en suspendre la commercialisation, le retirer du marché ou en retirer un lot déterminé, ainsi que de tout risque de rupture de stock sur un médicament ou produit sans alternative thérapeutique disponible ou en raison d'un accroissement significatif et imprévisible de la demande ».
Le V de l'article propose qu'un décret fixe les conditions (notamment de délai) dans lesquelles l'information est délivrée à l'agence.
*
M. Claude Evin a indiqué que cet article pose un problème rédactionnel. La mention des « entreprises » remplaçant celles des « établissements », il convient sans doute de transformer à divers endroits du code de la santé publique les « ils » en « elles ».
La commission a adopté l'article 21 sans modification.
Article 22
Autorisation d'importation par un particulier de médicament à usage humain
Le présent article vise à préciser les conditions d'importation de médicaments par un particulier.
Il vise à ce que les importations personnelles de médicaments, lorsqu'elles concernent des médicaments ayant fait l'objet d'une AMM dans un État membre de l'Union européenne ou des médicaments homéopathiques ayant fait l'objet d'un enregistrement dans un État membre soient autorisées a priori sans qu'une autorisation d'importation auprès des autorités françaises soit nécessaire.
Compte tenu des différences de prix entre pays, l'importation de médicaments peut poser de nombreux problèmes au pays importateur. En particulier, l'organisation d'un commerce parallèle de médicaments importés à bas prix peut avoir des effets négatifs sur les investissements en recherche et développement ainsi que sur la rentabilité des entreprises fabriquant les produits importés.
De plus, l'importation de médicaments a fait l'objet d'une réglementation stricte en raison de la nécessité d'assurer une sécurité sanitaire totale pour les patients. Il faut en outre distinguer l'importation en « transport personnel » par le particulier (le particulier rentre sur le territoire avec les médicaments), qui fait l'objet d'une réglementation plus souple, et des autres modes d'importation.
· La législation en vigueur
En raison des considérations mentionnées plus haut, le régime juridique applicable aux importations personnelles effectuées par les médicaments est assez strict.
L'état du droit est fixé par l'article L. 5124-13 du code de la santé publique dans sa rédaction issue de l'article 19 de la loi nº 2004-800 du 6 août 2004 relative à la bioéthique. L'importation sur le territoire douanier des médicaments à usage humain est soumise à une autorisation préalable délivrée par l'AFSSAPS. Cependant, l'autorisation de mise sur le marché ou l'enregistrement vaut autorisation. Conformément à l'article R. 5121-110 du code de la santé publique, « les particuliers ne peuvent importer un médicament qu'en quantité compatible avec un usage thérapeutique personnel pendant une durée de traitement n'excédant pas trois mois aux conditions normales d'emploi ou pendant la durée de traitement prévue par l'ordonnance prescrivant le médicament. Lorsqu'ils transportent personnellement ce médicament, ils sont dispensés d'autorisation ». Lorsque l'importation des médicaments n'est pas réalisée par transport personnel, les règles générales d'importation, prévues aux articles R. 5142-2 et R. 5142-14 du code de la santé publique, s'appliquent.
· La jurisprudence de la Cour de justice des communautés européennes
Comme le précise l'exposé des motifs du présent article, il s'agit de tirer les conséquences de la jurisprudence de la Cour de justice des communautés européennes (CJCE). L'arrêt de la deuxième chambre du 26 mai 2005 (C-212/03), « Commission des communautés européennes contre République française », arrêt relatif aux importations personnelles de médicaments, exige en effet la modification de la législation française.
À l'origine de l'affaire se trouve une plainte émanant d'un fabricant espagnol ayant fait l'objet de poursuites judiciaires en France. Il avait expédié à des patients français des médicaments homéopathiques alors même qu'il ne disposait pas d'autorisation d'importation délivrée par les autorités françaises. Il faut souligner que ces médicaments, destinés à l'usage personnel des patients, ne faisaient pas l'objet d'une revente en France.
Dans cette affaire, la Cour a jugé que les dispositions du droit français ayant pour objet de contrôler l'importation de médicaments d'un État membre par une personne ne sont pas conformes au droit communautaire, et notamment au principe de libre circulation des marchandises, alors même que les médicaments considérés ont reçu une autorisation de mise sur le marché dans l'État membre où ils ont été achetés mais pas en France. Plus précisément, la Cour a jugé qu'il était disproportionné, compte tenu du respect nécessaire de la liberté des échanges intracommunautaires, que la France applique une lourde procédure d'autorisation préalable à des médicaments, alors même que ces médicaments sont autorisés dans un autre pays membre et qu'ils ne font pas l'objet d'une exploitation commerciale en France. Cette décision de justice exige de la France une modification de son droit interne. En effet, à la suite de cette condamnation par la CJCE, la France est actuellement menacée du paiement d'une astreinte au titre de l'article 228 du traité CE. Rappelons qu'aux termes de l'article 228 du traité instituant la communauté européenne :
« 1. Si la Cour de justice reconnaît qu'un État membre a manqué à une des obligations qui lui incombent en vertu du présent traité, cet État est tenu de prendre les mesures que comporte l'exécution de l'arrête de la Cour de justice.
« 2. Si la Commission estime que l'État membre concerné n'a pas pris ces mesures, elles émet, après avoir donné à cet État la possibilité de présenter ses observations, un avis motivé précisant les points sur lesquels l'État membre concerné ne s'est pas conformé à l'arrêt de la Cour de justice.
« Si l'État membre concerné pas pris les mesures que comporte l'exécution de l'arrêt de la Cour dans le délai fixé par la Commission, celle-ci peut saisir la Cour de justice. Elle indique le montant de la somme forfaitaire ou de l'astreinte à payer par l'État membre concerné qu'elle estime adaptée aux circonstances.
« Si la Cour de justice reconnaît que l'État membre concerné ne s'est pas conformé à son arrêt, elle peut lui infliger le paiement d'une somme forfaitaire ou d'une astreinte.
« Cette procédure est sans préjudice de l'article 227. »
· La modification proposée
Le présent article propose deux modifications à l'article L. 5124-13 du code de la santé publique :
- d'une part, il propose de préciser dans la partie législative du code de la santé publique que l'autorisation préalable n'est pas obligatoire pour le particulier transportant personnellement un médicament, ce qui constitue une clarification bienvenue ;
- d'autre part, il est précisé que la procédure d'autorisation ne soit pas requise lorsque le particulier fait importer un médicament qui a déjà été autorisé dans l'Union européenne.
Le premier alinéa de la rédaction proposée pour l'article L. 5124-13 du code de la santé publique pose le principe qu'un particulier transportant personnellement des médicaments n'a pas à demander d'autorisation préalable.
Le deuxième alinéa vise le cas où un particulier procède à l'importation d'un médicament par une autre voie que le transport personnel. Le particulier n'est pas soumis à l'obligation de demander une autorisation préalable si ce médicament fait déjà l'objet d'une autorisation de mise sur le marché ou d'un enregistrement dans un État membre de l'Union européenne ou un État partie à l'accord sur l'Espace économique européen.
*
La commission a examiné un amendement de M. Bernard Depierre visant à renforcer la disponibilité des médicaments homéopathiques en permettant aux pharmaciens d'importer pour le compte du patient le médicament considéré.
Mme Maryvonne Briot a défendu l'amendement et précisé que, sans cette mention, le projet de loi limite considérablement le bénéfice de l'absence de l'autorisation préalable dans la mesure où le citoyen est obligé d'importer lui-même le produit souhaité.
La rapporteure s'est déclarée défavorable à cet amendement.
La commission a rejeté l'amendement.
La commission a ensuite adopté l'article 22 sans modification.
Article 23
Régime juridique des matières premières à usage pharmaceutique
Le présent article propose une nouvelle rédaction de l'article L. 5138-2 du code de la santé publique tendant à donner une nouvelle définition des matières premières à usage pharmaceutique.
Afin de garantir une sécurité sanitaire maximale des médicaments et des produits de santé en général, il est impératif de contrôler la qualité des composants des médicaments sur toute la chaîne, de la fabrication à la distribution. Ainsi, le considérant 19 de la directive précise qu'il « convient de garantir la qualité des médicaments à usage humain fabriqués ou disponibles dans la Communauté, en exigeant que les substances actives qui entrent dans leur composition soient conformes aux principes relatifs aux bonnes pratiques de fabrication. Il s'avère nécessaire de renforcer les dispositions communautaires relatives aux inspections et de mettre en place un registre communautaire portant sur les résultats de ces inspections. »
Les matières premières à usage pharmaceutique font l'objet du chapitre VIII du titre troisième du livre premier de la cinquième partie du code de la santé publique (partie relative aux produits de santé). Ce chapitre définit le régime juridique des matières premières à usage pharmaceutique en posant les principes suivants :
- les activités de fabrication, d'importation ou de distribution de matières premières à usage pharmaceutique sont soumise à une déclaration auprès de l'AFSSAPS ;
- les matières premières à usage pharmaceutique doivent répondre aux spécifications de la pharmacopée et être fabriquées et distribuées en conformité avec des bonnes pratiques définies par décision de l'AFSSAPS ;
- tout établissement de fabrication, d'importation ou de distribution de matières premières à usage pharmaceutique peut demander à l'AFSSAPS de certifier que l'établissement qui produit les matières premières respecte les bonnes pratiques définies plus haut.
Le présent article propose de modifier l'article L. 5138-2 du code de la santé publique, dont la rédaction en vigueur de résulte de l'ordonnance nº 2005-1087 du 1er septembre 2005. L'article, dans sa rédaction en vigueur, dispose que « les matières premières à usage pharmaceutique doivent répondre aux spécifications de la pharmacopée quand elles existent et être fabriquées et distribuées en conformité avec des bonnes pratiques dont les principes sont définis par décision de l'Agence française de sécurité sanitaire des produits de santé. »
Il est proposé que l'article L. 5138-2 du code de la santé publique établisse une définition des matières premières à usage pharmaceutique (paragraphe I de la rédaction proposée pour l'article L. 5138-2), crée une présomption d'usage pharmaceutique (paragraphe II de la rédaction proposée pour l'article L. 5138-2), impose au vendeur de ces matières premières de pouvoir justifier de la destination de ces matières premières (paragraphe III de la rédaction proposée pour l'article L. 5138-2) et, enfin, définisse les activités de production et de distribution des matières premières (paragraphe IV et V).
L'article 24 du présent projet (cf. infra) définit la composition des matières premières à usage pharmaceutique, et l'article 25 fixe les compétences d'inspection de l'AFSSAPS en ce qui concerne les matières premières à usage pharmaceutique.
Il est proposé que le I de l'article L. 5138-2 du code de la santé publique définisse les matières premières à usage pharmaceutique comme « tous les composants des médicaments », c'est-à-dire :
- la ou les substances actives du médicament ;
- les excipients (produits dépourvus de visée thérapeutique mais facilitant la préparation, la conservation ou l'administration du médicament considéré) ;
- les éléments de mise en forme pharmaceutiques (crème, gélule, sirop...).
Cette définition relativement large des composants du médicament permet d'inclure dans la réglementation applicable aux matières premières des éléments qui ne sont pas des principes actifs mais dont la qualité peut avoir un impact sur l'efficacité ou l'innocuité du médicament. Elle est inspirée de l'article 33 de la directive, qui modifie l'article 46 du code communautaire relatif aux médicaments à usage humain afin que le titulaire de l'autorisation de fabrication du médicament respecte les principes et lignes directrices relatifs aux bonnes pratiques de fabrication des médicaments et utilise des substances actives fabriquées conformément aux lignes directrices détaillées relatives aux bonnes pratiques de fabrication des matières premières ; il est expressément précisé que cette prescription est également applicable à certains excipients, dont la liste et les conditions spécifiques d'application sont arrêtées par une directive adoptée par la commission.
Il est ensuite proposé que le II de l'article L. 5138-2 du code de la santé publique établisse une présomption d'usage pharmaceutique lorsque les matières premières considérées (principes actifs d'un médicament, excipient et éléments de mise en forme pharmaceutique) sont cédées aux personnes ou organismes suivants :
- un établissement pharmaceutique mentionné à l'article L. 5124-1 ou L. 5124-2 du code de la santé publique ;
- une pharmacie à usage intérieur ;
- une officine de pharmacie ;
- un médecin ou un vétérinaire, ainsi que les personnes autorisées à préparer des autovaccins à usage vétérinaire.
La présomption de matières premières à usage pharmaceutique emporte l'application du régime juridique correspondant, notamment en ce qui concerne les procédés de fabrication, la distribution ou les compétences de l'AFSSAPS. Cependant, l'article dispose que cette présomption d'usage pharmaceutique peut être levée par la personne cédant ces matières premières par la présentation d'une attestation en ce sens émanant de l'acheteur.
Le III de la rédaction proposé pour l'article L. 5138-2 vise à prendre en compte le cas d'une matière première à usage pharmaceutique « par nature » (telles que définies par le I de l'article L. 5138-2) cédée à une personne autre que celles mentionnées par le II du même article. En effet, un établissement pharmaceutique peut acquérir une substance chimique susceptible d'être destinée à la fabrication d'un médicament pour en faire un autre usage (par exemple le nettoyage d'appareils ou de locaux). De plus, des matières premières à usage pharmaceutiques peuvent être cédées à d'autres personnes ou établissements, comme des intermédiaires. Or les fabricants doivent connaître l'usage final de la matière pour lui appliquer les bonnes pratiques et, le cas échéant, obtenir le certificat.
Dans ce cas de figure, afin d'établir ou non l'usage pharmaceutique de la matière première, le présent article impose au vendeur de justifier par tous moyens de la destination de ces matières premières. A cette fin, il peut demander à l'acheteur une attestation justifiant de leur destination.
Le IV et le V de l'article L. 5138-2 visent à définir les processus d'une part de fabrication de matières premières à usage pharmaceutique (IV) et d'autre part de distribution de ces matières (V).
La fabrication inclut non seulement la fabrication complète de la matière, mais aussi sa fabrication partielle et les divers procédés de division ou de conditionnement préalables à son incorporation dans un médicament et le stockage, en vue de sa vente. Cette définition correspond aux modifications introduites par l'article 34 de la directive, qui introduit un article 46 bis nouveau dans le code communautaire relatif aux médicaments à usage humain.
La distribution comprend les activités d'achat, de vente, de reconditionnement, de réétiquetage et de stockage.
Le réétiquetage peut être défini comme étant soit le retrait de l'étiquette du fabricant et l'apposition d'une nouvelle étiquette par le distributeur, mentionnant notamment ses coordonnées (le retrait de l'étiquette du fabricant ne doit cependant pas dissimuler la provenance de la matière première), soit l'apposition d'une étiquette supplémentaire sur le conditionnement de la matière première par le distributeur. Ce réétiquetage permet notamment une traçabilité des différents opérateurs.
Dans la majeure partie des cas, le réétiquetage ne se double pas de reconditionnement. Toutefois, certains distributeurs sont spécialisés dans l'achat de grandes quantités de matières premières pour une revente en petites quantités. Il procède alors au reconditionnement et au réétiquetage de la matière première.
*
La commission a adopté l'article 23 sans modification.
Article 24
Bonnes pratiques de fabrication et de distribution des matières premières
à usage pharmaceutique
Le présent article vise à réglementer la composition des matières premières à usage pharmaceutique et à introduire de bonnes pratiques de fabrication et de distribution des matières premières à usage pharmaceutique.
· Le droit en vigueur
L'article L. 5138-2 du code de la santé publique dispose que les matières premières à usage pharmaceutique doivent répondre aux spécifications de la pharmacopée (quand elles existent) et être fabriquées et distribuées en conformité avec des bonnes pratiques dont les principes sont définis par décision de l'Agence française de sécurité sanitaire des produits de santé.
· Les modifications introduites par la directive
Avant l'adoption de la directive dont la transposition en droit interne fait l'objet du présent projet de loi, l'article 46 du code communautaire relatif aux médicaments à usage humain prescrivait que le titulaire de l'autorisation de fabrication doit respecter les principes et lignes directrices de bonnes pratiques de fabrication des médicaments prévus par le droit communautaire. L'article 47 disposait que les principes et lignes directrices de bonnes pratiques de fabrication pour les médicaments sont adoptés sous forme d'une directive. Des lignes directrices détaillées conformes à ces principes sont publiées par la Commission et révisées en cas de besoin pour tenir compte des progrès scientifiques et techniques.
L'article 33 de la directive 2004/27, proposant de modifier l'article 46 du code communautaire relatif aux médicaments à usage humain, impose que le titulaire de l'autorisation de fabrication respecte les principes et lignes directrices relatifs aux bonnes pratiques de fabrication des médicaments ; en outre, le titulaire doit utiliser seulement en tant que matières premières des substances actives fabriquées conformément aux lignes directrices détaillées relatives aux bonnes pratiques de fabrication des matières premières. Cette prescription est également applicable à certains excipients, dont la liste et les conditions spécifiques d'application sont arrêtées par une directive adoptée par la commission. L'article 77 de la directive, modifiant l'article 111 du code communautaire permet aux autorités nationales compétentes de procéder à des inspections inopinées dans les locaux des fabricants de substances actives utilisées comme matières premières dans la fabrication des médicaments, lorsqu'elle considère qu'il y a des raisons de penser que les principes et les lignes directrices de bonnes pratiques de fabrication ne sont pas respectées. Ces inspections peuvent également avoir lieu à la demande d'un État membre, de la Commission ou de l'agence.
· Les modifications proposées par le présent article
Le présent projet propose de réécrire l'article L. 5138-3 du code de la santé publique, dont la rédaction en vigueur aménage la possibilité de demander à l'AFSSAPS de certifier que l'établissement qui produit les matières premières à usage pharmaceutique respecte les bonnes pratiques mentionnées à l'article L. 5138-2 du code de la santé publique. Il est proposé que la nouvelle rédaction de l'article L. 5138-3 comprenne deux alinéas.
Le premier alinéa vise à préciser que « les matières premières à usage pharmaceutique doivent répondre aux spécifications de la pharmacopée quand elles existent », disposition anciennement codifiée à l'article L. 5138-2 et qui est reprise à l'article L. 5138-3.
Comme le rappelle le site Internet de l'AFSSAPS, la pharmacopée, ouvrage réglementaire destiné à être utilisé par les professionnels de santé, définit notamment les critères de pureté des matières premières ou des préparations entrant dans la fabrication des médicaments et les méthodes d'analyses à utiliser pour en assurer leur contrôle. Le rôle de la pharmacopée est de participer à la protection de la santé publique en élaborant des spécifications communes et reconnues pour les matières premières à usage pharmaceutique. Il s'agit d'un référentiel scientifique régulièrement mis à jour. Il existe une pharmacopée européenne qui s'applique règlementairement à l'ensemble des États membres signataires de la convention pour l'élaboration de la pharmacopée européenne.
Le deuxième alinéa de l'article L. 5138-3 rappelle que le processus de fabrication des médicaments, qu'il s'agisse des substances actives ou des excipients, doit respecter des bonnes pratiques. A titre de rappel, les opérations de fabrication sont définies par le IV de l'article L. 5138-2 du code de la santé publique, dans sa rédaction modifiée par l'article 23 du présent projet (cf. supra). Pour la fabrication de médicaments, les établissements pharmaceutiques, les pharmacies à usage intérieur, les pharmacies d'officine, les médecins, les vétérinaires et les personnes autorisées à préparer des auto-vaccins à usage vétérinaire doivent utiliser en tant que matières premières à usage pharmaceutique, des substances actives fabriquées et distribuées conformément à des bonnes pratiques, y compris lorsqu'elles sont importées.
Les principes de ces bonnes pratiques seront définis conformément au droit communautaire par une décision de l'AFSSAPS, après avis de l'AFSSA (Agence française de sécurité sanitaire des aliments). Il est précisé que ce dispositif est également applicable aux excipients entrant dans la fabrication des médicaments à usage humain, dont la liste et les conditions spécifiques qui leur sont applicables sont fixées par décision de l'AFSSAPS conformément au droit communautaire.
*
La commission a adopté l'article 24 sans modification.
Article 25
Pouvoirs d'inspection de l'AFSSAPS en ce qui concerne les matières premières à usage pharmaceutique
Le présent article a pour objet de préciser les conditions dans lesquelles s'exercent les pouvoirs des inspecteurs de l'AFSSAPS en ce qui concerne les matières premières pharmaceutiques, en particulier s'agissant des inspections d'office des établissements procédant à des opérations sur les matières premières pharmaceutiques.
A cette fin, il propose une nouvelle rédaction de l'article L. 5138-4 du code de la santé publique et l'insertion d'un article L. 5138-5 nouveau dans le code de la santé publique.
· Les dispositions en vigueur
Le droit en vigueur, modifié par l'ordonnance nº 2005-1087 du 1er septembre 2005 relative aux établissements publics nationaux à caractère sanitaire et aux contentieux en matière de transfusion sanguine, repose sur un régime juridique de déclaration préalable, visé par l'article L. 5138-1 du code de la santé publique : « Toute activité de fabrication, d'importation ou de distribution de matières premières à usage pharmaceutique est soumise à une déclaration effectuée par l'établissement dans lequel s'exerce cette activité, auprès de l'Agence française de sécurité sanitaire des produits de santé. A cette déclaration doit être joint un dossier descriptif de cette activité, dont le contenu est fixé par décret en Conseil d'État. Toute modification des éléments constitutifs de la déclaration ou du dossier doit être communiquée à l'agence ».
A ce régime de déclaration préalable s'ajoute l'obligation que les matières premières à usage pharmaceutique répondent aux spécifications de la pharmacopée, quand elles existent. De plus, elles doivent être fabriquées et distribuées en conformité avec les bonnes pratiques, dont les principes sont définis par décision de l'AFSSAPS. Enfin, tout établissement peut demander à l'AFSSAPS de certifier que l'établissement qui produit les matières premières respecte les bonnes pratiques.
La rédaction en vigueur de l'article L. 5138-4 du code de la santé publique, issue de l'ordonnance nº 2000-916 du 19 septembre 2000, dispose que chaque demande présentée par un établissement de fabrication, d'importation ou de distribution de matières premières à usage pharmaceutique en vue d'obtenir le certificat mentionné à l'article L. 5138-3 du code de la santé publique (cf. infra) donne lieu au versement, au profit de l'AFSSAPS, d'un droit fixe dont le montant est fixé par décret dans la limite de 2 300 euros. Selon les informations communiquées à la rapporteure, actuellement l'AFSSAPS délivre bien des certificats aux fabricants de matières premières au titre de l'article L. 5138-4 du code de la santé publique. Cependant, elle ne perçoit aucune rémunération en raison de l'absence de décret d'application de cet article.
De plus, l'AFSSAPS peut effectuer des inspections d'office en vertu de ses pouvoirs généraux de contrôle prévus aux articles L. 5311-1 et L. 5133-2 du code de la santé publique.
· Les dispositions de la directive
L'article 77 de la directive, modifiant l'article 111 du code communautaire permet aux autorités nationales compétentes - en France, il s'agit de l'AFSSAPS - de procéder à des inspections inopinées dans les locaux des fabricants de substances actives utilisées comme matières premières dans la fabrication des médicaments. La directive n'autorise ces inspections que lorsque l'autorité compétente considère qu'il y a des raisons de penser que les principes et les lignes directrices de bonnes pratiques de fabrication ne sont pas respectés.
La directive prévoit la délivrance d'un certificat de bonnes pratiques à un fabricant qui respecte les principes et lignes directrices de bonnes pratiques de fabrication prévus par la législation européenne. Le respect de ces principes est vérifié au cours d'une inspection qui peut être effectuée sur demande du fabricant mais aussi à l'initiative de l'agence. La directive ne distingue pas le fait générateur de l'inspection.
· Les modifications proposées
Le I de l'article propose une nouvelle rédaction de l'article L. 5138-4 du code de la santé publique.
Il est d'abord proposé que la rédaction du premier alinéa de cet article du code de la santé publique oblige l'AFSSAPS, dans le cadre de ses pouvoirs d'inspection des opérations relatives aux matières premières pharmaceutiques (fabrication, reconditionnement et réétiquetage), à délivrer un « certificat de conformité » lorsqu'elle constate que les opérations respectent les bonnes pratiques mentionnées à l'article L. 5138-3 du code de la santé publique et visées par l'article 23 du présent projet.
Il est ensuite proposé de reprendre une partie des dispositions de l'article L. 5138-3 du code de la santé publique, qui fait l'objet d'une réécriture par l'article 24 du présent projet de loi (cf. supra). Le deuxième alinéa maintient la possibilité pour un établissement de demander à l'AFSSAPS d'établir un certificat de conformité aux bonnes pratiques, possibilité qui avait été introduite par l'article 4 de l'ordonnance nº 2005-1087 du 1er septembre 2005. Cette possibilité correspond au sixième alinéa de l'article 77 de la directive. Le modèle du certificat de conformité sera établi par l'AFSSAPS.
Le II de l'article n'est pas une mesure de transposition de la directive. Il propose de créer un article L. 5138-5 nouveau dans le code de la santé publique, afin de reprendre dans cet article une partie du contenu de l'article L. 5138-4 du code de la santé publique dans sa rédaction en vigueur. Il propose que toute inspection de l'AFSSAPS des établissements réalisant les activités relatives aux matières premières pharmaceutiques en vue de vérifier ou de certifier le respect des bonnes pratiques - qu'il s'agisse d'une inspection d'office ou sur la demande de l'établissement - donne lieu au versement d'un droit fixe au profit de l'agence.
Un décret fixera son montant, composé d'une part forfaitaire (dont le montant est limité par la loi à 2 000 euros) et d'une part variable. Le présent article fixe à 10 000 euros le montant total du droit fixe. Par rapport au montant maximum fixé par le droit en vigueur (2 300 euros), l'augmentation est très significative.
La mention « toute inspection, d'office ou sur demande » vise à soumettre le fabricant de matières premières à usage pharmaceutique au versement de la taxe à l'issue de toutes les inspections (quel que soit le fait générateur) ayant pour objet de vérifier et le cas échéant certifier le respect des bonnes pratiques, en application du principe d'égalité entre les fabricants.
Cette mention permet également d'éviter que l'AFSSAPS ne soit contrainte à engager des frais trop importants pour inspecter des fabricants de substance active, notamment dans les pays tiers, qui demanderaient des certificats de respect de bonnes pratiques sans être certains de pouvoir les respecter.
*
La commission a examiné un amendement de la rapporteure visant à diminuer le plafond du montant du droit fixe perçu par l'AFSSAPS en cas d'inspection des établissements en vue de vérifier le respect des bonnes pratiques relatives aux matières premières pharmaceutiques.
La rapporteure a indiqué que le projet de loi augmente excessivement ce montant, qui passerait de 2 300 euros à 10 000 euros. Il convient d'éviter une augmentation aussi drastique.
M. Claude Evin a souligné qu'il convient malgré tout de rémunérer convenablement l'AFSSAPS, même si le rapport du Sénat note que l'AFSSAPS dépend financièrement des ressources issues des services rendus de l'agence.
La commission a adopté l'amendement.
La commission a ensuite adopté l'article 25 ainsi modifié.
Article 26
Publicité de la synthèse des dossiers d'autorisation d'un nouveau médicament
Le présent article propose qu'une décision réglementaire fixe les conditions dans lesquelles l'AFSSAPS rend publiques les synthèses des dossiers d'autorisation des nouveaux médicaments auxquels elle délivre une autorisation de mise sur le marché (AMM).
La transparence du fonctionnement des agences et la publicité de leurs décisions constituent des pas importants vers ce qu'on a pu appeler la « démocratie sanitaire ». Le présent article ne révolutionne pas le droit en vigueur mais permettra au règlement de préciser les conditions dans lesquelles les synthèses des dossiers d'AMM sont rendues publiques, en intégrant les avancées de la directive.
Le premier problème posé par la publicité des travaux des agences sanitaires, particulièrement en ce qui concerne la délivrance des autorisations de mise sur le marché, est la nécessaire conciliation entre la transparence, réclamée notamment par les patients et leurs représentants, et la préservation de la confidentialité qui s'attache aux découvertes et procédés industriels, principe auquel sont légitimement attachés les exploitants des médicaments. Il semble en la matière que l'on s'oriente vers une quasi-transparence pour les décisions de pharmacovigilance. S'agissant des AMM, les impératifs de confidentialité limitent encore l'application du principe de transparence ; ils conduisent notamment à retarder dans le temps la mise à disposition au public des données précises de l'AMM.
Les conditions de présentation des informations au public, qu'il s'agisse des professionnels de santé ou du grand public, sont aussi problématiques. Faut-il des informations « brutes » ou faut-il des synthèses ? Ce problème concerne d'ailleurs trois types d'informations différentes : la synthèse de la décision d'AMM, la synthèse des travaux des experts et la publicité du fonctionnement de l'agence (ordre du jour et déclaration d'intérêts notamment).
S'agissant de la transparence des données, la mise à disposition d'informations aux patients ne risque-t-elle pas de mettre en porte à faux les professionnels de santé, particulièrement les médecins, sur lesquels une pression supplémentaire pourrait s'exercer en faveur de tel ou tel médicament, rendant leurs conditions d'exercice de plus en plus complexes ?
Il est ensuite dommage que les obligations nouvelles en matière de publicité des décisions s'appliquent au « flux », c'est-à-dire aux nouveaux médicaments auxquels sont délivrées des AMM. Qu'en est-il du « stock » - les AMM délivrées depuis des dizaines d'années ? Dans cette perspective, il y a lieu de se féliciter du lancement par l'AFSSAPS du projet « base AMM » visant à rendre accessible sous forme électronique d'ici fin 2008 la totalité des AMM délivrées en France (celles des années 1999 à 2006 étant déjà accessibles).
Enfin, les dispositions relatives à la transparence exigent des moyens supplémentaires. Les agences sanitaires - et en particulier l'AFSSAPS - pourront-elles assumer ces nouvelles missions ?
1. Les dispositions en vigueur
L'article L. 5311-1 du code de la santé publique, issu de la loi n° 98-535 du 1er juillet 1998 relative au renforcement de la veille sanitaire et du contrôle de la sécurité sanitaire des produits destinés à l'homme, dispose que l'AFSSAPS rend publique une synthèse des dossiers d'autorisation de tout nouveau médicament. Même si l'intérêt politique d'inscrire cette mesure dans le code de la santé publique est évident, la rapporteure note que d'un point de vue strictement juridique cette disposition législative aurait très bien pu faire l'objet d'une décision règlementaire, à moins de considérer que les conditions de fonctionnement interne d'une agence sanitaire appartiennent aux « principes fondamentaux » de la sécurité sociale mentionnés par l'article 34 de la Constitution.
2. Les dispositions de la directive et son application
Un des apports principaux de la directive est d'améliorer la transparence d'une part, des conditions dans lesquelles les AMM sont délivrées et, d'autre part, du fonctionnement des agences.
· L'autorisation de mise sur le marché
S'agissant des conditions de délivrance de l'AMM, l'article 19 de la directive modifie les points 3 et 4 de l'article 21 du code communautaire relatif aux médicaments à usage humain en disposant que les autorités compétentes (les agences) rendent l'AMM publiquement accessible, accompagnée du résumé des caractéristiques du produit pour chaque médicament qu'elles ont autorisé :
« 19) À l'article 21, les paragraphes 3 et 4 sont remplacés par le texte suivant :
« 3. Les autorités compétentes rendent sans retard l'autorisation de mise sur le marché publiquement accessible, accompagnée du résumé des caractéristiques du produit pour chaque médicament qu'elles ont autorisé. »
« 4. Les autorités compétentes rédigent un rapport d'évaluation et des commentaires sur le dossier quant aux résultats des essais pharmaceutiques, précliniques et cliniques du médicament concerné. Le rapport d'évaluation est mis à jour dès que de nouvelles informations qui s'avèrent importantes pour l'évaluation de la qualité, de la sécurité et de l'efficacité du médicament concerné deviennent disponibles.
« Les autorités compétentes mettent sans retard à la disposition du public le rapport d'évaluation et les raisons justifiant leur avis, après suppression de toute information présentant un caractère de confidentialité commerciale. Les motifs sont indiqués séparément pour chaque indication demandée. »
On peut également mentionner les articles 22 du code communautaire (publicité des conditions posées au demandeur d'une AMM), 125 (publicité des décisions d'AMM) et enfin 126 ter (fonctionnement des agences, cf. infra).
Compte tenu de ces dispositions, dès l'été 2005 un processus de préparation a été engagé au sein de l'AFSSAPS pour concevoir les formats et les contenus des documents à rendre accessibles. Des discussions ont été menées en parallèle avec les partenaires européens confrontés aux mêmes défis. Pour certains produits, la publication de rapports d'évaluation a été engagée par l'AFSSAPS sur une base volontaire dès juin 2004. La réflexion a conclu à la nécessité d'une approche progressive compte tenu de l'ampleur de la tâche et de la stabilité des ressources de l'AFSSAPS. La priorité a d'abord été donnée aux nouveaux médicaments princeps contenant des principes actifs nouveaux et à des variations d'AMM particulièrement significatives en termes d'enjeux de thérapeutique et de sécurité.
En application de l'article 21 du code communautaire (cf. supra), l'agence met désormais à la disposition du public, sur son site Internet, un rapport public d'évaluation (RAPPE) pour chaque AMM ou chaque modification majeure d'AMM. Le RAPPE recense les principales données scientifiques issues du dossier d'AMM, sur lesquelles l'agence, la commission d'AMM et les experts ont fondé leur analyse et leur avis, avis qui a conduit à la décision d'octroi de l'AMM ou de modification de l'AMM. Les génériques et les nouvelles formes pharmaceutiques ne donnant pas lieu à une nouvelle indication ne font pas l'objet d'un RAPPE. Le texte du rapport est soumis à la commission d'AMM pour approbation. Le RAPPE est assez complet puisqu'il comprend des données pharmaceutiques, des données toxicologiques, des données cliniques reprenant les études publiées, un développement sur l'analyse bénéfice/risques et une conclusion. Le RAPPE est mis en ligne après que l'AMM a été effectivement signée et notifiée à son titulaire.
En application de l'article 126 ter de la directive relatif au fonctionnement des agences du médicament (cf. infra), il a également été décidé de rendre publique une « fiche de synthèse » résumant les points-clé de l'évaluation et les débats éventuels : ces fiches posent les enjeux de la demande d'autorisation et rendent compte des débats intervenus au sein de la commission d'experts ; cependant, même si la mention d'opinions minoritaires apparaît, l'identité des experts émettant ces opinions n'est pas précisée.
L'AMM est accompagnée du résumé des caractéristiques du produit (RCP), qui précise notamment la dénomination du médicament, la composition qualitative et quantitative, la forme pharmaceutique ainsi que les données cliniques. Elle est également accompagnée de la notice destinée au patient, qui présente l'essentiel des informations du RCP dans un vocabulaire plus accessible.
· La transparence des travaux de l'agence
Un problème connexe à la publicité des rapports d'évaluation est celui de la transparence des travaux des agences du médicament. Conformément à l'article 85 de la directive, introduisant un article 126 ter nouveau dans le code communautaire relatif aux médicaments à usage humain, les États membres veillent à ce que l'autorité compétente (l'agence sanitaire) rende accessible au public son règlement interne et celui de ses comités, l'ordre du jour de ses réunions, les comptes rendus de ses réunions, assortis des décisions prises, des détails des votes et des explications de vote, y compris les opinions minoritaires.
L'AFSSAPS n'a pas attendu la transposition de la directive en droit interne pour se conformer à ces prescriptions. Lors de son audition le 25 octobre 2005 par la mission d'information sénatoriale sur le médicament, M. Jean Marimbert, directeur de l'AFSSAPS, a indiqué « que les comptes rendus [des commissions] seront établis par les secrétariats techniques et approuvés par les membres de chaque commission. L'expression des opinions dissidentes figurera au procès-verbal ainsi que leurs motivations et le résultat d'éventuels scrutins. Les premiers comptes rendus seront disponibles sur le site de l'agence dans le courant du mois de mars. »
Au début du mois d'avril 2005 a été publié le nouveau règlement intérieur de la commission d'AMM. Le 16 mars 2006 a été publié le premier compte rendu de la commission nationale de pharmacovigilance (compte rendu de la séance du 30 novembre 2005). A la fin du mois d'avril 2006, le premier compte rendu de commission d'AMM a été publié.
S'agissant de la commission d'AMM, le VI du règlement intérieur de l'AFSSAPS prévoit que les ordres du jour de la commission sont publiés quelques jours avant la réunion ; cependant, une visite du site Internet de l'AFSSAPS montre que cette disposition n'est pas encore appliquée. Les comptes rendus synthétiques des délibérations des commissions formulant un avis favorable sont publiés dès approbation du compte rendu. Au moment de la notification de la décision de délivrance de l'AMM, chaque dossier est complété du récapitulatif de l'instruction, y compris le résumé des points essentiels du débat. Il est prévu que l'AFSSAPS, au plus tard le 31 décembre 2006, procède à la réévaluation du contenu et des modalités de publication de ces comptes rendus.
3. Les modifications proposées
La modification proposée consiste à introduire dans le code de la santé publique une base légale visant à ce que des normes de nature réglementaire précisent les conditions dans lesquelles l'AFSSAPS rend publique une synthèse des dossiers d'autorisation de tout nouveau médicament. L'ajout des termes « dans des conditions déterminées par voie réglementaire » vise à permettre de transposer les ajouts du code communautaire, à savoir la publicité du résumé des caractéristiques du produit, de l'AMM et des éléments que doit contenir la synthèse.
*
La commission a examiné un amendement de la rapporteure prévoyant que l'AFSSAPS rend accessible les comptes rendus et l'ordre du jour de ses différents conseils et commissions.
La rapporteure a concédé que fixer avec précision le fonctionnement interne de l'AFSSAPS relève plutôt du règlement que de la loi. Néanmoins, en application de la directive, il convient d'inscrire le principe de transparence dans le code de la santé publique.
M. Claude Evin s'est interrogé sur le sens du terme « accessible », indiquant qu'en tout état de cause ces documents sont considérés comme des documents accessibles au sens de la législation relative à la communication des documents administratifs.
La commission a adopté l'amendement, que Mme Jacqueline Fraysse a déclaré vouloir cosigner.
La commission a ensuite adopté l'article 26 ainsi modifié.
La commission a examiné un amendement de Mme Jacqueline Fraysse visant à prévoir que l'AFSSAPS rend public l'ordre du jour de ses réunions, les comptes rendus de ses réunions, assortis des décisions prises, des détails des votes et des explications de vote, y compris les opinions minoritaires.
Mme Jacqueline Fraysse a précisé que cet amendement vise à transposer une disposition importante de la directive n° 2004/27 concernant la transparence des décisions prises par l'AFSSAPS en matière de délivrance d'AMM et de contrôle.
La rapporteure a indiqué que cet amendement est satisfait par l'amendement qu'elle a déposé à l'article 26 et que la commission a adopté.
La commission a rejeté l'amendement.
Article 27
Transmission d'échantillons à titre gratuit à l'AFSSAPS
Cet article vise à inscrire dans le code de la santé publique la possibilité pour l'AFSSAPS de demander aux entreprises la transmission d'échantillons de médicaments à titre gratuit.
Il propose de modifier la rédaction de l'article L. 5311-2 du code de la santé publique, article qui fixe les conditions dans lesquelles les missions de l'agence, définies à l'article L. 5311-1 du code de la santé publique, s'exercent.
· Le droit en vigueur
Les dispositions en vigueur prévoient que les inspecteurs de l'AFSSAPS ne peuvent prélever des échantillons qu'à l'occasion d'inspections. Cela limite considérablement le champ des contrôles et il a paru opportun d'élargir les possibilités, pour l'AFSSAPS, d'obtenir gratuitement des échantillons auprès des entreprises.
Il faut souligner que la directive dont la transposition fait l'objet du présent projet de loi ne comporte pas de mentions explicites relatives à la remise d'échantillons à titre gratuit aux agences sanitaires. L'article 77 de la directive, modifiant le point 1 de l'article 111 du code communautaire relatif aux médicaments à usage humain, autorise les agents de l'autorité compétente à prélever des échantillons, « notamment en vue d'analyses indépendantes par un laboratoire officiel pour le contrôle des médicaments ou par un laboratoire désigné à cet effet par un État membre ».
Le présent article, qui ne constitue pas stricto sensu une transposition de la directive, permet à l'agence de mener à bien les missions qui lui incombent, dont celles relatives au médicament. En vue de l'accomplissement de ses missions, l'AFSSAPS souhaite en effet pouvoir demander la transmission gratuite des échantillons de produits relevant de son champ de compétence pour analyse, sans avoir à nécessairement mandater une inspection en vue d'un prélèvement de ces échantillons. Or, si les industriels transmettent sans grande difficulté les médicaments chimiques, il n'en est pas de même pour certains médicaments biologiques, plus onéreux.
· Les modifications proposées
Le présent article propose d'introduire la possibilité pour l'agence de demander la transmission à titre gratuit d'échantillons de produits et objets mentionnés à l'article L. 5311-1 du code de la santé publique. Cette remise à titre gratuit est encadrée car, conformément à la rédaction du présent article, elle sera soumise aux conditions suivantes :
- elle ne peut être demandée qu'en application des compétences de l'AFSSAPS (définies par l'article L. 5311-2 du code de la santé publique) ;
- elle ne peut être demandée qu'à des fins d'analyse ;
- en outre, elle doit être formulée pour des raisons justifiées ;
- elle ne concerne que les médicaments et produits mentionnés à l'article L. 5311-1 du code de la santé publique.
*
La commission a adopté l'article 27 sans modification.
Article 28
Publicité et annualité des déclarations d'intérêts
Cet article vise à transposer en droit national l'obligation de déclaration d'intérêts prescrite par la directive n° 2004/27 et visant à la fois les personnes employées directement par les agences nationales et les personnes auxquelles les agences ont recours dans le cadre de la procédure de l'autorisation des médicaments.
Le présent article a pour objet d'étendre l'obligation de déposer une déclaration d'intérêts à tous les agents de l'AFSSAPS chargés d'accorder les autorisations ou ayant des fonctions de contrôle. Dans le droit en vigueur, seuls les membres de commissions et conseils relevant de l'AFSSAPS et les collaborateurs occasionnels (rapporteurs, experts externes) sont concernés par cette obligation. Il vise également à rendre cette déclaration d'intérêts annuelle.
La question de l'indépendance de l'expertise scientifique en matière de médicament est aussi centrale qu'ambiguë. En effet, elle peut apparaître comme la garantie de disposer de décisions fondées exclusivement sur des considérations médicales et scientifiques. Elle est donc un facteur essentiel de crédibilité et de légitimité à la fois pour les agences du médicament et les firmes pharmaceutiques.
Cependant, l'indépendance totale des experts, de facto impossible à assurer, est de surcroît peu souhaitable. En effet, il est assez constant que les meilleurs experts sur un sujet relatif au médicament soient en contact, de manière directe ou indirecte, selon des liens variés et à des degrés divers, avec les firmes pharmaceutiques.
De plus, la notion de conflits d'intérêts est subjective et difficile à cerner précisément. L'intérêt peut en effet être financier (comme le fait d'avoir en sa possession des actions d'un laboratoire) ou matériel ; c'est par exemple le cas lorsqu'un laboratoire soutient les recherches d'un expert. Il y a une certaine gradation dans la notion finalement assez floue et ambiguë de « conflits d'intérêts ». D'ailleurs, le nouveau règlement intérieur de la commission d'AMM distingue les « intérêts mineurs » des « intérêts importants ».
L'enjeu est donc le développement d'une démarche préventive efficace et transparente. Il faut d'abord connaître les intérêts de chacun afin d'éviter les conflits d'intérêts majeurs, et quand ils existent, pouvoir les contrôler afin d'éviter qu'ils ne mettent en danger la crédibilité et la qualité des décisions des agences. Cette question prend toute son acuité dans la mesure où l'AFSSAPS fait largement recours à l'expertise externe.
Cela pose plus globalement le problème de la reconnaissance de l'expertise, aussi bien en termes de rémunérations qu'en termes de déroulement de carrière pour les universitaires. Ainsi, devant la mission d'information mise en place par la commission des affaires sociales du Sénat sur les conditions de mise sur le marché et de suivi des médicaments, Mme Emmanuelle Wargon, adjointe au directeur de l'AFSSAPS, a rappelé que « l'agence emploie, pour l'AMM, une centaine d'experts internes - pharmaciens et médecins - et fait appel à un vivier d'environ 1 800 experts externes qui siègent à la commission ou sont rapporteurs d'un dossier. Les experts externes sont rémunérés exclusivement pour leurs travaux. Seuls, les présidents de commission et les médecins libéraux voient leur temps de présence dédommagé, à hauteur de 67 euros par vacation pour un maximum de douze vacations par mois pour les présidents et de quinze consultations par demi-journée de présence pour les libéraux. Les rapporteurs sont payés de une à cinq vacations par dossier. En 2005, 500 000 euros ont ainsi été versés aux présidents de commission et aux rapporteurs. »
Comme le précise le rapport de la commission d'enquête sénatoriale, « dans la mesure où la notion de conflits d'intérêts n'est pas définie par la loi, l'agence a entrepris de classer les divers types d'intérêts et d'identifier les situations conflictuelles. Cette classification, qui ne saurait être exhaustive au regard de la diversité des situations, présente l'avantage de la simplicité et repose sur les critères suivants :
- prise en compte du caractère actuel ou passé des intérêts ;
- degré d'implication de l'expert au sein de l'entreprise concernée par la procédure (participation au capital d'une entreprise, salariat ou participation à un organe décisionnel, prestations régulières, responsable d'une institution dépendant financièrement d'un laboratoire pharmaceutique) ;
- travaux effectués en relation avec le produit spécifique soumis à évaluation ou l'affaire traitée et nature de ces liens (par exemple, investigateur principal).
Cette classification est mise à la disposition des commissions afin de vérifier l'absence de conflits d'intérêts des membres d'une instance avec les dossiers inscrits à l'ordre du jour de la réunion ou avant de confier un dossier à un expert. Ce tableau a été diffusé à tous les experts de l'agence et est accessible sur le site Internet de l'agence. »
La rapporteure note donc que des progrès significatifs ont été accomplis dans ce domaine depuis quelques années.
Comme l'a indiqué, devant les membres de la mission d'information de la commission des affaires sociales du Sénat consacrée au médicament, le professeur François Chollet, président du conseil d'administration de l'AFSSAPS, le conseil d'administration de l'agence est régulièrement informé de la situation des experts. La publication des liens d'intérêts permet une meilleure gestion des conflits d'intérêts, contrôlés par un « groupe référent » composé d'experts indépendants des laboratoires. Il a également fait valoir que le rôle d'expert évolue au cours de la vie professionnelle : l'expert est souvent spécialisé dans un domaine donné au début d'une carrière, puis ses compétences deviennent plus transversales. Selon le professeur François Chollet, ces deux types d'expertises doivent être utilisés par l'agence : les experts transversaux plus expérimentés, indépendants des laboratoires et ayant conclu un contrat avec l'agence pourraient ainsi gérer une équipe d'experts plus jeunes et payés à l'acte.
Le régime juridique des dispositions visant à garantir l'indépendance et l'impartialité des personnes travaillant pour une agence sanitaire, fixé soit par la loi soit par le règlement intérieur, dépend du statut de la personne et de sa fonction. Il peut interdire à ces personnes de manière générale tout intérêt dans les entreprises contrôlées et/ou interdire aux personnes considérées de traiter une question dans laquelle elles auraient un intérêt direct ou indirect. Il peut imposer une déclaration d'intérêts, dont le caractère public constitue une certaine garantie. Cette déclaration peut être mise en ligne ou communiquée sur demande.
· Le droit existant prévoit déjà une déclaration d'intérêts pour certains personnels travaillant pour l'AFSSAPS
Quelles sont les personnes travaillant pour l'agence ? Il s'agit d'abord d'agents relevant du statut général des fonctionnaires ainsi que les personnels relevant de la fonction publique hospitalière. L'article L. 5323-2 du code de la santé publique autorise l'AFSSAPS à employer des agents contractuels de droit public ; l'article L. 5323-3 l'autorise à employer des agents contractuels de droit privé pour occuper des fonctions occasionnelles de caractère scientifique ou technique, alors même que ces agents continuent d'exercer par ailleurs à titre principal une activité professionnelle libérale.
· Les dispositions législatives en vigueur et leur application
La rédaction en vigueur de l'article L. 5323-4 du code de la santé publique, modifiée par l'article 31 de la loi nº 2002-303 relative aux droits des malades et à la modernisation du système de santé du 4 mars 2002 et l'article 12 de l'ordonnance nº 2005-1087 relative aux établissements publics nationaux à caractère sanitaire et aux contentieux en matière de transfusion sanguine du 1er septembre 2005, pose des garanties en la matière. Son septième alinéa dispose que « les personnes mentionnées aux deux alinéas précédents adressent au directeur général de l'agence, à l'occasion de leur nomination ou de leur entrée en fonctions, une déclaration mentionnant leurs liens, directs ou indirects, avec les entreprises ou établissements dont les produits entrent dans son champ de compétence, ainsi qu'avec les sociétés ou organismes de conseil intervenant dans ces secteurs. Cette déclaration est rendue publique et est actualisée à leur initiative dès qu'une modification intervient concernant ces liens ou que de nouveaux liens sont noués. »
Cette obligation de déclaration vise deux catégories de personnes : d'une part, les personnes collaborant occasionnellement aux travaux de l'agence ainsi que les autres personnes qui apportent leur concours aux conseils et commissions siégeant auprès d'elle, et d'autre part, les membres des commissions et conseils siégeant auprès de l'agence.
Cette déclaration n'est adressée au directeur de l'agence qu'à l'occasion de la nomination ou de l'entrée en fonction des personnes considérées. Sa portée est donc relativement limitée compte tenu des changements, notamment patrimoniaux, pouvant intervenir après la nomination ou l'entrée en fonction des personnes considérées. Dans ce cas, l'efficacité des mécanismes repose sur l'actualisation de la déclaration par les personnes considérées.
Quelle est l'application de cette disposition du code de la santé publique ?
Les auditions de la mission d'information du Sénat consacrée au médicament ont mis en évidence que cette obligation de déclaration d'intérêt n'était pas intégralement mise en œuvre, certains experts ne déclarant pas leurs liens avec les entreprises pharmaceutiques, comme l'a reconnu devant les membres de la mission M. Lionel Benaiche, vice-président du tribunal de grande instance de Nanterre. La mission sénatoriale note dans son rapport que « si l'AFSSAPS affiche le principe d'une publication annuelle des déclarations d'intérêts des membres des commissions et des groupes de travail, force est de constater qu'au début du mois de mai 2006, seules les déclarations 2004 sont disponibles. Cette situation ne peut que susciter des interrogations sur l'importance réellement attachée par l'agence à une politique de transparence en la matière. Par ailleurs, les auditions organisées par la mission d'information ont fait apparaître qu'environ 10 % des experts étaient en contravention par rapport à cette obligation de déclaration. »
Il faut noter que l'article 26 de la loi n° 2002-303 du 4 mars 2002 relative aux droits des malades et à la qualité du système de soins oblige les professionnels de santé qui ont des liens avec les laboratoires pharmaceutiques à en faire état lorsqu'ils s'expriment en public ou qu'ils publient. Cependant, cette disposition n'a pas fait l'objet du décret en Conseil d'État nécessaire à son application.
Selon les informations communiquées à la rapporteure, les déclarations d'intérêts sont des documents publics et soumis à la législation relative à la communication des documents administratifs (loi du 17 juillet 1978 portant diverses mesures d'amélioration des relations entre l'administration et le public et diverses dispositions d'ordre administratif, social et fiscal, modifiée par la loi du 12 avril 2000 relative aux droits des citoyens dans leurs relations avec les administrations).
· Le règlement intérieur de l'AFSSAPS
Le règlement intérieur de la commission d'AMM de l'AFSSAPS prévoit, en complément des dispositions législatives mentionnées ci-dessus, que « tous les membres de la Commission, les membres des groupes de travail et les experts/rapporteurs extérieurs remplissent sur l'honneur une déclaration mentionnant leurs liens, directs ou indirects, avec les entreprises ou établissements dont les produits entrent dans le champ de compétence de l'AFSSAPS, ainsi qu'avec les sociétés ou organismes de conseil intervenant dans ces secteurs. Ils s'engagent à actualiser leur déclaration d'intérêts au moins une fois par an ou, le cas échéant, lorsque de nouveaux liens sont noués ou en cas de modification des liens antérieurement déclarés ». Le règlement intérieur souligne la nécessité que les personnes considérées s'acquitte spontanément de cette obligation. Les déclarations d'intérêts des membres des instances consultatives de l'Agence et des experts nommés auprès d'elles, sont des documents accessibles au public. Ces déclarations peuvent être consultées par toute personne qui en fera la demande au directeur général.
· Les dispositions de la directive et du règlement européen
La directive n° 2004/27 prend en compte, pour la première fois dans une directive, le problème des éventuels conflits d'intérêts des personnels des agences nationales. L'article 85 de la directive introduit dans le code communautaire relatif aux médicaments à usage humain un article 126 ter nouveau, article dont le premier alinéa est relatif à l'indépendance des experts.
La première phrase du premier alinéa pose un principe général d'impartialité des « agents » des agences sanitaires chargés d'autoriser les médicaments. Il est précisé que les agents de l'autorité compétente chargés d'accorder les autorisations ne doivent pas avoir dans l'industrie pharmaceutique un intérêt, financier ou autre, qui « pourrait nuire à leur impartialité » ; cette interdiction s'étend aux experts et aux rapporteurs « chargés de l'autorisation et du contrôle des médicaments ». A ce stade, il est intéressant de noter que le principe posé par la directive ne s'étend pas à tout le personnel des agences sanitaires, mais uniquement aux agents de l'autorité compétente chargés d'accorder les autorisations aux experts et aux rapporteurs chargés de l'autorisation et du contrôle des médicaments.
La deuxième phrase du premier alinéa prévoit que les agents de l'agence chargés d'accorder les autorisations, les experts comme les rapporteurs, font chaque année une déclaration de leurs intérêts financiers. Si le rythme de la déclaration est bien précisé, il n'est pas fait mention des intérêts autres que financiers. La directive ne précise pas que la déclaration d'intérêts est rendue publique.
S'agissant de l'agence européenne du médicament, l'article 63-2 du règlement (CE) 726/2004 du Parlement européen et du Conseil du 31 mars 2004 impose une déclaration annuelle des intérêts financiers des membres des conseils d'administration, des membres des comités, des experts et des rapporteurs. En outre, ces personnes doivent, selon l'ordre du jour, déclarer leurs intérêts particuliers s'ils peuvent compromettre leur indépendance. Ces déclarations sont rendues accessibles au public.
· La modification proposée
Il est proposé de modifier le septième alinéa l'article L. 5323-4 du code de la santé publique, alinéa relatif à l'obligation de déclaration d'intérêts. Dans la rédaction en vigueur, cette déclaration doit mentionner les liens directes et indirects, non seulement avec les entreprises pharmaceutiques, mais aussi avec les sociétés de conseil. De plus, la déclaration rendue publique est actualisée à l'initiative des personnes considérées.
Cette obligation de déclaration s'applique aux personnes collaborant occasionnellement aux travaux de l'agence, aux autres personnes qui apportent leur concours aux conseils et commissions siégeant auprès d'elle et aux membres des commissions et conseils siégeant auprès de l'agence.
La modification proposée vise principalement à mieux délimiter le champ des personnes devant satisfaire à cette obligation de déclaration d'intérêts. Il est proposé qu'elle s'applique aux catégories de personnels suivantes :
- aux personnes mentionnées aux articles L. 5323-1 du code de la santé publique (personnes relevant du statut de fonctionnaires et personnel des établissements de santé employés par l'agence) ;
- aux personnes mentionnées à l'article L. 5323-2 du code de la santé publique (agents contractuels de droit public employés par l'agence) ;
- aux personnes mentionnés à l'article L. 5323-3 du code de la santé publique (agents contractuels de droit privé employés par l'agence) ;
- aux personnes collaborant occasionnellement aux travaux de l'agence, les autres personnes qui apportent leur concours aux conseils et commissions siégeant auprès d'elle et aux membres des commissions et conseils siégeant auprès de l'agence.
La modification proposée vise donc d'abord à étendre sensiblement le périmètre des personnes travaillant pour l'agence et soumis à l'obligation de déclaration d'intérêt.
Le présent article propose également que cette déclaration d'intérêt soit faite dès l'entrée en fonction ou la nomination, puis mise à jour annuellement.
Enfin, nuançant le principe de publicité des déclarations, il propose que seules les déclarations des personnes collaborant occasionnellement aux travaux de l'agence, des autres personnes qui apportent leur concours aux conseils et commissions siégeant auprès d'elle et des membres des commissions et conseils soient rendues publiques. Néanmoins, ces déclarations seront des documents communicables au sens de la législation relative aux documents administratifs.
Cette obligation de déclaration d'intérêts pesant sur les agents contractuels de l'agence vient renforcer un régime juridique déjà assez contraignant, puisque l'article L. 5223-4 du code de la santé publique interdit aux agents contractuels d'avoir un lien avec les entreprises et établissements contrôlés par l'agence.
*
La commission a adopté l'article 28 sans modification.
Chapitre II
Habilitation à prendre des ordonnaces
Article 29
Habilitation du gouvernement à prendre par ordonnances des dispositions dans le domaine du médicament
Le présent article propose que le gouvernement soit habilité à prendre par ordonnances des dispositions relevant de la loi dans le domaine du médicament, dans un délai de huit mois suivant la publication de la loi. Ces ordonnances seront prises suivant la procédure visée par l'article 38 de la Constitution.
La procédure des ordonnances, prévue par l'article 38 de la Constitution, permet au gouvernement de prendre des mesures relevant matériellement du domaine de la loi. L'utilisation de cette procédure doit être limitée dans le temps. Les ordonnances, prises en Conseil des ministres après avis du Conseil d'État, entrent en vigueur dès leur publication. Elles deviennent caduques si le projet de loi de ratification n'est pas déposé devant le Parlement avant la date fixée par la loi d'habilitation (cf. infra, commentaire du III du présent article).
Le recours aux ordonnances dans les domaines médical et sanitaire est assez fréquent, compte tenu de la technicité des sujets. Le recours aux ordonnances a augmenté très significativement depuis 2000, cet accroissement connaissant un pic en 2004 et 2005.
Selon des informations communiquées à la rapporteure, la version initiale du projet de loi comprenait environ 90 articles et transposait notamment une série de directives en date du 31 mars 2004 (les directives n° 2004/24/CE relative aux médicaments traditionnels à base de plantes, n° 2004/27/CE relative aux médicaments à usage humain et n° 2004/28/CE relative aux médicaments vétérinaires).
Le projet de loi a alors été jugé beaucoup trop conséquent pour qu'il soit discuté dans de bonnes conditions par le Parlement, surtout dans les délais assez restreints prévus pour un projet de loi de transposition de directive. La nature technique de la plupart des dispositions proposées accentuait cette lourdeur.
Il a donc été décidé d'alléger le texte tout en préservant sa cohérence. Le présent projet de loi a ainsi été limité aux dispositions transposant la directive n° 2004/27/CE (médicaments à usage humain). Le reste est renvoyé aux ordonnances prévues par le présent article.
Le I de l'article propose d'habiliter le gouvernement à prendre par ordonnance les dispositions de nature législative nécessaires à la transposition des directives ou de celles de leurs dispositions qui n'ont pas encore été transposées, ainsi que les mesures d'adaptation de la législation liées à cette transposition. Les mesures de transposition sont limitées aux cinq directives suivantes, classées par ordre chronologique.
Il s'agit d'abord de la directive 2002/98/CE du Parlement européen et du Conseil du 27 janvier 2003 établissant des normes de qualité et de sécurité pour la collecte, le contrôle, la transformation, la conservation et la distribution du sang humain, et des composants sanguins. La directive, modifiant la directive n° 2001/83/CE, établit des normes de sécurité et de qualité du sang et des composants sanguins tout au long de la filière transfusionnelle.
Il faut malheureusement souligner le retard pris à transposer une directive adoptée il y a plus de trois ans, même si la plupart des dispositions de la directive ont déjà été transposées par le décret n° 2006-99 du 1er février 2006 relatif à l'établissement français du sang (EFG) et à l'hémovigilance. D'après le contenu d'un projet d'ordonnance, les mesures faisant l'objet de l'ordonnance permettraient notamment de mieux délimiter les compétences respectives des établissements de santé et les établissements de transfusion sanguine en matière de distribution et de délivrance, modifieraient les sanctions pénales applicables en cas de non-respect des règles applicables à l'activité transfusionnelle et ajouteraient la délivrance de produits sanguins labiles aux actes ne pouvant être réalisés sans que soient réalisés les analyses biologiques et les tests relatifs aux maladies transmissibles.
Le présent article concerne aussi la directive n° 2003/15/CE du Parlement européen et du Conseil du 27 février 2003 modifiant la directive n° 76/768/CEE du Conseil concernant le rapprochement des législations des États membres relatives aux produits cosmétiques. L'ordonnance n° 2004-1148 du 28 octobre 2004, prise en application de la loi n° 2004-237 du 18 mars 2004 portant habilitation du gouvernement à transposer, par ordonnance, des directives communautaires et à mettre en œuvre certaines dispositions du droit communautaire, a déjà transposé quelques dispositions de la directive considérée. Cette ordonnance a été complétée par le décret n° 2004-1219 du 17 novembre 2004 et un arrêté du 17 novembre 2004.
Tout en maintenant la protection du secret commercial et du droit de propriété intellectuelle, l'ordonnance n° 2004-1148 a prévu la mise à la disposition du public (par tous moyens appropriés y compris électroniques) de certaines informations sur la composition des produits cosmétiques ainsi que sur les données existantes en matière d'effets indésirables pour la santé humaine résultant de l'utilisation des produits cosmétiques.
Les mesures qui devraient faire l'objet de l'ordonnance sont la transposition de l'article 1er de la directive, qui prévoit, sous certaines conditions, l'interdiction de mise sur le marché de produits cosmétiques dont la formulation finale ou les ingrédients, afin de satisfaire aux exigences de la directive, auraient fait l'objet d'une expérimentation animale. Le même article interdit la réalisation d'expérimentations animales portant sur des produits cosmétiques finis et leurs ingrédients.
Le présent article vise également la directive n° 2004/23/CE du Parlement européen et du Conseil du 31 mars 2004 relative à l'établissement de normes de qualité et de sécurité pour le don, l'obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissus et cellules humains. L'ordonnance permettrait notamment de renforcer le contrôle sur les établissements qui transforment, conservent, stockent et distribuent des tissus et cellules humains, de transposer dans la partie législative du code de la santé publique des dispositions de la directive prévoyant que chaque établissement désigne un responsable et que les produits en provenance de pays tiers sont soumis à autorisation après évaluation de leurs procédés de préparation. Elle rétablirait également les prérogatives de contrôle des agents des douanes dans les échanges intracommunautaires pour certaines catégories de produits sensibles limitativement énumérés et compléterait la liste des produits susceptibles de faire l'objet d'un contrôle douanier.
La directive n° 2004/24/CE du Parlement européen et du Conseil du 31 mars 2004 modifiant, en ce qui concerne les médicaments traditionnels à base de plantes, la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain, doit également être transposée. L'ordonnance instituerait la procédure d'enregistrement spécifique pour ces médicaments, créerait une taxe à verser à l'AFSSAPS par les demandeurs de cet enregistrement et étendrait à ces médicaments des dispositions relatives à la publicité, aux autorisations d'importation, à la prise en charge par la sécurité sociale et à certaines sanctions pénales applicables aux autres médicaments à usage humain.
Enfin, le présent article mentionne la directive n° 2004/28/CE du Parlement européen et du Conseil du 31 mars 2004 modifiant la directive 2001/82/CE instituant un code communautaire relatif aux médicaments vétérinaires. Cette ordonnance modifiera certaines dispositions du code de la santé publique relatives au médicament vétérinaire sur les points suivants : AMM, enregistrement du médicament homéopathique et médicaments génériques. Dans la majorité des cas, ces nouvelles mesures sont similaires à celles proposées par le présent projet de loi s'agissant du médicament à usage humain.
Le II de l'article propose d'autoriser le gouvernement à prendre, par ordonnances, les mesures relevant du domaine de la loi dans les domaines suivants.
Il s'agit d'abord (1° du II) d'adapter au droit communautaire les dispositions du code de la santé publique relatives :
- aux autorisations d'importation des médicaments à usage humain (modification de l'article L. 5124-13 du code de la santé publique afin notamment que l'autorisation temporaire d'utilisation soit considérée comme une autorisation d'importation) ;
- aux insecticides et acaricides destinés à l'homme ; l'ordonnance supprimera des dispositions spécifiques aux insecticides et acaricides appliqués sur l'homme ; d'après des lignes directrices européennes, ces produits sont en effet aujourd'hui considérés comme des médicaments à usage humain ;
- au régime juridique des aliments diététiques destinés à des fins médicales spéciales (ADDFMS) ; il s'agit de compléter l'art. L. 5137-1 du code de la santé publique pour définir les ADDFMS (définition donnée par la directive n° 1999/21/CE), prévoir leur utilisation sous contrôle médical et redéfinir la sous-catégorie d'ADDFMS pour laquelle la prescription médicale est obligatoire ;
- aux bonnes pratiques de fourniture et de délivrance.
Ensuite, une ordonnance (2° du II) harmonisera et complétera les dispositions pénales relatives aux produits mentionnés aux articles L. 5141-1 (médicaments vétérinaires) et L. 5311-1 (médicaments contrôlés par l'AFSSAPS) du code de la santé publique.
Une ordonnance sera également promulguée afin de « régir » les actions d'accompagnement des patients soumis à des traitements médicamenteux, conduites par les établissements pharmaceutiques, et définir les conditions de leur contrôle par l'AFSSAPS.
· Quels sont ces programmes d'accompagnement des patients ?
Initiés par le médecin lors de la consultation, les programmes d'accompagnement des patients - ou « programmes d'observance » - visent à favoriser l'observance du traitement par le patient ou à l'accompagner à travers différents dispositifs individualisés (« numéro vert », relance téléphonique, éducation personnalisée du patient, envoi d'infirmiers à domicile...).. Ils concernent surtout les médicaments « au long cours ».
Ces programmes ont donné lieu à des excès aux Etats-Unis, où le « coaching » des patients se développe de manière préoccupante dans un contexte de concurrence croissante, allant jusqu'à l'envoi de « contrôleurs » au domicile des patients. Ces programmes sont consacrés à un médicament et non à une pathologie. Il n'est pas sûr qu'ils véhiculent une information fiable et exhaustive sur la pathologie, information qui devrait mettre en évidence les différentes stratégies thérapeutiques existantes, les possibles effets indésirables, les conduites à tenir lors des éventuels traitements ainsi que les règles hygiéno-diététiques à suivre.
· Ces programmes doivent être encadrés
Si ces programmes ne sont pas assimilables stricto sensu à de la publicité, il convient néanmoins de les encadrer. La présente disposition ne correspond pas à une transposition de la directive. Cependant, la rapporteure estime indispensable que ces programmes d'observance ne se développent pas dans le silence de la loi et qu'un cadre juridique soit donné à ces programmes. Le présent projet de transposition de directive constitue, à cet égard, le vecteur législatif adéquat.
Cependant, le présent article ne donne aucune précision quant au régime juridique applicable à ces programmes d'observance. En particulier, l'habilitation ne précise pas si ces programmes devront faire l'objet d'une déclaration auprès de l'AFSSAPS, ou si l'agence sera chargée de les autoriser a priori.
Selon les informations communiquées à la rapporteure, l'ordonnance devrait introduire un régime d'autorisation préalable des programmes d'observance. Elle fixerait les critères que devront respecter ces programmes, afin de garantir qu'ils poursuivent un objectif de bon usage du médicament, qu'ils respectent les exigences éthiques en matière du consentement éclairé des patients, qu'ils soient effectués conformément aux données de l'AMM du médicament et qu'ils se conforment aux dispositions de la loi n° 78-17 modifiée du 6 janvier 1978 relative à l'informatique, aux fichiers et aux libertés. L'AFSSAPS serait l'autorité compétente en matière de délivrance, de suspension et de retrait de ces autorisations. L'ordonnance prévoirait également les sanctions en cas de mise en œuvre d'un programme d'observance en infraction avec cette réglementation.
Le débat en séance publique permettra au ministre chargé de la santé de décrire précisément les contours de l'ordonnance à venir.
Enfin, 4° du II, une ordonnance aura pour objet de permettre aux agents mentionnés au 1° de l'article L. 215-1 du code de la consommation (agents de la direction générale de la concurrence, de la consommation et de la répression des fraudes, de la direction générale des douanes et de la direction générale des impôts) de recourir à l'AFSSAPS dans l'exercice des pouvoirs d'enquête qui leur sont dévolus en application de l'article L. 5414-1 du code de la santé publique, en vertu duquel ces agents ont qualité pour rechercher et constater les infractions aux lois et règlements relatifs à certains médicaments et produits. Cette disposition ne constitue pas une transposition de la directive.
Conformément aux dispositions de l'article 38 de la Constitution, le III du présent article propose que les ordonnances soient prises dans un délai de huit mois suivant la publication de la loi. En outre, le III précise que le projet de loi de ratification de chacune des ordonnances sera déposé devant le Parlement au plus tard le dernier jour du deuxième mois à compter de la publication de l'ordonnance considérée.
*
La commission a examiné un amendement de Mme Jacqueline Fraysse visant à supprimer cet article.
Mme Jacqueline Fraysse a précisé que la sensibilité du sujet impose la transparence et le débat parlementaire. Certaines dispositions méritent un éclaircissement et un vrai débat public. Ainsi, le 3° du II de cet article, qui introduit une mesure qui n'apparaît pas dans la directive, concerne les actions d'accompagnements des traitements conduites par les laboratoires pharmaceutiques eux-mêmes. Il ouvre la possibilité d'engager des programmes d'observance de traitement par les laboratoires pour les traitements qu'ils ont eux-mêmes conçus ! Certes, c'est une activité qui peut s'avérer utile si elle est bien encadrée. Cependant, le recours à l'ordonnance prive les parlementaires d'un véritable débat sur cette question.
La rapporteure s'est déclarée défavorable à cet amendement. La technicité de certaines dispositions impose le recours aux ordonnances ; en outre, un amendement déposé au 3° du II répond aux inquiétudes exprimées.
M. Claude Evin a indiqué que, si le recours aux ordonnances se justifie effectivement sur certaines mesures techniques, tel n'est pas le cas, par exemple, des actions d'accompagnement des patients. Il s'agit en effet d'un sujet sensible, qui soulève des interrogations, et il serait légitime et nécessaire que le Parlement soit associé à la réflexion.
La commission a rejeté l'amendement.
La commission a ensuite examiné un amendement de la rapporteure visant à préciser que l'ordonnance prise en habilitation de la loi définira notamment les conditions dans lesquelles l'AFSSAPS autorisera le développement de ces programmes.
La rapporteure a indiqué que la question des programmes d'observance conduits par les laboratoires est préoccupante. Il est bon que la loi fixe leur régime juridique. Cependant, la rédaction de l'habilitation paraît excessivement générale. L'amendement propose donc que ces programmes fassent l'objet d'une autorisation préalable délivrée par l'AFSSAPS.
La commission a adopté l'amendement, puis l'article 29 ainsi modifié.
Article 30
Mayotte, Saint-Pierre-et-Miquelon, Terres australes et antarctiques françaises, Wallis, Futuna, Nouvelle-Calédonie et Polynésie française
Le présent article propose d'autoriser le gouvernement à prendre par ordonnances, sur le fondement de l'article 38 de la Constitution, les mesures nécessaires à l'extension et à l'adaptation des dispositions du chapitre IER de la présente loi, ainsi que de celles des ordonnances prises en application de son article 29 (cf. supra). Le recours aux ordonnances est régulier pour habiliter le gouvernement à étendre l'application de dispositions législatives aux collectivités d'outre-mer, à la Nouvelle-Calédonie et dans les terres australes et antarctiques françaises.
Le I de l'article propose d'autoriser le gouvernement à prendre par ordonnances les mesures visant à appliquer les dispositions visées par le chapitre premier de la présente loi et celles de l'article 29 (cf. supra) dans les circonscriptions territoriales suivantes : Mayotte, Saint-Pierre-et-Miquelon, Terres australes et antarctiques françaises, Wallis, Futuna, Nouvelle-Calédonie et Polynésie française.
Le II rappelle les procédures d'avis nécessitées par les différents statuts des collectivités d'outre-mer et de Nouvelle-Calédonie. Il s'agit donc de soumettre les projets d'ordonnance pour avis :
1°) lorsque leurs dispositions sont relatives à Mayotte, au conseil général de Mayotte ;
2°) lorsque leurs dispositions sont relatives à Saint-Pierre-et-Miquelon, au conseil général de Saint-Pierre-et-Miquelon dans les conditions prévues à l'article 28 de la loi n° 85-595 du 11 juin 1985 relative au statut de l'archipel de Saint-Pierre-et-Miquelon ;
3°) lorsque leurs dispositions sont relatives aux Terres australes et antarctiques françaises, au conseil consultatif ;
4°) lorsque leurs dispositions sont relatives à la Nouvelle-Calédonie, à l'institution compétente dans les conditions définies par la loi organique n° 99-209 du 19 mars 1999 relative à la Nouvelle-Calédonie ;
5°) lorsque leurs dispositions sont relatives à la Polynésie française, à l'institution compétente dans les conditions définies par la loi organique n° 2004-192 du 27 février 2004 portant statut d'autonomie de la Polynésie française ;
6°) lorsque leurs dispositions sont relatives aux îles Wallis et Futuna, à l'assemblée territoriale des îles Wallis et Futuna.
Conformément à l'article 38 de la constitution, le III de l'article propose que les ordonnances soient prises dans le délai de douze mois suivant la publication de la loi. En outre, le projet de loi de ratification de chacune des ordonnances prévues par le présent article devra être déposé devant le Parlement au plus tard le dernier jour du troisième mois à compter de la publication de l'ordonnance considérée.
*
La commission a examiné un amendement de Mme Jacqueline Fraysse visant à supprimer cet article.
Mme Jacqueline Fraysse a précisé que la sensibilité du sujet impose la transparence et le débat parlementaire.
La rapporteure s'est déclarée défavorable à cet amendement, les dispositions étendant l'application de textes législatifs à l'outre-mer étant régulièrement prises par ordonnance.
La commission a rejeté cet amendement.
La commission a adopté l'article 30 sans modification.
Puis la commission a adopté l'ensemble du projet de loi ainsi modifié.
*
En conséquence et sous réserve des amendements qu'elle propose, la commission des affaires culturelles, familiales et sociales demande à l'Assemblée nationale d'adopter le projet de loi portant diverses dispositions d'adaptation au droit communautaire dans le domaine du médicament - n° 3062.
Dispositions en vigueur ___ |
Texte du projet de loi ___ |
Propositions de la Commission ___ |
Projet de loi portant diverses dispositions d'adaptation au droit communautaire dans le domaine du médicament |
Projet de loi portant diverses dispositions d'adaptation au droit communautaire dans le domaine du médicament | |
Chapitre Ier |
Chapitre Ier | |
Dispositions relatives aux médicaments |
Dispositions relatives aux médicaments | |
Article 1er |
Article 1er | |
Code de la santé publique |
L'article L. 3110-3 du code de la santé publique est modifié ainsi qu'il suit : |
Sans modification |
Art. L. 3110-3. - Nonobstant les dispositions de l'article L. 1142-1, les professionnels de santé ne peuvent être tenus pour responsables des dommages résultant de la prescription ou de l'administration d'un médicament hors des conditions normales d'utilisation prévues par l'autorisation de mise sur le marché lorsque leur intervention était rendue nécessaire par l'existence d'une menace sanitaire grave et que la prescription ou l'administration du médicament avait été recommandée par le ministre chargé de la santé en application des dispositions de l'article L. 3110-1. |
I. - Les mots : « d'un médicament hors des conditions normales d'utilisation prévues par l'autorisation de mise sur le marché » sont remplacés par les mots : « d'un médicament en dehors des indications thérapeutiques ou des conditions normales d'utilisation prévues par son autorisation de mise sur le marché ou son autorisation temporaire d'utilisation ou bien d'un médicament ne faisant l'objet d'aucune de ces autorisations ». |
|
II. - Les mots : « avait été recommandée » sont remplacés par les mots : « a été recommandée ou exigée ». |
||
III. - Il est ajouté un alinéa ainsi rédigé : |
||
« Le fabricant d'un médicament ne peut davantage être tenu pour responsable des dommages résultant de l'utilisation d'un médicament en dehors des indications thérapeutiques ou des conditions normales d'utilisation prévues par son autorisation de mise sur le marché ou son autorisation temporaire d'utilisation ou bien de celle d'un médicament ne faisant l'objet d'aucune de ces autorisations lorsque cette utilisation a été recommandée ou exigée par le ministre chargé de la santé en application de l'article L. 3110-1. Il en va de même du titulaire de l'autorisation de mise sur le marché, de l'autorisation temporaire d'utilisation ou de l'autorisation d'importation du médicament en cause. Ces dispositions ne les exonèrent pas de l'engagement de leur responsabilité dans les conditions de droit commun en raison de la fabrication ou de la mise sur le marché du médicament. » |
||
Article 2 |
Article 2 | |
Art. L. 4113-6 - ................... Il ne s'applique pas non plus à l'hospitalité offerte, de manière directe ou indirecte, lors de manifestations de promotion ou lors de manifestations à caractère exclusivement professionnel et scientifique lorsqu'elle est prévue par convention passée entre l'entreprise et le professionnel de santé et soumise pour avis au conseil départemental de l'ordre compétent avant sa mise en application, et que cette hospitalité est d'un niveau raisonnable, reste accessoire par rapport à l'objectif principal de la réunion et n'est pas étendue à des personnes autres que les professionnels directement concernés. ........................................ |
Au troisième alinéa de l'article L. 4113-6 du code de la santé publique, les mots : « d'un niveau raisonnable, reste accessoire par rapport à l'objectif principal de la réunion » sont remplacés par les mots : « limitée à l'objectif principal de la manifestation ». |
Au ... ...mots : « , reste accessoire par rapport à l'objectif principal de la réunion » sont remplacés par les mots : « et limitée à l'objectif professionnel et scientifique principal de la manifestation ». Amendement n° 1 |
Article 3 |
Article 3 | |
L'article L. 5111-1 du code de la santé publique est ainsi modifié : |
Sans modification | |
I. - Le premier alinéa est remplacé par les dispositions suivantes : |
||
Art. L. 5111-1 - On entend par médicament toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l'égard des maladies humaines ou animales, ainsi que tout produit pouvant être administré à l'homme ou à l'animal, en vue d'établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions organiques. ........................................ |
« On entend par médicament toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l'égard des maladies humaines ou animales, ainsi que toute substance ou composition pouvant être utilisée chez l'homme ou chez l'animal ou pouvant leur être administrée, en vue d'établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions physiologiques en exerçant une action pharmacologique, immunologique ou métabolique. » |
|
Les produits utilisés pour la désinfection des locaux et pour la prothèse dentaire ne sont pas considérés comme des médicaments. |
II. - Il est ajouté un quatrième alinéa ainsi rédigé : |
|
« Lorsque, eu égard à l'ensemble de ses caractéristiques, un produit est susceptible de répondre à la fois à la définition du médicament et à celle d'autres catégories de produits régies par le droit communautaire ou national, il est, en cas de doute, considéré comme un médicament. » |
||
Article 4 |
Article 4 | |
Art. L. 5121-1 - On entend par : |
L'article L. 5121-1 du code de la santé publique est ainsi modifié : |
Alinéa sans modification |
....................................... |
I. - Le 5° est remplacé par les dispositions suivantes : |
I. - Non modifié |
5º Sans préjudice des dispositions des articles L. 611-2 et suivants du code de la propriété intellectuelle, spécialité générique d'une spécialité de référence, celle qui a la même composition qualitative et quantitative en principe actif, la même forme pharmaceutique et dont la bioéquivalence avec la spécialité de référence est démontrée par des études de biodisponibilité appropriées. La spécialité de référence et les spécialités qui en sont génériques constituent un groupe générique. En l'absence de spécialité de référence, un groupe générique peut être constitué de spécialités ayant la même composition qualitative et quantitative en principe actif, la même forme pharmaceutique et dont le profil de sécurité et d'efficacité est équivalent. Pour l'application du présent 5º, les différentes formes pharmaceutiques orales à libération immédiate sont considérées comme une même forme pharmaceutique et les différents sels, esters, éthers, isomères, mélanges d'isomères, complexes ou dérivés d'un principe actif sont considérés comme un même principe actif, sauf s'ils présentent des propriétés sensiblement différentes au regard de la sécurité ou de l'efficacité. Dans ce cas, des informations supplémentaires fournissant la preuve de la sécurité et de l'efficacité des différents sels, esters ou dérivés d'une substance active autorisée doivent être apportées ; ....................................... |
« 5° a) Sans préjudice des dispositions des articles L. 611-2 et suivants du code de la propriété intellectuelle, spécialité générique d'une spécialité de référence, celle qui a la même composition qualitative et quantitative en principes actifs, la même forme pharmaceutique et dont la bioéquivalence avec la spécialité de référence est démontrée par des études de biodisponibilité appropriées. Une spécialité ne peut être qualifiée de spécialité de référence que si son autorisation de mise sur le marché a été délivrée au vu d'un dossier comportant, dans des conditions fixées par voie réglementaire, l'ensemble des données nécessaires et suffisantes à elles seules pour son évaluation. Pour l'application du présent alinéa, les différentes formes pharmaceutiques orales à libération immédiate sont considérées comme une même forme pharmaceutique. De même, les différents sels, esters, éthers, isomères, mélanges d'isomères, complexes ou dérivés d'un principe actif sont regardés comme ayant la même composition qualitative en principe actif, sauf s'ils présentent des propriétés sensiblement différentes au regard de la sécurité ou de l'efficacité nécessitant que soient produites des données supplémentaires ; |
|
« b) groupe générique, le regroupement d'une spécialité de référence et des spécialités qui en sont génériques. Toutefois, une spécialité remplissant les conditions pour être une spécialité de référence, qui présente la même composition qualitative et quantitative en principes actifs et la même forme pharmaceutique qu'une spécialité de référence d'un groupe générique déjà existant, et dont la bioéquivalence avec cette spécialité est démontrée par des études de biodisponibilité appropriées, peut aussi figurer dans ce groupe générique, à condition que ces deux spécialités soient considérées comme relevant d'une même autorisation de mise sur le marché globale, définie par voie réglementaire. En l'absence de spécialité de référence, un groupe générique peut être constitué de spécialités ayant la même composition qualitative et quantitative en principes actifs, la même forme pharmaceutique et dont le profil de sécurité et d'efficacité est équivalent. » |
||
11º Médicament homéopathique, tout médicament obtenu à partir de produits, substances ou compositions appelés souches homéopathiques, selon un procédé de fabrication homéopathique décrit par la pharmacopée européenne, la pharmacopée française ou, à défaut, par les pharmacopées utilisées de façon officielle dans un autre Etat membre de la Communauté européenne. Un médicament homéopathique peut aussi contenir plusieurs principes ; ....................................... |
II. - Au 11°, les mots : « produits, substances ou composition appelés » sont remplacés par les mots : « substances appelées ». |
II. - Non modifié |
III. - Sont ajoutés un 14° et un 15° ainsi rédigés : |
III. - Alinéa sans modification | |
« 14° Médicament biologique, tout médicament dont la substance est produite à partir d'une source biologique ou en est extraite et dont la caractérisation et la détermination de la qualité nécessitent une combinaison d'essais physiques, chimiques et biologiques ainsi que la connaissance de son procédé de fabrication et de son contrôle ; |
« 14° Médicament biologique, tout médicament dont la substance active est ... ... son contrôle ; Amendement n° 2 | |
« 15° Sans préjudice des dispositions des articles L. 611-2 et suivants du code de la propriété intellectuelle, médicament biologique similaire, tout médicament biologique de même composition qualitative et quantitative en substance active et de même forme pharmaceutique qu'un médicament biologique de référence mais qui ne remplit pas les conditions prévues au a du 5° du présent article pour être regardée comme une spécialité générique en raison de différences liées notamment à la variabilité de la matière première ou aux procédés de fabrication et nécessitant que soient produites des données précliniques et cliniques supplémentaires dans des conditions déterminées par voie réglementaire. » |
Alinéa sans modification | |
Article 5 |
Article 5 | |
I. - L'article L. 5121-8 du code de la santé publique est modifié ainsi qu'il suit : |
I. - Alinéa sans modification | |
1° Les deux premiers alinéas sont remplacés par les dispositions suivantes : |
Alinéa sans modification | |
Art. L. 5121-8 - Toute spécialité pharmaceutique ou tout autre médicament fabriqué industriellement, ainsi que tout générateur, trousse ou précurseur qui ne fait pas l'objet d'une autorisation de mise sur le marché délivrée par la Communauté européenne en application du règlement (CEE) nº 2309/93 du Conseil du 22 juillet 1993 doit faire l'objet avant sa commercialisation ou sa distribution à titre gratuit, en gros ou en détail, d'une autorisation de mise sur le marché délivrée par l'Agence française de sécurité sanitaire des produits de santé. Cette autorisation peut être assortie de conditions adéquates. ....................................... |
« Toute spécialité pharmaceutique ou tout autre médicament fabriqué industriellement ou selon une méthode dans laquelle intervient un processus industriel ainsi que tout générateur, trousse ou précurseur qui ne fait pas l'objet d'une autorisation de mise sur le marché délivrée par la Communauté européenne en application du règlement (CE) n° 726/2004 du Parlement européen et du Conseil du 31 mars 2004 doit faire l'objet d'une autorisation préalable de mise sur le marché délivrée par l'Agence française de sécurité sanitaire des produits de santé. L'autorisation peut être assortie de conditions appropriées. |
Alinéa sans modification |
« Le demandeur de l'autorisation peut être dispensé de produire certaines données et études dans des conditions fixées par voie réglementaire. |
Alinéa sans modification | |
« Une autorisation de mise sur le marché ne peut être délivrée qu'à un demandeur établi dans un État membre de la Communauté européenne ou partie à l'accord sur l'Espace économique européen. |
Alinéa sans modification | |
« L'autorisation est délivrée pour une durée déterminée et peut ensuite être renouvelée, le cas échéant, sans limitation de durée, dans des conditions fixées par un décret en Conseil d'État. Ce décret détermine également les conditions dans lesquelles elle peut devenir caduque. » |
« L'autorisation ... ...déterminée de cinq ans et peut ... ... en Conseil d'État, sauf si l'Agence française de sécurité sanitaire des produits de santé décide pour des raisons justifiées ayant trait à la pharmacovigilance, de procéder à un nouveau renouvellement quinquennal. Ce décret ... ... caduque. » Amendements n°s 3 et 4 | |
L'accomplissement des formalités prévues au présent article n'a pas pour effet d'exonérer le fabricant ou, s'il est distinct, le titulaire de l'autorisation de mise sur le marché, de la responsabilité que l'un ou l'autre peut encourir dans les conditions du droit commun en raison de la fabrication ou de la mise sur le marché du médicament ou produit. |
2° Au dernier alinéa, les mots : « le fabricant ou, s'il est distinct » sont remplacés par les mots : « le fabricant et, s'il est distinct ». |
Alinéa sans modification |
II. - Les durées, déterminées par voie réglementaire, qui servent de référence pour la mise en œuvre du deuxième alinéa de l'article L. 5121-8 du code de la santé publique dans sa rédaction issue du I du présent article sont applicables dès lors que la demande d'autorisation de mise sur le marché du médicament ou de la spécialité de référence a été déposée postérieurement au 29 octobre 2005. |
II. - Non modifié | |
Article 6 |
Article 6 | |
L'article L. 5121-9 du code de la santé publique est modifié ainsi qu'il suit : |
Sans modification | |
Art. L. 5121-9 - L'autorisation prévue à l'article L. 5121-8 est refusée lorsqu'il apparaît que le médicament ou le produit est nocif dans les conditions normales d'emploi, ou qu'il n'a pas la composition qualitative et quantitative déclarée, ou que l'effet thérapeutique annoncé fait défaut ou est insuffisamment justifié par le demandeur. ....................................... |
I. - Au premier alinéa, les mots : « que le médicament ou produit est nocif dans les conditions normales d'emploi » sont remplacés par les mots : « que l'évaluation des effets thérapeutiques positifs du médicament ou produit au regard des risques liés à son utilisation n'est pas considérée comme favorable », et le mot : « justifié » est remplacé par le mot : « démontré ». |
|
II. - Les quatre derniers alinéas sont remplacés par les dispositions suivantes : |
||
Lorsque, pour certaines indications thérapeutiques, le demandeur peut démontrer qu'il n'est pas en mesure de fournir des renseignements complets sur l'efficacité et l'innocuité du médicament dans les conditions normales d'emploi, l'autorisation de mise sur le marché peut toutefois être délivrée, sous réserve du respect d'obligations spécifiques, dans l'un des cas suivants : - les indications prévues se présentent si rarement que le demandeur ne peut raisonnablement être tenu de fournir les renseignements complets ; - l'état d'avancement de la science ne permet pas de donner les renseignements complets ; - des principes de déontologie médicale interdisent de recueillir ces renseignements. |
« Toutefois, dans des circonstances exceptionnelles et sous réserve du respect d'obligations spécifiques définies par voie réglementaire, concernant notamment la sécurité du médicament, la notification à l'Agence française de sécurité sanitaire des produits de santé de tout incident lié à son utilisation et les mesures à prendre, l'autorisation de mise sur le marché peut être délivrée à un demandeur qui démontre qu'il n'est pas en mesure de fournir des renseignements complets sur l'efficacité et la sécurité du médicament dans des conditions normales d'emploi. Le maintien de cette autorisation est décidé par l'Agence sur la base d'une réévaluation annuelle de ces obligations et de leur respect par le titulaire. » |
|
Article 7 |
Article 7 | |
Après l'article L. 5121-9 du code de la santé publique, il est inséré un article L. 5121-9-1 ainsi rédigé : |
Sans modification | |
« Art. L. 5121-9-1. - Lorsqu'un médicament est autorisé dans un autre État membre de la Communauté européenne ou un État partie à l'accord sur l'Espace économique européen mais qu'il ne fait l'objet en France ni de l'autorisation de mise sur le marché prévue à l'article L. 5121-8, ni d'une demande en cours d'instruction en vue d'une telle autorisation, l'Agence française de sécurité sanitaire des produits de santé peut, pour des raisons de santé publique, autoriser la mise sur le marché de ce médicament dans des conditions prévues par voie réglementaire. » |
||
Article 8 |
Article 8 | |
Art. L. 5121-10 - Pour une spécialité générique définie au 5º de l'article L. 5121-1, l'autorisation de mise sur le marché peut être délivrée avant l'expiration des droits de propriété intellectuelle qui s'attachent à la spécialité de référence concernée. Le demandeur de cette autorisation informe le titulaire de ces droits concomitamment au dépôt de la demande. ........................................ |
L'article L. 5121-10 du code de la santé publique est ainsi modifié : |
Sans modification |
Le directeur général de l'agence procède à l'inscription de la spécialité générique dans le répertoire des groupes génériques au terme d'un délai de soixante jours, après avoir informé de la délivrance de l'autorisation de mise sur le marché de celle-ci le titulaire de l'autorisation de mise sur le marché de la spécialité de référence. Toutefois, la commercialisation de cette spécialité générique ne peut intervenir qu'après l'expiration des droits de propriété intellectuelle, sauf accord du titulaire de ces droits. |
I. - À la fin du troisième alinéa est inséré un alinéa ainsi rédigé : |
|
« Préalablement à cette commercialisation, le titulaire de l'autorisation de mise sur le marché de la spécialité générique informe le directeur général de l'agence des indications, formes pharmaceutiques et dosages de la spécialité de référence pour lesquels les droits de propriété intellectuelle n'ont pas expiré. » |
||
Aux seules fins d'en garantir la publicité, le directeur général de l'Agence française de sécurité sanitaire des produits de santé tient disponible au public la liste des titres de propriété intellectuelle attachés à une spécialité de référence si le titulaire de l'autorisation de mise sur le marché de cette spécialité la lui a communiquée à cet effet. Le laboratoire est seul responsable de l'exactitude des informations fournies. Les conditions de rémunération du service rendu par l'agence sont fixées par une décision de son conseil d'administration. |
II. - Les dispositions de la dernière phrase du dernier alinéa sont abrogées. |
|
Article 9 |
Article 9 | |
I. - Après l'article L. 5121-10 du code de la santé publique, il est inséré un article L. 5121-10-1 rédigé comme suit : |
Sans modification | |
« Art. L. 5121-10-1. - Une spécialité générique ne peut être commercialisée qu'à l'expiration d'une période de dix ans suivant l'autorisation initiale de mise sur le marché de la spécialité de référence. Toutefois, cette période est portée à onze ans si pendant les huit premières années suivant l'autorisation de la spécialité de référence, le titulaire de celle-ci obtient une autorisation pour une ou plusieurs indications thérapeutiques nouvelles considérées, lors de l'évaluation scientifique conduite en vue de leur autorisation, comme apportant un avantage clinique important par rapport aux thérapies existantes. |
||
« Les dispositions du présent article sont également applicables aux médicaments biologiques similaires et aux médicaments présentant des caractéristiques communes par rapport à un médicament de référence mais ne répondant pas à la définition du médicament générique en raison de différences portant sur un ou plusieurs éléments de cette définition nécessitant que soient produites des données supplémentaires dans des conditions déterminées par voie réglementaire. » |
||
II. - Le I du présent article n'est applicable que lorsque l'autorisation initiale de mise sur le marché de la spécialité de référence ou du médicament de référence a été délivrée au vu d'une demande déposée à compter du 30 octobre 2005. |
||
Article 10 |
Article 10 | |
Code de la propriété intellectuelle |
À l'article L. 613-5 du code de la propriété intellectuelle, il est ajouté un d rédigé comme suit : |
Sans modification |
Art. L. 613-5 - Les droits conférés par le brevet ne s'étendent pas : ........................................ |
||
« d) Aux études et essais requis en vue de l'obtention d'une autorisation de mise sur le marché pour un médicament, ainsi qu'aux actes nécessaires à leur réalisation et à l'obtention de l'autorisation. » |
||
Code de la santé publique |
Article 11 |
Article 11 |
Après l'article L. 5121-10-1 du code de la santé publique, il est inséré un article L. 5121-10-2 rédigé comme suit : |
Sans modification | |
« Art. L. 5121-10-2. - Pour un médicament biologique similaire défini au 15° de l'article L. 5121-1, l'autorisation de mise sur le marché peut être délivrée avant l'expiration des droits de propriété intellectuelle qui s'attachent au médicament biologique de référence. Le demandeur de l'autorisation informe le titulaire de ces droits concomitamment au dépôt de sa demande. |
||
« Lorsque l'Agence française de sécurité sanitaire des produits de santé a délivré une autorisation de mise sur le marché pour un médicament biologique similaire, elle en informe le titulaire de l'autorisation de mise sur le marché du médicament biologique de référence. |
||
« La commercialisation du médicament biologique similaire ne peut intervenir qu'après l'expiration des droits de propriété intellectuelle du médicament biologique de référence, sauf accord du titulaire de ces droits. |
||
« Préalablement à la commercialisation, le titulaire de l'autorisation de mise sur le marché du médicament biologique similaire informe le directeur général de l'Agence française de sécurité sanitaire des produits de santé des indications, formes pharmaceutiques et dosages du médicament biologique de référence pour lesquels les droits de propriété intellectuelle n'ont pas expiré. |
||
« Aux seules fins d'en garantir la publicité, le directeur général de l'Agence française de sécurité sanitaire des produits de santé tient disponible au public la liste des titres de propriété intellectuelle attachés à un médicament biologique de référence si le titulaire de l'autorisation de mise sur le marché de ce médicament la lui a communiquée à cet effet. Le laboratoire est seul responsable de l'exactitude des informations fournies. |
||
« Les dispositions du présent article s'appliquent également aux médicaments présentant des caractéristiques communes par rapport à un médicament de référence mais ne répondant pas à la définition du médicament générique en raison de différences portant sur un ou plusieurs éléments de cette définition nécessitant que soient produites des données supplémentaires dans des conditions déterminées par voie réglementaire. » |
||
Article 12 |
Article 12 | |
Art. L. 5121-12 - Les dispositions de l'article L. 5121-8 ne font pas obstacle à l'utilisation, à titre exceptionnel, de certains médicaments destinés à traiter des maladies graves ou rares lorsqu'il n'existe pas de traitement approprié : |
L'article L. 5121-12 du code de la santé publique est modifié comme suit : |
Sans modification |
a) Et que l'efficacité et la sécurité de ces médicaments sont fortement présumées, au vu des résultats d'essais thérapeutiques auxquels il a été procédé en vue d'une demande d'autorisation de mise sur le marché, et que cette demande a été déposée ou que le demandeur s'engage à la déposer dans un délai déterminé ; |
I. - Le b est remplacé par les dispositions suivantes : |
|
b) Ou que ces médicaments sont prescrits à des malades nommément désignés et, le cas échéant, importés dans ce but, sous la responsabilité de leur médecin traitant, dès lors que leur efficacité et leur sécurité sont présumées en l'état des connaissances scientifiques et qu'ils sont susceptibles de présenter un bénéfice réel. |
« b) Ou que ces médicaments, le cas échéant importés, sont prescrits, sous la responsabilité d'un médecin, à un patient nommément désigné et ne pouvant participer à une recherche biomédicale, dès lors qu'ils sont susceptibles de présenter un bénéfice pour lui et que, soit leur efficacité et leur sécurité sont présumées en l'état des connaissances scientifiques, soit une issue fatale à court terme pour le patient est, en l'état des thérapeutiques disponibles, inéluctable. Le médecin demandeur doit justifier que le patient, son représentant légal ou la personne de confiance qu'il a désignée en application de l'article L. 1111-6, a reçu une information adaptée à sa situation sur l'absence d'alternative thérapeutique, les risques courus, les contraintes et le bénéfice susceptible d'être apporté par le médicament. La procédure suivie est inscrite dans le dossier médical. » |
|
L'utilisation de ces médicaments est autorisée, pour une durée limitée, par l'Agence française de sécurité sanitaire des produits de santé, à la demande du titulaire des droits d'exploitation du médicament dans le cas prévu au a ou à la demande du médecin traitant dans le cas prévu au b du présent article. ........................................ |
II. - Au quatrième alinéa, le mot : « traitant » est remplacé par le mot : « prescripteur ». |
|
Article 13 |
Article 13 | |
Art. L. 5121-15 - Toute demande d'enregistrement mentionnée aux articles L. 5121-13 et L. 5121-14 donne lieu au versement, au profit de l'Agence française de sécurité sanitaire des produits de santé, d'un droit progressif dont le montant est fixé par décret dans la limite de 7 600 euros. ........................................ |
Au premier alinéa de l'article L. 5121-15 du code de la santé publique, après les mots : « L. 5121-13 et L. 5121-14 », sont insérés les mots : « ou toute demande de modification ou de renouvellement de cet enregistrement ». |
Sans modification |
Article 14 |
Article 14 | |
Art. L. 5121-16 - Toute demande d'autorisation de mise sur le marché doit être accompagnée du versement d'un droit progressif dont le montant est fixé par décret dans la limite de 25 400 euros. |
Au premier alinéa de l'article L. 5121-16 du code de la santé publique, après les mots : « demande d'autorisation de mise sur le marché », sont insérés les mots : « mentionnée à l'article L. 5121-8 ou toute demande de modification ou de renouvellement de cette autorisation. » |
Sans modification |
Article 15 |
Article 15 | |
Art. L. 5121-20 - Les modalités d'application du présent chapitre sont déterminées par décret en Conseil d'Etat, et notamment : |
L'article L. 5121-20 du code de la santé publique est modifié ainsi qu'il suit : |
Sans modification |
I. - Le 1° est remplacé par les dispositions suivantes : |
||
1º Les critères scientifiques justifiant le cas échéant l'exonération des études de biodisponibilité des spécialités génériques définies au 5º de l'article L. 5121-1, les modalités de création de groupes génériques en l'absence de spécialité de référence, ces groupes étant définis au 5º de l'article L. 5121-1, et la procédure d'inscription au répertoire des groupes génériques mentionnés à l'article L. 5121-10 ; |
« 1° Les critères scientifiques justifiant, le cas échéant, l'exonération des études de biodisponibilité des spécialités génériques définies au 5° de l'article L. 5121-1, la procédure d'inscription au répertoire des groupes génériques mentionné à l'article L. 5121-10, ainsi que les modalités de l'inscription dans un groupe générique existant d'une spécialité remplissant la condition pour être spécialité de référence et de la création de groupes génériques en l'absence de spécialité de référence ; » |
|
II. - Sont insérées au 2° les dispositions suivantes : |
||
2º (Alinéa abrogé) |
« Les conditions dans lesquelles des autorisations de mise sur le marché peuvent être considérées comme faisant partie d'une autorisation de mise sur le marché globale ; » |
|
III. - Le 3° est remplacé par les dispositions suivantes : |
||
3º Les règles concernant la présentation et la dénomination des médicaments et produits ; |
« 3° Les règles relatives à l'étiquetage, la notice et la dénomination des médicaments et produits mentionnés au présent chapitre ; » |
|
IV. - Le 4° est remplacé par les dispositions suivantes : |
||
4º Le contenu du dossier présenté à l'appui d'une demande d'autorisation de mise sur le marché prévu à l'article L. 5121-8 ; ....................................... |
« 4° Les modalités de présentation des demandes tendant à obtenir l'autorisation de mise sur le marché prévue à l'article L. 5121-8, le contenu du dossier présenté à l'appui de ces demandes, les conditions dans lesquelles le demandeur peut être dispensé de produire certains éléments du dossier et celles dans lesquelles interviennent les décisions accordant, modifiant, renouvelant, suspendant ou supprimant ces autorisations ainsi qu'après la délivrance de l'autorisation les modalités de son actualisation ; » |
|
V. - Le 6° est remplacé par les dispositions suivantes : |
||
6º Les conditions dans lesquelles interviennent les décisions accordant, modifiant, renouvelant, suspendant ou supprimant une autorisation de mise sur le marché ou un enregistrement de médicament homéopathique, ainsi que les règles de procédure applicables aux recours ouverts contre lesdites décisions ; ........................................ |
« 6° Les modalités de présentation des demandes tendant à obtenir l'enregistrement des médicaments homéopathiques prévu à l'article L. 5121-13, le contenu du dossier présenté à l'appui de ces demandes, ainsi que les conditions dans lesquelles interviennent les décisions accordant, modifiant, renouvelant, suspendant ou supprimant ces enregistrements ; » |
|
VI. - Le 8° est remplacé par les dispositions suivantes : |
||
8º Les conditions d'octroi, de suspension ou de retrait de l'autorisation permettant l'utilisation à titre exceptionnel de certains médicaments destinés à traiter des maladies graves ou rares lorsqu'il n'existe pas de traitement approprié selon les dispositions de l'article L. 5121-12 ; ........................................ |
« 8° Les modalités de présentation des demandes tendant à obtenir l'autorisation temporaire d'utilisation prévue à l'article L. 5121-12, le contenu du dossier présenté à l'appui de ces demandes, ainsi que les conditions dans lesquelles interviennent les décisions accordant, modifiant, renouvelant, suspendant ou supprimant ces autorisations ; » |
|
VII. - Le 11° est remplacé par les dispositions suivantes : |
||
11º Les règles applicables en cas de changement du titulaire de l'autorisation de mise sur le marché ; |
« 11° Les règles applicables en cas de changement du titulaire de l'autorisation de mise sur le marché ou du bénéficiaire de l'enregistrement de médicament homéopathique ; » |
|
12º Les modalités d'application des articles L. 5121-17 et L. 5121-18 relatifs à la taxe annuelle des médicaments et produits bénéficiant d'une autorisation de mise sur le marché ; |
VIII. - Le 12 °est abrogé. |
|
IX. - Le 13° est remplacé par les dispositions suivantes : |
||
13º Les règles applicables à la pharmacovigilance exercée sur les médicaments postérieurement à la délivrance de l'autorisation administrative de mise sur le marché prévue à l'article L. 5121-8, de l'autorisation temporaire d'utilisation prévue à l'article L. 5121-12 ou postérieurement à l'enregistrement des médicaments homéopathiques prévu à l'article L. 5121-13 ; ces règles fixent notamment les obligations de signalement incombant aux membres des professions de santé et aux entreprises exploitant un médicament ou un produit soumis aux dispositions du présent titre ; ........................................ |
« 13° Les règles applicables à la pharmacovigilance exercée sur les médicaments et sur les produits mentionnés à l'article L. 5121-1, notamment les obligations de signalement incombant aux membres des professions de santé et aux entreprises exploitant un médicament ou un produit soumis aux dispositions du présent titre ; » |
|
15º Les modalités de présentation des demandes tendant à obtenir l'enregistrement des médicaments homéopathiques prévu à l'article L. 5121-13, la nature du dossier ainsi que les règles relatives à l'étiquetage et à la notice de ces médicaments ; ....................................... |
X. - Le 15° est abrogé. |
|
Code de la sécurité sociale |
Article additionnel | |
Art. L. 161-38 - La Haute Autorité de santé est chargée d'établir une procédure de certification des sites informatiques dédiés à la santé et des logiciels d'aide à la prescription médicale ayant respecté un ensemble de règles de bonne pratique. ........................................ |
« Le premier alinéa de l'article L. 161-38 du code de la sécurité sociale est complété par la phrase suivante : » | |
« Elle veille à ce que les règles de bonne pratique spécifient que ces logiciels permettent de prescrire directement en dénomination commune internationale. » Amendement n° 5 | ||
Article 16 |
Article 16 | |
Art. L. 5122-4 - La publicité des spécialités définies au 5º de l'article L. 5121-1 doit mentionner l'appartenance à la catégorie des spécialités génériques. |
L'article L. 5122-4 du code de la santé publique est abrogé. |
Sans modification |
Article 17 |
Article 17 | |
L'article L. 5122-6 du code de la santé publique est ainsi modifié : |
Alinéa sans modification | |
I. - Au premier alinéa : |
I. - Non modifié | |
Art. L. 5122-6 - La publicité auprès du public pour un médicament n'est admise qu'à la condition que ce médicament ne soit pas soumis à prescription médicale, qu'il ne soit pas remboursable par les régimes obligatoires d'assurance maladie et que l'autorisation de mise sur le marché ou l'enregistrement ne comporte pas de restrictions en matière de publicité auprès du public en raison d'un risque possible pour la santé publique. ........................................ |
1° Avant les mots : « de restrictions » sont insérés les mots : « d'interdiction ou ». |
|
2° Les mots : « qu'il ne soit pas remboursable » sont remplacés les mots : « qu'aucune de ses différentes présentations ne soit remboursable ». |
||
3° Après les mots : « pour la santé publique » sont ajoutés les mots : « , notamment lorsque le médicament n'est pas adapté à une utilisation sans intervention d'un médecin pour le diagnostic, l'initiation, ou la surveillance du traitement ». |
||
II. - Le a est remplacé par les dispositions suivantes : |
||
a) Que le médicament ne soit pas soumis à prescription médicale et que son autorisation de mise sur le marché ou son enregistrement ne comporte pas de restriction en matière de publicité auprès du public en raison d'un risque possible pour la santé publique ; ....................................... |
« a) Que le médicament ne soit pas soumis à prescription médicale et que son autorisation de mise sur le marché ou son enregistrement ne comporte pas d'interdiction ou de restriction en matière de publicité auprès du public ; » |
|
La publicité auprès du public pour un médicament est nécessairement accompagnée d'un message de prudence et de renvoi à la consultation d'un médecin en cas de persistance des symptômes. |
III. - Le dernier alinéa est abrogé. |
III. - Supprimé Amendement n° 6 |
Article 18 |
Article 18 | |
L'article L. 5122-10 du code de la santé publique est ainsi modifié : |
Sans modification | |
I. - Les quatre premiers alinéas sont remplacés par les dispositions suivantes : |
||
Art. L. 5122-10 - Des échantillons gratuits ne peuvent être remis aux personnes habilitées à prescrire ou à dispenser des médicaments dans le cadre des pharmacies à usage intérieur que sur leur demande. |
« Des échantillons gratuits de médicaments ne peuvent être remis aux personnes habilitées à prescrire ou à dispenser des médicaments dans le cadre des pharmacies à usage intérieur que sur leur demande. |
|
Aucun échantillon de médicaments contenant des substances classées comme psychotropes ou stupéfiants, ou auxquels la réglementation des stupéfiants est appliquée en tout ou partie, ne peut être remis. |
« Ces échantillons ne peuvent contenir des substances classées comme psychotropes ou stupéfiants, ou auxquels la réglementation des stupéfiants est appliquée en tout ou partie. |
|
La remise d'échantillons de médicaments est interdite dans les enceintes accessibles au public à l'occasion de congrès médicaux ou pharmaceutiques. |
« Ils doivent être identiques aux spécialités pharmaceutiques concernées et porter la mention : "échantillon gratuit". |
|
Les échantillons doivent être identiques aux spécialités pharmaceutiques concernées et porter la mention : « échantillon gratuit ». |
« Leur remise directe au public à des fins promotionnelles, ainsi que leur remise dans les enceintes accessibles au public à l'occasion de congrès médicaux ou pharmaceutiques, est interdite. » |
|
Dans le cadre de la promotion des médicaments auprès des personnes habilitées à les prescrire ou à les délivrer, il est interdit d'octroyer, d'offrir ou de promettre à ces personnes une prime, un avantage pécuniaire ou un avantage en nature, à moins que ceux-ci ne soient de valeur négligeable. |
II. - Au dernier alinéa, après les mots : « de valeur négligeable », sont ajoutés les mots : « et ne soient relatifs à l'exercice de la médecine ou de la pharmacie ». |
|
Article 19 |
Article 19 | |
Art. L. 5122-16 - Sont définies par décret en Conseil d'Etat : ........................................ |
À l'article L. 5122-16 du code de la santé publique : |
Sans modification |
3º Les conditions dans lesquelles des échantillons gratuits peuvent être remis aux personnes mentionnées au premier alinéa de l'article L. 5122-10 ; |
I. - Au 3°, après les mots : « échantillons gratuits », sont insérés les mots : « de médicaments » ; |
|
4º Les modalités d'application de l'article L. 5122-15 et notamment la composition et les modalités de fonctionnement de la commission prévue au dernier alinéa de cet article. |
II. - Il est ajouté un 5° ainsi rédigé : |
|
« 5° Les mentions obligatoires des publicités pour les médicaments ainsi que les conditions dans lesquelles il peut y être dérogé, notamment lorsque ces publicités ont exclusivement pour objet de rappeler le nom, la dénomination commune internationale ou la marque des médicaments. » |
||
Article 20 |
Article 20 | |
L'article L. 5124-5 du code de la santé publique est ainsi modifié : |
Sans modification | |
Art. L. 5124-5 - Lorsqu'un médicament ou produit soumis à l'autorisation de mise sur le marché prévue à l'article L. 5121-8 est commercialisé, l'établissement pharmaceutique qui l'exploite communique, sans délai, la date de cette commercialisation à l'Agence française de sécurité sanitaire des produits de santé. |
I. - Les mots : « l'établissement pharmaceutique » sont remplacés par les mots : « l'entreprise ». |
|
II. - Les mots : « la date de cette commercialisation » sont remplacés par les mots : « les dates de commercialisation de chaque présentation de ce médicament ou produit ». |
||
Article 21 |
Article 21 | |
L'article L. 5124-6 du code de la santé publique est modifié ainsi qu'il suit : |
Sans modification | |
Art. L. 5124-6 - L'établissement pharmaceutique exploitant un médicament ou produit soumis aux dispositions du chapitre I du présent titre informe immédiatement l'Agence française de sécurité sanitaire des produits de santé de toute action qu'il a engagée pour en suspendre la commercialisation, le retirer du marché ou en retirer un lot déterminé. Il doit en indiquer la raison si celle-ci concerne l'efficacité du médicament ou produit ou la protection de la santé publique. Il doit en outre informer l'Agence française de sécurité sanitaire des produits de santé de tout risque de rupture de stock sur un médicament ou produit sans alternative thérapeutique disponible, dont il assure l'exploitation, ainsi que de tout risque de rupture de stock sur un médicament ou produit dont il assure l'exploitation, lié à un accroissement brutal et inattendu de la demande. |
I. - Les mots : « l'établissement pharmaceutique » sont remplacés par les mots : « l'entreprise ». |
|
II. - Le mot : « immédiatement » est supprimé. |
||
III. - Après les mots : « un lot déterminé », sont ajoutés les mots : « ainsi que de tout risque de rupture de stock sur un médicament ou produit sans alternative thérapeutique disponible ou en raison d'un accroissement significatif et imprévisible de la demande ». |
||
IV. - Au deuxième alinéa, le mot : « Il » est remplacé par le mot : « Elle ». |
||
V. - La dernière phrase est remplacée par les dispositions suivantes : « Les conditions, notamment de délai, dans lesquelles il est procédé aux informations prévues par le présent article ainsi que le contenu de ces informations sont déterminées par décret. » |
||
Article 22 |
Article 22 | |
Art. L. 5124-13 - L'importation sur le territoire douanier des médicaments à usage humain et l'importation et l'exportation des préparations de thérapie génique ou des préparations de thérapie cellulaire xénogénique mentionnées au 12º et au 13º de l'article L. 5121-1 sont soumises à une autorisation préalable délivrée par l'Agence française de sécurité sanitaire des produits de santé. ....................................... |
L'article L. 5124-13 du code de la santé publique est complété par deux alinéas rédigés ainsi qu'il suit : |
Sans modification |
« Une telle autorisation n'est pas requise pour le particulier qui transporte personnellement un médicament. |
||
« Lorsqu'un particulier procède à l'importation d'un médicament par une autre voie que le transport personnel, il n'est pas non plus soumis à l'obligation d'une autorisation préalable si ce médicament fait l'objet d'une autorisation de mise sur le marché au sens de l'article 6 de la directive 2001/83/CE du Parlement européen et du Conseil du 6 novembre 2001 ou d'un enregistrement au sens des articles 14 et 16 bis de la même directive dans un État membre de la Communauté européenne ou un État partie à l'accord sur l'Espace économique européen. » |
||
Article 23 |
Article 23 | |
L'article L. 5138-2 du code de la santé publique est remplacé par les dispositions suivantes : |
Sans modification | |
Art. L. 5138-2 - Les matières premières à usage pharmaceutique doivent répondre aux spécifications de la pharmacopée quand elles existent et être fabriquées et distribuées en conformité avec des bonnes pratiques dont les principes sont définis par décision de l'Agence française de sécurité sanitaire des produits de santé. |
« Art. L. 5138-2. - I. - On entend par matières premières à usage pharmaceutique tous les composants des médicaments au sens de l'article L. 5111-1, c'est-à-dire : |
|
« a) la ou les substances actives ; |
||
« b) le ou les excipients ; |
||
« c) les éléments de mise en forme pharmaceutique destinés à être utilisés chez l'homme ou chez l'animal ou à leur être administrés. |
||
« II. - L'usage pharmaceutique est présumé pour ces matières lorsqu'elles sont cédées à : |
||
« a) Un établissement pharmaceutique mentionné à l'article L. 5124-1 ou à l'article L. 5142-1 ; |
||
« b) Une pharmacie à usage intérieur ; |
||
« c) Une officine de pharmacie ; |
||
« d) Un médecin, à un vétérinaire ou à une personne autorisée à préparer des autovaccins à usage vétérinaire mentionnée à l'article L. 5141-12. |
||
« Il en va autrement lorsque la personne qui cède ces matières justifie d'une autre destination par la production d'une attestation émanant de l'acheteur. |
||
« III. - En vue d'établir, ou non, l'usage pharmaceutique d'une des matières premières mentionnées au I et cédées à une personne autre que celles énumérées au II, le vendeur doit pouvoir justifier de la destination de ces matières premières. À cette fin, il peut demander à l'acheteur une attestation justifiant de leur destination. |
||
« IV. - On entend par fabrication d'une matière première à usage pharmaceutique la fabrication complète ou partielle de cette matière première ainsi que les divers procédés de division ou de conditionnement préalables à son incorporation dans un médicament et le stockage, en vue de sa vente. |
||
« V. - On entend par distribution d'une matière première à usage pharmaceutique les activités d'achat, de vente, de reconditionnement, de réétiquetage et de stockage. » |
||
Article 24 |
Article 24 | |
L'article L. 5138-3 est remplacé par les dispositions suivantes : |
Sans modification | |
Art. L. 5138-3. - Tout établissement de fabrication, d'importation ou de distribution de matières premières à usage pharmaceutique peut demander à l'Agence française de sécurité sanitaire des produits de santé de certifier que l'établissement qui produit les matières premières respecte les bonnes pratiques mentionnées à l'article L. 5138-2. |
« Art. L. 5138-3. - Les matières premières à usage pharmaceutique répondent aux spécifications de la pharmacopée quand elles existent. |
|
Le contenu de ce certificat est fixé par décision de l'Agence française de sécurité sanitaire des produits de santé. |
« Pour la fabrication de médicaments, les établissements pharmaceutiques mentionnés à l'article L. 5124-1 ou à l'article L. 5142-1, les pharmacies à usage intérieur, les pharmacies d'officine, les médecins, les vétérinaires et les personnes autorisées à préparer des auto-vaccins à usage vétérinaire utilisent, en tant que matières premières à usage pharmaceutique, des substances actives fabriquées et distribuées conformément à des bonnes pratiques, y compris lorsqu'elles sont importées, dont les principes sont définis conformément au droit communautaire par décision de l'Agence française de sécurité sanitaire des produits de santé, après avis de l'Agence française de sécurité sanitaire des aliments. Ce dispositif est également applicable aux excipients entrant dans la fabrication des médicaments à usage humain, dont la liste et les conditions spécifiques qui leur sont applicables sont fixées par décision de l'Agence française de sécurité sanitaire des produits de santé conformément au droit communautaire. » |
|
Article 25 |
Article 25 | |
I. - L'article L. 5138-4 du code de la santé publique est remplacé par les dispositions suivantes : |
I. - Non modifié | |
Art. L. 5138-4. - Chaque demande présentée par un établissement de fabrication, d'importation ou de distribution de matières premières à usage pharmaceutique en vue d'obtenir le certificat mentionné à l'article L. 5138-3 donne lieu au versement, au profit de l'Agence française de sécurité sanitaire des produits de santé, d'un droit fixe dont le montant est fixé par décret dans la limite de 2 300 euros. |
« Art. L. 5138-4. - Lorsque dans le cadre de ses pouvoirs d'inspection, l'Agence française de sécurité sanitaire des produits de santé constate que la fabrication ou le reconditionnement et le réétiquetage en vue de la distribution des matières premières à usage pharmaceutique respecte les bonnes pratiques prévues à l'article L. 5138-3, elle délivre un certificat de conformité. |
|
Ce droit est recouvré selon les modalités prévues pour le recouvrement des créances des établissements publics administratifs de l'Etat. |
« Tout établissement réalisant une des activités mentionnées au premier alinéa peut demander à l'Agence de certifier qu'il respecte ces bonnes pratiques. |
|
« Le modèle du certificat de conformité est établi par l'Agence française de sécurité sanitaire des produits de santé. » |
||
II. - Après l'article L. 5138-4 du code de la santé publique, il est inséré un article L. 5138-5 ainsi rédigé : |
II. - Alinéa sans modification | |
« Art. L. 5138-5. - Toute inspection, d'office ou sur demande, par l'Agence française de sécurité sanitaire des produits de santé, des établissements réalisant les activités énoncées à l'article L. 5138-4 en vue de vérifier et, le cas échéant, de certifier le respect des bonnes pratiques mentionnées à l'article L. 5138-3, donne lieu au versement d'un droit fixe au profit de l'Agence dont le montant est fixé par décret dans la limite de 10 000 €. Ce droit fixe se compose d'une part forfaitaire ne pouvant excéder 2 000 € et d'une part variable tenant compte des différences de situation entre les établissements. |
« Art. L. 5138-5. - Toute ... ... limite de 7 500 €. Ce ... ... établissements. Amendement n° 7 | |
« Ce droit est recouvré selon les modalités prévues pour le recouvrement des créances des établissements publics administratifs de l'État. » |
Alinéa sans modification | |
Article 26 |
Article 26 | |
Art. L. 5311-1. - L'Agence française de sécurité sanitaire des produits de santé est un établissement public de l'Etat, placé sous la tutelle du ministre chargé de la santé. ........................................ |
À l'article L. 5311-1 du code de la santé publique, après les mots : « Elle rend publique une synthèse des dossiers d'autorisation de tout nouveau médicament », sont ajoutés les mots : « dans des conditions déterminées par voie réglementaire ». |
La première phrase du vingt et unième alinéa de l'article L. 5311-1 du code de la santé publique est remplacée par deux phrases ainsi rédigées : |
Elle rend publique une synthèse des dossiers d'autorisation de tout nouveau médicament. Elle organise des réunions régulières d'information avec des associations agréées de personnes malades et d'usagers du système de santé mentionnées à l'article L. 1114-1 sur les problèmes de sécurité sanitaire des produits de santé. |
« L'agence rend accessible les compte rendus et l'ordre du jour des réunions de ses commissions et conseils. Elle rend également accessible ses avis et décisions. » Amendement n° 8 | |
Article 27 |
Article 27 | |
Art. L. 5311-2. - En vue de l'accomplissement de ses missions, l'agence : ....................................... |
À l'article L. 5311-2 du code de la santé publique, le 5° est remplacé par les dispositions suivantes: |
Sans modification |
5º Est chargé du fonctionnement de la commission de la transparence. |
« 5° Pour la mise en œuvre du 1° au 4°, demande, à des fins d'analyse et pour des raisons justifiées, la transmission à titre gratuit d'échantillons de produits et objets mentionnés à l'article L. 5311-1. » |
|
Article 28 |
Article 28 | |
Art. L. 5323-4. - Les agents contractuels mentionnés à l'article L. 5323-2 et L. 5323-3 : ........................................ |
À l'article L. 5323-4 du code de la santé publique, le septième alinéa est remplacé par les dispositions suivantes : |
Sans modification |
Les personnes mentionnées aux deux alinéas précédents adressent au directeur général de l'agence, à l'occasion de leur nomination ou de leur entrée en fonctions, une déclaration mentionnant leurs liens, directs ou indirects, avec les entreprises ou établissements dont les produits entrent dans son champ de compétence, ainsi qu'avec les sociétés ou organismes de conseil intervenant dans ces secteurs. Cette déclaration est rendue publique et est actualisée à leur initiative dès qu'une modification intervient concernant ces liens ou que de nouveaux liens sont noués. |
« Les personnes mentionnées aux articles L. 5323-1, L. 5323-2 et L. 5323-3, ainsi que les personnes mentionnées aux deux alinéas précédents, adressent au directeur général, à l'occasion de leur nomination ou de leur entrée en fonctions, puis annuellement, une déclaration mentionnant leurs liens, directs ou indirects, avec les entreprises ou établissements dont les produits entrent dans le champ de compétence de l'Agence, ainsi qu'avec les sociétés ou organismes de conseil intervenant dans ces secteurs. Cette déclaration est actualisée à leur initiative dès qu'une modification intervient concernant ces liens ou que de nouveaux liens sont noués. La déclaration adressée par les personnes mentionnées aux deux alinéas précédents est rendue publique. » |
|
Chapitre II |
Chapitre II | |
Habilitation à prendre des ordonnances |
Habilitation à prendre des ordonnances | |
Article 29 |
Article 29 | |
I. - Dans les conditions prévues à l'article 38 de la Constitution, le Gouvernement est autorisé à prendre, par ordonnances, les dispositions relevant du domaine de la loi nécessaires à la transposition des directives ou de celles de leurs dispositions qui n'ont pas encore été transposées, ainsi que les mesures d'adaptation de la législation liées à cette transposition : |
I. - Non modifié | |
a) Directive 2002/98/CE du Parlement européen et du Conseil du 27 janvier 2003 établissant des normes de qualité et de sécurité pour la collecte, le contrôle, la transformation, la conservation et la distribution du sang humain, et des composants sanguins, et modifiant la directive 2001/83/CE ; |
||
b) Directive 2003/15/CE du Parlement européen et du Conseil du 27 février 2003 modifiant la directive 76/768/CEE du Conseil concernant le rapprochement des législations des États membres relatives aux produits cosmétiques ; |
||
c) Directive 2004/23/CE du Parlement européen et du Conseil du 31 mars 2004 relative à l'établissement de normes de qualité et de sécurité pour le don, l'obtention, le contrôle, la transformation, la conservation, le stockage et la distribution des tissus et cellules humains ; |
||
d) Directive 2004/24/CE du Parlement européen et du Conseil du 31 mars 2004 modifiant, en ce qui concerne les médicaments traditionnels à base de plantes, la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain ; |
||
e) Directive 2004/28/CE du Parlement européen et du Conseil du 31 mars 2004 modifiant la directive 2001/82/CE instituant un code communautaire relatif aux médicaments vétérinaires. |
||
II. - Dans les mêmes conditions, le Gouvernement est autorisé à prendre, par ordonnances, les mesures relevant du domaine de la loi requises : |
II. - Alinéa sans modification | |
1° Pour adapter au droit communautaire les dispositions du code de la santé publique relatives aux autorisations d'importation des médicaments à usage humain et celles du même code concernant les insecticides et acaricides destinés à l'homme, ainsi que celles définissant le régime juridique des aliments diététiques destinés à des fins médicales spéciales ; |
Alinéa sans modification | |
2° Pour harmoniser et compléter les dispositions pénales relatives aux produits mentionnés aux articles L. 5141-1 et L. 5311-1 du code de la santé publique ; |
Alinéa sans modification | |
3° Pour régir les actions d'accompagnement des patients soumis à des traitements médicamenteux, conduites par les établissements pharmaceutiques, et définir les conditions de leur contrôle par l'Agence française de sécurité sanitaire des produits de santé ; |
3° Pour fixer le régime juridique applicable aux actions d'accompagnement conduites par les établissements pharmaceutiques et visant des patients soumis à des traitements médicamenteux, et définir les conditions de leur autorisation préalable et de leur contrôle ... ... santé » Amendement n° 9 | |
4° Pour permettre aux agents mentionnés au 1° de l'article L. 215-1 du code de la consommation de recourir à l'Agence française de sécurité sanitaire des produits de santé dans l'exercice des pouvoirs d'enquête qui leur sont dévolus en application de l'article L. 5414-1 du code de la santé publique. |
Alinéa sans modification | |
III. - Les ordonnances prévues par le présent article sont prises dans le délai de huit mois suivant la publication de la présente loi. |
III. - Non modifié | |
Le projet de loi de ratification de chacune des ordonnances prévues par le présent article est déposé devant le Parlement au plus tard le dernier jour du deuxième mois à compter de la publication de cette ordonnance. |
||
Article 30 |
Article 30 | |
I. - Dans les conditions prévues à l'article 38 de la Constitution, le Gouvernement est autorisé à prendre, par ordonnances, les mesures nécessaires à l'extension et à l'adaptation des dispositions du chapitre Ier de la présente loi, ainsi que de celles des ordonnances prises en application de son article 29, à Mayotte, à Saint-Pierre-et-Miquelon, aux Terres australes et antarctiques françaises, aux îles Wallis et Futuna et, en tant qu'elles relèvent des compétences de l'État, en Nouvelle-Calédonie et en Polynésie française. |
Sans modification | |
II. - Les projets d'ordonnance sont soumis pour avis : |
||
1° Lorsque leurs dispositions sont relatives à Mayotte, au conseil général de Mayotte dans les conditions prévues à l'article L. 3551-12 du même code ; |
||
2° Lorsque leurs dispositions sont relatives à Saint-Pierre-et-Miquelon, au conseil général de Saint-Pierre-et-Miquelon dans les conditions prévues à l'article 28 de la loi n° 85-595 du 11 juin 1985 relative au statut de l'archipel de Saint-Pierre-et-Miquelon ; |
||
3° Lorsque leurs dispositions sont relatives aux Terres australes et antarctiques françaises, au conseil consultatif ; |
||
4° Lorsque leurs dispositions sont relatives à la Nouvelle-Calédonie, à l'institution compétente dans les conditions définies par la loi organique n° 99-209 du 19 mars 1999 relative à la Nouvelle-Calédonie ; |
||
5° Lorsque leurs dispositions sont relatives à la Polynésie française, à l'institution compétente dans les conditions définies par la loi organique n° 2004-192 du 27 février 2004 portant statut d'autonomie de la Polynésie française ; |
||
6° Lorsque leurs dispositions sont relatives aux îles Wallis et Futuna, à l'assemblée territoriale des îles Wallis et Futuna. |
||
III. - Les ordonnances prévues par le présent article sont prises dans le délai de douze mois suivant la publication de la présente loi. |
||
Le projet de loi de ratification de chacune des ordonnances prévues par le présent article est déposé devant le Parlement au plus tard le dernier jour du troisième mois à compter de la publication de cette ordonnance. |
AMENDEMENTS NON ADOPTÉS PAR LA COMMISSION
Article 2
Amendements présentés par M. Claude Evin :
· Supprimer cet article.
· Rédiger ainsi cet article :
« Au troisième alinéa de l'article L. 4113-6 du code de la santé publique, les mots : « d'un niveau raisonnable, reste accessoire par rapport à l'objectif principal de la réunion » sont remplacés par les mots : « d'un niveau raisonnable et limitée à l'objectif principal de la manifestation ».
(devenu sans objet)
Article 4
Amendement présenté par M. Claude Evin :
Dans la dernière phrase de l'alinéa 3 de cet article, substituer aux mots : « présentent des propriétés sensiblement différentes », les mots : « ont démontré des effets différents ».
Article 5
Amendements présentés par Mme Jacqueline Fraysse :
· Compléter l'alinéa 6 de cet article les deux phrases suivantes :
« L'attribution de l'autorisation de mise sur la marché engage l'entreprise à procéder à l'examen de sa spécialité en vue de son inscription sur la liste des spécialités remboursables visée à l'article L. 162-17 du code de la sécurité sociale. Si l'efficacité thérapeutique est attestée le Ministre chargé de la santé et de la sécurité sociale arrête l'inscription du médicament sur cette liste. »
· Après l'alinéa 6 de ce article, insérer l'alinéa suivant :
« Pour se voir délivrer l'autorisation, le demandeur doit obligatoirement présenter dans son dossier des essais cliniques comparatifs démontrant les avantages, les risques, les contraintes et l'efficacité de sa spécialité évaluée par comparaison avec les meilleures méthodes diagnostiques, thérapeutiques ou de prévention en usage. »
· Après l'alinéa 6 de ce article, insérer l'alinéa suivant :
« Tout dosage, forme pharmaceutique, voie d'administration et présentation supplémentaire, ainsi que toute modification et extension, doivent également obtenir une autorisation ou être inclus dans l'autorisation de mise sur le marché initiale. »
Après l'article 5
Amendements présentés par Mme Jacqueline Fraysse :
· Après l'article L. 5121-8 du code de la santé publique, il est inséré un article L. 5121-8-1 ainsi rédigé :
« Art. L. 5121-8-1. - L'impact environnemental de toute spécialité pharmaceutique faisant l'objet d'une demande d'autorisation de mise sur le marché, conformément à l'article L. 5121-8, est étudié et, au cas par cas, des dispositions particulières visant à le limiter sont envisagées. »
· I. Est créé avant le livre 1er de la 5ème partie du code de la santé publique un livre 1er rédigé comme suit :
« Livre Ier
« Conseil national du médicament
« Titre Ier
« Missions du Conseil national du médicament
« Art. L. 5110-1. - En collaboration avec le Haut Conseil de Santé publique, la mission du Conseil national du médicament consiste à fixer des objectifs de santé publique à l'ensemble de l'industrie pharmaceutique.
« Il est chargé, en collaboration avec l'ensemble des instances publiques missionnées à cet effet, de l'élaboration de toutes recommandations et propositions de réformes qui lui paraissent de nature à faciliter la mise en œuvre des objectifs précités.
« Les administrations de l'Etat, les établissements publics de l'Etat et les organismes chargés de la gestion des régimes obligatoires et complémentaires de l'assurance maladie sont tenus de communiquer au Conseil les éléments d'information et les études dont ils disposent pour l'exercice de ses missions. Le Conseil fait connaître ses besoins d'informations statistiques pour qu'ils soient pris en compte dans les programmes des travaux statistiques et d'études de ces administrations, organismes et établissements.
« Art. L. 5110-2. - Le Conseil national élabore un rapport annuel sur :
« 1° L'adéquation entre les besoins de santé et la production pharmaceutique
« 2° L'état de la recherche pharmaceutique et préconiser des orientations de recherche
« 3° Ses préconisations en matière d'aide financière publique à la recherche sélective en fonction de la satisfaction effective des besoins de santé
« Art. L. 5110-3. - En vue de l'accomplissement de ses missions, le Conseil :
« 1° Procède ou fait procéder à toutes les expertises médicales, sanitaires, sociales et économiques qu'il jugera utile d'engager.
« 2° Recueille toutes les données scientifiques et techniques nécessaires à ces missions
« 3° Participe à l'action européenne et internationale de la France
« Titre II
« Organisation du Conseil national du médicament
« Art. L. 5110-4. - Le Conseil national du médicament est composé d'un représentant de chaque groupe parlementaire de l'assemblée nationale et du sénat, de six représentants des directions et des salariés des laboratoires pharmaceutiques, de six représentants des chercheurs du secteur public et du secteur privé, d'un représentant de chaque syndicat représentatif des professionnels de santé, d'un représentant du Conseil de l'Ordre des pharmaciens, de trois personnalités qualifiées issues des conseils d'administration de la CNAMTS, de la MSA et de la CANAM, de trois représentants d'associations d'usagers des services de santé, et de trois personnalités qualifiées mandatées par la Conférence nationale de santé.
« Art. L. 5110-5. - Les ressources du Conseil sont constituées notamment par :
« 1° Un relèvement de 1 % de la contribution sociale de solidarité sur les bénéfices du secteur de l'industrie, des laboratoires et des officines pharmaceutiques
« 2° Des produits divers, dons et legs
« 3° Des taxes nouvelles sur les produits de l'alcool, du tabac, de l'industrie automobile, de l'industrie agro-alimentaire prévues à son bénéfice
« II. Les modalités d'application du présent livre sont déterminées par décret en Conseil d'Etat. »
· Après le premier alinéa de l'article L. 162-17 du code de la sécurité sociale, il est inséré un alinéa ainsi rédigé :
« Si les médicaments visés au précédent alinéa présentent un service médical rendu insuffisant ou nul déterminé selon des critères fixés par décret et après avis des commissions compétentes, l'autorisation de mise sur le marché leur est retirée et le ministre chargé de la santé et de la sécurité sociale procède au retrait du médicament sur la liste visée au premier alinéa. »
Article 7
Amendement présenté par Mme Jacqueline Fraysse :
Après les mots : « raisons de santé publique », rédiger ainsi la fin de l'alinéa 2 de cet article :
« justifiées et rendues publiques, autoriser la mise sur le marché provisoire de ce médicament pour une durée ne pouvant excéder un an, dans des conditions prévues par voie réglementaire. »
Article 9
Amendements présentés par Mme Jacqueline Fraysse :
· Dans la dernière phrase de l'alinéa 2 de cet article, après les mots : « un avantage clinique important », insérer les mots : « et à valeur thérapeutique ajoutée ».
· Compléter l'alinéa 2 de cet article par les mots : « démontré par des études comparatives indépendantes ».
Amendement présenté par M. Claude Evin :
Compléter cet article par l'alinéa suivant :
« La dernière phrase du troisième alinéa de l'article L. 5121-10 du code de la santé publique est supprimée. »
(retiré en commission)
Article 15
Amendements présentés par Mme Jacqueline Fraysse :
· Dans l'alinéa 7 de cet article, après les mots : « relatives à l'étiquetage », insérer les mots :
« les règles concernant la traduction du nom du médicament en braille sur l'emballage et l'obligation pour le détenteur de l'autorisation de mise sur le marché de veiller à ce que la notice d'information soit disponible, sur demande, dans des formats appropriés pour les aveugles et les malvoyants, ».
· Dans l'alinéa 9 de cet article, après les mots : « , le contenu du dossier présenté à l'appui de ces demandes, », insérer les mots :
« les modalités d'évaluation de l'impact environnemental des spécialités pharmaceutiques faisant l'objet de ces demandes, ».
· Après l'alinéa 18 de cet article, insérer l'alinéa suivant :
« comme l'obligation pour le titulaire de l'autorisation de mise sur la marché de conserver des rapports détaillés de tous les effets indésirables présumés survenus partout ou sa spécialité est exploitée, son obligation d'enregistrer toute présomption d'effet indésirable grave ayant été portée à son attention par un professionnel de santé et de la notifier aussitôt à l'autorité compétente de l'État membre sur le territoire duquel l'incident s'est produit, et au plus tard dans les quinze jours suivant la réception de l'information à l'ensemble des Etats membres, son obligation de veiller à ce que toute présomption d'effet indésirable grave et inattendu ainsi que toute présomption de transmission d'agents infectieux par un médicament sur le territoire d'un pays soient aussitôt l'Agence Européenne du médicament et les autorités compétentes des États membres dans lesquels le médicament est autorisé en soient informées, et au plus tard dans les quinze jours suivant la réception de l'information, ».
Article 22
Amendement présenté par M. Bernard Depierre :
Dans l'alinéa 3 de cet article, après les mots :
« Lorsqu'un particulier procède à l'importation d'un médicament par une autre voie que le transport personnel, »,
insérer les mots :
« y compris par l'intermédiaire d'une pharmacie, ».
Après l'article 26
Amendement présenté par Mme Jacqueline Fraysse :
L'article L. 5311-1 du code de la santé publique est complété par un alinéa ainsi rédigé :
« Elle rend public l'ordre du jour de ses réunions, les comptes rendus de ses réunions, assortis des décisions prises, des détails des votes et des explications de vote, y compris les opinions minoritaires. »
Article 29
Amendement présenté par Mme Jacqueline Fraysse :
Supprimer cet article.
Article 30
Amendement présenté par Mme Jacqueline Fraysse :
Supprimer cet article.
¬ Association des médicaments dérivés du sang et analogues (ADMSA) -M. Jérôme Erath, président, et Mme Bénédicte Garbil, consultante
¬ Agence française de sécurité sanitaire des produits de santé (AFSSAPS) -M. Jean Marimbert, directeur général, et Mme Elisabeth Hérail, chef du service des affaires juridiques et européennes
¬ Conseil national de l'ordre des pharmaciens - M. Jean Parrot, président, et Mme Isabelle Adenot, présidente du conseil central de la section A (pharmaciens titulaires d'officine)
¬ Direction générale de la santé (DGS) - M. Didier Houssin, directeur général, et Mme Hélène Sainte-Marie, sous-directrice de la politique des produits de la santé
¬ Association Générique même médicament (GEMME) - M. Pascal Brière, président du laboratoire Biogaran, Mme Isabelle Saint-Jean, directrice des affaires pharmaceutiques du laboratoire Ratiopharm, et M. Alain Djiane, directeur du développement du laboratoire Sandoz
¬ Laboratoire Servier - M. Pierre Montès, directeur des affaires pharmaceutiques France, Mme Corinne Moizan, directrice chargée des relations avec le Parlement
¬ La Mutualité française - M. Daniel Lenoir, directeur général, Mme Laure Lechertier, responsable du département politique du médicament, et M. Vincent Figureau, responsable du département relations extérieures
¬ Les entreprises du médicament (LEEM) - Mme Blandine Fauran, directrice juridique et fiscale, Mme Anne-Priscille Vlasto, chargée de mission, Mme Sophie Leroy, chargée de mission, et Mme Aline Bessis-Marais, responsable des affaires publiques
¬ Revue Prescrire - Mme Danielle Bardelay, pharmacienne, rédactrice de la rubrique « nouveau médicament » (fondatrice de la revue)
¬ Union fédérale des consommateurs (UFC) - Que choisir - M. Jacques Mopin, vice-président, et M. Alexandre Biosse Duplan, chargé de mission santé
SCHÉMA DE LA PROCÉDURE DE CODÉCISION SUIVIE POUR LA RÉVISION DE LA DIRECTIVE N° 2001/83/CE INSTITUANT UN CODE COMMUNAUTAIRE RELATIF AUX MÉDICAMENTS À USAGE HUMAIN
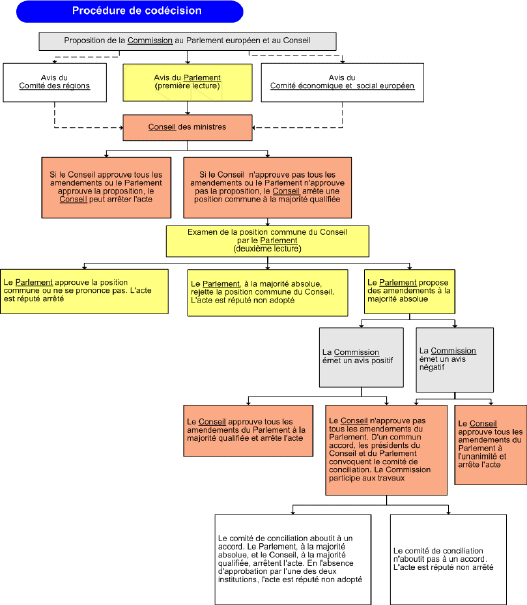
Source : Commission européenne
-------
N° 3238 - Rapport fait par Mme Cécile Gallez au nom de la commission des affaires culturelles sur le projet de loi (n°3062) portant diverses dispositions d'adaptation au droit communautaire dans le domaine du médicament
1 () Rapport de la mission d'information n° 382 de Mme Marie-Thérèse Hermange, au nom de la commission des affaires sociales du Sénat, sur les conditions de mise sur le marché et de suivi des médicaments, « Restaurer la confiance » (juin 2006).
2 () Aux termes du quatrième considérant de la directive n° 2004/27.
3 () Directive n° 2004/27/CE du 31 mars 2004 du Parlement européen et du Conseil modifiant la directive n° 2001/83/CE du 23 octobre 2001 instituant un code communautaire relatif aux médicaments à usage humain. Par simplification, cette directive sera ultérieurement dénommée : « la directive n° 2004/27 ».
4 () « Europe et médicament », revue Prescrire n° 252 (juillet-août 2004).
5 () Dans sa décision n° 2004-496 DC du 10 juin 2004 relative à la loi pour la confiance dans l'économie numérique, le Conseil constitutionnel a ainsi rappelé que « la transposition en droit interne d'une directive communautaire résulte d'une exigence constitutionnelle à laquelle il ne pourrait être fait obstacle qu'en raison d'une disposition expresse contraire de la Constitution ».
6 () Créé par la décision n° 75/320/CEE du Conseil du 20 mai 1975, le comité a notamment pour mission d'examiner toute question relative à l'application des directives concernant les spécialités pharmaceutiques. Il est composé de représentants des États membres et est présidé par un représentant de la Commission.
7 () Directive 2004/24/CE du Parlement européen et du Conseil du 31 mars 2004 modifiant, en ce qui concerne les médicaments traditionnels à base de plantes, la directive 2001/83/CE.
8 () Directive 2004/28/CE du Parlement européen et du Conseil du 31 mars 2004 modifiant la directive 2001/82/CE instituant un code communautaire relatif aux médicaments vétérinaires.
9 () Directive 2002/98/CE du Parlement européen et du Conseil du 27 janvier 2003 établissant des normes de qualité et de sécurité pour la collecte, le contrôle, la transformation, la conservation et la distribution du sang humain, et des composants sanguins, et modifiant la directive 2001/83/CE.
10 () Décret n° 2005-156 du 18 février 2005 relatif aux modifications d'autorisation de mise sur le marché de médicaments à usage humain et modifiant le code de la santé publique.
11 () En référence à un contentieux judiciaire américain (Bolar versus Roche) ayant conduit, en 1984, à l'adoption d'une réforme législative du statut des génériques.
12 () Cf. sur ce point, le rapport n° 1092 de M. Jean-Michel Dubernard au nom de la commission des affaires culturelles, familiales et sociales sur le projet de loi relatif à la politique de santé publique.
13 () Rapport général de la Commission européenne sur l'expérience acquise dans le fonctionnement des procédures communautaires d'autorisation de mise sur le marché des médicaments (COM 2001-606 final) du 23 octobre 2001.
14 () « Publicité directe aux consommateurs des médicaments d'ordonnance au Canada », conseil canadien de la santé, janvier 2006
15 () A propos du Vioxx, le rapport « Publicité directe aux consommateurs des médicaments d'ordonnance au Canada » du conseil canadien de la santé (janvier 2006) note que « dans les cinq ans où ce médicament s'est trouvé sur le marché, Merck a dépensé plus de 500 millions de dollars américains à en faire la publicité auprès du public américain. En l'an 2000, les dépenses de promotion populaire du Vioxx ont dépassé les dépenses de publicité de Pepsi-Cola. »
16 () Par simplification, cette directive sera ultérieurement dénommée : « la directive n° 2001/83 » ou encore « le code communautaire ».
17 () Par simplification, cette directive sera ultérieurement dénommée : « la directive n° 2004/27 ».
18 () Rapport de M. Jean-Pierre Door au nom de la mission d'information sur la grippe aviaire : mesures préventives, « Menace de pandémie grippale : préparer les moyens médicaux » (tome I), publié le 26 janvier 2006.
19 () Directive n° 85/374/CEE du Conseil du 25 juillet 1985 modifiée relative au rapprochement des dispositions législatives, réglementaires et administratives des États membres en matière de responsabilité du fait des produits défectueux.
20 () Rapport n° COM (2001) 404 - C5-0592/2001 - 2001/0253(COD).
21 () Rapport sur le fonctionnement des procédures communautaires d'autorisation de mise sur le marché des médicaments (COM 2001-606 final), publié par la Commission européenne, le 23 octobre 2001.
22 () Le mot « physiologique » est défini par le dictionnaire Robert comme : « ce qui concerne le fonctionnement d'un organisme vivant, d'un organe, d'une cellule ou les activités de l'organisme humain, se traduisant par des modifications physico-chimiques ».
23 () Arrêté du 23 avril 2004 fixant les normes et protocoles applicables aux essais analytiques, toxicologiques et pharmacologiques ainsi qu'à la documentation clinique auxquels sont soumis les médicaments ou produits mentionnés à l'article L. 5121-8 du code de la santé publique.
24 () Décret n° 2005-156 du 18 février 2005 relatif aux modifications d'autorisation de mise sur le marché de médicaments à usage humain et modifiant le code de la santé publique.
25 () Règlement (CE) n° 726/2004 du Parlement européen et du Conseil du 31 mars 2004 établissant des procédures communautaires pour l'autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant une Agence européenne des médicaments.
26 () Position commune arrêtée par le Conseil de l'Union européenne le 29 septembre 2003 en vue de l'adoption de la directive modifiant la directive n° 2001/83.
27 () Communication de la Commission au Parlement européen, conformément à l'article 251, paragraphe 2 du traité CE, concernant la position commune arrêtée par le Conseil en vue de l'adoption d'une directive du Parlement européen et du Conseil modifiant la directive n° 2001/83(SEC/2003/1082 final), 7 octobre 2003.
28 () Le Conseil d'État, saisi d'une demande d'annulation d'une décision d'inscription au répertoire des groupes génériques d'une spécialité en tant que générique d'un princeps, a en effet rappelé que l'identité d'indications thérapeutiques n'était pas au nombre des conditions d'identification des spécialités génériques (Conseil d'État, 23 juillet 2003, n° 246716, Société Lilly France).
29 () Loi n° 2003-1199 du 18 décembre 2003 de financement de la sécurité sociale pour 2004.
30 () Conseil d'État, arrêt d'Assemblée du 21 novembre 1958, Syndicat national des transporteurs aériens.
31 () Voir notamment sur ce point, les décisions du Conseil constitutionnel n° 69-57 L du 24 octobre 1969 et n°77-100 L du 16 novembre 1977.
32 () Loi organique n° 2001-692 du 1er août 2001 relative aux lois de finances.
33 () En référence à un contentieux judiciaire américain (Bolar versus Roche) ayant conduit, en 1984, à l'adoption d'une réforme législative du statut des génériques.
34 () Jugements du 12 octobre 2001 (Science Union Servier/AJC Pharma et Expanpharm) et du 25 janvier 2005 (Science Union et Servier/Biophelia).
35 () Rapport sur le fonctionnement des procédures communautaires d'autorisation de mise sur le marché des médicaments (COM 2001-606 final) du 23 octobre 2001.
36 () Position commune (CE) n° 61/2003 du 29 septembre 2003 arrêtée par le Conseil, statuant conformément à la procédure visée à l'article 251 du traité instituant la Communauté européenne, en vue de l'adoption d'une directive du Parlement européen et du Conseil modifiant la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain.
© Assemblée nationale