
________________________________________________________________________________________________________________
Enregistré à la Présidence de l'Assemblée nationale le 30 avril 2008.
RAPPORT D’INFORMATION
DÉPOSÉ
en application de l’article 145 du Règlement
PAR LA COMMISSION DES AFFAIRES CULTURELLES, FAMILIALES ET SOCIALES
en conclusion des travaux de la mission d’évaluation et de contrôle
des lois de financement de la sécurité sociale
sur
la prescription, la consommation et la fiscalité des médicaments
ET PRÉSENTÉ
PAR Mme Catherine LEMORTON,
Députée.
——
INTRODUCTION 9
I.- LA FRANCE SE CARACTÉRISE PAR UNE CONSOMMATION RECORD DE MÉDICAMENTS 11
A. LA FRANCE EST LE PREMIER PAYS CONSOMMATEUR DE MÉDICAMENTS EN EUROPE 11
1. Les Français détiennent le record européen de la consommation de médicaments 11
a) Ce constat est largement partagé 11
b) La France détient toujours le record en termes de quantité et de dépense de médicaments par habitant 12
c) Mais dans certaines classes de médicaments, les écarts de consommation avec les principaux pays européens ont tendance à se réduire 13
2. La consommation de médicaments est fortement concentrée sur les personnes relevant d’une affection de longue durée 15
a) Chaque Français consomme, en moyenne, une boîte de médicaments par semaine 15
b) Mais les affections de longue durée (ALD) concentrent la moitié de la consommation de médicaments 15
c) Au sein des ALD, la consommation est concentrée sur les affections qui nécessitent un traitement pharmaceutique innovant et coûteux 16
d) La durée de traitement des personnes relevant d’une ALD a tendance à s’allonger 16
e) L’augmentation des dépenses de médicaments pour les personnes relevant d’une ALD constitue l’essentiel de la croissance de la dépense totale de médicaments 16
3. Près d’un quart de la consommation de médicaments en ville résulte de prescriptions hospitalières et cette part devrait continuer de croître 17
a) La rétrocession hospitalière diminue mais la prescription en activité ambulatoire se développe 18
b) La prescription hospitalière exerce un effet d’entraînement sur la prescription en ville 18
c) Les effets de la sortie de la réserve hospitalière : l’exemple de l’érythropoïétine (EPO) 19
4. Les médecins de ville français sont de gros prescripteurs de médicaments 20
a) Les médecins français prescrivent beaucoup 20
b) La prescription est davantage orientée vers les spécialités récentes et comporte peu de génériques 21
c) Les médicaments prescrits ne sont pas toujours efficaces et la qualité des prescriptions n’est pas toujours optimale 22
B. LA CONSOMMATION ÉLEVÉE DE MÉDICAMENTS EST COÛTEUSE ET PEUT ENTRAÎNER CERTAINES CONSÉQUENCES SANITAIRES REGRETTABLES 22
1. Les dépenses de médicaments sont en forte croissance 22
a) Les dépenses de médicaments représentent une part importante du PIB 23
b) La part des dépenses de santé que consacre la France aux dépenses de médicaments est plus importante que dans la plupart des autres pays 23
c) Le rythme de croissance des dépenses de médicaments est élevé 24
d) Les nouveaux produits « tirent » la croissance du marché 25
e) Une boîte de médicaments vendue sur cinq est un produit générique 25
f) La tendance à l’augmentation des dépenses de médicaments devrait se poursuivre 27
2. Les médicaments remboursés représentent la quasi-totalité de la consommation et l’automédication est peu développée 27
a) 90 % des médicaments consommés font l’objet d’une prise en charge par l’assurance maladie 28
b) Il est encore trop tôt pour évaluer les effets de la franchise sur la consommation des médicaments 29
c) Les médicaments consommés sont, pour l’essentiel, soumis à prescription médicale obligatoire 31
d) L’automédication occupe une place limitée et stagne 32
e) Les effets des changements dans les conditions de remboursement sont différents selon qu’il s’agit d’une diminution du taux de remboursement ou d’un déremboursement 34
3. Le fort recours aux médicaments n’est pas toujours justifié et peut entraîner des effets sanitaires néfastes 35
a) La surconsommation médicamenteuse française n’apparaît pas pleinement justifiée au regard des indicateurs de morbi-mortalité 35
b) La surconsommation de médicaments peut avoir des effets négatifs en termes de santé publique 36
II.- AMÉLIORER L’ENCADREMENT DE LA VIE DU MÉDICAMENT ET RENFORCER LA SÉLECTIVITÉ DE L’ACCÈS AU REMBOURSEMENT 37
A. AMÉLIORER L’ENCADREMENT DE LA VIE DU MÉDICAMENT 37
1. Le circuit administratif du médicament est complexe 37
a) Le chemin est long, du fabricant au consommateur 37
b) En France, la régulation du médicament est répartie entre plusieurs acteurs 39
c) D’autres pays ont un système de régulation plus regroupé 39
2. Veiller au bon usage des procédures dérogatoires de mise sur le marché et de prescription 40
a) Veiller au bon usage de l’autorisation temporaire d’utilisation 40
b) Contrôler et évaluer les prescriptions hors autorisations de mise sur le marché (AMM) 41
B. RENFORCER LA SÉLECTIVITÉ DE L’ACCÈS AU REMBOURSEMENT 42
1. Renforcer les règles de l’admission au remboursement et de fixation du prix 42
a) Recourir au critère de l’intérêt de santé publique pour l’appréciation du service médical rendu (SMR) 42
b) Développer les essais cliniques contre comparateurs pour l’appréciation de l’amélioration du service médical rendu (ASMR) 43
c) Veiller à la mise en œuvre rapide de la nouvelle compétence médico-économique de la Haute Autorité de santé (HAS) 43
2. Améliorer le suivi des médicaments en pratique médicale réelle 45
a) Renforcer l’efficacité de la pharmacovigilance 45
b) Développer l’évaluation post-autorisation de mise sur le marché 46
c) Gérer de manière plus active la liste des médicaments remboursables 47
III.- FAIRE ÉVOLUER LES COMPORTEMENTS DES PRESCRIPTEURS ET DES CONSOMMATEURS 49
A. RENFORCER L’INFORMATION SUR LE MÉDICAMENT 49
1. Assurer l’indépendance et la transparence de l’expertise 49
2. Mettre en place une base publique d’information, exhaustive et gratuite, sur le médicament 50
B. AGIR SUR LES DÉTERMINANTS DE LA PRESCRIPTION 51
1. Améliorer la formation des médecins en pharmacologie et en économie de la santé 51
a) Réformer la formation initiale des médecins sur le médicament 51
b) Veiller à la montée en charge de la formation professionnelle continue et de l’évaluation des pratiques professionnelles 52
2. Rééquilibrer l’information des médecins sur le médicament 54
a) Maîtriser l’impact de la visite médicale 54
b) Développer l’information publique 56
c) Diffuser les logiciels certifiés d’aide à la prescription 58
3. Renforcer les actions de maîtrise médicalisée des dépenses de médicaments de l’assurance maladie 59
a) Développer l’analyse des prescriptions 59
b) Renforcer l’efficacité de la maîtrise médicalisée des dépenses de santé 60
c) Amplifier la communication de l’assurance maladie 62
d) Poursuivre la montée en charge des actions individuelles en direction des médecins 63
C. FAVORISER LE BON USAGE CHEZ LES CONSOMMATEURS 65
1. Développer et coordonner l’information du grand public sur les médicaments 65
a) Dissiper le malentendu entre les médecins et les patients sur la demande de médicaments 65
b) Renforcer l’information publique en direction des patients 66
2. Inciter à l’observance, encadrer les programmes d’accompagnement des patients et développer l’éducation thérapeutique 67
a) L’observance des traitements est un enjeu majeur 67
b) Encadrer strictement les programmes d’accompagnement des patients financés par l’industrie pharmaceutique 68
c) Favoriser le développement de programmes d’accompagnement des patients par l’assurance maladie et l’éducation thérapeutique des patients 69
d) Promouvoir sur internet l’information de qualité sur les médicaments 70
IV.- S’APPUYER SUR LE RÉSEAU DES PHARMACIES D’OFFICINE POUR PROMOUVOIR LES GÉNÉRIQUES ET DÉVELOPPER UNE AUTOMÉDICATION RESPONSABLE 73
A. DÉVELOPPER LE RÔLE DE CONSEIL DES PHARMACIENS D’OFFICINE 73
a) Le pharmacien d’officine de proximité doit pouvoir s’appuyer sur le dossier pharmaceutique pour assurer la sécurité de la dispensation 73
b) Le rôle des pharmaciens d’officine dans le conseil et l’accompagnement des patients doit être développé 74
B. PROMOUVOIR LES GÉNÉRIQUES 75
1. Contrer les stratégies de contournement des génériques 76
a) La France n’a comblé qu’une partie de son retard en matière de médicaments génériques 76
b) Les moyens de lutter contre les stratégies de contournement des génériques doivent être renforcés 77
2. Accroître la délivrance de génériques 78
a) Renforcer les actions conventionnelles avec les médecins et promouvoir la prescription en dénomination commune 78
b) Poursuivre la mobilisation des pharmaciens sur le droit de substitution et l’effort d’information des assurés 79
C. DÉVELOPPER UNE AUTOMÉDICATION RESPONSABLE 80
1. Définir l’automédication 80
a) Le droit français ne définit pas expressément l’automédication 80
b) L’automédication pourrait être définie comme un comportement 81
2. Maîtriser la mise devant le comptoir des officines de certains médicaments à prescription médicale facultative 82
a) Les conditions de mise en place sont en cours de définition 82
b) L’intérêt de la démarche n’est pas toujours bien compris et les conditions de mise en œuvre suscitent des inquiétudes 83
V.- FAVORISER LA MISE EN PLACE D’UNE FISCALITÉ PLUS SIMPLE ET STRUCTURANTE 85
A. VEILLER À L’INDÉPENDANCE DES ORGANISMES QUI PERÇOIVENT LES TAXES RÉMUNÉRANT DES SERVICES RENDUS 85
1. La Commission de la transparence est financée en quasi-totalité par une taxe payée par les laboratoires pharmaceutiques 85
2. Plus de la moitié du financement de l’AFSSAPS est assuré par cinq taxes payées par les laboratoires pharmaceutiques 86
3. Le mode de financement des organismes qui délivrent les autorisations et évaluent les médicaments doit permettre d’assurer leur indépendance 86
B. SIMPLIFIER, STABILISER OU RENDRE PLUS STRUCTURANTE LES TAXES AFFECTÉES À L’ASSURANCE MALADIE 87
1. Aménager les deux taxes destinées à maîtriser la dépense de médicaments remboursés par l’assurance maladie 88
a) Mobiliser la taxe sur les dépenses de promotion pour améliorer la régulation de la visite médicale 88
b) Simplifier la contribution à la clause de sauvegarde de l’objectif national de dépenses d’assurance maladie (ONDAM) pour la rendre effectivement applicable 88
2. Stabiliser les deux autres taxes destinées à procurer des ressources à l’assurance maladie 90
a) Maintenir la taxe sur les grossistes répartiteurs 90
b) Stabiliser la taxe sur le chiffre d’affaires 90
3. Assurer le recouvrement de la TVA sur les médicaments et évaluer les effets des déremboursements sur les recettes de taxes 91
CONCLUSION 93
LISTE DES PROPOSITIONS 94
TRAVAUX DE LA COMMISSION 105
ANNEXES 109
ANNEXE 1 : Composition de la mission 109
ANNEXE 2 : Liste des personnes auditionnées 111
ANNEXE 3 : Comptes rendus des auditions 115
ANNEXE 4 : Glossaire 397
ANNEXE 5 : Communication de la Cour des comptes concernant les taxes sur le mėdicament humain 401
ANNEXE 6 : Communication de la Cour des comptes concernant la consommation et la prescription des médicaments 431
La commission des affaires culturelles, familiales et sociales a demandé à la Mission d’évaluation et de contrôle des lois de financement de la sécurité sociale (MECSS) de consacrer son premier rapport de la législature à « la prescription, la consommation et la fiscalité des médicaments ». Cet intitulé traduit la volonté d’aborder le sujet du médicament selon une approche médico-économique équilibrée.
C’est une constante dans les travaux de la Mission que de prendre en compte à la fois les préoccupations de santé publique et les objectifs de maîtrise des finances sociales. La vocation de la MECSS est en effet de rechercher les voies et moyens de parvenir au plus haut niveau de protection sociale et de santé publique durablement soutenable et compatible avec les capacités de l’économie.
Dans ce cadre, la Mission a choisi de centrer son analyse sur la prescription et la consommation de médicaments en ville et sur la fiscalité spécifique applicable au secteur du médicament.
Au cours de ses travaux, la Mission a toujours veillé à éviter toute caricature ou stigmatisation des acteurs du médicament, qu’il s’agisse des laboratoires pharmaceutiques qui assurent la recherche, la fabrication et la fourniture des médicaments, des agences sanitaires et autorités de santé qui les régulent et formulent des recommandations, des médecins qui les prescrivent, des grossistes-répartiteurs qui les distribuent, des pharmaciens qui les dispensent, des patients qui les consomment, et de l’assurance maladie qui les rembourse. À ces acteurs, nombreux et aux intérêts multiples, complémentaires ou divergents, doit être ajouté l’État qui a pour mission d’assurer le pilotage du secteur du médicament dans le cadre fixé par le Parlement, notamment par les lois de financement de la sécurité sociale ainsi que celles relatives à la santé publique et à l’organisation des soins.
On comprendra que dans un tel contexte, il est difficile de définir les conditions permettant de concilier, d’une part, le bon usage des médicaments avec pour objectif de renforcer en permanence la qualité et l’efficience des soins et des prescriptions médicamenteuses et, d’autre part, le développement sur notre territoire du secteur économique stratégique que constituent les industries de santé, et tout particulièrement les laboratoires pharmaceutiques.
Tout au long de ses travaux, la MECSS a pu constater que les « mécanismes intimes » de la prescription et de la consommation des médicaments sont encore mal connus. Cela peut s’expliquer par le fait que les efforts de maîtrise médicalisée de la prescription et de la consommation de médicaments sont récents et que la recherche dans ce domaine est insuffisante.
Trois questions ont guidé les réflexions de la Mission :
– Le niveau de consommation de la France en médicaments est-il vraiment plus élevé que celui de pays voisins comparables, au point que l’on puisse parler d’une situation de surconsommation ?
– Comment promouvoir le bon usage des médicaments, faire évoluer les comportements des prescripteurs et des consommateurs ainsi que les stratégies thérapeutiques médicamenteuses et non médicamenteuses ?
– Comment améliorer la fiscalité du secteur du médicament ?
Pour tenter de répondre à ces questions complexes et formuler des propositions de réforme concrètes, opérationnelles et réalistes, la MECSS a bénéficié du concours de la Cour des comptes. Celle-ci a, en réponse à la demande qui lui avait été adressée lors de la législature précédente, remis deux communications à la Mission, la première concernant « les taxes sur le médicament humain », au mois de mai 2007, la seconde portant sur « la consommation et la prescription de médicaments », au mois de juillet 2007. Ces communications, dont il convient de remercier la Cour, sont publiées en annexe au présent rapport.
Par ailleurs, la Mission, avec la participation d’une magistrate de la Cour des comptes, a, durant quatre mois et demi, entendu, lors d’une trentaine de séances d’auditions publiques, soixante-quinze personnes représentant les principaux acteurs concernés par le médicament.
En outre, la MECSS a entendu des membres de l’Inspection générale des affaires sociales (IGAS) qui ont présenté deux rapports récents de leur administration concernant, d’une part, l’information des médecins sur le médicament, d’autre part, les programmes d’accompagnement des patients financés par l’industrie pharmaceutique.
La Mission a enfin entendu, le 12 février dernier, M. Éric Woerth, ministre du budget, des comptes publics et de la fonction publique, et Mme Roselyne Bachelot, ministre de la santé, de la jeunesse et des sports.
La Mission tient à remercier toutes les personnalités entendues dont la liste ainsi que les comptes rendus des auditions sont annexés au rapport.
La Mission a d’abord constaté que la France se caractérise par une consommation de médicaments record qui est la conséquence de comportements de prescription et de consommation difficiles à faire changer (I). La Mission formule ensuite dans les parties II à V de son rapport une série de propositions visant à développer un partenariat de santé favorisant le bon usage des médicaments.
*
I.- LA FRANCE SE CARACTÉRISE PAR UNE CONSOMMATION RECORD DE MÉDICAMENTS
Le médicament est l’élément le plus familier de notre consommation de soins. Chacun est, tout au long de sa vie, amené à consommer des médicaments, de manière plus ou moins contrainte. Mais le médicament est, encore aujourd’hui, souvent considéré comme le principal vecteur de la guérison. Le haut niveau de consommation français résulte de comportements de prescriptions et de consommation bien ancrés qu’il est difficile de faire changer.
A. LA FRANCE EST LE PREMIER PAYS CONSOMMATEUR DE MÉDICAMENTS EN EUROPE
Les comparaisons internationales sur la consommation de médicaments se sont développées depuis une vingtaine d’années. Dès les premières analyses, la situation de la France est apparue particulière, avec une dépense moyenne par habitant la situant au premier rang européen. La conjonction de forts volumes et du prix élevé des médicaments consommés, notamment dans les classes thérapeutiques majeures, ainsi que la faible propension à utiliser les réserves d’économie liées aux génériques semblent expliquer la situation particulière de la France au regard des autres pays européens.
Cette spécificité française continue à susciter des interrogations sur l’efficience du recours au médicament et sur les effets négatifs d’une telle surconsommation tant sur le plan sanitaire qu’économique.
1. Les Français détiennent le record européen de la consommation de médicaments
a) Ce constat est largement partagé
Dans sa communication à la MECSS sur la prescription et la consommation de médicaments, la Cour des comptes indique que « la France se caractérise par un niveau de consommation de médicaments supérieur à celui de ses voisins européens sans que cela se justifie par des indicateurs de morbidité ou de mortalité différents ».
De même, le Haut conseil pour l’avenir de l’assurance maladie, dans son avis du 29 juin 2006 sur le médicament, indique que « Les Français sont, avec les Américains, les premiers consommateurs de médicaments par habitant, en volume (doses journalières) comme en valeur relative. »
La Caisse nationale d’assurance maladie des travailleurs salariés (CNAMTS) note également, dans un Point d’information, du 13 mars 2008 que : « la France se caractérise par un recours élevé aux médicaments, avec à la fois des volumes de consommation importants et des coûts de traitements supérieurs à ceux de ses voisins européens. »
Les indicateurs de l’OCDE sur le panorama de la santé 2007 soulignent également que « La France est au tout premier rang pour la consommation de médicaments dans les pays de l’OCDE » et précise que « les dépenses de médicaments en France sont plus de 30 % supérieures à la moyenne des pays de l’OCDE. »
b) La France détient toujours le record en termes de quantité et de dépense de médicaments par habitant
Les données de l’OCDE concernent les médicaments prescrits et l’automédication, laquelle est plus ou moins développée selon les pays. Ces données sont exprimées en dollars américains, ajustées par la parité de pouvoir d’achat.
Alors que les dépenses de produits pharmaceutiques prescrits par habitant sont, en 2005, de 413 dollars en moyenne pour l’ensemble des pays de l’OCDE, la dépense s’élève en France à 554 dollars. Dans des pays à développement comparable, à l’exception des États-Unis (792 dollars) et du Canada (589), la dépense est moins élevée en Espagne (517), en Italie (509), en Allemagne (498), en Suisse (436), en Finlande (380), en Suède (351), en Irlande (320), aux Pays-Bas (318) et au Danemark (276).
Par ailleurs, la direction de la recherche, des études, de l’évaluation et des statistiques (DREES), dans une étude sur les cinq pays européens ayant les marchés du médicament les plus importants, note aussi que la France enregistre, en 2004, les ventes de médicaments par habitant les plus élevées (284 €), devant l’Allemagne (244 €, soit 14 % de moins qu’en France), le Royaume-Uni, l’Italie et l’Espagne (autour de 200 €, soit 30 % de moins que dans notre pays).
Mais la France se distingue surtout par le niveau beaucoup plus élevé des quantités vendues en officine par habitant, de plus de 50 % supérieur à la moyenne des autres pays. Le rapport est même de 1 à 2 avec l’Italie. Cette surconsommation quantitative est en partie compensée par un niveau de prix fabricant moyen inférieur d’environ 20 % à la moyenne des autres pays.
Chiffre d'affaires des ventes de médicaments par habitant,
quantités vendues par habitant et prix fabricant moyen, en 2004
Chiffre d’affaires des ventes aux officines par habitant |
Quantités d’unités standards vendues aux officines par habitant |
Prix fabricant moyen par unité standard | |
France Allemagne Royaume-Uni Italie Espagne Moyenne |
284 244 202 202 193 210 |
1 535 1 049 1 136 746 1 023 989 |
0,18 0,23 0,18 0,27 0,19 0,22 |
Source : IMS Health – calculs DREES.
Mais aux effets prix et quantités il faut ajouter l’effet structure. Or, la France se caractérise par une consommation plus forte de médicaments récents, innovants et plus chers que la moyenne des médicaments du marché et souvent génériqués.
La France continue donc à occuper le deuxième ou troisième rang mondial, après les pays d’Amérique du Nord, et la première place des pays européens pour la dépense moyenne de médicaments par habitant.
c) Mais dans certaines classes de médicaments, les écarts de consommation avec les principaux pays européens ont tendance à se réduire
Cependant, on note au niveau européen une tendance à la réduction des écarts, voire une inversion des écarts entre la France et les principaux pays européens pour certaines classes de médicaments. Dans les années quatre-vingt-dix, des analyses par classes thérapeutiques avaient mis en évidence des rapports de 1 à 2 ou 1 à 3, pour beaucoup de classes de médicaments, entre la France et le Royaume-Uni ou l’Allemagne, et des écarts moins significatifs avec les pays du Sud comme l’Espagne ou l’Italie. Des études récentes conduisent à nuancer cette analyse en faisant notamment apparaître que la France est aujourd’hui devancée par d’autres pays pour certaines classes thérapeutiques : par exemple, par le Royaume-Uni pour les anti-asthmatiques et les statines (utilisées contre le cholestérol), par l’Allemagne pour les anti-hypertenseurs ou encore par l’Espagne pour les inhibiteurs de la pompe à protons (IPP, utilisés contre les maladies digestives).
Cette situation est confirmée par les résultats d’une étude récente publiée par la CNAMTS, au mois de décembre 2007 (Points de repère n° 12), qui compare les consommations de la France et de ses principaux voisins (Allemagne, Royaume-Uni, Espagne et Italie) pour huit classes de médicaments couramment prescrits et représentant près de 30 % du volume global de la consommation de médicaments et 40 % des dépenses totales de médicaments de l’assurance maladie (8 milliards d’euros en 2006).
La CNAMTS relève que c’est en France que le montant moyen par habitant est le plus élevé des cinq pays européens pour ces huit classes de médicaments : 118 euros, soit 24 euros de plus que le deuxième pays – l’Italie – et 46 euros de plus que l’Allemagne. Cette situation résulte de la combinaison de deux facteurs : d’une part des volumes de consommation qui restent pour la plupart des classes dans le haut de la fourchette, même si la France n’occupe pas la première place pour toutes les classes ; d’autre part, des coûts moyens de traitement souvent plus élevés que dans les autres pays, induits par une structure de consommation différente, où les produits les plus récents et les plus chers occupent une place prépondérante au détriment de molécules plus anciennes et souvent génériquées.
La comparaison, en volumes d’unités standards (comprimé, cuillerée…), place la France au premier rang pour la consommation d’antibiotiques, d’antidiabétiques oraux, d’hypocholestérolémiants (mais au second rang pour les seules statines), d’antidépresseurs et de tranquillisants. Elle est au second rang pour la consommation d’antiasthmatiques et d’IPP et au troisième rang pour la consommation d’antihypertenseurs.
L’unité de mesure en volume de la consommation de médicaments
Trois unités peuvent être utilisées pour mesurer la consommation de médicaments en volume et effectuer les comparaisons internationales en matière de consommation de médicaments : le nombre de boîtes, le nombre d’unités standards ou le nombre de doses d’entretien quotidiennes. Les trois unités de mesure ont leurs avantages et leurs inconvénients, et le choix de l’une ou de l’autre conduit à des résultats un peu différents en termes de classement relatif des pays.
L’« unité » de conditionnement (la boîte) est l’indicateur le plus utilisé du fait de sa simplicité d’accès. Il comporte néanmoins de nombreux biais du fait de la diversité des tailles de boîtes et de dosage que l’on constate dans les différents pays.
L’« unité standardisée », qui est un indicateur produit par la société d’étude sur le secteur du médicament IMS Health, correspond à l’unité de prise contenue dans le conditionnement. L’unité standard est ainsi un comprimé, une gélule, ou encore une cuillerée, une bouffée ou une injection. Cette mesure permet de s’affranchir des problèmes posés par les différences de conditionnement et de formes pharmaceutiques (comprimés, gélules, sachets, solutions buvables, suspensions injectables). Néanmoins, elle ne permet pas, par exemple, de distinguer un comprimé à faible dosage d’un comprimé à fort dosage.
La « DDD » (defined daily dose) est la dose d’entretien quotidienne usuelle pour un médicament dans son indication principale pour un adulte. Elle permet donc de s’abstraire des différences de conditionnement et de dosage, mais ne permet pas de tenir compte du nombre d’unités de prise.
Il convient toutefois de souligner que ces comparaisons internationales de consommation ne prennent pas en compte les spécificités de chaque pays en termes de marché et de distribution des médicaments, mais aussi de prévalence des différentes maladies et des recommandations qui y ont cours.
2. La consommation de médicaments est fortement concentrée sur les personnes relevant d’une affection de longue durée
a) Chaque Français consomme, en moyenne, une boîte de médicaments par semaine
Chaque Français consomme en moyenne cinquante unités (boîtes, flacons…) de médicaments par an, soit une par semaine, ou encore 1 500 unités standards par an.
Cette consommation est fortement concentrée. Ainsi, 10 % des Français n’en consomment pas, mais les 5 % qui en consomment le plus acquièrent près de 300 boîtes par an. En outre, parmi les 10 millions de personnes âgées de plus de 65 ans, environ 1,5 million consomment quotidiennement 7 médicaments, ou plus, de classes thérapeutiques différentes.
b) Mais les affections de longue durée (ALD) concentrent la moitié de la consommation de médicaments
La moitié de la dépense de médicaments remboursée est imputable aux traitements des personnes relevant d’une affection de longue durée (ALD).
Fin 2004, on recensait près de 8 millions de personnes en ALD, dont 60 % de personnes âgées. Chaque année, plus d’un million de nouvelles admissions en ALD sont enregistrées (1 130 000 en 2004). Entre 1994 et 2004, le nombre de personnes en ALD a augmenté de 5,7 % par an, en moyenne. En 2004, les personnes en ALD représentaient 14 % des assurés mais contribuaient à hauteur de 60 % aux dépenses d’assurance maladie remboursées. Elles pourraient représenter 70 % de ces dernières en 2015.
On peut aussi rappeler que les dépenses de soins sont sept fois supérieures pour un patient en ALD (7 450 € en 2004) que pour un patient non ALD (1 050 €). Plus précisément, en 2002, les achats par les personnes en ALD de médicaments remboursés représentaient près de la moitié (49 %) des dépenses de médicaments remboursées à l’ensemble des patients. Pour les patients en ALD, la pharmacie est le premier poste de dépenses et représentait 43 % des dépenses remboursables de soins de ville contre 37,9 % pour le reste de la population.
Toujours en 2002, la dépense moyenne en pharmacie s’élevait à 1 351 € pour les personnes en ALD et 218 € pour les personnes ne relevant pas de ce régime ; ainsi la dépense est six fois plus élevée pour une personne en ALD.
En 2001, le nombre moyen de boîtes de médicaments remboursées aux patients relevant d’une ALD s’élevait à 110 contre 28 pour les personnes non ALD.
Ainsi, le niveau élevé des dépenses de remboursement par l’assurance maladie des médicaments consommés par les patients en ALD résulte, d’une part, de la consommation très supérieure chez ces patients par rapport aux autres et, d’autre part, du niveau de prise en charge élevé du fait de l’exonération du ticket modérateur. Le classement en ALD ouvre, en effet, droit à une prise en charge intégrale des frais de traitement de l’ALD, dans la limite des montants remboursables.
c) Au sein des ALD, la consommation est concentrée sur les affections qui nécessitent un traitement pharmaceutique innovant et coûteux
Au sein même de la consommation de médicaments par les patients en ALD, on observe une concentration de la dépense. Certaines ALD font, en effet, principalement l’objet d’une prise en charge au moyen de traitements pharmaceutiques lourds et de faibles dépenses en honoraires médicaux et analyses biologiques. Il s’agit en particulier des ALD relatives au VIH, à l’hémophilie, à la mucoviscidose, aux transplantations d’organes et à la paraplégie. Ces ALD concernent souvent des personnes jeunes. Elles se caractérisent par une augmentation importante des remboursements par patient du fait d’innovations pharmaceutiques qui ont modifié les traitements. C’est notamment le cas de la trithérapie qui s’est fortement diffusée comme traitement des malades atteints du VIH.
d) La durée de traitement des personnes relevant d’une ALD a tendance à s’allonger
Le recours à des médicaments innovants et coûteux et la tendance à l’allongement de la durée des traitements des personnes atteintes d’ALD contribuent aussi fortement à la hausse de la dépense de médicaments remboursée.
De fait, le progrès médical permet de traiter efficacement aujourd’hui des personnes qui hier n’avaient aucun espoir. Des maladies chroniques sont devenues compatibles avec une vie normale et longue. En dix ans, l’espérance de vie des patients relevant d’une ALD a augmenté de quatre ans, en moyenne. Elle est passée de 71 ans en 1994 à 75 ans en 2004.
e) L’augmentation des dépenses de médicaments pour les personnes relevant d’une ALD constitue l’essentiel de la croissance de la dépense totale de médicaments
Au total, la croissance de la dépense remboursable de médicaments résulte pour une grande part de l’augmentation des dépenses imputables aux personnes relevant d’une affection de longue durée. Entre 2000 et 2002, les dépenses de médicaments remboursées pour les patients relevant d’une affection de longue durée ont contribué pour plus des trois quarts (13 points) à la croissance de la dépense totale de médicaments du régime général d’assurance maladie (17 points), les dépenses pour les autres patients n’y ayant contribué que pour moins d’un quart (4 points).
Ce constat recoupe, en grande partie, celui selon lequel le marché est dominé par quelques classes thérapeutiques. Ainsi, sur 341 classes thérapeutiques comprenant des médicaments remboursables, 25 concentrent 50 % du chiffre d’affaires global.
Les principaux postes de dépenses de médicaments remboursables concernent l’appareil cardio-vasculaire (21 %), le système nerveux central (20 %), l’appareil digestif (16 %) et l’appareil respiratoire (15 %). Cette répartition du marché français correspond à peu près à celle observée sur le marché européen.
En revanche, la France se singularise par sa consommation élevée d’antibiotiques (notamment pour certaines familles d’antibiotiques les plus récentes), une utilisation plus élevée de psychotropes (notamment les benzodiazépines) que dans la quasi-totalité des autres pays européens, et une forte consommation de statines.
Aussi, alors que le vieillissement de la population s’accélère, les maladies chroniques ainsi que les polypathologies tendent à devenir le centre de gravité du système de santé et constituent un enjeu majeur en matière de consommation de médicaments. Cette évolution laisse penser que le dynamisme de la demande de médicaments devrait se poursuivre, voire s’accentuer durant les prochaines années, tant en ville qu’à l’hôpital.
La CNAMTS indique qu’en 2007 les dépenses de médicaments destinées aux pathologies lourdes telles que le cancer, le sida ou la polyarthrite rhumatoïde ont augmenté de 11 % et représentent 56 % de la croissance totale des dépenses de médicaments.
3. Près d’un quart de la consommation de médicaments en ville résulte de prescriptions hospitalières et cette part devrait continuer de croître
De nombreuses prescriptions établies par des médecins hospitaliers à l’intention de patients qui ne sont pas hospitalisés sont exécutées en ville. Il peut s’agir de prescriptions effectuées à l’issue d’un séjour dans un établissement hospitalier ou bien lors d’une consultation externe ou à l’occasion de soins dispensés dans un service d’urgence.
En 2004, 23 % des médicaments délivrés en ville et remboursés (soit 4,7 milliards d’euros) ont été prescrits par des médecins hospitaliers ou en centre de santé. Cette part a continué de progresser. En 2007, selon la CNAMTS, elle s’élevait à 25 %.
a) La rétrocession hospitalière diminue mais la prescription en activité ambulatoire se développe
Les médicaments délivrés par les pharmacies hospitalières destinées à soigner des malades non hospitalisés, c’est-à-dire la rétrocession hospitalière, représentaient en 2004 environ 40 % des prescriptions hospitalières exécutées en ville. Au début des années 2000, la rétrocession hospitalière a connu une forte croissance. Depuis 2005, en raison du nouveau cadre juridique de la rétrocession, défini par le décret du 15 juin 2004, et de la sortie de certains médicaments de la réserve hospitalière, le montant des médicaments rétrocédés à tendance à diminuer.
En 2004, les médicaments prescrits à l’occasion de l’activité ambulatoire, de consultations externes et de passages aux urgences représentaient aussi environ 40 % des prescriptions hospitalières exécutées en ville.
Enfin, environ 20 % du montant des médicaments prescrits par les médecins hospitaliers et délivrés en ville étaient directement liés à un séjour hospitalier. Il s’agit de médicaments que l’assuré hospitalisé est invité à aller chercher en pharmacie d’officine (comme les produits de contraste en radiologie).
b) La prescription hospitalière exerce un effet d’entraînement sur la prescription en ville
La prescription hospitalière est d’autant plus importante qu’elle acquiert aux yeux du patient et du médecin un statut particulier. En effet, le médecin de ville, qui a la charge de poursuivre le traitement, peut éprouver par la suite des difficultés à le modifier pour, par exemple, y substituer des génériques ou modifier la prescription. La prescription hospitalière peut ainsi induire un effet d’entraînement sur la prescription de ville, d’où l’importance essentielle de la qualité de la prescription hospitalière.
Or, il faut rappeler que faute d’enveloppe médicaments au sein de l’ONDAM, l’objectif national de dépenses d’assurance maladie, les dépenses de médicaments sont imputées sur l’enveloppe hospitalière ou sur l’enveloppe de ville.
Les dépenses de médicaments résultant de prescriptions hospitalières qui font l’objet d’une dispensation en ville ne sont donc pas prises en charge sur l’enveloppe hospitalière mais sont remboursées aux assurés sociaux par les caisses d’assurance maladie et imputées sur l’enveloppe de soins de ville. Les deux secteurs ne sont donc pas étanches et il y a bien une certaine porosité entre eux.
Les transferts d’achats de médicaments de l’hôpital vers la ville expliquent donc une partie de l’augmentation du chiffre d’affaires de la dispensation en ville. Ainsi, la CNAMTS, dans son Point d’information du 13 mars 2008, indique que les prescriptions hospitalières ont représenté, en 2007, près de la moitié (49 %) de la croissance des dépenses de médicaments de ville.
L’impact du transfert croissant de médicaments de l’hôpital vers la ville devrait se poursuivre, particulièrement sous l’effet du développement de la chirurgie ambulatoire et des soins à domicile, notamment pour certaines pathologies lourdes qui peuvent désormais être soignées à domicile (cancer, sida) après une sortie précoce de l’hôpital.
Force est cependant de constater que si le poids de la prescription peut passer du secteur hospitalier au secteur de ville, des mouvements inverses peuvent aussi exister. Des médecins de ville peuvent, par commodité ou dans le but de ne pas « dégrader » leur profil de prescriptions, conseiller au patient, dans certains cas, de se rendre à l’hôpital et, ainsi, être à l’initiative de prescriptions de médicaments (ou d’actes divers) qui apparaîtront comme hospitalières.
Plus généralement, ce constat renvoie à la nécessité de sortir du cloisonnement actuel du système de soins et de construire la continuité indispensable entre les soins ambulatoires, les soins hospitaliers et la prise en charge médico-sociale. La mise en place des agences régionales de santé (ARS) devrait favoriser cette approche globale.
c) Les effets de la sortie de la réserve hospitalière : l’exemple de l’érythropoïétine (EPO)
Au printemps 2005, l’érythropoïétine (EPO), utilisée dans le traitement de l’anémie chez les patients souffrant d’insuffisances rénales chronique et les adultes traités par chimiothérapie, est sortie de la réserve hospitalière.
À la suite de cette décision, les produits composés d’EPO ont contribué, pour chacune des années 2005 et 2006, à un point de croissance du marché des médicaments remboursables délivrés en ville, ce qui a fait de cette classe thérapeutique la plus dynamique du marché. En 2006, en l’absence de l’effet EPO, la croissance du marché officinal – qui a été de 1 % – aurait été nulle. En 2007, l’impact direct lié à la sortie de la réserve hospitalière semble avoir été stoppé.
L’évolution constatée est caractéristique des produits sortis de la réserve hospitalière : leur croissance, très élevée la première année, diminue ensuite rapidement, car le caractère spécifique des pathologies traitées entraîne une saturation du marché.
Cependant, la sortie de la réserve hospitalière s’est aussi accompagnée d’une augmentation de la taille du marché en volume : dans les deux mois qui ont suivi la sortie de la réserve hospitalière, les volumes ont atteint 3 millions de doses quotidiennes alors que les volumes à l’hôpital ne baissaient que de 2,3 millions. On a ainsi constaté un saut dans les volumes totaux ville + hôpital d’environ 700 000 doses. Selon la DREES, ce saut représente une croissance du marché de 20 % en volume. Après ce saut, les volumes ont continué d’augmenter selon la dynamique forte observée avant la sortie de la réserve hospitalière.
4. Les médecins de ville français sont de gros prescripteurs de médicaments
La prescription médicale est l’élément central de la consommation médicamenteuse. On le sait, globalement, les médecins français prescrivent beaucoup et pas toujours de la manière la plus rationnelle ou la plus utile qui devrait conduire à retenir le médicament le plus efficace et le moins coûteux, c’est-à-dire celui présentant le meilleur rapport coût-efficacité.
a) Les médecins français prescrivent beaucoup
Les comparaisons internationales soulignent une médicalisation plus forte qu’ailleurs des problèmes de société en France et un recours plus systématique aux médicaments dans la stratégie thérapeutique. En conséquence, le niveau de prescription des médecins est plus élevé que dans les autres pays européens. Cette situation ne semble avoir que peu évolué depuis vingt ans.
Selon l’étude réalisée en 2005 par IPSOS, à la demande de la CNAMTS sur le rapport des patients à l’ordonnance et aux médicaments dans quatre pays européens (France, Allemagne, Espagne et Pays-Bas), c’est en France que l’équation « consultation = ordonnance = médicaments » est la plus forte.
En moyenne, dans ces quatre pays, 80 % de la population consulte un médecin au moins une fois dans l’année. En revanche, si le niveau de consultation en France est proche de celui de deux des trois autres pays étudiés, il est nettement supérieur à celui du troisième. Ainsi, le nombre de consultations dans l’année est de 4,9 en France, contre 5,2 en Allemagne, 4,8 en Espagne mais il est de 3,2 aux Pays-Bas.
En France, l’accès aux médicaments est facile et sécurisé
La France compte environ 210 000 médecins dont 120 000 médecins libéraux qui se répartissent entre 54 000 spécialistes et 61 000 omnipraticiens (dont 54 000 généralistes).
La densité moyenne de médecins s’élève à 340 médecins pour 100 000 habitants. Elle est de 15 % supérieure à celle de la moyenne des pays de l’OCDE (289) et légèrement plus importante que celle de l’Union européenne (326). La densité moyenne de médecins libéraux s’élève en France à 88 pour 100 000 habitants. À partir de 2008, sous le double effet, d’une part, de la baisse du numerus clausus jusqu’au milieu des années 1990, et, d’autre part, des départs à la retraite de nombreux professionnels (la moyenne d’âge des médecins libéraux est de 50 ans), le nombre de médecins devrait commencer à baisser, pour diminuer de 15 % à 25 %, selon les diverses estimations et sous l’hypothèse du maintien d’un numerus clausus à 7 000, entre 2002 et 2025.
Le réseau de distribution et de dispensation des médicaments en ville est très encadré et développé. La recherche de la sécurité a conduit la France à instituer un monopole de la dispensation par des professionnels bien formés et indépendants. Cela interdit de fait, puisque la publicité sur les médicaments remboursés est prohibée, la vente de médicaments en dehors des officines, par correspondance ou par internet.
Afin d’assurer en tout lieu l’accès rapide, complet et sécurisé au médicament, un maillage dense du territoire a été institué. La dispensation des médicaments est assurée par un réseau de 23 000 pharmacies d’officine (soit en moyenne une officine pour cinq médecins) et un peu plus de 54 000 pharmaciens exerçant en officine (soit un peu moins d’un pharmacien d’officine pour deux médecins libéraux). La France compte ainsi, en moyenne, une officine pour 2 560 habitants (contre 3 300 habitants, en moyenne, en Europe).
En 2006, environ 9 400 présentations, dont 6 600 présentations remboursables et 2 800 non remboursables, ont été vendues au moins une fois. Ce nombre est plutôt supérieur à ce qu’on connaît dans les autres pays européens.
Le prix public moyen d’un médicament remboursable (prix réglementé) s’élevait à 9,10 euros, en 2005. Celui d’un médicament d’automédication (prix libre) s’élevait à environ la moitié (4,50 euros).
Par ailleurs, selon le rapport sur le médicament, de juin 2006, du Haut conseil pour l’avenir de l’assurance maladie, 90 % des consultations réalisées en France, se concluent par une ordonnance (en moyenne plus longue que dans d’autres pays), contre 83 % en Espagne, 72 % en Allemagne et 43 % aux Pays-Bas. Ce taux de 90 % est même jugé plus bas que la réalité par les médecins interrogés dans le cadre de l’enquête, pour lesquels ces 10 % de consultations sans ordonnance de médicaments renvoient à des situations non thérapeutiques, comme la délivrance d’attestations pour la pratique sportive.
La DREES, a publié, en novembre 2005, les résultats d’une enquête sur les consultations et visites des médecins généralistes libéraux menée en 2002 (Études et résultats n° 440). Le taux de consultation se terminant par une ordonnance est un peu inférieur au chiffre avancé par le Haut conseil pour la France. Selon cette enquête près de 80 % des consultations donnaient lieu, en 2002, à la prescription d’au moins un médicament. Et, en moyenne, les médecins prescrivent 2,9 médicaments et 6 boîtes de médicaments par consultation.
Quoi qu’il en soit, cette habitude de conclure presque systématiquement (dans au moins trois cas sur quatre) une consultation par une prescription n’est pas le seul fait d’une minorité de surprescripteurs, mais constitue une pratique courante et quasi généralisée. C’est bien le trait le plus marquant de ce que le Haut conseil pour l’avenir de l’assurance maladie dans son avis sur le médicament, de juin 2006, appelle le « modèle français de prescription ».
b) La prescription est davantage orientée vers les spécialités récentes et comporte peu de génériques
En outre, les médecins français sont très sensibles à la mise sur le marché des nouveaux médicaments et prescrivent plutôt des spécialités récentes qui sont en général plus coûteuses que les spécialités plus anciennes ou les génériques, encore peu prescrits par les médecins. À cet égard, force est de constater que la progression récente des génériques est davantage le résultat de l’exercice du droit de substitution par les pharmaciens que de la prescription des médecins. La situation a tout de même commencé à évoluer puisque, lorsque le médecin a le choix entre un princeps et un générique, il prescrit un générique dans un peu plus d’un tiers des cas.
c) Les médicaments prescrits ne sont pas toujours efficaces et la qualité des prescriptions n’est pas toujours optimale
Enfin, selon une étude de 2005 de la Fédération nationale de la Mutualité française, les médecins français prescrivent plus de médicaments à service médical rendu insuffisant (SMRI) : 2,2 lignes de prescription par habitant et par an, contre 1 ligne en Allemagne, 0,8 en Espagne, 0 au Royaume-Uni et au Canada.
On peut d’ailleurs ajouter que, selon cette même étude, 17 % des médicaments – prescrits ou non – vendus en France sont des médicaments à SMR insuffisant.
Enfin, la qualité des prescriptions n’est pas toujours optimale.
Les quelques études menées sur ce point, d’ailleurs de manière dispersée et sans cohérence d’ensemble, montrent que les indications pour l’utilisation des médicaments fixées par les autorisations de mise sur le marché (AMM) et les recommandations de l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) et de la Haute Autorité de santé (HAS) ne sont pas toujours respectées, de même que les posologies et les durées de traitement.
Ces études ponctuelles montrent que dans 30 % à 50 % des cas, les recommandations ne sont pas respectées et que de nombreuses prescriptions sont effectuées en dehors des indications prévues par l’AMM, ou entraînent des dépassements des posologies usuelles maximales recommandées et des dépassements de durée de traitement.
B. LA CONSOMMATION ÉLEVÉE DE MÉDICAMENTS EST COÛTEUSE ET PEUT ENTRAÎNER CERTAINES CONSÉQUENCES SANITAIRES REGRETTABLES
1. Les dépenses de médicaments sont en forte croissance
En 2006, les ventes de médicaments en ville ont représenté 3 milliards de boîtes vendues pour un montant de 20,4 milliards d’euros, soit environ 15 % de l’ONDAM (141,8 milliards d’euros), un tiers des dépenses de soins de ville (66,7 milliards d’euros) et la moitié de l’ensemble des prescriptions (médicaments, indemnités journalières, examens…) des médecins libéraux (41,6 milliards d’euros).
a) Les dépenses de médicaments représentent une part importante du PIB
Les données de l’OCDE portant sur l’année 2003, montraient que la France était le pays où la part des médicaments délivrés sur ordonnance et en automédication (donc à l’exclusion de la consommation pharmaceutique à l’hôpital) dans le PIB était nettement la plus forte. En 2003, la part du PIB consacrée par la France aux dépenses de médicaments était supérieure de 9 % à celle des États-Unis, de 30 % à celle de l’Allemagne, de 45 % à celle du Japon et de 73 % à celle du Royaume-Uni.
Dépenses de produits pharmaceutiques en % du PIB en 2003
France |
États-Unis |
Italie |
Canada |
Allemagne |
Japon |
Royaume-Uni |
2,11 |
1,94 |
1,86 |
1,67 |
1,62 |
1,45 |
1,22 |
Source : Eco santé OCDE 2005
Les nouvelles données « Eco santé » de l’OCDE portant sur l’année 2005 montrent toutefois que le pourcentage français a sensiblement baissé.
Dépenses de produits pharmaceutiques en % du PIB en 2005
France |
États-Unis |
Italie |
Canada |
Allemagne |
Japon |
1,8 |
1,9 |
1,8 |
1,7 |
1,6 |
1,5 |
Source : Eco santé OCDE 2007
Selon ces nouvelles statistiques, la part du PIB français consacrée aux dépenses d’achats de médicaments prescrits et à l’automédication reste à un niveau élevée mais proche de celle qu’y consacrent des pays comparables.
b) La part des dépenses de santé que consacre la France aux dépenses de médicaments est plus importante que dans la plupart des autres pays
La part des dépenses pharmaceutiques dans les dépenses de santé est en France supérieure à celle de nombreux pays de l’OCDE comparables. En 2003, les dépenses pharmaceutiques représentaient 20,9 % des dépenses de santé en France, contre 18,4 % au Japon, 16,9 % au Canada, 14,6 % en Allemagne, 12,9 % aux États-Unis, 11,4 % aux Pays-Bas, 10,5 % en Suisse, 9,8 % au Danemark et 9,4 % en Norvège (plus du double). Seules l’Italie (22,1 %) et l’Espagne (21,8 %) consacrent aux dépenses de médicaments une part des dépenses de santé proche de celle de la France.
c) Le rythme de croissance des dépenses de médicaments est élevé
Selon les données de l’OCDE, sur la période 1998-2003, la croissance annuelle moyenne des dépenses pharmaceutiques a été beaucoup plus forte en France (+ 5,8 %) qu’en Allemagne (+ 3,5 %), en Espagne (+ 3,4 %), en Italie (+ 3,2 %) et au Japon (+ 2,2 %). Elle a en revanche été sensiblement inférieure à celle des États-Unis (+ 9,6 %).
En France, sur la même période, la croissance annuelle des dépenses pharmaceutiques a été aussi nettement plus élevée (5,8 %) que la croissance de l’ensemble des dépenses de santé (3,5 %). Le rythme de croissance annuelle des dépenses pharmaceutiques est ainsi de 66 % supérieur à celui de l’ensemble des dépenses de santé. Il est aussi nettement supérieur à la croissance du PIB en valeur (+ 3,9 %).
Les données « Eco santé 2007 » de l’OCDE, concernant la période 1995-2005, fournissent de nouveaux éléments qui modifient sensiblement les résultats de la comparaison avec les pays comparables. Sur cette période, la croissance moyenne annuelle réelle des dépenses pharmaceutiques en France est ramenée à 3,1 % (au lieu de 5,8 % sur la période 1998-2003). Elle est ainsi du même ordre que celle de l’Allemagne (inchangée à 3,5 %) et de l’Italie (2,9 %) mais inférieure à celle de l’Espagne (3,4 %) et des États-Unis (7,1 %), pays dans lequel on constate un ralentissement de la progression, ces dernières années.
Par ailleurs, l’OCDE note que, depuis 1995, dans l’ensemble des pays membres de l’organisation, le montant moyen des dépenses pharmaceutiques par habitant a augmenté de plus de 50 % en valeur réelle et plus rapidement que l’ensemble des dépenses de santé.
Selon les données nationales, depuis 2003, la croissance des dépenses de médicaments en France s’est poursuivie à un rythme élevé (+ 6,4 % en 2004 et + 4,8 % en 2005), sauf en 2006.
Le ralentissement de la croissance du marché des médicaments remboursables en 2006 (+ 1,1 %) s’explique par les déremboursements ou les changements de taux de remboursement de certaines classes thérapeutiques, la montée en charge des génériques (notamment dans les classes très vendues), la diminution de 15 % des prix sur le répertoire générique et la réduction en volume des ventes de certains médicaments, laquelle résulte de la politique de maîtrise médicalisée des dépenses de l’assurance maladie.
Ce freinage aura toutefois été de courte durée, puisque, selon la CNAMTS, les remboursements de médicaments par le régime général d’assurance maladie ont à nouveau fortement augmenté en 2007, de + 4,8 %, soit au même rythme qu’en 2005.
La CNAMTS note toutefois, dans son Point d’information du 13 mars 2008, que, sous l’effet de la maîtrise médicalisée, du développement des génériques et des baisses de prix, la croissance enregistrée en 2007 a été deux fois moins élevée que celle observée au début des années 2000 (+ 8,8 % en 2001).
d) Les nouveaux produits « tirent » la croissance du marché
L’âge des produits a une incidence importante sur le dynamisme des ventes pharmaceutiques. Ce sont les nouveaux produits qui constituent l’essentiel de la croissance des dépenses de médicaments.
Nouveaux produits et nouvelles présentations
La nouveauté réside soit dans la mise sur le marché de nouveaux médicaments qui n’existaient pas l’année précédente soit dans une présentation générique ou encore dans de nouvelles présentations de produits qui existaient déjà et peuvent prendre la forme de nouvelles associations, de nouvelles formes d’administration, de contenances ou de dosages différents d’un même produit.
En 2006, les produits mis sur le marché depuis moins de dix ans, qui représentent 51 % des médicaments remboursables, ont contribué à 8,7 points de la croissance des ventes. Par ailleurs, ce sont les produits de moins de deux ans qui apportent la plus forte contribution et « tirent » le marché. Inversement, les médicaments qui ont été mis sur le marché il y a plus de dix ans ont un impact négatif sur les ventes globales (-7,7 points).
Par ailleurs, la CNAMTS indique qu’en 2007 les dépenses de médicaments de moins de trois ans ont représenté une dépense de près de 1,4 milliard d’euros et ont contribué pour 85 % à la croissance totale des dépenses de médicaments. Seulement un tiers de cette augmentation est lié à des innovations thérapeutiques importantes (ASMR de niveau 1,2 ou 3). Le reste des dépenses supplémentaires est imputable pour 45 % à des molécules qui ne présentent pas ou peu d’amélioration du service médical rendu et à hauteur de 25 % à des traitements transférés de l’hôpital vers la ville (en grande partie des anti-cancéreux).
En 2006, le dynamisme des nouvelles présentations s’explique surtout par l’arrivée de conditionnements de trois mois pour des produits déjà existants. Un décret du 16 décembre 2004 prévoit en effet, pour le traitement des affections de longue durée, la possibilité de délivrer des médicaments pour une période de trois mois.
e) Une boîte de médicaments vendue sur cinq est un produit générique
En 2006, plus d’une présentation remboursable sur trois était une présentation générique, recensée au répertoire de l’AFSSAPS. Les génériques représentaient 9 % des ventes globales de médicaments et 18 % du nombre total de boîtes vendues.
Cet écart provient des différences de prix entre médicaments princeps et génériques. Il faut en effet rappeler que, depuis février 2006, un médicament générique ne peut être commercialisé que si son prix est inférieur de 50 % au prix du princeps.
Médicaments génériques et princeps en 2006
Génériques |
Princeps génériqués |
Autres médicaments |
Total | |
Nombre de présentations (%) |
38 |
6,5 |
55 |
100 |
Part de marché 2006 (%) |
9 |
11 |
80 |
100 |
Taux de croissance 2006 (%) |
11,4 |
- 33,4 |
7,7 |
0,94 |
Sources : GERS, AFSSAPS, traitement DREES.
La croissance des ventes de génériques s’est poursuivie en 2006 à un rythme élevé (+ 11,4 %) quoique moins soutenu qu’en 2005 (+ 24,2 %).
Toutefois, la part des génériques dans le total des ventes reste encore modeste en France, en comparaison avec d’autres pays européens. Par exemple, aux Pays-Bas, en 2005, une boîte de médicaments vendue sur deux était une boîte de médicaments génériques.
La pénétration des génériques sur le marché du médicament est inégale : sur 348 classes thérapeutiques, seules 85 comptaient des présentations génériques, en 2006.
Mais les classes thérapeutiques dans lesquelles la part des génériques est la plus importante ont peu d’impact sur la croissance globale du marché. En effet, il s’agit pour la plupart de classes dans lesquelles les présentations génériques sont apparues depuis plusieurs années et qui semblent donc avoir atteint leur part de marché maximale. Elles traitent principalement les affections des appareils digestif, locomoteur et cardiovasculaire. S’y retrouvent également des anti-infectieux par voie générale.
Par ailleurs, la mise en œuvre, depuis 2003, du tarif forfaitaire de responsabilité (TFR), qui permet de rembourser les médicaments appartenant à un groupe générique sous TFR au prix des médicaments génériques, favorise la baisse des prix des princeps. En outre, dans les groupes soumis au TFR, les ventes de génériques se substituent progressivement aux princeps et, en deux ou trois ans, prennent le pas (c’est-à-dire représentent plus de la moitié des ventes) sur celles de ces derniers.
En 2006, les 126 groupes génériques soumis au TFR représentaient 2,2 % des ventes du marché global des médicaments et 12,4 % des ventes de l’ensemble des groupes génériques (soumis ou non au TFR).
f) La tendance à l’augmentation des dépenses de médicaments devrait se poursuivre
L’augmentation des dépenses de santé est le résultat des effets cumulés des deux types de causes. Les causes liées aux soins : les progrès du dépistage, de la médecine prédictive, des techniques et des pratiques médicales et les nouveaux médicaments. Les causes liées aux patients : l’évolution de la morbidité liée à la démographie et à l’émergence de pathologies nouvelles, les attentes et les exigences des usagers, les modes de consommation des soins.
Aussi, dans les années à venir, sous l’effet du vieillissement de la population et, si rien n’est fait, de l’augmentation des admissions en ALD conjuguée à l’allongement de la durée des traitements, la dépense de médicaments devrait continuer de croître. L’utilisation de nouveaux médicaments plus coûteux que les anciens, tirée par certains produits réellement innovants mais aussi par les politiques industrielles et commerciales des laboratoires qui poussent au renouvellement et à la diffusion rapide des produits pour contourner les mesures de maîtrise, pourrait aussi contribuer à pousser la dépense à la hausse. On peut s’interroger sur le point de savoir si cette tendance au renchérissement du coût des médicaments est inévitable.
Pour sa part, la CNAMTS prévoit une croissance des dépenses de santé et de médicaments de 50 %, d’ici 2015.
La croissance des dépenses de médicaments est par ailleurs l’une des principales causes de l’augmentation des dépenses de santé. Cependant, il faut souligner que la relation entre les deux termes est complexe. Ainsi, une augmentation accrue des dépenses pharmaceutiques pour soigner des maladies qui autrement nécessiteraient une hospitalisation et une intervention coûteuses, peut conduire à une réduction des dépenses globales de santé.
Quoi qu’il en soit, la forte consommation française de médicaments génère des coûts croissants qui sont très largement pris en charge par l’assurance maladie et les organismes de protection sociale complémentaire.
2. Les médicaments remboursés représentent la quasi-totalité de la consommation et l’automédication est peu développée
La prise en charge des médicaments par l’assurance maladie permet de garantir, en principe, l’accès de tous à tous les médicaments qui répondent à des exigences minimales de service rendu.
En 2006, le chiffre d’affaires du médicament remboursable s’est élevé à 18,1 milliards d’euros (en prix fabricant hors taxes). Ces dépenses représentent environ un tiers des dépenses de médecine de ville et « tirent » la croissance des soins de ville.
a) 90 % des médicaments consommés font l’objet d’une prise en charge par l’assurance maladie
Les médicaments pris en charge par l’assurance maladie représentent la quasi-totalité du marché puisque 90 % des unités et près de 93 % du marché (en chiffre d’affaires au prix fabricant) sont remboursables par les régimes sociaux.
Selon l’OCDE, en 2005, la part des dépenses totales de santé financées sur fonds publics est de 80 % en France, soit un niveau supérieur à celui de pays comparables (Allemagne et Italie 77 %, Espagne 71 %, Pays-Bas 66 %, Suisse 60 %, États-Unis 45 %), à l’exception du Royaume-Uni (87 %).
En revanche, la part des dépenses pharmaceutiques (délivrées sur ordonnance et en automédication) financées sur fonds publics en France s’élève à 69 %, soit un niveau un peu inférieur à celui de l’Allemagne et de l’Espagne (73 %), mais nettement supérieur à celui des États-Unis (24 %), de l’Italie (50 %) et des Pays-Bas (57 %).
Mais le financement public des produits pharmaceutiques (prescrits et d’automédication) est sensiblement moins important que pour les autres services médicaux, en France (69 % contre 86 %), comme dans les autres pays de l’OCDE. Cela traduit l’orientation commune aux différents pays consistant à assurer une meilleure prise en charge des dépenses hospitalières par des financements publics par rapport aux dépenses de santé de ville.
Le marché des médicaments remboursables
et médicaments non remboursables en 2005 et 2006
Médicaments non remboursables |
Médicaments remboursables au taux de : |
Total marché | ||||
15 % |
35 % |
65 % |
100 % | |||
Nombre de présentations en 2005 |
2 596 |
0 |
1 484 |
4 377 |
256 |
8 713 |
Nombre de présentations en 2006 |
2 839 |
92 |
1 217 |
5 019 |
280 |
9 447 |
Part de marché 2005 |
6,8 % |
0 % |
16,7 % |
70 % |
6,5 % |
100 % |
Part de marché 2006 |
8,0 % |
1,2 % |
12,4 % |
70,6 % |
7,8 % |
100 % |
Chiffre d’affaires 2006 (milliards d’€) |
1,56 |
0,23 |
2,44 |
13,81 |
1,53 |
19,57 |
Source : GERS, traitement DREES.
Le marché pharmaceutique français (médicaments non remboursables compris) se compose principalement de médicaments remboursés par la sécurité sociale à 65 %. En 2006, les présentations remboursées à 65 % représentent plus d’une présentation sur deux et concentrent 71 % des ventes. Ces présentations restent donc celles qui ont l’impact le plus dynamique même si leur contribution se réduit d’années en années.
Cette situation s’explique notamment par le fait que la quasi-totalité des médicaments qui obtiennent une autorisation de mise sur le marché sont admis au remboursement (96 %), et que 87 % le sont au taux de 65 %. Pourtant, plus de la moitié des médicaments évalués chaque année (58 % en 2005, 54 % en 2006) n’apportent pas d’amélioration du service médical rendu.
Environ 0,5 % des médicaments prescrits sont remboursés à 100 % mais ils représentent près de 10 % des dépenses de remboursement.
Par ailleurs, les médicaments dont le prix est supérieur à 30 euros la boîte représentent près de 15 % des 5 000 médicaments remboursés par l’assurance maladie. Ils correspondent à 5,5 % des médicaments prescrits mais génèrent 44 % des dépenses de remboursement de médicaments en 2006 (contre 40 % en 2005).
En outre, toujours en 2006, les médicaments de plus de 15 euros représentaient près de 67 % des dépenses de remboursement de médicaments (42 % en 2000 et 63,5 % en 2005).
Le marché des médicaments remboursables est dominé par un nombre restreint de produits et de classes thérapeutiques. En 2006, sur les 348 classes thérapeutiques comprenant des médicaments remboursables, 25 concentraient 49 % du chiffre d’affaires global.
Ces données sur les remboursements traduisent à nouveau la tendance à la concentration de la dépense de médicaments sur le traitement des ALD et sur des produits de plus en plus chers.
b) Il est encore trop tôt pour évaluer les effets de la franchise sur la consommation des médicaments
À partir du 1er janvier 2008, une franchise médicale s’applique sur les boîtes de médicaments, les actes paramédicaux et les transports sanitaires. Le montant de la franchise est de 50 centimes d’euro par boîte de médicaments ou toute autre unité de conditionnement (flacon…), de 50 centimes d’euro par acte paramédical et de 2 euros par transport sanitaire. La franchise est plafonnée à 50 euros par an, au total, par assuré.
La franchise sur les médicaments concerne tous les médicaments allopathiques ou homéopathiques et les préparations magistrales. Elle ne s’applique qu’aux médicaments remboursés par l’assurance maladie. Elle n’est donc pas applicable aux médicaments achetés sans prescription médicale et ne concerne pas l’automédication. En outre, dans le cadre de sa politique de prévention, l’assurance maladie a décidé de ne pas appliquer la franchise et de maintenir la prise en charge à 100 % du vaccin anti-grippal pour les personnes de plus de 65 ans et du vaccin contre la rougeole (ROR) pour les enfants de moins de 13 ans.
Le prix des médicaments remboursables payé en officine n’est pas affecté par la franchise. Le montant de la franchise est déduit du remboursement effectué par l’assurance maladie pour la boîte de médicaments achetée. En cas de tiers payant, la franchise est déduite sur un remboursement ultérieur. Il y a donc un décalage entre la date de l’acte et la date de prélèvement de la franchise. Dans la quasi-totalité des cas, les mutuelles et assurances complémentaires ne prévoient pas la prise en charge des franchises.
Cependant, environ un Français sur quatre est exonéré de la franchise : les bénéficiaires de la couverture maladie universelle complémentaire, les femmes enceintes et les enfants. Par ailleurs, les personnes en difficulté qui sont assujetties à la franchise peuvent demander à bénéficier d’une aide sociale financée par le fonds d’action sociale de la sécurité sociale.
Compte tenu du décalage des remboursements, la CNAMTS estimait que pour le mois de janvier 2008 il était difficile d’évaluer précisément l’impact éventuel des franchises sur les volumes de soins de ville. Pour le premier mois de l’année 2008, le montant de la franchise s’est, au total, élevé à 69 millions d’euros. Dans sa note d’actualité concernant le mois de février 2008, la CNAMTS souligne la tendance à la croissance modérée des dépenses de soins de ville remboursées par le régime général observée depuis plusieurs mois, notamment en raison d’un contexte épidémique plus favorable qu’en 2007. Dans ce contexte, le poste médicaments est en diminution sensible : - 3,6 % (en données corrigées des jours ouvrables et des variations saisonnières). La CNAMTS ajoute que « l’évolution du poste médicaments traduit en particulier la mise en place des franchises médicales depuis le 1er janvier ». Mais elle ne fournit pas davantage de précisions. Il faudra attendre de connaître les données du premier trimestre 2008 pour pouvoir procéder à une première évaluation de l’impact de la franchise sur la prescription et la consommation de médicaments.
L’instauration de la franchise vise notamment à responsabiliser les patients sur leur consommation de soins. Cependant, plusieurs personnes auditionnées par la Mission ont estimé que la franchise ne permettra pas d’infléchir durablement la consommation de médicaments. Leur prévision est qu’elle pourrait entraîner une réduction passagère liée à l’utilisation des médicaments stockés dans les armoires à pharmacie familiales, avant que la consommation ne retrouve le niveau et le rythme antérieur de progression. Par ailleurs, le risque de voir certains malades refuser ou retarder le recours aux soins et aux médicaments a été évoqué qui pourrait entraîner l’aggravation de certaines pathologies et, finalement, une consommation de soins accrue.
La MECSS souhaite que soit assuré l’égal accès aux médicaments et que soit prévu un suivi précis des effets de l’instauration de la franchise sur la prescription et la consommation de médicaments. La Mission souhaite que l’évaluation permette notamment de prendre la mesure d’éventuels retards dans le recours aux médicaments par les patients et de leurs conséquences sanitaires. La MECSS souhaite aussi que soient évalués les effets de la franchise sur le développement des médicaments génériques.
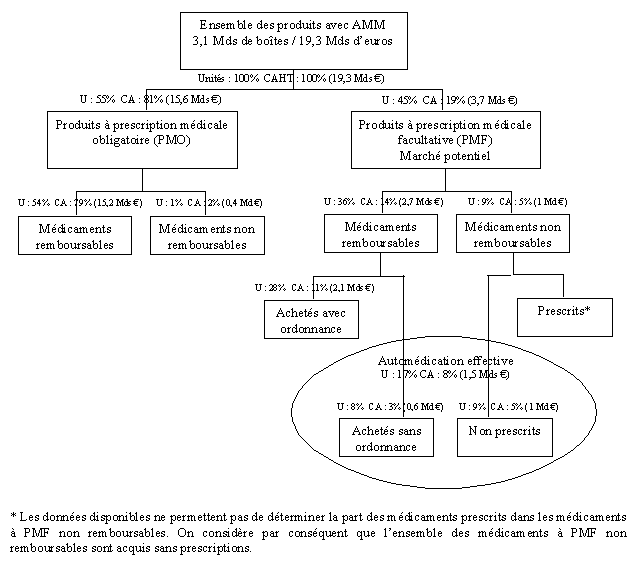
c) Les médicaments consommés sont, pour l’essentiel, soumis à prescription médicale obligatoire
Les médicaments consommés peuvent être soumis à prescription médicale obligatoire (PMO), faire l’objet d’une prescription médicale facultative (PMF) ou être autoconsommés, c’est-à-dire être directement achetés en officine par le consommateur, sans prescription médicale. L’essentiel des médicaments consommés en France est constitué de produits à prescription médicale obligatoire, lesquels sont pratiquement tous remboursables, puisque seulement 1 % des unités et 2 % du chiffre d’affaires ne le sont pas.
En 2005, les médicaments PMO ont représenté 55 % du marché total en volumes mais 81 % en valeur.
Les produits à prescription médicale facultative ont donc représenté 45 % du marché total en volume (1,4 milliard de boîtes) mais seulement 19 % du chiffre d’affaires (3,6 milliards d’euros). Cependant, l’immense majorité des produits de prescription médicale facultative est remboursable : 80 % en unités et 75 % en valeur.
Cette situation est spécifique à la France, puisque dans de nombreux pays on assimile totalement ou largement prescription médicale facultative et médicaments non remboursables.
Répartition du marché des médicaments selon l’obligation de prescription
et le taux de remboursement (chiffre d’affaires 2005 - prix fabricants hors taxes en millions d’euros)
Vignette |
Prescription obligatoire |
Prescription facultative |
TOTAL |
Non remboursable |
366 |
941 |
1 308 |
15 % |
0 |
344 |
344 |
35 % |
1 415 |
1 464 |
2 879 |
65 % |
12 618 |
867 |
13 486 |
100 % |
1 263 |
0 |
1 263 |
Total |
15 662 |
3 617 |
19 279 |
Source : données GERS ; exploitation secrétariat général du HCAAM
On peut ainsi constater que :
– d’une part, la prescription est le mode privilégié d’accès au médicament ;
– d’autre part, les médicaments prescrits sont presque toujours remboursables.
Cela traduit, fort logiquement, la préférence partagée par les médecins et les patients pour les médicaments prescrits et remboursables.
d) L’automédication occupe une place limitée et stagne
Le rapport sur la situation de l’automédication en France et les perspectives d’évolution, remis au gouvernement par MM. Alain Coulomb et Alain Baumelou, en janvier 2007, souligne que le marché de l’automédication français se distingue de celui des pays voisins européens par sa faible importance, en valeur comme en volume, et par sa faible dynamique.
En 2005, les médicaments d’automédication ont représenté près d’un cinquième des unités de médicaments vendues en ville (17 %) mais seulement moins d’un dixième du chiffre d’affaires total (8 %). Cet écart s’explique par le fait que les médicaments d’automédication sont en moyenne moins chers que les médicaments remboursables.
Source : rapport sur la situation de l’automédication en France et les perspectives d’évolution, janvier 2007.
En 2006, selon la DREES, les ventes de médicaments non remboursables, prescrits ou non, ont représenté 1,6 milliard d’euros de chiffre d’affaires. En outre, les médicaments non remboursables regroupaient un nombre important de présentations : 2 840 (en augmentation de près de 10 % en 2006), soit près d’un tiers du total des présentations de médicaments.
En 2007, selon les données d’IMS Health, le chiffre d’affaires de l’automédication s’est élevé à 1,9 milliard d’euros et a connu une progression de 4,4 % par rapport à 2006. L’automédication représentait 423 millions d’unités vendues (en augmentation de 4,1 %), soit une part de marché de 13,2 % en volume. Le prix public moyen des médicaments d’automédication s’élevait à 4,56 euros par boîte. Comme pour les produits remboursables, c’est l’innovation qui tire la croissance du secteur. Ainsi, les 95 nouveaux produits lancés en 2007 ont contribué à hauteur de 61 % à l’augmentation du chiffre d’affaires. En outre, les dix premières marques de médicaments d’automédication représentent un cinquième du chiffre d’affaires des produits à prescription médicale facultative non remboursables.
Cependant, le rapport Coulomb-Baumelou précité indique qu’entre 2000 et 2005, alors que le marché pharmaceutique total a évolué de 5,9 % par an en valeur et de 0,7 % en volume, le marché des médicaments PMF a stagné tant en valeur qu’en volume et que la part des PMF est en recul constant.
Cette stagnation des médicaments de PMF résulte, d’une part, d’un accroissement des ventes de médicaments remboursés au sein des PMF, d’autre part, d’une régression de l’automédication (- 2,1 % en volume et - 1 % en valeur).
La comparaison des cinq principaux marchés européens de l’automédication montre que la France est en dernière position après l’Allemagne, le Royaume-Uni, l’Italie et l’Espagne.
Au total, alors que les dépenses de médicaments en France sont parmi les plus élevées de l’OCDE, celles-ci concernent peu les produits d’automédication et relativement moins que dans les autres pays européens : 27 euros par personne et par an en France, contre 60 euros en Allemagne et 40 euros au Royaume-Uni et en Italie.
e) Les effets des changements dans les conditions de remboursement sont différents selon qu’il s’agit d’une diminution du taux de remboursement ou d’un déremboursement
La politique de remboursement est un élément stratégique de pilotage du marché des médicaments. Il est donc important de connaître, à partir des expériences passées, l’impact des changements de conditions de remboursement sur la consommation de médicaments.
En ce qui concerne les produits dont les taux de remboursement sont passés de 65 % à 35 %, la réduction du taux de remboursement n’a pas entraîné une réduction de la consommation des médicaments concernés, et leur évolution tendancielle n’a pas été modifiée. En outre, la réduction du taux de remboursement n’a pas conduit à des phénomènes de substitution entre produits de la même classe. La structure de consommation de ces médicaments a finalement peu changé. De plus, les prix des produits, qui ne sont pas libres du fait de leur caractère remboursable, ont varié de façon limitée en dehors des négociations régulières. Au total, la diminution du taux de remboursement n’a pas eu d’incidence sur la consommation. Ce constat est logique dans la mesure où les couvertures complémentaires garantissent le plus souvent le remboursement total des médicaments remboursables, quelle que soit la part remboursée par les régimes obligatoires. La demande de médicaments est donc relativement peu sensible à la diminution du taux de remboursement. Toutefois, dans certains cas, on note un effet de déport de la prescription vers d’autres médicaments mieux remboursés.
En revanche, en cas de déremboursement, la consommation des produits déremboursés diminue fortement. La diminution, mesurée par le chiffre d’affaires, est encore plus nette quand on tient compte de la politique de prix des laboratoires. En effet, en cas de déremboursement, les laboratoires peuvent fixer librement les prix des produits et, en général, choisissent d’augmenter fortement les prix de ces produits. Le plus souvent, cette augmentation des prix ne permet pas de maintenir la consommation de ces produits au niveau antérieur au déremboursement. On observe même que l’augmentation des prix, qui peut être forte (par exemple de 50 %, dès le mois suivant le déremboursement), peut entraîner l’accélération du cycle de vie du produit et son déclin. En outre, dans la plupart des cas, la diminution du chiffre d’affaires des produits déremboursés semble n’induire qu’un effet de substitution limité par des produits restant remboursés au sein des mêmes classes de médicaments. Toutefois, dans certains cas, on observe un effet de substitution plus marqué des produits déremboursés par des produits innovants non remboursés par la sécurité sociale. Dans ces cas, l’aspect innovant du produit l’emporte sur son caractère non remboursable et le nouveau produit concentre sur lui les consommations des anciens produits.
3. Le fort recours aux médicaments n’est pas toujours justifié et peut entraîner des effets sanitaires néfastes
À côté du défi financier que représente la forte consommation de médicaments, celle-ci recouvre aussi des enjeux de santé publique importants.
a) La surconsommation médicamenteuse française n’apparaît pas pleinement justifiée au regard des indicateurs de morbi-mortalité
Si, globalement, les indicateurs de santé de la France sont favorables, ils ne sont pas forcément meilleurs que ceux des pays comparables qui dépensent moins pour leur santé et consomment moins de médicaments.
Mme Roselyne Bachelot, ministre de la santé, de la jeunesse, des sports et de la vie associative, a ainsi remarqué, lors de son audition par la MECCS, le 12 février 2008 : « Tout le monde reconnaît que la consommation de médicaments par les Français n’est pas en corrélation avec nos performances en matière de morbidité et de mortalité. »
Cependant, les résultats d’une étude académique du Commonwealth Fund, publiée au mois de janvier 2008, et réalisée dans 19 pays à niveau de prise en charge de la santé comparable à la France, montre que notre pays est placé en première position pour la guérison des maladies curables. Il est bien évidemment difficile de distinguer ce qui, dans ce résultat, est imputable à l’organisation du système de soins, à la compétence des médecins, à la meilleure observance des traitements par les patients ou à d’autres facteurs comme l’importance de la consommation de médicaments.
b) La surconsommation de médicaments peut avoir des effets négatifs en termes de santé publique
Le mauvais usage du médicament peut se traduire par une quantité excessive, une qualité inadaptée ou encore par la non-observance du traitement prescrit par le médecin. Le mésusage peut entraîner des risques de iatrogénie et d’accidents liés à la prise de médicaments. Cela peut même conduire au développement de résistances et à une perte d’efficacité du médicament, comme dans le cas des antibiotiques.
Actuellement, il est estimé que la iatrogénie médicamenteuse est responsable de 130 000 hospitalisations par an, ce qui représente 5 à 10 % des hospitalisations au total. Or, il est considéré que 40 à 60 % de ces événements iatrogènes, qui sont d’origines diverses, sont évitables.
La population des plus de 65 ans est la plus exposée, notamment en raison du nombre important mais souvent nécessaire de médicaments qu’elle consomme. En effet, parmi les 10 millions de personnes âgées de 65 ans ou plus, environ 1,5 million consomment quotidiennement sept médicaments ou plus de classes thérapeutiques différentes.
Par ailleurs, en France, la résistance aux antibiotiques est un important problème de santé publique. Avec plus de 30 doses d’antibiotiques par jour pour 1 000 habitants, la France consomme deux fois plus d’antibiotiques que l’Allemagne et le Royaume Uni et trois fois plus que les Pays-Bas. Cette consommation est aussi la plus importante des pays européens pendant les mois d’hiver, période de recrudescence des affections hivernales. Elle augmente de 33 % à cette période contre 20 % au maximum dans les pays du nord (Suède, Danemark, Norvège). Cette forte consommation d’antibiotiques a une conséquence regrettable : la France est le pays européen où les phénomènes de résistances aux antibiotiques sont les plus élevés. Cette situation est susceptible d’entraîner une perte d’efficacité des antibiotiques et des pertes de chance de guérison pour les patients.
Partant de ce constat sur le modèle de surconsommation français de médicaments, la MECSS a souhaité formuler un ensemble cohérent de propositions concernant les différents acteurs de la chaîne du médicament pour faire évoluer les comportements de prescription et de consommation dans le sens du bon usage.
II.- AMÉLIORER L’ENCADREMENT DE LA VIE DU MÉDICAMENT ET RENFORCER LA SÉLECTIVITÉ DE L’ACCÈS AU REMBOURSEMENT
En France, compte tenu des caractéristiques du médicament, c’est-à-dire son utilité en matière de soins et sa dangerosité potentielle, la mise sur le marché ainsi que la distribution et la dispensation sont strictement encadrées. De même, en raison de la prise en charge collective d’une grande partie du coût des médicaments remboursables, ceux-ci relèvent d’un régime de prix administrés et l’admission au remboursement est décidée par l’État ainsi que le niveau de remboursement.
Des améliorations pourraient être apportées tant en matière de mise sur le marché qu’en ce qui concerne l’admission au remboursement et le suivi des médicaments en pratique médicale réelle.
A. AMÉLIORER L’ENCADREMENT DE LA VIE DU MÉDICAMENT
1. Le circuit administratif du médicament est complexe
a) Le chemin est long, du fabricant au consommateur
Le médicament n’est pas un produit comme les autres. En raison de sa nature et du fait qu’il est généralement pris en charge par la collectivité, il fait l’objet d’un encadrement de sécurité sanitaire et de conditions de mise sur le marché et de vente spécifiques.
Pour être commercialisé en France, un médicament doit avoir obtenu une autorisation de mise sur le marché (AMM), délivrée par l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) ou par l’Agence européenne d’évaluation du médicament (EMEA). L’AMM résume les caractéristiques du médicament, sa composition, les maladies qu’il est destiné à soigner ou à prévenir, son mode d'action et ses limites d'utilisation.
La Commission de la transparence, constituée, au sein de la Haute Autorité de santé (HAS), de médecins hospitaliers, de médecins généralistes et de pharmaciens, évalue ensuite, indication par indication, le service médical rendu (SMR) et l’amélioration du service médical rendu (ASMR) par le médicament.
La décision d’inscription sur la liste des spécialités remboursables est prise par le ministre en charge de la santé au vu de l’avis rendu par la Commission de la transparence.
Le prix des médicaments remboursables par l’assurance maladie est fixé par le Comité économique des produits de santé (CEPS) en fonction de l’amélioration du service médical rendu (ASMR) et après négociations avec les laboratoires qui les produisent.
Les taux de remboursement sont fixés par l’UNCAM, l’Union nationale des caisses d’assurance maladie. Ils varient en fonction du service médical rendu (SMR), tel qu’apprécié par la Commission de la transparence :
– médicaments irremplaçables et coûteux (vignette blanche et barrée) : 100 % ;
– médicaments dont le service médical rendu a été considéré comme majeur ou important (vignette blanche) : 65 % ;
– médicaments destinés au traitement des affections sans caractère habituel de gravité ou dont le service médical rendu n’a pas été considéré comme majeur ou important (vignette bleue) : 35 % ;
– médicaments destinés au traitement de certaines affections sans caractère de gravité et dont le service médical rendu est jugé insuffisant : 15 % (taux créé à titre provisoire jusqu’au 1er janvier 2008 pour certains médicaments antérieurement mieux remboursés mais désormais classés à SMR insuffisant à la suite de leur réévaluation).
La somme qui reste à la charge de l’assuré social après le remboursement par l’assurance maladie est le ticket modérateur. Celui-ci peut être remboursé par une mutuelle ou une assurance complémentaire.
En cas d'affection grave et de longue durée (ALD) inscrite sur une liste officielle de trente maladies, les médicaments destinés à soigner cette maladie peuvent être remboursés au taux de 100 %, quelle que soit leur catégorie. C’est également le cas pour d’autres maladies graves, lorsqu’elles sont déclarées « hors liste » par un médecin conseil de l’assurance maladie.
Le prix des médicaments non remboursables est libre et peut donc varier d’une pharmacie à l'autre.
Les médicaments disponibles sur ordonnance (médicaments à prescription médicale obligatoire : PMO) sont les médicaments présentant des difficultés d'emploi ou des risques en cas d'utilisation inappropriée. Ils sont inscrits sur deux listes distinctes : la liste I ou la liste II. Ils ne peuvent être obtenus que sur prescription d’un médecin, d’un dentiste ou d’une sage-femme.
En France, les médicaments sont tous vendus au public dans des pharmacies (officines et pharmacies hospitalières), qu’ils soient disponibles sur ordonnance ou non. Les médicaments PMO inscrits sur la liste I ne peuvent être délivrés qu’une seule fois par le pharmacien, sauf si le médecin mentionne expressément la possibilité d’un renouvellement sur son ordonnance. La délivrance des médicaments PMO inscrits sur la liste II peut être renouvelée pendant six mois, même si le médecin ne le mentionne pas. À chaque renouvellement, le pharmacien ne délivre que la quantité nécessaire à un mois de traitement, sauf dans le cas des contraceptifs.
Les médicaments dont la prescription par un médecin n’est pas obligatoire (les médicaments sans ordonnance ou à prescription médicale facultative : PMF), souvent utilisés dans le cadre de l’automédication, sont soumis aux mêmes règles de surveillance que les médicaments à prescription obligatoire.
Les médicaments à prescription médicale obligatoire sont en principe remboursables. Certains médicaments à prescription médicale facultative sont remboursables, d’autres ne le sont pas.
b) En France, la régulation du médicament est répartie entre plusieurs acteurs
Les acteurs intervenant dans la régulation du secteur du médicament sont nombreux. Les compétences sont éclatées et le circuit administratif du médicament remboursable est complexe.
La France a fait le choix, confirmé avec la loi du 13 août 2004 relative à l’assurance maladie, d’une organisation complexe de la régulation du secteur du médicament qui pose certains problèmes de pilotage et de coordination.
Dans une note de veille sur l’État et les agences, de janvier 2008, le Centre d’analyse stratégique (CAS) soulignait, en faisant référence aux agences sanitaires du type de l’AFSSAPS, à laquelle est délégué l’octroi des autorisations de mise sur le marché des médicaments, le risque de « tutelle inversée ». La note du CAS indique notamment : « Il est peut-être difficile pour les 300 fonctionnaires de la direction générale de la santé de coordonner et de piloter l’action des sept agences mentionnées quand les seuls effectifs cumulés des deux principales (l’Agence française de sécurité sanitaire de l’alimentation et l’Agence française de sécurité sanitaire des produits de santé) s’élèvent déjà à 2 000 agents. »
Par ailleurs, le conseil de modernisation des politiques publiques du 4 avril 2008 a décidé de regrouper les agences sanitaires nationales en pôles cohérents correspondant à leurs grandes missions. L’objectif est de simplifier les conditions de pilotage par l’État des agences, de renforcer leurs capacités d’expertise interne, de réduire les risques de redondance entre elles et de rendre plus lisible l’ensemble du dispositif.
Le même conseil de modernisation a également décidé de réorganiser l’administration centrale de la santé et de la solidarité et, à cet effet, de clarifier le rôle et la position de certaines missions et délégations, cette clarification pouvant aller jusqu’à la ré-internalisation de certaines missions au sein des directions d’administration centrales.
c) D’autres pays ont un système de régulation plus regroupé
L’organisation française de la régulation du médicament est proche de celle qui prévaut en Grande-Bretagne. En revanche, d’autres pays comme la Belgique ont fait le choix de confier à une instance unique le soin de veiller à la qualité, la sécurité et l’efficacité des médicaments, de leur conception à leur emploi, dans l’intérêt de la santé publique. Placée au niveau fédéral, la direction belge du médicament regroupe ainsi les fonctions recherche et développement, la mise sur le marché des médicaments, la vigilance sur leur utilisation, le contrôle de la production et de la distribution, ainsi que le contrôle de la publicité, de l’information et du bon usage des médicaments.
Quelle que soit l’organisation, l’objectif est d’assurer la sécurité des médicaments et leur bon usage. Cela suppose, en premier lieu, de veiller à l’indépendance des experts en charge de vérifier le respect des conditions de mise sur le marché des produits.
2. Veiller au bon usage des procédures dérogatoires de mise sur le marché et de prescription
Comme cela a été rappelé, en principe, pour être commercialisés, tous les médicaments doivent faire l’objet d’une autorisation de mise sur le marché. Toutefois, certains médicaments ne disposant pas d’AMM peuvent être exceptionnellement commercialisés en obtenant une autorisation temporaire d’utilisation (ATU). Par ailleurs, les médicaments disposant d’une AMM peuvent être prescrits en dehors des indications de l’autorisation.
Ces deux pratiques qui ont pour objet de contourner l’AMM et tendent à se développer devraient être mieux contrôlées.
a) Veiller au bon usage de l’autorisation temporaire d’utilisation
Une ATU peut être délivrée par l’AFSSAPS à des médicaments nouveaux qui sont autorisés à l’étranger ou encore en cours de développement. Elles sont délivrées aux spécialités destinées à traiter, prévenir ou diagnostiquer des maladies graves ou rares, lorsqu’il n’existe pas de traitement approprié, et que leur efficacité et leur sécurité d'emploi sont présumées en l'état des connaissances scientifiques.
Il existe deux types d’autorisation temporaire d’utilisation. L’ATU nominative est délivrée pour un seul malade nommément désigné, à la demande et sous la responsabilité du médecin prescripteur. L’ATU de cohorte concerne un groupe de patients, traités et surveillés suivant des critères définis dans un protocole d’utilisation thérapeutique et de recueil d’informations. Elle est délivrée à la demande du titulaire des droits d’exploitation, qui s'engage à déposer une demande d’AMM dans un délai fixé.
Ce dispositif, mis en place en France depuis 1994, concerne plusieurs centaines de spécialités pharmaceutiques et a permis le traitement par de nouveaux médicaments, plusieurs mois avant leur AMM, de plusieurs dizaines de milliers de patients en situation d’échec thérapeutique. En 2004, plus de 24 000 ATU nominatives concernant plus de 180 spécialités ont été octroyées par l’AFSSAPS.
En outre, le prix des médicaments disposant d’une autorisation temporaire d’utilisation est libre. Il est tout de même prévu la possibilité de récupérer tout ou partie de la différence entre le prix pratiqué sous le régime de l’ATU et le prix fixé par le CEPS après l’AMM. Cependant, pour certains médicaments, le délai pour passer de l’ATU à l’AMM est particulièrement long et peut atteindre sept ans.
La MECSS souhaite donc que la procédure dérogatoire de l’ATU ne soit pas utilisée pour contourner l’AMM et que le délai entre l’ATU et l’AMM soit le plus court possible.
b) Contrôler et évaluer les prescriptions hors autorisations de mise sur le marché (AMM)
Le résumé des caractéristiques du produit (RCP) définit les conditions d’utilisation du médicament. En principe, les prescriptions de médicaments en dehors des indications de l’AMM ne peuvent donc être admises au remboursement. Cependant, on estime que les prescriptions hors AMM pourraient représenter 15 % à 20 % du total des prescriptions. Les prescriptions hors AMM peuvent résulter d’erreurs de prescription mais aussi du délai de mise à jour de l’AMM en fonction de nouvelles données scientifiques ou du refus des laboratoires de modifier l’AMM pour éviter le coût des essais.
À l’hôpital, avec la mise en place de la tarification à l’activité, la prescription hors AMM de médicaments innovants et coûteux a été autorisée dans le cadre de protocoles thérapeutiques définis par l’AFSSAPS, la HAS ou l’Institut national du cancer (INca).
Depuis 2007, un dispositif dérogatoire analogue est prévu pour la médecine de ville. Lorsque, pour le traitement d’une affection de longue durée, il n’existe pas d’alternative appropriée, tout médicament utilisé hors AMM peut faire l’objet d’un remboursement pour une durée limitée et dans le cadre d’un avis ou d’une recommandation de la HAS pris après consultation de l’AFSSAPS.
Il convient de s’assurer que les nouvelles règles de prescription hors AMM ne sont pas utilisées par les laboratoires pour contourner l’AMM.
À cet effet, la MECSS souhaite que les prescriptions hors AMM en médecine de ville soient contrôlées et qu’il soit procédé à une évaluation de l’impact médico-économique de ces prescriptions, en particulier de celles effectuées dans le cadre du dispositif dérogatoire. La Mission considère que sur la base des résultats de l’évaluation, un renforcement de l’encadrement des prescriptions hors AMM pourrait, le cas échéant, être envisagé. La MECSS souhaite également que la HAS agisse auprès des médecins, aussitôt après que le médicament est mis sur le marché, afin d’éviter l’installation d’usages non conformes aux indications de l’AMM.
B. RENFORCER LA SÉLECTIVITÉ DE L’ACCÈS AU REMBOURSEMENT
1. Renforcer les règles de l’admission au remboursement et de fixation du prix
a) Recourir au critère de l’intérêt de santé publique pour l’appréciation du service médical rendu (SMR)
Actuellement, le système d’admission au remboursement des médicaments, fondé sur l’appréciation du SMR, est peu sélectif. La quasi-totalité des médicaments qui obtiennent une AMM sont admis au remboursement (96 %). En 2006, seules 16 spécialités ont été considérées comme ayant un SMR insuffisant au stade de la première inscription au remboursement, soit 3,5 % de l’ensemble des SMR attribués. En outre, la plupart des médicaments (87 % en 2006) se voient attribuer un SMR important qui ouvre droit à un remboursement à 65 %.
Cette situation est due au fait que le SMR est principalement apprécié en fonction de l’efficacité et des effets indésirables du médicament, c’est-à-dire de son intérêt clinique. Le critère d’admission au remboursement fondé sur l’intérêt de santé publique, c’est-à-dire l’intérêt pour la collectivité (impact du médicament sur l’état de santé de la population, réponse apportée à un besoin de santé publique et impact du médicament sur le système de santé) pourtant prévu pour l’appréciation du SMR, est peu utilisé en pratique. La prise en compte effective du critère d’intérêt de santé publique permettrait de donner une dimension collective à l’appréciation du SMR et à l’admission au remboursement et irait dans le sens d’une meilleure analyse médico-économique. Cette évolution devrait être aussi l’occasion de clarifier la notion de SMR insuffisant ; celui-ci devrait signifier soit que l’intérêt clinique du médicament est insuffisant, soit que l’intérêt qu’il présente pour la santé publique n’est pas suffisant pour justifier une prise en charge par la collectivité.
La concrétisation du projet de réforme de l’appréciation du service médical attendu (lors de l’évaluation initiale) ou rendu (lors d’une réévaluation) par le médicament qui prévoit trois niveaux de cotation – important, modéré et mineur – pourrait permettre d’aller dans le sens souhaité puisqu’il est prévu qu’en cas de service médical mineur, le médicament ne peut être inscrit sur la liste des médicaments remboursables.
M. Didier Houssin, directeur général de la santé, a indiqué, lors de son audition par la MECSS, partager le constat de l’insuffisante sélectivité de l’admission au remboursement des médicaments. Il a souhaité que la dimension médico-économique et l’intérêt de santé publique soient mieux pris en compte par la Commission de la transparence. En outre, après avoir souligné que la Commission de la transparence se distingue trop peu de la Commission d’AMM, il a évoqué la possibilité, d’une part, de transférer la compétence actuelle de la Commission de la transparence à la Commission d’AMM, d’autre part, de confier à la HAS ou à une nouvelle structure l’appréciation de l’intérêt de santé publique.
La MECSS souhaite une meilleure sélectivité dans l’admission au remboursement des médicaments. À cet effet, elle préconise le recours au critère de l’intérêt de santé publique pour l’appréciation du SMR, dans l’attente d’une réforme plus globale.
Par ailleurs, la Mission souhaite que des contrôles puissent être effectués en ce qui concerne le respect des règles relatives aux indications remboursables et aux indications non remboursables pour lesquelles l’ordonnance doit, en principe, porter la mention « NR » (non remboursable).
b) Développer les essais cliniques contre comparateurs pour l’appréciation de l’amélioration du service médical rendu (ASMR)
L’appréciation de l’amélioration du service médical rendu détermine le niveau de prix du médicament mais aussi l’admission au remboursement pour les médicaments qui n’apportent pas d’amélioration du service médical rendu mais qui apportent une économie dans le coût du traitement médicamenteux. Or, actuellement, l’appréciation de l’ASMR est le plus souvent fondée sur la seule comparaison du médicament avec un placebo et dans moins de la moitié des cas sur une comparaison avec des comparateurs. Cependant, une majorité des médicaments qui sont évalués par la Commission de la transparence n’apportent pas d’amélioration du service médical rendu (58 % en 2005, 54 % en 2006).
La MECSS, dans le même esprit de privilégier l’admission au remboursement des médicaments qui apportent une réelle ASMR, demande que l’appréciation de l’ASMR soit fondée non seulement sur une comparaison avec un placebo mais également sur des essais cliniques contre comparateurs, lorsqu’ils existent, afin de mesurer la valeur ajoutée thérapeutique. En outre, dans un but de transparence et d’analyse des remboursements de médicaments selon leur ASMR, la Mission souhaite que soit établie une liste des médicaments classés par niveau d’ASMR.
c) Veiller à la mise en œuvre rapide de la nouvelle compétence médico-économique de la Haute Autorité de santé (HAS)
La loi de financement de la sécurité sociale pour 2008 a doté la HAS d’une compétence médico-économique.
Cet élargissement de la compétence de la HAS doit lui permettre de développer l’appréciation du rapport coût-efficacité et la transversabilité de l’approche. La HAS, à la différence de l’AFSSAPS, peut désormais procéder à des comparaisons entre le médicament et les autres possibilités thérapeutiques (chirurgie, radiothérapie…). Le médicament sera replacé dans le cadre d’une stratégie globale de prise en charge, et la HAS devrait pouvoir, au regard des évaluations médico-économiques et des analyses sur la complémentarité des stratégies thérapeutiques, recommander ou non une stratégie thérapeutique médicamenteuse.
Cela suppose que la Haute Autorité de santé mette rapidement en place les outils qui lui permettront de développer sa capacité d’analyse médico-économique. La HAS a déjà engagé la réflexion sur ce sujet. Celle-ci pourrait utilement s’inspirer des expériences étrangères, notamment celles de l’Allemagne et du Royaume-Uni qui consistent à définir des priorités concernant l’admission au remboursement et la définition du panier de médicaments pris en charge.
La HAS considère que l’intégration de la dimension économique dans la décision de santé ne peut être durablement acceptée que si elle s’inscrit dans une démarche globale de prise en compte des dimensions collectives. Pour désigner cette approche globale de l’évaluation en santé, qui intégrerait les aspects économiques, sociaux et éthiques, elle envisage de recourir à la notion d’évaluation du « service rendu à la collectivité » (SERC). Préalablement à la montée en charge de sa nouvelle compétence médico-économique, la HAS a engagé des concertations sur ce thème, au premier trimestre 2008, et elle a prévu de constituer un réseau de partenaires pour développer la culture de l’évaluation et un groupe d’experts pour définir les méthodes d’évaluation du SERC.
En outre, la HAS a prévu de mener, en 2008, des analyses médico-économiques dans le cadre de la réévaluation de plusieurs classes de médicaments : les IEC (utilisés dans le traitement de l’hypertension et de l’insuffisance cardiaque) et les sartans (diabète), les IPP (acidités de l’estomac) et les statines (cholestérol). Des évaluations de type SERC seront aussi engagées concernant les hormones de croissance, certaines injections intra-vitréennes pour lutter contre la dégénérescence maculaire liée à l’âge et la prise en charge médicamenteuse de la maladie d’Alzheimer.
Par ailleurs, un programme d’étude pluriannuel porte sur la prise en charge par les assurés de leur santé et le recours au médecin. Dans ce cadre, doivent notamment être abordées les questions relatives à la responsabilisation du patient et au développement des alternatives aux prescriptions médicamenteuses.
Afin de mettre en œuvre sa nouvelle compétence médico-économique, la HAS a annoncé, en mars 2008, la mise en place d’une nouvelle organisation qui vise à favoriser l’approche transversale et globale des sujets.
La HAS est désormais organisée pour assumer ses deux missions principales qui consistent à :
– donner un avis sur le panier de biens et services (médicaments, actes et dispositifs) et sur les actions de santé publique, en associant une approche médicale et une approche économique ;
– mettre en œuvre des actions d’amélioration de la qualité des soins, selon une approche mieux intégrée allant de la recommandation aux actions de mise en œuvre.
À cet effet, deux nouvelles directions sont mises en place :
– La direction de l’évaluation médicale, économique et de santé publique (DEMESP) est chargée de l’évaluation des médicaments, actes et dispositifs, ainsi que de la production de recommandations et de rapports d’orientation en santé publique. Le développement de l’évaluation médico-économique est l’une des priorités de cette nouvelle direction ;
– La direction de l’amélioration de la qualité et de la sécurité des soins (DAQSS) se voit confier la gestion de l’ensemble des dispositifs dédiés à l’amélioration des pratiques : certification, indicateurs, évaluation des pratiques professionnelles et accréditation, recommandations de bonnes pratiques et prise en charge des ALD. L’objectif est de favoriser l’optimisation du panier de biens et l’amélioration effective de la qualité et de la sécurité des soins.
La MECSS considère que la HAS, désormais dotée d’une nouvelle compétence médico-économique, devrait participer à la redéfinition régulière des contours du panier de médicaments pris en charge. Elle souhaite que la HAS conduise une réflexion sur les moyens les plus efficaces de réduire la surprescription qualitative et quantitative, les usages irrationnels et injustifiés de médicaments ainsi que la valorisation de la qualité des pratiques. Dans cet esprit, la Mission souhaite que la HAS mène rapidement ses travaux sur le développement des alternatives à la prescription de médicaments. Elle souhaite aussi que la HAS évalue mieux l’impact de ses recommandations et mette en place des tableaux de bord pour mesurer l’évolution des comportements de prescription des médicaments. La MECSS souhaite également que la HAS puisse disposer d’un corps de délégués de santé pour promouvoir le bon usage des médicaments
2. Améliorer le suivi des médicaments en pratique médicale réelle
Le suivi des médicaments après leur commercialisation reste insuffisant et les conséquences qui sont tirées de l’observation en vie réelle des médicaments sont souvent partielles et tardives.
a) Renforcer l’efficacité de la pharmacovigilance
Avant de délivrer l’AMM, l’AFSSAPS effectue une évaluation de la balance bénéfice-risque du médicament. Après la mise sur le marché, l’AFSSAPS doit surveiller le risque et le bon usage du médicament. Cette mission de pharmacovigilance de l’agence revêt une importance accrue depuis que la loi du 26 février 2007 transposant la directive européenne 2004/27/CE sur le médicament a prévu la possibilité de renouvellement de l’AMM sans limitation de durée (l’AMM est, en principe, délivrée pour cinq ans).
En fonction du résultat des réévaluations, l’AFSSAPS peut décider le retrait ou la suspension de l’AMM ou prévoir des restrictions d’indications. Cependant, parfois les décisions de l’Agence sont tardives (jusqu’à quatre ans après les résultats de l’enquête montrant les effets indésirables) ou limitée à de simples restrictions d’indications alors que l’efficacité des médicaments n’est pas avérée. En outre, la transparence sur les études de pharmacovigilance n’est pas toujours assurée ou les résultats des études sont publiés tardivement. Aussi, l’information en direction des prescripteurs n’est pas non plus toujours assurée ni le contrôle de l’application des restrictions d’indications.
En conséquence, la MECSS souhaite, d’une part que les résultats des études de pharmacovigilance soient publiés rapidement et que les décisions de suspension ou de retrait d’AMM soient prises sans retards, d’autre part que l’information sur les restrictions d’indications soit systématisée et que le contrôle de leur application soit assuré.
b) Développer l’évaluation post-autorisation de mise sur le marché
Au-delà de la stricte pharmacovigilance, les études post-AMM peuvent aussi contribuer à la réalisation de l’objectif de sécurité des médicaments. Mais, les dispositions récentes visant à renforcer les obligations dans ce domaine sont peu appliquées et l’intervention publique reste trop réduite.
Depuis fin 2005, les laboratoires sont tenus d’intégrer des plans de gestion des risques (PGR) dans le dossier d’AMM pour certains produits (nouveaux médicaments, médicaments génériques en cas de risque identifié sur le princeps, extension d’AMM avec changements significatifs des conditions d’emploi). Ces plans peuvent prévoir, à la demande de l’AFSSAPS, des études post-AMM et/ou un plan de minimisation des risques.
Des études post-AMM peuvent aussi être demandées par le CEPS en application de l’accord-cadre conclu avec le LEEM – les entreprises du médicament – en 2003, visant à les développer et à assurer leur financement. Mais, en l’absence d’un dispositif de sanction applicable, les dispositions de cet accord sont encore peu appliquées. L’article 42 de la loi de financement de la sécurité sociale pour 2008 qui visait justement à prévoir un dispositif de sanction, a été annulé par le Conseil constitutionnel pour un motif de forme. Cette disposition devra être reprise rapidement.
Pour sa part, l’AFSSAPS développe un programme d’études pharmaco-épidémiologiques mais ces dernières ne sont pas toujours publiées. Par ailleurs, l’activité du groupement d’intérêt scientifique « évaluation épidémiologique des produits de santé », qui réuni le ministère de la santé, la CNAMTS et l’INSERM, reste limitée. Dans certains cas, la difficulté d’accéder à des données nominatives peut ralentir voire empêcher la réalisation des études.
La MECSS souhaite, d’une part, que l’action publique soit plus active en matière d’évaluation post-AMM et que soit relancée l’activité du groupement d’intérêt scientifique « évaluation épidémiologique des produits de santé », d’autre part, que soit instauré un dispositif de sanction pour défaut ou retard de réalisation d’études post-AMM.
c) Gérer de manière plus active la liste des médicaments remboursables
Les conséquences des réévaluations des médicaments ne sont pas non plus toujours tirées dans les meilleurs délais en ce qui concerne les conditions de remboursement.
Depuis 1999, l’ensemble des médicaments remboursables a fait l’objet d’une réévaluation du SMR. Sur les 4 490 spécialités réévaluées par la Commission de la transparence, 835, soit près de 20 %, ont présenté un SMR insuffisant pour justifier leur remboursement par la sécurité sociale. L’AMM de certaines spécialités a été retirée.
S’agissant des spécialités dont l’AMM a été maintenue, le ministre en charge de la santé n’a pas toujours suivi l’avis de déremboursement de la HAS. Pour certaines spécialités, il a bien été décidé de suivre l’avis de la HAS et de dérembourser les spécialités concernées. En revanche, pour d’autres, le remboursement a été maintenu, à titre provisoire, mais le taux de remboursement est passé à 35 % ou 15 % et, dans certains cas, une baisse de prix pouvant aller jusqu’à 20 % leur a été aussi appliquée. De ce fait, on ne tire pas tout le parti des réévaluations et les économies potentielles qui devraient en résulter ne sont que partiellement réalisées.
Or, l’objectif d’affectation en priorité des financements collectifs à la prise en charge des traitements les plus performants devrait conduire à une gestion plus active de la liste des produits remboursables. La nouvelle compétence médico-économique de la HAS devrait favoriser une telle évolution.
La MECSS souhaite donc que les recommandations de la HAS consécutives à la réévaluation des SMR des médicaments remboursables soient mieux suivies et appliquées avec davantage de célérité.
III.- FAIRE ÉVOLUER LES COMPORTEMENTS DES PRESCRIPTEURS ET DES CONSOMMATEURS
La juste prescription doit permettre d’une part, d’améliorer la qualité des soins et la santé publique, d’autre part, de mieux maîtriser les dépenses de médicaments. Dans ces domaines, il semble qu’il y ait des marges de progrès considérables. Mais, le renforcement de l’efficience des prescriptions et la responsabilisation des consommateurs passent par une démarche partenariale. Seul le développement d’un partenariat de santé peut favoriser le bon usage des médicaments. Cela suppose la mobilisation et la participation de tous les acteurs pour faire évoluer les comportements de chacun, les prescripteurs comme les consommateurs.
A. RENFORCER L’INFORMATION SUR LE MÉDICAMENT
1. Assurer l’indépendance et la transparence de l’expertise
Pour assurer leur mission d’évaluation, l’AFSSAPS et la Haute Autorité de santé recourent à des experts extérieurs. Afin d’assurer la crédibilité de l’expertise, qui est essentielle tant pour les patients que pour les professionnels de santé, il est indispensable d’assurer la transparence des procédures et d’éviter les conflits d’intérêts. Or, les pratiques dans ces domaines restent perfectibles et il faut soutenir tout ce qui favorise la transparence des processus d’évaluation et de décision dans le domaine du médicament.
La loi du 26 février 2007 de transposition de la directive communautaire 2004/27 CE a fixé de nouvelles obligations pour les agences en matière de transparence. Elle prévoit le principe d’une déclaration annuelle des experts et agents. Mais cette obligation n’est pas toujours respectée. Certaines déclarations sont faites avec retard et les actualisations ne sont pas systématiques. C’est notamment le cas pour les déclarations d’intérêts de certains experts extérieurs de la Commission de la transparence qui joue un rôle déterminant en matière d’appréciation du SMR et de l’ASMR du médicament, alors même que ces éléments conditionnent très largement l’admission au remboursement et le prix du médicament. L’indépendance des experts de la Commission de la transparence est donc cruciale.
La MECSS considère que la confiance dans l’expertise est une question centrale et qu’il y a lieu d’être très vigilant en matière d’indépendance des experts et de gestion des conflits d’intérêts. Elle demande, en conséquence, à l’AFSSAPS et la HAS, de veiller, avec la plus grande rigueur, au respect des règles en vigueur.
Dans le même esprit, la MECSS souhaite que l’AFSSAPS et la HAS appliquent strictement et complètement les dispositions de la loi du 26 février 2007 concernant la transparence de l’expertise qui prévoit la publication du règlement intérieur des commissions, des ordres du jour et des comptes rendus, assortis des décisions prises et du détail des votes et des explications de vote, y compris les opinions minoritaires. La Mission souhaite que ces différents documents soient systématiquement publiés dans les meilleurs délais et, en particulier, les ordres du jour et les comptes rendus de la Commission de la transparence, lesquels ne sont toujours pas mis en ligne sur le site internet de la HAS. Par ailleurs, la MECSS souhaite que la HAS et l’AFSSAPS développent leur politique d’ouverture au public des travaux d’expertise. Cela pourrait se traduire par l’organisation de séances publiques, par exemple pour des réunions de la Commission de la transparence.
2. Mettre en place une base publique d’information, exhaustive et gratuite, sur le médicament
Actuellement, en dépit des dispositions législatives prises au début de la décennie, il n’existe aucune base d’information publique et exhaustive sur les médicaments. Les bases auxquels peuvent accéder les prescripteurs et les consommateurs, sont partielles, incomplètes et certaines sont privées.
La base AMM de l’AFSSAPS comprend essentiellement les relevés des caractéristiques des produits dont l’AMM a été délivrée après le 1er janvier 2002. Lors de son audition par la MECSS, le 4 octobre dernier, M. Jean Marimbert, directeur général de l’AFSSAPS, a indiqué que sur les 16 000 produits autorisés, dont 11 000 à 12 000 sont effectivement commercialisés, 5 000 RCP avaient été mis en ligne et que, pour la fin de 2008, l’ensemble de la production serait téléchargé sur la base médicament de l’agence. Toutefois, cette base ne comporte pas d’indication sur le service médical rendu, alors qu’il s’agit d’une information essentielle pour les prescripteurs.
De même, les deux bases privées (Vidal et Claude Bernard) qui sont payantes, et la base publique Thesorimed, qui a succédé à la base Thériaque, ne comprennent pas toutes les données utiles à la complète information de l’ensemble des partenaires de santé et, notamment, des prescripteurs.
La base Thériaque, initiée il y a plus de vingt ans par le Centre national interhospitalier d’information sur le médicament (CNHIM), est la plus complète. Mais, le GIE Système d’information sur les produits de santé (SIPS) qui est chargé de gérer et de financer la base a connu des difficultés liées, notamment, au désengagement du CNHIM du GIE, au mois d’août 2007. Et, contrairement à ce que prévoyait le projet initial, l’AFSSAPS a toujours refusé de s’associer au groupement.
La nouvelle base Thesorimed qui reprend les informations contenues dans la base Thériaque et en comprendra d’autres est en cours de finalisation et devrait être accessible gratuitement au public, c’est-à-dire à tous les professionnels de santé et au grand public, au mois d’avril 2008. Thesorimed est d’ores et déjà accessible, avec un abonnement payant, aux établissements hospitaliers. Elle sera interopérable avec les logiciels d’aide à la prescription (LAP) de médecine de ville, comme cela est déjà possible pour les LAP hospitaliers. La base comprendra une fonction d’analyse d’ordonnance qui permettra notamment de connaître les interactions médicamenteuses. Le GIE SIPS, chargé de la gérer, associe désormais la CNAMTS (40 % des voix), la Mutualité sociale agricole (MSA, 30 %) et le Régime social des indépendants (RSI, 30 %).
La HAS, pour sa part, développe une base de données comportant les fiches de transparence établies par la Commission de la transparence. Ces fiches ont pour but de guider les praticiens dans leurs prescriptions et favorisent le bon usage du médicament. Elles font la synthèse des travaux des experts sur le service médical rendu et l’amélioration du service médical rendu et visent à faire le point sur une classe thérapeutique ou sur une stratégie thérapeutique.
L’assurance maladie met aussi en ligne des informations sur les médicaments, notamment sur les taux de remboursement.
Cette dispersion des données ne peut perdurer. Il est souhaitable que les informations des trois bases soient regroupées dans une base unique, facilement accessible. Parallèlement, les bases privées pourront se développer dans le cadre de la certification des sites internet dédiés à la santé mise en œuvre par la HAS.
La MECSS considère que la création d’une base publique d’information sur les médicaments, indépendante, exhaustive, gratuite, accessible à tous les acteurs du système de santé et interopérable avec les logiciels d’aide à la prescription est indispensable. Elle veillera tout particulièrement à la réalisation de ce projet que la ministre en charge de la santé, s’est engagée, lors de son audition par la Mission, à faire aboutir pour la fin de 2009.
B. AGIR SUR LES DÉTERMINANTS DE LA PRESCRIPTION
La formation et l’information des médecins sur le médicament sont essentielles pour influencer le niveau et la qualité des prescriptions.
1. Améliorer la formation des médecins en pharmacologie et en économie de la santé
La formation est un élément déterminant de l’amélioration de l’usage du médicament dans le système de santé, en particulier en médecine de ville. Les actions de formation doivent s’inscrire dans un continuum : formation initiale, formation médicale continue et évaluation des pratiques professionnelles (EPP).
a) Réformer la formation initiale des médecins sur le médicament
Le constat est souvent fait des insuffisances de la formation médicale initiale aux questions thérapeutiques, celle-ci étant essentiellement orientée vers l’apprentissage clinique. L’enseignement de la pharmacologie est peu développé. Cette matière est peu enseignée (environ 80 heures) et il est considéré que les cours sont dispensés trop tôt dans le cursus universitaire. L’enseignement en économie de la santé est aussi insuffisant. Les futurs médecins ne sont pas informés du coût des thérapeutiques et ils sont mal informés des moyens de financement de la solidarité nationale. Il est pourtant indispensable que les jeunes médecins aient une vision des grands enjeux médico-économiques du système de santé. Et l’initiation à la politique du médicament est un moyen de préparer au bon usage de celui-ci.
Depuis septembre 2007, une clarification des compétences entre la direction générale de la santé (DGS) et la direction de l’hospitalisation et de l’organisation des soins (DHOS) a été apportée dans le pilotage de la formation médicale. La DHOS a désormais une vision d’ensemble de la formation initiale et de la formation continue.
La DHOS qui a, avec le ministère de l’enseignement supérieur et de la recherche, la responsabilité directe de la formation initiale des médecins, a engagé une réflexion pour revoir les volumes horaires et l’organisation des enseignements, notamment des médecins généralistes dans le cadre de la nouvelle filière universitaire. Il s’agit d’un enjeu majeur car ce sont les généralistes, dont le rôle est renforcé dans le parcours de soins, qui sont au quotidien les premiers prescripteurs de médicaments.
La MECSS souhaite que les réflexions engagées visant à réformer la formation médicale initiale des médecins, au-delà de la réforme en cours de la première année de médecine, aboutissent rapidement et permettent de renforcer les enseignements en pharmacologie, en pratiques thérapeutiques, notamment médicamenteuse, et en économie de la santé de manière à mieux intégrer la prescription en dénomination commune internationale et les problématiques de santé publique. La Mission souhaite aussi que soit limitée l’influence des laboratoires pharmaceutiques sur les étudiants en médecine, notamment par le biais des leaders médicaux et lors des stages en milieu hospitalier.
b) Veiller à la montée en charge de la formation professionnelle continue et de l’évaluation des pratiques professionnelles
L’obligation de formation continue des médecins a été instaurée, en 1995, il y a treize ans, et l’obligation d’évaluation des pratiques professionnelles (EPP), en 2004, il y a quatre ans. Force est de constater que ces obligations sont encore peu respectées. Cela s’explique notamment par le fait que l’organisation qui a été retenue pour leur mise en œuvre est complexe et tarde à se mettre en place. En outre, le financement de la formation professionnelle continue reste essentiellement assuré par les firmes pharmaceutiques et les risques de conflits d’intérêts subsistent.
Selon le rapport présenté en janvier 2006 par l’Inspection générale des affaires sociales (IGAS) sur l’organisation juridique, administrative et financière de la formation professionnelle continue des professions médicales et paramédicales, l’essentiel de la formation professionnelle continue (plus des trois quarts) est financé par les firmes pharmaceutiques : entre 300 et 600 millions d’euros par an en 2005. Les financements institutionnels publics et privés sont très modestes : 70 millions d’euros pour les financements conventionnels de la CNAMTS et 5 millions d’euros pour le fonds d’assurance formation des professions libérales.
L’organisation de la formation médicale continue (FMC) a été confiée à trois conseils nationaux de la formation professionnelle continue (CNFMC) spécialisés pour les médecins libéraux, les médecins hospitaliers et les médecins salariés. Afin d’harmoniser le fonctionnement et les procédures (barème de FMC et cahier des charges uniques), un comité de coordination a été créé. Pour l’évaluation des pratiques professionnelles, la compétence est partagée entre la HAS et la profession. La HAS est chargée de la définition des méthodes d’évaluation et de l’agrément des organismes d’EPP, et pour les médecins libéraux, les unions régionales des médecins libéraux (URML) habilitent les médecins prestataires d’EPP qui sont formés par la HAS. L’éclatement de la gestion de la formation professionnelle continue et de l’EPP est source de complexité pour les acteurs et notamment pour les organismes de formation. L’IGAS, dans le rapport susmentionné, a recommandé, dans un but de simplification, d’efficacité et d’économie, d’adosser les CNFMC à la HAS.
En outre, les dispositions visant à éviter les conflits d’intérêts ne sont pas suffisantes tant en ce qui concerne la délivrance des agréments aux organismes de formation par la CNFMC – qui regroupent notamment des représentants des organisations syndicales et des organismes de formation – que pour l’encadrement des pratiques des entreprises pharmaceutiques en matière de financement, de qualité et d’indépendance des formations. Le code de bonnes pratiques conclu entre le LEEM et les CNFMC est dépourvu de caractère contraignant.
Sur les cinq priorités définies par le CNFMC, en 2006, pour les cinq prochaines années, une seule concerne le médicament : la iatrogénèse.
La MECSS souhaite que la mise en place du dispositif de formation médicale continue des médecins et d’EPP soit rapidement achevée afin que celui-ci puisse réellement monter en charge. La Mission demande que soit étudiée la possibilité d’adosser les CNFMC à la HAS et que le ministère de la santé, la HAS et l’assurance maladie soient associés à la décision sur les thèmes prioritaires de FMC et d’EPP. Elle préconise de renforcer les conditions d’agrément des organismes de formation (cahier des charges plus précis et référentiel de qualité) afin d’assurer leur indépendance à l’égard de l’industrie pharmaceutique, de confier le contrôle du respect de l’agrément à des organismes habilités par la HAS et de prévoir des sanctions en cas de non-respect. Par ailleurs, la Mission souhaite qu’un dispositif de sanction soit prévu en cas de non-respect des obligations de formation continue et d’EPP. La MECSS souhaite aussi que des formations associant médecins et pharmaciens soient développées de même que la formation à l’écoute et à la gestion de la relation avec les patients. Enfin, la Mission souhaite que soit étudiée la possibilité d’un renforcement des financements institutionnels de la FMC et que soit créé un fonds regroupant les financements publics et privés.
2. Rééquilibrer l’information des médecins sur le médicament
La qualité de l’information sur le médicament est un enjeu majeur tant en termes de qualité des soins que de maîtrise des dépenses.
Les médecins s’estiment globalement bien informés sur le médicament. Mais ils trouvent l’information surabondante, éprouvent des difficultés à l’ordonner et la hiérarchiser et font aussi état de manques et de besoins non satisfaits. Par ailleurs, et peut-être surtout, le trait le plus marquant est, comme pour la formation médicale continue, le poids déterminant de l’industrie pharmaceutique dans le financement de l’information des médecins sur le médicament. Les firmes pharmaceutiques consacrent des moyens très importants à la promotion de leurs produits et jouent, par le biais de la visite médicale, un rôle essentiel dans l’information des médecins sur les médicaments. À côté de l’action des fabricants de médicaments, l’action publique est insuffisante pour contrebalancer la première.
Il est donc indispensable que les pouvoirs publics définissent une stratégie cohérente de communication sur le médicament. En effet, la promotion du bon usage des médicaments conditionne les bonnes pratiques de prescription.
a) Maîtriser l’impact de la visite médicale
Selon le rapport de l’IGAS de septembre 2007 sur l’information des médecins généralistes sur le médicament, l’industrie pharmaceutique consacre au moins 3 milliards d’euros à la promotion de ses produits, dont les trois-quarts à la visite médicale (VM). Le poids important de la visite médicale des prescripteurs dans les dépenses promotionnelles s’explique notamment par le fait que la publicité pour les médicaments remboursables, qui représentent l’essentiel de la consommation, est interdite en direction des patients.
Le reste de l’action marketing de l’industrie pharmaceutique est consacré à la publicité à caractère général et en direction des prescripteurs (13 %) – notamment dans la presse médicale, aux congrès (9 %) et aux échantillons (2 %).
L’intensité de la visite médicale est particulièrement forte en France. Les moyens affectés à la VM représentent 14 % du chiffre d’affaires ou encore 25 000 euros par médecin généraliste, c’est-à-dire l’équivalent de 285 heures d’activité du médecin (environ un mois et demi de travail).
Près de 23 000 visiteurs médicaux, soit 22 % des effectifs de l’industrie pharmaceutique, effectuent en moyenne 330 visites de 8 à 9 minutes, par an et par généraliste, ce qui représente l’équivalent d’environ une semaine et demi de travail. Le nombre de visiteurs médicaux a doublé en vingt ans. Ils sont en partie (20 % à 30 %) rémunérés en fonction de l’expansion des prescriptions et de l’atteinte des objectifs (volume de prescription, part de marché…).
La visite médicale est, en fait, financée par la collectivité à travers les prix administrés du médicament. Selon l’IGAS, la visite médicale « s’avère un moyen coûteux d’apporter de l’information aux généralistes. » En outre, le contenu informatif de la VM est souvent biaisé et pousse à la prescription, en particulier des médicaments nouveaux, pas toujours plus efficaces mais souvent plus chers. Une étude réalisée par l’Institut de recherche et documentation en économie de la santé (IRDES), menée à la fin des années 1990, a bien montré la corrélation forte qui existe entre l’investissement promotionnel des firmes et le nombre de lignes de prescription des médicaments concernés. En outre, la VM ne semble pas être le meilleur vecteur pour promouvoir le bon usage.
Les tentatives d’encadrement de la VM au moyen de la certification par la HAS, en application de la charte de la visite médicale qui a été conclue entre le LEEM et le CEPS en décembre 2004, ne semblent pas avoir profondément modifié la situation. Les organismes certificateurs n’ont pas les moyens nécessaires à la vérification du respect des prescriptions de la charte et aucun dispositif de sanction n’est prévu en cas de non-respect. On peut même craindre que la certification par la HAS donne à la visite médicale une caution dont elle ne disposait pas auparavant.
Cependant, force est de reconnaître qu’à défaut de moyen d’information institutionnel concurrent aussi efficace, la visite médicale organisée par les firmes pharmaceutiques est appréciée d’un grand nombre de médecins généralistes qui considèrent que, malgré les biais qui peuvent l’affecter, elle est un moyen d’information utile. Le contact, en face à face, permet d’apporter directement aux praticiens, dans son cabinet, de l’information sur des médicaments nouveaux qui peuvent être sophistiqués et complexes à utiliser. À certains égards, la visite médicale permet de pallier les insuffisances de la formation médicale continue et de l’information sur les médicaments. Encore peut-on observer que les nouvelles générations de médecins, mieux informés des nouvelles connaissances scientifiques et médicales, semblent moins intéressées par la VM et recourent davantage à d’autres sources d’information, comme l’Internet.
L’IGAS, dans le rapport précité, a recommandé un « désarmement » promotionnel qui pourrait se traduire par une réduction progressive de moitié des dépenses de promotion. Selon son calcul, cela permettrait de mobiliser progressivement plus de 1 à 1,5 milliard d’euros d’économie sur les dépenses de promotion du médicament à l’avantage des laboratoires (baisse des charges de promotion) et des pouvoirs publics (prix et remboursements moins élevés). Pour y parvenir, le rapport de l’IGAS propose d’augmenter la taxe sur la promotion en élargissant son champ et de fixer des objectifs quantitatifs de VM pour les classes de médicaments où la promotion est manifestement excessive.
D’ores et déjà, parallèlement à la régulation qualitative de la VM par la charte et la certification, les firmes pharmaceutiques se sont engagées conventionnellement dans un commencement de régulation quantitative. Le LEEM et le CEPS ont signé un accord qui prévoit la réduction sur trois ans (- 6 % en 2006, - 10 % en 2007 et - 12 % en 2008) du nombre de visites médicales pour quatre classes de médicaments : les statines, les sartans, les médicaments de l’asthme et les antibiotiques fluoroquinolones. Les pourcentages de réduction se cumulent, de sorte que, fin 2008, le nombre de contacts pour ces classes devrait avoir diminué de 28 % par rapport à 2005. Toutefois, les effets de cet accord devraient être limités puisque les classes retenues représentaient, en 2005, seulement 20 % du nombre de VM. Les résultats de la première année ont conduit le CEPS à décider, en 2007, une baisse de prix de 3 % pour quatre produits. Ce système constitue un premier pas positif.
La MECSS estime que la qualité de la visite médicale peut être encore améliorée et que la qualité doit primer sur la quantité. À cet égard, il serait souhaitable de s’assurer que les visiteurs médicaux remettent systématiquement le relevé des caractéristiques du médicament et la fiche de transparence. La MECSS souhaite aussi que les organismes certificateurs disposent des moyens juridiques nécessaires pour vérifier le respect de la charte et qu’un dispositif de sanction soit instauré.
En outre, la Mission souhaite que la politique contractuelle de réduction du nombre de contacts engagée entre le CEPS et les firmes pharmaceutiques soit poursuivie et étendue. Elle souhaite, notamment, d’une part, que l’abus de visite médicale soit défini dans la charte, d’autre part, que des objectifs quantitatifs plus ambitieux soient progressivement fixés qui, pour être équitable et éviter de créer un avantage concurrentiel, s’appliquent à toutes les firmes.
Par ailleurs, la MECSS souhaite que des études comparatives soient menées permettant de mettre en évidence les différences de comportement de prescription et de respect du bon usage entre les médecins gros consommateurs de VM et les médecins qui les refusent.
b) Développer l’information publique
Les médecins disent avoir besoin d’une information objective, claire, complète, mais synthétique et adaptée aux exigences de leur exercice quotidien. Ils souhaitent disposer des informations utiles concernant les modalités pratiques de prescription (forme, posologie) et susceptibles de servir de support à leurs choix thérapeutiques (place de la molécule dans la stratégie thérapeutique, comparaison avec les autres molécules…). L’information doit être simple mais non simpliste et doit permettre de satisfaire le prescripteur pressé comme le prescripteur curieux.
Les grands groupes pharmaceutiques obéissent à des stratégies industrielles et commerciales internationales, voire mondiales, et déploient leur communication à tous les niveaux et par tous les canaux possibles, les plus efficaces. Par ailleurs, la communication des firmes est très intense et diffuse. Elle intervient à tous les niveaux de la vie du prescripteur, lors des études initiales, durant les stages, à l’occasion de l’exercice de l’activité professionnelle et pendant la formation médicale continue. Il serait souhaitable que cette présence très forte de la communication des firmes pharmaceutique puisse être équilibrée par une information d’intérêt général essentiellement orientée sur le bon usage. D’autant que, selon les praticiens, la crédibilité de l’information venant de l’industrie pharmaceutique est faible ou très faible (80 %), alors que la crédibilité de l’information provenant des organismes publics (HAS et AFSSAPS) est bonne ou très bonne (90 %).
L’information publique sur le médicament destinée aux médecins devrait avoir une place plus importante. Cela correspond au souhait de voir se développer l’information par les pouvoirs publics, par des media diversifiés. Or, l’information publique est souvent dispersée, sous-utilisée et peu adaptée aux pratiques médicales. Le « bruit » promotionnel et la disproportion entre les moyens publics et ceux des laboratoires sont tels que l’information publique est peu audible. L’information publique devrait donc davantage recourir à certaines des techniques utilisées par les firmes pharmaceutiques.
La loi du 13 août 2004 a donné un rôle primordial à la HAS en matière d’information médicale des professionnels de santé et du public. L’AFSSAPS dispose, pour sa part, d’une compétence spécifique en matière de sécurité sanitaire et de contrôle de la publicité. Quant à la compétence en matière de bon usage, elle est partagée entre ces deux organismes. Il en résulte que chacun d’eux publie plusieurs types de documents d’information au contenu scientifique et pratique variable. En dépit d’efforts réels de simplification des messages et d’amélioration de leur lisibilité, la dualité des structures et la multiplicité des supports compliquent l’accès à l’information et ne favorisent pas autant qu’il le faudrait la sécurité et la qualité des prescriptions.
À cet égard, on peut rappeler que la loi de financement de la sécurité sociale pour 2008 a donné à la HAS le droit d’émettre des avis médico-économiques sur les stratégies de soins, de prescription ou de prise en charge les plus efficientes.
La MECSS souhaite faciliter l’accès à l’information sur les médicaments, notamment en qui concerne leur bon usage, et renforcer les moyens d’actions de la HAS en ce domaine. À cet effet, la Mission préconise de faire de la HAS l’émetteur unique d’information sur le bon usage des médicaments. Elle préconise aussi de renforcer sa capacité d’analyse et d’évaluation des stratégies promotionnelles et de communication de l’industrie pharmaceutique ainsi que des prescriptions. Cela pourrait se traduire, par exemple, par la création d’un observatoire de la prescription et d’un réseau de médecins chargés de l’observation de la visite médicale. Il serait notamment utile que la HAS établisse une recommandation sur le bon usage de la visite médicale. En outre, la MECSS souhaite, par mesure de cohérence, que la mission de contrôle de la publicité soit transférée de l’AFSSAPS à la HAS. Elle demande que l’accessibilité – notamment sur le site internet de la HAS, et la lisibilité de l’information sur les médicaments soient encore améliorées et que soient multipliés les documents synthétiques et pratiques du type fiches de bon usage du médicament (qui indiquent souvent le coût du traitement). Par ailleurs, la MECSS souhaite que la HAS soit dotée de la capacité de diffuser de l’information sur le médicament aux médecins, en face à face. À cet effet, la HAS devrait pouvoir disposer d’un corps de « délégués de santé » spécialement formés et certifiés par la HAS. Ce corps, constitué à partir des délégués de l’assurance maladie, serait copiloté par la HAS et la CNAMTS et géré administrativement par la CNAMTS.
c) Diffuser les logiciels certifiés d’aide à la prescription
L’objectif des logiciels d’aide à la prescription (LAP) est de faciliter le travail du prescripteur pour lui permettre d’améliorer la qualité et la sécurité de la prescription tout en diminuant le coût du traitement à qualité égale. Les LAP sont utilisés depuis plusieurs années dans les établissements hospitaliers. Des LAP de médecine de ville existent aussi. Mais ils sont encore peu utilisés, surtout dans leur fonction administrative (édition de feuilles d’ordonnance…) et peu dans leur fonction d’aide à la prescription.
Afin de fiabiliser les LAP, la loi du 13 août 2004 a confié à la HAS, la mission de déterminer la procédure de certification des logiciels.
La HAS a mis en place un référentiel de certification. Elle a aussi décidé que, pour être certifiés, les LAP doivent utiliser une information de qualité et, en conséquence, des bases de données conformes à une charte de qualité. Les LAP doivent tout d’abord fournir l’information sur leur concepteur et la nature de leur financement. Les LAP doivent aussi permettre, notamment, la prescription en dénomination commune internationale (DCI), l’affichage du SMR, de l’ASMR, de l’éventuelle TFR et de l’inscription du médicament dans un groupe générique, des interactions médicamenteuses, de l’historique des traitements, de messages d’alerte, des conditions de remboursement ainsi que du coût par lignes de prescriptions et du coût total, afin de permettre l’optimisation du coût. Par ailleurs, les LAP sont susceptibles d’être utilisés par les praticiens pour les aider dans la démarche d’évaluation de leurs pratiques professionnelles.
Depuis dix-huit mois, les éditeurs de logiciels ont accompli un important travail de mise à niveau.
Cependant, la mise en œuvre de la certification des LAP est retardée par la redéfinition, en cours, du référentiel afin, notamment, de tenir compte de la nouvelle disposition de la loi de financement de la sécurité sociale pour 2008 prévoyant l’affichage des prix des médicaments au moment de la prescription. Une fonction de comparaison de prix devrait être intégrée dans les LAP. Les modalités d’adhésion à la charte qualité devraient être précisées au mois de mai 2008. Les éditeurs de logiciels pourront ensuite adhérer à la charte et la certification des LAP devrait débuter au mois de juin 2008.
Actuellement, la certification des LAP n’est pas obligatoire. L’IGAS, dans le rapport précité, préconise de la rendre obligatoire.
La MECSS souhaite que la certification des logiciels d’aide à la prescription soit obligatoire et que la procédure de certification des LAP en médecine de ville soit mise en œuvre rapidement afin que tous les logiciels sur le marché soient certifiés à la fin de l’année 2009. La Mission souhaite aussi que soit étudiée la possibilité d’adjoindre aux LAP une fonction d’aide à la décision thérapeutique. Compte tenu de l’impact de la prescription hospitalière sur la prescription et la consommation de ville, la MECSS souhaite également que le processus de certification des LAP hospitaliers soit engagé rapidement.
3. Renforcer les actions de maîtrise médicalisée des dépenses de médicaments de l’assurance maladie
La maîtrise médicalisée des dépenses vise à inciter les médecins à réduire leurs prescriptions sur certains postes jugés prioritaires et, dans ce sens, les actions de communication de l’assurance maladie contribuent à la réalisation des objectifs conventionnels et individuels fixés et au développement du bon usage. Les diverses actions menées sur ces thèmes ont commencé à produire des résultats, mais les réserves d’économies liées au développement du bon usage sont encore importantes.
La réalisation du potentiel d’optimisation des prescriptions suppose de renforcer les outils d’analyse des prescriptions, d’amplifier la maîtrise médicalisée et de développer les outils de communication adaptés.
a) Développer l’analyse des prescriptions
La conduite d’une politique d’amélioration de la sécurité et de la qualité des prescriptions de médicaments ainsi que de maîtrise des coûts suppose de pouvoir contrôler, analyser et évaluer les prescriptions.
Actuellement, le système d’information de la CNAMTS ne permet pas de procéder à une analyse systématique de toutes les prescriptions. Il ne permet que des contrôles limités du bon usage (par rapprochement de la prescription et de la pathologie) effectués à partir de l’historique des remboursements des prescriptions d’un praticien lorsque la prescription est spécifique à une pathologie ou en matière d’affections de longue durée. En 2005, la Commission nationale informatique et libertés (CNIL) a, en effet, autorisé la CNAMTS à exploiter les données rendues anonymes du fichier des ALD. La CNAMTS peut notamment contrôler le respect de l’ordonnancier bizone qui prévoit que seules les prescriptions de médicaments liées à l’ALD doivent être mentionnées dans la partie haute de l’ordonnance pour ouvrir droit au remboursement à 100 %.
La MSA a mis en place, depuis 2004, un système d’information médicalisée, dénommé ARCHIMED, généralisé à l’ensemble des services du contrôle médical. Lorsqu’une anomalie de prescription est repérée, un courrier est adressé au praticien, et le cas échéant, un entretien confraternel est organisé. Un courrier suffit d’ailleurs en général à modifier les habitudes de prescription et le succès repose sur l’implication du praticien. Le dispositif est donc conçu pour offrir aux médecins un retour sur leurs pratiques. Il a été enrichi de fonctions « observatoire du médicament », « gestion du risque » et de modules concernant la maîtrise médicalisée (polymédication des personnes âgées, interactions, génériques, respect de l’ordonnance bizone…).
La MECSS souhaite que les caisses d’assurance maladie poursuivent le développement de leur capacité d’analyse des prescriptions, à l’instar du système d’information médicalisée ARCHIMED de la MSA. Les caisses devraient ainsi pouvoir alerter les prescripteurs en cas de primo-prescription non conforme au bon usage ou de polymédication présentant des risques iatrogènes.
L’extension, voire la généralisation, du dispositif d’analyse des prescriptions est en effet une des conditions du renforcement de l’efficacité de la maîtrise médicalisée des dépenses d’assurance maladie.
b) Renforcer l’efficacité de la maîtrise médicalisée des dépenses de santé
La convention médicale du 12 janvier 2005 et l’avenant n° 12, de mars 2006, ont fixé des objectifs de diminution des prescriptions (antibiotiques, statines, inhibiteurs de la pompe à protons, anti-ulcéreux, hypnotiques, lutte contre la iatrogénie), ou de limitation d’augmentation (sartans) ou d’augmentation de prescriptions (génériques).
La Cour des comptes, dans ses communications à la MECSS, dresse un bilan sévère des actions conventionnelles de maîtrise médicalisée. Elle considère que les objectifs qui ont été fixés en montants auraient dû être fixés en volume, afin de neutraliser les effets prix (baisses de prix décidées par le CEPS), de structure (arrivée sur le marché de nouveaux génériques) et de substitution (prescription du médecin dans le répertoire ou exercice du droit de substitution par le pharmacien). En outre, certains objectifs sont imprécis et il n’est pas précisé comment ils doivent être atteints (développement des génériques, limitation ou meilleur ciblage des prescriptions). Par ailleurs, les modalités de calcul des objectifs sont contestables. La méthode de calcul la plus favorable, c’est-à-dire la moins exigeante, a été systématiquement retenue pour les différents paramètres : base de référence, tendance d’évolution, calcul de l’évolution. Cela a conduit à surestimer les économies attendues.
En dépit de ce mode de calcul favorisant quelque peu artificiellement le respect des objectifs conventionnels, ces derniers n’ont pas été atteints et l’impact sur les prescriptions a été très faible.
En 2005, seulement 17 millions d’euros d’économies ont été générés par l’infléchissement des prescriptions sur les trois classes de médicaments visées par la convention médicale (antibiotiques, statines et psychotropes). Si l’on tient compte de la pénétration des génériques, les économies de maîtrise médicalisée s’élèvent à environ 70 millions d’euros (à comparer aux 340 millions d’économies attendues). Toutefois, une stabilisation du nombre de nouveaux traitements par statines a été observée alors que l’effectif de malades à risque cardiovasculaire augmente.
En 2006, les économies nettes des effets prix, générication et déremboursement, notamment liées au plan médicament, s’élèvent à 120 millions d’euros au lieu de 222 millions d’euros d’économies attendues. Cela confirme que la maîtrise médicalisée contribue à modifier les prescriptions des médecins, mais dans des proportions moindres que ce qui était attendu.
L’avenant n° 23, de juin 2007, à la convention fixe des objectifs de réduction de la iatrogénie et prévoit la déclinaison individuelle des objectifs nationaux en ce qui concerne les génériques, les IPP, les antibiotiques et le respect de l’ordonnancier bizone. Mais les objectifs individuels ne sont pas opposables et la convention ne précise pas comment évaluer leur respect.
L’article 43 de la loi de financement de la sécurité sociale pour 2008 a apporté plusieurs améliorations. Désormais, les engagements conventionnels de maîtrise médicalisée des dépenses de prescription de médicaments devront être exprimés en volume, indépendamment de toute évolution tarifaire. Par ailleurs, les médecins peuvent conclure avec les caisses locales d’assurance maladie des contrats comportant des objectifs individuels, notamment de prescription, d’amélioration des pratiques, de formation et d’information. Ces contrats individuels prévoiront des contreparties financières liées à l’atteinte des objectifs par le praticien.
La CNAMTS a publié, au mois d’octobre 2007, une étude comparative des pratiques en France et dans quatre autres pays européens (Allemagne, Espagne, Italie et Royaume-Uni) concernant la consommation et les dépenses liées à neuf classes de médicaments qui représentent plus d’un tiers de la consommation de médicaments et 40 % des dépenses. Il en ressort que la France est en tête de la consommation pour 6 des 9 classes étudiées. Elle apparaît surtout comme la plus dispendieuse avec le montant moyen par habitant le plus élevé des cinq pays européens pour ces 9 classes de médicaments : 130 euros, soit 32 euros de plus que le deuxième pays, l’Italie. Selon la CNAMTS, ce sont plusieurs centaines de millions d’euros qui pourraient être économisés si la France avait une consommation et des coûts moyens similaires à ceux de ses voisins. Si l’on prend les trois principales classes étudiées (hypertenseurs, IPP et statines), le différentiel de la France par rapport à l’Allemagne, c’est-à-dire avec la même structure de consommation que l’Allemagne, atteint 1,5 milliard d’euros : 640 millions d’euros sur les hypertenseurs et les sartans, 430 millions d’euros sur les IPP et 400 millions d’euros sur les statines.
La consommation moyenne de statines (anti-cholestérol) est de 50 % supérieure à celle de l’Allemagne (en dose consommée par habitant, par jour). Et cet écart ne peut s’expliquer par des différences d’état de santé de la population puisque la prévalence des facteurs de risques cardio-vasculaire est la même dans les deux pays.
La marge de progrès en matière de maîtrise médicalisée apparaît donc importante. À cet égard, on peut indiquer que la MSA a décidé, pour 2007, de passer d’une logique d’obligation de moyens à une obligation de résultats en fixant un montant national d’économies à réaliser par régions.
La MECSS souhaite que les efforts de maîtrise médicalisée soient poursuivis, amplifiés et étendus à de nouvelles classes de médicaments. La Mission souhaite également, d’une part, que la politique de publication d’accords de bon usage des soins soit relancée, d’autre part, que les dispositions issues de la dernière loi de financement de la sécurité sociale, et notamment celle concernant l’individualisation des objectifs de maîtrise médicalisée et d’amélioration des pratiques, soient rapidement mises en œuvre par les partenaires conventionnels. Elle souhaite que cette action s’appuie en particulier sur un effort accru des caisses d’assurance maladie en matière de communication sur la démarche qualité et le bon usage.
c) Amplifier la communication de l’assurance maladie
Les caisses d’assurance maladie concourent à l’information et à la communication sur le bon usage du médicament et viennent appuyer la réalisation des objectifs de maîtrise médicalisée.
Les campagnes de communication sur les antibiotiques, ciblées à la fois sur les prescripteurs et les consommateurs, ont donné des résultats encourageants. La campagne menée depuis 2002 a notamment pour objectif de lutter contre les phénomènes de résistance aux antibiotiques qui sont un problème majeur de santé publique. Elle a contribué à la baisse sensible de la consommation d’antibiotiques, surtout chez les enfants de 0 à 5 ans qui sont la cible prioritaire du programme. Depuis 2002, le nombre de prescriptions pendant la période d’hiver a diminué de 4,5 %, en moyenne, et l’exposition aux antibiotiques a été réduite de près de 20 %. Cela prouve l’efficacité d’une action médico-économique coordonnée entre les différents acteurs et soutenue dans la durée. Mais la France continue de consommer deux fois plus d’antibiotiques que l’Allemagne et le Royaume-Uni et trois fois plus que les Pays-Bas. La marge de progression du bon usage et d’économies qui pourraient en résulter apparaît donc, là aussi, importante.
On peut également citer l’efficacité de l’action de maîtrise médicalisée et de communication de l’assurance maladie concernant les psychotropes qui a permis de réduire les remboursements des anxiolitiques et des hypnotiques. Il reste que le niveau de consommation des Français est encore, en moyenne, neuf fois plus élevé qu’en Allemagne, quatre fois plus élevé qu’en Espagne et le double de celui du Danemark ou la Norvège.
Une autre action coordonnée a été engagée par l’assurance maladie pour lutter contre la iatrogénie médicamenteuse, notamment chez les personnes âgées. Cette action s’appuie sur la diffusion de documents d’information, notamment les recommandations de l’AFSSAPS sur la iatrogénie et de la HAS sur la polymédication, auprès des prescripteurs et des patients, ainsi que sur le développement des entretiens confraternels.
L’assurance maladie diffuse aussi d’autres documents d’information claire et synthétique comme la « lettre aux médecins » et la « lettre aux pharmaciens » ou encore des supports mémos, brefs, clairs et synthétiques, sous le double timbre de la CNAMTS et de l’AFSSAPS, à visée de bonnes pratiques ou médico-économique et faisant apparaître le coût de traitement mensuel. Dans l’objectif de réduction des dépenses, ces fiches apparaissent particulièrement utiles. À titre d’exemple, on peut citer le cas du coût du traitement mensuel par statines qui peut, selon le médicament prescrit, aller du simple, avec la molécule la moins chère, au triple, avec la molécule la plus chère. S’agissant d’une classe de médicament très consommée et avec une forte dynamique potentielle de développement, on mesure, là encore, la marge de progrès possible en termes d’économies tout en assurant la même qualité de traitement.
La MECSS souhaite que l’assurance maladie amplifie ces actions de communication et développe des campagnes sur de nouveaux thèmes ciblés.
d) Poursuivre la montée en charge des actions individuelles en direction des médecins
L’assurance maladie développe aussi des actions individuelles auprès des médecins, notamment par le biais des délégués de l’assurance maladie (DAM) qui visent à contrebalancer l’influence de la visite médicale des fabricants de médicaments.
La mise en place des DAM a commencé en 2005 par redéploiement d’effectifs au sein du régime général d’assurance maladie. Leur nombre est actuellement de 950 et devrait être porté à 1 400 à la fin de 2009. Ce nombre peut être comparé aux près de 24 000 visiteurs médicaux des 200 laboratoires pharmaceutiques. Pour 2007, les DAM avaient un objectif de 300 000 visites de praticiens.
Les DAM reçoivent une formation théorique (300 heures) et pratique (900 heures) et devraient être progressivement tous certifiés. Ils viennent en appui des actions de communication de l’assurance maladie pour faire connaître la convention médicale et sensibiliser les médecins aux objectifs de maîtrise médicalisée. Les DAM visent ainsi à favoriser la réalisation de l’important potentiel d’optimisation des prescriptions. À cet effet, ils apportent notamment aux praticiens un retour d’information sur leur pratique de prescription de médicaments au moyen de profils personnalisés et d’éléments de comparaison. L’image des DAM semble avoir évolué positivement, même si, il faut le reconnaître, des résistances subsistent. S’ils ont souvent été perçus, au départ, comme des contrôleurs, les praticiens considèrent aujourd’hui qu’ils peuvent leur apporter un retour d’information utile sur leurs pratiques. Ils sont aussi considérés comme un moyen de rompre l’isolement de la pratique individuelle et d’engager un dialogue dans un cadre différent de celui de la démarche, principalement commerciale, de la visite médicale. La visite des DAM peut aussi permettre de préparer dans de meilleures conditions un éventuel entretien confraternel avec un médecin contrôleur de la caisse d’assurance maladie.
Les premiers éléments d’évaluation de l’activité des DAM semblent démontrer leur efficacité. Par exemple, la diminution de la consommation des antibiotiques est plus forte, lorsque la visite des DAM aux praticiens est répétée tous les mois. Cela permet de vérifier que le contact direct, en face à face, est un des leviers les plus efficaces pour agir sur les comportements individuels. L’industrie pharmaceutique l’a bien compris qui utilise ce moyen depuis longtemps et dispose d’outils d’analyse très fins du rapport coût-efficacité de l’investissement promotionnel que constitue la visite médicale.
Pour sa part, la MSA utilise son réseau, dense, de délégués cantonaux pour relayer les actions qu’elle mène en matière de médicaments. Dans son plan d’action pour 2006 et 2007, elle a notamment prévu de développer le dialogue médical de groupe.
La MECSS considère que les contacts en face à face avec les médecins doivent être développés et estime que l’action conduite par les DAM est intéressante et a produit de premiers résultats positifs. Elle souhaite cependant que, dans un but de rationalisation et d’efficacité, le corps des délégués soit placé sous le contrôle de la HAS qui a une compétence essentiellement médicale et a vocation à piloter les actions de communication visant à optimiser les prescriptions et à promouvoir le bon usage des médicaments. Dans cette optique, elle souhaite un renforcement de la formation médicale des délégués de santé et que tous les délégués soient certifiés par la HAS. Par ailleurs, elle souhaite que soient développés, en coordination entre la HAS et l’assurance maladie, les outils permettant aux médecins d’évaluer leur pratique ainsi que les communications personnalisées sous la forme de courriers, de contacts téléphoniques ou d’entretiens confraternels, en particulier lorsque des anomalies de prescriptions (polymédication excessive, risques iatrogéniques, primo-prescription trop longue, choix de médicaments chers, non-respect de l’ordonnancier bizone…) sont constatées.
C. FAVORISER LE BON USAGE CHEZ LES CONSOMMATEURS
1. Développer et coordonner l’information du grand public sur les médicaments
a) Dissiper le malentendu entre les médecins et les patients sur la demande de médicaments
Certes, l’acte de prescription est le fait du praticien. Mais celui-ci est soumis aux attentes de prescription, voire à la pression du patient. Cette pression est d’ailleurs ressentie par les praticiens français comme étant plus forte que dans d’autres pays européens. Pourtant, l’étude réalisée par IPSOS-santé, publiée en octobre 2005, sur le rapport des Français et des Européens à l’ordonnance et aux médicaments montre que la perception de cette demande de médicaments par les praticiens ne semble pas correspondre aux attentes réelles des patients. Il existerait donc un malentendu entre médecins et patients sur la demande de médicaments. Dans certains cas, la surprescription de médicaments résulterait d’une surévaluation de l’attente du patient.
Les Français, champions d’Europe de la consommation de médicaments, sont ouverts à des évolutions et imaginent sans peine de nouvelles pratiques. Ils sont prêts à rompre avec le système de l’ordonnance reine et de la prescription quasi-systématique de médicaments. Les patients sont nombreux à déclarer être davantage demandeurs d’écoute, d’explications, de réassurance, de conseils hygiéniques et de messages d’éducation en santé susceptibles de les aider à modifier leurs habitudes et leur comportement, plutôt que de traitements médicamenteux. De fait, pour les patients comme pour les médecins, l’échange est la première attente, bien avant l’ordonnance et la prescription médicamenteuse. Huit patients sur dix déclarent qu’ils auraient confiance dans un médecin qui ne leur prescrirait pas de médicament en fin de consultation ou qui saurait remplacer certains médicaments par des conseils utiles. Pour les patients, la prescription, éventuelle, de médicaments n’est qu’un des éléments de la prise en charge.
Force est de reconnaître que la priorité donnée à la prescription de médicaments résulte souvent de l’habitude d’un schéma de consultation. Elle est aussi, parfois, une facilité permettant d’apporter une réponse immédiate à une souffrance sociale et à un besoin psycho-social qui pourrait nécessiter une prise en charge différente ou plus lourde et plus compliquée et surtout supposant de disposer de davantage de temps que lors d’une consultation normale. Le principe de précaution et le manque de formation à l’échange, à l’éducation thérapeutique et au conseil peuvent aussi expliquer le réflexe du recours au médicament. Dans certains cas, la brièveté de la consultation peut pousser à la surprescription de médicaments et aussi favoriser l’apparition de phénomènes de dépendance aux médicaments.
L’information du grand public et des patients sur le médicament peut contribuer à réduire ce hiatus, limiter l’appétence pour les médicaments, modifier les comportements des patients et inciter à la bonne observance des traitements. Elle doit inciter le patient à devenir acteur de sa propre santé et à rechercher un nouvel équilibre entre le conseil médical et la prescription de médicaments.
La MECSS souhaite que les études médicales et la formation médicale continue permettent de valoriser le dialogue médecin-patient et les alternatives à l’ordonnance et au « tout médicament ».
b) Renforcer l’information publique en direction des patients
Malgré des améliorations récentes, l’information publique sur les médicaments en direction des patients est insuffisante et mal coordonnée.
Outre la diffusion de documents d’information sur support papier, notamment par le biais des pharmacies, le régime général d’assurance maladie et la MSA ont mené des campagnes grand public qui ont produit des effets positifs, par exemple pour les antibiotiques et les génériques. L’assurance maladie a d’ailleurs reconduit pour la période 2008-2010 son programme sur les antibiotiques lancé en 2002. Les caisses recourent aussi, depuis peu, à des techniques de communication directe auprès des assurés qui ont montré leur efficacité. En outre, les courriers d’information personnalisée envoyés aux assurés et les contacts téléphoniques ciblés sur certains thèmes comme les médicaments génériques ont permis de réaliser des économies importantes. Les sites internet des caisses mettent aussi à disposition des informations pratiques. Dans ce domaine, un effort de simplification a été fait, mais les sites pourraient encore être améliorés pour faciliter l’accès aux informations utiles.
L’AFSSAPS diffuse des documents pratiques à destination du grand public : « Vous et votre traitement » et « Questions/réponses ». Elle développe, et c’est un des objectifs de la convention d’objectifs et de gestion, un partenariat avec les associations de patients, lesquelles sont notamment invitées à procéder à la relecture des documents produits par l’agence. Elle joue aussi un rôle essentiel dans le contrôle a priori de la publicité destinée au grand public pour ce qui concerne les médicaments d’automédication. L’AFSSAPS a diffusé des avertissements concernant la contrefaçon des médicaments et l’achat de médicaments par internet. Le site internet de l’agence, désormais plus accessible, reste perfectible : même si l’information de l’agence intéresse principalement les professionnels, il serait souhaitable qu’un accès par catégories de public soit prévu.
Le site de la HAS comporte désormais des accès spécifiques aux informations dédiées aux différentes catégories de public, ce qui en facilite l’utilisation. Les patients atteints d’une ALD peuvent ainsi accéder aux guides patients (par exemple « Vivre avec un diabète de type 2 »). Cependant, la nature des informations auxquelles peut accéder le grand public est peu différente de celles accessibles aux professionnels de santé. Les informations à destination du grand public devraient faire l’objet d’un retraitement pour les simplifier et améliorer leur compréhension.
L’Institut national de prévention et d’éducation pour la santé (INPES) peut aussi jouer un rôle important en matière de consommation de médicaments. Les campagnes de prévention et d’éducation à la santé que mène l’institut peuvent notamment contribuer à freiner le recours aux médicaments, en favorisant l’éducation à la santé et la prévention, le développement des aptitudes individuelles, la création d’environnements favorables à la santé et en luttant contre la banalisation du médicament. Encore faut-il observer que les relations entretenues entre la prévention et le médicament sont ambivalentes puisque, dans certains domaines, les actions de prévention peuvent contribuer à accroître la consommation de médicaments. On peut citer le cas de la lutte contre le tabagisme.
La MECSS souhaite que les actions d’information institutionnelle du grand public sur les médicaments soient davantage développées et mieux coordonnées. L’INPES devrait être le diffuseur de l’information sur le médicament en direction du grand public. Par ailleurs, la MECSS souhaite également que soient multipliés les actions ciblées et les contacts directs avec les patients, notamment à l’initiative des caisses d’assurance maladie. Elle demande que l’information en matière d’achat de médicaments sur internet ou par correspondance et de contrefaçon soit développée et davantage visible. Elle estime utile que les associations de patients soient davantage associées à la définition et à l’évaluation des actions d’information sur le médicament des différents acteurs institutionnels. L’INPES pourrait aussi contribuer, grâce à une action dans la durée, à installer dans l’opinion le thème du bon usage du médicament et à responsabiliser les patients en modifiant leur regard sur la prise en charge de leur santé. La Mission souhaite enfin que l’ergonomie des sites internet des organismes soit améliorée. Le site de la CNAMTS, dénommé ameli, devrait être aussi enrichi de données ciblées et de conseils, notamment concernant les pathologies chroniques. Ces compléments pourraient être accessibles sur des sites spécialisés par pathologies.
2. Inciter à l’observance, encadrer les programmes d’accompagnement des patients et développer l’éducation thérapeutique
a) L’observance des traitements est un enjeu majeur
Le médicament n’est, et ne sera jamais, un produit de consommation ordinaire : on ne peut attendre de bénéfices d’un médicament que s’il est correctement utilisé ; son usage comporte toujours des risques.
Pourtant, bien que mal évaluée, la non-observance thérapeutique est fréquente. Selon le rapport de l’IGAS d’août 2007 sur l’encadrement des programmes d’accompagnement des patients associés à un traitement médicamenteux, financés par les entreprises pharmaceutiques, la non-observance concernerait 30 % à 50 % des patients voire 90 % des personnes atteintes d’affections chroniques à un moment donné de leur maladie. La non-observance entraîne des effets délétères pour la santé du patient (diminution de l’efficacité du traitement, risques de complications et de rechutes plus graves), parfois pour son entourage (risques contagieux) et pour la société tout entière (résistance aux antibiotiques, mauvaise couverture vaccinale et prévalence plus importante de l’agent infectieux). Les enjeux de la non-observance sont également financiers tant pour le financeur public (surcoûts de prise en charge liés à l’augmentation de la consommation de soins en ville ou à l’hôpital) que pour les entreprises pharmaceutiques (manque à gagner lié au mésusage). L’industrie pharmaceutique américaine estime que le manque à gagner lié au mésusage s’élève à 30 milliards de dollars par an.
L’observance des traitements est donc une question centrale. Elle relève au premier chef du praticien qui a prescrit le traitement. Cependant, faute de temps, le suivi par le praticien n’est pas toujours bien assuré. Il y a donc un véritable besoin d’accompagnement de certains patients dans l’observance de leur traitement.
Depuis quelques années, les initiatives des firmes pharmaceutiques se multiplient pour accompagner les patients et, depuis peu à l’initiative de l’assurance maladie, pour développer l’éducation thérapeutique.
b) Encadrer strictement les programmes d’accompagnement des patients financés par l’industrie pharmaceutique
Les firmes pharmaceutiques tentent de développer des programmes d’aide à l’observance ou d’accompagnement des patients. Ces programmes répondent à plusieurs évolutions de l’industrie pharmaceutique : la sophistication et la complexité d’administration de certains produits, notamment sortis de la réserve hospitalière, le changement dans les stratégies de communication des firmes qui cherchent à compenser la pression sur la visite médicale et à contourner l’interdiction de la publicité directe auprès du public pour les spécialités remboursables, la volonté des firmes de se différencier des concurrents et de développer des interventions à distance pour fidéliser leurs clients.
La plupart des programmes concernent aujourd’hui des médicaments injectables. Ils visent à former le patient à l’auto-injection et associent diverses actions : formation et assistance par téléphone, brochures d’information, DVD d’auto-formation, courriers, visites.
En l’absence de cadre juridique clair – les initiatives tendant à encadrer ces programmes par la loi n’ayant pu aboutir, l’AFSSAPS a autorisé quelques programmes qui sont, en réalité, davantage des programmes d’éducation des patients que d’accompagnement ou d’aide à l’observance. Ces programmes présentent des risques et ouvrent une brèche dans la nécessaire protection renforcée du public qui ne doit pas être soumis à une pression promotionnelle pour les médicaments prescrits.
La MECSS souhaite qu’un cadre législatif définisse et limite très strictement les programmes d’accompagnement ou d’aide des patients associés à un traitement médicamenteux, financés par les entreprises pharmaceutiques. Seuls des programmes de courte durée et limités à l’apprentissage d’une technique d’administration particulièrement complexe concernant un médicament présentant un intérêt thérapeutique important (ASMR 1 ou 2) pourraient être autorisés. L’autorisation pourrait être délivrée aux opérateurs par la HAS à condition de satisfaire à des exigences (compétence, indépendance, qualité, objectivité, non-sélectivité…).
c) Favoriser le développement de programmes d’accompagnement des patients par l’assurance maladie et l’éducation thérapeutique des patients
Depuis 2007, l’assurance maladie peut mettre en place des programmes d’accompagnement des patients atteints de maladies chroniques visant à leur apporter des conseils en termes d’orientation dans le système de soins et d’éducation à la santé (article 91 de la loi de financement de la sécurité sociale pour 2007).
L’assurance maladie a lancé, au mois de mars 2008, avec l’autorisation de la CNIL, un service d’accompagnement pour les personnes atteintes de maladies chroniques dénommé sophia. La création de ce service s’inscrit aussi dans le cadre de la politique de gestion du risque de l’assurance maladie et correspond à un des objectifs du plan ministériel 2007-2011 d’amélioration de la qualité de vie des personnes atteintes de maladies chroniques, qui prévoit de « développer un accompagnement personnalisé des malades ». L’assurance maladie cherche ainsi à instaurer un nouveau type de relation avec ses assurés et à développer un véritable partenariat de santé.
Le service est, dans un premier temps, mis en place dans dix départements. Il est proposé à 136 000 patients diabétiques pris en charge à 100 %, mais concerne aussi les 6 000 médecins traitants. Il s’agit d’un service volontaire et gratuit qui vise à relayer l’action des médecins traitants en proposant aux patients des services de conseil, d’écoute et d’information. Le service comprend des outils d’information pratiques et pédagogiques (notamment le magazine « sophia et vous »), un accompagnement téléphonique par du personnel paramédical de l’assurance maladie, formé et expérimenté (principalement des infirmières) et des services disponibles sur internet. L’accompagnement doit être adapté aux besoins de chaque patient. Le principe est de créer des contacts réguliers toutes les six semaines. Les médecins traitants participants au programme seront rémunérés par un forfait annuel par patient.
De manière générale, il est souhaitable de développer l’éducation thérapeutique des patients par l’intermédiaire des médecins au premier chef, notamment dans le cadre des réseaux de soins, mais aussi d’autres acteurs, tout particulièrement l’assurance maladie qui, en tant qu’assureur, peut s’impliquer dans la gestion et la prévention du risque.
L’éducation thérapeutique du patient-acteur de sa santé est une discipline relativement jeune. Le potentiel de mieux-être, d’amélioration de la santé et, finalement, d’économies que recèlent l’éducation thérapeutique et une bonne hygiène de vie est difficile à évaluer, mais il est probablement important. Les effets se mesurent à moyen et long terme. À cet égard, on peut évoquer l’importance des marges potentielles de progrès concernant certains problèmes de santé publique comme le tabagisme, l’obésité ou le stress.
Il est donc essentiel de mieux utiliser le levier de l’éducation thérapeutique et du conseil en hygiène de vie pour faire évoluer les comportements, en amont de la prescription et de la consommation de médicaments. L’enjeu est d’améliorer l’état de santé et d’optimiser les soins, c’est-à-dire de choisir le mode d’intervention le plus efficace.
La MECSS considère qu’il appartient en priorité aux acteurs institutionnels d’apporter des réponses adaptées aux besoins d’aide à l’observance et d’accompagnement des patients. Elle souhaite que l’assurance maladie poursuive et amplifie ses efforts dans ce domaine, en coordination avec l’AFSSAPS et la HAS et en concertation avec les médecins et les associations de patients. Au-delà, la MECSS souhaite que la HAS réfléchisse à la définition d’une stratégie de développement de l’éducation thérapeutique.
d) Promouvoir sur internet l’information de qualité sur les médicaments
Les patients vont de plus en plus chercher de l’information sur internet. Aujourd’hui, environ un patient sur cinq consulte un site internet pour rechercher de l’information médicale ou de santé. Dans certains cas, la recherche d’information sur internet a pour objet de pallier l’insuffisance des informations délivrées par le professionnel de santé. Les patients sont à la recherche de plus de conseil et de plus de dialogue. Ils cherchent à mieux comprendre leur maladie et leur traitement mais aussi à communiquer avec d’autres malades. Ils estiment être ainsi plus à même de partager une décision concernant leur santé et pouvoir mieux se prendre en charge. Mais cette évolution peut modifier la relation avec le médecin. Moins de la moitié des internautes parlent à leur médecin de leurs recherches d’informations sur internet. Certains n’osent pas, car ils ne savent pas comment aborder la question, d’autres ne souhaitent pas donner au médecin l’impression de le remettre en cause. Pourtant, l’échange entre le médecin et le patient peut permettre à ce dernier de progresser dans la connaissance de sa maladie et d’être davantage acteur de sa santé. Cela peut être aussi l’occasion pour le médecin d’enrichir son expertise de la pathologie, de développer un dialogue constructif susceptible de déboucher sur des conseils hygiéniques ou comportementaux et de conseiller au patient des sites de qualité.
La qualité de l’information sur les médicaments diffusée sur internet revêt donc une importance croissante. La loi du 13 août 2004, répondant aux recommandations du plan d’action eEurope 2002 visant notamment à développer les usages d’internet en matière de santé, a confié à la HAS la mission de déterminer les règles de bonnes pratiques devant être respectées par les sites français d’information de santé. Le contrôle systématique et continu de la qualité de l’information médicale délivrée par les sites n’apparaissant pas réaliste, la HAS a établi une recommandation destinée aux patients pour les aider à chercher une information sur internet et à identifier des sites santé en évitant les pièges d’internet.
Par ailleurs, la HAS a confié à une organisation non gouvernementale, Health On The Net (HON), l’attribution d’un label HON aux sites qui respectent les huit principes d’un code de bonnes conduites (HONcode). Ces principes concernent la qualification des rédacteurs, l’indication du caractère complémentaire des informations à la relation médecin-patient, la confidentialité des informations personnelles soumises par les visiteurs du site, la mention des dates et sources des données en ligne, la justification des affirmations sur les bienfaits et inconvénients des produits ou traitement, le professionnalisme et l’accessibilité de l’information, la transparence du financement, la séparation de la publicité de la politique éditoriale. Le label ne garantit pas la qualité du contenu, mais garantit la transparence et l’éthique du fonctionnement du site. Health On The Net est une fondation à but non lucratif dont le siège est situé à Genève. Fin 2007, HON était présente dans 72 pays. Environ 5 700 sites étaient certifiés HON dont plus de 300 sites français. La certification est délivrée pour un an et il est procédé à une réévaluation annuelle systématique.
Le recours à l’information sur internet, notamment en santé et sur les médicaments, va continuer de se développer. Une étude de Médiamétrie publiée en mars 2008, montre que 80 % des internautes vont chercher, chaque jour, des informations de toutes natures sur internet. Internet est donc un outil d’information quotidien qui pourrait être mieux utilisé, pour informer de façon ciblée les patients sur leur pathologie, le bon usage et l’observance des traitements, les accompagner de manière interactive et favoriser leur expression et leurs échanges (blogs, forums).
La MECSS souhaite que les sites certifiés soient tenus de faire figurer sur leur page d’accueil des liens internet vers les sites institutionnels d’information en santé. Par ailleurs, la MECSS souhaite que la HAS, d’une part, évalue la mise en œuvre de la certification des sites d’information en santé, d’autre part, mène des études sur la demande et la consommation d’information en santé et les possibilités d’améliorer l’accès du grand public à une information simple et de qualité sur les médicaments.
IV.- S’APPUYER SUR LE RÉSEAU DES PHARMACIES D’OFFICINE POUR PROMOUVOIR LES GÉNÉRIQUES ET DÉVELOPPER UNE AUTOMÉDICATION RESPONSABLE
A. DÉVELOPPER LE RÔLE DE CONSEIL DES PHARMACIENS D’OFFICINE
a) Le pharmacien d’officine de proximité doit pouvoir s’appuyer sur le dossier pharmaceutique pour assurer la sécurité de la dispensation
Les pharmacies d’officine jouent un rôle essentiel dans la consommation et le bon usage des médicaments. Les 23 000 pharmacies d’officine disposent du monopole de vente des médicaments et assurent la qualité et la sécurité de la délivrance des médicaments. Avec le développement de la contrefaçon des médicaments (des matières premières ou des produits finis) et des possibilités d’achats par internet, le rôle de sécurisation de la dispensation assuré par le pharmacien d’officine apparaît particulièrement nécessaire.
Par ailleurs, les officines qui élaboraient ou préparaient les médicaments sont devenues, principalement, des dispensateurs de médicaments fabriqués par l’industrie pharmaceutique. Et il s’agit, le plus souvent, de médicaments remboursables : environ 80 % du chiffre d’affaires des officines correspond à des produits pris en charge par l’assurance maladie. Par ailleurs, les pharmaciens d’officine sont souvent les professionnels de santé les plus proches et les plus faciles à contacter. Ils bénéficient d’une forte confiance et sont le premier recours susceptible de rendre des services adaptés et personnalisés pour ensuite orienter le patient vers d’autres professionnels de santé. Chaque jour, 4,5 millions de personnes entrent dans une officine.
Afin de favoriser la coordination, la qualité, la continuité des soins et la sécurité de la dispensation des médicaments, la loi du 30 janvier 2007 a prévu la création d’un dossier pharmaceutique (DP). Il a pour objet d’enregistrer tous les médicaments, prescrits ou non, remboursables ou non, délivrés au patient dans n’importe quelle officine au cours des quatre derniers mois. Il comporte uniquement l’identification, la quantité et la date de délivrance des médicaments. Le prescripteur, le prix et le lieu de délivrance ne sont pas indiqués.
Le DP est ouvert avec l’accord du patient. Tout pharmacien d’officine peut ainsi consulter et alimenter le DP du patient au moment de la dispensation en utilisant la carte Vitale du patient (mais sans utiliser son numéro de sécurité sociale) et sa carte professionnelle. Le DP n’est donc consultable que par le pharmacien, en présence du patient. Le DP est destiné à alimenter le futur dossier médical personnel mais il restera distinct de lui. Le DP doit permettre au pharmacien de déceler les risques de redondances ou d’interactions indésirables entre des médicaments, d’améliorer ainsi son conseil et de faciliter le suivi thérapeutique.
Afin de tester le dispositif, une expérimentation, autorisée par la CNIL, a été lancée dans six départements, au second semestre 2007. Le 14 février 2008, la CNIL a autorisé la prolongation de l’expérimentation pour six mois et son extension à deux nouveaux départements et 2 000 officines dans les autres départements. L’Ordre des pharmaciens, qui finance le dispositif, envisage la généralisation du DP à l’ensemble de la France au mois de septembre 2008.
Dans les départements pilotes, 14 % des officines concernées sont entrées dans le dispositif. 168 000 dossiers ont été ouverts, ce qui correspond à un taux d’acceptation par les patients de 80 %. Seulement 20 % le refusent. Le DP est considéré comme fiable et facile d’utilisation. Il peut être un vecteur efficace pour renforcer la confiance entre le patient et le pharmacien. Le patient peut refuser l’utilisation du DP et demander sa fermeture à tout moment. Il peut aussi en demander une copie papier pour compléter l’information de son médecin ou en cas d’hospitalisation. En effet, le médecin peut consulter « l’historique des remboursements » du patient (précédemment appelé Web-médecin), qui est géré par la CNAMTS, mais cet historique ne comporte que les médicaments présentés à l’assurance maladie pour remboursement.
La MECSS souhaite un renforcement de la vigilance à l’égard des contrefaçons de médicaments. Elle demande, d’une part, que toutes les mesures soient prises pour assurer la traçabilité des lots de médicaments afin de faciliter le repérage et le retrait des lots contrefaits, d’autre part que les actions d’inspection auprès des fabricants et grossistes soient multipliées. Elle souhaite aussi un renforcement de la lutte contre les circuits non contrôlés d’importation, de distribution et de commercialisation ainsi que la relance de la réflexion engagée sur la création d’un site internet permettant de sécuriser l’offre de dispensation à distance de médicaments par le circuit des pharmaciens d’officine. Par ailleurs, dans l’attente de la mise en œuvre du dossier médical personnel, la MECSS souhaite le déploiement national, aussi rapidement que possible, du dossier pharmaceutique et la publication du décret nécessaire à cette généralisation.
b) Le rôle des pharmaciens d’officine dans le conseil et l’accompagnement des patients doit être développé
La dispensation du médicament ne se limite pas à la simple délivrance. Le code de la santé publique prévoit notamment que le pharmacien doit aussi fournir aux patients les informations et les conseils nécessaires au bon usage des médicaments. À cet effet, la convention nationale des pharmaciens du 23 mars 2006 comporte des dispositions concernant le suivi et l’accompagnement pharmaceutique des patients, l’observance des traitements et le bon usage des médicaments. Un sondage d’IPSOS-santé sur « les Français et leur pharmacien », effectué en janvier 2008, montre que ce rôle de conseil du pharmacien est reconnu par les patients. D’ailleurs, le recours aux pharmacies est souvent déconnecté de l’acte d’achat de médicaments ; dans près de la moitié des cas, la personne entre dans la pharmacie pour demander au pharmacien un conseil au sujet d’un problème de santé ou des informations sur un médicament consommé. Différentes études montrent d’ailleurs que les patients sont généralement très fidèles à leur pharmacien, ce qui est favorable à la création d’un lien continu entre le patient et le pharmacien.
Le dossier pharmaceutique est un outil pratique qui peut favoriser l’évolution vers un meilleur accompagnement du patient. Le DP contribue, en effet, à renforcer le rôle du pharmacien dans le système de soins et dans la prise en charge individualisée du patient. Il permet un approfondissement de la relation du pharmacien avec le patient et rend possible la mise en place d’un véritable suivi thérapeutique à l’officine.
Cette évolution est d’ailleurs souhaitable. Avec le développement de médicaments de plus en plus complexes, notamment sortis de la réserve hospitalière et destinés à traiter des maladies graves, tels que le cancer et le sida, le besoin de conseils, d’aide à l’observance et d’accompagnement thérapeutique des patients devrait augmenter. Le pharmacien d’officine, dont la formation doit être adaptée aux nouvelles exigences de la dispensation, sera de plus en plus sollicité. Cette évolution a d’ailleurs été amorcée avec l’exercice du droit de substitution pour les médicaments génériques. Elle sera encore accentuée et d’autant plus nécessaire avec le développement, d’une part du maintien à domicile des personnes âgées souvent atteintes d’affection de longue durée et polymédiquées, d’autre part de l’automédication. Par ailleurs, l’évolution de l’exercice de la médecine vers une pratique plus collégiale pouvant associer, à l’instar d’autres pays comme le Royaume-Uni, plusieurs catégories de professionnels médicaux et paramédicaux (médecins, pharmaciens, infirmières, diététiciens…) devrait également conduire à développer le rôle de conseil et d’accompagnement du pharmacien d’officine. À cet égard, on peut citer l’exemple de la nouvelle convention des infirmières qui donne à celles-ci le droit de prescrire certains dispositifs médicaux.
La MECSS souhaite que soient définis, en concertation avec les représentants de la profession, les voies et moyens d’une valorisation du rôle des pharmaciens d’officine et d’une optimisation du service pharmaceutique qui passe par un renforcement de leur mission en matière de conseil, d’éducation thérapeutique, d’observance, d’aide et d’accompagnement des patients.
La politique de développement des génériques a été engagée en France en 1996, avec retard par rapport à l’Allemagne qui a commencé la promotion des génériques dès les années 1980.
En outre, on peut rappeler que la France se caractérise par un phénomène qui ne se retrouve pas dans les autres pays européens : un déplacement des prescriptions vers les produits les plus récents non génériqués, plus onéreux et dont le service médical rendu est souvent équivalent. Par exemple, au Royaume-Uni, 83 % des prescriptions d’IPP sont réalisées dans le répertoire des génériques contre 50 % en France.
Or, les médicaments génériques ont la même efficacité que les médicaments princeps mais ils sont environ 30 % à 40 % moins chers. La politique de développement des génériques ne vise donc pas un objectif direct de santé publique, mais a pour objet une meilleure maîtrise des dépenses de médicaments. Il s’agit, à efficacité thérapeutique identique, de favoriser la prescription des médicaments au plus faible coût.
Le marché des génériques qui représente déjà 10 % du marché mondial des médicaments devrait se développer rapidement dans les prochaines années. En France, avec l’arrivée, entre 2004 et 2007, de nombreux brevets de médicaments dans le domaine public, la politique de développement des génériques revêt une grande importance. On estime que les molécules concernées représenteraient une économie potentielle de plus de 3,8 milliards d’euros pour l’assurance maladie. La question est donc de savoir comment réaliser ce potentiel.
1. Contrer les stratégies de contournement des génériques
a) La France n’a comblé qu’une partie de son retard en matière de médicaments génériques
La France, en dix ans, a rattrapé une partie de son retard sur ses principaux voisins européens. La part des génériques, en volume, dans les médicaments remboursables est passée de 5,4 % en 2000 à 17,7 % en 2006. Cela reste toutefois inférieur au niveau atteint par l’Allemagne et le Royaume-Uni, où la part des ventes de génériques dans l’ensemble des ventes de médicaments était déjà supérieure à 20 % en 1996 ou 1997. En 2006, en France, un peu plus d’une boîte de médicaments vendue sur six est générique, contre une boîte sur deux aux Pays-Bas, au Danemark et en Allemagne. Au Royaume-Uni, les génériques représentent 60 % des boîtes et 24 % du chiffre d’affaires. En France, les génériques représentaient 9,9 % du chiffre d’affaires des médicaments.
Fin mai 2007, le taux de pénétration dans le répertoire des génériques s’élevait à 74,5 % (cela signifie que la délivrance du générique est acceptée dans les trois quarts des cas), soit une hausse de 4,5 points par rapport à fin 2006. La France se situe ainsi à un niveau comparable à celui de ses voisins européens. Le répertoire des génériques est la liste des spécialités princeps généricables et des génériques qui lui sont attachés. C’est dans ce répertoire que peut s’exercer le droit de substitution des pharmaciens.
L’assurance maladie rembourse environ 440 millions de boîtes de génériques par an, soit cinq fois plus qu’en 1999. La CNAMTS estime qu’au total, en 2006, plus d’un milliard d’euros d’économies a été réalisé grâce au développement des médicaments génériques. La pénétration des génériques a contribué à ces économies à hauteur de 600 millions d’euros. Le reste des économies est lié aux baisses de prix appliquées dans le répertoire (400 millions d’euros) et à la mise en place des tarifs forfaitaires de responsabilité (70 millions d’euros).
La poursuite de la progression des génériques se heurte maintenant à la diminution de la part du répertoire dans l’ensemble des médicaments. Fin 2007, le répertoire des génériques comprenait 211 molécules.
b) Les moyens de lutter contre les stratégies de contournement des génériques doivent être renforcés
Les firmes pharmaceutiques mettent en place des stratégies de contournement des génériques pour éviter que ceux-ci ne viennent se substituer au médicament princeps au moment où son brevet tombe dans le domaine public. Afin de bloquer la mise sur le marché des génériques ou limiter la substitution du générique au princeps, les laboratoires cherchent à accroître la durée de la protection du brevet, notamment en étendant les indications du médicament princeps, ou en diversifiant leur gamme de produits par la création de nouveaux dosages, d’associations de molécules ou de nouvelles présentations.
La principale stratégie de contournement de génériques consiste à mettre sur le marché avant la générication d’un produit fabriqué par le laboratoire (ou même d’un produit concurrent) un produit très similaire (un me too). Alors que le produit en voie d’être génériqué n’est plus du tout promu, le nouveau produit, à force de promotion par les visiteurs médicaux auprès des médecins, viendra remplacer l’ancien produit et captera la part de marché durant plusieurs années, sans risque d’être génériqué.
Cette stratégie a, par exemple, parfaitement fonctionné pour Inexium d’AstraZeneca qui a été mis sur le marché dans le but de remplacer le Mopral (génériqué en 2004) mais aussi ses concurrents.
La stratégie la plus récente de contournement de génériques consiste à fabriquer des associations de molécules avec une molécule dont le brevet tombe dans le domaine public et une autre qui est encore protégée par un brevet. L’association permet de conserver une part de marché de la molécule génériquée tout en captant une autre part de marché de la molécule associée.
On peut citer l’exemple du laboratoire MSD qui a lancé, fin 2005, le produit Fosavance, dans le but de reprendre la part de marché de Fosamax qu’il commercialisait mais dont le brevet était menacé. La substitution du nouveau produit à l’ancien a très bien fonctionné. Fosamax est aujourd’hui substitué à 60 %.
Afin de contrer ces stratégies de contournement des génériques, plusieurs modifications de la législation ont été récemment apportées. D’une part, la création, en 2005, de l’autorisation de mise sur le marché globale (AMM globale) vise à empêcher l’obtention d’une protection supplémentaire en cas d’extension de gamme des médicaments princeps. Par ailleurs, la définition du médicament générique et le champ du répertoire des groupes génériques ont été étendus.
Cependant, le droit actuel ne permet pas de limiter les associations de médicaments. Or, ces associations, qui permettent d’accroître la durée de protection des données mais ne visent pas à apporter une réelle innovation thérapeutique, augmentent les risques de surmédication.
Par ailleurs, la principale difficulté que rencontrent les génériqueurs est le manque d’information sur la date d’expiration des brevets.
La MECSS souhaite que soit encouragé l’élargissement du répertoire des génériques et que soit prévue la possibilité d’empêcher la prolongation de la protection des données en cas d’association de médicaments n’apportant pas de réelle innovation thérapeutique. La MECSS souhaite également l’instauration, pour les laboratoires exploitants des spécialités pour lesquelles ils détiennent des brevets, d’une obligation de déclarer au CEPS les titres considérés et leur date d’échéance. En outre, la MECSS souhaite qu’il soit apporté une attention particulière aux conditions permettant d’assurer la viabilité économique de la fabrication de génériques et le développement de ce marché en France.
2. Accroître la délivrance de génériques
a) Renforcer les actions conventionnelles avec les médecins et promouvoir la prescription en dénomination commune
Le développement des génériques suppose d’inciter les médecins à prescrire, soit directement un générique, soit un princeps dans le répertoire des génériques pour que le pharmacien puisse exercer le droit de substitution, soit, enfin, un médicament en dénomination commune internationale (DCI), ce qui facilite l’exercice du droit de substitution.
L’accord conventionnel du 5 juin 2002 prévoyait qu’en contrepartie de la revalorisation du tarif de la consultation à 20 euros les médecins devaient prescrire 25 % de leurs lignes de prescription en DCI ou directement en génériques. La prescription en DCI a progressé mais l’objectif n’a pas été atteint. Elle est passée de 4,2 %, en 2002, à 11,2 %, fin 2006. En outre, elle reste beaucoup moins fréquente chez les spécialistes (4,1 % fin 2006). En revanche, l’objectif de prescription dans le répertoire a été largement atteint.
La convention des médecins du 12 janvier 2005 met l’accent sur le développement des génériques. Les délégués de l’assurance maladie, dans le cadre du retour personnalisé d’information des médecins sur leurs pratiques, doivent notamment les informer de leur niveau de prescription de génériques.
Par ailleurs, l’avenant n° 12, du 3 mars 2006, à la convention prévoit une amplification de l’effort en matière de prescription dans le répertoire des génériques, notamment sur la classe thérapeutique des statines, des IPP et des inhibiteurs de l’enzyme de conversion (IEC) et des sartans.
Enfin, l’avenant n° 23, du 29 mars 2007, fixe un objectif collectif de prescription d’IPP dans le répertoire de 75 % (en nombre de boîtes) et une déclinaison par médecin avec un retour d’information personnalisé.
La MECSS souhaite que la mobilisation pour la prescription de génériques soit poursuivie et que la formation initiale et continue des médecins intègre mieux l’enseignement en DCI.
b) Poursuivre la mobilisation des pharmaciens sur le droit de substitution et l’effort d’information des assurés
Les pharmaciens sont les premiers promoteurs des génériques auprès des assurés. Le développement des génériques résulte, en effet, en grande partie, de l’exercice du droit de substitution par les pharmaciens d’un générique à un médicament princeps dans le répertoire des génériques prescrit par le médecin.
Les pharmaciens jouent donc un rôle clé dans ce domaine et leur mobilisation sur ce thème est essentielle. L’accord conventionnel du 6 janvier 2006 a fixé un objectif de pénétration des médicaments génériques à 70 % du répertoire pour la fin 2006 (61,4 % en 2005). Cet objectif a été décliné pour chaque officine, en fonction du taux initial de délivrance de médicaments génériques. L’objectif a été globalement atteint au niveau national mais des différences subsistent selon les pharmacies : les pharmacies de petite taille ou situées dans les grandes villes délivrent moins de génériques. En 2007, les délégués de l’assurance maladie ont effectué 24 000 visites chez les pharmaciens afin de présenter l’accord ainsi que les profils personnalisés par officine et un numéro de la « lettre aux pharmaciens » de l’assurance maladie a été consacré à ce sujet.
Les campagnes de communication de l’assurance maladie qui ont pour but de convaincre les patients d’accepter les génériques contribuent aussi au développement de ces produits. L’assurance maladie a multiplié les contacts directs avec les assurés. Plus de 400 000 courriers leur ont été envoyés. Des lettres ciblées destinées, d’une part à informer les assurés qui consomment peu ou pas de génériques, d’autre part à remercier ceux qui ont modifié leur comportement en faveur des génériques, ont été envoyées.
La montée en charge du dispositif « tiers payant contre génériques » qui prévoit la suppression de la dispense d’avance de frais en cas de refus de la substitution d’un générique au princeps a également favorisé le développement de la substitution. Ce dispositif est actuellement appliqué dans plus de la moitié des départements (55).
La MECSS considère que des marges de progrès dans la délivrance des médicaments génériques subsistent. Afin de les réaliser, elle souhaite l’accentuation de la mobilisation des pharmaciens et des DAM sur ce thème et le développement de la communication de l’assurance maladie en direction des assurés. Elle souhaite également que le dispositif du « tiers payant contre génériques » soit généralisé à l’ensemble des départements qui n’atteignent pas les objectifs fixés par les accords conventionnels.
C. DÉVELOPPER UNE AUTOMÉDICATION RESPONSABLE
On peut rappeler qu’en France, l’automédication occupe une place limitée. Elle représente 17 % des médicaments vendus, en volume, et 8 % du chiffre d’affaires, soit 1,6 milliard d’euros, en 2005. En outre, la part de marché des médicaments consommés en automédication est stagnante. Rappelons aussi que l’automédication comprend les produits qui ne peuvent pas être remboursés et la part des produits remboursables de prescription médicale facultative (PMF) qui ne sont pas présentés au remboursement parce qu’ils ont été achetés sans ordonnance (un quart des PMF).
L’automédication correspond au souhait d’autonomie des patients qui sont, pour une bonne part d’entre eux, de mieux en mieux informés ou, parfois, croient l’être. L’automédication est l’expression de la volonté du patient de se prendre en charge lui-même pour soigner son affection. En décidant de recourir à l’automédication pour de petits maux de santé, le patient s’inscrit dans une démarche responsable.
Le développement d’une automédication responsable et maîtrisée devrait permettre de mieux responsabiliser les patients et au médecin de redéployer une partie son temps médical vers d’autres activités, par exemple de prévention ou d’éducation thérapeutique.
a) Le droit français ne définit pas expressément l’automédication
Le droit français ne définit pas explicitement les médicaments à prescription médicale facultative ni l’automédication.
On peut rappeler que tous les médicaments, qu’ils soient soumis à prescription médicale obligatoire (PMO) ou à prescription médicale facultative (PMF), doivent, pour être vendus en officine, avoir fait l’objet de la délivrance d’une autorisation de mise sur le marché (AMM) par l’AFSSAPS.
Le code de la santé publique dispose que la prescription est obligatoire pour toutes les spécialités inscrites sur une liste (liste I, liste II, stupéfiants). Les listes I et II comprennent en particulier « les médicaments susceptibles de présenter directement ou indirectement un danger pour la santé » ou « contenant des substances dont l’activité ou les effets indésirables nécessitent une surveillance médicale » (article L. 5132-6). La liste I comprend les médicaments présentant les risques les plus élevés.
Mais le code de la santé publique ne fixe pas de définition spécifique pour les spécialités de prescription médicale facultative. Ces dernières représentent, par défaut, toutes les spécialités ne présentant pas les critères d’inscription sur une des listes citées.
La réglementation européenne est un peu plus précise. Elle prévoit, en effet, que les médicaments non soumis à prescription sont « ceux qui ne répondent pas aux critères énumérés » que doivent respecter les médicaments soumis à prescription médicale (article 72 du code communautaire relatif aux médicaments à usage humain). Le code communautaire ajoute que c’est l’autorité d’enregistrement qui, en délivrant l’AMM, décide du statut du médicament.
L’Organisation mondiale de la santé (OMS) donne, dans une recommandation relative à l’automédication publiée en 2000, une définition plus explicite encore puisqu’elle considère que l’automédication consiste en la sélection et l’utilisation de médicaments par les personnes pour traiter les maux et symptômes qu’elles ont elles-mêmes identifiés. Dans cette recommandation, l’OMS ajoute, d’une part, que les médicaments d’automédication sont ceux qui ne nécessitent pas de recours à une prescription médicale, qui sont produits, distribués et vendus dans le but d’une utilisation à l’initiative du consommateur et sous sa propre responsabilité, d’autre part, que le conditionnement et les informations sur le produit figurant sur la boîte et la notice doivent être adaptés pour permettre une correcte automédication.
Actuellement, tous les produits dont le principe actif ne figure pas sur une des listes prévues par le code de la santé publique peuvent donc faire l’objet d’une automédication, c’est-à-dire être achetés sans prescription médicale. Or, l’accès au remboursement et l’obligation de prescription médicale ne sont pas superposables, puisque certains médicaments ne sont disponibles que sur prescription médicale sans être remboursables par l’assurance maladie. En conséquence, le champ des médicaments d’automédication recouvre des produits non remboursables mais aussi des produits qui sont remboursables lorsqu’ils font l’objet d’une prescription médicale.
b) L’automédication pourrait être définie comme un comportement
Le rapport sur l’automédication remis, en janvier 2007, par MM. Alain Coulomb et Alain Baumelou à M. Xavier Bertrand, alors ministre de la santé et des solidarités, estime que l’automédication doit être considérée comme un comportement et non comme une catégorie de produits.
En conséquence, il propose de définir « l’automédication comme le fait pour un patient d’avoir recours à un ou plusieurs médicaments de prescription médicale facultative (PMF) dispensé(s) dans une pharmacie et non effectivement prescrit(s) par un médecin ».
On peut noter que cette définition exclut l’utilisation des médicaments à prescription obligatoire stockés dans « l’armoire à pharmacie familiale ».
L’Ordre des pharmaciens, dans son livre blanc sur le bilan et les perspectives de la pharmacie d’officine en France, publié au mois de janvier 2008, estime qu’il serait préférable d’utiliser l’expression de « médication officinale » plutôt que celle d’automédication. La première traduirait mieux le caractère sécurisé de la dispensation à l’officine.
2. Maîtriser la mise devant le comptoir des officines de certains médicaments à prescription médicale facultative
Le gouvernement a annoncé sa volonté d’autoriser la mise à disposition de médicaments non soumis à prescription devant le comptoir des pharmacies dans des conditions devant permettre d’assurer la sécurité de la dispensation et dans le respect du monopole des pharmaciens. À cet égard, on peut rappeler que, lors de la remise du rapport présenté par M. Jacques Attali, au nom de la commission pour la libération de la croissance française, au mois de janvier 2008, le Président de la République a indiqué qu’il n’était pas favorable à la mise en vente de médicaments dans les grandes surfaces.
a) Les conditions de mise en place sont en cours de définition
En décembre 2007, quatre groupes de travail ont été créés pour préparer le cadre de cette évolution. À l’issue de la réunion de concertation, organisée le 15 janvier 2008, la ministre de la santé a fixé les principes du libre accès de certains médicaments dans les officines, avec pour objectif un démarrage effectif au premier semestre 2008.
Le principe du libre choix, pour le pharmacien, d’organiser ou non dans son officine l’accès direct à des médicaments PMF a été retenu. L’AFSSAPS doit établir la liste de spécialités qui devront respecter certains critères. Il est prévu de commencer avec une première liste d’environ 200 spécialités qui sera progressivement élargie. Le conditionnement devra être adapté à la posologie et à la durée de traitement et, à terme, muni d’un dispositif en vue de s’assurer qu’il n’a pas été ouvert. Le contenu de la notice devra être adapté pour la bonne information du patient. Les spécialités ne devront pas avoir fait l’objet d’une restriction de publicité dans leur AMM.
Les spécialités en accès direct devront être présentées dans un espace réservé et clairement identifié. L’objectif est d’éviter toute confusion ou mélange avec des produits n’ayant pas d’AMM.
Les prix devront être clairement affichés et l’ensemble des acteurs devra s’engager dans une modération des prix. Un « observatoire des prix » de ces médicaments sera créé pour suivre leur évolution.
Dans sa rédaction actuelle, le code de la santé publique interdit le libre accès à des médicaments, qu’ils soient à prescription obligatoire ou facultative, remboursables ou non. Un décret en Conseil d’État devrait donc être publié au printemps 2008 pour permettre de démarrer le libre accès à certains médicaments PMF dans les officines avant l’été.
Ces principes paraissent de nature à pouvoir sécuriser le démarrage de la mise à disposition devant le comptoir des produits à PMF. Le déploiement national du dossier pharmaceutique pourrait aussi y contribuer.
b) L’intérêt de la démarche n’est pas toujours bien compris et les conditions de mise en œuvre suscitent des inquiétudes
Cependant, à ce stade, selon deux sondages Ipsos santé, réalisés aux mois de janvier et février 2008, l’idée d’un libre accès de certains médicaments dans les pharmacies ne recueille qu’une approbation minoritaire des patients (62 % des personnes se disant plutôt pas favorables – 30 % – ou pas du tout favorables – 32 %) et des pharmaciens (84 % y sont défavorables). Certains pharmaciens expriment la crainte que les médicaments PMF finissent un jour par être vendus dans les grandes surfaces, que le libre accès ne fasse pas baisser le prix des médicaments et ne permette pas à la sécurité sociale de faire des économies ni à responsabiliser le patient et provoque des comportements dangereux. Ils évoquent aussi le manque d’information ou de place dans la pharmacie et le manque d’intérêt de la part des patients.
Force est de constater qu’il reste à lever certaines inquiétudes et à convaincre les principaux acteurs de l’intérêt de cette démarche.
La MECSS souhaite que la mise à disposition devant le comptoir des produits à prescription médicale facultative s’effectue dans le respect du libre choix des pharmaciens. La Mission souhaite aussi qu’il soit veillé au respect de l’engagement de modération sur les prix des médicaments non remboursables et à ce que la dispensation soit sécurisée et accompagnée du conseil du pharmacien. Par ailleurs, la MECSS souhaite, d’une part, que soient étudiées les conditions de la prise en charge des médicaments concernés par les organismes de protection sociale complémentaire, d’autre part, qu’il soit procédé à une évaluation de la mise en avant des comptoirs des médicaments à prescription médicale facultative.
V.- FAVORISER LA MISE EN PLACE D’UNE FISCALITÉ PLUS SIMPLE ET STRUCTURANTE
La MECSS a souhaité compléter son approche sanitaire du médicament en se penchant sur la fiscalité applicable au secteur, stratégique, du médicament. La fiscalité est, en effet, un levier essentiel qui peut contribuer à la compétitivité de notre industrie et favoriser le développement des activités de recherche et de production de médicaments sur notre territoire.
Or, au fil du temps, la fiscalité spécifique du médicament s’est de plus en plus complexifiée. Elle est, aujourd’hui, non seulement complexe mais aussi instable et peu structurante.
Les entreprises qui fabriquent ou qui distribuent les médicaments sont redevables de onze taxes, à rendement très inégal. En 2006, le montant total de ces taxes s’est élevé à 1 021 millions d’euros, ce qui représente environ 4 % du chiffre d’affaires de l’industrie pharmaceutique réalisé en France.
On peut répartir ces taxes en deux catégories, d’une part les taxes qui rémunèrent des services rendus par les organismes régulateurs (environ 60 millions d’euros en 2006), d’autre part les taxes affectées à la sécurité sociale (962 millions d’euros en 2006).
Par ailleurs, on peut ajouter la TVA sur les médicaments (1,1 milliard d’euros en 2006) dont l’essentiel de la ressource est affecté à l’assurance maladie.
A. VEILLER À L’INDÉPENDANCE DES ORGANISMES QUI PERÇOIVENT LES TAXES RÉMUNÉRANT DES SERVICES RENDUS
La Haute Autorité de santé et l’AFSSAPS, qui jouent un rôle essentiel en matière de régulation du médicament, sont en partie financées par des taxes pour services rendus.
1. La Commission de la transparence est financée en quasi-totalité par une taxe payée par les laboratoires pharmaceutiques
La Haute Autorité de santé perçoit une taxe destinée à couvrir le coût de l’évaluation du service médical rendu par le médicament. Elle est d’un montant forfaitaire pour chaque dossier présenté, mais variable selon que la demande concerne l’inscription initiale sur la liste des médicaments remboursables ou une modification ou un renouvellement de l’inscription.
Cette taxe est de faible rendement puisqu’elle a rapporté 4,2 millions d’euros en 2006, ce qui correspond à près de 7 % des recettes de la HAS. Mais elle couvre 90 % du coût de fonctionnement de la Commission de la transparence (rattachée à la HAS) qui est chargée de l’évaluation du SMR des médicaments. Cette taxe ne génère aucun contentieux.
2. Plus de la moitié du financement de l’AFSSAPS est assuré par cinq taxes payées par les laboratoires pharmaceutiques
L’AFSSAPS perçoit cinq taxes : une taxe est calculée sur le chiffre d’affaires, les quatre autres sont liées au nombre de dossiers de demandes traités par l’agence. En 2006, au total, les cinq taxes ont rapporté à l’AFSSAPS 55 millions d’euros, ce qui représente 55 % de son budget total, lequel s’est élevé à près de 100 millions d’euros.
La première est la taxe sur le chiffre d’affaires des médicaments disposant d’une AMM. Cette taxe vise à financer l’ensemble des activités de sécurité sanitaire de l’agence. Elle est payable selon un barème qui comprend neuf tranches. En 2006, elle a été payée par 400 entreprises et a rapporté 19,3 millions d’euros, ce qui représente 19 % des ressources de l’AFSSAPS.
Les quatre autres taxes sont :
– le droit progressif par demande d’autorisation de mise sur le marché (nouveaux principes actifs ou nouvelles indications) ou de modification ou de renouvellement ; il varie, selon le type de demande de 700 à 25 000 euros ; en 2006, il a rapporté à l’AFSSAPS environ 30 millions d’euros, ce qui représente 30 % des ressources de l’agence ;
– le droit pour chaque demande de visa ou de renouvellement de visa de publicité (5,6 millions d’euros) ;
– le droit d’enregistrement des médicaments homéopathiques (0,1 million d’euros) ;
– le droit pour les dossiers de demande d’importation parallèle (0,06 million d’euros).
Hormis la taxe sur le chiffre d’affaires, le rendement des autres taxes est instable et varie d’une année sur l’autre, en fonction de la politique industrielle et commerciale des laboratoires.
3. Le mode de financement des organismes qui délivrent les autorisations et évaluent les médicaments doit permettre d’assurer leur indépendance
Le financement du contrôleur par le contrôlé peut présenter un risque de conflit d’intérêts. Cette question a été fréquemment évoquée lors des auditions auxquelles a procédé la Mission. M. Éric Woerth, ministre du budget, des comptes publics et de la fonction publique, ainsi que la Cour des comptes, les différents directeurs en charge de la tutelle des organismes concernés de même que les dirigeants des organismes ont estimé que le financement par des taxes pour services rendus d’une partie non négligeable des besoins de la HAS et de l’AFSSAPS ne posait pas de réel problème, dans la mesure où le dispositif est transparent.
La Cour des comptes estime toutefois préférable que la collecte de la taxe sur le chiffre d’affaires des spécialités soit assurée par la direction générale des impôts (fusionnée début avril 2008 avec la direction générale de la comptabilité publique au sein de la direction générale des finances publiques) en même temps que la TVA. Il conviendrait toutefois de s’assurer qu’un éventuel changement des modalités de collecte ne vienne pas compliquer les formalités à accomplir par les entreprises.
La MECSS comprend l’avantage qu’il y a à financer par des taxes pour services rendus une partie des besoins de la HAS et de l’AFSSAPS. Toutefois, la Mission souhaite qu’il soit veillé à ce que les conditions de financement des organismes concernés permettent d’assurer leur indépendance à l’égard de l’industrie pharmaceutique.
B. SIMPLIFIER, STABILISER OU RENDRE PLUS STRUCTURANTE LES TAXES AFFECTÉES À L’ASSURANCE MALADIE
Depuis plusieurs années, dans le cadre de la politique de maîtrise des dépenses de médicaments et de renforcement des ressources de l’assurance maladie, cinq taxes ont été créées dont les produits lui sont affectés. En 2006, elles ont représenté 962 millions d’euros.
En outre, depuis 2006, l’assurance maladie bénéficie du reversement du produit de la TVA sur les médicaments : 1,1 milliard d’euros.
Au total, en 2006, ce sont ainsi près de 2,1 milliards d’euros de taxes sur le médicament qui ont abondé les ressources de l’assurance maladie.
Taxes sur le médicament affectées à l’assurance maladie, en 2006
(en millions d’euros)
TVA nette sur le médicament |
1 142 |
Taxe sur le chiffre d’affaires |
375 |
Taxe sur les grossistes répartiteurs |
375 |
Taxe sur la promotion |
212 |
Contribution de sauvegarde |
0 |
Total |
2 104 |
1. Aménager les deux taxes destinées à maîtriser la dépense de médicaments remboursés par l’assurance maladie
a) Mobiliser la taxe sur les dépenses de promotion pour améliorer la régulation de la visite médicale
La taxe sur les dépenses de promotion des médicaments remboursables (375 millions d’euros) est calculée à partir de quatre tranches de barème applicables aux dépenses de promotion. L’assiette est constituée des rémunérations et des charges sociales et fiscales afférentes, des frais des visiteurs médicaux et des frais de publication et d’achats d’espace publicitaire (rémunérations, frais, publication). Des taux progressifs (en 2006 : 19 %, 29 %, 36 % et 39 %) sont appliqués en fonction de quatre tranches de barème qui tiennent compte du rapport entre les dépenses de promotion et le chiffre d’affaires en spécialités remboursables. Plus le poids relatif de l’effort de promotion dans le chiffre d’affaires est élevé, plus le taux de contribution est important. Elle est acquittée par les entreprises sur une base déclarative.
L’impact réel de la taxe sur les dépenses de promotion est incertain. La Cour des comptes estime qu’« il est vraisemblable que son effet régulateur est faible ». En outre, la taxe sur les dépenses de promotion est source de contentieux concernant la définition des différents éléments pris en compte pour son calcul (frais professionnels, dépenses de fabrication des objets promotionnels, dépenses de publication et d’achats d’espaces publicitaires…) et la date d’application des changements de doctrine de l’administration fiscale.
M. Éric Woerth, ministre du budget, des comptes publics et de la fonction publique, a indiqué, lors de son audition par la Mission, qu’une réflexion était engagée concernant la réforme de la taxe sur la promotion et de la taxe sur le chiffre d’affaires, réflexion qui pourrait se traduire par des dispositions insérées dans le projet de loi de financement de la sécurité sociale pour 2009.
La MECSS souhaite que la sécurité juridique de la taxe sur la promotion soit assurée et que les textes nécessaires à la clarté du dispositif soient publiés dans les meilleurs délais. Elle souhaite aussi qu’une réflexion soit engagée avec l’industrie du médicament et le CEPS en vue d’améliorer la régulation quantitative (objectifs quantifiés) et qualitative (analyse des effets de la certification) de la visite médicale et, le cas échéant, de renforcer la taxe sur la promotion.
b) Simplifier la contribution à la clause de sauvegarde de l’objectif national de dépenses d’assurance maladie (ONDAM) pour la rendre effectivement applicable
La contribution à la clause de sauvegarde de l’ONDAM a été créée en 1999. Elle est destinée à recouvrer une partie du dépassement constaté entre la croissance du chiffre d’affaires de l’ensemble des entreprises pharmaceutiques et un taux de progression (taux K) défini, chaque année, en loi de financement de la sécurité sociale. Elle est due seulement par les entreprises qui n’ont pas conclu de convention avec le CEPS et dont le chiffre d’affaires réalisé au titre des spécialités remboursables dépasse le taux de progression de l’ONDAM. Les entreprises ayant conclu une convention avec le CEPS sont exonérées du paiement de cette contribution et s’acquittent, en contrepartie, du paiement de remises conventionnelles.
Le taux K est donc le taux de progression à partir duquel le calcul de la contribution est effectué. Ce taux a ainsi évolué : 2 % en 2000, 3 % en 2001 et 2002, 4 % en 2003, 3 % en 2004, 1 % de 2005 à 2007, 1,4 % en 2008.
En cas de dépassement, un taux de contribution croissant (50 %, 60 % et 70 %) est appliqué en fonction de l’importance du dépassement du chiffre d’affaires par rapport à l’objectif fixé pour l’ONDAM (inférieur à 0,5 %, entre 0,5 % et 1 %, au-dessus de 1 %). Plus le dépassement est important, plus le taux de la contribution est élevé. Le montant de la contribution est réparti entre les entreprises redevables et en trois parts qui prennent en compte le niveau du chiffre d’affaires (30 %), la progression du chiffre d’affaires (40 %) et les frais de promotion (30 %).
Le dispositif est particulièrement complexe. Il articule un seuil de déclenchement, une taxation collective et une valorisation individuelle compliquée qui prend donc en compte, notamment, les dépenses de promotion, lesquelles sont déjà assujetties à la taxe sur la promotion. D’autre part, les URSSAF qui sont chargées du recouvrement rencontrent des difficultés pour déterminer les entreprises redevables et des difficultés pratiques de calcul qui engendrent de nombreuses opérations de régularisation. En outre, le dispositif ne permet, en théorie, de récupérer qu’une partie du dépassement et on ne peut pas considérer qu’il correspond à la logique d’enveloppe fermée ou de crédits limitatifs que le rapport Attali, susmentionné, propose d’instaurer.
En fait, la contribution à la clause de sauvegarde de l’ONDAM est de rendement nul. Elle a perdu tout objectif de rendement et aucune entreprise ne l’acquitte. De fait, elle n’est plus qu’un dispositif d’incitation à la contractualisation des laboratoires avec le CEPS.
Il faut en effet rappeler que les entreprises conventionnées, en application de l’accord-cadre conclu entre le CEPS et le LEEM, s’engagent à verser des remises conventionnelles. En 2006, celles-ci ont représenté 507 millions d’euros, soit environ 2 % du chiffre d’affaires de l’industrie pharmaceutique.
La clause de sauvegarde conserve donc son utilité : elle sert d’étalon puisque les remises ne peuvent excéder, globalement, le montant de la clause de sauvegarde que l’ensemble des entreprises auraient payé si elles n’avaient pas été conventionnées.
On peut ajouter que le champ d’application de la clause de sauvegarde de l’ONDAM a été étendu en 2006 aux médicaments faisant l’objet d’une rétrocession hospitalière mais que les textes d’application de cette mesure n’ont pas encore été publiés.
La MECSS souhaite, d’une part, que le dispositif de clause de sauvegarde de l’ONDAM soit simplifié afin de rendre possible, le cas échéant, son application effective, d’autre part, que les textes d’application de ladite contribution aux spécialités pharmaceutiques faisant l’objet d’une rétrocession hospitalière soient rapidement publiés.
2. Stabiliser les deux autres taxes destinées à procurer des ressources à l’assurance maladie
a) Maintenir la taxe sur les grossistes répartiteurs
La taxe sur les grossistes-répartiteurs, aussi dénommée « contribution sur les ventes directes », est due par les entreprises de vente en gros de médicaments (grossistes-répartiteurs) et par les fabricants de médicaments qui vendent directement en gros. La taxe est calculée, d’une part, sur le chiffre d’affaires au taux de 1,9 %, d’autre part, sur la progression du chiffre d’affaires au taux de 2,25 %. En 2006, elle a rapporté 375 millions d’euros.
En outre, la loi de financement de la sécurité sociale pour 2007, a créé une contribution exceptionnelle de régulation due par les mêmes entreprises et calculée sur la même assiette avec des taux respectifs de 0,21 % et 1,5 %. Le rendement prévu était de 37 millions d’euros.
Le projet de loi de financement de la sécurité sociale pour 2008 prévoyait de proroger pour 2008 cette contribution exceptionnelle en portant le taux applicable au chiffre d’affaires à 0,22 % et en laissant inchangé à 1,5 % le taux applicable à la progression du chiffre d’affaires. Le rendement attendu était de 50 millions d’euros. La contribution a finalement été remplacée par une réduction des marges des grossistes, d’impact financier équivalent.
b) Stabiliser la taxe sur le chiffre d’affaires
La taxe sur le chiffre d’affaires est due par les entreprises qui exploitent des médicaments remboursables. Instaurée en 2004, à titre exceptionnel, pour compenser la baisse des recettes résultant de la modification des tranches du barème de la taxe sur la promotion, elle a été reconduite depuis, tous les ans, par les lois de financement de la sécurité sociale. Son taux, initialement fixé à 0,525 % en 2004, a été porté à 0,6 % en 2006. Le taux de la contribution a été fixé, « à titre exceptionnel », à 1,76 % en 2007 et à 1 % en 2008.
La loi de financement de la sécurité sociale pour 2008 a, en outre, supprimé l’abattement à la taxe sur le chiffre d’affaires créé par la loi du 26 février 2007 d’adaptation du droit communautaire dans le domaine du médicament, à la suite de la décision prise par le Conseil stratégique des industries de santé. Cette suppression est la conséquence de la réforme du crédit d’impôt recherche (CIR) prévu par la loi de finances pour 2008. La réforme du CIR est en effet bien plus favorable pour les entreprises pharmaceutiques que l’abattement.
En 2006, la taxe sur le chiffre d’affaires a rapporté 375 millions d’euros.
La taxe sur le chiffre d’affaires est utilisée comme un moyen de régulation. Le taux est adapté en fonction de l’évolution des ventes de médicaments. Elle est un des outils disponibles pour assurer la régulation du secteur, à côté des réductions de prix et des remises conventionnelles.
La fluctuation du taux de la taxe et le fait que celui-ci soit connu tardivement, au moment de la promulgation de la loi de financement de la sécurité sociale, font l’objet de critiques, les entreprises se plaignant de manquer de visibilité pour établir leurs prévisions budgétaires.
La MECSS estime qu’il serait souhaitable de donner une meilleure visibilité aux entreprises pharmaceutiques en matière de taxe sur le chiffre d’affaires. Elle demande qu’une réflexion soit engagée avec l’industrie pharmaceutique, par exemple dans le cadre du Conseil stratégique des industries de santé, visant à fixer un taux pluriannuel de taxe sur le chiffre moins élevé en contrepartie d’un renforcement des remises conventionnelles.
3. Assurer le recouvrement de la TVA sur les médicaments et évaluer les effets des déremboursements sur les recettes de taxes
Depuis 2006, la sécurité sociale reçoit, en compensation des exonérations de charges sociales, un panier de taxes – parmi lesquelles la TVA sur les médicaments – dont une grande partie du produit est affectée à l’assurance maladie.
Le taux de la TVA applicable aux médicaments remboursables s’élève à 2,1 % et celui applicable aux médicaments non remboursables à 5,5 %. En 2006, le montant de la TVA nette déclarée par les entreprises du médicament s’est élevé à 1,1 milliard d’euros.
Les contrôles opérés ont été productifs puisqu’ils ont permis de récupérer 53 millions d’euros en 2006, soit près de 5 % de la recette nette de TVA sur les médicaments.
Les décisions de déremboursement sur les recettes de TVA et des autres taxes sur les médicaments ont des conséquences sur les prix des médicaments et les volumes vendus et remboursés. Mais on n’a qu’une connaissance partielle de ces effets. Ainsi, on sait que le déremboursement d’un médicament conduit à une diminution des dépenses d’assurance maladie et met fin au versement des taxes dues au titre des médicaments remboursables, mais on ne peut en établir un bilan précis.
La MECSS souhaite, d’une part, que l’administration fiscale poursuive une politique active de contrôle de la TVA sur les médicaments, d’autre part, que l’impact des déremboursements sur les recettes de TVA et des autres taxes sur le médicament soit évalué.
*
La MECSS a souhaité s’appuyer sur un constat chiffré, étayé et précis de la consommation de médicaments en France et a cherché à prendre la mesure des différents facteurs expliquant le haut niveau de prescription et de consommation de médicaments en ville qui caractérise notre pays.
Les propositions qu’elle formule visent à promouvoir le bon usage des médicaments et à renforcer l’efficience des prescriptions. Elles concernent tous les acteurs intéressés et leur mise en œuvre suppose une mobilisation de chacun d’entre eux dans le cadre d’un partenariat actif de santé. Le pari de la responsabilisation de tous peut être gagné si les enjeux sont bien compris, la démarche éclairée et partagée. Notre souhait est que le présent rapport puisse y contribuer.
Les mesures proposées seront d’autant plus efficaces si elles s’inscrivent dans l’évolution de l’organisation du système de soins avec le décloisonnement de la médecine de ville, de l’hôpital et du médico-social que devrait notamment permettre la création des agences régionales de santé, un exercice plus en équipe de la médecine de ville, le développement de la prévention, du conseil et de l’accompagnement des patients que doit favoriser l’évolution vers de nouveaux modes de rémunération des professionnels médicaux et paramédicaux.
À l’issue de cette réflexion, la MECSS souhaite en effet souligner le rôle déterminant de la prévention, de l’éducation en santé, de l’éducation thérapeutique et du conseil pour faire évoluer les comportements de prescription et de consommation des médicaments.
*
La MECSS, comme le prévoit l’article LO 111-9-3 du code de la sécurité sociale, notifiera les préconisations du présent rapport au Gouvernement et aux organismes de sécurité sociale concernés, lesquels seront tenus d’y répondre dans un délai de deux mois, et assurera le suivi de ses conclusions.
I. AMÉLIORER L’ENCADREMENT DE LA VIE DU MÉDICAMENT ET RENFORCER LA SÉLECTIVITÉ DE L’ACCÈS AU REMBOURSEMENT
A. Veiller au bon usage des procédures dérogatoires de mise sur le marché et de prescription des médicaments
1. Veiller à ce que l’autorisation temporaire d’utilisation (ATU) ne soit pas utilisée pour contourner l’autorisation de mise sur le marché (AMM)
2. Veiller à ce que le délai entre l’ATU et l’AMM soit le plus court possible
3. Contrôler, évaluer et encadrer la prescription hors AMM
B. Renforcer les règles de l’admission au remboursement et à la fixation du prix des médicaments
4. Recourir au critère de l’intérêt pour la santé publique (ISP) pour l’appréciation du service médical rendu (SMR) qui détermine le taux de remboursement du médicament
5. Contrôler le respect des règles relatives aux indications remboursables ou non (mention « NR » sur l’ordonnance)
6. Rendre obligatoire les essais cliniques contre comparateurs avant l’appréciation de l’amélioration du service médical rendu (ASMR) par la Commission de la transparence
7. Établir un classement des médicaments par niveau d’ASMR
8. Veiller à la mise en œuvre rapide de la nouvelle compétence médico-économique de la Haute Autorité de santé (HAS)
9. Faire participer la HAS à la redéfinition régulière du panier de médicaments pris en charge
10. Demander à la HAS d’étudier les moyens de réduire la surprescription, développer le bon usage et les alternatives à la prescription médicamenteuse
11. Demander à la HAS d’évaluer l’impact de ses recommandations et de mettre en place des indicateurs et tableaux de bord pour mesurer l’évolution des comportements de prescription
12. Évaluer les effets de la franchise sur la prescription et la consommation de médicaments ainsi que sur le développement des médicaments génériques
C. Améliorer le suivi des médicaments en pratique médicale réelle et gérer de manière plus active la liste des médicaments remboursables
13. Veiller à publier rapidement les résultats des études de pharmacovigilance et prendre sans retards les décisions de suspension ou de retraits d’AMM
14. Rendre systématique l’information sur les restrictions d’indications et assurer le contrôle de leur application
15. Relancer l’activité du groupement d’intérêt scientifique « évaluation épidémiologique des produits de santé » afin de développer l’évaluation post-AMM
16. Instaurer un dispositif de sanction pour défaut ou retard d’étude post-AMM
17. Appliquer avec davantage de célérité les recommandations de la HAS consécutives à la réévaluation des SMR
II. FAIRE ÉVOLUER LES COMPORTEMENTS DES PRESCRIPTEURS ET DES CONSOMMATEURS
A. Renforcer l’information sur le médicament
18. Veiller à l’indépendance des experts de l’AFSSAPS et de la HAS, notamment des membres de la Commission de la transparence
19. Assurer la transparence et la publicité des travaux d’expertise de l’AFSSAPS et de la HAS
20. Mettre en place, dans le meilleur délai, une base publique, exhaustive et gratuite d’information sur les médicaments
B. Agir sur les déterminants de la prescription
Réformer la formation initiale des médecins sur le médicament
21. Réformer la formation médicale initiale des médecins afin de renforcer les enseignements en pharmacologie, en pratiques thérapeutiques, notamment médicamenteuse, en économie de la santé et en prescription en dénomination commune internationale (DCI)
22. Limiter l’influence des laboratoires pharmaceutiques sur les étudiants en médecine, notamment lors des stages en milieu hospitalier
Veiller à la montée en charge de la formation médicale continue et de l’évaluation des pratiques professionnelles
23. Achever la mise en place des dispositifs de formation médicale continue (FMC) et d’évaluation des pratiques professionnelles (EPP)
24. Étudier la possibilité d’adosser les conseils nationaux de la FMC à la HAS
25. Associer le ministère de la santé, la HAS et l’assurance maladie à la définition des thèmes prioritaires de FMC et d’EPP
26. Renforcer les conditions d’agrément des organismes de formation : cahier des charges plus précis, référentiel de qualité, indépendance à l’égard de l’industrie pharmaceutique, contrôle, sanctions
27. Instaurer un dispositif de sanctions en cas de non-respect des obligations de formation continue et d’EPP
28. Développer les formations associant médecins et pharmaciens
29. Développer les formations à l’écoute et à la gestion de la relation avec les patients ainsi qu’aux alternatives au médicament
30. Créer un fonds regroupant les financements publics et privés et étudier la possibilité d’un renforcement des financements institutionnels de la FMC
Rééquilibrer l’information des médecins sur le médicament
Maîtriser l’impact de la visite médicale
31. S’assurer de la remise systématique par le visiteur médical (VM) au médecin du relevé des caractéristiques du produit (RCP) et de la fiche de transparence
32. Définir l’abus de VM, doter les organismes certificateurs de la visite médicale des moyens juridiques nécessaires afin de leur permettre de contrôler le respect de la charte de la VM, instaurer un dispositif de sanction en cas de non-respect de la charte
33. Demander au Comité économique des produits de santé (CEPS) d’étendre et renforcer la régulation quantitative de la visite médicale
34. Mener des études sur l’impact de la visite médicale sur les comportements de prescription
Développer l’information publique
35. Faire de la HAS l’émetteur unique d’information sur le bon usage des médicaments
36. Transférer de l’AFSSAPS à la HAS la mission de contrôle de la publicité sur les médicaments
37. Renforcer les capacités de la HAS en matière d’analyse et d’évaluation des stratégies promotionnelles de l’industrie pharmaceutique et des prescriptions : création d’un observatoire de la prescription adossé à un réseau de médecins
38. Demander à la HAS d’établir une recommandation sur le bon usage de la visite médicale
39. Améliorer la lisibilité du site internet de la HAS et développer la diffusion de documents d’information pratique indiquant le coût des traitements
40. Doter la HAS de la capacité de diffuser de l’information sur les médicaments aux médecins en face à face : création d’un corps de « délégués de santé », spécialement formés et certifiés par la HAS, notamment pour promouvoir le bon usage des médicaments ; ce corps, constitué à partir des délégués de l’assurance maladie, serait copiloté par la HAS et la CNAMTS et géré administrativement par la CNAMTS
Diffuser les logiciels certifiés d’aide à la prescription
41. Rendre obligatoire la certification d’aide à la prescription (LAP), mettre en œuvre rapidement la procédure de certification des logiciels en médecine de ville afin que tous les logiciels proposés sur le marché soient certifiés à la fin de l’année 2009
42. Étudier la possibilité d’adjoindre aux LAP une fonction d’aide à la décision thérapeutique
43. Engager, sans tarder, le processus de certification des LAP hospitaliers
Renforcer les actions de maîtrise médicalisée de l’assurance maladie
Développer l’analyse des prescriptions
44. Développer les capacités d’analyse des prescriptions des caisses d’assurance maladie
45. Étendre au régime général le système, en vigueur à la MSA, d’alerte du prescripteur en cas de primo-prescription non conforme au bon usage ou de polymédication présentant des risques iatrogènes
Renforcer l’efficacité de la maîtrise médicalisée
46. Amplifier les efforts de maîtrise médicalisée et les étendre à de nouvelles classes de médicaments
47. Relancer la politique de publication d’accords de bon usage des soins
48. Veiller à ce que les partenaires conventionnels mettent en œuvre rapidement les dispositions issues de la loi de financement de la sécurité sociale pour 2008 concernant l’individualisation des objectifs de maîtrise médicalisée et d’amélioration des pratiques
49. Renforcer les actions de communication des caisses d’assurance maladie sur la démarche qualité et le bon usage des médicaments
Poursuivre la montée en charge des actions individuelles sur les médecins
50. Développer, en coordination entre la HAS et l’assurance maladie, les outils permettant aux médecins d’évaluer leur pratique ainsi que les communications personnalisées de l’assurance maladie sous la forme de courriers, de contacts téléphoniques ou d’entretiens confraternels
C. Favoriser le bon usage chez les consommateurs
Développer et coordonner l’information du grand public sur les médicaments
51. Faire de l’Institut national de prévention et d’éducation pour la santé (INPES) le pilote de la diffusion de l’information sur le médicament en direction du grand public
52. Développer la communication de l’INPES sur le bon usage des médicaments et la prise en charge de sa santé
53. Renforcer la communication sur les alternatives au médicament, sur la contrefaçon de médicaments, ainsi que la vente de médicaments par correspondance et par internet
54. Améliorer la coordination des actions de communication en direction du grand public et multiplier les actions ciblées et les contacts directs avec les patients à l’initiative des caisses d’assurance maladie
55. Associer davantage les associations de patients à la définition et l’évaluation des campagnes sur le médicament
56. Enrichir et améliorer l’ergonomie des sites internet d’information institutionnelle sur le médicament
57. Mettre en place des sites d’information institutionnelle spécialisés par pathologies
Inciter à l’observance, encadrer les programmes d’accompagnement des patients et développer l’éducation thérapeutique
Encadrer strictement les programmes d’accompagnement des patients financés par l’industrie pharmaceutique
58. Définir un cadre législatif qui limite très strictement les programmes d’accompagnement ou d’aide des patients associés à un traitement médicamenteux, financés par les entreprises pharmaceutiques : seuls des programmes de courte durée et limités à l’apprentissage d’une technique d’administration particulièrement complexe concernant un médicament présentant un intérêt thérapeutique important (ASMR 1 ou 2) pourraient être autorisés par la HAS à condition de satisfaire à des exigences de compétence, d’indépendance, de qualité, d’objectivité, de non-sélectivité…
Favoriser le développement de programmes d’accompagnement des patients par l’assurance maladie et l’éducation thérapeutique des patients
59. Demander à la HAS de définir, en coordination avec l’AFSSAPS et l’assurance maladie, une stratégie de développement de l’éducation thérapeutique
60. Amplifier les efforts de l’assurance maladie en matière d’aide à l’observance et d’accompagnement des patients, en coordination avec l’AFSSAPS et la HAS et en concertation avec les médecins et les associations de patients
Promouvoir l’information de qualité sur les médicaments sur internet
61. Prévoir, dans le cadre de la certification des sites internet d’information en santé, l’obligation de faire figurer, sur la page d’accueil, des liens internet vers les sites institutionnels d’information en santé, tels que ceux de la HAS, l’AFSSAPS et l’INPES
62. Demander à la HAS d’évaluer la mise en œuvre de la certification des sites d’information en santé
63. Demander à la HAS de mener des études sur la demande et la consommation d’information en santé et les possibilités d’améliorer l’accès du grand public à une information simple et de qualité sur les médicaments
III. S’APPUYER SUR LE RÉSEAU DES PHARMACIES D’OFFICINE POUR PROMOUVOIR LES GÉNÉRIQUES ET DÉVELOPPER UNE AUTOMÉDICATION RESPONSABLE
A. Développer le rôle de conseil des pharmaciens d’officine
Le pharmacien d’officine de proximité doit pouvoir s’appuyer sur le dossier pharmaceutique pour assurer la sécurité de la dispensation
64. Renforcer la vigilance à l’égard des contrefaçons de médicaments, assurer la traçabilité des lots de médicaments afin de faciliter le repérage et le retrait des lots contrefaits, multiplier les actions d’inspection auprès des fabricants et grossistes
65. Renforcer la lutte contre les circuits non contrôlés d’importation, de distribution et de commercialisation
66. Étudier la création d’un site internet qui permette de sécuriser l’offre de dispensation à distance de médicaments par le circuit des pharmaciens d’officine
67. Réaliser le déploiement national du dossier pharmaceutique et publier le décret nécessaire, aussi rapidement que possible, dans l’attente de la mise en œuvre du dossier médical personnel
Développer le rôle de conseil et d’accompagnement des patients des pharmaciens d’officine
68. Valoriser le rôle des pharmaciens d’officine et optimiser le service pharmaceutique en matière de conseil, d’éducation thérapeutique, d’observance, d’aide et d’accompagnement des patients
B. Promouvoir les génériques
Renforcer les moyens de lutter contre les stratégies de contournement des génériques
69. Encourager l’élargissement du répertoire des génériques
70. Prévoir la possibilité d’empêcher la prolongation de la protection des données en cas d’association de médicaments n’apportant pas de réelle innovation thérapeutique
71. Instaurer, pour les laboratoires exploitant des spécialités dont ils détiennent les brevets, une obligation de déclarer au CEPS les titres considérés et leur date d’échéance
Accroître la délivrance de génériques
Renforcer les actions conventionnelles avec les médecins et promouvoir la prescription en dénomination commune
72. Amplifier la mobilisation de tous les acteurs, notamment des parties à la convention médicale, pour la prescription de génériques et renforcer l’action des DAM sur ce thème
73. Développer l’enseignement en DCI, en formation initiale et en formation médicale continue
Poursuivre la mobilisation des pharmaciens sur le droit de substitution et l’effort d’information des assurés
74. Amplifier la mobilisation des pharmaciens pour l’exercice du droit de substitution
75. Développer la communication de l’assurance maladie en direction des assurés sur le thème des médicaments génériques
76. Généraliser à l’ensemble des départements qui n’atteignent pas les objectifs fixés par les accords conventionnels le dispositif du « tiers payant contre génériques »
C. Développer une automédication responsable
Maîtriser la mise devant le comptoir des officines de certains médicaments à prescription médicale facultative
77. Définir l’automédication
78. Respecter le libre choix des pharmaciens pour la mise à disposition devant le comptoir des produits à prescription médicale facultative (PMF)
79. S’assurer que la dispensation des médicaments exposés devant le comptoir soit sécurisée et accompagnée du conseil du pharmacien
80. S’assurer du respect de l’engagement de modération sur les prix des médicaments non remboursables
81. Étudier les conditions de la prise en charge des médicaments concernés par les organismes de protection sociale complémentaire
82. Développer la communication institutionnelle sur l’automédication et veiller à la qualité de l’information délivrée par l’industrie pharmaceutique sur les produits à PMF concernés
83. Évaluer les effets de la mise devant le comptoir des officines de certains médicaments à prescription médicale facultative
IV. FAVORISER LA MISE EN PLACE D’UNE FISCALITÉ PLUS SIMPLE ET STRUCTURANTE
A. Veiller à l’indépendance des organismes qui perçoivent les taxes rémunérant des services rendus
84. Veiller à ce que le financement de l’AFSSAPS et de la HAS par des taxes pour services rendus permette d’assurer l’indépendance des organismes à l’égard de l’industrie pharmaceutique
B. Simplifier, stabiliser ou rendre plus structurante les taxes affectées à l’assurance maladie
Aménager les deux taxes destinées à maîtriser la dépense de médicaments remboursés par l’assurance maladie
Mobiliser la taxe sur les dépenses de promotion pour améliorer la régulation de la visite médicale
85. Publier les textes nécessaires pour assurer la sécurité juridique de la taxe sur la promotion
86. Engager une réflexion avec l’industrie du médicament et le CEPS ayant pour objet d’améliorer la régulation quantitative (objectifs quantifiés) et qualitative (analyse des effets de la certification) de la visite médicale et, le cas échéant, de renforcer la taxe sur la promotion
Simplifier la contribution à la clause de sauvegarde de l’ONDAM pour la rendre effectivement applicable
87. Simplifier le dispositif de clause de sauvegarde de l’ONDAM afin de rendre possible, le cas échéant, son application effective
88. Publier les textes d’application de ladite contribution aux spécialités pharmaceutiques faisant l’objet d’une rétrocession hospitalière
Stabiliser les deux autres taxes destinées à procurer des ressources à l’assurance maladie
89. Maintenir la taxe sur les grossistes répartiteurs
90. Engager une réflexion avec l’industrie pharmaceutique, par exemple dans le cadre du Conseil stratégique des industries de santé, visant à fixer un taux pluriannuel de taxe sur le chiffre d’affaires moins élevé en contrepartie d’un renforcement des remises conventionnelles
Assurer le recouvrement de la TVA sur les médicaments et évaluer les effets des déremboursements sur les recettes de taxes
91. Demander à l’administration fiscale de développer les contrôles du paiement de la TVA sur les médicaments
92. Évaluer l’impact des déremboursements sur les recettes de TVA et des autres taxes sur le médicament
*
La commission des affaires culturelles, familiales et sociales a examiné le rapport d’information, présenté par Mme Catherine Lemorton, rapporteure, en conclusion des travaux de la mission d’évaluation et de contrôle des lois de financement de la sécurité sociale (MECSS), sur la prescription, la consommation et la fiscalité des médicaments, au cours de sa séance du mercredi 30 avril 2008.
Un débat a suivi l’exposé de la rapporteure.
M. Georges Colombier, après avoir remercié Mme la rapporteure, ainsi que les deux coprésidents de la Mission, MM. Pierre Morange et Jean Mallot, qui ont assisté cette dernière dans son travail, a souligné combien le rapport d’information, dont le titre, « Médicaments : prescrire moins, consommer mieux », constitue à lui seul un programme très positif et démontre encore une fois l’utilité de la MECSS.
Il convient maintenant de veiller à l’application des quatre-vingt-douze propositions du rapport.
M. Pierre Morange, coprésident de la MECSS, président, a rappelé que la philosophie de la MECSS n’est pas de se contenter de déclarations pertinentes, mais bien de voir chacune de ses recommandations suivie d’effet, comme de nombreux exemples en attestent.
M. Jean-Pierre Door a estimé que ce rapport d’information, non seulement est excellent, mais répond également à l’ambition ancienne de la MECSS de se pencher sur le sujet, important, du médicament.
Concernant, d’abord, le constat, l’analyse de l’impact des affections de longue durée (ALD) dans la consommation de médicaments constituera une excellente base pour l’élaboration du prochain rapport de la MECSS sur les ALD.
De même, en retraçant toute la chaîne du médicament, qui va du chercheur au patient, le rapport fait parfaitement ressortir les points qui peuvent être améliorés, qu’il s’agisse de la consommation médicamenteuse des particuliers, de la prescription des professionnels de santé, de l’intervention des grossistes répartiteurs, de la qualité de la visite médicale ou encore de l’attractivité du territoire auprès de l’industrie pharmaceutique.
S’agissant, ensuite, des propositions d’action, il convient de souligner, en particulier, celles relatives à la formation des prescripteurs, à l’information et l’accompagnement des patients ou au déploiement des logiciels certifiés d’aide à la prescription auprès des professionnels de santé ainsi qu’au dialogue à développer avec l’industrie pharmaceutique, le tout afin d’éviter une trop grande consommation.
Ce qu’il faut, c’est mieux soigner sans trop consommer.
M. Jean Mallot, coprésident de la MECSS, a tenu également à souligner la qualité du rapport d’information présenté par Mme Catherine Lemorton, que les coprésidents de la MECCS n’ont fait qu’assister dans son travail.
Dans sa première partie, le rapport comporte une somme de données objectives qui seront utiles à tous, tant pour argumenter en la matière que pour imaginer, éventuellement, d’autres propositions. Parmi ces données, il convient notamment de souligner le poids économique extrêmement fort du secteur du médicament dont les ventes en ville ont représenté, en 2006, 20,4 milliards d’euros, soit environ 15 % de l’ONDAM, chiffres qui montrent l’importance de ce secteur d’activité pour la maîtrise médicalisée des dépenses de santé. Les propositions formulées soulignent à cet égard tout l’intérêt pour la santé publique d’une meilleure prescription, donc d’une meilleure consommation et d’un meilleur état de santé des Français.
Concernant les propositions, il a semblé d’abord nécessaire de clarifier les compétences au sein du paysage institutionnel du médicament, qui a beaucoup évolué au cours des quinze ou vingt dernières années. Ce paysage n’est en effet pas toujours très lisible avec autant d’intervenants que l’AFSSAPS, le CEPS, l’INPES, la CNAMTS (Caisse nationale d’assurance maladie des travailleurs salariés), la HAS et d’autres encore, d’autant que chacun dans son rôle a un impact en matière d’autorisation de mise sur le marché, d’appréciation de l’amélioration du service médical rendu et de remboursement par l’assurance maladie. Toutes ces questions concernent la santé publique en général, et il serait souhaitable que les propositions formulées, qui susciteront certainement des réactions, soient suivies d’effet.
Dans cette nébuleuse, la HAS devrait disposer d’un rôle plus important en ce qui concerne les aspects médico-économiques. L’orientation médico-économique de la prescription, soulignée à juste titre par plusieurs personnes auditionnées, doit en effet permettre de redonner tout son rôle à la puissance publique – ce qui explique d’ailleurs la volonté de créer des délégués de santé venant, en quelque sorte, contrebalancer le rôle des visiteurs médicaux –, et la HAS semble la mieux placée pour incarner l’intérêt général en cette matière.
Il est encore trop tôt pour apprécier l’impact des franchises médicales sur la consommation de médicaments, le nécessaire développement de la prévention, le recours aux génériques ou encore la responsabilisation du patient. Il y a lieu d’être, à ce stade, très dubitatif concernant cet impact. Mais les évaluations du nouveau dispositif permettront d’y voir plus clair.
Il convient, par ailleurs, de mettre l’accent, d’une part, sur les propositions formulées en matière de formation des professionnels de santé, en particulier des médecins – c’est en effet une des clés du problème –, et d’autre part, sur la fiscalité, question que le rapport analyse parfaitement sans toutefois avancer de propositions bien précises, étant entendu qu’une réflexion est en cours en la matière.
S’il ne ressort pas des travaux de la MECSS que le poids de la fiscalité spécifique au secteur des médicaments soit excessif ou qu’il nuise à la compétitivité des entreprises françaises sur le marché international ou à l’implantation de laboratoires pharmaceutiques sur le territoire national, les critiques concernant la complexité de cette fiscalité ont conduit à avancer des propositions afin de la simplifier, de la rendre plus structurante et, surtout, de la stabiliser, car les chefs d’entreprise pharmaceutique ont besoin de connaître à l’avance les règles applicables en la matière.
En tout état de cause, le travail considérable qui a été accompli, à la suite notamment des nombreuses et intéressantes auditions, va maintenant se poursuivre par le biais aussi bien des réactions qu’il suscitera que par la mise en œuvre des préconisations présentées au sein des institutions concernées. La MECSS veillera, en effet, à ce que ses recommandations soient suivies d’effet.
M. Pierre Morange, coprésident de la MECSS, président, s’est félicité du travail extrêmement positif ainsi réalisé, qui ne fait que justifier la pertinence de la MECSS, dont la composition paritaire l’exonère de tout esprit polémique et lui donne la capacité à aborder, dans un excellent esprit, les problèmes de façon exhaustive et à formuler des propositions pragmatiques et concrètes. Elle a su mettre en œuvre la logique de l’évaluation et du contrôle, dont il est tant question depuis quelques années.
La MECSS aura à cœur, dans le cadre de la discussion du projet de loi de financement de la sécurité sociale pour 2009, de veiller à la traduction, par voie législative ou réglementaire, de ses préconisations afin que celles-ci soient appliquées effectivement dans le système de soins.
La commission a autorisé, en application de l’article 145 du Règlement, le dépôt du rapport d’information en vue de sa publication.
ANNEXE 1 : COMPOSITION DE LA MISSION
Présidents
M. Jean Mallot
M. Pierre Morange
Membres
Mme Martine Billard
M. Philippe Boënnec
Mme Martine Carrillon-Couvreur
M. Georges Colombier
M. Rémi Delatte
M. Jean-Pierre Door
Mme Jacqueline Fraysse
Mme Catherine Génisson
M. Maxime Gremetz
Mme Danielle Hoffman-Rispal
M. Olivier Jardé
Mme Catherine Lemorton
M. Claude Leteurtre
Mme Geneviève Levy
M. Jean-Luc Préel
Mme Marisol Touraine
ANNEXE 2 : LISTE DES PERSONNES AUDITIONNÉES
Pages
27 septembre 2007
11 heures – Mme Rolande Ruellan, présidente de la sixième chambre de la Cour des Comptes, M. Michel Braunstein, conseiller maître, et Mme Stéphanie Bigas, conseiller référendaire 115
4 octobre 2007
9 h 30 – Mme Anne-Marie Brocas, directrice de la recherche, des études, de l’évaluation et des statistiques (DREES) au ministère du travail, des relations sociales et de la solidarité 131
10 h 30 – M. Jean Marimbert, directeur général de l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS), et M. Michel Pot, secrétaire général 140
11 h 30 – M. le Professeur Laurent Degos, président de la Haute autorité de santé (HAS), M. François Romaneix, directeur, M. le Professeur Gilles Bouvenot, membre du collège de la HAS et président de la commission de la transparence, et M. Étienne Caniard, membre du collège de la HAS et président de la commission qualité et diffusion de l'information médicale 151
18 octobre 2007
9 h 30 – M. Bertrand Fragonard, président du Haut Conseil pour l'avenir de l'assurance maladie, et M. Pierre-Jean Lancry, vice-président 157
10 h 30 – M. Didier Houssin, directeur général de la santé au ministère de la santé, de la jeunesse et des sports, et Mme Danièle Golinelli, adjointe à la sous-directrice politique des pratiques et des produits de santé 165
11 h 30 – M. Frédéric Van Roekeghem, directeur général de l’Union nationale des caisses d’assurance maladie (UNCAM) et de la Caisse nationale d'assurance maladie des travailleurs salariés (CNAMTS) et M. Jean-Pierre Roblet, directeur de l’offre de soins, M. Yves Humez, directeur général de la Mutualité sociale agricole (MSA), et M. Pierre-Jean Lancry, directeur de la santé, M. Dominique Liger, directeur général du Régime social des indépendants (RSI), et M. Philippe Ulmann, directeur de la politique de santé et gestion du risque 174
8 novembre 2007
9 h 30 – M. Dominique Libault, directeur de la sécurité sociale au ministère de la santé, de la jeunesse et des sports, et M. Lionel Joubaud, chef du bureau produits de santé 183
10 h 30 – M. Noël Renaudin, président du Comité économique des produits de santé 194
11 h 30 – M. Jean-Martin Cohen-Solal, directeur général adjoint de la Fédération nationale de la mutualité française, Mme Laure Lechertier, responsable du département politique du médicament, et M. Vincent Figureau, responsable du département relations extérieures 206
22 novembre 2007
9 h 30 – M. le Professeur Robert Nicodème, membre du Conseil national de l'Ordre des médecins, vice-président de la section formation et compétences médicales 216
10 h 30 – M. Pierre Levy, secrétaire général de la Confédération des syndicats médicaux français, M. Jean-Louis Caron, secrétaire général du Syndicat des médecins libéraux, M. Félix Benouaich, président de l’Alliance intersyndicale des médecins indépendants de France accompagné de M. Jean-Gabriel Brun, vice-président, et M. Martial Olivier-Koehret, président de MG France 226
11 h 30 – M. Alain Rouché, directeur santé de la Fédération française des sociétés d'assurances, et M. Gilles Johanet, président du comité maladie-accidents, M. Michel Charton, directeur technique santé individuelle d’AXA France, M. Henri Laurent, directeur général de SwissLife prévoyance et santé, et M. Laurent Doubrovine, directeur assurance de personnes des Assurances générales de France (AGF) 233
6 décembre 2007
9 h 30 – M. Jean Parrot, président du Conseil national de l’ordre des pharmaciens 243
10 h 30 – M. Philippe Gaertner, président de la Fédération des syndicats pharmaceutiques de France, accompagné de M. Jean-Pierre Lamothe, premier vice-président, président de la commission économie de l'officine, M. Claude Japhet, président de l'Union nationale des pharmaciens de France, et Mme Marie-Josée Augé-Caumon, membre du conseil d'administration de l'Union des syndicats de pharmaciens d'officine 256
11 h 30 – M. Christian Lajoux, président du LEEM – Les entreprises du médicament – accompagné de M. Claude Bougé, directeur général adjoint 266
20 décembre 2007
9 h 30 – Mme Marie-Noëlle Banzet, vice-présidente des laboratoires Servier, M. Éric Ducourneau, secrétaire général des laboratoires Pierre Fabre, et M. Christian Lajoux, président directeur général de Sanofi Aventis France, accompagné de M. Philippe Cheng, directeur de la stratégie 278
10 h 30 – Mme Anne Baille, présidente des Laboratoires Ranbaxy pharmacie génériques, Mme Marie-Josèphe Baud, présidente directrice générale de Sandoz, Mme Catherine Bourrienne-Bautista, déléguée générale de GEnériques Même MEdicaments (GEMME), M. Maurice Chagnaud, président directeur général du Laboratoire Teva Classics, et M. Gilles Chaufferin, directeur général délégué adjoint des Laboratoires Boiron 287
11 h 30 – M. Christophe Weber, président de Laboratoires internationaux de recherche (LIR), M. Jean-Christophe Tellier, président de Novartis Pharma, M. Louis Couillard, président directeur général de Pfizer France, et Mme Sabine Dandiguian, présidente directrice générale de Janssen-Cilag France 298
17 janvier 2008
9 h 30 – Mme Annie Podeur, directrice de l'hospitalisation et de l'organisation des soins au ministère de la santé, de la jeunesse et des sports, M. Bernard Ortolan, président du conseil national de la formation médicale continue des médecins libéraux, M. Alain Beaupin, président du conseil national de la formation médicale continue des médecins salariés, et M. Dominique Bertrand, président du conseil national de la formation médicale continue des médecins hospitaliers 312
10 h 30 – Mme Nathalie Tellier, chargée de mission à l’Union nationale des associations familiales et membre du bureau du Collectif interassociatif sur la santé, M. Jacques Mopin, administrateur de l’Union fédérale des consommateurs - Que choisir, accompagné par M. Christophe Le Guehennec, chargé de mission santé, et M. Thierry Saniez, délégué général de la Confédération de la consommation, du logement et du cadre de vie 324
11 h 30 – M. Bertrand Garros, président de l’Institut national de prévention et d'éducation pour la santé, et M. Philippe Lamoureux, directeur général 331
31 janvier 2008
9 h 30 – M. Pierre-Louis Bras, inspecteur général, et M. Aquilino Morelle, inspecteur, membres de l’Inspection générale des affaires sociales (IGAS) 338
10 h 30 – M. Philippe Brunet, directeur du cabinet du commissaire européen en charge de la santé 345
11 h 30 – M. Bruno Toussaint, directeur de la rédaction de la revue Prescrire 354
12 février 2008
9 h 30 – M. Jacques Sauret, directeur du Groupement d’intérêt public du dossier médical personnel (GIP-DMP) 363
10 h 30 – M. Éric Woerth, ministre du budget, des comptes publics et de la fonction publique 373
11 h 30 – Mme Roselyne Bachelot, ministre de la santé, de la jeunesse et des sports 381
ANNEXE 3 : COMPTES RENDUS DES AUDITIONS
Audition de Mme Rolande Ruellan, présidente de la sixième chambre de la Cour des comptes, M. Michel Braunstein, conseiller maître, et Mme Stéphanie Bigas, conseillère référendaire, sur les deux communications de la Cour des comptes à la MECSS concernant la prescription, la consommation et la fiscalité des médicaments.
M. Pierre Morange, coprésident : Je vous remercie, mesdames, monsieur, d’avoir répondu à notre invitation et vous félicite, madame la présidente, pour votre récente nomination à la tête de la sixième chambre de la Cour des comptes.
La Mission d’évaluation et de contrôle des lois de financement de la sécurité sociale a demandé, fin 2005, à la Cour des comptes d’effectuer un travail préalable sur la prescription, la consommation et la fiscalité des médicaments. Celle-ci nous a remis deux communications, l’une sur la fiscalité du médicament au mois de mai dernier et l’autre sur la prescription et la consommation des médicaments au mois de juillet. Je vous donne la parole, madame la présidente, pour la présentation de ces rapports.
Mme Rolande Ruellan : Pour répondre à la demande de la Mission d’évaluation et de contrôle des lois de financement de la sécurité sociale, la Cour des comptes vous a fait parvenir, fin 2005, les extraits de ses rapports annuels sur la sécurité sociale concernant le médicament. Les deux communications de la Cour des comptes, qui vous ont été transmises récemment, permettent d’approfondir et de vérifier les évolutions en ce domaine.
Je présenterai en premier le rapport sur la consommation et la prescription des médicaments, dont l’objet était d’analyser les facteurs pouvant expliquer la surconsommation de médicaments en France.
Trois séries de facteurs explicatifs ont été isolées : le circuit de la mise sur le marché et de l’admission au remboursement, qui est insuffisamment sélectif, les comportements de prescription des médecins, trop faiblement encadrés, et les comportements de consommation des patients, dont l’information est encore insuffisante. Le champ de l’enquête a été limité aux médicaments délivrés en ville, à l’exception de l’automédication, qui se limite en France à 6 % de la consommation. Je signale, par ailleurs, que, dans le rapport annuel 2007 de la Cour des comptes qui est paru ce mois-ci, est analysé l’achat de médicaments à l’hôpital.
La situation française est bien connue. Notre pays est au premier rang en Europe pour le niveau des médicaments prescrits et vendus, sans que cela se justifie par des indicateurs de morbidité ou de mortalité particuliers. La consommation est très concentrée. Elle concerne les affections de longue durée (ALD) et les personnes âgées, 10 % des assurés sociaux consommant 47 % des médicaments remboursés. La classe qui détient le record des médicaments consommés reste les antibiotiques, malgré une baisse due à la campagne, très efficace, conduite par la Caisse nationale d’assurance maladie des travailleurs salariés – la CNAMTS. On constate toujours une surconsommation d’antibiotiques générant des résistances – comme les céphalosporines –, de psychotropes, de statines, d’IPP – inhibiteurs de la pompe à protons – de veinotoniques et de vasodilatateurs.
Le niveau de prescription est élevé puisque 90 % des consultations de généralistes comportent une prescription de médicaments. On note de grandes disparités de comportement entre départements et entre catégories de praticiens pour un même médicament.
Les enjeux de santé publique sont importants avec des problèmes d’affections iatrogènes et l’impact sur les dépenses d’assurance maladie est lourd puisque le coût des médicaments dans les soins de ville augmente très fortement chaque année.
L’une des premières raisons de la situation atypique de la France, de cette autre exception française, est que le circuit de la mise sur le marché et de l’admission au remboursement est insuffisamment sélectif, que ce soit lors de la première inscription des médicaments ou après leur commercialisation. La Cour des comptes avait déjà dénoncé ce manque de sélectivité en 2004.
Les autorisations de mise sur le marché (AMM) sont maintenant essentiellement délivrées par l’Agence européenne du médicament (EMEA). Celles délivrées par l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) concernent surtout des génériques et des me too. La revue Prescrire a noté que plus de 85 % des dossiers examinés n’apportent rien de nouveau. Toutefois la procédure d’AMM n’a pas pour objet de faire des comparaisons : si les trois critères de l’efficacité, de l’innocuité et de la qualité du médicament sont réunis, l’AMM est délivrée.
S’agissant de l’admission au remboursement, la Cour a constaté que la réforme des critères de l’évaluation initiale prévue depuis 2004 n’est toujours pas intervenue. Les quatre niveaux de service médical rendu (SMR) qui déterminent les taux de remboursement – 65 %, 35 % ou 0 – ne permettent pas une grande sélectivité, comme le prouve le fort taux – 86 % – d’attribution de SMR important en 2006.
L’intérêt de santé publique n’est pas suffisamment pris en compte, alors qu’il a été défini par la Haute autorité de santé, la HAS. Il en résulte une confusion entre les notions de SMR insuffisant et d’inefficacité du produit. La Cour recommande donc une nouvelle fois la réforme des critères d’admission selon un schéma qui permette de prendre en compte l’intérêt de santé publique.
L’amélioration du service médical rendu (ASMR) doit être appréciée pour chaque indication, sur la base de comparaisons avec des alternatives thérapeutiques. Elle détermine le niveau de prix. Les ASMR de niveau V doivent diminuer le coût du traitement pour être admis. Une majorité de décisions de la commission de la transparence débouche sur des ASMR V.
La Cour regrette que les essais cliniques entre comparateurs ne soient pas obligatoires et qu’il n’existe pas de liste de médicaments classés par niveau d’ASMR. Elle recommande de remédier à ces insuffisances.
En 2004, la Cour avait déjà noté qu’il n’y a pas d’analyse médico-économique permettant de rapporter l’efficacité des médicaments à leur coût. Des pays voisins, dont la Grande-Bretagne et l’Allemagne, introduisent un critère économique dans leurs décisions de prise en charge des médicaments. La CNAMTS a reçu le pouvoir de fixer le taux de remboursement mais pas la liste des médicaments ni leur prix. Son pouvoir est donc illusoire, alors qu’elle est habilitée à inscrire les actes médicaux dans la classification commune de ces derniers, la CCAM. La Cour considère que le financeur devrait avoir plus de place dans la procédure d’admission au remboursement. La CNAMTS pourrait s’appuyer sur des analyses médico-économiques, que la HAS est prête à faire, pour intervenir en ce domaine.
La Cour s’est à nouveau intéressée au suivi des médicaments en vie réelle pour constater que la réévaluation de la balance bénéfice-risque est très timide. Elle a notamment déploré l’insuffisance de réactivité en pharmacovigilance, en observant que les décisions de suspension ou de retrait interviennent tardivement, alors qu’elles sont plus rapides dans d’autres pays. Cela lui a paru d’autant plus dommage que la loi du 26 février 2007 permet de renouveler l’AMM sans limitation de durée au bout de cinq ans, ce qui impose une plus grande vigilance aux difficultés pouvant surgir pendant l’utilisation des médicaments.
Je déplore que la communication adressée à la MECSS se soit retrouvée dans la presse fin août, alors qu’elle comprenait des exemples que nous avions jugés utiles de vous signaler et qui ne sont pas publiés dans le rapport annuel de la Cour des comptes sur la sécurité sociale.
La réglementation communautaire impose, depuis novembre 2003, d’intégrer des plans de gestion des risques dans le dossier d’AMM pour certains produits. Ces plans peuvent prévoir des études post-AMM, que la Cour avait déjà demandées en 2004.
L’AFSSAPS et le Comité économique des produits de santé (CEPS) demandent la réalisation de telles études mais peu vont jusqu’à leur terme. La Cour s’interroge sur la manière de rendre ces procédures plus efficaces et suggère de prévoir des sanctions financières contre les entreprises qui ne réalisent pas, ou ne réalisent pas dans les délais impartis, les études post-AMM demandées par l’AFSSAPS.
Le suivi des médicaments est également apparu peu réactif. Les opérations de réévaluation entreprises entre 1999 et 2001 n’ont pas été suivies d’effet suffisamment rapidement : il y a eu des baisses de prix et la création d’un ticket modérateur à 15 %, présenté comme une marche d’escalier vers le déremboursement. La Cour constate que les économies attendues en ont été réduites.
De manière générale, et la Cour n’est pas la seule à le dire, la transparence des travaux d’évaluation des médicaments n’est pas suffisamment assurée. La Cour a examiné la question des déclarations d’intérêt des agents et des experts, la publication des rapports d’évaluation d’AMM – les fameux RAPPE –, ainsi que celle des ordres du jour, des comptes rendus et du règlement intérieur des commissions, toutes informations rendues obligatoires par la loi du 26 février 2007 qui a assuré la transposition de la directive communautaire 2004/27/CE. Si la loi date de 2007, la directive remonte à 2004 et les établissements concernés auraient dû se préparer plus vite. On constate encore des délais très longs et des insuffisances dans la publication. La Cour recommande aussi de renforcer la transparence des groupes de travail de l’AFSSAPS et d’améliorer la gestion des conflits d’intérêt.
La forte consommation de médicaments est également due au fait que les prescriptions ne sont que faiblement encadrées.
Deux méthodes de contournement de l’AMM, qui ont leurs justifications, peuvent conduire à des abus : la procédure d’autorisation temporaire d’utilisation (ATU) et les prescriptions hors AMM.
L’ATU nominative ou cohorte est normalement accordée par l’AFSSAPS dans l’intérêt des malades. La Cour n’en conteste pas le bien fondé, dès lors que cela ne sert pas de procédure dilatoire pour éviter de faire des demandes d’AMM. Les risques dus à la liberté des prix des médicaments en ATU ont été pris en compte dans la loi de financement de la sécurité sociale de 2007 mais l’article correspondant est d’une telle complexité que l’on est en droit de se demander s’il sera jamais appliqué.
Les prescriptions hors AMM sont plus problématiques car, quoique normalement interdites, elles sont nombreuses. Il faudrait mieux les contrôler. La prescription hors AMM est autorisée à l’hôpital selon des protocoles thérapeutiques définis par l’AFSSAPS, la HAS et l’Institution national du cancer, l’INca. En outre, la loi de financement de la sécurité sociale pour 2007 a tenté d’encadrer la pratique en ville, qui représente entre 15 et 20 % des prescriptions : elle est permise pour des ALD ou des maladies rares sur la base de recommandations de la HAS. Il faudra veiller à ce que les autorisations dérogatoires soient utilisées uniquement dans l’intérêt médical du patient mais la Cour n’a pas de recul pour porter une appréciation sur la mise en œuvre de ce texte.
La Cour s’est également intéressée à la formation et à l’information des médecins.
Elle n’a pas procédé à une enquête spécifique sur la formation initiale, d’une part, parce que cela n’est pas de la compétence de la sixième chambre de la Cour et, d’autre part, parce que le Sénat a rédigé un rapport sur le sujet. Ce dernier montre que la place accordée au médicament dans les études médicales est extrêmement réduite et que le ministre de la santé joue un rôle trop limité dans la définition des enseignements.
S’agissant de la formation continue, la Cour n’a pas, comme elle le souhaitait, fait d’investigation particulière car l’Inspection générale des affaires sociales (IGAS) venait de réaliser un rapport. Elle a fait quelques études complémentaires centrées sur les questions du médicament. Il en ressort que la formation médicale continue (FMC) qui se met en place au bout de dix ans, avec beaucoup de difficultés, fait l’objet d’une organisation complexe qui ne permet pas d’éviter les conflits d’intérêts. Elle continue d’être essentiellement financée par l’industrie pharmaceutique, avec laquelle les fonds publics ne pourront jamais rivaliser.
Un code de bonnes pratiques signé entre « Les entreprises du médicament » (LEEM), les comités nationaux de FMC et le ministre de la santé en novembre 2006 a admis le financement par l’industrie, encadré par des procédures d’agrément des organismes de formation, mais elles sont dépourvues de tout caractère contraignant. Les cahiers des charges devront être précisés et il faudra peut-être prévoir des sanctions si les prescriptions de ces derniers ne sont pas respectées. Un suivi devra être organisé de cette procédure engagée depuis déjà longtemps.
L’évaluation des pratiques professionnelles (EPP) prévue par la loi de 2004 est une autre approche qui devrait permettre de progresser sur l’encadrement des pratiques de prescription. Son organisation est très complexe. Alors que ce sont souvent les mêmes organismes qui font de la formation et de l’évaluation, ces deux aspects obéissent à des cahiers des charges et à des procédures parallèles.
Au total, ni le ministère en charge de la santé, ni l’assurance maladie, qui pourtant finance, ni la HAS n’ont les moyens de définir des priorités en matière de FMC ou d’EPP, ce qui empêche d’en faire un moyen d’action sur les prescriptions. Sur les cinq priorités définies pour la FMC en 2006, une seule concerne le médicament : la iatrogénie.
S’agissant de l’information des médecins, la Cour est revenue sur les bases de données de médicament pour regretter qu’il n’existe pas encore une base publique d’accès gratuit, exhaustive, objective, regroupant toutes les données administratives et médicales : AMM, dénomination commune internationale (DCI), SMR, ASMR, taux de remboursement, prix. La Cour a analysé les trois bases existantes, dont deux sont privées et une publique. La base publique Thériaque est la plus proche de l’optimum mais son avenir est menacé par la mésentente avec les partenaires du GIE Système d’information sur les produits de santé (SIPS). La Cour reste convaincue de la nécessité d’une base d’accès gratuite et indépendante.
La principale source d’information des médecins reste la visite médicale dont l’Institut de recherche et documentation en économie de la santé (IRDES) a démontré l’influence sur le comportement des prescripteurs. La HAS doit, en vertu de la loi du 13 août 2004 relative à l’assurance maladie, certifier la visite médicale afin d’en garantir la conformité à la charte de la visite médicale (VM) signée en décembre 2004 entre le LEEM et le CEPS. La Cour est restée perplexe devant la définition de l’objectif premier de la VM, qui est « d’assurer la promotion des médicaments auprès du corps médical et de contribuer au développement des entreprises du médicament. ». Le souci d’informer sur la qualité et d’éviter le mésusage arrive en second. Il faudra rester vigilant. Le CEPS doit arrêter les classes thérapeutiques pour lesquelles il estime qu’une réduction de la VM est nécessaire et prévoir des sanctions en cas de non-respect. Il est trop tôt pour apprécier l’effet que cela peut avoir.
L’information publique délivrée aux médecins souffre d’une trop grande dispersion entre la HAS et l’AFSSAPS. Les publications de cette dernière sont riches mais trop complexes et d’un accès parfois difficile. Celles de la CNAMTS sont plus synthétiques et claires : lettres aux médecins et aux pharmaciens, supports mémo. L’assurance maladie a également développé des entretiens confraternels et les visites des délégués à l’assurance maladie, les DAM, qui ont bien démarré et devront s’amplifier.
Les outils d’aide à la prescription doivent être certifiés par la HAS. Le processus est en cours de mise en forme. Le problème est la base de données sur le médicament à laquelle les organismes certificateurs devront adhérer.
L’assurance maladie, de son côté, développe une analyse des prescriptions des médecins mais celle-ci est limitée par la méconnaissance, en dehors des ALD, des pathologies. Elle inclut également des objectifs de maîtrise médicalisée sur des postes prioritaires : antibiotiques, statines, anxiolytiques, génériques, IPP, inhibiteurs de l'enzyme de conversion de l’angiotensine (IEC), Sartans. La Cour montre que le bilan est en demi-teinte, surtout si on retire des économies réalisées ce qui ne relève pas de la modification des comportements mais de la politique de baisse des prix, y compris les progrès des génériques – qui sont souvent le fait des pharmaciens – ou de déremboursement. L’impact financier de la baisse des volumes est toujours nettement inférieur aux objectifs.
Enfin, les accords de bon usage des soins, les fameux AcBUS, sur lesquels était fondé beaucoup d’espoir, ont eu un impact modeste, puisqu’il n’y en a qu’un qui a été appliqué, mais il l’a été avec succès : le test de dépistage rapide de l’angine. L’usage des antiagrégants plaquettaires a été retardé.
La Cour s’est également penchée sur les actions sur les comportements des patients.
L’information grand public sur le médicament est insuffisante, malgré les efforts déployés par la CNAMTS, la Mutualité sociale agricole (MSA), l’AFSSAPS et la HAS, qui ont des sites internet. La CNAMTS a conduit une seule campagne de communication sur les antibiotiques, qui a eu beaucoup de succès. De manière générale, l’information produite est éparpillée et peu lisible.
Là encore, une place importante est laissée à l’information privée, qui dépend de l’industrie. Les programmes d’aide à l’observance ou d’accompagnement des patients, développés pas les laboratoires, devront être strictement encadrés. Ils sont actuellement soumis à la commission de la publicité de l’AFSSAPS, qui en a approuvé huit, qui sont plus des programmes d’éducation du patient. Une annexe d’une recommandation de l’agence européenne – sans portée réglementaire – permet d’intégrer ces programmes dans les plans de gestion des risques. Suite au retrait d’un article du projet qui est devenu la loi du 26 février 2007 habilitant le Gouvernement à encadrer ces programmes par ordonnance, une proposition de loi est en cours d’élaboration. La Cour ne peut qu’insister sur le fait que le besoin d’accompagnement des patients ne doit pas être abandonné à l’industrie.
Concernant les génériques, la Cour a noté leur développement rapide ces dernières années mais n’a pas fait, cette fois-ci, d’étude très approfondie des stratégies de contournement des laboratoires : procédures contentieuses afin de défendre des brevets en justice, tendance à étendre les indications ou à diversifier les présentations pour retarder l’entrée dans le domaine public des médicaments.
La transposition en droit interne d’une directive de 2004, qui a introduit la notion d’AMM globale et une extension de la définition du générique, permettra peut-être de limiter certaines dérives. Cependant la définition de l’AMM globale est actuellement sujette à discussion.
Par ailleurs, le droit actuel ne permet pas de limiter les associations de médicaments, si bien qu’on peut fabriquer un nouveau médicament à partir de deux qui n’avaient pas beaucoup d’intérêt, la combinaison des deux n’en ayant pas davantage.
Les génériqueurs, de leur côté, butent sur la difficulté de connaître la date d’expiration des brevets.
Pour encourager la délivrance des génériques, plusieurs mesures ont été prises : accords conventionnels avec les médecins pour développer la prescription dans le répertoire et en DCI – dénomination commune internationale – ; droit de substitution et accord conventionnel en faveur des pharmaciens.
Auprès des patients, la Caisse primaire d’assurance maladie de Paris a initié un refus de tiers payant en cas de refus de la substitution. La loi de financement de la sécurité sociale pour 2007 a permis d’étendre cette mesure par convention à tous les assurés, y compris à ceux bénéficiant de la couverture maladie universelle (CMU). Elle n’est pas encore généralisée. Début 2007, seuls seize départements l’ont mise en œuvre.
Mme Catherine Lemorton, rapporteure : Je vous remercie, madame Ruellan, pour le panorama complet de la situation que vous avez dressé. En ma qualité de pharmacienne, je puis en mesurer l’exhaustivité.
J’aimerais savoir quelle est la formation, notamment pharmacologique, des délégués de l’assurance maladie ?
Par ailleurs, quelles sanctions peuvent être envisagées à l’encontre des contournements des génériques, d’une part, par les laboratoires qui jouent sur la forme galénique ou la présentation orodispersible ou micronisée pour que leurs médicaments ne soient pas génériqués et, d’autre part, par les médecins qui ne jouent toujours pas le jeu des génériques ?
Mme Rolande Ruellan : La Cour n’a pas fait d’investigation spécifique sur le rôle et la formation des délégués de l’assurance maladie.
Mme Stéphanie Bigas : Aucun bilan de l’action des délégués de l’assurance maladie (DAM) et aucune enquête sur la satisfaction des médecins ne sont encore disponibles à l’assurance maladie. La formation des délégués de l’assurance maladie est un problème qui suscite effectivement des inquiétudes car ce sont souvent des personnes reconverties qui font ce métier. Un premier bilan du ressenti des médecins sur les compétences et l’information dispensée par ces délégués serait donc important, mais il n’a pas encore été réalisé.
Mme Rolande Ruellan : Cela ne fait que deux ans que la CNAMTS a mis en place la visite des DAM. Par ailleurs, le système SESAM-Vitale a entraîné une réduction de ses effectifs et la reconversion des liquidateurs en interlocuteurs des médecins. La Cour a prévu d’aller voir ce que sont devenus ses effectifs, en termes quantitatifs et qualitatifs.
M. Pierre Morange, coprésident : Je précise que la télétransmission des feuilles de soins électroniques atteint désormais un taux de couverture de 70 %, et qu’elle a permis le non-remplacement d’un départ à la retraite sur deux, ce qui s’est traduit par une diminution du nombre de salariés au sein de l’assurance maladie de quelque 2 200 en 2005 et 1 400 en 2006, générant une économie de quelque 160 millions d’euros par an.
M. Michel Braunstein : L’action des délégués de l’assurance maladie vise surtout à alerter les médecins sur leur consommation par rapport à leurs collègues d’une même caisse primaire d’assurance maladie. Un certain nombre de CPAM se sont fortement impliquées dans le recueil de données en ce domaine. Celle de la Sarthe, par exemple, qui est citée dans le rapport de la Cour de 2005, fournit chaque année, depuis deux ans, des informations sur la consommation de chaque médecin en prescriptions médicales, en nombre de visites et en nombre d’indemnités journalières autorisées et situe chaque médecin par rapport à tous ses collègues de la CPAM, en le classant entre le premier et le dixième décile.
La CNAMTS s’emploie actuellement à généraliser ce travail dans toutes les CPAM. Le travail des DAM consiste à informer le médecin sur sa position par rapport à ses collègues.
Aucune étude d’ensemble n’a encore été faite sur l’activité des DAM puisque leur création est toute récente.
Mme Catherine Lemorton, rapporteure : Les DAM, qui interviennent aussi auprès des pharmaciens, ont effectivement une vision comparative entre professionnels de santé. Ils essaient de voir, par exemple, pourquoi telle pharmacie vend 80 % de génériques, alors que telle autre n’en délivre que 40 % pour la même population.
Il faut savoir aussi que, quand les médecins de la direction régionale des affaires sanitaires et sociales (DRASS) et de la direction départementale des affaires sanitaires et sociales (DDASS) interviennent chez leurs collègues qui exercent dans les cliniques ou les hôpitaux, ils sont très souvent mal reçus par ces derniers. Dès que l’intervention des DAM, qui ne sont pas médecins, dépasse le simple comparatif, leurs rapports avec les médecins doivent être difficiles. Cela méritera une évaluation. Cela étant, les visiteurs médicaux, bien que non médecins, sont bien accueillis par les médecins. On peut imaginer que, après formation, les DAM puissent faire un travail d’information objective identique au leur.
M. Michel Braunstein : Il suffit de lire la revue de presse quotidienne de la CNAMTS pour se rendre compte de la sensibilité très forte des médecins à ces visites et à leur contenu.
Mme Rolande Ruellan : Les articles de la presse spécialisée ont fait part du mécontentement des médecins à l’égard des caisses, allant même jusqu’à parler de harcèlement à leur égard ! Les DAM se présentent plus en informateurs qu’en contrôleurs. Mais la frontière peut paraître ténue.
Nous ne critiquons pas les médecins. Nous considérons qu’ils sont mal informés et mal outillés, et nous constatons que, quand les caisses font de gros efforts, cela produit des résultats. L’art et la manière, jointes à de bons supports, ne peuvent qu’être convaincants.
Concernant les stratégies de contournement, la Cour fonde ses espoirs sur la notion d’AMM globale, mais ce concept n’est pas encore clairement défini. Appliquer des sanctions semble difficile. Il semble préférable d’agir en amont, au stade de la délivrance des AMM.
M. Pierre Morange, coprésident : Vous avez souligné à plusieurs reprises, dans votre présentation la complexité des structures chargées, d’une part, de la formation et de l’information – HAS, CNAMTS, AFSSAPS – et, d’autre part, de l’analyse médico-économique – CNAMTS, Commission de la transparence, CEPS –, et parfois leur manque de coordination. Pouvez-vous nous faire part des réflexions de la Cour à ce sujet ?
Mme Rolande Ruellan : Nous avons recommandé que toutes les structures impliquées dans le médicament travaillent ensemble, de façon à mieux se répartir les tâches. En théorie, c’est simple : l’AFSSAPS est compétente pour la mise sur le marché des médicaments, le contrôle de la publicité et la pharmacovigilance ; la HAS, pour décider de l’admission au remboursement. Les deux instances édictent des recommandations de bonne pratique, réalisent des travaux et diffusent des publications.
Nous avons recommandé, sans préciser qui doit faire exactement quoi, qu’elles se coordonnent un peu plus et qu’elles aient des sites un peu plus accessibles. Comme elles interviennent toutes les deux, avec le CEPS et la CNAMTS, sur le médicament, il faut qu’elles se répartissent les travaux et les modalités d’information en direction à la fois des médecins, du public et de tous les intervenants du système de santé.
Des clarifications sont peut-être à apporter dans les textes mais, à partir du moment où le législateur a voulu séparer les métiers en relation avec le médicament, chaque structure a sa légitimité. Seul un travail en commun, sous l’égide de l’administration, peut permettre de remédier au fait qu’elles ont tendance à empiéter les unes sur les autres. Nous souhaitons que le ministère de la santé se préoccupe du sujet.
Mme Catherine Lemorton, rapporteure : La Cour a-t-elle réalisé une étude sur l’éducation sanitaire, notamment à l’école ? J’ai découvert très récemment que, dans un livre de lecture du cours préparatoire, on trouvait une publicité pour un sirop commercialisé sous AMM, qui avait échappé aux enseignants. Cela est de nature à impulser des comportements d’hyper-consommation dès le plus jeune âge.
La Cour a-t-elle étudié, par ailleurs, l’impact des publicités des médicaments sous AMM déremboursés, qui, de la même manière, font rentrer le médicament dans la vie quotidienne des personnes ?
La libre publicité des médicaments est sur le point d’être accordée au niveau européen. La France y est opposée mais le droit communautaire prend quelquefois le pas sur le droit français.
Mme Rolande Ruellan : La Cour n’a pas fait d’étude en direction de l’Éducation nationale. Les questions soulevées pourraient peut-être faire l’objet d’un approfondissement de la part de la MECSS.
Mme Stéphanie Bigas : Nous n’avons pas dissocié le cas des enfants du reste de la population. En revanche, nous avons fait une étude concernant la certification des sites d’information sur la santé. La HAS a reçu la mission de s’assurer que l’information mise à disposition du public, et qui est pléthorique faute d’information publique sur le sujet, présente un minimum de garanties. La Cour montre dans le rapport que la HAS a des difficultés pour mette en place cette procédure de certification et ne sait pas très bien par quel bout s’y prendre face à la multiplicité des sites et au renouvellement permanent de l’information. Dernièrement, elle songeait à déléguer cette fonction mais sans trop savoir comment.
M. Jean Mallot, coprésident : Dans le rapport, la Cour dénonce le fait que les décideurs ne sont pas les payeurs et que les payeurs ne sont pas les utilisateurs. Une meilleure coordination entre les structures se révèle souhaitable. L’État ne devrait-il pas avoir un rôle plus directif afin que chacun joue pleinement son rôle et que l’intérêt général soit mieux assuré ?
Mme Rolande Ruellan : Il est ressorti de plusieurs enquêtes que le ministère chargé de la santé, notamment la direction générale de la santé (DGS), ne remplissait pas suffisamment son rôle de pilote et de coordinateur des agences. Le ministère est un peu débordé maintenant par ces puissants féodaux, d’autant plus qu’il les finance peu. Quand les agences ne sont pas financées par l’industrie du médicament pour services rendus, elles le sont par l’assurance maladie. S’étant retiré du financement, l’État s’est peut-être un peu trop retiré en même temps de l’impulsion. Les audits sur la DGS et la direction de l’hospitalisation et de l’organisation des soins (DHOS) auxquels a procédé la Cour le montrent.
Nous avons insisté sur l’importance de disposer d’une base de données publique d’accès gratuit. L’État ne doit pas se désintéresser de cette réalisation. Or la base Thériaque est un véritable panier de crabes. Suite à une mésentente, la CNAMTS a décidé de réduire son financement et le partenaire ne veut plus, dès lors, rester dans l’affaire. De plus le nom « Thériaque » est réservé.
Mme Stéphanie Bigas : Nous n’avons pas eu les dernières évolutions – peut-être les choses se sont-elles débloquées ! – mais le principal problème est de savoir qui va conserver le travail réalisé. Si le CNIHM – le Centre national hospitalier d’information sur le médicament – se désengage du GIE-SIPS, on ne sait pas qui va conserver les droits de propriété sur la base et les applications nécessaires pour l’utilisation et l’exploitation de la base.
M. Pierre Morange, coprésident : Nous allons vous demander de présenter le second rapport, madame Ruellan, sur la fiscalité du médicament.
Mme Rolande Ruellan : On dénombre onze taxes d’importance très inégale, qui ont rapporté un milliard d’euros en 2006, représentant 4 % du chiffre d’affaires des industries.
Elles ont deux grandes finalités : d’une part, la rémunération d’un service rendu et, d’autre part, l’apport de ressources à l’assurance maladie.
La taxe perçue par la HAS, pour l’inscription d’un médicament sur la liste des spécialités pharmaceutiques remboursables ou la liste des médicaments agréés à l’usage des collectivités publiques, couvre 90 % du coût de fonctionnement de la commission de la transparence.
Certaines personnes sont parfois choquées que le contrôleur soit payé par le contrôlé. C’est une pratique assez courante. Toutes les hautes autorités de contrôle indépendantes qui œuvrent dans le domaine financier sont financées par les contrôlés. D’ailleurs, en 1945, le contrôle général de la sécurité sociale était censé être financé par les caisses. Cela ne s’est jamais fait parce que les partenaires sociaux et l’État n’avaient pas des relations suffisamment confraternelles pour assumer ce financement.
L’AFSSAPS perçoit cinq taxes qui concernent, d’une part, les demandes d’AMM, que ce soit un premier examen, un renouvellement ou une modification, et, d’autre part, le contrôle de la publicité, la pharmacovigilance. Elles représentent 55 % du budget de l’AFSSAPS.
Le nombre de dossiers d’AMM soumis à l’AFSSAPS baisse du fait de la montée en charge du dispositif d’AMM européenne. Les médicaments passant par l’agence française sont surtout des génériques et des me too. Les nouvelles molécules, les nouveaux principes actifs et les nouvelles indications, eux, sont traités au niveau européen. L’AFSSAPS participe à l’instruction des dossiers de l’agence européenne et est rémunérée à cette fin mais c’est en dehors de cette question de taxe.
Il n’y a pas de problème particulier de recouvrement des taxes de la HAS et de l’AFSSAPS. La Cour a simplement noté qu’il n’y avait eu aucun contrôle effectué sur l’assiette. Les deux agences ne sont pas des percepteurs. C’est pourquoi la Cour a de nouveau préconisé que la collecte de la taxe annuelle sur le chiffre d’affaires soit recouvrée par la direction générale des impôts (DGI) avec la TVA. Mais sa recommandation n’a pas eu beaucoup de succès chez les intéressées.
Les taxes affectées à l’assurance maladie sont plus importantes.
Elles ont deux finalités essentielles : maîtriser la dépense de médicament, et procurer des recettes à l’assurance maladie.
La première taxe destinée à maîtriser la dépense de médicaments est celle sur les dépenses de promotion des médicaments. Elle pose un énorme problème d’assiette suscitant des contestations contentieuses. Son champ n’est pas bien précisé. Une circulaire à son sujet tarde à être publiée.
En fait, c’est son principe même qui est contesté. Elle est ancienne, puisqu’elle a été instaurée en 1983 et est toujours aussi mal supportée par les laboratoires.
La Cour a suggéré que l’on en évalue l’impact sur les dépenses de promotion des laboratoires – a-t-elle un caractère dissuasif et limitatif ? – et d’examiner sa cohérence et sa complémentarité avec des dispositifs plus nouveaux : la charte de la visite médicale et la certification par des organismes accrédités de la conformité de la visite médicale à ladite charte. Néanmoins cette dernière est en cours de mise en œuvre et ne permet donc pas de faire cet examen.
La seconde taxe destinée à maîtriser la dépense de médicaments est la contribution de la clause de sauvegarde de l’ONDAM, appelée parfois, en raccourci, clause de sauvegarde, créée par la loi de financement de la sécurité sociale pour 1999. Elle est calculée d’une façon extrêmement complexe. Elle s’applique quand l’évolution du chiffre d’affaires hors taxe en France en spécialités remboursables – sauf médicaments orphelins – est supérieure au fameux taux K fixé en loi de financement de la sécurité sociale, et qui est, depuis plusieurs années, à 1 %, et ne concerne que les entreprises qui n’ont pas passé convention avec le CEPS, lesquelles sont très minoritaires. La taxe est ensuite répartie entre les laboratoires en fonction de trois éléments : le chiffre d’affaires, la progression de celui-ci et les dépenses promotionnelles.
La complexité de la méthode de calcul est accrue du fait que celle-ci doit tenir compte du montant versé au titre de la taxe sur les dépenses de promotion, lequel est connu avec retard. Le périmètre des débiteurs est très difficile à établir, du fait de la mobilité du tissu industriel. Les recouvreurs, qui sont les URSSAF de Paris et de Lyon, sont obligés de faire des enquêtes presque policières pour connaître le fichier éventuel des débiteurs, et sont donc en relation avec le CEPS.
En outre, cette taxation déclenchée sur la base d’une évolution du chiffre d’affaires global du secteur peut avoir pour résultat qu’une entreprise non conventionnée dont le chiffre d’affaires progresse plus que le taux K échappe à toute taxation si le chiffre d’affaires des produits remboursables n’a pas excédé ce taux, et inversement.
Depuis plusieurs années, cette taxe n’a aucun rendement. Son seul objet est finalement d’inciter au conventionnement avec le CEPS.
Depuis la loi de financement de la sécurité sociale pour 2006, elle est applicable aux spécialités rétrocédées. Mais les textes d’application n’ont pas été publiés.
La première taxe destinée à procurer des recettes à l’assurance maladie est celle sur les grossistes répartiteurs, due par les entreprises de vente en gros et les entreprises assurant l’exploitation d’une ou plusieurs spécialités quand elles vendent en gros. Elle est assise sur le chiffre d’affaires hors taxe réalisé en France sur les spécialités remboursables hors médicaments orphelins.
La loi de financement de la sécurité sociale pour 2007 a créé une contribution exceptionnelle de régulation assise sur le chiffre d’affaires hors taxe pour l’année civile 2006 réalisé en France auprès des pharmacies d’officine, mutualistes et due par les mêmes. Son rendement initialement de 50 millions d’euros a été ramené à 37 millions du fait de la baisse du taux de 0,28 à 0,21 % au cours de la discussion parlementaire.
La seconde taxe destinée à procurer des recettes à l’assurance maladie est celle sur le chiffre d’affaires, créée par la loi de financement de la sécurité sociale pour 2004 et pérennisée par la loi du 13 août 2004. Elle est appliquée au chiffre d’affaires hors taxe de toutes les entreprises qui exploitent des médicaments ayant une AMM, en dehors des génériques – à l’exception de ceux qui sont remboursés sur la base d’un tarif forfaitaire de responsabilité (TFR) – et des médicaments orphelins. Son taux est passé de 0,6 % à 1,76 % en 2006 et est revenu à 1 % en 2007.
La Cour a observé, sans trouver d’explication, qu’on avait, dans la même loi, la baisse du taux de la taxe sur le chiffre d’affaires et la création d’une contribution exceptionnelle applicable aux grossistes répartiteurs.
Enfin, la TVA brute collectée par les commerçants en gros en produits pharmaceutiques fait partie du panier fiscal affecté par la loi de finances pour 2006 à la compensation des exonérations de cotisations générales sur les bas salaires. La plus grosse part va à l’assurance maladie.
Depuis la loi de financement de la sécurité sociale pour 2005, ces quatre taxes – grossistes répartiteurs, clause de sauvegarde, taxe sur le chiffre d’affaires et taxe sur les dépenses de promotion – sont recouvrées, non plus par l’ACOSS, qui n’était pas très outillée pour procéder aux recouvrements, mais par les deux URSSAF de Paris et de Lyon.
Le coût de recouvrement est estimé à 325 000 euros en 2005. En 2006, le plan de contrôle des deux URSSAF a rapporté 126 000 euros pour trente et une entreprises contrôlées.
Les entreprises contestent ces contrôles et jusqu’à la compétence des inspecteurs du recouvrement. La loi de financement de la sécurité sociale pour 2004 a dû valider les opérations de contrôle appuyées sur le motif de l’irrégularité de l’agrément des inspecteurs. Cependant des actions sont pendantes devant la cour administrative d’appel de Paris.
Le rendement des taxes affectées à l’assurance maladie est de 961 millions d’euros en 2006 – à comparer au milliard rapporté par l’ensemble des taxes. Ce rendement est très sensible à la hausse des taux puisque la majoration de la taxe sur le chiffre d’affaires de 0,6 % à 1,76 % en 2006 a provoqué une augmentation de 40 % du produit global des taxes.
La fiscalité du médicament souffre d’une grande complexité due à des règles qui varient d’une taxe à l’autre, alors qu’elles visent les mêmes entreprises, à la mobilité du secteur – fusions, cessions, disparitions, remariages –, et à la tendance procédurale des débiteurs.
La conclusion de la Cour est modeste car elle n’a voulu ni faire perdre des ressources à l’assurance maladie, ni empiéter sur un domaine qui est éminemment de la compétence du Parlement. Elle estime cependant indispensable de revoir la pertinence de ces taxes, d’en réduire le nombre, d’en stabiliser et d’en simplifier le mode de calcul. Tout ce qui est compliqué est mal appliqué, est source de contentieux et de conflits et aboutit finalement à des pertes d’argent.
Mme Catherine Lemorton, rapporteure : Je veux revenir sur les ressources de l’AFSSAPS, qui accorde les AMM.
Selon la revue Prescrire, entre 80 et 85 % des nouveaux produits mis sur le marché avec AMM n’ont pas un service médical rendu supérieur à ceux qui existent déjà, et ont même parfois un SMR insuffisant. Ne pensez-vous pas qu’il y ait un rapport entre ces pourcentages et le fait que l’AFSSAPS n’ait pas de financement indépendant des laboratoires ?
Mme Rolande Ruellan : Franchement non. À partir du moment où ce sont des taxes qui sont prévues par la loi, il n’y a, dans la tête des experts et des agents de l’Agence, aucun rapport entre leurs décisions et le financement.
Mme Catherine Lemorton, rapporteure : Je regrette que, quand un médicament sous DCI fait l’objet, après un suivi post-AMM, d’un retrait du marché dans d’autres pays, la France tarde à en faire autant.
Mme Rolande Ruellan : Je vous renvoie aux réponses que nous avons publiées à ce sujet dans le rapport de la Cour sur la sécurité sociale qui est paru il y a quelques semaines. L’AFSSAPS n’a pas été satisfaite de notre travail. Quand nous lui avons reproché de tarder à prendre des décisions de suspension et de retrait de médicament, cela a été peu apprécié.
M. Pierre Morange, coprésident : Pouvez-vous, madame Ruellan, nous fournir des précisions sur les effets des déremboursements ? Il semble que vous ayez eu quelques difficultés à en mesurer tout l’impact.
Pouvez-vous également affiner le chiffrage de l’économie potentielle attendue de l’arrivée sur le marché de nouveaux médicaments génériques ?
Mme Rolande Ruellan : Les effets financiers des économies réalisées du fait des déremboursements sont difficiles à séparer de l’impact de la maîtrise médicalisée. L’administration, comme la CNAMTS, ne les distinguent pas toujours clairement dans leur bilan. De plus, certains plans médicament, prévus pour s’étaler sur plusieurs années, combinent plusieurs mesures qui interagissent les unes avec les autres.
M. Michel Braunstein : Les chiffres indiqués dans le rapport proviennent de la direction de la sécurité sociale (DSS). La Cour n’a pas procédé à des vérifications particulières.
Mme Stéphanie Bigas : Les chiffres de la DSS cités dans le rapport distinguent l’effet prix, l’effet structure et l’effet volume.
Mme Rolande Ruellan : Quant aux nouveaux génériques, la Cour n’a pas fait de prévisions sur les économies futures liées à la croissance de ce marché. C’est une idée d’étude intéressante, compte tenu du nombre de brevets qui vont tomber dans le domaine public.
Il sera d’autant plus important d’éviter les contournements. L’AMM obéit à des critères très objectifs et ne permet pas actuellement de faire des comparaisons et des sélections. Il faudra donc être très vigilant au niveau de l’admission au remboursement. Cela nécessitera de la part de la commission de la transparence et ensuite du ministre, puisque c’est lui qui décide de l’inscription sur la liste, une politique sélective en la matière.
M. Jean Mallot, coprésident : Existe-t-il un mécanisme qui permet d’avoir la conviction que la charge sur l’entreprise de la taxe sur les dépenses de promotion des médicaments n’est pas répercutée in fine sur le consommateur lors de la fixation des prix ?
Mme Rolande Ruellan : Les prix des médicaments remboursés sont réglementés. Ils sont d’ailleurs de plus en plus souvent fixés en référence aux niveaux de prix européen, voire international. Par contre, les prix des médicaments non remboursés étant libres, les laboratoires font ce qu’ils veulent.
M. Pierre Morange, coprésident : Nous vous remercions, mesdames, monsieur.
*
Audition de Mme Anne-Marie Brocas, directrice de la recherche, des études, de l'évaluation et des statistiques (DREES) au ministère du travail, des relations sociales et de la solidarité.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale. Avant de donner la parole à notre rapporteure, je vous propose de présenter brièvement les missions qui sont les vôtres dans le domaine du médicament qui fait actuellement l’objet des travaux de notre mission.
Mme Anne-Marie Brocas : En ce qui concerne le médicament, la DREES conduit deux types d’approche.
La première est une approche comptable, à travers l’analyse des comptes de la santé, qui sont des satellites des comptes de la nation et qui permettent de décrire l’ensemble des dépenses du secteur de la santé. Nous développons ainsi des analyses sur l’évolution de chacun des secteurs, en particulier sur le poste des médicaments pour lequel nous analysons les évolutions des prix et des volumes et nous recherchons quels sont les financeurs. Il y a donc d’un côté l’analyse des dépenses et de l’autre celle des recettes. C’est là que l’on trouve les données qui font l’objet de nombreux commentaires, c’est-à-dire celles qui ont trait à la part prise en charge par la sécurité sociale et par les organismes complémentaires et à celle qui reste à la charge des malades.
Cette année, nous avons mené des travaux sur la rétropolation afin d’établir de manière cohérente des séries chiffrées concernant les 55 dernières années. L’intérêt est de tracer une perspective historique afin de voir comment, au sein des dépenses de santé, la structure entre les différents postes a été déformée au fil du temps.
Cependant nous avons aussi une seconde approche, à partir des données issues des remontées statistiques des administrations ou des enquêtes, en particulier des informations émanant des banques de données propres au secteur du médicament.
À partir de là, la DREES analyse chaque année l’évolution du marché des médicaments remboursables. Depuis deux ans, à la suite d’une préconisation du Conseil national de l’information statistique (CNIS), elle s’efforce également d’analyser les chiffres relatifs aux types de médicaments qui font l’objet d’une rétrocession de la part des hôpitaux. Ce travail est encore récent mais on peut espérer que l’on disposera à l’avenir d’analyses plus détaillées.
Toujours à partir des données statistiques de l’industrie du médicament, des comparaisons sont effectuées entre le marché français et les marchés étrangers afin de mieux comprendre les ressorts propres à l’évolution de ce secteur en France.
À ces études générales s’ajoutent des analyses plus précises sur un produit ou sur une classe thérapeutique, comme l’étude conduite récemment sur les statines.
On le voit, il s’agit d’un travail d’études et d’analyse, mais il n’appartient pas pour autant à la direction que je dirige d’entrer dans le champ des préconisations en matière de politiques publiques.
Mme Catherine Lemorton, rapporteure : Je vous remercie pour cette présentation synthétique du périmètre de votre action.
Quelles sont les relations de la DREES avec l’Agence française de sécurité sanitaire des produits de santé, l’AFSSAPS, et avec la Haute Autorité de santé, la HAS ? Des données sont elles échangées avec ces organismes ? Travaillez-vous de manière interactive ? Votre direction leur apporte-t-elle des informations pour qu’elles les traitent ? À l’inverse, alimentent-elles vos travaux ?
Mme Anne-Marie Brocas : L’AFSSAPS dispose d’une base de données administrative puisque c’est elle qui est à l’origine des autorisations de mise sur le marché (AMM), qui donne son avis au titre de la commission de la transparence et qui recueille les déclarations administratives obligatoires des laboratoires. Jusqu’à présent, il était difficile de mobiliser ces informations, le CNIS l’avait d’ailleurs noté dans un rapport de 2005. La DREES a désormais passé une convention avec l’Agence afin de disposer de cette base de données, qu’elle utilisera à l’avenir.
Pour l’heure, les bases que la DREES peut le plus facilement mobiliser sont celles qui sont produites par l’industrie du médicament sur l’activité des laboratoires pharmaceutiques : le GERS – le groupement pour l’élaboration et la réalisation de statistiques – et l’IMS - Intercontinental Marketing Services. Ces données ne transitent pas par l’AFSSAPS. Les organismes qui les collectent sont totalement indépendants. La DREES paye pour pouvoir les utiliser.
Pour sa part, la HAS n’est pas une source de données, même si chacun constate bien évidemment les effets de ses actions, en particulier de la production des références de bonnes pratiques médicales, lorsque sont conduites des études sur certains secteurs ou sur le remboursement.
En fait, pour caractériser les relations de la DREES avec ces organismes on peut dire que les études que réalise la DREES, qui montrent des inflexions pour certaines classes thérapeutiques spécifiques, permettent à la Haute Autorité de santé, mais aussi à d’autres acteurs comme les caisses d’assurance maladie ou la direction du ministère de la santé chargée du médicament, de mesurer l’impact de telle ou telle action.
M. Pierre Morange, coprésident : La DREES rencontre-t-elle des difficultés particulières pour collecter les informations nécessaires à l’exercice de ses missions ? Quelles mesures concrètes, de nature législative ou réglementaire, conviendrait-il d’adopter afin qu’elle dispose de l’ensemble des données ?
Mme Anne-Marie Brocas : La démarche de collecte des données n’est pas entravée par des obstacles juridiques. Cela ne signifie pas que des progrès sont impossibles et la DREES s’efforce d’en réaliser, notamment en ce qui concerne le secteur des médicaments à l’hôpital, pour lequel elle manque d’informations, les données qu’elle collecte par les bases de données privées étant très focalisées sur la médecine ambulatoire. Si, en ville, grâce aux systèmes des prix administrés, elle dispose d’informations sur les prix et sur les volumes, à l’hôpital en revanche les prix peuvent varier de façon importante puisque les établissements hospitaliers sont libres de négocier l’achat des médicaments avec les laboratoires pharmaceutiques. Dans ce dernier cas, les remontées d’informations sont donc plus compliquées.
Il est vrai que, depuis deux ans, des données sur les volumes et sur les prix sont collectées dans les remontées sur les dépenses hospitalières mais nous n’avons pu présenter pour l’instant de publications que sur des données globales car nous ne disposons pas d’un recul suffisant pour apprécier le degré de fiabilité de ces informations. Il serait pourtant extrêmement utile de pouvoir mener des comparaisons sur les prix et sur les volumes entre les établissements hospitaliers, mais aussi entre la pratique hospitalière et la pratique de ville.
M. Pierre Morange, coprésident : Ces remontées récentes d’informations sont-elles le fruit d’une collecte de données macroéconomiques et macrosanitaires ou s’agit-il de l’addition des informations provenant de chaque structure hospitalière ?
Mme Anne-Marie Brocas : C’est bien d’une addition d’informations qu’il s’agit, qui permettront, à l’avenir, de se livrer à des analyses assez fines. Cependant, au préalable, une itération est nécessaire pour s’assurer de la fiabilité des données.
M. Pierre Morange, coprésident : La DREES dispose-t-elle de remontées d’informations quant aux effets de la politique des achats groupés ?
Mme Anne-Marie Brocas : Non. C’est l'un des sujets qui doivent être étudiés.
Ces données doivent aussi permettre d’analyser la rétrocession hospitalière, qui consiste à ce que des médicaments soient revendus aux malades par les hôpitaux qui sont eux-mêmes remboursés par la sécurité sociale. Dans les données qui émanent des hôpitaux, a été individualisée la part de cette rétrocession et la DREES dispose d’informations sur les types de médicaments qui en font l’objet, sur les volumes et sur les dépenses. Une analyse historique montre que le point culminant de cette rétrocession a été atteint en 2004, avec un montant global d’environ 2,5 milliards d’euros. Depuis lors, diverses mesures ont été prises et l’on observe une décrue.
Le premier exercice en la matière a porté uniquement sur les centres hospitaliers universitaires (CHU) et sur les centres de lutte contre le cancer. Sans surprise, il a été constaté que les antirétroviraux faisaient le plus fréquemment l’objet une rétrocession dans les CHU et les traitements contre le cancer dans les centres de lutte contre cette maladie.
Cette année, la DREES a pu étudier les informations en provenance de l’ensemble des centres hospitaliers et des cliniques privées. Pour l’instant, elle dispose de données sur les masses globales de dépenses et a réalisé une analyse sur les types de médicaments qui constituent les postes de dépenses les plus importants à l’hôpital, dans les centres de lutte contre le cancer et dans la rétrocession des hôpitaux.
Mme Catherine Lemorton, rapporteure : La DREES a-t-elle également étudié l’impact, en termes de volumes et de prix, de la sortie de la réserve hospitalière ? Il faut rappeler, en effet, que la rétrocession concerne souvent des médicaments pour les traitements lourds comme la trithérapie ou l’interféron, qui sont initialement prescrits à l’hôpital mais que le malade peut ensuite se procurer en ville.
Mme Anne-Marie Brocas : On mesure l’impact de ces traitements dans les études annuelles sur le marché du médicament. Ce sont en effet souvent des molécules très coûteuses qui font l’objet de cette sortie de la réserve hospitalière et les effets sur la dépense en ville sont donc forts.
M. Pierre Morange, coprésident : Les médicaments sortis de la réserve hospitalière représenteraient 20 % de la dépense des médicaments délivrés en ville. Est-il possible de confirmer ce chiffre ? Ces dépenses en ville, mais dont l’origine se trouve dans une prescription hospitalière, ont un impact sur l’évolution de l’objectif national des dépenses d’assurance maladie (ONDAM) ambulatoire. Peut-on le mesurer ?
Mme Anne-Marie Brocas : La DREES n’a pas réalisé de chiffrage mais elle regarde comment répondre précisément à cette question. Pour l’instant, elle se contente d’observer l’impact sur le marché global mais sans ventilation par produit.
Elle analyse la contribution à l’augmentation globale des dépenses des dix classes thérapeutiques qui y ont joué le rôle le plus important. Ce ne sont pas toujours les mêmes et l’on voit bien apparaître, en fonction des années, certaines molécules qui font l’objet d’une plus large diffusion. Ainsi, en 2005, nous avons constaté un fort impact de l'érythropoïétine, l’EPO, à hauteur de 1 % sur une croissance totale de 6,7 %. C’est très important !
Dans la mesure où l’on souhaite pouvoir distinguer ce qui incombe à la ville de ce qui relève de l’hôpital, la DREES doit vérifier si les outils dont elle dispose lui permettent de réaliser cette analyse.
Mme Catherine Lemorton, rapporteure : Au-delà du confort que représente pour le malade le fait de pouvoir se procurer à la pharmacie de son quartier le médicament qui est sorti de la réserve hospitalière, il est important de vérifier si le fait de lui permettre d’aller voir son généraliste une fois qu’il est muni de la prescription hospitalière initiale est en soi un gage d’économie pour l’assurance maladie, s’agissant de traitements qui coûtent souvent plus de 1 500 € par mois.
L’assurance maladie affirme que les médicaments coûtent de plus en plus cher en médecine de ville, mais il ne faut pas oublier que les sorties de la réserve hospitalière alourdissent le panier de soins en ville.
Mme Anne-Marie Brocas : Pour le vérifier, il faudrait analyser l’évolution de quelques molécules significatives au cours des dernières années.
On peut d’ailleurs rapprocher cette question de celle, plus générale, de la dynamique du marché du médicament : si l’on se demande ce qui tire ce marché vers le haut, on constate, année après année, que la dépense est tirée par les molécules les plus récentes. En moyenne, plus de la moitié de la croissance des dépenses est imputable aux molécules de moins de cinq ans. Et il y a bien là un lien avec la sortie de la réserve hospitalière, qui vise souvent des molécules récentes et coûteuses.
M. Pierre Morange, coprésident : Vous avez rappelé, à juste titre, que chaque site hospitalier a sa propre stratégie d’élaboration des prix ; mais que se passe-t-il lors de la sortie de la réserve hospitalière ? Il paraîtrait logique que l’effet volume entraîne une diminution du prix de vente en ville. Avez-vous des informations à ce propos ?
Mme Anne-Marie Brocas : La DREES ne dispose pas d’éléments de comparaison des prix, mais c’est un objectif qu’elle poursuit, car il paraît essentiel de disposer de telles données pour se comporter en acheteur avisé. À partir des données dont elle dispose, elle s’efforcera de fournir un éclairage pour une ou deux molécules.
Chaque année, un faible nombre de classes thérapeutiques explique les mouvements du marché, mais ces derniers sont en partie contrebalancés par la montée en charge des génériques. Celle-ci demeure sans doute insuffisante, mais elle évite une trop forte polarisation du marché : si les nouvelles molécules expliquent 50 % de la croissance aujourd’hui, elles en représentaient 80 % au début des années 2000. On observe, outre les effets du volume de vente et du prix des génériques, que l’existence de ces derniers conduit souvent les laboratoires à aligner le prix du princeps.
M. Pierre Morange, coprésident : Lors de l’audition de la semaine passée, il a été demandé aux représentants de la Cour des comptes s’ils disposaient d’informations sur les économies globales réalisées grâce aux génériques. Depuis 2002, de nombreuses molécules à fort taux de rentabilité – les blockbusters – sont tombées dans le domaine public et ce mouvement va se poursuivre jusqu’en 2010. Les économies potentielles réalisables sur les médicaments récemment tombés dans le domaine public ont été estimées à plus de trois milliards d’euros. Pouvez vous confirmer ce chiffre ? Avez-vous fait des prévisions d’économies pour l’assurance maladie pour les deux ou trois années à venir ?
Mme Anne-Marie Brocas : Aucune analyse prospective n’a été menée sur ce point.
En revanche, des analyses rétrospectives de la DREES sur la contribution des génériques à l’évolution des dépenses montrent que leur effet est très marqué.
Elle s’est également intéressée à la place des médicaments génériques dans les pays étrangers et a constaté que la France se caractérise par l’étroitesse de son répertoire de médicaments génériques et par une part des génériques dans le volume global des ventes jusqu’à cinq fois inférieures à ce que l’on constate chez nos voisins européens.
Mme Catherine Lemorton, rapporteure : La DREES a-t-elle comparé l’importance du phénomène de contournement des génériques dans les différents pays ? C’est sans doute un des éléments qui expliquent que le taux de pénétration ne progresse guère en France. On sait en effet que certains laboratoires, dès lors que leur princeps tombe dans le domaine public, en sortent une autre forme galénique, par exemple en remplaçant un effervescent par un orodispersible, sans que le service médical rendu ne soit amélioré. Ce phénomène a-t-il été évalué ? Alors que les pharmaciens ont beaucoup fait pour le développement des génériques, ces contournements font un peu mal au cœur…
Mme Anne-Marie Brocas : La DREES ne dispose pas des outils nécessaires et ne compte pas assez de médecins dans ses équipes pour mener des études aussi pointues, qui relèvent sans doute davantage de la HAS ou de l’AFSSAPS. Toutefois il n’est pas certain que les choses se présentent de façon différente chez nos voisins car les stratégies des laboratoires y sont sans doute identiques.
M. Pierre Morange, coprésident : Avez-vous mené des études sur les conséquences économiques et sanitaires des déremboursements ? Ces derniers entraînent-ils des modifications dans la stratégie de prescription au profit de médicaments remboursés, avec les effets financiers que l’on imagine ?
Mme Anne-Marie Brocas : Les déremboursements ont un impact comptable. On observe ainsi une nette rupture dans l’évolution des dépenses à la suite des mesures prises en 2006, la croissance du poste médicament n’ayant été que de 0,9 % alors qu’elle était comprise entre 5 et 7 % depuis une dizaine d’années.
En 2004, afin de mettre en lumière la stratégie des laboratoires à la suite de déremboursements et les effets de ces derniers sur la prise en charge des malades, la DREES a mené une étude sur quelques molécules ayant fait l’objet d’un déremboursement : anti-diarrhéiques, enzymes anti-inflammatoires, vasodilatateurs cérébraux et périphériques, anti-acides. En fait, cette étude a montré qu’il n’y avait pas de réponse unique à votre question et que les choses variaient en fonction du produit. Ainsi, pour les vasodilatateurs, la structure de consommation a peu changé, le prix a varié de façon limitée et il n’y a pas eu de transfert vers d’autres produits. En revanche, pour les antiacides, a été observé un effet de substitution par des produits remboursés. L’évolution tient donc à la nature même des produits et aux pathologies traitées et l’on ne peut pas parler de transferts systématiques vers des produits plus coûteux.
Mme Catherine Lemorton, rapporteure : Les vasodilatateurs sont fréquemment prescrits chez des personnes âgées, pour des affections de longue durée (ALD) prises en charge à 100 %. C’est sans doute parce que les complémentaires ont accepté de compléter le remboursement qu’il n’y a pas eu de changement de comportement dans les prescriptions. En revanche, pour les veinotoniques, le déremboursement a entraîné un transfert vers des produits de contention veineuse, plus onéreux.
Mme Anne-Marie Brocas : La DREES n’a pas étudié les veinotoniques. Sur les cinq classes thérapeutiques auxquelles elle s’est intéressée, elle a constaté une forte diminution globale de la consommation des produits déremboursés, mais avec des variations selon les classes, sans doute pour des motifs comme celui que la rapporteure a indiqué, qui tiennent en particulier aux types de malades et à la prise en charge assurancielle.
Mme Catherine Lemorton, rapporteure : Les déremboursements affectent fortement l’homéopathie, dont le taux de remboursement, qui est actuellement de 35 %, tomberait à 8 % avec la franchise de 50 cents par tube de granules. Si aucun système assuranciel ne vient prendre en charge cette différence, il y a un risque de transfert vers l’allopathie et de surcoût, comme celui qui a été observé en 2003 et 2004. La DREES dispose-t-elle d’une analyse à ce propos ?
Mme Anne-Marie Brocas : Non, ce n’est pas un sujet que la DREES a étudié spécifiquement.
M. Jean Mallot, coprésident : Les laboratoires interviennent dans plusieurs États et la réglementation est de plus en plus européenne. Les comportements envers les génériques varient selon les pays et les effets de contournement ne sont pas les mêmes. Est-il possible d’en savoir davantage à ce propos afin que l’on puisse mieux comprendre d’où viennent les problèmes ?
Mme Anne-Marie Brocas : Les exercices de comparaisons sont assez globaux. Ils montrent que la France se caractérise par un volume de médicaments consommés très supérieur à celui de ses voisins et que les prix y sont plutôt inférieurs.
S’agissant des volumes, si l’on regarde les ventes moyennes de médicaments aux officines, comptabilisées en unité standard par habitant, on obtient un chiffre de 750 pour l’Italie, de 1 000 pour l’Allemagne, l’Espagne et le Royaume-Uni, et de 1 500 pour la France.
Quant au prix moyen par unité standard, il est de 0,18 à 0,19 € en France, en Espagne et au Royaume-Uni mais il atteint 0,23 € en Allemagne, où le système de forfait par classe ne couvre que 60 % du marché, les prix étant libres par ailleurs. Le prix moyen est encore plus élevé en Italie puisqu’il atteint 0,27 €.
Le volume de médicaments vendus en France conduit à placer notre pays en tête de tous les pays européens pour la dépense de médicament par habitant. Ainsi, le chiffre d’affaires des ventes aux officines par habitant est de 284,40 € en France, contre 244 € en Allemagne et environ 200 € dans les autres pays.
On constate également qu’en France 8 ou 9 consultations sur 10 débouchent sur une prescription de médicaments alors que ce taux tombe à 1 sur 2 au Royaume-Uni.
Une analyse plus qualitative montre que la répartition par classe thérapeutique varie beaucoup selon les pays. Ainsi, la France est en tête pour les antibiotiques, tandis qu’en Allemagne d’autres médicaments sont beaucoup plus vendus. Les Allemands pourraient donc se poser à propos d’autres produits les questions que nous nous posons quant à la pertinence de la prescription des antibiotiques ou des antidépresseurs.
Au Royaume-Uni, on observe une forte consommation de médicaments peu consommés en France, comme ceux qui sont destinés à la lutte contre l’obésité ou au sevrage tabagique, ce qui s’explique par une action forte les pouvoirs publics en la matière.
M. Jean-Marie Rolland : Outre les études selon les classes thérapeutiques, la DREES procède-t-elle à des analyses en fonction du service médical rendu (SMR), par exemple sur la substitution de produits de contention et de conseils d’hygiène à la prescription de veinotoniques ?
Par ailleurs, la DREES a-t-elle déjà eu l’occasion de travailler sur la possibilité récemment offerte aux professionnels paramédicaux de prescrire eux-mêmes un certain nombre de produits, en particulier du petit matériel ?
Mme Anne-Marie Brocas : La DREES n’a pas conduit d’études prenant en compte le SMR. Cela relève plutôt de l’AFSSAPS ou d’autres organismes. Elle ne dispose pas non plus d’analyses sur la substitution aux prescriptions de veinotoniques. Ce phénomène de substitution, étudié à l’occasion de certains déremboursements, varie beaucoup en fonction de la classe thérapeutique envisagée.
L’autorisation de prescrire du petit matériel vient d’être donnée aux professionnels paramédicaux et la DREES n’a pu l’étudier pour l’instant. De manière plus prospective, il semble que l’évolution démographique des différentes professions conduira probablement à un déplacement du partage des compétences.
M. Pierre Morange, coprésident : La DREES dispose-t-elle d’informations sur l’implantation dans les pays voisins de logiciels d’aide à la prescription, dont la diffusion est encore embryonnaire en France ?
Mme Anne-Marie Brocas : La DREES n’a pas travaillé sur ce sujet. En revanche, il a été constaté que, par des mécanismes très différents, en particulier par l’action des associations de médecins et par le conventionnement, la prescription médicale est bien davantage orientée en Allemagne et au Royaume-Uni qu’elle ne l’est dans notre pays.
En la matière, l’organisation de la pratique médicale joue un rôle très important : un très gros cabinet de groupe anglais n’a rien à voir avec un généraliste français. En Allemagne, la situation est encore différente avec une forte présence des paramédicaux au sein des cabinets. Il est plus facile pour des cabinets importants que pour un médecin isolé de recourir à un logiciel d’aide à la prescription.
M. Pierre Morange, coprésident : La philosophie du système de santé mais aussi les modes de financement des médecins diffèrent beaucoup entre ces trois pays.
Mme Anne-Marie Brocas : Effectivement, mais l’organisation concrète de la pratique est également importante, de même que le rôle particulier que joue l’hôpital dans notre pays.
Toutefois cela ne contredit par l’enquête de la DREES sur les prescriptions des médecins généralistes et leurs déterminants en France. Il n’a pas été trouvé de différences majeures selon que l’on exerce en cabinet de groupe ou de manière individuelle. Peut-être cela tient-il au fait que, en France, ouvrir un cabinet de groupe consiste surtout à mettre en commun une secrétaire et des logiciels.
M. Georges Colombier : En Italie, on consomme donc moins de médicaments, mais les prix y sont plus élevés. A-t-on pour autant constaté que cela induisait des différences en termes de santé ?
Mme Anne-Marie Brocas : On dit souvent que le système français est le meilleur. Il faut pour le vérifier disposer des statistiques relatives à l’espérance de vie. Elles seront communiquées à la MECSS. Cela étant, on a du mal à établir une corrélation effective entre consommation de médicaments et de soins d’une part, espérance de vie d’autre part.
M. Pierre Morange, coprésident : Je vous remercie d’avoir répondu de façon aussi précise à l’ensemble de nos questions et je vous invite à faire parvenir à la Mission toutes les précisions, commentaires et propositions susceptibles de nourrir sa réflexion.
Mme Anne-Marie Brocas : J’ai pris bonne note des questions auxquelles il ne m’a pas été possible de répondre de façon complète et je ne manquerai pas de vous faire parvenir les compléments de réponse par écrit.
*
Audition de MM. Jean Marimbert, directeur général de l’AFSSAPS, et Michel Pot, secrétaire général.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale.
Mme Catherine Lemorton, rapporteure : Pouvez-vous définir le périmètre des missions de l’AFSSAPS et préciser ses relations, en termes d’échanges d’information, avec la Haute Autorité de santé et la Direction de la recherche, des études, de l’évaluation et des statistiques.
M. Pierre Morange, coprésident : Comment pourrait-on harmoniser les différentes missions qui sont imparties à ces organismes ?
M. Jean Marimbert : L’AFSSAPS est l’héritière de l’Agence du médicament. C’est une autorité sanitaire déléguée. Elle a reçu une très large délégation de l’État pour accomplir des tâches d’évaluation des produits de santé, notamment l’évaluation du bénéfice/risque, avant la mise sur le marché, et après la mise sur le marché.
L’Agence exerce également des missions de puissance publique, dans la mesure où elle est chargée de prendre des décisions publiques de gestion du bénéfice/risque : octroi des autorisations de mise sur le marché pour les médicaments, suspension, retrait, modification du régime du médicament au fil des modifications qui peuvent se produire dans sa vie, en raison de l’évolution de la connaissance scientifique et des informations fournies par les conditions réelles d’utilisation de ce médicament, par des milliers, voire des millions de personnes ; en effet, le rapport bénéfice/risque ne peut être qu’approché au moment de l’AMM.
Autre mission de puissance publique : l’inspection des sites. Il peut s’agir des sites de production, des laboratoires, des essais cliniques en France ou à l’étranger, des sites où se déploient d’autres types d’activités, comme la thérapie cellulaire.
Dernière mission, qui a pris de l’importance ces dernières années : le contrôle de qualité des produits en laboratoire. Il est bon que des laboratoires publics puissent pratiquer des contrôles, soit de façon aléatoire, soit de façon plus ciblée, en fonction d’une analyse de risques, en fonction de signaux qui leur parviennent par ailleurs, pour vérifier que les produits contiennent la quantité normale de substances actives, que leur qualité pharmaceutique est bonne et éventuellement qu’ils ne comportent pas de substances toxiques, comme on l’a vu dans certains cosmétiques importés.
Ces missions s’exercent au travers de quatre séries de métiers : l’évaluation avant et après l’AMM, l’inspection, le contrôle en laboratoire et la production d’informations touchant au bénéfice/risque du médicament.
J’ai été nommé à la direction de l’AFSSAPS en février 2004, quelques mois avant le vote de la loi sur l’assurance maladie, qui a créé la Haute Autorité de santé. Le législateur a décidé de transférer les fonctions d’évaluation du service rendu à la HAS, fonctions qui étaient jusqu’alors rattachés à l’Agence, après l’avoir été au ministère de la santé. Il a donc fallu construire des articulations avec la HAS sur deux points principaux.
Le premier a été le suivi post AMM. Les études post AMM sont utiles pour mieux informer, à partir des données de la vie réelle, pour apprécier le rapport bénéfice/risque du médicament – notamment le suivi de la toxicité des produits – et la réalité du service rendu, ce dernier domaine relevant de la commission de la transparence qui fait partie de la HAS.
Au deuxième semestre 2005 a été mis en place un mécanisme de coordination : d’un côté les équipes AMM et du suivi post AMM, de l’autre les équipes de la commission de la transparence. Elles se réunissent très régulièrement, environ tous les quinze jours, pour échanger sur les dossiers et les besoins d’études post AMM en fonction des produits concernés par l’évaluation. En cas d’évaluation française, les produits obtiennent l’AMM et passent ensuite à la transparence. En cas d’évaluation européenne, il faut anticiper les besoins en études post AMM avant même l’obtention de l’AMM européenne.
L’idée est de ne pas faire de demandes dispersées aux laboratoires et d’éviter que ces études ne fassent double emploi. On essaie de configurer des études post AMM susceptibles d’atteindre les deux types de résultats : les données sur le bénéfice/risque actualisées en vie réelle et les données sur le service rendu et l’intérêt de santé publique ; ou bien l’on articule les demandes d’études de manière qu’il n’y ait pas de chevauchements.
Cette première collaboration est complétée par des réunions avec la Direction générale de la santé, qui ont lieu deux ou trois fois par an. On n’examine pas l’ensemble des dossiers post AMM, mais on se concentre sur quelques grands dossiers qui représentent un enjeu de santé publique ; Accomplia ou Gardazil, par exemple.
Le deuxième aspect de cette coordination avec la HAS concerne la diffusion de l’information dans le domaine des produits de santé. Il faut remarquer que le terme de « bon usage » a plusieurs significations. Pour le sens commun, c’est l’usage sûr ; c’est la manière d’utiliser le produit en maximisant ses avantages et en minimisant ses risques. Toutefois c’est aussi le bon usage du point de vue de la valeur thérapeutique : l’usage du médicament, dans une stratégie thérapeutique où il n’y a pas nécessairement que le médicament. C’est enfin le bon usage du point de vue du payeur public, dans le sens du meilleur coût/bénéfice.
L’idée a été d’échanger avec la Haute Autorité de santé pour produire des documents qui soient les plus complémentaires possibles et, dans certains cas, produire des documents sous double timbre, comme ce fut le cas pour Accomplia.
Aujourd’hui, le principal problème pour l’information publique du médicament est quantitatif. Il est lié à la capacité de produire une bonne information et de l’actualiser.
On peut citer l’exemple de coopération, hors AMM, avec la HAS et l’Institut national du cancer – INca. Lorsque le décret sur la tarification à l’activité – T2A – est sorti, il a été prévu que les hôpitaux devraient passer avec les agences régionales d'hospitalisation – ARH – des conventions portant notamment sur le bon usage des médicaments. Le ministère s’est aperçu que, pour que ces conventions réussissent, il fallait disposer de référentiels de bon usage. Le plus simple qui existe, et qui est actualisé, c’est l’AMM.
Cependant il y a de très nombreuses prescriptions hors AMM, notamment à l’hôpital et en cancérologie. Il était donc nécessaire de produire des référentiels publics qui permettent de distinguer, à l’intérieur du hors AMM, ce qui est scientifiquement acceptable, car, même si l’on n’a pas le niveau de preuve de l’AMM, on dispose de l’expérience clinique des soignants, et ce qui est scientifiquement moins acceptable, voire pas du tout. On s’est alors rendu compte qu’aucune des institutions prise isolément n’avait les moyens de produire très rapidement l’ensemble des référentiels dont on avait besoin. On s’est donc réparti la tâche de façon pragmatique : la HAS commence à produire des référentiels sur les dispositifs médicaux ; l’INca des référentiels hors AMM en cancérologie ; l’AFSSAPS des référentiels de médicaments hors cancérologie, avec un mécanisme de relecture réciproque entre les équipes d’experts des différentes institutions.
M. Pierre Morange, coprésident : Au-delà de cette répartition des tâches, existe-t-il une base de données commune ? Est-ce que, sur un seul et même site, se trouve l’ensemble des données qui colligent l’ensemble des domaines qui ont été répartis en fonction de cette ventilation ? A-t-on couvert, 70, 80 % ou 100 % de ce champ entre les trois secteurs qui ont été rappelés ?
M. Jean Marimbert : Votre première interrogation renvoie à la question de la base de données sur le médicament. C’est un sujet lancinant, au sens où il est abordé dans un certain nombre de rapports parlementaires ou de la Cour des comptes depuis cinq ou six ans.
Il a été décidé de mettre en place une base publique du médicament. Celle-ci sera opérationnelle à la fin de 2008. Les médicaments représentent environ 16 000 produits autorisés dont 11 000 à 12 000 effectivement commercialisés. Pour chaque produit, existe un mode d’emploi et un relevé des caractéristiques, avec la particularité qu’ils ont été autorisés au fil du temps. On se trouve donc devant une masse d’informations dont la grande majorité n’est pas informatisée, se présentant parfois sous forme de pelures, qui ne sont pas très exploitables pour faire une base de données.
Un tel projet est très lourd : il consiste à reprendre les relevés des caractéristiques de produits, à les faire transcrire dans une forme informatiquement utilisable. Il faut les faire relire et valider par les pharmaciens de l’organisme public et, petit à petit, les mettre en ligne.
M. Michel Pot : Il y a 5 000 produits en ligne et 3 000 validés et en interaction avec les laboratoires pour vérification.
M. Jean Marimbert : D’ici fin 2008, les données de la base seront reprises sous forme informatique, en mode « base de données ». Pour y parvenir, il a fallu conduire, notamment en décembre 2005, des négociations budgétaires qui ont été âpres mais fructueuses.
Cette base est fondée sur des données de l’AMM. Évidemment, il serait utile d’utiliser cette base dans une logique d’intérêt public plus globale, dans un travail commun avec la HAS, pour y greffer des données autres que celles de l’AMM, en particulier sur le service médical rendu, ce qui profiterait aux médecins.
M. Michel Pot : Pour greffer des données autres, il faut qu’elles existent et qu’elles soient accessibles. Le problème se pose de la même façon pour la HAS : l’historique du service rendu remonte à un certain temps, où les données n’étaient pas informatiquement utilisables et donc pas immédiatement disponibles. On ne pourra donc pas les intégrer où les renvoyer à des liens tant qu’elle n’aura pas fait le même travail sur le service rendu.
Mme Catherine Lemorton, rapporteure : Quelles seront les données collectées et mises à la disposition des professionnels de santé à la fin de l’année 2008 ? Ces derniers auront-ils accès à la fiche de transparence du médicament, qui leur fait souvent défaut lorsqu’un visiteur médical d’un laboratoire vient au cabinet ou à la pharmacie, à l’AMM, au SMR ? Pourront-ils savoir si une étude post AMM est en cours ? Par qui ? De quelles informations pourront-ils disposer sur le prix ? Pourront-ils faire des comparaisons avec les prix des autres médicaments dans la même classe thérapeutique ?
M. Michel Pot : Non, le chantier consiste d’abord à mettre en ligne les données dont dispose l’AFSSAPS : les AMM, les annexes aux AMM, les résumés des caractéristiques du produit – RCP. Les fiches de transparence ne sont pas à l’AFSSAPS, c’est la HAS qui est en train de les informatiser.
M. Pierre Morange, coprésident : Il n’y a donc pas de travail d’articulation entre l’AFSSAPS, la HAS et l’INca pour la constitution d’une base de données commune qui permettrait de balayer le sujet du bénéfice/risque, la logique du SMR et le domaine de la pathologie cancéreuse ?
M. Michel Pot : L’AFSSAPS s’est engagée à mettre en ligne ses données et la HAS les siennes. Le jour où ce sera fait, il ne sera pas difficile de créer des liens sur les fiches de médicaments de l’AFSSAPS renvoyant aux fiches de transparence de la HAS – et inversement. Techniquement il n’y aura aucun problème. Le problème est d’abord de constituer la base de ses propres données.
M. Georges Colombier : Pouvez-vous nous préciser ce qu’est une fiche de transparence.
M. Jean Marimbert : Il faut distinguer l’AMM, qui consiste à évaluer le rapport bénéfice/risque du médicament et à décider d’autoriser sa mise sur le marché, et l’évaluation du service médical rendu – on dit plutôt maintenant du service médical « attendu » – qui consiste à comparer la quantité d’effets des médicaments, dont le rapport bénéfice/risque est déjà établi et positif, et à mesurer la valeur ajoutée thérapeutique.
La commission de la transparence est chargée de cette deuxième tâche. À partir de ce travail sur les produits qu’elle évalue, elle publie des fiches de transparence qui indiquent le service rendu par le médicament, ce dernier étant classé dans une des cinq catégories de SMR. Le niveau de remboursement et la fixation du prix se fondent sur cette évaluation de l’amélioration du service rendu.
Mme Catherine Lemorton, rapporteure : En ce qui concerne les médicaments génériques, comment peut-on expliquer la délivrance d’une autorisation de mise sur le marché à des laboratoires dont on sait que le princeps est génériqué ? Y a-t-il une raison cachée ? On peut citer l’exemple de l’AMM qui a été délivré à une Predmisolone orodispersible de même dosage que celle qui existait auparavant sous forme effervescente. Tous les professionnels de santé sont d’accord pour dire qu’elle n’a rien apporté de plus, si ce n’est qu’on ne peut plus génériquer le princeps.
M. Pierre Morange, coprésident : Je précise que la Predmisolone est un corticoïde, avec un effet anti-oedémateux et anti-inflammatoire.
M. Jean Marimbert : Globalement, le générique s’est développé ces cinq dernières années dans notre pays, même si on est parti de très bas.
Il n’y a pas de raison cachée au niveau de l’AFSSAPS, qui travaille dans l’intérêt de la santé publique, et qui cherche à être la plus transparente possible. C’est d’ailleurs la première agence d’Europe qui ait publié, début 2006, des comptes rendus de la commission de pharmacovigilance.
Je n’ai pas d’explication à fournir immédiatement concernant le cas de la Predmisolone, mais je vais me renseigner pour la donner à la Mission. Il faut néanmoins remarquer que, dans l’octroi de l’AMM des génériques, l’AFSSAPS est soumise à la législation européenne et doit respecter la définition européenne du générique qui est transposée dans la loi française.
Les critères sont intangibles : même composition et même forme thérapeutique. Si un produit ne remplit pas ces critères, on ne peut pas le traiter comme un générique, lequel bénéficie d’un régime très allégé. Dans le cas d’un générique, on n’exige pas des études cliniques, simplement la preuve de leur bioéquivalence, au stade de l’évaluation de l’AMM. Dans le cas contraire, on doit demander au laboratoire qui dépose le dossier d’apporter des éléments de preuve clinique, dans la mesure où il ne peut pas se situer par référence aux études cliniques faites pour un produit princeps.
Par ailleurs, au stade de l’AMM, on n’a pas à juger. On ne peut pas refuser l’autorisation parce que tel produit n’apporte pas un plus thérapeutique par rapport au précédent. La seule question à laquelle il faut répondre est : a-t-il plus d’efficacité que de risques ? La législation sur l’AMM n’empêche donc pas, en France comme ailleurs, l’entrée sur le marché des me too, c'est-à-dire des produits qui ne sont pas plus efficaces que les précédents et qui en sont très proches, même si on ne peut pas parler de génériques.
C’est en aval qu’on peut réguler les me too, au stade du travail de la transparence, de l’évaluation, de la comparaison entre médicaments. L’autorité publique peut très légitimement fixer un taux de remboursement ou un niveau de prix tenant compte du fait que le médicament n’apporte pas grand-chose par rapport aux générations précédentes ou à d’autres médicaments existants.
Il serait illégal, en revanche, de refuser une AMM sous prétexte qu’il existe déjà quatre ou cinq médicaments équivalents. Par ailleurs, les profils des patients sont très variés et ceux-ci ne réagissent pas de la même manière à des médicaments de la même classe, très proches sur le plan pharmaceutique et pharmacologique. L’un réussira à une personne, mais pas à une autre. Attention donc à une stratégie dans laquelle, sous prétexte que tel médicament n’apporte pas plus que les précédents, il ne faut pas autoriser sa mise sur le marché. On risque de réduire la palette thérapeutique des médecins.
Mme Catherine Lemorton, rapporteure : Quelle instance peut décider qu’il y a ou non contournement du générique, phénomène connu de tous les professionnels de santé, et décider de ne pas délivrer d’autorisation ?
M. Jean Marimbert : Il existe effectivement des stratégies de contournement. En 2005-2006, on s’est demandé si l’on pouvait modifier la définition du code de la santé publique sur les génériques, afin de faire figurer, dans la même catégorie du répertoire des médicaments génériques établi par l’Agence, davantage de produits. Ces tentatives ont échoué, le Conseil d’État ayant jugé que l’élargissement de la définition qui lui était proposé était contraire à la définition du générique, telle que prévue par la directive européenne.
Il peut exister des médicaments qui sont thérapeutiquement très proches du générique sans pouvoir être qualifiés de génériques, à cause de ce problème de définition. Il ne paraît pas exclu de réfléchir, dans ces cas-là, à la possibilité de permettre la substitution pour ces médicaments qui, à proprement parler, ne remplissent pas tous les critères très précis du générique. Mais il faudrait alors veiller à bien cadrer le système et poser une condition d’équivalence thérapeutique.
L’AFSSAPS est concernée par les polémiques sur la sécurité des génériques. Certains praticiens sont opposés à la substitution dans un domaine particulier, par exemple celui des épileptiques, où ils considèrent que la marge thérapeutique est beaucoup trop étroite. Des sociétés savantes françaises, et même étrangères, ont pris position contre la substitution.
Toute réflexion sur l’élargissement du répertoire, sur l’élargissement du champ de la substitution, doit prendre évidemment en compte les impératifs de sécurité. Si le générique s’est assez bien développé ces cinq dernières années, c’est parce que la population a globalement confiance dans le générique. Il ne faut pas porter atteinte à cette confiance par des accidents ou des manœuvres mal maîtrisées.
Il est tout à fait compréhensible qu’on veuille réfléchir à la façon de contrer les stratégies de contournement, mais on aura du mal à le faire dans le cadre de la notion de répertoire des génériques à proprement parler, en raison du caractère très strict de la définition communautaire. La politique du générique doit être une politique de confiance et il faut être très attentif aux enjeux de substitution qui peuvent parfois poser, même marginalement, des problèmes de sécurité.
M. Pierre Morange, coprésident : S’agissant des bases de données, il faut sortir d’une approche scientifique, le prescripteur n’étant pas du tout dans la même démarche, dans la mesure où il s’attache à la simplicité d’utilisation. Une base de données commune aboutirait à la création d’un logiciel d’aide à la prescription, que tout le monde appelle de ses vœux. En 2008, cela sera-t-il possible ? Aujourd’hui, 5 000 principes pharmaceutiques ont été informatisés. Est-ce que cela a été fait selon un classement alphabétique ou par ordre décroissant de prescription, ce qui serait plus utile ?
Par ailleurs, l’AFSSAPS a un rôle en matière de traçabilité sanitaire et, notamment, de fiabilité du médicament. Est-ce que l’ensemble des génériques qui sont vendus en France et en Europe est élaboré sur la plate-forme occidentale ? Certains sont-ils élaborés sur d’autres plates formes, notamment asiatiques ? On a en effet constaté que les unités de production de certains génériqueurs installés à l’Est du massif continental européen ont parfois montré le caractère aléatoire de la qualité de leurs productions.
M. Michel Pot : À partir de 1999, l’Agence a commencé à archiver électroniquement ses fiches électroniques et à les entretenir. Ce sont elles qui sont sur le site in extenso, c’est-à-dire avec les RCP. Le site comporte 5 000 premières spécialités avec leur composition pharmaceutique. Il n’y a pas de base exploitable facilement dans la mesure où, effectivement, le travail n’est que chronologique. En revanche le travail de reprise est fait en priorité sur des spécialités commercialisées.
S’agissant des logiciels d’aide à la prescription, la stratégie actuelle consiste à dire que pour être certifiés qualité, ces logiciels doivent être reliés à des bases de données médicamenteuses, lesquelles, qu’elles soient publiques ou privées, ont signé une charte de bonne conduite définie dans des travaux menés conjointement par la HAS et l’AFSSAPS. Cette charte, qui a été publiée, a été élaborée avec des professionnels des bases. Ces bases doivent répondre à un certain nombre de critères, notamment en termes d’exhaustivité, en particulier sur les RCP, les fiches de transparence.
Dès lors que l’AFSSAPS sera à même de fournir ces données qui seront aisément absorbables par n’importe quelle base et que la HAS sera à même de le faire de son côté, la question sera réglée par le biais de l’adhésion des bases, quelles qu’elles soient, à la charte qui a été définie entre l’AFSSAPS et la HAS.
M. Jean Marimbert : Pour faire une bonne base de données de données de médicaments, il faut d’abord faire les investissements préalables et mettre en forme utilisable pour une base de données tout le corpus existant. À partir de là, l’imagination peut être au pouvoir en matière de coopération, pour regrouper les données les plus utilisables et les plus accessibles pour les praticiens et les professionnels de santé, voire pour le public.
On peut citer l’exemple des dispositifs médicaux. L’AFSSAPS a des contacts avec la direction de la sécurité sociale depuis quelques mois. L’Agence dispose d’une source de données sur les dispositifs médicaux liée à la déclaration des dispositifs médicaux de trois des quatre classes ; elle concerne la mise sur le marché. La direction de la sécurité sociale souhaiterait que l’Agence mette dans cette base, dans un avenir le plus proche possible, des éléments sur l’inscription des dispositifs médicaux sur la liste des produits et prestations – LPP, c’est-à-dire l’équivalent d’éléments sur le service rendu. Le but est que soient accessibles sur une même base des données sur l’amont, c’est-à-dire sur la mise sur le marché du produit, et des données sur le statut du produit par rapport à la prise en charge. Cela signifie qu’à partir du moment où l’on dispose d’un outil à un endroit donné, on peut lui faire remplir un rôle qui dépasse les missions, dans une optique de bonne organisation du système.
M. Pierre Morange, coprésident : Pouvez-vous revenir à la question de la fiabilité des génériques, leur traçabilité et le problème de la sécurisation quand la chaîne de production implique des sites lointains de l’extrême Est ?
M. Jean Marimbert : Il faut être conscient que le fait d’utiliser des génériques, même produits sur le territoire national, pose des questions de traçabilité. À partir du moment où un médecin prescrit un médicament et ne s’oppose pas à sa substitution, un générique peut être substitué à ce médicament. En cas d’effets indésirables, on peut s’interroger sur la cause : est-ce que ces effets sont dus au produit princeps ou au générique ? Le pharmacien le sait, le patient aussi, mais pas le médecin. Des discussions ont eu lieu en commission de pharmacovigilance sur ce sujet. Il faut y travailler.
Ce qui est plus préoccupant, c’est que la chaîne de fabrication du générique est de plus en plus étendue. Certaines étapes sont de plus en plus souvent effectuées à l’étranger, parfois sur des sites très lointains. La fabrication des matières premières pharmaceutiques utilisées dans les génériques est à 80-90 % extra-européenne : Inde, Chine, Brésil et Amérique du Sud. Par ailleurs, les essais de bioéquivalence destinés à vérifier que le produit générique se diffuse dans l’organisme de manière comparable au produit princeps sont de plus en plus souvent effectués ailleurs, notamment sur des sites asiatiques. Dans ces pays, il y a des gens qui travaillent très bien, mais il y a aussi de vilains canards. Cela oblige à consacrer une partie de son temps à aller inspecter dans ces pays lointains, en particulier en Inde et en Chine, pour le compte de l’AFSSAPS ou de l’OMS. Il est évident que le contrôle de la qualité de l’amont de la chaîne, dans ces pays-là, dépasse les forces de n’importe quelle agence individuelle, même la FDA américaine.
M. Pierre Morange, coprésident : Il faut rappeler le cas d’équipes d’experts qui étaient passées dans des territoires asiatiques. Ils avaient constaté que les chaînes de production étaient conformes aux normes exigées, mais, quelques semaines après, les mauvaises habitudes étaient reprises. Cela pose des problèmes, notamment pour les pays qui commandent des médicaments à bas prix sur la base d’un référentiel théoriquement correct. Ce n’est pas parce qu’ils sont pauvres que leur population doit absorber des génériques mal élaborés.
M. Jean Marimbert : On ne sera jamais à l’abri de ce qu’on pourrait appeler « le syndrome de Potemkine » : on met en place des décors accrocheurs qu’on enlève après le passage des hautes personnalités. Reste que, pour relever ce défi, une coopération de plus en plus accrue est nécessaire entre les services d’inspection des différents pays. De nombreuses agences font des contrôles, mais, ce qui compte, c’est qu’au niveau européen et mondial, des échanges d’informations aient lieu et que lorsqu’un problème est repéré par l’une d’entre elles, elle le fasse savoir aux collègues des autres pays.
Pour la deuxième fois, une réunion a été organisée avec une vingtaine d’agences. Parmi les thèmes identifiés on peut citer : les moyens d’améliorer la coopération pour lutter contre la contrefaçon, qui se trouve à nos portes, et la lutte contre la fraude dans le domaine des essais cliniques.
Autre aspect : le contrôle en laboratoire.
Depuis sept ou huit ans, l’Agence s’est mise à opérer des contrôles de routine sur les génériques, quelle que soit leur origine. C’est le moyen de détecter d’éventuels défauts de qualité sur ces produits.
Mme Catherine Lemorton, rapporteure : Comment l’AFSSAPS travaille-t-elle avec les centres de pharmacovigilance ? Les pharmaciens sont amenés à participer à des recueils de données. Comment l’Agence procède-t-elle pour les recueillir et les traiter ?
Que pense l’Agence des ventes par Internet de médicaments soumis à AMM, au sens français ?
M. Jean Mallot, coprésident : Les taxes que perçoit l’AFSSAPS constituent une part importante de ses ressources. Quelles sont vos propositions pour assurer une meilleure cohérence du financement avec les objectifs que poursuit l’Agence ? Ne faudrait-il pas simplifier le dispositif, qui est assez complexe ? Enfin, comment simplifier et améliorer le système de recouvrement de ces taxes, lequel représente une charge pour l’Agence ?
M. Jean Marimbert : L’AFSSAPS a tenu une conférence de presse au mois de mai dernier, avec M. Jean Parrot, le président du conseil national de l’Ordre des pharmaciens, sur la contrefaçon, qui n’est plus un phénomène réservé aux pays en voie développement. À cette occasion, il a été rappelé que la loi française ne permettait pas de réguler la vente des médicaments sur Internet. Le droit européen est un peu plus nuancé en la matière. En conséquence, on ne peut pas garantir la qualité des médicaments vendus par Internet. Il ne faut donc pas aller sur Internet pour acheter des médicaments.
Malgré tout, Internet est très présent aujourd’hui. Certaines personnes ont même pris l’habitude d’acheter certains types de médicaments, par des filières qui ne sont contrôlées qu’épisodiquement par des sondages, au coup par coup. Il y a donc lieu de s’interroger sur la législation en la matière.
M. Pierre Morange, coprésident : On peut se demander si une simple mesure législative serait suffisante pour encadrer la vente des médicaments sur Internet. J’invite l’AFSSAPS à transmettre à la MECCS d’éventuelles suggestions, qui pourraient, le cas échéant, être intégrées, par voie d’amendement, dans le prochain projet de loi de financement de la sécurité sociale.
M. Jean Marimbert : Il faut aborder la question au niveau international et européen. On peut provoquer le débat au niveau européen sans même attendre une hypothétique régulation communautaire. Il conviendrait également de réfléchir à la manière d’organiser, sur le territoire français, la vente par Internet.
Trop souvent on s’adresse à des sites derrière lesquels il n’y a pas d’opérateurs pharmaceutiques sérieux, des sites dont les messages ne sont pas contrôlés, qui n’ont aucune accréditation et n’ont adhéré à aucune charte régissant la qualité de l’information délivrée au public. Certains pays voisins ont commencé à mettre en place des systèmes dans lesquels la vente sur Internet est possible, mais avec des opérateurs ayant pignon sur rue et répertoriés, offrant des garanties pharmaceutiques, dans des conditions de transparence et vérifiables par l’utilisateur. Une réflexion mériterait d’être lancée en ce sens.
Le système français de maillage territorial des centres régionaux de pharmacovigilance (CRPV), qui n’est pas très répandu en Europe, suscite de l’intérêt lors des débats internationaux. Les collègues étrangers s’intéressent à l’articulation entre une autorité nationale publique jouant un rôle de coordination et de mise en œuvre, l’AFSSAPS, et les centres régionaux qui constituent un appui. Ces centres de pharmacovigilance sont logés dans des CHU ; ainsi, les pharmacologues sont en relation avec les cliniciens. L’Agence a veillé, avec la direction des hôpitaux, à ce que, dans le cadre de la T2A, les missions des CRPV soient clairement identifiées comme des missions d’intérêt général.
Les CRPV sont assez fortement représentés au sein du comité technique de pharmacovigilance et de la commission de pharmocovigilance. On s’appuie en permanence sur eux. Par exemple, lorsqu’on met sur le marché un produit, on peut décider qu’en raison de certains risques identifiés lors des études cliniques, il faut mettre en place un plan de gestion des risques. Si, dans le cadre de ce plan, on prévoit un suivi renforcé de pharmacovigilance, on mandate généralement un centre régional qui deviendra pilote sur ce produit. Il sera chargé d’assurer la synthèse du suivi de tous les signalements qui remontent vers les centres, d’en faire rapport devant le comité technique de pharmacovigilance, puis devant la commission de pharmacovigilance, d’où l’importance de ce maillage et de ce travail avec les CRPV.
En ce qui concerne le financement de l’AFSSAPS, il y a fort à craindre d’un système de budgétisation totale de l’Agence, dans le contexte budgétaire d’aujourd’hui. Certes, le système actuel a des faiblesses. Une toute petite minorité de gens prétend que, parce que l’Agence collecterait elle-même des prélèvements obligatoires ayant pour la majorité la qualification de taxes fiscales prélevées par l’agent comptable d’un établissement public administratif national, au lieu de l’être par le receveur percepteur territorial, son financement deviendrait impur. Ce raisonnement est difficile à comprendre. En revanche, le système actuel comporte beaucoup d’avantages.
L’Agence dispose d’une ressource directement accessible ; c’est elle qui prélève les taxes pour le compte de l’État. Le taux de recouvrement des mandats émis est de 99,97 %, un quart de point supérieur au taux de recouvrement des impôts directs. De ce point de vue, le système est donc raisonnablement efficace.
Ensuite, il est beaucoup plus pratique que l’établissement public administratif perçoive directement, car l’assiette des prélèvements dépend de paramètres opérationnels liés aux AMM, par exemple la classification des AMM dont dispose l’Agence et dont ne disposerait pas un receveur. La vérification de ces paramètres en interne, au sein de l’établissement public, est donc très simple.
Enfin, personne n’a pu sérieusement soutenir que, sur la durée, le fait que trois quarts de ses ressources soient issues de prélèvements obligatoires sur les industries de santé – taxe sur le chiffre d’affaires perçue par l’établissement ou droits fixes perçus sur des dossiers – ait pu influencer significativement la ligne de santé publique de l’Agence. Je suis prêt à rendre compte du moindre des choix de l’Agence sur ce point-là, depuis mon arrivé à la direction de l’AFSSAPS, il y a quatre ans.
Le système de recouvrement fonctionne assez bien. Il est assez simple. Il a assuré, sur la durée, un financement correct des besoins d’exploitation et des besoins d’investissement de l’Agence. Or une agence des produits de santé ne doit pas être paupérisée. Elle doit pouvoir investir dans son système d’information, dans ses laboratoires.
Quand l’État a considéré qu’il y avait un fonds de roulement trop important, il en a repris l’essentiel, ce qui était normal. Quand les dépenses d’intérêt public sont inférieures aux ressources publiques affectées au fonctionnement de l’Agence, l’État peut les reprendre pour d’autres usages.
Il faut donc bien réfléchir avant de transformer ce système.
M. Pierre Morange, coprésident : Vous validez donc le système en place. Nous vous remercions de transmettre à la MECSS toute information complémentaire ou suggestion de réforme et de bien vouloir répondre aux questions qui vous seront adressées par écrit à la suite de cette audition.
*
Audition de MM. Laurent Degos, président de la Haute Autorité de santé (HAS), François Romaneix, directeur, Gilles Bouvenot, membre du collège de la HAS et président de la commission de la transparence, et Étienne Caniard, membre du collège de la HAS et président de la commission qualité et diffusion de l’information médicale.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale.
Mme Catherine Lemorton, rapporteure : La MECSS s’intéresse à la politique du médicament dans le cadre de la maîtrise médicalisée des soins. Il serait donc intéressant que vous nous indiquiez quel est le périmètre d’action de la Haute Autorité de santé et quelles relations celle-ci entretient avec l’AFSSAPS.
M. Laurent Degos : La HAS est une autorité indépendante – ce qui implique de se plier aux règles de la transparence, sur le plan tant des résultats que des méthodes, le médicament étant au centre de tous les acteurs de la santé –, dont le caractère scientifique signifie, ce qui fait sa force, qu’elle ne travaille pas dans l’arbitraire, mais sur des bases scientifiques, conformément aux méthodes de l’evidence based medecine, c’est-à-dire la médecine par la preuve. Afin de renforcer cette indépendance, outre le fait qu’un guide de gestion des conflits d’intérêt existe, les déclarations de conflit d’intérêt, de la part aussi bien des experts, des membres du collège que des responsables de la HAS, sont rendues publiques, et un groupe extérieur, présidé par M. Vigouroux, conseiller d’État, intervient en matière de déontologie et de règlement de tels conflits.
Le périmètre d’action de la Haute Autorité comporte trois grands domaines.
Tout d’abord, l’évaluation des technologies de santé, ce qui consiste à évaluer les médicaments, les dispositifs et les actes, dans le dessein d’aider le décideur – dans le cadre du service médical rendu et de l’amélioration de celui-ci – ainsi que les professionnels – dans le cadre du bon usage ;
Ensuite, les recommandations de bonne pratique médicale en matière de santé publique et de sécurité des soins ;
Enfin, l’action, puisque la HAS dispose des moyens de certifier à la fois les établissements hospitaliers et, par le biais de l’évaluation des pratiques professionnelles, tous les médecins, et qu’elle joue également un rôle en matière de prise en charge des maladies chroniques et de certification de l’information, laquelle va jusqu’à la certification de la visite médicale, ce que la France est seule à pratiquer.
Dans cet esprit, quatre objectifs guident la Haute Autorité.
Le premier est celui de la transversalité, c’est-à-dire le souci d’une vision globale du médicament au sein de la stratégie médicale, ce qui permet de préconiser des recommandations, et, en matière d’action, d’intervenir au niveau de la pratique professionnelle, de l’information et de la prise en charge à 100 %. La vision globale permet ainsi de mettre en lumière certains problèmes, par exemple le fait qu’un médicament aux effets modestes, mais au service médical rendu (SMR) suffisant, entrant dans une médication d’affection de longue durée (ALD), soit automatiquement pris en charge à 100 %.
Le deuxième objectif tient à une approche plus globale de la qualité, qui permet de replacer le médicament dans son contexte organisationnel et économique, sans oublier l’aspect sécurité des soins. Outre le fait que l’aspect médico-économique ne rencontre plus d’obstacles d’ordre culturel de la part des médecins, l’appréciation du coût du médicament peut répondre à différents critères : une approche coût/utilité comme en Grande-Bretagne – combien d’années de vie difficiles le patient est-il prêt à sacrifier pour une année de vie en bonne santé ? – ; une approche subjective – combien le patient est-il prêt à payer pour que le médicament soit pris en charge ? – ; ou une approche nouvelle coût/efficacité, telle celle sur laquelle les Allemands travaillent, mais à condition de l’aborder de manière séquentielle, c’est-à-dire en examinant d’abord l’aspect médical et, ensuite seulement, l’aspect économique.
Le troisième objectif porte sur l’évaluation continue. En France, dès que l’AFSSAPS autorise la mise sur le marché d’un médicament, la HAS détermine si celui-ci doit être ou non remboursé et à quel prix, alors qu’à l’étranger les listes ne sont pas positives, mais négatives – c’est-à-dire qu’après deux ou trois ans de vie, il peut être décidé que tel médicament n'est plus remboursé – et le prix d’un médicament y est d’emblée fixé par l’industriel. Encore faut-il, une fois la mise sur le marché intervenue, surveiller le médicament : tel est l’objet des études post-AMM.
Le quatrième objectif, enfin, a trait au bon usage du médicament, ce qui oblige la HAS à être lisible – par le biais d’édition de fiches courtes et très simples – et visible – en faisant connaître son action dans différents organes de presse –, effort de lisibilité et de visibilité auquel la Cour des comptes a rendu hommage. Dans le même esprit, il revient à la Haute Autorité de changer la culture des médecins dans leur pratique, et de réfléchir aux moyens de permettre une appropriation par tous les acteurs. C'est sur la base de l’avis de tous les professionnels que, par exemple, un plan d’action en matière de psychotropes, qui sera conduit par la direction générale de la santé, a été défini.
Mme Catherine Lemorton, rapporteure : Quels rapports la Haute Autorité entretient-elle avec l’AFSSAPS ?
M. Laurent Degos : La HAS, contrairement à l’AFSSAPS, a une vision globale du médicament. Elle peut donc procéder à une comparaison entre le médicament et les autres méthodes thérapeutiques – chirurgie, radiothérapie, etc. – et elle est consubstantielle à la solidarité, en ce sens qu’elle décide, face à un produit mis sur le marché national, s’il doit ou non être remboursé – travail qui, aux États-Unis, est effectué par les assurances. En revanche, il y a chevauchement pour tout ce qui concerne le bon usage et la pratique.
M. Pierre Morange, coprésident : Quand la HAS, associée à l’AFSSAPS, pourra-t-elle fournir un guide informatique opérationnel tant au prescripteur qu’à l’assuré ?
M. Étienne Caniard : La certification des logiciels d’aide à la prescription suit la même démarche que toutes les missions sur le médicament lancées par la HAS, à savoir être à l’écoute des besoins des prescripteurs – faire en sorte, par exemple, que l’ergonomie corresponde à la pratique des médecins.
L’étude des trois bases de données existantes – la mission donnée par la loi à l’AFSSAPS de constituer une base de données n’ayant jamais pu être totalement remplie – a montré qu’à quelques ajustements près elles répondent aux caractéristiques, notamment d’exhaustivité et de rapidité de mise à jour, que la HAS attend des logiciels d’aide à la prescription. L’action de la Haute Autorité a simplement consisté à publier une charte de qualité, au début du mois de septembre dernier. Ainsi, un logiciel ne sera certifié que s’il a été fait appel à une base de données médicamenteuses dont les éditeurs auront signé la charte.
Reste à savoir si l’utilisation d’un logiciel certifié modifiera réellement les pratiques et si les médecins seront suffisamment incités à utiliser un logiciel certifié plutôt qu’un logiciel non certifié. Ensuite, on pourra, le cas échéant, envisager d’instaurer une obligation de certification des logiciels.
M. Pierre Morange, coprésident : Reste que, actuellement, la dimension médico-économique n’est pas prise en compte dans les logiciels d’aide à la prescription. Quand sera-t-il possible d’accéder aux fiches de transparence ?
M. Gilles Bouvenot : Du fait de la demande pressante des ministres en charge de la santé, priorité a été donnée, à la fin des années quatre-vingt-dix, à la réévaluation des médicaments dits à SMR insuffisant, au détriment des réévaluations habituelles de classe. Comme il ne pouvait être question durant ces années de produire des fiches de transparence sur des produits qui pouvaient être déremboursés, ce n'est qu’une fois ce travail accompli, après que plus de 890 produits eurent été réévalués, que la HAS a pu reprendre son activité de réévaluation quinquennale des produits et de rappel pour réévaluation de classe, et donc d’édition de fiches de transparence.
Cependant, si les fiches de transparence apportent des informations, il n'est pas sûr qu’elles soient lues. C’est pourquoi l’accent a été mis, à la demande du Parlement, de la Cour des comptes et de l’IGAS, sur les fiches « bon usage du médicament ». La philosophie de la HAS n’étant pas de se contenter de lancer des actions, mais également de mesurer leur impact, les dix premières fiches publiées en 2007 – leur nombre devrait atteindre quinze par an –, feront l’objet d’une telle mesure d’impact, sans que cela se fasse au détriment de la production des fiches de transparence – celle sur les médicaments de la maladie d’Alzheimer est prête –, qui restent au cœur de la mission de la Haute Autorité, laquelle est de situer le médicament dans le cadre d’une stratégie globale de prise en charge.
En tout cas, à chaque fois qu’une dérive de prescription d’un médicament ou un risque de mésusage est possible, ou encore qu’un médicament est capable de modifier de façon très sensible l’organisation du système de soins, tels les nouveaux traitements de la polyarthrite qui ont fait exploser le nombre de séances en hôpital de jour, une fiche de bon usage sera éditée.
M. Pierre Morange, coprésident : On estime que 12,5 % de la population française consomment 47 à 48 % des dépenses d’assurance maladie. Avez-vous publié des fiches de bon usage des médicaments pour le traitement des maladies chroniques ?
M. François Romaneix : Au mois de juin 2007, 62 % des ALD étaient couvertes et 76 %, cancer inclus, par des fiches de bon usage. Le pourcentage de 100 % sera atteint, avec l’aide de l’INca, fin 2008.
M. Laurent Degos : La réflexion de la Haute Autorité de santé repose toujours sur l’étude des besoins des professionnels et des patients. À côté de la fiche de bon usage, de la fiche de transparence et des guides pour le médecin et pour le patient, existent également les fiches synthétiques qui préconisent des recommandations. Par exemple, si elles décrivent, pour l’hypertension, tous les moyens thérapeutiques existants, elles recommandent de commencer par les thiazides, qui coûtent moins cher. Cette action reste cependant discrète, faute pour la HAS d’avoir, jusqu’à maintenant, une compétence médico-économique.
M. Étienne Caniard : L’enjeu est moins de disposer de fiches exhaustives que d’offrir un outil qui permette au médecin de s’approprier celles-ci dans sa pratique quotidienne. Plutôt que de donner une date à laquelle l’ensemble du champ sera couvert, mieux vaudrait parler de date à laquelle seront mis à disposition les outils permettant au médecin une approche globale.
M. Pierre Morange, coprésident : Estimez-vous souhaitable de rendre obligatoire la certification des logiciels d’aide à la prescription et faudrait-il prévoir, dans cette hypothèse, une disposition réglementaire ou législative ?
M. Étienne Caniard : La loi du 13 août 2004 oblige, selon son interprétation actuelle, à mettre en place une procédure de certification, non à utiliser des logiciels certifiés. Dans ces conditions si le dispositif qui sera mis en place dans les prochaines années fonctionne, c’est-à-dire si une vraie discrimination dans l’utilisation des logiciels intervient et si le marché suit, l’obligation de certification ne sera pas nécessaire. Dans le cas contraire, il faudra passer à la seconde étape.
Mme Catherine Lemorton, rapporteure : Étant rappelé que les travaux de la MECSS visent à prendre en compte les enjeux financiers mais aussi les préoccupations de santé publique, il serait intéressant de savoir si des fiches de transparence relatives aux médicaments non remboursés sont également éditées par la HAS ou par d’autres instances.
M. Laurent Degos : La compétence de la Haute Autorité est limitée aux médicaments remboursés. L’évaluation des médicaments non remboursés manque à l’heure actuelle. La HAS, par exemple, ne peut être impliquée en matière d’automédication, faute de lien avec un quelconque remboursement.
Mme Catherine Lemorton, rapporteure : Avez-vous mené des études comparatives sur la prescription de médicaments en France et dans les autres pays européens. Des liens ont-ils été tissés avec des instances étrangères ? A-t-on pu établir une corrélation entre le rôle que jouent les laboratoires pharmaceutiques dans la vie médicale – lesquels dépensent en France, en dépenses de promotion, 8 500 euros par médecin – et la surconsommation médicamenteuse, apparemment propre à notre pays ?
M. Laurent Degos : Tisser des liens avec des organismes similaires à la HAS a été mon tout premier souci. Les liens avec l’Allemagne et la Grande-Bretagne ont été ainsi étendus au Danemark et à l’Irlande. Il vaut mieux, en effet, lutter à plusieurs que tout seul, ne serait-ce justement que vis-à-vis de l’industrie pharmaceutique dont la pression se fait sentir tant dans le domaine de la visite médicale que dans celui de la régulation européenne. Celle-ci est en effet soumise à un point tel à l’influence de l’industrie que le guide diabète, par exemple, que Bruxelles voulait imposer a été rejeté par la Haute Autorité qui refuse une telle déviation de la vision des problèmes.
M. Étienne Caniard a fait remarquer que si le volume global des dépenses de promotion du médicament est comparable entre les différents pays, la part de la visite médicale dans le total est, en revanche, beaucoup plus élevée en France. Elle y représente en effet 75 % des dépenses promotionnelles contre 68 à 70 % en Europe et 57 ou 58 % aux États-Unis, les différences de stratégie de communication s’expliquant tant par la faiblesse de contre-pouvoirs à la visite médicale que par les spécificités culturelles de chacun des pays.
M. Pierre Morange, coprésident : Je vous remercie pour ces précisions et vous demande de faire parvenir à la Mission toute suggestion ainsi que de répondre aux questions complémentaires qui vous seront adressées par écrit.
*
Audition de MM. Bertrand Fragonard, président du Haut Conseil pour l'avenir de l'assurance maladie, et Pierre-Jean Lancry, vice-président.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale pour cette audition qui s’inscrit dans le travail qu’effectue notre mission sur la prescription, la consommation et la fiscalité du médicament. Je donne sans plus tarder la parole à notre rapporteure.
Mme Catherine Lemorton, rapporteure : Comment expliquez-vous que le comité d’alerte se soit manifesté aussi tardivement alors que le seuil de déclenchement fixé à 0,75 % de dépassement de l’ONDAM – objectif national de dépenses d’assurances maladie – avait été largement atteint ?
M. Bertrand Fragonard : Vous devriez poser cette question aux responsables de ce comité. Ils ont en fait procédé en deux temps, en émettant d’abord une première appréciation puis un avis. La loi fixe comme date butoir le 1er juin, mais c’est au moment où le comité considère qu’existe un risque sérieux de dépassement de l’ONDAM de 0,75 % qu’il peut intervenir. Fallait-il qu’il le fasse quelques semaines plus tôt ? Je l’ignore, d’autant que le Haut Conseil se garde bien d’intervenir dans la vie conjoncturelle.
Cela étant, je connais les membres du comité d’alerte, qui sont compétents et indépendants, et je n’ai aucune raison de considérer qu’ils n’ont pas rempli leur fonction de façon pertinente et qu’ils n’ont pas agi conformément à l’esprit et à la lettre de la loi.
Mme Catherine Lemorton, rapporteure : Le seuil des 0,75 % avait été dépassé bien avant le mois de juin.
M. Bertrand Fragonard : Je ne suis pas sûr que l’on puisse dire cela. On n’a pas, notamment pour l’hôpital, une visibilité suffisante et assez précoce pour pouvoir prendre position plus tôt. L’avis définitif comporte d’ailleurs un certain nombre de réserves et c’est un des points qui fait contentieux avec les médecins libéraux, qui considèrent que l’on connaît mieux les risques de dépassement de l’ONDAM liés à leur pratique et que l’on est plus flou en ce qui concerne l’hospitalisation. Cela a peut-être joué dans l’idée qu’il fallait attendre quelques semaines de plus, mais prendre position au mois de mars aurait sans doute été prématuré.
Mme Catherine Lemorton, rapporteure : Vous avez observé lors de votre audition par la commission des affaires culturelles, familiales et sociales que l’on était passé de 5,4 à 9 visites par an chez le médecin, pouvez-vous préciser sur quelle durée ?
M. Bertrand Fragonard : Sur 25 ou 30 ans, mais l’état de santé des Français ne s’étant pas dégradé, au contraire, au cours de cette période, cela signifie que nous sommes entrés dans une société dans laquelle le recours au médecin est devenu un trait culturel. Il est encore plus fréquent au Japon, mais ce mouvement est significatif dans notre pays, d’autant que dans neuf cas sur dix la consultation débouche sur la prescription de médicaments. Cette évolution a une influence majeure sur les comptes de la sécurité sociale.
Mme Catherine Lemorton, rapporteure : Pensez-vous qu’en obligeant, dans le cadre du parcours de soins, à passer devant le médecin traitant avant de se rendre chez un spécialiste, la réforme de 2004 a entraîné une augmentation du nombre des visites ? Certains syndicats de médecins ont dit à ce propos qu’il ne fallait plus parler de « médecins traitants » mais de « médecins sous-traitants »…
M. Bertrand Fragonard : Il ne faut pas surestimer les effets du parcours de soins sur la pratique médicale. Dans les faits, beaucoup de Français avaient déjà un médecin de famille et les analyses sur les parcours spontanés, avant les contraintes tarifaires, montrent que le recours spontané aux spécialistes n’était pas très fréquent. La réforme ne pouvait donc pas avoir un effet radical sur les comportements. On a craint qu’elle entraîne une augmentation du volume des consultations, la visite chez le médecin traitant apparaissant souvent superfétatoire. Or, dans les faits, on observe plutôt une légère diminution du nombre des recours au médecin, sans qu’on puisse véritablement l’imputer au parcours de soins. En fait, c’est l’annonce même de la réforme qui a eu des effets psychologiques sur les médecins et sur les patients.
Il y a eu tout au plus un peu de crispation pour certaines spécialités, comme la dermatologie, qui ont amené les partenaires conventionnels à adopter quelques mesures de compensation.
M. Pierre Morange, coprésident : Si la philosophie générale du bouclier sanitaire est d’aboutir à une nouvelle répartition de la prise en charge, le Haut Conseil a-t-il également réfléchi aux effets que ce bouclier aurait sur les médicaments ? Quelles pourraient en être les conséquences sur le plan budgétaire et sur les comportements ?
M. Bertrand Fragonard : Le bouclier n’a pas vocation à influencer la pratique médicale. Il s’agit d’un réajustement, mais ce n’est pas cela qui modèle la prescription. Dans son avis de juin 2006 sur la prescription des médicaments, le Haut Conseil a exprimé sa conviction que, dans un marché très ouvert où l’on admet les médicaments vite et bien et à des prix plutôt raisonnables, la vraie question est celle de la pression que l’on exerce pour contenir la prescription.
Le constat est connu et les communications de la Cour des Comptes à la MECSS le confirment : nous sommes dans un pays où l’on a heureusement accès aux progrès thérapeutiques liés à l’innovation pharmaceutique et où l’on accepte le principe d’un marché ouvert. Or un marché ouvert fortement solvabilisé crée un contexte propice aux abus de prescription. Dès lors, la prescription n’est pas à l’optimum en quantité puisque presque chaque consultation débouche sur une ordonnance, en général de trois lignes et dont le coût unitaire progresse. En moyenne, chaque fois que l’on va voir un généraliste, cela conduit à 50 euros de prescriptions pharmaceutiques. La France est un pays gros consommateur de médicaments. Dans d’autres pays un grand nombre de consultations ne se traduisent pas par une ordonnance. Ce phénomène tient sans doute à la culture française, c’est-à-dire à la formation initiale des médecins.
La prescription n’est pas non plus à l’optimum en termes de qualité. Sans même parler du risque que fait courir la prescription de certains médicaments, nous constatons de nombreuses prescriptions hors autorisation de mise sur le marché (AMM), qui ne sont pas totalement maîtrisées, et, surtout, hors répertoire. L’économie du médicament repose sur le princeps, sur le générique et sur les médicaments dits « me too », c’est-à-dire des produits assimilables, de la même classe thérapeutique et qui ont probablement la même aptitude à soigner les malades.
Quand on arrive au terme d’un brevet, la générication est possible, elle entraîne une baisse du prix, qui a été plus prononcée depuis les récentes décisions gouvernementales puisque le générique est introduit à 50 % du prix du princeps et que l’on fait baisser le prix de ce dernier de 15 %. On attend donc de l’arrivée du générique une baisse prononcée du coût de la prescription. Or on n’obtient pas ce résultat car la prescription se fait souvent en dehors du répertoire. Le laboratoire responsable du princeps a tendance à limiter l’effort de promotion puisque cela lui rapporte moins, mais, faute de contacts avec les médecins, les génériqueurs n’ont pas de politique de promotion, si ce n’est en direction des pharmaciens. À l’inverse, les laboratoires qui proposent des me too exercent une pression forte sur les prescripteurs. Ainsi, alors que chacun sait que l’on pourrait soigner davantage en utilisant le répertoire – princeps ou générique –, on observe une augmentation préoccupante de la prescription hors répertoire.
C’est pour cela que nous avons insisté sur la convergence des prix, c'est-à-dire sur la nécessité de revoir les prix des me too au moment où l’on génériquait un produit. Cette option a été retenue dans la lettre d’orientation que le ministre a envoyée au mois d’octobre au président du Comité économique des produits de santé (CEPS).
Il n’y a pas encore eu de modélisation pour voir quelle pourrait être l’étendue du répertoire. C’est un exercice difficile, car la notion de me too n’est pas aussi précise que celle de générique et l’on ne peut pas considérer que tous les produits sont équivalents. Le répertoire représente actuellement 20 % des prescriptions, on ignore s’il pourrait atteindre 30 % ou 40 %, mais il est certain qu’il y a une marge de progression, donc un gisement considérable, probablement de plusieurs centaines de millions d’euros. C’est pour cela que depuis le mois d’octobre le président du CEPS s’efforce de revoir les prix des me too qui sont à l’origine légèrement inférieurs au prix princeps mais qui, après la générication, valent plus que le générique mais aussi que le princeps. Il faut faire converger le prix des me too vers cette référence qu’est le prix de marché, l’enjeu étant de repérer le gisement et de l’exploiter.
Même si l’avenant 23 à la convention médicale fixe un objectif de prescription dans le répertoire pour certaines classes de médicaments, faire évoluer la prescription nécessite beaucoup de temps. Aussi, l’impatience gagnant les gestionnaires, il faut contourner cette résistance en ajustant la politique des prix, c’est l’objet du processus de convergence. C’est un sujet essentiel car si l’on ne fera pas varier ainsi le volume et la qualité de la prescription, on agira au moins sur son prix.
M. Jean Mallot, coprésident : Vos comparaisons internationales, par exemple sur la formation initiale des médecins, vous ont-elles permis de repérer de bonnes idées qui permettraient d’améliorer la situation dans notre pays ?
M. Bertrand Fragonard : La formation initiale et continue et l’évaluation des pratiques sont des enjeux fondamentaux.
Le rapport complexe à la consultation chez le généraliste renvoie à des contenus humains qui n’ont rien à voir avec la mécanique financière que je viens d’exposer. La pratique médicale est le fait de professionnels compétents mais qui évoluent peu et qui sont peu confrontés aux autres. C’est précisément ce qui rend importantes la formation continue et l’évaluation des pratiques. Toutefois il y a aussi une vie collective du corps médical et c’est pour cela que la maîtrise médicalisée a tendance à faire changer les habitudes de prescription.
Une politique brutale de déremboursement de médicaments aurait sans doute des effets plus rapides, mais elle n’est ni souhaitable ni envisageable. La maîtrise médicalisée progresse lentement, mais elle va dans le bon sens. Éclairer la pratique médicale, ne serait-ce qu’en la décrivant, rendre les référentiels de la Haute Autorité de santé (HAS) plus précis, mieux les diffuser, permet de faire évoluer la prescription. Cependant, cette politique ne saurait se substituer à une politique des prix et d’admission sur le marché à des conditions de remboursement raisonnables.
M. Jean Mallot, coprésident : Il faut donc se demander comment accélérer les effets de la maîtrise médicalisée et comment gagner en efficacité.
M. Bertrand Fragonard : Dans un cadre conventionnel on est aussi tenu par le rythme de ses partenaires. Les directeurs des caisses ont sans doute d’autres pistes, mais quand on a fait le choix, que je trouve positif, de la maîtrise médicalisée on est tenu d’avancer au rythme auquel on élabore les instruments. N’oublions pas que la maîtrise médicalisée a décollé à partir du moment où l’on a pu coder les médicaments.
Il n’est pas facile d’aller plus vite. Dans la mesure où les médecins sont les ordonnateurs, on peut certes essayer de réduire la pression des laboratoires, mais lorsque, en dépit des efforts en faveur de la maîtrise médicalisée, on constate que la croissance des prescriptions est forte, il est légitime que l’assureur récupère une partie du surcroît sur les laboratoires, sur les grossistes-répartiteurs et sur les pharmacies d’officine.
M. Pierre Morange, coprésident : Nous allons en revenir aux questions de notre rapporteure, mais il serait intéressant que nous disposions de plus d’éléments sur les comparaisons internationales en ce qui concerne les schémas organisationnels, l’articulation entre les différentes structures qui régulent le marché du médicament, la constitution des prix et la fiscalité, dont les communications de la Cour des Comptes à la MECSS soulignent la très grande complexité et le manque d’efficacité dans notre pays.
Mme Catherine Lemorton, rapporteure : Pensez-vous que le mode de rémunération actuel des médecins par le paiement à l’acte a des effets sur la prescription excessive de médicaments ?
Considérez-vous par ailleurs que la place que prennent les visiteurs médicaux dans l’information des médecins a une influence sur les prescriptions hors répertoire ?
M. Bertrand Fragonard : Aucune analyse n’a été menée sur ce sujet. On ignore donc si la rémunération à l’acte entraîne une inflation des recours au médecin, des indemnités journalières et des prescriptions. Il faut par ailleurs prendre en compte le contexte de la démographie médicale : lorsqu’un médecin a trop de clients, les défauts éventuels de la tarification à l’acte s’effacent.
Le projet de loi de financement de sécurité sociale pour 2008 introduit la possibilité très intéressante de mener des expérimentations, pendant cinq ans, d’engagements des médecins sur des objectifs contractuels individualisés d’amélioration de leurs pratiques et l’on verra si la réorganisation de la prise en charge a un effet sur le recours au médecin et sur la prescription d’indemnités journalières et de médicaments.
Les comparaisons internationales sont très difficiles car les contextes ne sont pas les mêmes. Certains pays appliquent des méthodes plus énergiques, par exemple avec des enveloppes fermées de prescriptions et d’honoraires.
M. Pierre-Jean Lancry : En effet, on ne parvient pas vraiment à comparer, même si l’on voit bien que nous consommons beaucoup plus que les autres. On a pensé pendant un moment qu’il y avait un effet d’offre car 9 000 médicaments, dont 6 700 remboursables, sont vendus en France. Mais on n’observe pas ce phénomène en Allemagne, où il y a pourtant deux fois plus de médicaments. La comparaison ne donne donc pas de résultats très nets.
Il est vrai que les contextes sont très différents puisqu’on a observé que les patients n’utilisaient pas les mêmes formes galéniques. Les Anglais refusent les suppositoires tandis que, dans le pourtour méditerranéen, l’injection est considérée comme une preuve de l’importance de la pathologie. En France, on a tout !
Une étude assez ancienne des comportements des prescripteurs a toutefois permis de mettre en évidence certains éléments, en particulier qu’il y aurait un lien entre l’activité d’un médecin et la prescription par acte : plus le médecin fait d’actes, plus il prescrit à chaque acte, peut-être pour se couvrir dans la mesure où il consacre moins de temps à son patient, peut-être aussi pour rassurer celui-ci.
M. Bertrand Fragonard : Ce n’est pas un phénomène majeur.
On peut bien évidemment s’intéresser de près à quelques très gros prescripteurs, mais ce dont on parle, ce sont des habitudes globales de prescription. Les modifications d’organisation ont donc plus d’effet que des processus administratifs ou que d’éventuels changements du mode de rémunération. Personne ne propose d’aller vers une rémunération par capitation. Tout au plus peut-on valoriser des comportements différents par des rémunérations différentes.
Il est évident que les visiteurs médicaux ont une influence sur la prescription hors répertoire : ils sont là pour promouvoir leurs produits. Notre pays a entrepris d’encadrer cette pratique par une charte, qui est suivie, qui a une influence, mais qui n’empêche pas la promotion. On essaye aussi de la contenir au moyen d’une taxation spécifique.
Cependant, d’autres influences s’exercent sur le médecin. Ainsi, celui qui élabore les référentiels doit guider la pratique médicale, ce qui renvoie aux problèmes d’élaboration et d’appropriation des référentiels. Une autre influence est celle du gestionnaire du risque et la Caisse nationale d’assurance maladie s’efforce de peser dans ce débat. Enfin, on n’a pas encore exploité la possibilité d’une gestion collective par les médecins libéraux. S’ils discutaient davantage, s’ils se situaient dans une analyse collégiale des pratiques, il est certain que les choses évolueraient plus rapidement.
C’est parce que la visite médicale a tendance à se porter vers le me too au moment de l’apparition du générique qu’il nous a semblé nécessaire de compléter tout ceci par une politique des prix. Car il n’est pas gênant que le médecin prescrive le me too si son prix est voisin de celui du répertoire. C’est tout l’enjeu de la convergence.
Et si l’on a fait beaucoup d’efforts en faveur du droit de substitution, les caisses étant allées jusqu’à subordonner le tiers-payant à l’acceptation du générique, pour que la substitution joue, encore faut-il que la prescription soit faite dans le répertoire. Pour cela, la visite médicale ne doit pas être le seul élément qui détermine la prescription.
M. Pierre Morange, coprésident : Quel est votre sentiment quant aux différents schémas d’organisation qui existent en Europe ?
Que pensez-vous par ailleurs d’une éventuelle extension des compétences de la Haute Autorité de santé (HAS) à la formulation de recommandations prenant en compter le critère médico-économique ?
M. Bertrand Fragonard : Le débat a été assez vif en 2004. Donner des responsabilités différentes à la HAS serait une bonne chose, à condition qu’elle ne soit pas perçue comme obsédée par la volonté de contenir les prix et de limiter les actes. Il faut donc qu’elle conserve son autorité d’émetteur de référentiels et qu’elle montre que certains processus de prise en charge thérapeutique sont plus efficaces et plus économiques que d’autres. C’est sur des grands chantiers comme l’éducation thérapeutique ou la prise en charge des malades chroniques que la Haute Autorité peut adosser une approche plus économique. Toutefois il faut aussi qu’elle garde présente à l’esprit l’importance des liens de confiance avec la communauté médicale et hospitalière.
Mme Catherine Lemorton, rapporteure : Un sondage publié l’an dernier montre que 78 % des généralistes ignorent l’existence de la charte de la visite médicale.
On sait par ailleurs que la formation initiale des médecins est particulièrement centrée sur l’hôpital. Existe-t-il précisément, au sein des hôpitaux, une obligation de prescrire des génériques ? Car si un jeune médecin n’a eu que des spécialités à prescrire à l’hôpital, on peut penser qu’il aura ensuite le réflexe de faire de même dans son cabinet.
M. Pierre-Jean Lancry : Certains centres hospitaliers universitaires, enseignent les thérapeutiques avec les dénominations communes internationales, les DCI, d’autres ne le font pas. Il est certain qu’un jeune médecin qui a appris les noms de spécialités les prescrit plus facilement par la suite. Par ailleurs, les prescriptions à l’hôpital portent souvent sur des produits innovants et onéreux, sous brevet, pour lesquels il n’est pas possible de prescrire des génériques. Pour les produits qui sont davantage de routine, il est assez fréquent qu’on ne se préoccupe pas des DCI car, en raison du mécanisme d’achat par appel d’offres, le médecin dispose d’une liste de produits arrêtés par son comité économique du médicament. Qui plus est, les produits déjà anciens sont vendus à un prix dérisoire sur le marché hospitalier car les laboratoires savent très bien que l’on suscite la prescription de ville par la prescription hospitalière.
En Grande-Bretagne, les gros cabinets de généralistes disposent d’un pharmacien conseil qui, même s’il ne dispose pas du droit de substitution, est chargé de voir ce qu’il est possible de faire à partir de la prescription. Cependant cette dépense ne peut être amortie que dans le cadre d’un cabinet de groupe.
M. Pierre Morange, coprésident : Quelle appréciation le Haut Conseil porte-t-il sur les logiciels d’aide à la prescription ?
M. Bertrand Fragonard : Nous n’avons pas eu à étudier cette question. Le Haut Conseil s’est borné à constater qu’il y a un peu de retard dans l’agrément des logiciels et que l’approbation des référentiels est lente.
Mme Catherine Lemorton, rapporteure : Le système des ententes préalables pour les médecins « déviants » peut-il s’appliquer aux médicaments ?
M. Jean Mallot, coprésident : J’aimerais aussi connaître l’appréciation que vous portez sur les effets de la probable institution des franchises.
M. Bertrand Fragonard : L’article relatif à la mise sous entente préalable a été introduit en 2004, mais pour un champ limité, en particulier les indemnités journalières. L’article 26 du projet de loi de financement de la sécurité sociale pour 2008 propose d’étendre la procédure à l’ensemble des prescriptions.
Il est d’ores et déjà possible, lorsque l’on dispose d’éléments concordants sur la prescription d’indemnités journalières et de médicaments, d’évaluer le comportement global d’un médecin.
Par ces dispositions, on cherche d’abord à rappeler à l’ordre une petite minorité dont le comportement n’a, statistiquement, rien à voir avec la pratique courante. Seulement quelques dizaines de praticiens sont concernés – 150 cette année – et l’économie attendue est donc marginale. Mais on entend aussi adresser un message global aux médecins et à l’opinion. En abaissant le seuil de repérage on changerait d’exercice car il s’agit pour l’instant d’une procédure d’exception, qui vise des médecins qui sont huit ou dix fois au-dessus de la moyenne, certains allant jusqu’à prescrire 25 000 indemnités journalières dans l’année.
S’agissant des franchises, il faut d’abord savoir si elles seront réassurables. La question semble avoir été tranchée, la notion de contrats responsables exerçant une pression sur les assureurs pour qu’ils ne prennent pas en charge les franchises.
Les franchises poursuivent à la fois un objectif de responsabilisation et un objectif financier.
L’institution d’une franchise non réassurable de 0,50 euro par boîte ne saurait avoir un effet majeur par rapport au prix des médicaments. La modestie relative des sommes en jeu - 850 millions d’euros, tous secteurs confondus - montre bien qu’il n’y a pas de déremboursement massif. D’ailleurs, on ne cherche pas à avoir un effet fort sur les prix mais à adresser un signal. On peut par exemple se dire que, dès lors que la franchise éveillera l’attention, les patients vérifieront s’ils n’ont pas déjà ce médicament chez eux avant de l’acheter. En revanche, il est peu probable que le comportement du prescripteur s’en trouve modifié. L’effet sur les comportements est difficile à apprécier. Il va de soi qu’un déremboursement majeur influe sur la consommation. La seule expérience dont on dispose en la matière est celle du Kentucky, où on a constaté que, quand on diminuait radicalement le taux de remboursement, la consommation diminuait et l’état de santé se dégradait. Toutefois s’il y a un effet structurant quand on réduit de 25 % le taux de remboursement, ce ne sera pas le cas avec des franchises de faible montant comme c’est le cas pour le médicament avec une franchise de 0,50 € par boîte.
Les franchises sont une des approches possibles pour stabiliser le taux de prise en charge de la sécurité sociale, mais il en existe bien d’autres, par exemple l’augmentation du ticket modérateur. Tout dépend en fait de l’importance de l’effort supplémentaire que l’on entend demander aux assurés sociaux. À cet égard, l’émotion suscitée par les franchises est peut-être excessive. Je n’avais d’ailleurs pas non plus compris l’émotion qu’avait provoquée l’annonce du forfait journalier de 18 euros à l’hôpital, mesure logique et de faible incidence financière.
M. Pierre Morange, coprésident : Il est vrai que cet effet est marginal, surtout si on le compare au reste à charge pour l’accueil en établissement pour personnes âgées dépendantes, qui n’est couvert par aucun organisme complémentaire.
M. Bertrand Fragonard : Le Haut Conseil, où l’on cherche un accord, y compris avec des organisations qui sont très rétives à l’idée que l’on touche au taux de remboursement, a quand même admis un certain nombre d’éléments. Il a par exemple évoqué la piste d’un déremboursement des médicaments à vignette bleue à prescription facultative.
M. Pierre Morange, coprésident : Merci pour cette analyse très fine du sujet.
*
Audition de M. Didier Houssin, directeur général de la santé au ministère de la santé, de la jeunesse et des sports, et Mme Danièle Golinelli, adjointe à la sous-directrice politique des pratiques et des produits de santé.
M. Pierre Morange, coprésident : Nous avons le plaisir d’accueillir M. Didier Houssin, directeur général de la santé au ministère de la santé, de la jeunesse et des sports, accompagné de deux collaboratrices.
Mme Catherine Lemorton, rapporteure : Monsieur le directeur, nous aimerions connaître les domaines de compétence de la direction générale de la santé en matière de médicaments, en particulier délivrés en ville. Comment est-il géré ? Comment expliquer la surconsommation de médicaments en France par rapport aux autres pays européens ?
M. Didier Houssin : Merci de votre invitation.
La direction générale de la santé a un rôle assez général en matière de santé publique. Son intervention sur le champ du médicament se fait essentiellement à travers la notion d’intérêt de santé publique. Bien sûr, elle joue un rôle en matière de réglementation et de discussion au niveau européen sur les aspects réglementaires, mais c’est son rôle en matière de santé publique qui est le plus caractéristique.
Elle cherche à s’assurer que certaines populations qui n’ont pas accès à certains médicaments puissent y accéder, et à faire en sorte que l’on prenne en compte, dans le processus qui conduit au remboursement du médicament la notion d’intérêt de santé publique. Nous partageons tout à fait l’analyse qui a été faite par la Cour des comptes en ce domaine : cette prise en compte est sans doute tout à fait insuffisante aujourd’hui.
Par ailleurs, elle est amenée, pour des raisons de sécurité sanitaire, à travers sa participation au Comité économique des produits de santé (CEPS), à promouvoir le développement des études post AMM – autorisation de mise sur le marché –, qui permettent d’apprécier la manière dont les médicaments fonctionnent dans la vie réelle. Sur ce point, la Cour des comptes a souligné qu’il y avait beaucoup de progrès à faire.
Je ne parlerai pas des aspects qui touchent au médicament en matière de sécurité sanitaire : tout ce qui concerne la préparation aux plans de défense contre le terrorisme ou les menaces de grande ampleur. En ce domaine, la direction générale de la santé a un rôle assez spécifique d’identification, de stockage. De la même façon, elle a un rôle particulier en cas de problèmes majeurs de santé publique générés par l’utilisation de certains médicaments ou l’absence de médicaments ; je pense aux problèmes de vaccinations dans le cas de certaines épidémies. Mais je crois que ce n’est pas l’objet de vos travaux.
Je me concentrerai donc sur la question du médicament dans la pratique habituelle, en particulier sur le point souligné par la Cour des comptes, la surconsommation de médicaments en situation de médecine libérale.
La Cour a en effet remarqué que le circuit qui conduit à l’admission au remboursement des médicaments est insuffisamment sélectif. Nous partageons totalement cette analyse. Selon nous, la commission de la transparence se distingue trop peu aujourd’hui de la commission de l’AMM. Elle s’appuie d’ailleurs sur les dossiers d’AMM et son travail duplique celui de la commission de l’AMM. Elle est composée de professionnels de santé de nature assez voisine de ceux qui peuplent la commission de l’AMM, ce qui explique en grande partie cet état de fait. Nous serions donc très favorables à ce que la composition de la commission de la transparence soit nettement modifiée, dans un sens beaucoup plus tourné vers la prise en compte de l’intérêt de santé publique. Quelle sera la population cible pour l’usage de ce médicament ? Comment mieux prendre en compte l’impact de ce médicament en termes de mortalité, morbidité, lié à l’usage de ce médicament ? Et surtout, comment mieux prendre en compte en compte la dimension médico-économique ? En effet l’équilibre financier du système d’assurance maladie est pour la direction générale de la santé un objectif majeur en termes de santé publique. C’est ce dispositif qui garantit aujourd’hui l’égalité d’accès aux soins.
Il est exact que la surconsommation de médicaments a un grand impact, qui ne fait que s’accentuer, sur le plan économique. Nous voudrions donc que la dimension médico-économique soit beaucoup mieux prise en compte par la commission de la transparence, par exemple lorsqu’un médicament représente un intérêt sanitaire marginal par rapport à un produit existant, mais qu’il revient beaucoup plus cher.
A-t-on besoin d’une commission de la transparence, dans son fonctionnement actuel ? On pourrait imaginer qu’il suffirait d’élargir un peu le champ de vision de la commission de l’AMM pour qu’elle puisse s’occuper du service médical attendu. La commission de l’AMM remplirait ainsi le rôle que joue aujourd’hui la commission de la transparence. Pourquoi pas, à la condition qu’au sein de la HAS, ou à côté, on crée une structure qui concentrerait son activité autour de la notion de santé publique.
Pour alimenter cette réflexion et cette décision sur l’intérêt de santé publique, il faut des gens qui aient une compétence en matière de santé publique et une compétence médico-économique. Il faudrait aussi que cette structure puisse s’appuyer sur des données. Or aujourd’hui, la commission de la transparence n’a pas beaucoup de données complémentaires par rapport à la commission de l’AMM. Si on voulait que l’intérêt de santé publique soit mieux pris en compte, il faudrait que, très en amont, avec les industriels, lors du dépôt de la demande d’AMM, on puisse disposer d’études beaucoup plus précises pour répondre aux questions suivantes : quelle est la population cible ? Comment se présentera la dimension médico-économique ? Comment aborder la question vis-à-vis des produits existants par ailleurs ? Toutes ces questions sont assez peu documentées actuellement.
Voilà vers quoi nous aimerions aller. Je ne méconnais pas les difficultés que nous pouvons rencontrer dans ce sens. Dans le court terme, il est sûr que la manière dont sera composée et présidée la commission de la transparence sera déterminante. Nous souhaitons voir celle-ci s’infléchir d’ores et déjà de plus en plus vers la prise en compte de l’intérêt de santé publique.
Autre question : la prescription peu encadrée. Aujourd’hui, un des facteurs de surconsommation réside très certainement dans le fait que la mécanique de l’exercice libéral est très propice à l’accentuation de la prescription. On pourrait imaginer que dans une mécanique différente de l’exercice médical, qui permettrait un meilleur équilibre entre la prise en compte des aspects de santé publique, de prévention et la prise compte des aspects de soins, le moteur qui conduit à une prescription importante serait freiné.
Des questions importantes se posent s’agissant du mode de rémunération des médecins. La prescription à l’acte est un facteur déterminant de l’accentuation importante de la prescription de médicaments. Si l’on veut s’attaquer à la racine de la surconsommation, il faudra s’intéresser à la manière dont s’organise la relation entre le médecin et le malade, en particulier au mode de rémunération de l’exercice médical.
Autre aspect de la surconsommation : la formation et l’information des médecins.
Je ne suis pas sûr que, dans ce domaine, on puisse attendre des miracles. Les médecins sont aujourd’hui exposés à une information envahissante, multiforme. Il est donc très difficile de décider d’accentuer l’information sur les médicaments. Des progrès ont été faits, mais c’est un travail de longue haleine, qui passera peut-être par la formation des médecins pour qu’au fil des années, la dimension de santé publique et la dimension médico-économique soient prises en compte. Actuellement, dans sa prescription, un médecin se demande rarement si tel médicament est plus cher que tel autre. C’est une question très difficile.
La direction générale de la santé joue par ailleurs un rôle de coordinateur entre les très nombreux acteurs : AFSSAPS, HAS, assurance maladie, Comité économique des produits de santé. Je pense notamment aux études post AMM, qui ne sont pas assez développées en France. Nous avons créé un comité de liaison pour avoir une approche la plus rassemblée possible dans ce domaine. Des sujets très « chauds » se sont fait jour dernièrement, comme le vaccin contre le virus HPV – responsable du cancer de l’utérus.
M. Pierre Morange, coprésident : Vous parlez du groupe d’intérêt scientifique d’évaluation épidémiologique des produits de santé ?
M. Didier Houssin : Ce groupe d’intérêt scientifique a été en effet créé pour favoriser le développement des études post AMM et permettre l’accès aux bases de données. En effet, on ne dispose pas en France d’accès facile à une base de données publique permettant de conduire aisément des études post AMM. Mais il se trouve que le GIS ne nous a pas permis de percée spectaculaire dans ce domaine.
Mme Danièle Golinelli : Trois études ont effectivement été mises en œuvre à travers ce groupe d’intérêt scientifique, mais celui-ci n’a pas fonctionné comme nous l’aurions souhaité, pour des raisons techniques. Nous avons interrogé la Caisse nationale d'assurance maladie des travailleurs salariés (CNAMTS) à ce propos. Il faut reconnaître que le recueil des données fait par les caisses d’assurance maladie n’est pas destiné, au départ, à renseigner des études épidémiologiques. Il faudrait des moyens assez considérables pour modifier l’orientation et faire en sorte que ce recueil puisse être utilisé dans le cadre de ces études. C’est un frein important.
M. Pierre Morange, coprésident : Dans la prochaine audition, nous écouterons à la fois les représentants de la Caisse nationale d'assurance maladie des travailleurs salariés (CNAMTS), de la Mutualité sociale agricole (MSA) et du Régime social des indépendants (RSI). Il existe tout de même un dispositif d’information avec des logiciels et des structures informatiques qui s’inscrit dans cette perspective. Et il ne serait pas inintéressant de vous adresser à d’autres partenaires. La MSA et le RSI peuvent fournir des bases de données. Ils ont certainement développé de façon plus précoce une structuration ancienne et mené des expériences intéressantes à cet égard. La MSA a, par exemple, développé un outil d’analyse des prescriptions dénommé ARCHIMED.
M. Didier Houssin : Étant donné les difficultés auxquelles on se heurte du côté de la CNAMTS, il serait peut-être utile – c’est l’une des préconisations du rapport en cours de finalisation de M. Bernard Bégaud, établi à la demande de la direction générale de la santé et l’AFSSAPS, intitulé « La France face au défi de l’évaluation des médicaments après leur mise sur le marché et la gestion des risques » – de se tourner vers la MSA, sans doute plus accessible, techniquement, s’agissant de l’accès à ses bases de données. Cela dit, il existe aujourd’hui d’autres méthodes.
M. Pierre Morange, coprésident : Vous nous avez dit que trois études avaient débouché sur une mise en œuvre. De quoi s’agissait-il ?
Mme Danièle Golinelli : En raison de ces importantes difficultés, nous n’avions pas prévu de faire plus de quatre ou cinq études par an. Il s’agit d’études de suivi des médicaments, dans leurs conditions réelles de prescription. Elles doivent servir à fonder la réflexion de la commission de la transparence lorsque celle-ci doit réévaluer les médicaments et apprécier le service médical rendu au bout de quelques années de consommation – cinq années, en l’occurrence.
M. Pierre Morange, coprésident : Est-il possible, dans ce délai de cinq ans, d’apprécier l’ensemble des molécules à disposition ?
M. Didier Houssin : Cela n’est pas impossible, mais il faut bien reconnaître que jusqu’à présent, peu de choses ont pu être mises en route. Le processus est de toutes façons long et coûteux. On peut d’ailleurs s’interroger sur la pertinence de ce type de méthode. D’autres méthodes existent en matière de pharmacoépidémiologie, qui seraient peut-être plus fructueuses et plus rapides. Je pense aux études cas témoins.
Les études post AMM consistent à suivre une population dans la vraie vie, et à compter ce qui s’est passé au terme d’un certain délai. D’autres types d’études consistent à partir de l’hypothèse qu’il peut survenir telle ou telle pathologie : par exemple la sclérose en plaques chez des sujets jeunes, après une vaccination. On peut alors construire une étude cas témoins, à partir d’un groupe de témoins et d’un groupe de patients traités par le vaccin. Cela permet de faire émerger beaucoup plus rapidement le sur-risque qui pourrait être lié à l’utilisation du vaccin. On peut faire la même chose s’agissant de médicaments qu’on soupçonne de comporter des risques cardiovasculaires, ou de diabète, etc.
Une telle approche, plus ciblée sur des hypothèses de départ, a l’avantage d’être plus rapide et d’éviter de s’engager dans des études à très grande échelle, alors même qu’on redoute tel ou tel inconvénient. Aujourd’hui, peu de personnes ou de groupes sont capables de faire des enquêtes cas témoins, mais elles vont sûrement se développer.
Mme Catherine Lemorton, rapporteure : Je reviens sur la prescription peu encadrée de la médecine libérale. Pour l’instant, les prescripteurs sont essentiellement informés sur les médicaments par les laboratoires eux-mêmes, par la visite médicale. Pensez-vous que les délégués de l’assurance maladie, les DAM, peuvent avoir une action positive en termes de prescription de médicaments ? Pensez-vous que leur formation doive être améliorée ?
M. Didier Houssin : L’industrie pharmaceutique a une grande force motrice, à travers la visite médicale, très professionnalisée, très présente et très équipée. D’où une promotion considérable de la prescription. L’assurance maladie a tenté de réorienter certains de ses agents, qui sont chargés d’éclairer les médecins sur leur pratique. C’est une bonne idée. Cela permet aux médecins, à partir de documents relativement simples, de se situer, de comparer leur pratique de prescription avec celle de leurs collègues, et éventuellement de la modifier.
Est-ce que les DAM ont été convenablement formés ? Il est un peu tôt pour le dire, car ils ne sont installés que depuis deux ans. Il serait intéressant de tenter d’évaluer l’impact de leur action et de voir, éventuellement, ce qu’il faudrait faire pour améliorer leur efficacité.
M. Pierre Morange, coprésident : Avez-vous réfléchi sur les schémas organisationnels ? Avez-vous un éclairage européen sur des modèles qui pourraient s’avérer plus efficients ?
M. Didier Houssin : Le sujet est vaste. On peut certainement s’interroger à propos de l’information sur le médicament. Mais ces questions ne sauraient être totalement retirées à l’AFSSAPS, qui se trouve au point de départ de l’usage du médicament et doit jouer un rôle majeur en la matière.
Le rôle de la HAS est plutôt orienté vers la stratégie thérapeutique et l’insertion du médicament dans un ensemble thérapeutique. Il faut donc organiser la complémentarité, ce qui n’est pas très facile. J’ai d’ailleurs souligné à plusieurs reprises que la question de l’indépendance de la HAS m’avait semblé problématique.
M. Jean Mallot, coprésident : La multiplication d’agences plus ou moins indépendantes, de commissions, a abouti à une certaine dilution des responsabilités et à la dépossession de l’État de son rôle d’arbitre. Il faut trouver un équilibre. N’est-on pas allé un peu loin dans la dispersion, la perte de lisibilité du dispositif et la perte de responsabilités ?
M. Didier Houssin : Oui et non. La création des agences a été un processus réactionnel dans le domaine de la sécurité sanitaire, la réponse à des problèmes. De ce point de vue, l’État s’est renforcé, il s’est doté de bras armés capables d’agir et a amélioré le service qu’il pouvait rendre à la population.
Aujourd’hui cependant, la situation est un peu parcellisée, avec une collection d’agences qui, de l’extérieur, peut sembler compliquée. Il n’est pas exclu qu’un jour on décide de rassembler ce qui est épars, dans un ordre un peu différent.
S’agissant du médicament, la question se pose entre l’AFSSAPS, la HAS, l’assurance maladie et le CEPS : faut-il instituer un dispositif plus simple ? S’agissant d’autres domaines intéressant la sécurité sanitaire, la question peut également se poser. Je ne suis pas sûr qu’il faille une agence unique, en raison des spécificités. Dans les années à venir, on aura à prendre conscience de cette diversité et il faudra trouver des arrangements.
Mme Catherine Lemorton, rapporteure : Comment envisagez-vous de sanctionner les laboratoires qui tardent à produire des études post AMM ?
M. Didier Houssin : Ces études sont difficiles, longues et coûteuses. On ne peut pas s’attendre à des résultats immédiats, mais il est vraisemblable que des laboratoires traînent un peu les pieds en s’appuyant sur les difficultés qui se posent en termes de méthode. Serons-nous capables de discerner ceux qui ont effectivement des difficultés et ceux qui traînent les pieds ? Nous pensons qu’il faut prévoir un dispositif de sanctions. La question est de déterminer les critères qui permettront de décider de ces sanctions.
Néanmoins j’aurais tendance à dire que, plutôt que de sanctionner, il faudrait faire un gros effort d’anticipation pour régler le problème des études post AMM. Il faudrait aborder d’ores et déjà la question du médicament dans la vraie vie et celle de l’intérêt de santé publique.
M. Jean Mallot, coprésident : Quels sont les leviers sur lesquels on peut jouer pour faire évoluer les comportements des consommateurs ?
M. Didier Houssin : Le consommateur a une grande part de responsabilité, en effet. Si on analysait, par une approche humaine et sociale, les mécanismes intimes de la prescription, on trouverait chez l’usager une accentuation du désir de consommation au fil des années. Cependant on trouverait aussi chez le médecin le sentiment que la prescription est devenue, au plan concret, son unique mode de réponse. Dans une situation de diagnostic difficile, ce peut être le moyen de reporter le problème, et lorsqu’un patient vient en demandant un scanner, il le prescrit.
M. Pierre Morange, coprésident : Quel est le domaine de compétence de l’AFSSAPS en matière de traçabilité et de sécurité sanitaire, s’agissant des filières de production de médicaments dans certaines zones où le contrôle est plus malaisé et où les capacités et les critères de vérification et d’expertise ne sont pas les mêmes que sur le sol français ? Y a-t-il des mesures à prendre ? Car le risque ira toujours croissant.
M. Didier Houssin : Je partage votre analyse. Nous avons eu l’occasion d’évoquer ces questions avec M. Jean Marimbert.
Dans les années à venir, nous allons être confrontés à un problème majeur en termes de risques sanitaires liés aux médicaments, du fait du développement de certaines organisations ou pratiques. Je pense notamment aux opérateurs des pays émergents, qui vont prendre de l’importance dans le domaine de la fabrication des médicaments génériques. Cela rend plus difficile les contrôles et la vérification de la traçabilité, ne serait-ce que celle des matières premières.
Autre problème : celui des circuits de distribution. On voit se développer à l’étranger des importations parallèles de médicaments. La France est encore relativement préservée, en raison de son circuit d’officines, de son monopole pharmaceutique et d’une industrie pharmaceutique solide. Mais cela ne va pas durer. Sans compter le problème d’Internet.
Voilà pourquoi il me semble que nous allons devoir gérer des risques sanitaires liés aux médicaments mal faits, non intentionnellement ou intentionnellement.
M. Pierre Morange, coprésident : Sans parler des mécanismes de contrefaçon industrielle.
M. Didier Houssin : Au moment de la présidence allemande, j’avais beaucoup insisté pour que le thème de la contrefaçon du médicament soit mis à l’ordre du jour. Le sujet est abordé au niveau européen et il concerne la Commission. Ce sera l’un des enjeux de l’Europe dans les années à venir que de prévenir les risques liés à la sécurité du médicament. En l’occurrence je vise ses aspects de base, à savoir le produit lui-même.
M. Pierre Morange, coprésident : Il est évident que la question est de dimension européenne. La France se trouve encore dans une bulle sécurisée, mais celle-ci peut se fragiliser à tout moment. A-t-on planifié les travaux sur cette question au niveau européen ?
Mme Danièle Golinelli : Pour le moment, il n’y a pas de réflexion très formalisée sur la contrefaçon. Néanmoins, ce sujet est évoqué dans le cadre du Forum pharmaceutique européen d’une manière générale.
M. Didier Houssin : Nous n’avons pas encore été confrontés à des situations dramatiques. Il n’est pas facile de mettre, d’avance, des dispositifs en place. Toutefois viendra un jour où on se reprochera de n’avoir pas su anticiper. Il faudrait, au niveau européen, être plus prégnant sur ce sujet.
Mme Danièle Golinelli : Des réseaux ont tout de même été mis en place. L’AFSSAPS y participe. Il s’agit de réseaux de systèmes d’inspection dans les États membres. Un groupe se réunit régulièrement pour mettre en commun un certain nombre d’éléments relatifs à la contrefaçon du médicament.
M. Pierre Morange, coprésident : L’AFSSAPS a mis en œuvre plusieurs actions en coopération avec un certain nombre d’États de différents continents, afin de faire partager cette culture de sécurité sanitaire. Une démarche similaire a-t-elle été envisagée à l’échelle européenne, pour sécuriser certains sites de production très fragiles ?
M. Didier Houssin : L’AFSSAPS, dans le cadre de ses relations avec l’Agence européenne du médicament et avec les autres agences nationales, travaille sur ce type de projet. Je me demande si la question ne devrait pas être envisagée à un niveau encore plus large.
En matière de sécurité sanitaire s’agissant du médicament, mais aussi du dépistage des épidémies, la formation est un enjeu majeur au niveau mondial : formation des cadres, des dirigeants de laboratoires, des agences nationales. Le pôle de Lyon que la France anime autour de l’OMS pourrait être un lieu de formation de ces cadres pour les pays émergents et en voie de développement. Mais la question doit être abordée à l’échelon de l’OMS et au niveau mondial.
Mme Danièle Golinelli : L’OMS s’est penchée sur ces problèmes de contrefaçon. Elle a mis en place il y a deux ans le Groupe Impact, dans lequel sont représentés les pays ainsi que l’Union européenne, qui contribue à ses travaux. Il y a donc d’assez nombreuses réflexions sur le sujet. L’idée était d’aboutir à des instruments assez contraignants. Mais ensuite, se posent des difficultés de mise en œuvre dans les différents pays.
Mme Catherine Lemorton, rapporteure : Que pensez-vous du fait que l’AFSSAPS collecte les taxes sur les laboratoires ? Pourquoi ne serait-ce pas l’URSSAF ou l’administration fiscale qui s’en chargerait ? Je m’interroge en effet sur l’indépendance de décision de l’agence.
M. Didier Houssin : On voit très bien les réflexions que cela peut susciter : l’agence risque de ne plus être indépendante des laboratoires. Dès lors qu’elle est financée par tous les laboratoires, je ne pense pas que le problème se pose de manière aussi aiguë. Malgré tout, il faut une limite. L’État doit jouer son rôle et contribuer.
Autant l’indépendance des experts est un sujet majeur sur lequel nous avons beaucoup travaillé ces derniers mois, autant le financement d’une agence par l’ensemble des laboratoires ne me paraît pas un élément à même de compromettre l’indépendance de celle-ci.
Aujourd’hui, nous nous heurtons à de grandes difficultés en matière d’expertise, comme dans d’autres pays. Aux États-Unis, l’affaire a pris des proportions importantes. On y a écrit de nombreux ouvrages et de nombreux articles dans les revues médicales sur la manière dont l’articulation se faisait entre les laboratoires et les experts. C’est pourquoi la direction générale de la santé a produit un projet de rapport : « Indépendance et valorisation de l’expertise », lequel est actuellement examiné par l’ensemble des agences sanitaires et des organismes de recherche apparentés, comme l’INSERM. L’idée est de valoriser des mécanismes permettant de renforcer encore l’indépendance de l’expertise, par exemple : code de déontologie ; contrôle de l’indépendance des experts une fois qu’ils auraient déclaré d’éventuels conflits d’intérêts. Cela fait partie des propositions que nous ferons au ministre.
Se pose aussi la question de la valorisation de l’expertise. On constate aujourd’hui, qu’il s’agisse de la carrière des chercheurs ou des enseignants chercheurs, que le travail d’expertise, qui peut-être très lourd et représenter une grande responsabilité par les conséquences qu’il risque d’avoir en termes de santé publique, n’est pas considéré comme très sérieux, à l’égal d’une publication originale ou d’un brevet. Nous sommes donc en train de travailler avec le Conseil national des universités et la Conférence des présidents d’université et des organismes de recherche pour faire en sorte que, petit à petit, sur des critères qui restent à définir, le travail d’expertise sanitaire soit mieux valorisé dans le secteur public.
M. Pierre Morange, coprésident : Monsieur le directeur général, je vous remercie. Bien sûr, de nombreuses questions n’ont pas pu vous être posées. Nous serons donc très attentifs aux propositions que vous pourriez nous faire et que nous pourrions intégrer au cadre législatif.
*
Audition de MM. Frédéric Van Roekeghem, directeur général de l’Union nationale des caisses d’assurance maladie (UNCAM) et de la Caisse nationale d’assurance maladie des travailleurs salariés (CNAMTS), Jean-Pierre Roblet, directeur de l’offre de soins, Yves Humez, directeur général de la Mutualité sociale agricole (MSA), et Pierre-Jean Lancry, directeur de la santé, Dominique Liger, directeur général du Régime social des indépendants (RSI), et Philippe Ulmann, directeur de la politique de santé et gestion du risque.
M. Pierre Morange, coprésident : Je souhaite la bienvenue aux représentants de la CNAMTS, de la MSA et du RSI, et je passe immédiatement la parole à madame la rapporteure.
Mme Catherine Lemorton, rapporteure : Je vous demanderai d’abord, messieurs, pourquoi les caisses d’assurance maladie acceptent de rembourser des médicaments ou des me too mis sur le marché afin de contourner les génériques, surtout lorsque leur prix est supérieur à celui de ces derniers. La décision est-elle prise de façon collégiale au niveau des trois caisses ?
M. Frédéric Van Roekeghem : En la matière, Mme Rolande Ruellan a déjà pu préciser lors d’une audition précédente, ici même, qu’il n’existe pas de possibilité juridique de ne pas rembourser un médicament dès lors que celui-ci a reçu l’autorisation de mise sur le marché – l’AMM – par l’AFSSAPS et que le service médical rendu a été évalué par la commission de la transparence. Dans ce contexte, le Comité économique des produits de santé (CEPS) négocie les prix, et il n'est pas fondé à refuser d’attribuer un prix. À partir du moment où la discussion aboutit au sein du CEPS, au sein duquel nous sommes tous représentés, les procédures réglementaires font obligation au directeur général de l’UNCAM de fixer un taux, sa seule marge étant de ne pas fixer de taux aux médicaments à SMR insuffisant, faute de texte précis en la matière. La décision est alors renvoyée au ministère de la santé.
Mme Catherine Lemorton, rapporteure : La promotion des génériques auprès de la population représente un travail énorme. Leur contournement par des spécialités à SMR égal est décourageant.
M. Frédéric Van Roekeghem : Notre pays a fait le choix politique de proposer au remboursement tous les médicaments, ce qui présente des avantages – aucun frein n'est mis à l’arrivée de médicaments –, mais également des inconvénients. Reste la question de la lisibilité de la politique tarifaire, notamment avec les remises arrière.
M. Pierre Morange, coprésident : Quelle appréciation portez-vous sur les nouveaux horizons qui s’ouvrent à la Haute Autorité de santé avec l’article 29 du projet de loi de financement de la sécurité sociale pour 2008, qui prévoit de lui attribuer une capacité à formuler des avis et des recommandations d’ordre médico-économique ? Quelles en seront les conséquences sur les différentes missions de la CNAMTS et des autres partenaires assurantiels ?
M. Frédéric Van Roekeghem : Le problème ne tient pas à l’élargissement des missions de la HAS en matière médico-économique, évolution que nous soutenons, mais à sa capacité à rendre des avis non pas fondés sur une approche globale, mais qui soient lisibles et applicables sur le terrain. L’autorité allemande, l’IQWIG, par exemple, a émis un avis très clair sur les médicaments anticholestérol en privilégiant des médicaments moins onéreux que d’autres, tout en étant aussi efficaces. De même, la Grande-Bretagne, vient d’aborder le sujet de l’efficacité thérapeutique des médicaments pour les patients en Alzheimer en phase avancée. Là encore, on voit la distance qui nous sépare des systèmes voisins.
M. Yves Humez : Nous sommes, pour notre part, des opérateurs du remboursement engagés dans la maîtrise médicalisée. À cet égard, il nous faut des dispositifs simples, clairs et applicables.
M. Pierre-Jean Lancry : Le CEPS ne prend pas en compte les études médico-économiques, mais il s’intéresse à l’impact budgétaire. Selon la formule du président du Comité : « Si au prix que vous demandez, le produit est efficace, il le sera encore plus au prix que je vous donne. »
Néanmoins, si l’article 29 du projet de loi de financement de la sécurité sociale pour 2008 est voté, se posera alors la question du lien entre le travail de la HAS et celui du CEPS, car, théoriquement, c’est ce dernier qui a en charge les aspects médico-économiques du médicament.
M. Pierre Morange, coprésident : C’est bien pour que les choses soient parfaitement claires en ce domaine, que l’article en question a, me semble-t-il, été écrit.
M. Dominique Liger : Nous sommes d’accord avec nos collègues s’agissant de l’opposabilité des décisions de la HAS...
M. Pierre Morange, coprésident : Il ne s’agit que d’avis ou de recommandations.
M. Dominique Liger : ...mais je ne vois pas, pour le moment, comment le système pourrait fonctionner.
M. Pierre Morange, coprésident : Sans faire un plaidoyer pro domo, l’avantage du dispositif est de permettre à la HAS de formuler des avis et recommandations sur des bases scientifiques, tout en prenant en compte l’incidence médico-économique du médicament. Il faut bien aux responsables politiques et assurantiels un éclairage objectif et indépendant afin de pouvoir prendre une décision.
M. Frédéric Van Roekeghem : Avant de poser la question de l’opposabilité, il faut être certain de l’efficacité du système et, en particulier, de l’applicabilité des recommandations. C'est pourquoi nous privilégions plutôt une approche pragmatique qui permette assez vite une application concrète, vu l’ampleur du déficit.
M. Philippe Ulmann : Dans les autres pays, on assiste – avec NICE en Grande-Bretagne ou encore avec l’IQWIG en Allemagne – à la mise en place d’agences qui ont la capacité de faire des recommandations à partir d’une approche de type micro-économique et fondées sur une comparaison coût/efficacité d’un médicament par rapport à d’autres. Une approche globale risquerait de déboucher sur des recommandations trop générales pour avoir une réelle utilité.
Néanmoins, ces agences s’appuient sur une expertise publique. Or, pour des raisons budgétaires, la capacité de la France à mobiliser des équipes indépendantes de chercheurs ou d’universitaires est très limitée. On reproche à l’industrie pharmaceutique de ne publier que les études qui l’arrangent, mais les pouvoirs publics ne se sont jamais dotés des moyens d’une contre-expertise publique.
La question porte donc à la fois sur l’applicabilité et sur l’éventuelle opposabilité de la démarche. L’expérience a montré que des référentiels potentiellement opposables entraînent des changements de comportement.
Mme Catherine Lemorton, rapporteure : Pour en venir à la formation continue des médecins, la MSA et le RSI ont-ils adopté la même démarche que le régime général avec les délégués de l’assurance maladie- les DAM ?
M. Dominique Liger et M. Yves Humez : Non.
Mme Catherine Lemorton, rapporteure : Quelle est la formation des DAM et quelle est leur mission auprès des cabinets médicaux ? Sont-ils encadrés ?
M. Frédéric Van Roekeghem : Le système des DAM a été mis en place à partir de 2005 pour faire connaître aux différents professionnels de santé la nouvelle convention médicale, notamment les engagements en matière de maîtrise médicalisée. Ces personnels, recrutés par redéploiement au sein de l’assurance maladie, ont vu progressivement leur nombre et leur formation évoluer. Alors qu’au début ils étaient 450, ils devraient être environ 950 à la fin de l’année, l’objectif étant de certifier 350 d’entre eux fin 2007, tous devant, au final, répondre au standard applicable dans ce domaine.
Les matières abordées ayant un contenu médical, il n’était pas souhaitable de laisser cette activité se développer de façon trop aléatoire. C'est pourquoi une équipe a été constituée, dédiée aux produits de santé et comportant trente personnes dirigées par un pharmacien-conseil. Toutes les visites des DAM font l’objet d’un cahier des charges. Les laboratoires pharmaceutiques soumettent d’ailleurs la Caisse à un examen extrêmement attentif pour des raisons à la fois d’image du produit et de respect de la concurrence. Dans ces conditions, devoir à la fois analyser la littérature internationale, notamment anglo-saxonne, s’assurer des conditions des AMM, et vérifier que toutes les réglementations sont respectées lorsque les DAM se rendent sur place, nécessite une organisation extrêmement précise.
La formation des délégués repose sur un dispositif de formation de 300 heures théoriques et méthodologiques avec une activité professionnelle encadrée de 900 heures, sachant que nous avons « benchmarké » avec les visiteurs médicaux. En outre, le processus de validation de la certification professionnelle repose sur cinq outils de validation de compétences, et la décision a été prise de professionnaliser la fonction de manager de DAM.
Il existe, par ailleurs, une articulation entre les DAM et les praticiens-conseils dans certaines entreprises du secteur privé de manière que lorsque les premiers sont confrontés à une question à contenu médical important, ils puissent la renvoyer aux seconds. La médicalisation de l’action de l’assurance maladie reposera d’ailleurs de plus en plus sur un rapprochement entre les équipes médicales et administratives. Nous avons ainsi créé, fin 2005, en accord avec le médecin-conseil national, des équipes pluridisciplinaires.
M. Pierre Morange, coprésident : Le nombre de 950 DAM sera-t-il un maximum ?
M. Frédéric Van Roekeghem : Notre convention d’objectif et de gestion prévoit la suppression de 4 500 emplois, après les 3 000 de la période 2004-2005, mais nous avons obtenu l’accord de l’État de pouvoir augmenter jusqu’à 1 400 à la fin 2009 le nombre de nos DAM par redéploiement au sein des caisses – où le métier de DAM est maintenant considéré comme valorisant –, sans qu’il s’agisse pour autant d’arriver à saturation comme les 200 laboratoires avec leurs 23 000 visiteurs.
Pour ce qui est du taux de refus de visite, il est largement inférieur à celui d’autres systèmes car nous essayons de nous positionner en tant que promoteur non pas du produit mais du bon usage des soins et des ressources communes.
M. Pierre Morange, coprésident : Existe-t-il une volonté de coordination entre vos trois caisses afin d’assurer le meilleur rapport coût/efficacité des soins ? Qu’en est-il, par exemple, avec le dispositif ARCHIMED de la MSA ?
M. Yves Humez : Le logiciel ARCHIMED est un outil approprié à notre problématique puisque les 8 % de la population que nous assurons sont répartis sur tout le territoire, contrairement au régime général qui s’adresse à l’ensemble des assurés. Ce logiciel nous permet, par exemple, d’observer les prescriptions abusives, et, à cet égard, je considère que nos médecins-conseils ont une action complémentaire à celle menée par les DAM. Cela n’empêche pas que nous partagions, au sein du collège des directeurs, les mêmes enjeux en matière de maîtrise médicalisée.
M. Frédéric Van Roekeghem : La MSA et le RSI disposent d’équipes médicales et administratives intégrées ce qui, historiquement, n’est pas le cas du régime général. Nous avons cependant arrêté une orientation de notre réseau qui privilégie un tel rapprochement. En outre, la MSA dispose d’un système interne informatique avec des niveaux d’accès différents et sécurisés, ce que la Caisse nationale d’assurance maladie doit mettre progressivement en place. Ce qui est possible au sein du RSI et de la MSA ne l'est donc pas toujours au sein du régime général, mais nous nous voyons régulièrement pour confronter nos points de vue sur des enjeux importants. L’historique des remboursements, par exemple, est commun aux trois caisses.
Mme Catherine Lemorton, rapporteure : Les chiffres de la consommation des médicaments sont-ils comparables entre les trois régimes ?
M. Frédéric Van Roekeghem : Les populations concernées ne sont pas tout à fait comparables. Or si l’on veut comparer, il faut le faire à « patientèle » comparable. Pour notre part, nous menons des travaux sur la segmentation des patients.
M. Yves Humez : D’autres paramètres, notamment territoriaux, sont à prendre en compte, et c'est en les croisant que l’on pourra mettre en évidence les meilleures pratiques et s’aligner sur elles.
M. Philippe Ulmann : Une réunion des directeurs doit avoir lieu à la fin du mois justement pour essayer de mettre en œuvre des comparaisons entre les régimes. La population visée par le RSI consomme, par exemple, beaucoup de médicaments – c’est le second poste de nos dépenses après celui de l’hospitalisation –, mais comme la consommation de soins est un peu moins forte dans notre régime que dans les autres, il faut relativiser cette donnée.
M. Frédéric Van Roekeghem : En matière de générique, la MSA était pour sa part apparue un peu meilleure que le régime général.
M. Yves Humez : La MSA dispose de sept délégués dans chaque canton. C’est un moyen de communication important qui explique que nos campagnes de prévention aient un bon retour, par exemple en matière de génériques.
M. Pierre Morange, coprésident : De nombreux brevets de molécules blockbuster sont tombés récemment dans le domaine public. Une étude a-t-elle été faite sur le gisement d’économies potentielles recélé par les molécules qui tomberaient dans les trois prochaines années dans le domaine public ? Par ailleurs, où en est-on du dossier pharmaceutique ?
M. Frédéric Van Roekeghem : Des évolutions sont intervenues au sein de la CNAMTS depuis 2004. En particulier, ont été mis en œuvre, d’une part, un suivi des actions grâce à un système de reporting performant – nous avons ainsi été capables de surveiller les ventes de Tamiflu au jour le jour –, et, d’autre part, un mécanisme d’anticipation permettant de prévoir, par exemple, quels seront les prochains blockbusters.
M. Pierre Morange, coprésident : Disposez-vous de chiffres en la matière ?
M. Frédéric Van Roekeghem : De mémoire uniquement. Nous vous les fournirons par écrit.
Le problème ne tient pas seulement à la tombée des molécules dans le groupe générique, mais à l’effet de fuite qui en découle. Dès lors qu’une molécule est génériquée, le mix produit se révèle beaucoup plus coûteux en France qu’ailleurs en Europe, du fait de cet effet de fuite important.
M. Pierre-Jean Lancry : Dans l’ensemble, les génériques ont un coût de 30 % supérieur en France.
M. Pierre Morange, coprésident : Pourquoi ?
Mme Catherine Lemorton, rapporteure : Du fait des marges arrière.
M. Frédéric Van Roekeghem : Nous avons tout de même baissé récemment le prix du générique de façon importante. Par ailleurs, la CNAMTS a proposé d’expérimenter, dans le cadre du CEPS, une sorte de mise en concurrence de médicaments génériques dans certaines classes.
M. Pierre Morange, coprésident : Qu’en est-il du dossier pharmaceutique ?
M. Frédéric Van Roekeghem : Nous ne sommes pas pour notre part en charge de ce dossier. Néanmoins sa mise en place est soutenue notamment par le fonds d’aide à la qualité des soins de ville, qui a dégagé une enveloppe financière à cet effet. Ce dossier nous semble complémentaire de l’historique des remboursements.
Mme Catherine Lemorton, rapporteure : Avez-vous constaté des changements de comportement de la part des médecins après les visites des DAM ?
M. Frédéric Van Roekeghem : Pour prendre l’exemple des statines, qui ont donné lieu à des visites, mais également, comme tous les cas d’intervention plus médicalisée, à des entretiens confraternels, le taux de croissance de la consommation des statines a été notablement infléchi suite au plan médicament de 2005. Au Royaume-Uni au contraire, où les statines dosées à dix milligrammes ont été mises en vente libre, leur consommation, qui était en 2002 bien en deçà de la nôtre, l’a aujourd’hui dépassée. Pour autant, les patients qui, en France, ont besoin de ces médicaments ne sont pas pénalisés, puisque le taux de couverture par les statines des patients diabétiques, par exemple, continue de croître.
M. Jean-Pierre Roblet : Nous procédons à un suivi des visites à la fois quantitatif et qualitatif afin de déterminer leur impact sur les prescriptions et leurs conséquences financières. Que l’étude soit menée en interne ou en externe, comme avec IPSOS pour les pharmaciens, les résultats montrent que l’objectif de la convention, qui a été le fait générateur de la maîtrise en encourageant les engagements individuels, est beaucoup plus partagé qu’il y a cinq ou dix ans.
M. Frédéric Van Roekeghem : Une enquête BVA menée sur Paris en janvier 2006 auprès des généralistes, des spécialistes et des pharmaciens a montré que 83 % d’entre eux jugeaient que la démarche DAM était plutôt novatrice, 77 % qu’elle était légitime, 70 % qu’elle faisait réfléchir, 70 % qu’elle les intéressait, et 61 % qu’elle les incitait à adapter leur comportement. Il nous faut néanmoins amener ces DAM au meilleur standard de qualité et faire évoluer le contenu de la visite. Nous attendons à cet égard l’avis de la HAS pour médicaliser la visite portant sur les statines.
M. Jean Mallot, coprésident : On ne peut dissocier le prescripteur et le patient. Les caisses se préoccupent-elle de cette interface ?
M. Pierre Morange, coprésident : Nous avons été frappés par l’extrême difficulté de la communication entre l’AFSSAPS, la HAS et l’assurance maladie pour constituer une base de données commune. Où en êtes-vous sur ce sujet stratégique qui vise à mettre à la disposition des prescripteurs comme, éventuellement, de la population, des données certifiées ?
M. Frédéric Van Roekeghem : On ne peut déployer des politiques de gestion du risque efficaces si elles ne sont pas déployées à la fois sur l’offre de soins et sur les patients qui sont les porteurs du risque. Cependant, autant l’action en direction des prescripteurs doit être impérativement coordonnée, autant chacun des régimes est responsable de ses assurés. C'est en ce sens qu’un accord est intervenu très tôt au sein du collège des directeurs : nous agissons sur les mêmes thèmes, mais chacun est responsable du développement de ses actions. Le RSI et la MSA ont ainsi lancé des actions qui ont inspiré la CNAMTS.
M. Yves Humez : Changer les comportements des patients est un travail sur le long terme. À cet égard, nous redoutons toute inversion de politique qui viendrait couper l’élan engagé. Il ne faudrait pas, par exemple – hypothèse qui est évoquée – changer la donne avec les ARS – agences régionales de santé – en bouleversant la dynamique que nous avons lancée.
M. Pierre Morange, coprésident : Qu’en est-il des bases de données ?
M. Frédéric Van Roekeghem : Nous n’en partageons pas avec l’AFSSAPS et avec la HAS, mais il faut bien reconnaître que la question n’a pas été posée.
M. Philippe Ulmann : Nos trois régimes font partie d’un GIP et, dès l’origine, il avait été proposé à l’AFSSAPS de nous rejoindre, ce qui n’a pu se faire.
M. Pierre Morange, coprésident : Le projet est en cours ?
M. Frédéric Van Roekeghem : La question n’a pas été reposée. En tout cas, l’assurance maladie est très attachée à cette base de données publique. Il faut simplement la remettre sur la bonne voie, c’est-à-dire apurer les retards et faire saisir les données par des personnels adaptés.
M. Pierre Morange, coprésident : Un agenda existe-t-il ?
M. Frédéric Van Roekeghem : Oui. Une fiche pourra vous être fournie sur le sujet. De manière générale, il serait souhaitable de se rapprocher notamment de l’AFSSAPS qui dispose d’une base plus détaillée que la nôtre. Ce serait un gain en termes de productivité et d’efficacité.
M. Jean Mallot, coprésident : Une évolution conduisant à des regroupements serait certainement rassurante. Il semble cependant que la stratification se poursuive.
M. Frédéric Van Roekeghem : Il est sûr que la tendance dans notre pays est à la constitution d’institutions de plus en plus nombreuses, sans pour autant supprimer celles présentes antérieurement. Il faut cependant tenir aussi compte des spécificités et de l’histoire. En tout cas, la création du RSI est un exemple de diminution du nombre des institutions.
M. Pierre Morange, coprésident : Le premier thème des réflexions de la MECSS portait justement sur la mutualisation, et le RSI est la matérialisation réussie de ses préconisations, alors que des esprits chagrins avaient jugé cette création impossible. Au-delà de la nécessité de respecter l’histoire, le bon sens veut que la mutualisation et les technologies permettent une harmonisation des procédures afin d’améliorer la rationalité.
Messieurs, je vous remercie. Les questions qui n’ont pas pu vous être posées vous seront adressées par écrit. Nous serons également très attentifs aux propositions que vous pourriez faire.
*
Audition de MM. Dominique Libault, directeur de la sécurité sociale (DSS) au ministère de la santé, de la jeunesse et des sports, et Lionel Joubaud, chef du bureau produits de santé.
M. Pierre Morange, coprésident : Monsieur Libault, je vous souhaite la bienvenue à l’Assemblée nationale pour cette audition qui s’inscrit dans le travail qu’effectue notre mission sur la prescription, la consommation et la fiscalité du médicament. Je donne sans plus tarder la parole à notre rapporteure.
Mme Catherine Lemorton, rapporteure : Pouvez-vous nous rappeler quelles sont les missions de la direction de la sécurité sociale ?
M. Dominique Libault : La direction de la sécurité sociale a pour mission de piloter l’ensemble des questions de sécurité sociale, telles qu’elles se posent à l’État depuis 1945, date de mise en place de celle-ci et de création de la direction. Le champ de la sécurité sociale n’a pas beaucoup changé depuis 1945. Il prend en compte quatre risques : maladie-maternité, vieillesse-veuvage, famille, accidents du travail. Ce pilotage porte sur une masse financière qui se monte pour 2008 à 420 milliards d’euros.
La mission de la direction de la sécurité sociale est triple.
La première est financière et consiste à éclairer les décideurs sur la situation financière de la sécurité sociale et les mesures de nature à remédier à d’éventuels problèmes financiers : pilotage du projet de loi de financement de la sécurité sociale (PLFSS), préparation des rapports de la commission des comptes de la sécurité sociale.
La deuxième mission est de nature plus juridique. Elle consiste à porter les politiques relevant de la sécurité sociale : politiques de la vieillesse, d’accès aux soins, de la famille, de la santé au travail, ainsi que de la maîtrise de la dépense. L’une des raisons d’être de la direction de la sécurité sociale, à laquelle je tiens beaucoup, est de trouver l’équilibre adéquat entre les solutions à apporter aux problèmes financiers et la préservation, voire le développement, des politiques sociales qui, pour moi, sont des éléments fondamentaux de notre pays.
La troisième mission de la direction de la sécurité sociale est d’assurer la tutelle du service public de la sécurité sociale, c’est-à-dire des différentes caisses – Caisse d’assurance maladie des travailleurs salariés (CNAMTS), Caisse nationale des allocations familiales (CNAF), Caisse nationales d’assurance vieillesse des travailleurs salariés (CNAVTS) – et de les piloter à travers les conventions d’objectifs et de gestion.
L’ONDAM, l’objectif national des dépenses de l’assurance maladie, que nous proposons au Gouvernement, comprend des arbitrages entre les différentes sphères de l’assurance maladie qui peuvent se traduire par des sous-ONDAM. Le médicament n’a pas de sous-ONDAM. Néanmoins, il s’inscrit dans une logique et une cohérence globales. Quand on propose des mesures, on cherche un équilibre entre ce qui peut porter sur le médicament, comme sur les rémunérations en ville ou les frais à l’hôpital.
La première question que l’on est en droit de se poser est de savoir quelles sont les marges de manœuvre en matière de prix des médicaments. Certains documents issus de l’industrie pharmaceutique montrent que cette dernière pense que l’on pourrait avoir une progression très élevée de la dépense de médicaments dans les prochaines années. Je ne peux pas partager ce point de vue compte tenu des finances sociales de la France et de la nécessité de rééquilibrer les comptes sociaux. Dans la loi de financement de la sécurité sociale, des prévisions pluriannuelles sont faites. Si l’on veut parvenir en 2010 au rééquilibrage de l’assurance maladie comme le souhaitent les pouvoirs publics, il faut, même avec une politique de ressources dynamique, une croissance, une masse salariale et des recettes satisfaisantes, qu’il y ait une maîtrise de la dépense se traduisant par un ONDAM pas très élevé, en tout cas moins élevé que ce que serait la tendance spontanée des dépenses de santé dans notre pays.
La politique du médicament remboursé doit s’inscrire dans cette stratégie.
Quand on entend les représentants de l’industrie du médicament, on a l’impression qu’ils sont les premiers à souffrir de ces plans. Cela pose la question de la juste mesure. Or on constate que la consommation de médicaments est très forte en France par rapport à d’autres pays et que son poids dans le PIB est plus important. Cela laisse penser qu’il y a encore des marges de manœuvre, tout en veillant à préserver l’accessibilité de tous au médicament, c’est-à-dire à assurer la pérennité de la sécurité sociale, avec des niveaux de remboursement satisfaisants. Cela exige, d’abord, de payer correctement le prix du médicament et, ensuite, de ne pas avoir une consommation excessive par rapport aux besoins de santé de la population. Nous essayons, à travers des propositions de nature législative ou réglementaire, d’édicter des règles du jeu incitant la chaîne du médicament à s’organiser en ce sens.
M. Jean Mallot, coprésident : Considérez-vous que l’État dispose des outils nécessaires pour obtenir l’équilibre dont vous parlez ? Sinon, quels outils faudrait-il ?
M. Dominique Libault : C’est une question pertinente que nous nous posons en permanence. La plupart des personnes que vous avez entendues – dont j’ai lu attentivement les auditions – ainsi que les rapports publiés sur le sujet, notamment celui du Haut conseil pour l’avenir de l’assurance maladie, soulignent que la spécificité de la consommation de médicaments en France est liée à des facteurs pluriels, dont certains sont culturels, liés en particulier à la formation des médecins et à l’éducation à la santé de la population. Agir sur ces facteurs demande des actions dans la durée, dont les résultats ne sont pas immédiats. Il est plus facile de faire une action sur les prix des médicaments à travers le Comité économique des produits de santé qu’une action sur les volumes ou les prescriptions.
Un certain nombre d’outils ont été mis en place, notamment lors de la réforme de 2004. D’autres sont proposés dans le PLFSS pour 2008.
Le bilan de ces outils est mitigé. Je ne peux pas dire que, à ce stade, il y ait des évolutions importantes des comportements. Je suis frappé de voir qu’un certain nombre des résultats que nous avons obtenus ne sont pas toujours dus à l’action directe des médecins. L’un des succès indéniables en matière de changement de comportement, à savoir l’achat de génériques, a ainsi été obtenu en passant par le biais des pharmaciens.
On arrive à un résultat quand il y a une dimension très forte de santé publique, comme cela a été le cas pour les antibiotiques. Quand la prescription d’un médicament est néfaste pour la santé publique, il y a plus d’accroche et de capacité de réponse. Sur des sujets plus économiques, il est plus difficile de faire changer les comportements.
Parmi les outils dont on dispose, il y a, notamment, les délégués de l’assurance maladie et la Haute Autorité de santé (HAS). Dans le projet de loi de financement de la sécurité sociale pour 2008, le rôle médico-économique de la HAS a été renforcé afin d’améliorer le dialogue entre celle-ci et les autres acteurs, notamment l’assurance maladie.
Néanmoins il y a encore beaucoup à faire pour rendre plus efficaces les différents outils qui ont été mis en place. Sans doute en faudra-t-il d’autres, mais il faut d’abord rendre plus opérationnels ceux qui existent déjà et qui ne sont pas sans intérêt.
Mme Catherine Lemorton, rapporteure : Est-ce vous qui déclenchez le Comité d’alerte quand vous sentez un dérapage des comptes ? Êtes-vous en amont ou en aval ? Comment expliquez-vous que le Comité d’alerte ait déclenché l’alerte très tard en 2007, alors que les volumes des médicaments en médecine de ville ont dépassé l’ONDAM dès le mois de mars ?
M. Dominique Libault : La direction de la sécurité sociale n’avise pas le Comité d’alerte. Celui-ci se tient au courant et dispose d’un certain nombre de moyens d’information. Il peut demander des auditions des administrations : la DSS, qui est l’une de ses sources d’information importantes, mais aussi, bien entendu, la Caisse nationale d’assurance maladie. Au vu de ces auditions et de ses informations, le Comité d’alerte décide en toute liberté et indépendance du moment où il juge bon de déclencher l’alerte.
Les prévisions en matière d’assurance maladie sont très compliquées. Il y a eu des signaux de dépassement assez tôt dans l’année, mais l’information la plus intéressante est celle de la consommation en date de soins qui est connue avec deux ou trois mois de retard par rapport à l’événement. Pour connaître les premiers mois de l’année - janvier, février -, il faut attendre deux ou trois mois. Personnellement, je n’ai rien à dire de particulier sur le moment où le Comité d’alerte a déclenché son alerte.
Mme Catherine Lemorton, rapporteure : Quand vous définissez l’ONDAM de l’année à venir, avez-vous tous les éléments pour chiffrer en fonction des nouveaux médicaments de ville qui vont arriver sur le marché, qu’il s’agisse des molécules qui vont tomber dans le domaine public ou des nouvelles molécules promues par les laboratoires ?
M. Dominique Libault : Nous avons une certaine visibilité dans le domaine du médicament, car les processus sont longs. Le Comité économique des produits de santé a une visibilité encore meilleure, puisqu’il suit la chaîne du médicament en permanence.
Cela étant, dire qu’il y a un lien mécanique entre cette visibilité et la construction de l’ONDAM serait excessif. D’abord, il n’y a pas de sous-objectif du médicament en tant que tel. Ensuite, dans ce domaine, comme dans les autres, il faut essayer d’appréhender à la fois l’impact de nouvelles stratégies thérapeutiques ou de nouvelles décisions d’admission au remboursement ou d’amélioration des droits sociaux, et les possibilités d’économies et de maîtrise des dépenses. La construction de l’ONDAM est opérée à partir de l’équilibre entre ces deux points de vue. C’est un exercice difficile.
Nous nous sommes efforcés, dans les annexes du PLFSS pour 2008, notamment dans l’annexe numéro 7, de mieux documenter cet équilibre entre la partie économie et la partie dépenses nouvelles.
On ne peut évidemment pas prétendre que la construction de l’ONDAM soit précise au point de permettre de connaître à l’avance le coût de l’innovation.
Je rappelle également que, même si nous connaissons les molécules qui peuvent arriver, la discussion des prix, souvent, n’a pas eu lieu. Une chose est de connaître qu’une molécule va tomber dans le domaine public, une autre est de connaître son impact réel sur les comptes de l’assurance maladie : cela suppose d’avoir une vue sur le prix et sur la force de pénétration du médicament, c’est-à-dire sa vitesse de substitution aux produits antérieurs. C’est un exercice assez complexe.
M. Pierre Morange, coprésident : Le rapport de la Cour des comptes montre que la fiscalité du médicament est quelque peu anarchique. Les règles qui la définissent ont un caractère changeant, peu propice à l’établissement d’une stratégie de court, moyen et long termes. Or l’industrie pharmaceutique a besoin, du fait de ses contraintes et de sa spécificité, de planifier ses investissements, notamment en recherche et développement. Quand les règles changent tous les ans, il est impossible d’établir une stratégie. Une stabilisation de celles-ci serait nécessaire.
En ce qui concerne la prescription et la consommation des médicaments, une information objective s’impose. Il conviendrait à ce sujet de mieux définir les missions et les actions des différentes agences et des différents acteurs, tant publics que privés, chargés de celle-ci.
Enfin, avez-vous des informations sur la décision qu’a rendue ou que doit rendre le tribunal de grande instance au sujet de la base Thériaque permettant d’alimenter les logiciels à la prescription ?
M. Dominique Libault : Je comprends la demande faite par l’industrie pharmaceutique d’avoir une politique du médicament stable et visible à long terme.
M. Pierre Morange, coprésident : Demande appuyée par la Cour des comptes…
M. Dominique Libault : La politique du médicament a fait de grands progrès depuis quinze ans, et l’industrie pharmaceutique le reconnaît. La politique conventionnelle mise en place à compter de 1994 est très stable dans son fonctionnement, ses modalités et ses objectifs et établit un rapport satisfaisant entre ceux qui fixent le prix des médicaments et l’industrie pharmaceutique.
Les critiques de la Cour des comptes concernant la fiscalité – j’ai lu avec attention l’audition de Mme Rolande Ruellan –, portent sur l’addition d’un certain nombre de taxes complexes. Chacune est due, en fait, à un épisode de l’histoire, voire de la négociation.
Faut-il simplifier ou stabiliser, même si c’est complexe ? J’entends plutôt un message de stabilité. Les taxes sont certes complexes, mais tout le monde y est habitué.
M. Pierre Morange, coprésident : Simplification et stabilisation ne sont peut-être pas incompatibles.
M. Dominique Libault : Je n’en suis pas sûr.
D’une part, nous avons affaire à des entreprises qui ont de très bons services juridiques qui n’hésitent pas à faire beaucoup de procédure à chaque nouvelle définition d’assiette ou autre. Nous sommes donc assez prudents sur les redéfinitions. La taxe de la promotion pharmaceutique a donné lieu à beaucoup de contentieux, une difficulté consistant, par exemple, à savoir si le véhicule de fonction des visiteurs médicaux était ou non un avantage en nature et s’il devait ou non être compris dans l’assiette. Cette taxe est aujourd’hui à peu près stabilisée.
Redéfinir quelque chose, c’est aussi poser à nouveau un certain nombre de problèmes et créer une incertitude juridique. Chaque fois que l’on crée du droit, on crée aussi de l’incertitude.
Je n’ai pas de dogme en la matière, mais il est clair qu’il faut choisir entre stabilité et simplification.
D’autre part, il faut bien voir que la condition de la stabilité de la fiscalité, c’est l’équilibre de l’assurance maladie. Si l’on n’atteint pas l’équilibre, on se retrouve dans un déséquilibre. Les pouvoirs publics s’agitent, le Parlement interpelle le Gouvernement en faisant valoir qu’il est insupportable de reporter nos dépenses de médicament d’aujourd’hui sur les générations futures, et prône des mesures.
L’hôpital n’est pas un bon « client » pour réaliser des économies de court terme dans un plan d’assurance maladie. Il n’y a pas non plus beaucoup d’enthousiasme à baisser les honoraires. Le médicament est l’un des segments où il est plus facile de faire des économies de court terme. C’est pourquoi je dis fréquemment aux responsables de l’industrie du médicament qu’ils sont ceux qui ont le plus intérêt à ce qu’on ne fasse pas de plan de maîtrise parce qu’ils risquent d’être mis à contribution et de pâtir de l’instabilité de la politique en ce domaine.
La condition de la stabilité de la politique du médicament, que ce soit sur la fiscalité ou sur la dépense, c’est, je le répète, l’équilibre de l’assurance maladie. Donc, vous devriez, mesdames, messieurs les parlementaires, être les premiers promoteurs de cet équilibre, c’est-à-dire avoir des objectifs compatibles avec les perspectives financières de la France. Fixer des objectifs ONDAM de 2 % au-dessus du PIB n’est pas très sérieux.
M. Pierre Morange, coprésident : Nous entendons bien le message selon lequel la rationalisation des moyens dans le secteur hospitalier ne peut générer que des marges de manœuvre à long terme. Ce propos, que l’on tient depuis un certain nombre d’années, n’aboutit cependant qu’à favoriser les mesures de court terme.
M. Dominique Libault : La direction de la sécurité sociale est très favorable à une politique de moyen et de long termes sur l’assurance maladie. Nous avons développé les éléments pluriannuels dans la loi organique relative aux lois de financement du 2 août 2005. Nous sommes prêts à continuer et je sais que le Parlement est dans la même disposition d’esprit. Plus nous aurons une vision globale et cohérente dans la durée, mieux ce sera pour l’assurance maladie et pour tous les partenaires de celle-ci. Je reconnais la légitimité de la demande de l’industrie pharmaceutique sur ce point.
Sur votre deuxième question, concernant l’information sur le médicament, je partage votre diagnostic. Il y a beaucoup d’acteurs qui font de l’information sur le médicament et leur coordination n’est pas toujours parfaite, loin de là. Dans la loi relative à l’assurance maladie du 13 août 2004, on a essayé de donner un rôle de coordination à la Haute autorité de santé, qui ne s’est pas totalement concrétisé jusqu’à présent.
Je serai, personnellement, très attentif aux préconisations de votre mission sur un tel sujet. Chaque agence fait valoir qu’elle a une légitimité ou des données qui justifient sa présence sur ce terrain, ce qui n’est pas complètement faux. L’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) a des données particulières.
M. Pierre Morange, coprésident : L’Inspection générale des affaires sociales (IGAS), dans son récent rapport sur l’information des médecins sur les médicaments recommande que la HAS soit l’interlocuteur unique pour dispenser l’information à destination des prescripteurs, afin de clarifier le rôle et l’articulation avec l’AFSSAPS. Quel est votre avis à ce sujet ?
M. Dominique Libault : Pour parler franchement, j’aimerais être plus convaincu de la capacité de la HAS à divulguer des messages simples et lisibles avant de lui donner ce rôle. C’est l’une des missions qu’on lui avait données en 2004 quand elle s’est substituée à l’Agence nationale d’accréditation et d’évaluation en santé (ANAES). Les messages de cette dernière étaient d’une telle complexité qu’ils étaient difficilement lisibles pour le généraliste de base. La HAS a commencé ce travail avec des fiches produits mais n’a pas encore la simplicité requise.
Actuellement, l’information est donnée soit par des gens qui ne sont pas des autorités scientifiques aussi établies que la HAS mais qui ont plus l’habitude de passer des messages de vulgarisation compréhensibles, soit par des autorités scientifiques qui n’ont pas encore tout à fait la capacité de faire passer l’information scientifique en messages simples. Je comprends la difficulté de l’exercice. Comme l’information est donnée sous le label HAS, elle est entourée de toutes les précautions propres aux scientifiques qui rechignent à simplifier à l’extrême et à exclure telle ou telle figure.
Tout cela est en construction. Je n’ai pas encore d’avis déterminé, mais je serai attentif, là aussi, à vos recommandations.
M. Lionel Joubaud : Je répondrai à votre troisième question, monsieur le président, sur le contentieux portant sur la base Thériaque. Les divergences entre les deux membres du GIE – groupement d’intérêt économique – qui sert de support à la base, en particulier entre la CNAMTS et le Centre national hospitalier d’information sur le médicament (CNHIM), a entraîné, au cours de l’été, le retrait de ce dernier, un changement de statut et un changement de nom. Elle s’appelle désormais la base Thesorimed. La DSS n’a pas d’information sur l’issue du contentieux mais peut vous rassurer : la base continue à être mise à jour. Des retards ont été pris du fait des turbulences engendrées par les divergences mais les gestionnaires s’attachent à le rattraper et la base est toujours financée par la CNAMTS.
M. Pierre Morange, coprésident : Quand la base sera-t-elle finalisée ?
M. Lionel Joubaud : Elle est déjà finalisée. Le retard concerne les mises à jour. Les gestionnaires prévoient qu’il sera comblé d’ici à la fin de l’année.
M. Jean Mallot, coprésident : Le contentieux n’est pas encore tranché ?
M. Lionel Joubaud : A ma connaissance non.
Mme Catherine Lemorton, rapporteure : Quel est votre avis sur l’incidence du mode de rémunération à l’acte des médecins libéraux de ville en France ? Y a-t-il un lien entre ce paiement à l’acte et la surconsommation de médicaments dans notre pays ?
Une charte de la visite médicale a été signée, mais plus de 60 % des médecins généralistes libéraux n’en ont jamais eu connaissance. Il est prévu à cette occasion la présentation de la fiche de transparence du produit. Or peu de visiteurs médicaux l’ont sur eux.
M. Dominique Libault : Sur la question de savoir s’il y a un rapport entre le paiement à l’acte et la surprescription, je serai, là aussi, assez humble. J’ai lu beaucoup d’avis à ce sujet. Certains disent que, dans d’autres pays où le paiement est également à l’acte, il ne s’accompagne par comme chez nous d’une surprescription ; le rapport n’est pas aussi simple que cela. D’autres évoquent le fait que le paiement à l’acte n’a pas tout à fait les mêmes effets en période de surdémographie médicale où le médecin peut être tenté de répondre plus à la demande de son patient et en période, comme actuellement, de pénurie de médecins où ceux-ci n’ont pas besoin d’aller chercher le patient, et sont plus autonomes et plus capables de réguler les demandes de ce dernier.
Beaucoup de médecins faisaient valoir à une époque que, s’ils ne prescrivaient pas à leurs patients les médicaments qu’ils demandaient ou ne leur accordaient pas l’arrêt de travail qu’ils sollicitaient, ils iraient en voir un autre. On observe que les patients ne changent pas de médecin traitant tous les jours. Le raisonnement selon lequel, le système étant tellement libéral, les patients peuvent aller voir ailleurs, n’empêche pas de faire un peu d’éducation et d’expliquer pourquoi il n’y a pas forcément besoin d’une ordonnance aussi longue.
Ce qui explique la surprescription, c’est moins le paiement à l’acte que le fait qu’il n’y a pas, dans le système financier, d’incitations à délivrer de bonnes prescriptions ou à faire évoluer les comportements d’une façon plus cohérente avec les besoins de santé publique ou de l’assurance maladie.
En prévoyant l’instauration dans le projet de loi de financement de la sécurité sociale pour 2008, d’un contrat individuel – dont les finalités restent à définir –, on se donne la possibilité d’expérimenter des tarifications annexes qui peuvent intéresser aussi le médecin et faire évoluer son comportement. Cela me semble une voie d’avenir.
Concernant la seconde question, il est certain que la charte de la visite médicale est insuffisamment connue. Il faut davantage l’installer dans le paysage. Si la fiche de transparence n’est pas en possession du visiteur, c’est dommage. Il faut de plus en plus renforcer les exigences déontologiques de la visite médicale qui contribue à l’information des médecins.
Mme Catherine Lemorton, rapporteure : Y a-t-il un comparatif, entre les pays européens notamment, de la pénétration d’un nouveau médicament en fonction de l’investissement fait sur la visite médiale ?
M. Dominique Libault : Des études ont été réalisées sur le taux de pénétration d’une nouvelle molécule en France. Elles montrent que ce taux est beaucoup plus rapide en France que dans beaucoup d’autres pays.
Deux explications sont possibles : la médecine libérale, d’une part, la sécurité sociale, d’autre part. Ce qui caractérise fondamentalement notre pays, c’est qu’il n’y a pas de problèmes financiers pour accéder très rapidement à de nouvelles molécules. Quand les médicaments sont inscrits au remboursement, il y a une diffusion rapide de ces derniers.
Distinguer l’effet de la sécurité sociale par rapport à l’effet de promotion est beaucoup plus complexe.
M. Lionel Joubaud : À ma connaissance, il n’y a pas eu d’étude établissant un lien.
M. Jean Mallot, coprésident : Parmi les outils à votre disposition, il en est deux principaux : le déremboursement et le prix du médicament. Quel est leur impact sur la prescription et la consommation ?
M. Dominique Libault : Si l’on veut faire place à de nouveaux médicaments, il faut être capable d’avoir une certaine fluidité du panier remboursé. Donc il est assez logique que certains produits sortent, à un moment donné, du remboursement.
Si je précise ce point, c’est parce que, bien qu’elle ne soit pas nouvelle, cette stratégie soulève encore des questions. Une partie de l’industrie pharmaceutique y reste hostile, alors que cette manière de procéder me semble indispensable pour faire place à l’innovation dans le remboursement.
Il ne faut pas renoncer à la fluidité du panier de soins, mais il faut également faire une plus grande place à l’automédication, sujet repris récemment par Mme Roselyne Bachelot, ministre de la santé, de la jeunesse et des sports. Certains médicaments qui ne sont pas prioritaires dans le remboursement doivent pouvoir avoir une vie après le déremboursement et les gens doivent pouvoir se les procurer. Le jour où l’on aura progressé sur la possibilité de donner une vie au médicament après remboursement, ce sera plus facilement vécu.
M. Pierre Morange, coprésident : La DSS a-t-elle des informations sur l’incidence financière, notamment sur le poste médicament de l’assurance maladie, du déremboursement d’une bonne centaine de médications et du transfert de remboursement sur d’autres thérapeutiques ?
M. Dominique Libault : Nous pourrons vous donner un document sur l’impact financier des déremboursements. Sur les effets de substitution, nous n’avons pas d’étude générale, mais des éléments d’appréciation. Nous avons regardé ce qu’il en est sur certains champs, et il ressort de notre analyse qu’il y a bien un impact positif.
Cela étant, comme je l’ai déjà indiqué, étant donné qu’il y a un remboursement très fort des médicaments par la base et les complémentaires, il existe une certaine indifférence en France par rapport au prix des médicaments. Ce dernier, même très élevé, n’est pas un facteur de diminution de la consommation.
M. Jean-Marie Rolland : L’apparition d’une nouvelle classe thérapeutique bouleverse aussi les habitudes de soins. On n’opère plus, par exemple, de l’ulcère de l’estomac parce que sont arrivés, dans les années 1980, de nouveaux médicaments qui permettent de traiter cette maladie. Un grand nombre de médicaments existent pour traiter l’hypertension artérielle. Ils coûtent de plus en plus cher, mais entraînent une diminution des accidents vasculaires cérébraux et des séquelles graves et un allongement de l’espérance de vie. Si les hôpitaux psychiatriques ont été en grande partie vidés de leurs patients, c’est grâce à l’apparition de neuroleptiques retard et à l’aménagement de modes de prise en charge ambulatoire. Est-on capable aujourd’hui d’évaluer les effets de ces évolutions, à la fois sur l’économie générale et sur la classe thérapeutique ?
M. Dominique Libault : Vous avez raison de souligner qu’il existe des stratégies par le médicament qui permettent d’éviter des hospitalisations, mais cela n’apparaît pas dans les comptes et il n’y a pas d’automaticité parce que les financements et les personnes sont distincts. Le sujet est très complexe. Il y a également plus de spécialisations du fait du vieillissement de la population.
Il est central de mesurer l’effort à faire sur tel ou tel secteur au regard des stratégies thérapeutiques et des besoins en santé publique. Nous devons en effet progresser dans la bonne allocation des ressources dans notre système contraint.
Une des raisons pour lesquelles on a mis la commission de la transparence au sein de la HAS, est de travailler davantage sur les stratégies thérapeutiques afin que le collège puisse donner son avis et nous éclairer sur celles-ci. Tout cela est actuellement en germe.
M. Pierre Morange, coprésident : Il apparaît très malaisé de réaliser une étude qui prenne en compte les sorties et les entrées liées à l’apparition de nouvelles thérapeutiques. Un récent rapport de la CNAMTS étudie trois postes médicamenteux – les inhibiteurs de la pompe à protons, les statines et les antihypertenseurs – et fait état de marges de manœuvre financières importantes, puisque la somme totale avoisinerait le milliard et demi d’euros, les économies potentielles pour chaque poste indiqué variant entre 400 et 600 millions d’euros.
Autant il peut être aisé de dresser le bilan de l’effet de la suppression d’une hospitalisation en cas d’ulcère grâce aux antiulcéreux de dernière génération, autant il est malaisé de suivre les processus plus fins de prise en compte thérapeutique, comme celui entraîné par le passage, pour les antihypertenseurs, les diurétiques et les beta-bloquants aux inhibiteurs de l’enzyme de conversion et aux sartans. A-t-on globalisé le coût de chaque stratégie pour chaque pathologique ?
M. Dominique Libault : Non.
M. Pierre Morange, coprésident : Il serait intéressant que, dans le cadre des nouvelles dispositions prévues par le PLFSS pour 2008, les analyses médico-économiques réalisées par la HAS ciblent ces trois postes et permettent une analyse globale des coûts induits par les anciennes stratégies – qui avaient fait la démonstration de leur efficacité, mais entraînaient des effets secondaires ou une surveillance importante – et des coûts des nouvelles stratégies, qui sont plus onéreuses, mais nécessitent peut-être moins de surveillance ou s’accompagnent d’effets secondaires moins importants. De tels éléments sont-ils, à votre connaissance, maîtrisés ?
M. Dominique Libault : Ils ne sont pas maîtrisés, mais votre question montre tout l’intérêt que la HAS s’oriente vers une stratégie médico-économique. Cela me semble un thème tout à fait intéressant pour le démarrage des travaux de celle-ci.
Mme Catherine Lemorton, rapporteure : Je vous poserai une dernière question sur le thème de l’égalité d’accès aux soins et aux médicaments, à laquelle vous avez déclaré être vigilant.
Les premiers déremboursements concernaient des vitamines, des tonifiants et des revitalisants, alors qu’ils portent maintenant sur des médicaments qui ont leur efficacité, comme la pseudoephedrine. On peut considérer que leur SMR est insuffisant mais, dans l’exemple que j’ai cité, le médicament apportait du confort aux personnes souffrant de petites pathologies respiratoires. Les classes modestes ne peuvent plus y accéder, car il est devenu trop cher. Il faut savoir que les laboratoires multiplient par deux, voire par trois, le prix hors taxe une fois que les médicaments sont déremboursés. C’est compréhensible : ils se rattrapent sur le prix par rapport à la perte de volume, puisque ces médicaments ne sont plus prescrits.
Mme Roselyne Bachelot, ministre de la santé, de la jeunesse et des sports a déclaré que ces médicaments seront mis « en libre-service » devant les comptoirs des pharmacies. Or, au bout d’un moment, il y aura forcément transfert sur des produits remboursés. Je crains donc que les déremboursements massifs des médicaments précédemment prescrits pour les pathologies respiratoires génèrent un transfert sur les antibiotiques dont les prescriptions vont augmenter. Quel est votre avis ?
M. Dominique Libault : La direction de la sécurité sociale n’intervient pas du tout dans la détermination de la liste des médicaments à service médical rendu insuffisant. Cette liste est établie par une autorité scientifique et nous la respectons.
Par ailleurs, il y a de plus en plus de médicaments qui sont pris en charge à 100 % par la sécurité sociale du fait de la croissance des affections de longue durée, les ALD. Si l’on reste dans le système tel qu’il est, on sait que ces dernières vont concentrer une part croissante des capacités de ressources de l’assurance maladie obligatoire. Si l’on se projette à quinze ou vingt ans, on pourrait craindre que les médicaments soient remboursés pour les maladies longues et le soient moins pour les maladies courantes. Cela nécessite une réflexion sur la part des médicaments remboursés à 100 %, à 65 %, etc.
Cela étant, la maîtrise du 100 % est indispensable si l’on veut garder une part importante de médicaments courants pour des pathologies bénignes, importants dans le vécu des gens, qui soient bien pris en charge par la sécurité sociale.
Le remboursement est de plus en plus opéré par l’assurance de base et une assurance complémentaire, puisque, heureusement, 90 % des assurés sociaux ont une complémentaire. Il faut également continuer à travailler pour les gens qui n’en ont pas, en particulier, par le biais de la couverture maladie universelle complémentaire, la CMUc. On travaille beaucoup également sur l’aide à la complémentaire santé, qui ne fonctionne pas encore suffisamment bien pour solvabiliser au-delà de la CMUc. L’équilibre entre le remboursement à 100 % et le maintien d’un bon remboursement pour des médicaments courants est une question qui nous interpelle.
M. Pierre Morange, coprésident : Nous aurions une foule d’autres questions à vous poser. Nous nous permettrons de vous les transmettre par écrit.
Je vous remercie, messieurs.
*
Audition de M. Noël Renaudin, président du Comité économique des produits de santé (CEPS).
M. Pierre Morange, coprésident : Monsieur Renaudin, je vous souhaite la bienvenue à l’Assemblée nationale pour cette audition qui s’inscrit dans le travail qu’effectue notre mission sur la prescription, la consommation et la fiscalité du médicament et je vous donne la parole pour une brève présentation de l’organisation et des missions du Comité économique des produits de santé.
M. Noël Renaudin : Le Comité économique des produits de santé est un petit organisme. Ses services permanents regroupent quatorze personnes à temps plein et le comité, lui-même, qui est collégial, rassemble dix personnes, avec voie délibérative. Outre le président et le vice-président, il comprend quatre représentants de l’État – le directeur de la sécurité sociale ou son représentant, le directeur général de la santé ou son représentant, le directeur général des entreprises au ministère de l’économie, des finances et de l’emploi ou son représentant et le directeur général de la concurrence, de la consommation et de la répression des fraudes ou son représentant – et, depuis la réforme de l’assurance maladie de 2004, quatre représentants des payeurs : trois représentants de l’assurance maladie obligatoire – deux de la Caisse nationale d’assurance maladie des travailleurs salariés (CNAMTS) et un représentant commun au régime des agriculteurs, la Mutualité sociale agricole (MSA), et au régime social des indépendants (RSI) – et un représentant des organismes d’assurances complémentaires, délégués par l'Union nationale des organismes d'assurance maladie complémentaire (UNOCAM).
La mission principale du comité est de fixer les prix des médicaments remboursables ou les tarifs des dispositifs médicaux pris en charge par l’assurance maladie. Il est à cette fin organisé en deux sections – une section du médicament et une section des dispositifs médicaux – composées pour une part des mêmes personnes qui se réunissent à des moments différents, avec des ordres du jour différents et une configuration éventuellement différente. La fixation des prix comprend la fixation initiale et également l’évolution des prix : augmentation ou diminution.
De façon secondaire, bien que cela l’occupe pas mal de temps, le comité est chargé par la loi d’une mission générale de régulation – mais il ne s’agit pas vraiment de régulation – du marché du médicament remboursable en conventionnant les entreprises dans le cadre de la contribution de sauvegarde, organisée par la loi, aux termes de laquelle, lorsque les ventes de médicaments ont dépassé, par rapport à l’année précédente, un taux fixé par le Parlement – le taux K –, les entreprises présentes sur ce marché sont astreintes à une contribution dont elles peuvent être exonérées si elles ont passé des conventions avec le comité.
En pratique, toutes les entreprises passent des conventions avec le CEPS et ces dernières donnent lieu au versement de ristournes qui sont la contrepartie de la taxe qui n’est pas payée. Il y a une différence importante entre les deux systèmes en ce sens que la taxe a, comme toute taxe, des règles nécessairement simples pour ne pas dire simplistes, alors que la convention permet de répartir la contribution des entreprises de façon plus conforme aux orientations de la politique du Gouvernement. Par exemple, dans le système conventionnel, on exonère la croissance des génériques car cela paraît idiot de faire payer les entreprises sous prétexte qu’elles ont vendu plus de génériques alors qu’on les y encourage, et on exonère pendant un certain temps les produits les plus innovants en reconnaissance de leur caractère innovant. Le système conventionnel présente d’autres spécificités par rapport à la taxe de droit commun, mais ce sont là les deux principales.
Cette deuxième mission est accessoire par rapport à la fixation des prix, puisqu’elle n’a pas une grosse influence sur les grands équilibres de l’assurance maladie, mais elle alimente un débat conventionnel avec les entreprises, qui présente une certaine utilité.
En couplage avec cette deuxième mission, le comité est chargé d’observer le marché du médicament, de rendre compte au Gouvernement des observations qu’il peut faire et, éventuellement, de l’alerter, sans se mettre, bien entendu, à la place du comité d’alerte, si la situation lui semble appeler des mesures correctrices, y compris réglementaires ou législatives.
Au fil des ans, la loi a dévolu au comité un certain nombre d’autres activités. Il a été chargé, en particulier, de négocier la charte de la visite médicale avec l’industrie pharmaceutique. Celle-ci est maintenant remise entre les mains de la Haute Autorité de santé (HAS), à qui la loi confie le soin de la faire appliquer et de veiller à la certification des entreprises au regard de celle-ci en agréant les organismes certificateurs. La HAS, à partir de la charte, a établi le référentiel de certification qui sert aujourd’hui aux entreprises pour se mettre en conformité avec celle-ci.
Il revient également au CEPS de sanctionner les entreprises dont les publicités ont été interdites par le directeur de l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS). Le comité a le pouvoir – et le devoir – de prononcer, le cas échéant, des sanctions pécuniaires à l’encontre des entreprises en infraction.
Le comité exerce ses missions en appliquant le code de la sécurité sociale, lequel contient très peu d’indications sur la fixation des prix, mais des indications importantes. Il y est, en particulier, précisé qu’on ne peut pas inscrire un médicament ou un dispositif médical qui n’apporte pas d’amélioration du service rendu s’il n’entraîne pas une économie. Autrement dit, pour inscrire un médicament, il faut ou bien qu’il y ait une amélioration du service rendu – dans ce cas, il peut être aussi cher ou plus cher –, ou bien, s’il n’y a pas d’amélioration, qu’il soit moins cher. C’est une règle de bon sens mais la France est presque le seul pays à l’avoir.
Il est également indiqué, dans le code de la sécurité sociale, que les prix sont normalement fixés par convention entre le CEPS et les entreprises. Ils peuvent être fixés autrement, notamment de façon unilatérale par le comité qui a un pouvoir réglementaire de ce point de vue. Il ne le fait jamais à l’inscription car cela n’aurait pas de sens, puisque personne ne peut forcer une entreprise à commercialiser un médicament. Donc, ou bien on se met d’accord sur un prix et c’est la voie conventionnelle, ou bien on ne se met pas d’accord et le produit n’est pas inscrit. En revanche, la possibilité de fixer les prix de façon unilatérale s’exerce parfois, bien que très rarement, lorsqu’il s’agit de modifier les prix de médicaments ou de dispositifs déjà inscrits au remboursement.
Concrètement, les entreprises qui demandent l’inscription d’un produit déposent un dossier composé de deux parties principales : une partie médico-technique destinée principalement à la commission de la transparence de la HAS et une partie économique destinée au CEPS. Les produits sont d’abord évalués par la commission de la transparence qui se prononce sur le service médical rendu, sur l’amélioration de celui-ci et sur la population ciblée par le médicament, puis le comité désigne ou non un rapporteur, selon la complexité du sujet. Celui-ci peut être soit un agent des caisses d’assurance maladie, soit un fonctionnaire, soit un retraité et doit avoir pour qualités principales d’être indépendant de l’industrie et d’être intéressé par le sujet à la fois sous l’angle pharmaceutique et sous l’angle économique. Il présente le dossier au comité qui vote. Le jeu consiste à réunir à la fois une majorité du CEPS en faveur de l’inscription, ce qui ne va pas de soi, et l’accord de l’entreprise. On considère qu’un prix sur lequel on a réussi à mettre d’accord une majorité du comité, constitué de la manière collégiale que j’ai décrite, et l’entreprise a des chances d’être un bon prix.
Mme Catherine Lemorton, rapporteure : Quand il apparaît qu’il y a un contournement de générique, comme cela s’est produit pour un médicament de la classe thérapeutique des anti-ulcéreux, comment se fait-il, alors que le service médical rendu (SMR) n’est pas supérieur, qu’il obtienne un prix plus important ?
M. Noël Renaudin : On s’est rendu compte de l’existence de ce problème il y a sept ou huit ans seulement. Un ou deux produits sont rentrés dans la classe sans qu’on s’en aperçoive pour ainsi dire. Ces problèmes peuvent être réglés éventuellement par des baisses de prix ultérieures. C’est ce qu’on a fait dans un certain nombre de cas.
M. Pierre Morange, coprésident : Pouvez-vous nous faire un bilan de ces baisses de prix au fur et à mesure de la vie du médicament et nous donner une idée des économies générées ?
M. Noël Renaudin : Une doctrine, issue des orientations ministérielles, prévaut clairement en la matière : lorsqu’un produit faisant l’objet d’une demande de remboursement présente un caractère de détournement de générique – c’est-à-dire est susceptible de se substituer à un produit qui est génériqué ou en passe de l’être –, on distingue deux cas, en dehors de celui, très rare, où ce produit est assez fortement innovant, c’est-à-dire avec une amélioration du service médical rendu (ASMR) 3, 2 ou 1, auquel cas il est traité comme une innovation.
Premièrement, dans le cas où le produit ne présente pas d’amélioration du service médical rendu, il peut être inscrit à un prix tel qu’il ne coûte pas plus cher que le générique. Cette règle est mise en œuvre de façon régulière. L’exemple qui concerne le marché le plus élevé est celui des antihistaminiques H1. Nous avons inscrit le Xyzall, qui est de la lévocétirizine, c’est-à-dire un lévogyre de Zyrtec, à un prix tel que, en prix public, c’est-à-dire chez le pharmacien, il coûte autant que le Zyrtec.
Ce qu’il faut bien comprendre, c’est que l’État est neutre en la matière. Il lui importe peu que les ventes soient faites par un génériqueur ou par le fabricant du princeps. Ce qui l’intéresse, c’est le prix. Si un médicament princeps est vendu au même prix que le médicament générique, cela nous est égal. Nous considérons que ces médicaments ont le droit de vivre dès l’instant qu’ils ne sont pas plus chers.
Le second cas est celui où le médicament a une ASMR 4, ce qui correspond au médicament que vous aviez en tête, madame la rapporteure, quand vous avez posé votre question : l’esomeprazole, vendu sous le nom d’Inexium. Les orientations ministérielles dans ce cas sont les suivantes : nous devons nous débrouiller pour que le produit soit inscrit à un prix tel, ou avec un échéancier de prix tel, que sa prescription ne coûte pas plus cher que la prescription du princeps remplacé. L’Inexium remplace le Mopral, qui est génériqué par l’Omeprazol. Les médecins prescrivent peu l’Omeprazol. Par contre, il est substitué au Mopral chez les pharmaciens, selon un taux assez important. Nous avons fait une hypothèse hardie, tablant sur une substitution des prescriptions de Mopral à 60 % les dix-huit premiers mois et à 80 % au bout de deux ou trois ans, si bien que nous avons pris, pour le prix public d’Inexium, la moyenne pondérée entre le prix de Mopral et le prix de l’Omeprazol.
Si l’Inexium est objectivement plus cher à la consommation que certains autres inhibiteurs de la pompe à protons (IPP), cela vient du fait qu’il existe deux catégories d’IPP : Mopral et les autres. Une indication essentielle des inhibiteurs de la pompe à protons est la protection contre les dégâts éventuellement commis par les anti-inflammatoires non stéroïdiens. Dans cette indication, le Mopral n’a d’autorisation de mise sur le marché (AMM) qu’à la pleine dose – 20 milligrammes –, alors que les autres IPP ont l’indication à la demi-dose. C’est pourquoi ils coûtent moins cher.
Pour en revenir à l’explication de la doctrine ministérielle, le prix d’Inexium a été fixé sur la base d’une hypothèse ambitieuse qui s’est avérée juste par la suite.
Le prix d’Inexium s’est ensuite à nouveau trouvé décalé par rapport au prix pondéré Mopral-Omeprazol parce que, entre-temps, on a baissé l’ensemble du répertoire générique, c’est-à-dire princeps et génériques. Début 2006, la diminution a été de 15 %, et parfois plus, et elle a concerné Mopral et Omeprazol, de sorte que les prescriptions de Mopral ont à nouveau coûté moins cher que celles d’Inexium. Cet écart s’est maintenu un certain temps, je suis prêt à le reconnaître, mais nous y avons remédié l’année dernière en organisant une nouvelle baisse assez importante du prix d’Inexium dans le cadre de ce qu’on a appelé les baisses de cohérence qui correspondent à une autre orientation ministérielle selon laquelle, lorsque, dans une classe pharmaco-thérapeutique, coexistent des médicaments ayant perdu leur brevet et des médicaments l’ayant encore, on doit s’efforcer de rapprocher les prix des médicaments qui restent brevetés des prix des médicaments qui ont perdu leur brevet, afin de ne pas laisser subsister des écarts trop importants dans des classes que l’on pourrait considérer comme des classes d’équivalence.
Procéder à des baisses de prix au fur et à mesure que les produits vieillissent est une activité permanente du comité, et celui-ci les actualise tous les ans.
Les chiffres des quatre dernières années sont assez présents à mon esprit parce que, comme vous vous en souvenez sans doute, le Gouvernement a demandé au comité de procéder à de fortes baisses de prix dans le cadre de ce qu’on a appelé « le plan médicament » qui accompagnait la réforme de l’assurance maladie. En 2004, il a été demandé de réaliser 354 millions d’euros d’économies au moyen de baisses de prix des médicaments sous brevet en trois ans. Cette demande a été renforcée en 2006, où il a fallu accroître l’effort de 200 millions d’euros, ce qui portait les baisses à 550 millions sur trois ans. À mi-année, le comité a été prié, après l’alerte donnée par le Comité d’alerte, d’en ajouter encore 100 millions.
En pratique, les économies dues à des baisses de prix ont été d’environ 160 millions d’euros en 2005, après 28 ou 30 millions réalisées dès 2004. Il s’y est ajouté 190 millions d’euros en 2006. Elles devraient s’élever à 250 millions en 2007.
M. Jean Mallot, coprésident : Comment sont mesurées les économies ?
M. Noël Renaudin : On multiplie la baisse par boîte en prix public par le taux de remboursement réel de l’assurance maladie et par le nombre de boîtes vendues dans l’année. Cela permet d’obtenir l’impact réel de la baisse.
M. Pierre Morange, coprésident : Vous mesurez l’impact strict.
M. Noël Renaudin : Absolument. On ne compte pas deux fois les économies, et la mesure se fait médicament par médicament.
Je donnerai un exemple. On a baissé de manière assez forte le prix du COX-2 subsistant, Celebrex. On a alors mesuré un certain niveau d’économie. Puis les ventes ont beaucoup baissé. Nous avons compté une déséconomie, c’est-à-dire une économie négative, compte tenu du calcul que je viens d’indiquer.
M. Pierre Morange, coprésident : Il ressort du point d’information mensuel publié par l’assurance maladie le 19 octobre 2007 que, en matière de consommation d’inhibiteurs de la pompe à proton, la France se situe au deuxième rang de la consommation des pays européens derrière l’Espagne. Or bien que la France ait une consommation moins élevée que l’Espagne, ses dépenses sont plus importantes. Ce phénomène est dû au fait que les Espagnols consomment à peu près 85 % d’IPP génériqués, donc moins coûteux, alors que leur part en France n’est que de 50 %. Cette tendance à la prescription des médicaments les plus récents et les plus coûteux tend à s’accentuer. Avec la même consommation d’IPP qu’actuellement, et avec un coût moyen par habitant comparable à celui de ses voisins, la France pourrait réaliser une économie de l’ordre de 430 millions d’euros. Quelles réflexions vous inspire cette analyse ?
M. Noël Renaudin : Elle m’inspire deux réflexions.
Le point établi par l’assurance maladie apporte plusieurs informations.
La première, qui est très importante, est que, en France, les médecins prescrivent plus cher. Quand il y a un mix-produit dans une classe de médicaments, qu’il s’agisse des antihypertenseurs, des IPP ou autres, alors même que les prix de chacun des composants sont plutôt plus bas en France et que le taux de substitution est satisfaisant, les médecins prescrivent volontiers hors du répertoire, c’est-à-dire des molécules pour lesquelles il ne peut pas y avoir de substitution. Le mix-produit prescrit est plus cher. Cette tendance appelle indéniablement des mesures d’orientation de la prescription.
La note d’information de l’assurance maladie met en avant un autre point, sur lequel je suis moins d’accord, à savoir que les prix des génériques en Espagne sont beaucoup moins chers qu’en France. C’est exact pour les génériques d’omeprazole et de lansoprazole – autre IPP déjà génériqué en Espagne, alors qu’il ne le sera en France qu’en décembre –, mais on ne peut pas en tirer de conclusion générale car il faut raisonner sur l’ensemble du marché des génériques. On peut toujours trouver – et c’est un jeu qui fait fureur – un pays où un médicament est moins cher, et quelquefois beaucoup moins cher, qu’en France. En revanche, quand on regarde le marché, il est difficile de trouver un pays dans lequel la moyenne des prix des médicaments est plus basse qu’en France. J’ai entendu les choses les plus invraisemblables au sujet des génériques. On a dit, par exemple, qu’en prix sortie d’usine, ils étaient six fois moins chers au Royaume-Uni qu’en France.
La réalité est que les génériqueurs en France ne gagnent pas d’argent – sauf deux, et encore ils n’en gagent pas beaucoup –, alors qu’ils ont les mêmes conditions d’approvisionnement que leurs concurrents anglais, espagnols ou allemands. Les grands génériqueurs indiens comme le grand génériqueur israélien Teva ne s’approvisionnent pas différemment que pour le marché français ou le marché anglais. Néanmoins, les génériqueurs français ne gagnent pas d’argent. Dès lors, on ne peut pas soutenir que les prix des génériques sont trop élevés en France.
Cela étant, ils sont un peu plus chers en prix public parce qu’on a fait le choix – choix imposé dans la mesure où il n’y a aucune sensibilité au prix, et on ne souhaite pas qu’il y en ait, chez les assurés – de rémunérer les pharmaciens pour la vente des génériques. Cela consomme un peu de marge, mais pas au point, de mon point de vue, d’entraîner un réel problème, même en prix public. Il faut avoir une vue d’ensemble sur les prix des génériques.
C’est ma réponse au miroitement d’une possible économie de 430 millions d’euros. Un tel raisonnement donne l’impression que tout le monde est tombé sur la tête et qu’il n’y a qu’à être un peu intelligent pour économiser 400 millions d’euros sur les IPP et – pourquoi pas ? – 350 millions sur les antihypertenseurs, et 600 millions sur les statines. Ce n’est pas possible. Pas de cette manière, en tout cas.
En revanche, on peut certainement – et on doit – économiser de l’argent en posant des règles en matière de prescription.
M. Pierre Morange, coprésident : Comment peut-on expliquer que les prix des génériques soient moins élevés en Espagne ? Est-ce dû à la localisation des sites de production ?
M. Noël Renaudin : Je précise à nouveau que, si les prix des génériques d’omeprazole et de lansoprazole sont moins chers en Espagne qu’en France, ce n’est pas un cas général. Ensuite, les différences de prix ne proviennent jamais d’une différence de sites de production. Elles résultent éventuellement des conditions de marché.
En France, il y a une règle – qui vaut ce qu’elle vaut – selon laquelle la décote du générique par rapport au princeps est à peu près constante – car c’est plus simple. Actuellement, le générique coûte chez nous 50 % de moins que le princeps.
Dans l’ensemble, ce système fonctionne assez bien parce que nos princeps, surtout en fin de vie lorsque le générique arrive, sont plutôt moins chers que dans le reste du monde.
Sur les marchés où il existe, pour des raisons diverses, des éléments de concurrence par les prix, ces derniers peuvent baisser éventuellement beaucoup plus. La production de l’omeprazole ne coûte pratiquement rien, ni en France, ni en Grande-Bretagne, ni en Espagne. Donc les différences de prix ne proviennent pas de la production mais du fait que les entreprises essaient de gagner de l’argent. Or il est connu que les génériqueurs français n’en gagnent pas beaucoup et même que la plupart en perdent. La fixation des prix des génériques à la moitié des prix des princeps donne un niveau général des prix plutôt bon.
Comme nous n’avons aucun élément de concurrence par les prix sur aucune molécule, sur certaines molécules très concurrencées, on peut trouver, ici ou là, des prix vertigineusement plus bas. Le paracétamol est vendu cinq fois moins cher qu’en France sur tel ou tel marché ou sur internet.
M. Pierre Morange, coprésident : Les médicaments génériques vendus en France sont-ils tous produits en Europe ou proviennent-ils d’autres sites de production ?
M. Noël Renaudin : Ils sont actuellement très majoritairement, c’est-à-dire dans une proportion de 90 %, produits en Europe, mais évidemment pas exclusivement.
Les deux grands génériqueurs qui représentent une bonne partie du marché en France sont MERCK génériques et Biogaran. Ce dernier, à ma connaissance, ne produit qu’en Europe, c’est-à-dire en France dans les usines Servier ou, en Hongrie où Servier a fait des usines de production importantes. MERCK génériques est installé en Europe, en particulier en Allemagne.
L’un des deux génériqueurs indiens, Ranbaxy, avait hérité du portefeuille Rhône-Poulenc et fabriquait en France, ce qui lui coûtait très cher. Il est en train de retourner vers ses approvisionnements normaux indiens, qui sont de bonne qualité, contrairement aux produits chinois pour lesquels nous avons quelques craintes car ils sont nettement moins propres, mais ils finiront par le devenir. Les usines indiennes sont visitées par la FDA, Food and drug administration, comme les usines françaises.
Pour l’instant, la production de génériques est majoritairement européenne et il est souhaitable que cela dure.
M. Pierre Morange, coprésident : Vous avez évoqué la prime à la nouveauté et les mécanismes de transfert de la prescription sur des molécules plus récentes, suite à ce rapport de l’assurance maladie…
M. Noël Renaudin : Nous manquons cruellement d’un dispositif crédible d’aide à la prescription. Le système français est le meilleur du monde en matière d’évaluation et de mise sur le marché des médicaments. C’est le plus rationnel. C’est aussi le seul qui permette d’éviter les me too, qui peuvent atteindre, voire dépasser, le prix des médicaments déjà existants ; le code de la sécurité sociale ne le permet pas. Mais notre système est radicalement insuffisant, une fois que les médicaments sont là, pour que la prescription soit rationnelle.
La raison en est à la fois historique, culturelle et réglementaire. La France a adopté un système d’assurance maladie universelle, dans lequel personne ne paie. On ne veut pas que les médecins soient responsabilisés financièrement sur leurs prescriptions. Dans d’autres pays, ceux-ci ont des enveloppes, l’accès aux spécialistes est long, les restes à charge peuvent être élevés pour les assurés, et les uns et les autres sont sensibilisés aux prix. En France, lorsque vous allez chez le médecin, vous savez qu’il ne sera pas empêché de vous prescrire le meilleur médicament, sous prétexte qu’il a déjà atteint la limite de son budget. Et c’est une bonne chose. Reste qu’il nous manque une mécanique médico-économique d’orientation de la prescription chez les médecins.
Aujourd’hui, nous disposons d’un certain nombre d’instruments d’ordre médical, comme les fiches de transparence qui indiquent que, dans telle situation pathologique, il faut tel ou tel médicament, tel ou tel cheminement thérapeutique. C’est plutôt bien fait. Nous disposons aussi d’un début d’orientation d’ordre économique, lorsque la CNAMTS demande que les médecins prescrivent dans le répertoire. Mais ce n’est pas suffisant.
Notre système est très bien pour inscrire les médicaments qui ne sont pas meilleurs, ce qui permet de faire des économies ; il ne permet pas de payer les médicaments les plus innovants à un prix significativement différent de celui accepté dans les autres pays de l’Union européenne. Mais nous n’avons pas de système médico-économique qui réponde à la question : les prix des médicaments étant ceux-ci, comment peut-on utiliser ces médicaments de façon rationnelle ? La seule institution qui en soit capable et qui ait la légitimité pour le faire est la HAS.
Voici un exemple pour illustrer mon propos : dans l’hypertension, la catégorie la plus moderne (le système rénine/angiotensine) est composée de deux classes de médicaments : les inhibiteurs d’enzymes de conversion, ou IEC, qui sont génériqués et qui coûtent 20 centimes par jour ; et les inhibiteurs de récepteurs à l’angiotensine II, les sartans, dont aucun n’est génériqué et qui coûtent deux ou trois fois plus cher que les IEC. Les sartans ont eu, à l’époque, une ASMR 3, qui était sans doute justifiée. En effet, 20 %, des patients qui consomment des IEC toussent de façon très désagréable, et les sartans évitent la toux. Toutefois, il existe aujourd’hui de très bonnes initiations au traitement qui se font directement par les sartans. Les médecins auraient tort de se gêner : ils sont sûrs que le traitement aura de l’effet sur l’hypertension et qu’en outre, le patient ne toussera pas. Mais qui peut dire aujourd’hui aux médecins : c’est mal de commencer par un sartan, il faudrait commencer par un IEC. Je ne peux pas le dire. La CNAMTS non plus.
M. Jean-Marie Rolland : N’est-ce pas le rôle des délégués de l’assurance maladie (DAM) ?
M. Noël Renaudin : La CNAMTS n’en a pas la légitimité. Cela dit, les sartans seront bientôt génériqués. Il en est de même des statines et des inhibiteurs de la pompe à protons. S’il n’y avait que cela, demain, on pourrait dormir sur nos deux oreilles, mais ce n’est évidemment pas le cas, et la dépense de médicaments ne baisse pas. En effet, alors que ces grandes classes de médicaments décroissent, le marché s’enrichit de médicaments horriblement coûteux, des médicaments de « niches », pour des populations cibles assez restreintes.
Notre système consiste à séparer, au niveau de la HAS, et sur des critères purement médicaux, ce qui apporte quelque chose, de ce qui apporte significativement quelque chose – et dont l’absence entraînerait une perte de chances inacceptable pour un groupe de malades – et de ce dont on pourrait éventuellement se passer. Néanmoins, une fois cela fait, on n’a plus de liberté sur le prix.
Il faudrait que quelqu’un puisse dire : à ce prix-là, nous demandons que vous, médecins, réserviez la prescription à telle sous partie de la population cible. On ne peut pas faire autrement. Mais c’est beaucoup plus difficile.
Autre exemple exagérément simple : nous dépensons 400 millions d’euros en érythropoïétine, utilisée pour compenser certains dégâts de la chimiothérapie ou pour les dialyses. Il ne viendrait à l’idée de personne de dire que ce n’est pas un bon médicament, mais il y a peut-être quelque chose à dire sur les taux au-dessous desquels il est légitime d’en prescrire, et au-dessus desquels il serait légitime d’attendre. On voit bien que ce n’est pas la CNAMTS qui peut le dire. Il faut que cela soit dit par des personnes ayant une légitimité et une responsabilité médicales pour le faire. La loi va peut-être le préciser, mais je pense qu’un des enjeux importants de la période qui vient sera que la HAS fasse la promotion, non pas du bon usage du médicament, mais de son usage rationnel.
Mme Catherine Lemorton, rapporteure : Je rejoins M. Jean-Marie Rolland et sa remarque sur les délégués de l’assurance maladie. Il y a un an et demi, ils avaient été désignés pour surveiller une des classes de statines, le Crestor, un des médicaments beaucoup plus cher…
M. Noël Renaudin : Non, le Crestor coûte exactement le même prix que la simvastatine générique. Le Comité économique des produits de santé applique les orientations des ministres.
Mme Catherine Lemorton, rapporteure : Vous avez sans doute raison. Le risque était que les médecins aillent immédiatement au plus fort dosage, qui était évidemment plus cher. Et les DAM avaient pour mission de faire de l’information auprès des médecins.
M. Noël Renaudin : La Commission de la transparence s’était prononcée sur le Crestor. Le dosage le plus élevé devait être peu utilisé. Il faut savoir que le Crestor a été inscrit en deux temps : dans un premier temps, on a inscrit un dosage élevé, pour une population très restreinte, avec un contrat bien ficelé entre l’entreprise et le CEPS ; dans un second temps, est arrivé le Crestor 5 mg, qui est devenu une statine de plein exercice, comparable aux autres, utilisable sans risque à la place des autres. Et c’est ce dosage à 5 mg dont nous avons fixé le prix, conformément aux orientations du ministre, au prix de la simvastatine générique.
Je suis totalement indifférent au fait que les médecins prescrivent du Crestor ou du Zokor substitué par la simvastatine. Je préfère même qu’ils prescrivent du Crestor, car c’est l’équivalent d’une substitution à 100 %. Un petit débat est né entre l’assurance maladie et nous-mêmes. Chaque position se défend. L’assurance maladie a une préférence pour le générique. À prix égal, je n’en ai pas.
M. Pierre Morange, coprésident : Si je comprends bien, vous êtes relativement serein s’agissant de ces grandes classes de molécules qui vont tomber dans le domaine public. Avez-vous pu établir une prospective des économies générées potentiellement sur les cinq prochaines années ? Je ne vous demande pas l’information tout de suite, mais il serait utile que vous nous fournissiez des éléments assez précis sur le sujet.
S’agissant des thérapeutiques onéreuses, quelle stratégie est envisagée, notamment au niveau européen ? Les populations cibles sont peu nombreuses sur le sol national, mais la dépense pourrait être mutualisée sur le plateau continental européen, d’autant qu’il s’agit de populations solvables, ce qui permettrait de faire baisser le prix des produits.
M. Noël Renaudin : Je pense que c’est le marché qui décidera. Cela étant, notre stratégie consiste à dire et à redire en France et à l’étranger aux entreprises multinationales qu’elles sont sur une voie impossible et qu’elles vont devoir changer de modèle. Certaines commencent à s’en rendre compte et à faire ce que nous disons depuis un certain temps : les grands pays développés, qui ont fait la fortune des entreprises de médicaments depuis trente ans, commencent, sans exception, à toucher le plafond. Et ce d’autant plus vite qu’aux États-Unis, l’inégalité d’accès à ces médicaments innovants devient un vrai problème. Le jour où les Américains, qui ont été la principale ressource de l’industrie pharmaceutique, vont ressentir la nécessité de faire accéder les plus pauvres ou les personnes âgées aux médicaments innovants, ils ne pourront pas les payer à ces prix-là. Et alors, il me semble inévitable que les prix s’assagiront.
M. Pierre Morange, coprésident : Le marché est donc adossé à la solvabilité des puissances occidentales, solvabilité qui est plafonnée. Ne pourrait-on pas envisager la création d’une dynamique occidentale ou européenne, qui anticipe cette évolution inéluctable du marché et qui, jouant sur les volumes, fasse baisser les coûts, ce qui bénéficierait au plus grand nombre de patients ? Car les médicaments innovants, très onéreux, représentent tout de même des chances de vie.
M. Noël Renaudin : Je crois que c’est trop tôt. Les grands pays d’Europe du Nord ne fixent pas les prix. Ils ont tort et je pense qu’ils y viendront, mais, pour l’instant, ils résistent. Nous n’avons donc pas d’interlocuteurs dans ces pays-là. Nous ne pouvons pas nous adresser aux Italiens, parce qu’il faudrait discuter avec toutes les régions italiennes. Nous pourrions discuter avec les Espagnols, et nous avons commencé à le faire, mais cela ne va pas très loin.
Cela dit, la France a tout de même une influence sur le niveau général des prix. En effet, les entreprises peuvent très difficilement se passer d’elle pour de tels produits. Nous ne pesons que 5 % ou 6 % du marché mondial, un peu plus pour les produits très chers parce que nous avons un système généreux. Néanmoins, c’est d’autant plus intéressant pour les entreprises qu’un certain nombre de pays suivent ce qui se passe en France et s’en inspirent pour fixer les prix de remboursement.
D’une certaine manière, donc, la France pèse sur les prix. Mais je ne pense pas que nous puissions le faire en nous entendant avec nos collègues.
Mme Catherine Lemorton, rapporteure : Nous avions beaucoup d’autres questions que nous allons vous adresser. Merci beaucoup.
*
Audition de M. Jean-Martin Cohen Solal, directeur général adjoint de la Fédération nationale de la mutualité française, Mme Laure Lechertier, responsable du département politique du médicament, et M. Vincent Figureau, responsable du département relations extérieures.
M. Pierre Morange, coprésident : Madame, messieurs, bienvenue à l’Assemblée nationale.
Mme Catherine Lemorton, rapporteure : Merci d’avoir répondu à notre invitation. Pensez-vous que l’admission des médicaments au remboursement est suffisamment sélective ? De votre point de vue, la franchise de 50 centimes sur le prix des médicaments aura-t-elle un impact positif sur le volume de produits consommés ? Risque-t-elle de créer une inégalité dans l’accès aux soins ?
M. Jean-Martin Cohen-Solal : Merci de nous avoir invités. Je vous prie d’accepter les excuses de M. Daniel Lenoir, qui a eu un empêchement de dernière minute.
Le sujet du médicament nous préoccupe et notre fédération y travaille depuis de très longues années. Le poste « médicament », pour les mutuelles que nous représentons, est le premier poste de dépenses, soit 34 % de celles-ci. Si cette proportion est stable, son volume croît chaque année de 5 %. C’est donc un sujet majeur, en termes à la fois quantitatifs et qualitatifs.
Depuis très longtemps, la mutualité communique sur le médicament. Nous avons été les premiers à parler des génériques. Cela paraissait assez étonnant à l’époque, on s’interrogeait même sur notre légitimité à en parler comme sur l’intérêt des génériques. Or il y a deux jours, un sondage a révélé que, maintenant, 80 % des Français y sont favorables.
S’agissant de l’admission des médicaments au remboursement, la mutualité a proposé, lors de son congrès de Toulouse en 2003, la création d’une haute autorité de santé. Nous avons donc été très été satisfaits de sa création par la loi du 13 août 2004. Nous attendions, avec les autres organismes chargés du médicament, que cette Haute Autorité de santé (HAS) procède à un classement et donne un avis très précis sur la nécessité ou non d’admettre le remboursement. Nous avons regretté que le Gouvernement ne suive pas les avis de la HAS jusqu’au bout et maintienne un remboursement minimum pour certains médicaments à service médical rendu insuffisant ; il s’agissait des veinotoniques. La mutualité, qui représente 38 millions de personnes protégées, a pris alors une décision collective consistant à ne pas rembourser les médicaments remboursés à hauteur de 15 %, les médicaments à vignette orange. Elle suivait en cela l’esprit de la recommandation de la HAS. Cela a provoqué une forte baisse de la prescription, donc de la consommation de ces médicaments en 2006 et 2007.
Pour nous, il est effectivement important que la HAS puisse faire un choix et donner un avis sur l’utilité médicale majeure de tel ou tel produit pharmaceutique. Le poste du médicament croît tous les ans de façon importante. La presse évoque presque quotidiennement le problème de la surconsommation des médicaments en France. Il vaudrait mieux utiliser ces sommes importantes à des médicaments vraiment innovants et vraiment efficaces. Pour cela, nous faisons confiance aux organisations qui sont chargées de le faire.
Nous nous inquiétons de toutes les manœuvres de contournement comme les me too, de l’insuffisance de développement de la politique du générique – même si cela a beaucoup évolué – et de la politique des marges arrières qui nous semble contestable. Un rapport d’IMS Health a montré, la semaine dernière, que la différence de prix des médicaments génériques par rapport aux médicaments princeps n’était pas supérieure à 40 % en France contre 60 à 80 % dans les pays scandinaves probablement, en particulier, à cause des marges arrières. On peut donc aller plus loin dans la politique du générique, ce qui permettrait de consacrer le juste prix aux médicaments.
Nous disposons d’un tableau qui, en tant que professionnel de santé, me paraît intéressant. Il est fait sur la base de molécules phares. S’agissant des antiulcéreux, la différence de prix entre le Mopral princeps – 28 comprimés de 20 mg – et l’Oméprazol générique – 28 comprimés de 20 mg – est de 41,5 %. C’est la même molécule et l’efficacité thérapeutique est la même. Cela représente des sommes non négligeables, qui devraient être mieux utilisées dans le système de santé. Le cas des statines est encore plus marquant : entre le Tahor, le plus prescrit et le plus cher, et la prévastatine en générique, sous une même présentation et le même conditionnement, la différence est de 61,5 % : 37,81 euros et 14,57 euros. Je pourrais vous laisser ce tableau, s’il vous intéresse. Il porte sur des médicaments totalement comparables en termes d’efficacité, sinon des génériques purs. Des économies non négligeables pourraient donc être réalisées, sans diminuer en rien la qualité de la santé publique.
La mutualité a clairement exprimé son opposition au principe même des franchises. Nous estimons que ce n’est pas une bonne solution. Quant à la fixer à 50 centimes par boîte de médicament, cela nous semble étonnant à plusieurs titres.
Premièrement, ce n’est pas l’assuré qui décide de la prescription, c’est le médecin. J’exerce encore comme médecin libéral, je fais des ordonnances, je prescris et je choisis les médicaments pour le patient.
Deuxièmement, l’idée de la franchise est de responsabiliser financièrement le patient en disant que ce dernier va se rendre compte du coût du médicament et qu’il va réfléchir. Mais le prescripteur connaît-il lui-même le coût du médicament qu’il indique sur l’ordonnance ? Je veux bien faire le pari que non. On peut faire l’effort et chercher sur le Vidal le prix du médicament, mais ce n’est pas spontané. Jamais on ne responsabilise le prescripteur sur le prix des médicaments, des analyses ou des examens. Plutôt que de culpabiliser le patient sur la consommation de médicaments, on ferait mieux de sensibiliser le prescripteur à son rôle économique.
Plus généralement, à la mutualité, nous pensons que le corps médical dans son ensemble n’intègre pas suffisamment le fait qu’il est aussi un acteur économique. Quand on fait une prescription, quelle qu’elle soit, on génère une dépense sur des fonds collectifs ou sur des fonds privés, souvent mutualisés. Cette prescription a donc un coût que la collectivité subit. Il ne semblerait pas aberrant d’aller vers des modes de prescription qui permettent au prescripteur de le percevoir. Je pense notamment à des logiciels d’aide à la prescription.
M. Pierre Morange, coprésident : Le principe de la mention du prix des médicaments dans les logiciels d’aide à la prescription a été inscrit, par voie d’amendement, dans le projet de loi de financement de la sécurité sociale (PLFSS) pour 2008. Cette disposition sera définitivement adoptée après le vote de la Haute assemblée.
M. Jean-Martin Cohen-Solal : Nous avions bien noté que c’était l’un des points positifs du PLFSS 2008 voté, en première lecture, par l’Assemblée nationale. Si l’on veut, en effet, que les logiciels d’aide à la prescription soient efficaces, il faut faciliter la vie des médecins et intégrer les prix dans les logiciels. Sinon, ce sera un vœu pieux.
M. Pierre Morange, coprésident : Ce n’est pas un vœu pieux, si on se réfère au texte du PLFSS. De toute façon, le dispositif s’inscrira dans le cadre de logiciels qui devront être certifiés.
M. Jean-Martin Cohen-Solal : En dernier lieu, de nombreuses dépenses de médicaments sont réglées en tiers payant. Lorsque l’assuré ira à la pharmacie, il ne paiera pas la franchise, qui sera seulement imputée sur ses remboursements futurs. Pour que cette franchise soit perceptible, il faudrait supprimer le tiers payant, ce qui serait un recul social et technique. Enfin, en cas de traitements répétitifs, la franchise peut représenter un coût non négligeable de 50 euros par an, et la mesure peut alors être vécue, en effet, comme source d’inégalité.
M. Pierre Morange, coprésident : Faites-vous la même analyse s’agissant du forfait hospitalier ?
M. Jean-Martin Cohen-Solal : À l’origine, le forfait hospitalier a été instauré pour compenser les dépenses de bouche des patients hospitalisés. Puis le système a dévié. Son montant a tellement augmenté qu’on ne peut soutenir aujourd’hui qu’il ne sert qu’à compenser les dépenses de bouche.
Nous craignons que la franchise ne subisse le même sort. En effet, si la loi l’a instituée, elle n’en a pas fixé le montant, qui est d’ordre réglementaire ; et nous avons peur que, par la suite, on utilise ce mécanisme pour remplir les caisses de l’assurance maladie.
M. Pierre Morange, coprésident : Je considère que le forfait hospitalier, qui a connu des augmentations conséquentes, n’est plus pertinent. Il faut plutôt considérer le reste à charge. Je pense notamment aux personnes âgées dépendantes accueillies en établissements d’hébergement. En effet, il n’y a aucune prise en charge, ce qui pose le problème de l’inégalité d’accès aux soins.
M. Jean-Martin Cohen-Solal : On peut craindre la multiplication de ces mesures.
M. Pierre Morange, coprésident : Il faudrait que les principes républicains d’équité et d’égalité d’accès aux soins soient préservés.
M. Jean-Martin Cohen-Solal : En outre, la franchise n’est pas structurante. Elle ne modifie pas l’organisation du système de soins, ni du médicament. Ce n’est qu’une mesure comptable.
Mme Catherine Lemorton, rapporteure : Elle ne répond pas non plus aux problèmes d’excès de consommation de médicaments et de mauvais encadrement de la prescription. Une telle mesure ne s’inscrit pas dans une vision de la politique de maîtrise des dépenses de médicaments. Nous constatons que celle-ci est aujourd’hui quasiment inexistante.
Nous constatons également une disparité au niveau organisationnel : trois instances différentes, l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS), la Haute Autorité de santé (HAS) et le Comité économique des produits de santé (CEPS), et par ailleurs, des pouvoirs de négociation conférés aux nombreux industriels, d’où parfois un manque de cohérence dans l’accès au marché des médicaments.
M. Pierre Morange, coprésident : Nous avons bien compris votre position, mais l’objet de la MECSS est bien de veiller à la rationalisation de l’utilisation des deniers publics dans le domaine sanitaire et social et de préserver ces conditions d’accès républicain. Sa composition paritaire lui permet de s’exonérer de prises de position politiques. On y prend acte des positions de chacun, mais ce n’est pas une tribune.
Mme Catherine Lemorton, rapporteure : L’objectif des franchises, auxquelles je suis opposée, est tout de même de réduire la consommation de médicaments, dès lors qu’ils sont pris en charge par l’assurance maladie. Cependant, on remarque que, dès lors qu’ils ne sont plus remboursés, on en facilite l’accès. Pourtant, ainsi que Mme Roselyne Bachelot, ministre de la santé, de la jeunesse et des sports, l’a répété dans l’hémicycle, il y a la volonté de favoriser la vente des médicaments devant le comptoir des pharmacies, quasiment en libre-service. Voilà la réalité.
Quel regard la mutualité porte-t-elle sur la formation continue des médecins ?
M. Jean-Martin Cohen-Solal : La formation initiale en matière de thérapeutique courante est notoirement insuffisante. La formation médicale continue, malgré un certain nombre de textes allant dans le bon sens, n’a pas pris l’essor qu’elle aurait dû avoir dans un pays comme le nôtre. La place prise par l’industrie pharmaceutique compense, de facto, un manque de formation médicale continue organisée et cohérente.
Il est inévitable que l’industrie pharmaceutique fasse en sorte de développer la vente de ses produits. Selon les chiffres récents de l'Inspection générale des affaires sociales (IGAS), 25 000 euros sont dépensés par généraliste, chaque année, pour la visite médicale, laquelle mobilise 23 000 visiteurs médicaux. Au total, c’est une somme de 3 milliards d’euros qui est dépensée pour la visite médicale, ce qui est sans commune mesure avec les pays voisins. Cela est dû notamment au fait que la formation médicale continue est mal organisée.
Quelles en sont les raisons ?
La mutualité française considère que le mode d’exercice libéral des médecins et le paiement exclusif à l’acte ne favorisent pas la formation médicale continue. En outre, on n’aide pas les médecins à se former régulièrement. Pour eux, avoir accès aux dernières données de la science demande un effort important. Il est beaucoup plus simple qu’un visiteur médical vienne les leur exposer, entre deux patients. Cela s’intègre facilement à leur quotidien.
On ne pourra pas isoler ce problème de la formation médicale continue si l’on ne prend pas en compte l’organisation générale du système. Le président de la mutualité a regretté que la médecine libérale en France soit toujours organisée selon la Charte de 1927, alors que la médecine a fondamentalement évolué. Moi-même, je me suis aperçu que, depuis trente-quatre ans que je suis sorti de la faculté, s’agissant d’une pathologie simple comme celle de l’ulcère de l’estomac, les diagnostics avaient changé plusieurs fois et les thérapeutiques avaient été profondément modifiées. Face à une telle évolution, un médecin, seul dans son cabinet, a du mal à s’informer. Rien n’est fait pour le favoriser. Il faudrait donc que les organismes publics, notamment la HAS, dont c’est l’une des missions, fassent davantage pour améliorer la formation médicale continue. La HAS doit faire en sorte que les informations des guides de bonne pratique qu’elle diffuse soient aisément assimilables et intégrables dans le quotidien des médecins ; les schémas thérapeutiques doivent être simples et compréhensibles.
M. Pierre Morange, coprésident : Le réseau mutualiste a décidé collectivement de ne pas rembourser certains médicaments. Quelle a été l’incidence financière de ces mesures sur la comptabilité de la mutualité française en termes d’économies ?
M. Jean-Martin Cohen-Solal : Si les mutuelles avaient décidé de rembourser les médicaments à 15 %, elles auraient dû augmenter leurs cotisations de deux points.
M. Pierre Morange, coprésident : J’ai du mal à comprendre ce raisonnement, sachant que ces médicaments étaient auparavant remboursés à 35 %.
M. Jean-Martin Cohen-Solal : Plus précisément, faire passer le remboursement de ces médicaments de 35 % à 15 % ou 20 % aurait abouti à une augmentation des cotisations de deux points.
Ce choix a été difficile à expliquer à nos adhérents. Il nous a fallu faire beaucoup d’efforts de communication, notamment au sein de la presse mutualiste, pour leur faire comprendre pourquoi nous ne remboursions plus ces médicaments. Nous leur avons dit que nous suivions en cela les avis de la Haute Autorité de santé.
Ce que l’on a pu faire pour les médicaments, à 15 %, ceux à vignette orange, n’est pas possible pour les médicaments à 35 %, à vignette bleue. En effet, les organismes d’assurance complémentaire ne connaissent que les pourcentages de remboursement et ne peuvent pas faire un tri entre les médicaments à service médical rendu insuffisant (SMRI) et à SMR important.
Mme Laure Lechertier : En fait, vous nous interrogez sur l’impact de ces mesures administratives sur le ticket modérateur. Nous avons fait un tableau récapitulatif sur 2006, qui permet de constater les économies réalisées : 267 millions d’euros grâce au déremboursement de certains médicaments à SMRI ; 41 millions d’euros, grâce à la baisse du taux de remboursement et à une baisse de prix de 12 %, etc. Tout cela rentre dans une gestion dynamique du panier de soins, en vue de financer des thérapeutiques de qualité.
M. Pierre Morange, coprésident : L’économie de 300 millions d’euros réalisée par la mutualité au travers de mesures de déremboursements a-t-elle porté exclusivement sur le médicament ou sur une prise en charge plus globale sur les frais d’optique, les frais dentaires ou autres ? Est-ce que ces économies étaient proportionnelles aux pourcentages de dépenses que vous avez évoqués ? Est-ce que le poste des médicaments, qui représente 34 % de vos dépenses, a donné lieu à des économies à hauteur de 34 % de ces 300 millions d’euros ?
M. Jean-Martin Cohen-Solal : Les dépenses ne sont pas affectées de cette façon-là. Parallèlement, il y a eu des baisses de remboursement des médicaments à 35 %, et de nouvelles charges sur les mutuelles. Globalement, l’augmentation des dépenses des mutuelles suit l’augmentation des dépenses de santé de l’assurance maladie obligatoire. En l’occurrence, c’est l’augmentation des cotisations qui a pu être ainsi modulée.
Mme Catherine Lemorton, rapporteure : L’analyse que je vais développer pour le déremboursement des veinotoniques vaut pour les déremboursements d’autres classes. On sait que le déremboursement des veinotoniques s’est traduit par un transfert sur la contention, qui est dans la classe des dispositifs et non des médicaments. Je ne le regrette pas, car la contention est bien plus efficace que les veinotoniques. Avez-vous pris en compte ce transfert ?
M. Jean-Martin Cohen-Solal : Non ! Certaines mutuelles prennent en charge les bas de contention, d’autres non. Effectivement, il y a eu un transfert, mais, objectivement, les dépenses de moyens de contention ne sont pas du tout du même niveau que les dépenses de veinotoniques. Comme nous ne disposons pas de données suffisamment fines, nous n’avons pas pu chiffrer ce transfert.
On a dit aussi que ce déremboursement avait provoqué une augmentation de la consommation d’autres médicaments, comme les antalgiques. Mais, là encore, nous sommes incapables d’apprécier s’il y a bien eu un transfert ou si, tout simplement, nous sommes face à un phénomène culturel, celui des Français vis-à-vis du médicament. Dans son discours au Sénat, le Président de la République a rappelé que 90 % des visites chez le médecin en France se terminent par une ordonnance, contre 40 à 60 % dans les pays du Nord : 40 % aux Pays-Bas.
On peut enfin penser que, face aux problèmes de jambes lourdes, l’exercice physique, la contention, le fait de surélever les pieds, sont souvent au moins aussi efficaces que des médicaments.
M. Pierre Morange, coprésident : Vous avez dit que la baisse du remboursement à 15 % des médicaments à 35 % se serait traduite pour vous par une augmentation des cotisations de deux points. Confirmez-vous les chiffres du rapport du Haut conseil pour l’avenir de l’assurance maladie, qui s’était penché sur le système assurantiel en général ? Celui-ci évoquait le haut niveau de l’assurance obligatoire français et remarquait que le transfert d’un point d’assurantialité obligatoire sur les complémentaires se traduirait par une augmentation de 4 % des primes de ces complémentaires.
M. Jean-Martin Cohen-Solal : C’est le rapport que nous avons effectivement en tête. C’est le débat qui aura lieu sans doute dans les prochains mois sur la répartition régime obligatoire/complémentaires.
M. Pierre Morange, coprésident : C’est vous qui avez fait ce calcul ?
M. Jean-Martin Cohen-Solal : Non, ce sont les services du Haut conseil.
M. Pierre Morange, coprésident : La philosophie est différente, entre les complémentaires et le régime obligatoire qui prend en compte la réalité sanitaire et ne fait pas de sélection.
M. Jean-Martin Cohen-Solal : C’est tout le débat sur le rôle des complémentaires, qui sont des assurances santé de droit privé, avec des contrats différents et des risques importants de transfert de l’obligatoire vers le complémentaire. La mutualité pourra contribuer à ce débat.
Mme Catherine Lemorton, rapporteure : On sait que des médecins travaillent dans les centres de soins de la mutualité. Comment est assurée leur formation continue ? Est-elle différente de celle des médecins libéraux, seuls dans leur campagne ou dans leur cabinet et qui ont du mal à s’informer ? Les médecins de la mutualité ont-ils d’autres comportements lorsqu’ils font des prescriptions ?
M. Jean-Martin Cohen-Solal : C’est une très bonne question. Dans les établissements mutualistes, nous faisons en sorte de privilégier une formation de meilleur niveau, en équipe, et avec une dimension médico-économique. Pour nous, la santé publique passe par une adéquation des moyens aux besoins. Le fait d’intégrer des données médicales et économiques pour mieux utiliser des ressources collectives consacrées à la santé doit faire partie de la base de la réflexion du corps médical.
Sans que ce soit parfait, nous essayons de faire des efforts pour dégager du temps et des moyens afin de former les médecins. Nous essayons également de faire passer des informations spécifiques aux médecins mutualistes. Néanmoins nous n’avons pas d’éléments fiables montrant que la prescription est très différente dans les établissements mutualistes. Quoi qu’il en soit, l’effort de formation est intégré dans l’exercice. Nous nous sommes notamment battus pour que les médecins fassent des prescriptions de génériques ou en dénomination commune internationale (DCI).
M. Pierre Morange, coprésident : Vous avez évoqué les marges arrière. La mutualité a-t-elle mené des réflexions ou fait des propositions sur ce sujet ?
M. Jean-Martin Cohen-Solal : Nous considérons comme particulièrement choquantes les marges arrière sur les génériques, tout comme les remises sur un certain nombre de médicaments. Il y a une certaine opacité s’agissant du calcul du prix des médicaments. Le système des marges arrière est aberrant. J’ai d’ailleurs noté que le Président de la République s’était prononcé pour leur suppression dans la grande distribution ; je pense qu’il visait le principe même des marges arrière. Cela me semble la bonne attitude et j’espère qu’elle concernera aussi le domaine du médicament.
Nous avons constaté, concernant les marges arrière pour les génériques, qu’il devait y avoir une limite de 15 % ; qu’il devait y avoir un contrôle de la Direction générale de la concurrence, de la consommation et de la répression des fraudes (DGCCRF), mais qu’un moratoire avait été institué. Nous souhaitons que ce moratoire soit levé et, surtout, qu’on aille vers la suppression du système des marges arrière. Ce dernier explique en partie, à notre sens, que la différence de prix entre les médicaments princeps et les médicaments génériques en France ne soit pas aussi importante que dans d’autres pays.
Nous souhaitons donc que l’on revienne sur le problème des marges arrière.
M. Pierre Morange, coprésident : Pour les génériques ?
M. Jean-Martin Cohen-Solal : Oui, car il faut qu’il y ait une vérité des prix et que la baisse des prix, notamment des génériques, bénéficie au consommateur.
Il en est de même des remises de prix, qui bénéficient à l’assurance maladie obligatoire, mais pas aux complémentaires, ni à l’assuré. C’est un problème de vérité des prix du médicament.
Mme Laure Lechertier : Nous ne souhaitons pas voir se développer les mécanismes qui déconnectent un prix facial d’un prix réel.
M. Jean-Martin Cohen-Solal. Nous souhaitons une vraie transparence des prix, et que les payeurs paient le prix réel.
Mme Catherine Lemorton, rapporteure : Il faut rappeler l’historique des marges arrière et la volonté de favoriser la pénétration des génériques. Certes, il convient de revenir sur les marges arrière qui ont forcément une incidence sur le prix des génériques, mais cela doit se faire obligatoirement avec la prescription en DCI.
M. Jean-Martin Cohen-Solal : C’est notre position depuis plusieurs années. Cela nous ramène à notre propos sur la formation médicale initiale et continue. Il est exact que les médecins n’ont pas appris la prescription en DCI. Les professionnels de santé ont en tête des noms de produits ; il faut leur mettre en tête des noms de DCI, ce qui est tout de même plus simple. Vous avez donc raison, et je pense que le mouvement prendra progressivement de l’ampleur, tout comme pour les génériques.
M. Pierre Morange, coprésident : Cela nous ramène à la réflexion sur les logiciels d’aide à la prescription, qui sont devenus indispensables.
Mme Laure Lechertier : Avec une base de données publique, permettant cette prescription directe, ce qui n’est pas le cas aujourd’hui.
M. Jean-Martin Cohen-Solal : Ces logiciels devraient permettre à la fois la prescription en DCI et le calcul des coûts.
M. Vincent Figureau : Vous avez évoqué l’avancée permise par l’Assemblée nationale sur la DCI. Cependant cette avancée avait déjà eu lieu dans la loi portant diverses dispositions d’adaptation au droit communautaire sur le médicament du 26 février 2007. C’était d’ailleurs la résultante d’une de nos propositions qui avait été reprise par la rapporteure de l’Assemblée nationale, Mme Cécile Gallez, adoptée à l’unanimité et conservée ensuite au Sénat. Par ailleurs, la commission des affaires sociales du Sénat a adopté hier un amendement mettant en place une banque de données publique sur le médicament. Nous nous en réjouissons, car c’était, là encore, une de nos propositions.
M. Jean-Martin Cohen-Solal : Je veux préciser que nous publions des documents sur le médicament, ainsi qu’un certain nombre de rapports. Nous envoyons également tous les ans à nos responsables et aux élus un mémento du médicament.
En conclusion, je m’étonne qu’aujourd’hui, malgré toute l’information diffusée sur les problèmes de surconsommation en France, on ait du mal à passer vraiment à l’acte. Il serait bon qu’il y ait quelque contrôle s’agissant du lobby pharmaceutique. Dans le domaine de la formation continue et de la visite médicale, les médecins n’ont pas de contre-pouvoir pour s’opposer aux laboratoires, qui font par ailleurs leur métier. Nous souhaitons qu’il y ait de plus en plus de médicaments innovants. La mutualité n’est pas contre l’industrie pharmaceutique, contrairement à ce qui a été dit. Simplement, il faut consacrer la juste ressource aux médicaments innovants et à la recherche, aux dépens, notamment, de la promotion. Nous souhaitons également que les élus prennent des décisions pour modifier vraiment la politique du médicament en France, qui a besoin d’être modernisée.
Mme Catherine Lemorton, rapporteure : Quelle est votre position sur les délégués de l’assurance maladie ?
M. Jean-Martin Cohen-Solal : C’est une bonne chose, même si j’ai lu dans le compte rendu de votre audition du directeur de la CNAMTS, qu’il en est prévu 950 pour la fin de 2007, alors qu’il y a 23 000 visiteurs médicaux.
Dans le mode de fonctionnement des professionnels de santé, le contact humain est beaucoup plus simple que le contact écrit. Reste à savoir si, aujourd’hui, l’assurance maladie a les moyens d’affecter autant de personnes pour aller discuter avec ces professionnels.
La formule permet également aux médecins de mieux comprendre l’assurance maladie, qui n’est pas leur adversaire, et d’améliorer, de façon générale, les relations entre les payeurs et les professionnels de santé. On ne fera évoluer le système que s’ils parviennent à travailler ensemble.
M. Pierre Morange, coprésident : Nous avions encore toute une série de questions à vous poser, que nous nous permettrons de vous adresser prochainement. Je vous remercie.
*
Audition de M. le Professeur Robert Nicodème, membre du Conseil national de l'Ordre des médecins, vice-président de la section formation et compétences médicales.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale pour cette audition qui s’inscrit dans le travail qu’effectue notre mission sur la prescription, la consommation et la fiscalité des médicaments. Je donne sans plus tarder la parole à notre rapporteure.
Mme Catherine Lemorton, rapporteure : En effet, notre mission se penche sur le problème du médicament en France. Et c’est à dessein que je parle de « problème » car nous savons bien qu’il existe, en particulier par rapport à nos voisins européens, une hyperconsommation qui a des conséquences sur les comptes de la sécurité sociale. Le but de cette étude est donc d’en comprendre les raisons pour, si possible, trouver des solutions.
Nous tenions tout particulièrement à auditionner l’Ordre des médecins – le tour de l’Ordre des pharmaciens viendra bientôt –, tout simplement parce qu’ils sont les principaux prescripteurs de médicaments.
Pourriez-vous en premier lieu nous indiquer quelle est la durée moyenne d’une consultation chez un médecin généraliste ?
M. Robert Nicodème : Je suis particulièrement sensible à votre invitation et il est particulièrement intéressant pour un médecin qui a plus de trente années d’expérience de parler devant vous du médicament.
En moyenne, les médecins généralistes effectuent trois à quatre consultations par heure. Leur activité est de vingt consultations par jour, cinq jours par semaine.
Mme Catherine Lemorton, rapporteure : J’avais pour ma part entendu parler d’une durée de sept minutes pour une consultation…
M. Robert Nicodème : Les données varient considérablement en fonction des territoires. S’il est facile d’établir une moyenne nationale grâce aux données de l’assurance maladie, on se rend compte que dans certaines régions de moindre densité médicale, on arrive jusqu’à 40 ou 50 consultations par jour.
Mme Catherine Lemorton, rapporteure : Savez-vous combien, toujours en moyenne, les médecins reçoivent de visiteurs médicaux chaque année ?
M. Robert Nicodème : Non et j’ignore si quelqu’un le sait. Il est vrai que le nombre de visites est important, mais les médecins savent les réguler eux-mêmes : ils reçoivent deux ou trois visiteurs par semaine ou un par jour, en début de consultation. Néanmoins il faut aussi savoir que les laboratoires ont recours à des visiteurs différents pour présenter les mêmes médicaments.
Mme Catherine Lemorton, rapporteure : Les généralistes sont-ils confrontés à des difficultés dans leur formation continue ? Le Conseil de l’Ordre s’est-il penché sur cette question ? Pouvez-vous nous présenter un tableau de cette formation et des voies choisies par les médecins libéraux ?
M. Robert Nicodème : Les ordonnances Juppé de 1996 avaient prévu la mise en place d’un Conseil national de la formation médicale continue (FMC), mais celui-ci a fait long feu en raison de problèmes de financement et de difficultés à trouver des référents.
La loi du 9 août 2004 relative à la politique de santé publique a institué trois conseils nationaux : un pour les médecins libéraux, un autre pour les médecins salariés et un troisième pour les médecins hospitaliers. Ils doivent être chapeautés par un comité de coordination, mais nous attendons toujours les décrets d’application pour mettre en place les conseils régionaux de la FMC, qui seront adossés aux conseils régionaux de l’Ordre, ce dernier en assurant le financement.
L’objectif est d’améliorer la formation médicale continue et l’évaluation des pratiques professionnelles, qui seront obligatoires dès le début de l’année prochaine pour chaque médecin, qu’il soit libéral, salarié ou hospitalier.
M. Jean Mallot, coprésident : Comment sont déterminées les priorités en matière de formation ? On comprend que les médecins souhaitent eux-mêmes se former dans un certain nombre de domaines, mais la puissance publique, y compris l’Ordre des médecins, doivent aussi pouvoir faire passer leur priorité au sein du système de formation.
M. Robert Nicodème : Les priorités varient selon les spécialités. Globalement, il faut veiller à l’actualisation des connaissances en matière de diagnostic et de thérapeutiques, qu’il s’agisse d’inciter à renoncer à certaines techniques de prise en charge qui n’ont pas véritablement prouvé un bénéfice pour le patient ou qui ont été à l’origine d’effets indésirables, ou de mettre en place de nouvelles activités et de nouveaux modes de prise en charge.
Bien évidemment, les priorités concernent aussi le médicament.
M. Jean Mallot, coprésident : Peut-on imaginer que les autorités qui ont en charge ce secteur décident que, pour une période donnée, la formation des médecins se concentrera sur tel ou tel aspect, par exemple pour les amener à modifier leurs habitudes de prescription ?
M. Robert Nicodème : La prescription de médicaments évolue. Par exemple, la prescription des nouveaux anti-inflammatoires connus sous le nom de Coxibs a été complètement modifiée après la mise en évidence d’événements indésirables. De même, à la suite de l’intervention de la Haute Autorité de santé (HAS), la stratégie thérapeutique a beaucoup évolué en ce qui concerne le traitement hormonal substitutif (THS) de la ménopause. Il s’agit bien là d’orientations ciblées.
S’agissant encore de la formation continue, on peut s’étonner que soient désormais appelées à coexister une formation continue conventionnelle, rémunérée au motif qu’elle correspond aux thèmes fixés par l’assurance maladie, et une formation continue obligatoire non rémunérée, bien qu’elle poursuive en fait les mêmes objectifs.
M. Pierre Morange, coprésident : Pourriez-vous nous indiquer concrètement à combien d’heures de formation correspondent ces FMC obligatoires et conventionnelles ? Ce qui nous intéresse est de savoir, au-delà des textes, quelle est la formation continue dont bénéficient effectivement les médecins sur le terrain.
J’aimerais aussi connaître votre sentiment sur les logiciels d’aide à la prescription.
M. Robert Nicodème : La durée de la FMC est codifiée par le Conseil national sous forme de crédits de formation. Je ne dispose pas ici des chiffres exacts mais je vous les ferai parvenir. Chaque médecin doit recevoir un certain nombre de ces crédits pour valider sa FMC obligatoire. Le système impose également une évaluation, collective ou individuelle, des pratiques professionnelles. Ces obligations ne sont pas très lourdes.
M. Pierre Morange, coprésident : Au-delà du montant des crédits de formation, ce qui nous intéresse est de savoir à quoi ils correspondent dans les faits, même si nous sommes conscients qu’il faut s’attendre à une montée en charge de ce nouveau dispositif.
M. Robert Nicodème : Ces informations existent et je vous les adresserai également.
Nous ferons par la suite l’évaluation de ce dispositif.
Il existe en effet des logiciels de prescription, dont le plus connu est édité par Vidal. D’autres logiciels jouent un rôle beaucoup plus précis et leur diffusion est plus confidentielle. C’est par exemple le cas de ceux qui calculent la dose de traitement d’antivitamine K et d’anticoagulant en fonction des résultats biologiques de la coagulation sanguine ou de ceux qui déterminent le risque cardio-vasculaire pour savoir s’il faut ou non traiter. L’ensemble de ces logiciels sont édités soit par le groupe Vidal soit par l’industrie pharmaceutique.
S’agissant plus particulièrement du médicament et de la prescription, j’observe que les structures en place travaillent toujours avec un temps de retard. Ainsi, l’industriel qui lance une nouvelle molécule mène des études rigoureuses au vu desquelles il demande une autorisation de mise sur le marché (AMM) européenne qui est ensuite validée par la France. De la sorte, l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) et la HAS étudient les programmes validés par les industriels mais elles-mêmes ne produisent aucune étude sur le médicament. Pourtant, pour rigoureuses qu’elles soient, ces études sont parfois éloignées de la vraie vie en ce qu’elles retiennent des critères d’exclusion importants, dans la mesure où, pour apprécier les états d’un médicament donné, il est légitime d’éviter les interférences. Or, dans la vraie vie les interférences sont fréquentes.
En ce qui concerne la toxicité du médicament, il existe un certain nombre d’événements indésirables. Reprenons l’exemple des Coxibs, qui ont été un des groupes de médicaments les plus vendus ces dernières années et qui ne le sont pratiquement plus, le dernier venant d’être retiré du marché. En fait, les évènements indésirables ont été observés, à un moment où le produit était déjà très largement utilisé, alors même que, pour un coût élevé, son service médical rendu (SMR) était comparable à celui des autres anti-inflammatoires.
N’oublions pas qu’il n’y a guère chaque année qu’un ou deux médicaments vraiment innovants mais qu’il en sort des dizaines qui appartiennent à des classes thérapeutiques existantes. Les centres de pharmacovigilance travaillent précisément à l’amélioration du service médical rendu (ASMR), qui est notée de 1 à 5, et je rends hommage au travail accompli à Toulouse par le professeur Montastruc, qui avait levé le lièvre des Coxibs deux ans et demi avant tout le monde.
Pour l’instant, aucun logiciel ne mentionne l’ASMR, alors que ce serait de nature à aider le prescripteur.
Ce qui gêne aussi le prescripteur c’est le trop grand nombre de médicaments par classe thérapeutique. Si l’on prend l’amoxicilline, pénicilline qui a été un grand progrès il y a plus de vingt-cinq ans et qui marche toujours très bien, il y en a plus de cinquante sur le marché. Les hôpitaux ont fait le ménage : au sein d’une classe, ils choisissent un seul produit. Dans le secteur libéral, on a les 50 amoxicillines et on demande aux médecins de faire le choix. Il me semble que, sans retirer pour autant les autres produits du marché, l’assurance maladie pourrait quand même décider que c’est tel médicament qui rend le meilleur service et qui a le meilleur rapport coût-efficacité. Un logiciel qui serait élaboré par une structure indépendante pourrait y aider.
Vous m’avez également interrogé sur l’articulation avec la HAS. Celle-ci marque incontestablement un grand progrès au plan professionnel : elle permet aux médecins de disposer de recommandations claires qui les aident. Cela étant, il en manque beaucoup. Par exemple quand on pose la question de savoir s’il faut donner des hypolipémiants pour faire baisser le taux de cholestérol chez les personnes de plus de 80 ans, personne n’apporte de réponse. Un grand nombre de patients concernés sont sous statines, avec une bonne efficacité sur le taux de cholestérol, mais quand on en discute de façon scientifique au plus haut niveau, personne n’est capable de dire si c’est ou non ce qu’il faut faire.
Même si l’on ne dispose pas de référence scientifique solide, il me semble que la Haute Autorité de santé pourrait donner une orientation, par exemple en disant que lorsqu’une personne va bien après 80 ans, même avec 3 grammes de cholestérol, il n’est pas nécessaire de la traiter. Cela apporterait une véritable aide et ferait diminuer la prescription de médicaments.
Mme Catherine Lemorton, rapporteure : On sait que les industries pharmaceutiques investissent chaque année environ 25 000 euros par médecin dans le cadre de la visite médicale. Pensez-vous que celle-ci est trop prégnante dans les cabinets ? Un récent rapport de l’Inspection générale des affaires sociales (IGAS) est assez critique à l’égard de cette pratique ; c’est un sujet qui nous intéresse tout particulièrement et nous aimerions avoir votre avis à ce propos.
M. Robert Nicodème : Les grandes études scientifiques sur les médicaments sont faites par l’industrie. Les grands congrès mondiaux qui font autorité dans les spécialités n’existent que par l’industrie. Bien entendu, les visiteurs médicaux s’appuient sur ces références, c’est-à-dire sur les travaux réalisés par leurs entreprises et par les interventions favorables à leurs produits effectuées à l’occasion des congrès. Or, si les progrès sont indéniables, il est évident que le coût pourrait être beaucoup amélioré au bénéfice de l’assurance maladie.
On dit souvent que la prescription médicamenteuse est une prescription technique : il y a un examen, un diagnostic et l’on prescrit le médicament qu’il faut. Il n’y a donc là rien de magique. Pourtant, une revue sérieuse titrait récemment sur « l’art de transformer les perceptions en prescriptions »… On retrouve là toute la dimension non scientifique de la prescription médicale, qui correspond à une demande des patients, pour lesquels il y a en effet une part de magie ou d’irréel, ainsi qu’une représentation de la maladie et du médicament qui peuvent induire une demande. Et c’est peut-être ainsi que, lorsqu’une personne âgée est en pleine forme mais a un peu de cholestérol, le médecin est tenté de prescrire une statine. C’est là qu’intervient l’effet de la visite médicale, qui a montré tous les bénéfices que pouvait avoir un médicament donné, qui a donné un grand nombre d’arguments scientifiques que le médecin a, consciemment ou inconsciemment, intégré et transformé en prescription.
D’ailleurs la visite médicale marche très bien : plus il y en a, plus on prescrit. Qui plus est – on rejoint là la question sur la HAS –, il n’existe pas de contre-pouvoir à cette action de l’industrie pharmaceutique. Cela serait pourtant possible, par une meilleure articulation entre la HAS, l’AFSSAPS et le conseil scientifique de la Caisse nationale d’assurance maladie des travailleurs salariés (CNAMTS). Ainsi pourrait-on, si ce n’est produire des recommandations scientifiques, au moins aider les médecins en leur donnant des orientations dans les domaines qui ne sont pas scientifiquement démontrés. Ce serait une bonne chose et un logiciel pourrait y aider.
M. Pierre Morange, coprésident : On voit bien qu’il y a là un triptyque avec d’une part la recommandation sur la prescription médicale, et l’idée d’y intégrer le SMR, d’autre part la formation, à propos de laquelle vous constatez que l’on part d’assez bas, ce qui pourrait nous amener à formuler des préconisations, et enfin l’évaluation. Comment articuler ces trois idées afin d’être véritablement opérationnel en combinant satisfaction des besoins des patients et rationalisation des moyens ?
M. Robert Nicodème : La formation se met en place, les médecins se forment. On a d’ailleurs une médecine de soins habituels de très bonne qualité : la population est prise en charge et le tissu médical assume ses responsabilités à l’égard de la santé publique.
Il faut en fait distinguer trois types de formation. La première porte sur les connaissances pures et l’on peut regretter de ce point de vue que les référentiels ne s’appuient que sur les travaux des laboratoires, alors qu’ils pourraient aussi se fonder sur des groupes d’experts, pour peu qu’on les réunisse et qu’ils parviennent à des conclusions claires.
Le second type de formation porte sur l’activité du médecin dont le contenu doit progresser afin de mieux assurer son rôle de santé publique. Ainsi, compte tenu de la raréfaction de certains spécialistes, un généraliste doit se mettre à niveau en pédiatrie ou en gynécologie.
Le troisième niveau est celui de la coordination des soins, du travail en équipe : le médecin doit être capable d’organiser des soins, de demander des avis. C’est ce qui se fait déjà à travers les « groupes de pairs ».
Pour l’évaluation il est également nécessaire de comparer l’activité du médecin avec des référentiels quand il en existe ou avec celle de ses confrères qui se trouvent dans la même situation, c’est aussi le rôle des groupes de pairs.
Mme Catherine Lemorton, rapporteure : Quels pourraient être selon vous les contre-pouvoirs à la visite médicale des laboratoires ? Ce rôle pourrait-il être tenu par les délégués de l’assurance maladie, dès lors qu’on leur donnerait un autre cahier des charges, une autre mission et une autre formation ?
M. Robert Nicodème : C’est à tort que j’ai utilisé l’expression « contre-pouvoir », mieux vaut parler de : « autres références ».
Les délégués de l’assurance maladie font un excellent travail : ils constatent qu’un médecin consomme plus que les confrères de son secteur, qu’il prescrit plus de transports en ambulance ou plus de médicaments de tel ou tel type par rapport à la moyenne, mais ils n’apportent pas de références et leurs relevés des écarts d’activité ne comportent pas de dimension technique. En fait, ils constatent mais ils n’apportent pas de solution ; ils sensibilisent les médecins, mais ils ne développent pas d’argumentation. C’est toute la différence avec la visite médicale : le laboratoire présente un argumentaire scientifique en faveur de son produit.
À l’université, nous avons fait depuis quelques années un énorme travail de formation à la lecture critique des articles scientifiques. Nous apprenons aux internes à regarder quels sont exactement les critères de jugement de l’étude. En fait, un seul critère doit prévaloir, celui de la morbidité-mortalité : quand on donne une statine à une personne de 80 ans, ce qui est intéressant ce n’est pas de voir si son taux de cholestérol va chuter mais si elle va vivre une ou deux années de plus qu’elle n’aurait vécu sans le médicament. Le taux de cholestérol est un indicateur technique, ce n’est pas un résultat pour la santé.
M. Jean Mallot, coprésident : Verriez-vous un intérêt à ce que, en prévoyant des contreparties, on aille vers une sorte de contractualisation avec chaque médecin sur des objectifs précis, notamment en termes de prescription ? Quels pourraient être les obstacles à une telle démarche ? Comment la mettre en œuvre concrètement ?
M. Robert Nicodème : L’idée de l’assurance maladie d’octroyer quelques avantages aux médecins en contrepartie d’une réduction des coûts a fait l’objet d’un courrier du Conseil national de l’Ordre rappelant qu’on ne peut pas contractualiser l’activité des médecins sur la base d’une diminution des coûts car ils doivent demeurer indépendants afin de se consacrer à leur objectif principal qui est le soin apporté aux patients. Nous sommes d’accord pour que l’on s’efforce de réduire les surcoûts mais nous refusons une contractualisation sur la base d’un objectif purement financier.
Nous nous sommes beaucoup intéressés à cette question de l’implication personnelle de chaque médecin dans une amélioration globale de la prescription. Il faut en particulier prendre en considération le fait que près de la moitié des internes qui sortent d’une formation en médecine générale se tournent vers les urgences, la gériatrie, la médecine polyvalente, voire vers des remplacements, mais qu’ils ne veulent pas s’installer en ville car ils trouvent que les contraintes de l’exercice libéral sont trop importantes. C’est en particulier le cas des jeunes filles, qui sont désormais la moitié des internes et qui jugent souvent l’exercice libéral incompatible avec la maternité. Cet état de fait va aggraver la crise de la démographie médicale.
Dans ces conditions, si on impose aux médecins, outre la permanence des soins et le risque de la responsabilité médico-légale – sujet sur lequel il faudra bien revenir un jour –, des objectifs financiers dans leur façon de travailler, je crains fort que l’on ne déstabilise totalement cette profession. Il faut donc faire très attention.
M. Jean Mallot, coprésident : Dans ces conditions, sur quels objectifs faudrait-il contractualiser ?
M. Robert Nicodème : Aux termes de l’article 8 du code de déontologie médicale, « le médecin doit limiter ses prescriptions et ses actes à ce qui est nécessaire à la qualité, à la sécurité et à l’efficacité des soins. ». Il faut en rester là et je pense que cette audition doit avant tout nous amener à insister sur la nécessité de trouver des références dans des domaines qui sont mal balisés pour la prescription des médicaments.
Mme Catherine Lemorton, rapporteure : Nous aimerions quand même comprendre pourquoi, en France, 90 % des consultations débouchent sur une prescription de médicaments, contre 45 % aux Pays-Bas et moins de 75 % en Allemagne.
Pour notre part, nous avons d’ores et déjà identifié deux pistes et nous aimerions savoir si vous considérez que c’est bien dans cette direction qu’il faut aller ou s’il existe à vos yeux d’autres explications à ce décalage.
En premier lieu, dans la mesure où nous avons observé l’importance de la visite médicale des laboratoires pharmaceutiques dans notre pays, il serait intéressant de savoir si elle est aussi prégnante dans les autres pays d’Europe.
En second lieu, on peut se demander s’il n’y a pas là une conséquence du paiement à l’acte, qui conduit à une inflation du nombre des consultations, donc à la nécessité de chercher à répondre rapidement à la demande du patient. Pour reprendre l’exemple des statines, il paraît en effet plus facile, plus simple et plus rapide de les prescrire que de prendre le temps nécessaire, après un premier dépistage de cholestérol, pour rappeler l’importance de l’hygiène diététique et de l’exercice physique.
M. Robert Nicodème : Nous sommes d’accord sur le constat de la consommation de médicaments.
Cela tient tout d’abord au fait que, en France, le médicament est globalement bien remboursé et qu’il ne coûte presque rien au patient.
Par ailleurs, sa prescription n’est finalement pas très encadrée. Du fait de la visite médicale, c’est souvent le plus cher et le dernier médicament qui est prescrit, bien que l’amélioration du service médical rendu ne soit pas très importante car, je l’ai souligné, le médecin ne la connaît pas.
Vous évoquez le paiement à l’acte, mais il n’est absolument plus nécessaire de s’en prendre à lui, car, aujourd’hui, tous les jeunes médecins généralistes veulent être salariés. En effet, ils préfèrent très largement être praticiens hospitaliers, avec le salaire correspondant, les gardes payées et un repos compensatoire, qu’exercer en ville ! C’est bien pourquoi nous sommes persuadés qu’il est nécessaire d’aller vers des maisons médicales avec un statut particulier pour les médecins, qui viendraient par exemple y travailler deux ou trois jours et qui y prendraient leurs gardes de nuit.
S’agissant du médicament, ce sont les règles de prise en charge qui font défaut. En Angleterre, on n’opère pas un patient qui a besoin d’une intervention cardiaque tant qu’il n’a pas arrêté de fumer. En France, lorsqu’un patient obèse présente un taux trop élevé de cholestérol, on ne lui demande pas d’arrêter de boire, de fumer et de trop manger avant de lui donner un médicament. Cela n’est pas dans notre culture : d’un point de vue éthique nous considérons qu’il faut le protéger contre le risque d’accident cardio-vasculaire en lui donnant le médicament. Mais, en fait, les choses sont plus compliquées que cela : quand on dit que ce patient ne fait pas les efforts nécessaires parce qu’il a des problèmes psychologiques, force est de remarquer que ces problèmes peuvent l’empêcher d’arrêter de fumer mais pas de consulter, de suivre un régime mais pas de prendre ses médicaments. Quand on parle d’êtres humains, il est parfois difficile de raisonner en termes statistiques…
M. Pierre Morange, coprésident : Merci pour ces propos empreints d’humanisme et de bon sens.
Quel est votre sentiment en ce qui concerne la vente des médicaments sur Internet ?
M. Robert Nicodème : Le problème tient à la sécurité sanitaire. Nous avons la chance que soit assurée en France une bonne sécurité sur les aliments et sur les médicaments. En va-t-il de même des médicaments génériques vendus sur Internet, qui sont fabriqués moins cher dans des pays émergents ?
Il faut d’abord se demander s’ils sont efficaces, c’est-à-dire s’ils contiennent effectivement la molécule parfaite nécessaire. De ce point de vue, il me semble que, dès lors qu’il y a prescription médicale et prise en charge par l’assurance maladie, l’action d’un médicament devrait être vérifiée.
Je suis moins inquiet quant à une éventuelle toxicité de ces produits, tout simplement parce que les effets indésirables se voient et parce que les centres de pharmacovigilance les repèrent, alors qu’il est plus difficile d’apprécier l’efficacité d’un médicament censé traiter une maladie chronique.
M. Pierre Morange, coprésident : L’AFSSAPS, qui délivre la certification, doit s’assurer que la molécule est effectivement présente, mais on peut imaginer que certains produits passent à travers les mailles du filet.
M. Robert Nicodème : Dans un autre domaine, chez Airbus, de nombreuses pièces sont produites à l’étranger mais les ingénieurs se rendent sur place pour vérifier le processus de fabrication et le contrôle qualité s’exerce au moment où les pièces arrivent en France.
M. Pierre Morange, coprésident : L’AFSSAPS effectue également des contrôles sur les sites de production et à l’arrivée des médicaments. Mais par rapport au volume des ventes, c’est la fréquence et la régularité des contrôles qui peut poser problème.
Mme Catherine Lemorton, rapporteure : Dans la mesure où les Français sont hyperconsommateurs de médicaments, pensez-vous que ce soit une bonne idée de mettre en libre-service ce que l’on appelle les « médicaments conseils », surtout sachant qu’il ne s’agit plus seulement de vitamines et de revitalisants et que, dans le cadre des accords commerciaux imposés par les laboratoires, on va ainsi trouver bientôt sur les présentoirs des pharmacies des produits comme la pseudo-éphédrine ?
Cette mesure vous paraît-elle de nature à réguler la consommation des médicaments dans le cadre du cabinet du médecin ?
M. Robert Nicodème : Le médicament a une dimension un peu magique et nous sommes de plus en plus dans une société d’addiction. Ainsi, on trouve aujourd’hui des personnes qui ont besoin de vitamines dès le matin pour se sentir en forme, qui mangent peu le midi, qui prennent quelque chose dans l’après-midi pour éviter le coup de barre et qui prennent à nouveau un produit pour trouver le sommeil le soir. Ainsi, aux fonctions naturelles d’une personne qui se porte comme un charme, s’ajoutent tout au long de la journée des produits plus ou moins inefficaces. Cette habitude anglo-saxonne n’est pas bonne et il faut donc éduquer les patients afin qu’ils comprennent que, dans un pays où on s’alimente correctement, on n’a pas besoin de médicaments pour vivre normalement.
Toutefois ce qui nous préoccupe plus particulièrement, c’est la surconsommation de médicaments remboursés et il faut donc la replacer dans le cadre de notre système de prise en charge globale. Quand on s’interroge sur l’efficacité de ce système, on est bien obligé de constater que nous avons d’excellents résultats, par exemple en ce qui concerne la longévité. Si les Françaises vivent plus longtemps que leurs voisines, c’est peut-être parce qu’elles sont mieux soignées et parce qu’on leur donne plus de médicaments.
Le taux de consommation de psychotropes est de 20 % en France contre 6 % en Allemagne. Ces produits ont non seulement un rôle personnel mais aussi un rôle social : un certain nombre de personnes, en particulier des cadres et des intellectuels, seraient incapables de mener une vie normale s’ils n’en prenaient pas. La question n’est donc pas de savoir si l’on en consomme trop mais s’il est bon pour la population d’en prendre.
Pour conclure, je souhaite à nouveau insister sur la nécessité de donner des références, même si elles ne sont pas scientifiques et si elles émanent d’un groupe d’experts : nous avons en France suffisamment de personnes compétentes pour y parvenir.
Mme Catherine Lemorton, rapporteure : Merci beaucoup. Je vous sais gré en particulier d’avoir mentionné le professeur Montastruc car je fais partie du même réseau de pharmacovigilance.
M. Pierre Morange, coprésident : Merci.
*
Audition de MM. Pierre Levy, secrétaire général de la Confédération des syndicats médicaux français, Jean-Louis Caron, secrétaire général du Syndicat des médecins libéraux, Félix Benouaich, président de l’Alliance intersyndicale des médecins indépendants de France accompagné de Jean-Gabriel Brun, vice-président, et Martial Olivier-Koehret, président de MG France.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue. Quel est votre sentiment à propos de la surconsommation de médicaments en France ? Que préconisez-vous pour rationaliser la consommation ?
M. Pierre Levy : Avant d’évoquer la surconsommation, il convient de parler de la prescription. La Confédération des syndicats médicaux français (CSMF) a toujours prôné une prescription fondée sur la qualité. Elle l’a prouvé avec la convention médicale de 1993 et les références médicales opposables, qui ont généré des économies notables et continuent d’être appliquées par des médecins et des étudiants en médecine. Toutefois, après le succès fugace de la maîtrise médicalisée, sont survenues dix années de maîtrise comptable qui se sont soldées par un échec total.
Parmi les dispositions du projet de loi de financement de la sécurité sociale (PLFSS) pour 2008, les contrats individuels liant la rémunération du médecin aux économies accomplies sur ses prescriptions seraient tout à fait délétères pour la profession.
La convention médicale de 2005, dispositif plus récent de maîtrise médicalisée, a permis de dégager 1,3 milliard d’économies en 2005-2006, du jamais vu. Les médecins libéraux ont manifestement joué le jeu, et nous sommes étonnés que le système soit modifié. Nous pensons simplement que des marges de progression demeurent et que les référentiels devraient être revus périodiquement car ils ne sont pas toujours adaptés à l’exercice de la médecine ambulatoire.
M. Pierre Morange, coprésident : La Mission d’évaluation et de contrôle des lois de financement de la sécurité sociale (MECSS) n’est pas un tribunal. Nous avons pour seul objectif d’optimiser l’utilisation des deniers publics dans le domaine de la protection sanitaire et sociale. C’est pourquoi nous souhaitons que vous nous donniez votre sentiment sur les dysfonctionnements du système que vous constatez au quotidien dans votre pratique de professionnels de santé et de prescripteurs. Nous tournons toujours autour du même sujet : la formation initiale et continue, les logiciels d’aide à la prescription, les recommandations de la Haute Autorité de santé (HAS) et les processus d’évaluation.
M. Pierre Levy : La dernière étude de la Caisse nationale d’assurance maladie des travailleurs salariés (CNAMTS) montre que le dynamisme des prescriptions est en grande partie dû au développement des affections de longue durée (ALD).
Les gains imputables à la maîtrise médicalisée reposent sur trois socles.
Premièrement, l’information doit être diffusée vers les patients et les professionnels de santé, dans le cadre conventionnel. Je reçois régulièrement de ma commission paritaire locale d’excellents documents statistiques sur les prescriptions.
Deuxièmement, nous espérons que le dossier médical personnel (DMP) sera recentré sur sa vocation : un outil de partage de l’information, même s’il doit rester la propriété exclusive du patient. Mais, comme avec le livret de santé, il existe un risque d’échec majeur, compte tenu de la possibilité d’un masquage des informations par le médecin traitant, sous la pression du patient.
Troisièmement, la formation initiale et continue est cruciale, de même que l’évaluation des pratiques. Elles sont maintenant entrées dans les mœurs mais le retard est considérable et nous attendons encore le décret de mise en place des conseils régionaux de la formation médicale continue. Les médecins se désespèrent car la formation médicale continue est obligatoire depuis 1995. L’information émane d’abord de la Haute Autorité de santé (HAS). L’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) continue d’émettre des recommandations. Il est aberrant d’éliminer l’industrie pharmaceutique de la réflexion et de l’information ; la charte de la visite médicale, élaborée en concertation avec tous les acteurs, prouve que l’industrie pharmaceutique s’engage dans une démarche de qualité. Enfin, la charte de la formation médicale continue doit être respectée et ce volet doit bénéficier de financements.
M. Jean-Louis Caron : Le médecin prescripteur intervient en bout de chaîne. Il a été formé essentiellement à l’hôpital, avec des médicaments princeps. Il est soumis à des informations diverses, plus ou moins objectives, de la part des délégués de l’assurance maladie (DAM), qui passent une ou deux fois par an, des délégués médicaux, qui leur rendent visite cinq ou six fois par semaine, voire davantage, ou par le biais de courriers des caisses.
Les médecins sont pour la plupart de grands professionnels mais il leur est difficile de faire le tri entre ces informations contradictoires, d’autant que la prescription est rédigée à l’issue d’une négociation avec le patient. Les médecins ne sont pas formés pour acquérir l’esprit critique nécessaire ; ils se le forgent au fil de la pratique. J’appelle de mes vœux une formation et une information neutres, objectives, pédagogiques, accessibles aux médecins et d’une intensité équivalente à celle de la visite médicale, sur des disciplines comme la pharmacologie ou la iatrogénie, à partir d’un fonds dédié, constitué en partie par l’industrie.
M. Félix Benouaich : Je m’exprimerai en tant que représentant syndical de médecins, mais aussi en tant que médecin libéral en exercice.
Le médecin prescripteur est le dernier maillon de la chaîne : il n’agit aucunement sur la validité du médicament ni sur son prix, fixé en fonction du coût de la recherche. Nous reconnaissons la qualité des nouveaux médicaments et, une fois la molécule amortie, nous acceptons volontiers les génériques, bénéfiques à la sécurité sociale.
La prescription est délivrée uniquement en fonction de la santé du patient. Si des antibiotiques sont prescrits pour soigner une maladie virale, c’est que les pressions sont énormes, tout particulièrement de la part des parents lorsqu’un enfant présente de la fièvre. La nouvelle convention a débouché sur une maîtrise médicalisée qui a essentiellement porté sur les antibiotiques. Les résultats sont sans doute insuffisants mais la courbe de croissance s’est infléchie et même inversée : la consommation d’antibiotiques a baissé de 17 %, ce qui représente 13 millions de traitements inutiles évités.
Une prescription doit systématiquement être accompagnée d’explications, à condition que le médecin s’en donne le temps ; sinon, il se décharge en prescrivant n’importe quoi. Mais comment demander à un médecin de prendre beaucoup de temps, alors qu’il doit faire face à tellement de charges ? Il faudrait que nous ayons moins de patients à soigner, afin de gagner autant en travaillant moins, mais nous sommes aussi tributaires de la demande des malades, des épidémies et du stress engendré par les problèmes socioéconomiques.
L’information que nous dispensons est essentielle mais il faut d’abord que nous la recevions. Les DAM, opérationnels depuis peu, présentent un handicap : ils sont ressentis comme des inquisiteurs. Les médecins de la sécurité sociale devraient nouer des contacts plus fréquents avec les médecins de ville, afin d’entretenir des relations confraternelles.
Dans notre cursus initial, il manque une formation en économie et en gestion car nous prescrivons sans savoir ce que recouvre une ordonnance, en termes de coûts des médicaments et des examens complémentaires.
Le masquage d’informations sur le DMP ne doit pas être autorisé. Cela dit, si une femme a subi une interruption volontaire de grossesse (IVG) à dix-huit ans, il serait inutile de lui imposer de l’indiquer dans son dossier.
Est-il envisageable d’instituer un statut de médecin salarié ? Dans quelles conditions ? Quid de la semaine de trente-cinq heures compte tenu des gardes et des heures supplémentaires ? Quelles seront les conditions de départ à la retraite ? Quand j’ai demandé à M. Jean-Marie Spaeth, président du conseil de la CNAMTS, s’il souhaitait la fonctionnarisation des médecins, il m’a répondu qu’il n’était pas fou…
M. Martial Olivier-Koehret : Si nous en sommes là, c’est que les politiques du médicament menées depuis dix ou vingt ans n’ont pas abouti aux résultats escomptés. Tout a été essayé : les sanctions et la maîtrise médicalisée n’ont abouti qu’à une augmentation généralisée des prescriptions. Seules trois orientations ont produit des effets : le déremboursement, mais je n’y suis pas favorable ; la campagne « Les antibiotiques, c’est pas automatique ! », qui a entraîné une baisse de 19 % des ventes, même si les dépenses de médicaments continuent de croître, compte tenu de la mise sur le marché de nouveaux produits ; le médecin référent, qui renforce la relation entre le médecin et son patient.
Cette augmentation pose-t-elle un problème sanitaire ? La France manque cruellement de statistiques sérieuses sur les conséquences des prescriptions ; cela participe de la désorganisation du secteur, marqué par l’absence de gouvernance.
L’article 8 du code de déontologie médicale dispose :
« Dans les limites fixées par la loi, le médecin est libre de ses prescriptions qui seront celles qu’il estime les plus appropriées en la circonstance.
« Il doit, sans négliger son devoir d’assistance morale, limiter ses prescriptions et ses actes à ce qui est nécessaire à la qualité, à la sécurité et à l’efficacité des soins.
« Il doit tenir compte des avantages, des inconvénients et des conséquences des différentes investigations et thérapeutiques possibles. »
Comment faire pour respecter ces principes ? Le système d’information souffre de procéder pour l’essentiel de l’industrie pharmaceutique. Ce que les organismes nous proposent ne correspond pas à nos besoins. Les médecins britanniques, par exemple, disposent d’une base de données immédiatement accessible avec le National British formulary. En France, il existe une base indépendante mais la CNAMTS tente de la fermer. Les déterminants de la prescription des médecins généralistes tournent autour de la discussion du couple médecin-patient ; les autres paramètres n’influent pas.
La loi du 13 août 2004 relative à l’assurance maladie a créé un couple entre le médecin traitant et le patient. Cependant MG France n’a pas signé la convention médicale de 2005 parce que le DMP, tel qu’il a été présenté, ne contient pas l’ensemble du retour d’information de la part des autres prescripteurs. De surcroît, le système pousse au développement de l’automédication, comme si la surconsommation ne posait un problème sanitaire que lorsque les médicaments sont remboursés. Par exemple, la pathologie qui suscite le plus d’automédication est l’infection urinaire, avec des traitements qui font le lit de l’insuffisance rénale et de la dialyse, faute de prise en charge correcte. Je ne suis pas opposé à l’automédication mais je ne comprends pas que l’information soit jugée inutile dès que l’on sort du champ des médicaments remboursés.
Les logiciels médicaux en sont encore à la préhistoire de l’informatique. En l’état actuel de désorganisation du système de soins et du parc logiciel, je doute qu’une décision quelconque du législateur puisse améliorer la situation en la matière.
Les déremboursements entraînent des économies et réduisent l’activité des médecins généralistes. Cependant, ils occultent la nécessité de définir le rôle du premier recours et ils sont contradictoires avec le choix effectué par tous les autres pays occidentaux pour stabiliser la consommation médicamenteuse : faciliter l’accès des patients au premier recours.
M. Jean-Gabriel Brun : Le généraliste est le principal prescripteur car il intervient en dernière ligne, après le spécialiste et l’hôpital. Une mesure très simple consisterait à demander à l’hôpital et au spécialiste de prescrire en dénomination commune internationale (DCI).
Mme Catherine Lemorton, rapporteure : Je poserai quelques questions très courtes.
Les franchises sur les transports sanitaires et surtout sur les médicaments vont-elles pousser les Français à diminuer leur consommation de médicaments ?
Quand une classe de médicaments est déremboursée, constatez-vous un transfert de prescriptions sur une autre classe ? Si oui, êtes-vous en mesure de le chiffrer ?
Que pensez-vous du dossier pharmaceutique expérimenté dans six départements ?
M. Pierre Levy : L’enjeu consiste-t-il à réduire la consommation de médicaments ou à optimiser les soins ? Le déremboursement est clairement un obstacle au recours au médecin, ou plutôt une incitation au report des soins.
Les déremboursements entraînent effectivement un transfert de prescriptions sur d’autres classes.
Comme l’historique des remboursements, le dossier pharmaceutique est un outil complémentaire au DMP mais il ne saurait s’y substituer. Il ne faudrait surtout pas tirer prétexte des difficultés à mettre sur pied le DMP pour se contenter du dossier pharmaceutique et de l’historique de remboursement.
Le renouvellement de la prescription hospitalière est un gros problème. Quand un médecin hospitalier conseille à un patient de poursuivre son traitement, le petit médecin généraliste de banlieue aura du mal à le convaincre de l’interrompre.
M. Jean-Louis Caron : Les franchises entraîneront certainement des économies mais aussi la pénalisation des traitements longs pour les maladies chroniques.
Il est vrai que les déremboursements provoquent des transferts : les prescriptions d’un spray bien connu ont explosé parce qu’il est resté remboursé, contrairement à tous les autres, dont la distribution stagne ou régresse.
Si les sources d’informations restent dissociées, comme c’est actuellement le cas, notamment avec le dossier pharmaceutique et le Web-médecin, nous ne saurons plus où rechercher les éléments dont nous avons besoin. Nous ne pouvons donc nous contenter de succédanés qui retarderaient la mise sur pied du DMP.
M. Félix Benouaich : S’agissant des franchises, pour ceux qui sont à l’aise financièrement, un déremboursement de cinquante ou cent euros par an passe inaperçu ; il ne pose un problème qu’à ceux qui n’ont pas les moyens. La gratuité n’est pas source d’économies mais elle est nécessaire pour que certains malades accèdent aux soins. Or, en tant que médecins, c’est notre seule préoccupation.
Quant aux déremboursements, ils provoquent évidemment des transferts de prescriptions vers des traitements équivalents.
M. Martial Olivier-Koehret : La question des franchises et des déremboursements concerne au premier chef le législateur. La France arrive au deuxième rang mondial en matière de prélèvements obligatoires ; il n’est donc guère compréhensible que vous demandiez aux gens de payer quand ils sont malades, mais c’est à vous de l’assumer lorsque vous retournez vers vos électeurs.
Les franchises auront un impact sur le niveau des remboursements mais cela signifie que des patients, faute de moyens financiers, n’auront pu accéder aux soins. De surcroît, elles vont désorganiser les soins en générant du second recours différé, et cela coûtera plus cher encore. Les franchises procèdent d’une approche idéologique.
Le déremboursement est une hypocrisie complète car les médicaments concernés sont bon marché : pour financer une ampoule d’antimitotique à 9 000 euros l’unité, il faudra économiser sur beaucoup de boîtes de fortifiants ou de gouttes pour le nez !
Le dossier pharmaceutique est un très bon outil pour le pharmacien, tout comme l’historique des remboursements est parfait pour l’assurance maladie. Toutefois, nous autres prescripteurs avons besoin de disposer d’une synthèse d’informations sur les actes de chaque intervenant, du biologiste au radiologue.
Mme Catherine Lemorton, rapporteure : Un article du projet de loi de financement de la sécurité sociale pour 2008 prévoit que les logiciels d’aide à la prescription doivent permettre d’afficher le montant total de la prescription. Cela aidera-t-il les médecins à prescrire, notamment pour les patients socialement défavorisés ?
Puisque les patients seront poussés à l’automédication, ne pourrait-elle pas être intégrée dans le DMP, d’autant que celui-ci met du temps à voir le jour ?
M. Pierre Levy : Le coût de l’ordonnance interpelle le médecin, et pas seulement pour les personnes défavorisées. En vertu du code de déontologie, nous devons systématiquement prescrire à bon escient et au meilleur coût. Il serait utile aussi que les hôpitaux disposent d’un tel outil pour que les internes prennent conscience de ce qu’ils prescrivent.
M. Jean-Gabriel Brun : C’est déjà le cas !
M. Pierre Levy : Je constate que la médecine ambulatoire a accompli 1,3 milliard d’économies et que l’hôpital est resté à zéro.
La CSMF s’est élevée contre l’automédication, qui comporte des risques. Avec des vitamines, ce n’est pas bien grave ; avec des anti-inflammatoires ou des molécules innovantes, il y a des effets secondaires. En outre, cela peut retarder le diagnostic d’une maladie grave. Il est donc indispensable que le médecin puisse connaître l’automédication de son patient.
M. Jean-Louis Caron : Le coût d’une ordonnance serait une information intéressante, sous réserve que je puisse le comparer avec d’autres options, afin de faire mieux économiquement tout en conservant la même qualité médicale de prescription.
Nous sommes favorables à l’automédication car les Français possèdent un niveau de connaissance suffisant pour se prendre en charge, par exemple, face à un état grippal, qui, de toute façon, passe tout seul au bout de cinq jours. Mais il convient d’encadrer l’automédication en proposant une formation aux patients, à l’école et pendant d’autres moments de la vie. Les pouvoirs publics doivent aussi organiser des campagnes d’information. Cela dit, l’exposition des produits en libre-service devant le comptoir pose problème, eu égard aux effets secondaires. L’industrie pharmaceutique doit produire des boîtes adaptées à l’automédication, avec des quantités limitées. Et nous sommes favorables à l’intégration de l’automédication dans le DMP.
J’ajoute que nous sommes également favorables au masquage d’informations, sur la demande du patient.
M. Félix Benouaich : L’information sur le coût de l’ordonnance serait en effet utile. Dans un second temps, il faudrait ajouter une dimension comparative.
L’automédication doit être intégrée dans le DMP. Reste à s’entendre sur la signification exacte de l’automédication, mais ce n’est pas le sujet.
M. Martial Olivier-Koehret : Le logiciel que j’utilise me permet déjà de connaître le prix de la boîte de médicaments ; il faut donc aller plus loin. Les logiciels médicaux sont importants sur les plans sanitaire et économique mais ils ne sont pas pilotés ; vous pourriez faire preuve d’innovation en créant un agrément, fondé sur des critères comme l’information relative à la posologie ou à l’aide à la prescription. Pour que nous fassions bien notre métier, nous devons être en mesure de soigner nos patients indépendamment de leur capacité financière, avec le seul souci de traiter leur état de santé. Aujourd’hui, la même angine ne coûte pas le même prix suivant l’endroit où elle est soignée.
M. Jean-Gabriel Brun : À l’hôpital, quand je prescris un traitement onéreux, on m’apporte un formulaire en trois exemplaires sur lequel je dois spécifier le prix des médicaments, le nombre de jours et la posologie. C’est le pharmacien qui délivre ces médicaments, par périodes très courtes. L’hôpital a donc déjà répondu partiellement à votre question. En revanche, le système pêche au niveau des services des urgences, qui travaillent rapidement, et des cliniques, où les médecins remplissent leurs feuilles blanches comme ils l’entendent.
M. Pierre Levy : Je précise que je partage pleinement l’avis de mes confrères sur les logiciels d’aide à la prescription.
M. Pierre Morange, coprésident : Le sujet n’est pas clos ; nous restons ouverts aux remarques et suggestions que vous pourrez nous transmettre par écrit.
*
Audition de MM. Alain Rouché, directeur santé de la Fédération française des sociétés d'assurances, et Gilles Johanet, président du comité maladie-accidents, Michel Charton, directeur technique santé individuelle d’AXA France, Henri Laurent, directeur général de SwissLife prévoyance et santé, et Laurent Doubrovine, directeur assurance de personnes des Assurances générales de France (AGF).
M. Pierre Morange, coprésident : Madame, messieurs, nous sommes heureux de vous accueillir pour vous entendre sur le thème « la prescription, la consommation et la fiscalité du médicament ».
M. Jean Mallot, coprésident : De par vos fonctions, chacun dans son domaine, vous avez certainement une opinion sur le sujet. J’aimerais que vous nous livriez votre analyse et vos propositions.
M. Gilles Johanet : Inutile de détailler la singularité de la consommation française de médicaments, que ce soit en termes de volumes ou de coût. Je remarquerai cependant que la politique engagée il y a une quinzaine d’années pour remplacer le volume par la valeur et faire que le prix français du médicament rejoigne les standards européens et américains, a échoué. L’écart de prix négatif s’est considérablement réduit, mais l’écart de volume positif ne s’est absolument pas réduit. Cela signifie qu’il y a bien des raisons de fond, des causes majeures à l’origine de cette singularité française.
J’insisterai sur une seconde caractéristique française, qui est l’extrême opacité du système de prescription et de consommation. Nous n’avons pas de dispositif de connaissance et de suivi de la prescription hospitalière, a fortiori de la prescription hospitalière par médecin prescripteur. En 1998, lorsque je suis arrivé à la direction de la Caisse nationale d’assurance maladie des travailleurs salariés (CNAMTS), un groupe travaillait depuis dix-huit mois sur l’individualisation de la prescription. Neuf ans plus tard, je ne doute pas qu’il continue de travailler avec acharnement, mais les résultats font partie de ce qu’on appelle les « variables discrètes ».
Autre singularité française : l’assurance maladie complémentaire n’a pas le droit de savoir ce qu’elle rembourse. C’est un problème par rapport à la loi du 13 août 2004 qui instaure un partenariat entre l’assurance maladie obligatoire et l’assurance maladie complémentaire, partenariat symbolisé par la création de l’Union nationale des caisses d’assurance maladie (UNCAM) et de l’Union nationale des organismes d’assurance complémentaire (UNOCAM). Et c’est un obstacle à toute recherche de performance s’agissant des contrats de complémentaire santé, puisqu’il n’est pas possible de sélectionner ce qui sera remboursé, en dehors des expérimentations Babusiaux, sur lesquelles je reviendrai, et d’autorisations acquises à titre exceptionnel.
Autre facteur d’opacité : nous n’avons pas de bilan de la politique conventionnelle suivie entre le Comité économique des produits de santé (CEPS) et les laboratoires, que ce soit par classe de médicaments ou par laboratoire. Il n’y a pas d’articulation entre les baisses de prix décidées par le CEPS et les remises. Une mesure très grossière de ces décisions de baisse ou de remise nous fait penser que l’impact financier des remises est environ trois fois plus élevé que celui des baisses de prix. Toutefois la différence principale est ailleurs : les baisses de prix sont définitives et profitent de façon juste et équitable à l’assurance maladie obligatoire et à l’assurance maladie complémentaire ; les remises de prix sont par essence contractuelles, précaires et ne profitent qu’à l’assurance maladie obligatoire, alors que la cause de la remise, c’est-à-dire un dérapage des consommations, a été supportée financièrement par les complémentaires autant que par l’obligatoire ; enfin, elle aboutit à faire en sorte que le taux de prise en charge officiel de certains médicaments soit très différent de leur taux de prise en charge réel.
Je veux revenir sur les propos tenus devant la MECSS par M. Noël Renaudin, président du CEPS, qui ont été repris par la presse, sur l’apparition et la multiplication de médicaments « de niche » horriblement chers, ce qu’on pourrait traduire par « médicaments à spectre étroit et à prix élevé ». Les opérateurs que nous sommes vont se retrouver devant cette alternative : soit maintenir l’opacité actuelle, et le dérapage est garanti ; soit mettre fin au dérapage, et l’on est devant la perspective d’une traçabilité individuelle des prescriptions et des consommations.
La question se pose alors d’un partenariat entre l’assurance maladie obligatoire (AMO) et l’assurance maladie complémentaire (AMC). Est-ce que nous acceptons que l’AMC devienne un acheteur avisé, en commençant par exemple par le médicament ? Un tel partenariat n’est pas incompatible avec nos réflexions sur d’éventuels transferts entre l’AMO et l’AMC, qui pourraient porter sur les médicaments à 35 %. Mais cette recherche de performance impliquerait qu’on ait accès au code identifiant de présentation (CIP) des médicaments, en commençant par exemple par les codes des médicaments remboursés par l’AMO à 35 %. Cela reviendrait à reprendre et à accepter, pour cette fois, l’amendement proposé par M. Yves Bur, lors de l’examen du projet de loi de financement de la sécurité sociale (PLFSS) pour 2005.
Une autre question se pose, celle du desease management (soutien à la prise en charge thérapeutique des patients atteints de maladies chroniques). Nous pensons qu’un desease management encadré peut être positif. C’est ce que font certaines sociétés d’assistance liées avec les sociétés d’assurance. Ainsi Mondial Assistance, qui est liée au AGF, fait du desease management auprès de patients atteints d’ostéoporose, et cela marche bien. Le fait de dire que c’est l’intérêt financier des laboratoires n’épuise pas le sujet, dans la mesure où cela peut avoir aussi une utilité médicale pour les patients. C’est en tout cas une piste à explorer.
Je vais évoquer un dernier point sur l’évolution de l’offre : l’automédication.
Celle-ci est très en retard en France. Elle ne se développera jamais et n’aura jamais d’incidence financière pour l’assurance maladie obligatoire tant que 85 % des médicaments en automédication seront également en prescription médicale facultative.
Je termine en remarquant que le remboursement des médicaments représente un poste de dépenses important pour les complémentaires maladies, et qu’il est en croissance forte.
M. Alain Rouché : Pour les complémentaires, la problématique d’accès aux données de soin est très importante. Nous ne connaissons que globalement le taux de remboursement des médicaments. Cette opacité nous empêche d’avoir une véritable efficacité et de mener une véritable politique de gestion des risques.
Nous nous battons depuis de nombreuses années. En 2003, est paru le rapport de M. Christian Babusiaux sur l’accès des assureurs complémentaires aux données de santé des feuilles de soins électroniques. Depuis, la situation a évolué puisque des expérimentations ont démarré ou vont démarrer dans les jours qui viennent. Je pense que Michel Charton, d’AXA, évoquera l’expérimentation qui a débuté dans le département de l’Hérault et Henri Laurent celle de Swisslife. Pour nous, cela est fondamental pour développer des assurances santé répondant mieux aux besoins des assurés et plus efficaces en termes de gestion des risques.
Je vais illustrer l’intérêt de l’accès aux données : en cas de décision de baisse du taux de remboursement, on pourrait se dispenser d’augmenter les cotisations de nos adhérents pour faire face au transfert de charge vers la complémentaire et décider d’un reste à charge de 30 points. Aujourd’hui, on n’a pas d’autre solution que de tout prendre en charge ou de ne rien prendre en charge par taux de remboursement de la sécurité sociale.
Aujourd’hui, nous prenons en charge quelques médicaments non remboursables. Nous devons alors demander à nos assurés de récupérer une facture chez le pharmacien et de nous la transmettre pour que nous puissions procéder au remboursement.
Cette problématique d’accès aux données est absolument essentielle pour les complémentaires. Nous espérons qu’au terme des expérimentations Babusiaux cet accès aux données sera généralisé et définitivement reconnu aux complémentaires.
M. Pierre Morange, coprésident : Quelle est la part du médicament pour les complémentaires, dans le cadre d’un contrat standard ?
M. Michel Charton : De 15 à 40 % selon les cas, car il y a beaucoup de contrats. Pour l’entrée de gamme, c’est 40 %.
M. Alain Rouché : La question des baisses de prix et des remises est très importante pour nous. Nous sommes favorables aux baisses de prix, dans la mesure où les complémentaires en bénéficient, tandis qu’en cas de remises, seule l’assurance maladie obligatoire en bénéficie. Récemment, l’UNOCAM a fait un petit calcul concernant le Gardazil utilisé contre le cancer de l’utérus. Il est théoriquement remboursé à 65 % par l’assurance maladie, mais, si l’on tient compte du processus de remise, les complémentaires en remboursent 53 %, et non 35 %.
M. Michel Charton : Nous avons remarqué que tous nos clients étaient persuadés que le médicament était un produit gratuit. La pratique du tiers payant est devenue universelle. Il est désormais absolument nécessaire de faire évoluer ce concept du médicament gratuit et remboursé systématiquement à 100 % si l’on veut amener nos clients à faire des choix.
Cette part de 15 à 40 % est très importante. Elle est beaucoup plus élevée que pour les remboursements dentaires. La raison en est que l’on n’arrive pas à accéder aux données. Hors remises, l’AMC rembourse à peu près 8 milliards d’euros de dépenses de médicaments. Il s’agit d’une somme considérable, et il est évident que si l’on trouvait les mécanismes qui permettent de réguler ce marché et de mettre en œuvre une gestion des risques, l’intérêt financier serait tout à fait notable.
L’expérimentation Babusiaux menée par AXA a été engagée en mars 2003. Il nous a fallu quatre ans pour commencer à faire passer des flux dans ce système nouveau. Le rapport de M. Babusiaux a proposé de donner aux organismes complémentaires un droit d’accès aux données de santé en respectant certaines conditions. Il a tracé les voies de ces conditions, mais il a fallu mettre des rails derrière. On s’est alors aperçu qu’il était très complexe de vouloir tout protéger et tout sécuriser. AXA a travaillé pendant pratiquement un an pour préparer le dossier à soumettre à la Commission nationale de l’informatique et des libertés (CNIL), avec l’aide de la FFSA dans la mesure où nous nous appuyons sur sa maîtrise d’ouvrage. Il nous a fallu ensuite un an de discussions avec la CNIL pour obtenir, en plusieurs étapes, les autorisations nécessaires. Nous avons ainsi commencé nos développements techniques à partir de 2006. Cependant la complexité est telle que les premiers flux n’ont eu lieu que depuis quelques semaines.
Ces changements sont donc lents, progressifs et coûteux. Nous sommes obligés de répercuter ces coûts de fonctionnement sur nos clients. Plus on crée des usines compliquées pour connaître enfin une information dont on pense qu’elle est vraiment nécessaire, plus on génère de frais généraux. C’est un élément à prendre en compte, s’agissant de la façon dont on pourra accéder demain aux données. Certes, on pourra mutualiser certains coûts, mais ils resteront importants et inévitables en matière de décryptage et de sécurité.
Que faire de tout cela ? Il existe de nombreuses pistes de travail. Demain, AXA ayant sa chaîne de traitement spécifique pourrait proposer à ses clients de faire du tout générique et du me too. Cette idée n’est pas aberrante ; nous l’avons mise en pratique pour l’optique. C’est peut-être plus compliqué pour le médicament, mais ce n’est pas infaisable.
On pourrait également décider de ne plus rembourser tel ou tel type de médicaments remboursés par l’AMO à 35 % et à 15 %, voire des médicaments à service médical rendu faible remboursés à 65 % ; et compenser éventuellement par de l’automédication.
On pourrait définir des gammes de produits correspondant aux vrais besoins de certaines populations. Pourquoi mettre dans un contrat qui s’adresse aux jeunes familles la prévention ou le traitement de l’ostéoporose ? Pourquoi laisser le remboursement des anticonceptionnels microdosés dans des contrats destinés aux personnes de plus de cinquante-cinq ans ?
L’accès aux données nous permettrait très concrètement de travailler sur les classes de médicaments. Tel est précisément le cas dans l’expérimentation AXA. Nous avons besoin d’un contrôle de la prescription dans l’immédiat, mais l’idée est de permettre à nos clients de bénéficier d’un remboursement qui serait automatiquement fait à partir des données transmises du poste du professionnel de santé, sans passer par la facturette dont parlait M. Alain Rouché. Cela est très important. La seule façon de développer cette pratique est d’entrer dans un processus de dématérialisation, car le coût de gestion d’une facturette papier est de 2 à 10 euros ; tandis que le flux électronique revient à 0,13 euro. Toutes les tentatives des uns et des autres consistant à mettre un peu d’automédication, un peu de médicaments non remboursés dans nos contrats aboutissent à des faibles taux d’utilisation. Cela est d’ailleurs heureux, sinon nous aurions bien du mal à en supporter les coûts de gestion et à les faire payer à nos clients.
Un processus fluide de dématérialisation et d’accès aux données permettrait de faire baisser de dix points le rapport sinistres/primes, c’est-à-dire de baisser de dix points le prix de la complémentaire santé à service rendu au moins équivalent, si ce n’est supérieur.
M. Henri Laurent : Nous sommes convaincus que la maîtrise du coût du médicament, qui représente 30 % des coûts des assureurs, passe par la mise en place de conditions qui permettent de sortir de cette logique du remboursement systématique. Les assureurs pourraient avoir des politiques de remboursement diversifiées. On pourrait imaginer d’offrir des contrats dont le prix varie en fonction des niveaux de remboursement du médicament ou d’autres prescriptions. On pourrait imaginer des stratégies marketing en ciblant les médicaments qu’on va rembourser en fonction des populations auxquelles on s’adresse.
Swisslife a conduit une expérience en ce domaine. Elle a mis en place la « carte blanche » ; l’objectif est de prendre des initiatives en matière de prévention. Elle a institué un comité d’éthique indépendant, qui l’aide à constituer les listes de médicaments qui seront remboursés, même s’ils ne le sont pas dans le régime général. Cela dit, si l’on sait rembourser des médicaments qui ne le sont pas par le régime général, on ne sait pas ne pas rembourser des médicaments qu’il rembourse, mais dont l’intérêt est douteux. Swisslife a mis également en place des consultations pharmaciens avec ses assurés, qui ont la possibilité de rencontrer leur pharmacien sur des questions d’observance.
D’après nos sondages, nos assurés considèrent que l’assureur est légitime dans ses démarches, non seulement de remboursement, mais aussi d’accompagnement, voire de conseil. Si nous avions la possibilité de mener des politiques de remboursement plus nuancées, nous pourrions mieux maîtriser nos dépenses et limiter l’évolution de nos primes d’assurance, tout en offrant à nos assurés des services qui les amèneraient à consommer de façon plus efficace. Tout cela passe bien entendu par la possibilité de connaître la nature des médicaments que nous remboursons.
Swisslife participe également aux expérimentations. Nous avons choisi la voie du consentement exprès de l’assuré. Si on peut démontrer que l’assuré a donné de manière explicite à son assureur son accord sur la transmission de l’information médicale qui le concerne, on échappe aux restrictions réglementaires. C’est ce que nous sommes sur le point d’expérimenter dans la région de Cambrai.
M. Laurent Doubrovine : Je ne peux qu’appuyer ce qu’ont dit mes collègues. Militer pour une meilleure transparence sur le marché du médicament n’est pas défendre un intérêt particulier, celui des assureurs complémentaires, mais l’intérêt général. L’opacité actuelle de ce marché est à l’origine d’inefficacités monstrueuses, qui se répercutent sur le coût de la prise en charge de la santé.
Nous ne participons pas, à ce stade, aux expérimentations Babusiaux, mais nous les soutenons. Nous avons la prétention de penser que notre métier consiste à donner de la valeur ajoutée à nos clients, laquelle ne peut pas s’exprimer sur un marché sur lequel nous ne disposons d’aucune variable ni d’aucune liberté. Le marché de la santé en général, et celui du médicament en particulier, sont très frustrants de ce point de vue. Nous nous contentons donc des quelques domaines dans lesquels nous pouvons exercer notre créativité, comme la prévention par la prise en charge de vaccins, du sevrage tabagique, par le conseil et l’orientation des clients, la négociation avec certaines officines sur les médicaments non remboursés.
Nous sommes très frustrés par cette situation, qui pourrait facilement évoluer si le problème de l’accès aux données de soin était résolu, ce qui devient crucial.
M. Jean Mallot, coprésident : Avez-vous des éléments complémentaires sur les expérimentations ? Avez-vous des idées d’économies potentielles ?
M. Pierre Morange, coprésident : Il faudrait que vous quantifiiez certains points. Une réflexion est en cours sur un éventuel transfert de l’AMO vers l’AMC ; celui-ci pourrait se répercuter sur les primes d’assurance. Il serait pertinent que vous nous présentiez un catalogue précis de propositions, qui pourraient déboucher sur des mesures législatives ou réglementaires.
Mme Catherine Lemorton, rapporteure : Quand une classe thérapeutique est déremboursée dans sa totalité, assistez-vous à une baisse de votre participation au remboursement de médicaments ?
Pensez-vous que les franchises sur les médicaments feront baisser la consommation des médicaments en France ?
M. Alain Rouché : Mes collègues assureurs constatent mieux que moi l’impact des déremboursements sur nos résultats. Il est clair qu’ils jouent également sur la part des complémentaires.
M. Henri Laurent : C’est mécanique !
M. Alain Rouché : Nous avons observé, certaines années, une augmentation beaucoup plus modérée que précédemment sur le poste médicaments. C’est une observation globale.
M. Henri Laurent : En cas de déremboursement total !
M. Alain Rouché : Dans un premier temps, nous avons émis des réserves concernant les franchises, à commencer par le fait que ce sont les médecins qui prescrivent et que nous ne voyons pas quel impact ces franchises pourraient avoir sur le comportement des patients.
Ces franchises médicales sont différentes de nos franchises d’assureurs. Lorsque l’aile de la voiture est froissée, le conducteur peut décider ou non de la faire réparer. C’est un peu plus difficile quand on se trouve dans une logique de soins et de prescriptions par les médecins.
Dans un second temps, nous avons revendiqué de pouvoir rembourser ces franchises sans que nos contrats ne soient pénalisés fiscalement. L’idée était de laisser le choix aux assurés, et non de vouloir à tous crins rembourser les franchises. Il me semblerait très préoccupant pour notre avenir d’assureurs complémentaires d’accepter toute une série de contraintes sans que nous ne puissions proposer à nos assurés toute une série de solutions. Il est tout aussi responsabilisant de dire à un assuré que s’il désire que les franchises lui soient remboursées, il verra sa cotisation augmenter de tant, mais qu’il peut choisir un contrat qui ne prévoit pas cette prise en charge.
Par ailleurs, je doute un peu que ces franchises aient un impact sur la consommation de médicaments. Mais soyons sans a priori et contentons-nous d’observer ce qu’il en sera.
M. Gilles Johanet : J’ajoute qu’il s’agit là d’une réforme partielle qui accroît encore la complexité du système et la difficulté que peut avoir le consommateur à le comprendre et à le maîtriser en partie. Il existerait ainsi des franchises à 0,50 euro, à 1 euro ou à 2 euros dont le remboursement serait irresponsable, et la franchise à 18 euros dont le remboursement serait responsable ? Cela ne signifierait-il pas que l’assuré, en aucun cas, ne peut être considéré comme responsable de sa consommation d’actes lourds, mais qu’il serait exclusivement responsable de sa consommation de médicaments ? Ce serait d’ailleurs cohérent avec une autre approche, sémantique celle-là, qui amène à répéter que les Français sont les premiers consommateurs de médicaments du monde, mais jamais à dire que les médecins français sont les premiers prescripteurs de médicaments du monde !
Le débat sur la franchise est ancien. Il a été posé nettement en 1993, et je l’ai vécu. À l’époque, la CNAMTS avait proposé une franchise fondée sur la maîtrise médicalisée, qui n’était pas encore la politique officielle des pouvoirs publics. Cette approche a amené la CNAMTS à proposer que la franchise soit modulée et beaucoup plus forte à partir du cinquième médicament – et non pas à partir du premier médicament – sur une ordonnance. Il y a en effet consensus des experts pour constater que, à partir de cinq médicaments sur une ordonnance, on ne contrôle plus les interactions médicamenteuses. Cette mesure a d’ailleurs été adoptée à l’époque par Mme Simone Veil, alors ministre en charge de la santé… pendant 24 heures, ce qui lui laisse son caractère entièrement novateur pour l’avenir…
M. Henri Laurent : Selon certaines études, notamment américaines, une franchise médicale est efficace à partir d’un montant d’environ 20 % de la dépense. En l’occurrence, nous sommes très loin d’un tel montant. Personnellement, je ne crois pas du tout que les franchises auront un effet sur la consommation de médicaments, même si elles ont un effet mécanique – faible – sur leur coût.
M. Michel Charton : L’effet des franchises sera mineur et très limité dans le temps. Il y aura très vite un effet d’accoutumance à des sommes aussi faibles.
Par ailleurs, s’aperçoit-on des déremboursements ? Le problème vient du fait que lorsque l’on dérembourse une classe de médicaments, on en rembourse une autre. Le Gardazil, c’est 0,5 point sinistrable de plus sur nos contrats. J’en ai discuté avec les laboratoires ; il faut dire que nous commençons à parler avec l’industrie pharmaceutique depuis quelque temps, depuis qu’elle sait que nous pourrions avoir accès aux données de soin ; nous commençons même à réfléchir à des modèles de régulation différents de ceux de la distribution actuelle. Il faut dire que les grands groupes comme AXA interviennent sur le monde entier, et que les enjeux financiers sont de plusieurs milliards d’euros.
Nous ne sommes pas capables de déterminer quel est l’impact sur nos comptes de l’abandon total du remboursement d’une classe thérapeutique. Il y a en permanence 11 000 références de médicaments et lorsqu’on en dérembourse trois, on en rembourse trois autres.
On ne mesure jamais l’effet de substitution du déremboursement. Nous ne pouvons pas le faire, puisque la seule information à laquelle nous accédons est l’indication PH 2, PH 4, PH 7, etc., et le régime obligatoire ne le fait jamais. Toutes les mesures de déremboursement sont évaluées, à condition de fonctionnement égale, sans que jamais le principe de substitution ne soit évoqué. Et ce quelle que soit la substitution, médicamenteuse ou non.
Voilà pourquoi les prévisions et les résultats d’évaluation ne peuvent être fiables. Nous croyons davantage en notre capacité à orienter le client et à le faire payer plus cher si cela est utile. C’est plus efficace qu’un système qui cherche à réguler l’ensemble des processus.
M. Laurent Doubrovine : Après l’instauration du forfait de 1 euro sur la consultation, nous avons observé très temporairement une tout petite diminution de la croissance de la consommation. Mais celle-ci s’est vite effacée, au bout de six mois ou d’un an. Il faut avoir par ailleurs en tête qu’un système complexe coûte très cher à gérer. Le complexifier encore coûte sans doute au moins aussi cher que ce que cela permet d’économiser.
M. Pierre Morange, coprésident : Que pensez-vous du principe des logiciels d’aide à la prescription ?
M. Gilles Johanet : C’est une ambition que nous avons depuis très longtemps et qui se heurte à une difficulté : l’absence d’harmonisation des bases de données, sur laquelle on finira par progresser. À première vue, les principes d’adoption et de diffusion de logiciels d’aide à la prescription sont une bonne chose. Cependant, si cela devait augmenter le handicap majeur qui affecte notre système de soins, ce serait une mauvaise chose.
Je m’explique : dans notre système de soins, l’amont est fort peu régulé, et l’aval l’est beaucoup. On demande aux généralistes de faire preuve d’une vertu romaine et d’une vigueur permanente pour dire « non ». Il me semble très innovant, et très français, de bâtir son chiffre d’affaires sur le fait de dire « non » à ses clients !
On propose de donner aux généralistes le prix des médicaments, pour qu’ils se rendent compte, etc., mais, psychologiquement, on sait qu’ils vont très mal le prendre ; cela revient en effet à leur dire que, maintenant qu’ils connaissent les prix, ils sont encore plus responsables qu’avant.
Penchez-vous sur certaines décisions du CEPS. Vous constaterez qu’un me too, médicament à amélioration du service médical rendu mineure (ASMR 4), peut obtenir un prix de 20 % supérieur au médicament qu’il est censé remplacer ! L’existence d’un logiciel d’aide à la prescription crée, en aval, pour le généraliste, une responsabilité qu’on aurait peut-être pu lui éviter. On ne peut pas tout reporter sur les généralistes. C’est exactement la même problématique que pour les affections de longue durée (ALD), dont les généralistes ne supportent plus le système.
M. Pierre Morange, coprésident : Je vous remercie.
*
Audition de M. Jean Parrot, président du Conseil national de l’ordre des pharmaciens.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale pour cette audition qui s’inscrit dans le travail qu’effectue notre mission sur la prescription, la consommation et la fiscalité du médicament. Je donne sans plus tarder la parole à notre rapporteure.
Mme Catherine Lemorton, rapporteure : Je vous remercie, monsieur Parrot, d’avoir répondu à l’invitation de la Mission.
Les études qui ont été réalisées montrent que les Français consomment plus de médicaments que la plupart de leurs partenaires européens. Je vous poserai donc la question que je pose à tous les acteurs de la santé entendus par la MECSS : pensez-vous que l’application de la franchise de cinquante centimes par boîte de médicaments permettra une diminution de la consommation en France ?
M. Jean Parrot : Je précise que l’Ordre des pharmaciens ne fait pas partie de la chaîne économique du médicament. Un certain nombre d’éléments relatifs à l’économie du médicament sont de la responsabilité des syndicats pharmaceutiques ou des autres acteurs de la chaîne du médicament. Cela étant, ayant la responsabilité de l’éthique et de l’ensemble de la gouvernance de la profession, nous sommes en mesure d’apporter un éclairage professionnel sur la question que vous soulevez.
L’application d’une franchise sur les boîtes de médicaments peut inciter les patients à réduire leur consommation, tout en sachant que ces derniers, comme les pharmaciens d’officine, ne sommes pas les prescripteurs. C’est le médecin qui est l’arbitre de la quantité de boîtes qui résulte de son ordonnance.
On pourrait penser que, si l’on propose une boîte de 90 unités de médicament, une seule franchise s’appliquera au lieu de trois pour trois boîtes de 30, mais cette approche est biaisée par de nombreux facteurs qui n’ont pas été perçus par les parlementaires au moment où la mesure a été présentée à l’Assemblée nationale.
Premièrement, les boîtes de 90 ne sont pas aujourd’hui réellement mises sur le marché pour la totalité des molécules destinées au traitement des maladies chroniques. Par ailleurs, il arrive aujourd’hui que les génériques présentés en boîtes de 30 soient moins chers pour l’assurance maladie que la boîte de 90 de génériques. En outre, souvent, seul le médicament princeps est vendu en boîtes de 90. Cela est très pervers car le pharmacien doit, théoriquement, telle que la loi a été votée, délivrer la boîte de 90. Or, s’il le fait, cela ne sera pas source d’économies pour l’assurance maladie puisque le prix de la boîte de 90, qui est la boîte de princeps, sera, en fin de compte, plus chère que trois boîtes de génériques du produit équivalent. C’est un premier biais par rapport au but économique recherché.
Nous en voyons un autre, dont nous avons informé les membres des commissions des affaires sociales des deux assemblées – et Mme la rapporteure, en tant que confrère en activité, y sera sensible – : sur l’ordonnance d’un patient chronique, la totalité du traitement, ne pourra pas toujours être donnée pour trois mois. Dans le cadre de l’observance et du suivi que les pharmaciens font, au mois le mois, pour accompagner les patients dans la prise de leur traitement chronique et vérifier qu’ils séquencent bien la totalité de leurs médicaments et ne « font pas leur marché » dans l’ordonnance en prenant un médicament et pas l’autre, cela leur compliquera encore plus la tâche.
Nous avons souhaité – et la profession, dans son ensemble, est très demanderesse – la mise en place du fameux dossier pharmaceutique – DP – qui est en expérimentation, depuis six mois, dans six départements. Grâce au dossier pharmaceutique, le pharmacien pourra désormais connaître, au moment de la dispensation, tous les médicaments que le patient a pris, toutes pharmacies confondues, dans les quatre mois précédents ; ainsi le biais lié au recours aux boîtes de 90 disparaîtra.
Lors de la mise en place du DP, nous avions prévu de faire uniquement le relevé de trois mois de traitement. A cause des boîtes de 90, nous avons porté ce délai à quatre mois pour bénéficier d’un mois supplémentaire, afin d’être sûrs que ne nous échappe pas la dispensation d’une boîte de 90.
Comme vous le voyez, madame la rapporteure, les réponses à votre question peuvent être multiples. C’est seulement après un temps d’application d’une, deux, voire trois années, que nous saurons si le but recherché, qui est de davantage responsabiliser le patient, sera atteint.
M. Pierre Morange, coprésident : Pouvez-vous dresser un premier bilan d’étape après six mois d’expérimentation du dossier pharmaceutique ? A-t-il déjà permis d’éviter des interactions médicamenteuses ? Quand pensez-vous étendre le dispositif à l’ensemble des pharmacies ?
M. Jean Parrot : La phase d’expérimentation sur les six départements choisis s’est bien passée. Les pharmacies d’officine concernées ont créé plus de 100 000 dossiers pharmaceutiques. Il faut savoir que, dès qu’un dossier est créé, il est automatiquement consulté lors de toute visite du patient dans une pharmacie équipée. Par le simple fait de la convergence dans le système informatique de la carte du patient et de la carte de professionnel de santé du pharmacien, le dossier apparaît à l’écran en trois secondes. Le pharmacien n’a pas à faire une requête spéciale.
Aujourd’hui, 300 pharmacies sont raccordées et entre 1 000 et 1 200 nouveaux dossiers sont créés chaque jour. Globalement, l’expérience est très positive.
Nous avons cependant rencontré quelques soucis que nous n’avions pas imaginés.
Dans l’expérimentation menée, nous ne devions pas, à l’origine, être les seuls acteurs. Il devait y avoir des communications communes – au besoin avec les moyens des grands medias – pour que la population soit informée de la création des différents dossiers électroniques de santé : le DP au niveau des pharmaciens, et le DMP, le dossier médical personnel, qui devait déboucher sur la mise en place d’un portail destiné à informer les patients. Vous connaissez les aléas qu’a connus le DMP. Cela ne remet pas en cause la philosophe globale du dispositif ni la nécessité d’avoir un jour un dossier électronique pour la totalité des consommations des biens de santé, mais cela nous a laissés seuls. Résultat : nous ne pouvons communiquer qu’avec chaque officine équipée. Nous ne pouvons pas faire de communication globale car les pharmaciens seraient submergés de demandes auxquels les pharmaciens, s’ils ne sont pas équipés, ne pourraient répondre et créerait une certaine frustration chez les patients qui ne pourraient accéder au dispositif présenté par les medias. Ce n’est pas le scénario que nous avions imaginé puisque nous avions même prévu un plan de communication commun avec le groupement d’intérêt public (GIP) du DMP.
La deuxième difficulté a été que, dans le cadre de la mise en place du DP, la Commission nationale de l’informatique et des libertés (CNIL) nous a demandé de respecter un certain nombre de règles pour la protection des données individuelles des patients. C’est certainement nécessaire, mais cela a considérablement compliqué l’architecture du DP et son déploiement. Là où nous avions prévu de passer relativement peu de temps avec chaque patient pour expliquer la création et la mise en place du dossier pharmaceutique, la CNIL nous a imposé tout un protocole qui nous oblige à y consacrer plus de temps.
Notre troisième souci vient des difficultés rencontrées par le DMP et du relais médiatique qui en a été fait. Nous devons parfois donner plus d’explications au patient pour le convaincre que ce que nous proposons n’est pas le DMP et que le DP est un dossier professionnel. Cela a nécessité un investissement plus important que prévu.
Mais tout cela ne remet pas en cause le principe général, et le DP fonctionne bien.
Pour répondre à la demande de la CNIL, tous les patients ont la liberté, s’ils le souhaitent, de se rétracter ou même de dire qu’ils ne veulent pas que tel médicament figure sur leur dossier. Dans ce dernier cas, comme le DP reste quand même un dossier professionnel, et non un dossier patient, il y a une trace mentionnant que le dossier est incomplet. Lors de toute dispensation ultérieure faite à ce patient, le pharmacien pourra lui demander si le médicament qu’il lui dispense ce jour-là présente un risque par rapport à ce qu’il n’a pas souhaité porter à la connaissance de l’ensemble des confrères. Le taux de refus de DP est d’environ 15 % et le taux de dossiers incomplets est de 0,002 %.
Les refus se fondent sur des critères variables. Quand on étudie la pyramide des âges, on voit un petit pic correspondant à la population, plutôt féminine, qui débute son âge adulte : les jeunes filles commencent à avoir une vie sexuelle active et ne souhaitent pas que leur famille puisse, éventuellement, savoir le choix de moyen contraceptif qu’elles ont fait.
M. Pierre Morange, coprésident : Cette crainte n’a effectivement aucun fondement. Le DP est un dossier professionnel basé sur la confidentialité.
M. Jean Parrot : Cette crainte n’a effectivement absolument aucun fondement, mais nous devons respecter la liberté des patients.
Il y a un autre pic correspondant à une population relativement jeune, active et, en général, assez instruite, qui estime qu’elle est capable de se gérer toute seule et n’envisage pas de déposer un dossier. Cette tendance ressort dans des départements très urbains. Nous l’avons constatée surtout dans le Rhône.
Pour essayer d’affiner ces observations, nous avons déposé une requête auprès de la CNIL pour passer par une deuxième phase intermédiaire, avant de procéder à un déploiement national.
Nous avons décidé de mettre en place la première phase pilote avec six partenaires qui ont aujourd’hui des dossiers permettant de faire du tiers payant à l’officine, et qui sont nos partenaires les plus importants au niveau du traitement des données pour l’assurance maladie. Le DP a été introduit par les éditeurs dans les logiciels de gestion d’officine (LGO) et nous avons à peu près une vingtaine de partenaires sur toute la France qui font les séquençages nous permettant d’avoir des relations de flux avec l’assurance maladie pour les remboursements de tiers payant.
M. Pierre Morange, coprésident : Ces partenaires sont-ils tout à la fois hébergeurs et transmetteurs ?
M. Jean Parrot : Ils sont transmetteurs.
Les hébergeurs qui existent dans le cadre des LGO sont soit des équipes professionnelles créées par les pharmaciens, soit des outils réalisés de façon indépendante. Des propositions ont même été faites au niveau des consortiums de banques. Mais cela concerne le flux financier, qui est complètement différent du flux DP. Pour ce dernier, nous avons créé une ligne spéciale, directe et sécurisée et nous avons traité avec un hébergeur national – SANTEOS – après avoir lancé un appel d’offres européen. Le flux DP est complètement sécurisé et indépendant.
Sur une vingtaine d’éditeurs LGO, ceux qui ont choisi de nous accompagner dès le départ sont au nombre de six. En réalité, il n’y en a que cinq qui ont vraiment fait l’effort de bien intégrer le DP pour qu’il fonctionne. Si nous décidions un déploiement national du DP, il n’y aurait donc que six éditeurs de LGO capables, du jour au lendemain, de répondre à la demande des pharmaciens dans toute la France. Nous ne sommes pas sûrs que les autres auraient fait l’effort, durant les six mois où ils sont restés dormants, d’adapter leurs logiciels au DP. À mon avis, ils ne seraient pas capables de soutenir la concurrence avec ceux qui ont commencé il y a six mois.
Nous avons demandé à la CNIL une autorisation pour un nouveau déploiement permettant de toucher 5 % de l’effectif des pharmaciens dans tous les départements afin, d’une part, de former des référents DP dans chaque département qui pourront ensuite aider à la formation de leurs confrères et, d’autre part, d’obliger tous les petits fabricants de LGO qui, parfois, agissent seulement dans un ou deux départements, à adhérer au système DP. Nous avons gardé chez SANTEOS un département test pour que tous les LGO entrent en phase test dès le mois de janvier 2008 – dès que la CNIL nous aura donné sa deuxième autorisation – pour être sûrs qu’ils soient tous prêts quand nous ferons le déploiement national – certainement vers le mois de juin ou juillet 2008.
Parmi les LGO qui ont démarré avec nous dans la première phase d’expérimentation, certains sont très gros. L’un d’entre eux représente pratiquement 30 % du parc. Son niveau actuel d’équipement ainsi que celui de la moitié des pharmaciens qui utilisent son LGO, c’est-à-dire environ 2 000, lui permettraient, dès l’autorisation de la CNIL, de réaliser la mise en œuvre du DP par un téléchargement nocturne pour ces 2 000 pharmacies. Tous les pharmaciens qui utilisent le logiciel LGPI de Pharmagest savent que c’est possible. Néanmoins, un basculement aussi rapide n’est pas souhaitable parce que les pharmaciens ne maîtriseraient pas tout le process à suivre pour bien recueillir le consentement du patient et mettre en place le DP.
Nous allons nous employer à former des formateurs dans tous les départements, puis nous continuerons à faire un accompagnement très progressif. Nous voudrions éviter qu’il y ait un effet pic et que cela retombe ensuite. Il y a un temps d’investissement important au moment de la formation et de l’intégration du dossier pharmaceutique.
M. Pierre Morange, coprésident : Pour quand prévoyez-vous la couverture nationale par le DP ?
M. Jean Parrot : La couverture nationale se fera au minimum sur deux ans. D’une part, le parc informatique de nos confrères devra être revu pour moitié parce qu’il n’est pas assez performant. D’autre part, tous les pharmaciens n’ont pas accès à l’ADSL. Il y a encore des zones géographiques en France qui ne sont pas raccordées. Nous avons des contacts avec le ministère concerné pour qu’un effort particulier soit fait en la matière. Par ailleurs, il faut un ADSL sécurisé.
M. Pierre Morange, coprésident : Fin 2009, la couverture devrait donc être à peu près complète ?
M. Jean Parrot : Oui, mais il faut savoir que cela va demander un investissement lourd à la profession. Puisqu’il s’agit d’un outil informatique professionnel, la profession a choisi de le prendre en charge elle-même, comme elle l’a toujours fait pour tout développement informatique.
M. Pierre Morange, coprésident : Avez-vous pu mesurer des premiers bénéfices du dossier pharmaceutiques dans les six départements expérimentaux ?
M. Jean Parrot : Oui, mais notre évaluation est limitée puisque nous nous sommes engagés à ne jamais recueillir de données individuelles patients et à ne pas les traiter de façon nominative. Ce que nous analysons, c’est la quantité de médicaments en cours de dispensation et qui, à un moment donné, ont été stoppés et retirés de la dispensation. Cela signifie que, alors que le pharmacien était en train de faire sa dispensation, sa banque de données lui a signalé qu’il y avait une interaction entre ce qu’il était en train de faire et le dossier patient qu’il avait sur son écran ; il a donc retiré le médicament qu’il avait introduit.
Sur les 100 000 dossiers traités, il y a eu 112 retraits de médicaments.
Nous allons voir avec la CNIL si nous pouvons faire une approche plus fine et connaître l’âge du patient, ou la tranche d’âge dans laquelle il se situe, et, si possible, la classe thérapeutique du médicament.
M. Pierre Morange, coprésident : Avez-vous interrogé les services d’urgence des départements expérimentaux afin de voir si la mise en place du DP avait entraîné une diminution des hospitalisations pour interaction médicamenteuse ?
M. Jean Parrot : C’est aujourd’hui prématuré. Nous ne pourrons mesurer l’impact du DP sur la iatrogénie évitable que lorsque le réseau sera déployé.
Mais ce qui est notable aujourd’hui, c’est qu’on a l’assurance que, dans 112 cas, un médicament en cours de dispensation a été retiré. L’anecdote comptoir qui va avec ce progrès, c’est que des pharmaciens ont appelé leur référent local pour exprimer leur satisfaction d’avoir éviter une interaction médicamenteuse.
M. Jean Mallot, coprésident : Quelle est la structure qui met en place le dossier pharmaceutique et le pilote ?
M. Jean Parrot : En 2004, tous les ordres professionnels de santé ont reçu pour mission supplémentaire de s’assurer de la sécurité de leurs actes. L’Ordre des pharmaciens a donc estimé qu’il lui revenait d’assurer la sécurisation des dispensations. Quand le DMP a été créé, nous avons souhaité nous y intégrer pour créer un outil professionnel de sécurisation en vue de l’abondement thérapeutique.
M. Jean Mallot, coprésident : L’Ordre des pharmaciens est effectivement en première ligne dans ce projet.
M. Jean Parrot : En effet et les cotisations des pharmaciens ont permis de développer, au sein des services de l’Ordre, un département entièrement consacré au DP, et de recruter les personnels nécessaires à sa mise en place.
Mme Catherine Lemorton, rapporteure : Les médecins peuvent-ils ou pourront-ils accéder au DP au moment où ils prescrivent ?
Comme il est prévu de favoriser l’automédication familiale, donc de faciliter l’accès des patients aux médicaments conseil, qui ne sont pas dénués d’interactions médicamenteuses ni d’effets secondaires, pensez-vous qu’il serait souhaitable d’intégrer ces médicaments dans le DP ?
M. Jean Parrot : Il est malheureusement impossible pour le moment aux médecins d’accéder directement au DP puisque ce dernier est un outil professionnel conçu et organisé au travers d’outils de nos fabricants de logiciels en vue du remboursement par le tiers payant.
Deux éléments favorables ont permis la création du DP. Le premier est le faible nombre de LGO travaillant avec nous – une vingtaine –, dont deux représentant 60 % du marché. Nous savions que nous pouvions monter quelque chose avec eux parce qu’ils n’étaient pas trop émiettés. Le second élément est que tous les médicaments sont codés, tous les pharmaciens ont une carte professionnelle et tous les patients une carte Vitale. De plus, tous les pharmaciens sont équipés en informatique parce qu’ils font tous du tiers payant. Tous les critères étaient donc réunis pour pouvoir créer un outil professionnel.
La seule chose possible vis-à-vis des médecins – et c’est ce qui se fait déjà sur le terrain –, c’est de faire, à la demande du patient, une copie de son dossier, qu’on lui remet en mains propres. Le seul accès du médecin au DP pour le moment est une copie papier.
Compte tenu de la façon dont notre outil est conçu aujourd’hui, il faut, pour qu’un médecin ait accès au DP, que le DMP existe et que le DP y soit intégré. Malgré les difficultés du projet DMP, j’ai plaidé en ce sens auprès de Mme la ministre de la santé, auprès des responsables du GIP-DMP et auprès de la mission d’information de l’Assemblée nationale sur le DMP. Dès l’instant où nous avons l’accord du patient, il faut que nous puissions mettre les données DP dans un outil consultable par les autres professionnels de santé.
Les médicaments conseil figurent déjà dans le DP. Les LGO ne prenaient jusqu’alors en compte que les médicaments remboursables puisque les flux qu’ils traitaient étaient destinés à l’assurance maladie. Ils ont élargi le champ des données collectées et intègrent désormais la totalité des médicaments.
Nous nous étions limités aux médicaments qui commencent par le code 3, mais, comme nous acceptons maintenant les médicaments d’importation, lesquels commencent par un code différent, nous travaillons avec les sociétés informatiques pour voir comment intégrer d’autres codes et, en même temps, comment intégrer les dispositifs médicaux.
Nous aurons également à consentir des efforts en 2009 pour voir comment on peut connecter les pharmacies hospitalières, puisque celles-ci le demandent. Nous reviendrons donc certainement devant le Parlement l’année prochaine pour vous demander une autorisation supplémentaire afin de raccorder les pharmacies hospitalières. Nous nous heurterons alors à une petite difficulté car, si les pharmaciens de ville ont payé leur déploiement et leur intégration au dispositif, il nous faudra, dans le cas des pharmaciens hospitaliers, passer des contrats avec les services hospitaliers. Je me suis déjà rapproché de la Fédération hospitalière de France pour lui présenter le sujet et réfléchir à une solution.
M. Pierre Morange, coprésident : Il ressort des travaux de la mission d’information sur le DMP, présidée par M. Jean-Pierre Door, qui vous a auditionné, que le délai de mise en œuvre de ce dispositif pourrait être de quatre à sept ans et que les masses financières qu’il serait nécessaire de mobiliser pourraient s’élever entre 4 et 5 milliards d’euros. Or vous avez indiqué qu’il était indispensable que le dossier pharmaceutique puisse être connecté avec le futur DMP pour lui donner sa cohérence.
J’ai proposé, en attendant que ce dernier soit mis en place, de cibler la population qui en a besoin de façon primordiale, c’est-à-dire les personnes atteintes d’une affection de longue durée, qui nécessitent une polythérapie et pèsent pour plus de 50 % dans le budget de l’assurance maladie. On pourrait mettre à disposition de chacun de ces patients une clé USB qui serait connectable sur le PC de l’ensemble des intervenants de la chaîne de santé. Un tel dispositif aurait l’avantage d’éviter d’être confronté, non seulement aux difficultés liées au problème de l’hébergeur et du transmetteur qui bloque dans le cadre du DMP, mais également aux réserves de la CNIL sur le plan de la confidentialité. Il pourrait être rapidement mis en place et aurait un coût moindre, tournant autour de 300 millions d’euros, en évaluant une clé USB entre 20 et 30 euros. Le directeur de la sécurité sociale et l’assurance maladie se sont déclarés intéressés par ce dispositif. Qu’en pensez-vous ?
M. Jean Parrot : Le problème posé par le recours à une clé USB est celui de la lisibilité de son contenu par les différents partenaires de santé. Quand vous recherchez quelque chose sur une clé USB, cela demande beaucoup de temps et n’est pas facile, surtout dans le cas d’un patient en ALD ayant des partenaires de santé multiples. Cela ne facilitera pas forcément la tâche lors des consultations. Le chargement sera certainement bien fait, mais il n’est pas sûr que l’outil soit facile à utiliser.
Dans le cadre des auditions auxquelles nous avons été conviés, il nous a souvent été demandé comment nous voyions le montage qui pourrait être fait par rapport à l’outil DP. Nous pensons qu’ils serait utile d’inviter et d’aider tous les professionnels à monter leur propre système : radiologistes, biologistes, pharmaciens, médecins généralistes, hôpitaux, en particulier dans le cadre de la montée en charge de la tarification à l’activité. Ce serait un premier outil vertical.
On pourrait très bien imaginer, au-dessus, un DMP de deux niveaux.
Le premier devrait être un outil partagé entre les professionnels de santé. Les médecins généralistes ne sont pas les seuls intéressés. Les biologistes souhaiteraient également connaître le traitement du patient quand ils font une glycémie. Inversement, les pharmaciens aimeraient connaître les derniers résultats des analyses quand ils doivent délivrer des médicaments anticoagulants. Des liens transprofessionnels doivent être établis. Les responsables du GIP-DMP ont déjà recensé ce que chacun a besoin de connaître dans le métier de l’autre. On pourrait utiliser toutes ces données pour construire ce premier étage.
On pourrait imaginer un deuxième niveau qui serait l’outil patient. Chaque profession, en accord avec les associations de patients, y mettrait toutes les données qui lui sont utiles.
Je vous donne un exemple concernant les pharmaciens.
Il est normal qu’ils donnent au patient la lisibilité des médicaments qu’il achète mais ce dernier n’a pas besoin d’avoir les dates de péremption de ceux-ci ni des détails sur des retraits de lots qui ne le concernent pas. Or il est prévu, dans notre outil professionnel, dès que cela sera possible avec l’industrie pharmaceutique et dès que le système le permettra, de procéder en même temps à une surveillance des spécialités afin de se garantir au maximum contre les entrées en France de médicaments frauduleux.
La contrefaçon des médicaments est un fléau mondial aujourd’hui. Les pays européens qui nous entourent devraient normalement se sentir sécurisés. Or ils ne le sont pas parce que, dans nombre d’entre eux, les importations de médicaments ne sont pas sous la responsabilité pharmaceutique.
En Angleterre, elles peuvent ainsi être faites par tout importateur détenant une licence d’importation. Il y a plus de 3 000 importateurs dans ce pays, qui peuvent importer des médicaments comme du thé ou des chaussures. Ils font rentrer des conteneurs provenant d’Asie ou d’ailleurs et les produits sont ensuite déversés sur les marchés. Or, aujourd’hui, les capacités des contrefacteurs sont telles que la dernière saisie faite en Angleterre correspondait à des produits d’un laboratoire français et portait le même numéro de lot que les médicaments vendus en France au même moment, ce qui a obligé le laboratoire français ainsi que l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) à réagir de façon très rapide : ils ont dû retirer et mettre en quarantaine tous les médicaments portant ce numéro de lot en France. Un patient français, qui aurait lu dans un quotidien que tel numéro de lot de tel médicament que lui-même prenait – et qui, de plus, n’était pas un médicament banal – était retiré en Angleterre, n’aurait pas compris qu’il soit laissé en vente en France. Il a été nécessaire de tout bloquer.
En France, la distribution des médicaments est entièrement contrôlée par les pharmaciens. L’Europe, qui nous présentait autrefois comme très archaïque, étudie maintenant notre modèle.
Pour qu’un lot change de place dans l’usine qui le fabrique, un pharmacien responsable de l’usine signe et transmet à un autre pharmacien des affaires réglementaires. Quand le lot est déclaré conforme, le pharmacien industriel le met en quarantaine et attend, avant de le libérer, d’avoir reçu tous les certificats attestant que le lot est libérable. Le lot est alors stocké, soit chez un dépositaire, soit dans le laboratoire et ne peut être vendu qu’à un grossiste répartiteur, un pharmacien engageant sa signature dans le bon de commande.
Ainsi le pharmacien industriel vend à un pharmacien dépositaire, lequel vend à un grossiste. Le grossiste peut également acheter directement au laboratoire. Le lot part ensuite, soit dans une pharmacie hospitalière avec le numéro d’enregistrement d’un pharmacien hospitalier, soit dans une pharmacie d’officine avec le numéro du pharmacien d’officine. Tout cela est relativement transparent parce qu’on ne passe pas notre temps à échanger nos numéros. Les adresses et les certifications sont telles que, si une pharmacie demande à un laboratoire de lui envoyer des médicaments, ce dernier vérifie à la fois l’adresse de son établissement pharmaceutique et son référencement à l’intérieur du site de l’Ordre pour s’assurer qu’elle est bien une officine et qu’elle est ouverte et enregistrée. Ce n’est qu’après toutes ces vérifications qu’elle recevra ses médicaments. Sinon, elle ne reçoit rien.
Grâce à notre système – aussi archaïque qu’il puisse paraître –, il semblerait qu’on n’ait pas eu d’importations frauduleuses jusqu’à présent. Mais nous allons vérifier tout cela.
M. Pierre Morange, coprésident : Sur la sécurité de la chaîne pharmaceutique, vous prêchez des convaincus, mais il était important de rappeler devant les médias que la profession de pharmacien, notamment d’officine, qui a souvent été traitée de ringarde, est la gardienne de la sécurité en matière de médicaments.
Le développement de la contrefaçon de médicaments est préoccupant puisqu’on évoque, notamment en Afrique et dans le Sud-Est asiatique, des pourcentages allant de 35 % à 45 %.
M. Jean Parrot : Le taux de médicaments contrefaits dans les flux médicamenteux dans les pays en voie de développement atteint ces chiffres. Dans des pays dits de haute sécurité, comme les États-Unis d’Amérique, on l’estime entre 10 et 15 %. Cela est dû en partie au fait qu’aux États-Unis, les médicaments sont très chers et qu’environ 50 millions d’Américains n’ont pas de protection sociale. Pour trouver les médicaments les moins chers, ils vont sur Internet où la moitié des médicaments proposés sont contrefaits.
Le Canada est confronté au même problème, d’autant que les réglementations diffèrent selon les États. Le Manitoba a imaginé des pharmacies virtuelles et même des pharmacies situées dans les étages des immeubles, qui ne voient jamais les médicaments car ceux-ci viennent de l’étranger et repartent directement vers les États-Unis. Elles n’enregistrent que les flux financiers. La loi au Manitoba est telle aujourd’hui qu’un médicament qui rentre et qui est destiné à l’étranger n’a pas à être certifié ni ses normes enregistrées. C’est scandaleux.
La contrefaçon est devenue un énorme marché dans le monde, surtout celle des médicaments. Il est beaucoup plus facile et moins cher aujourd’hui pour un contrefacteur de faire des bouts de papier de couleurs différentes et d’y mettre un blister avec pratiquement rien du tout que de fabriquer un faux médicament. Cela nécessite encore moins d’investissement en matière première et la rentabilité est bien meilleure.
Pour revenir au DP, il est un domaine sur lequel nous voulons aussi agir. Nous voulons essayer, également avec l’industrie pharmaceutique, de mettre en place, en quelques années, un marquage de chaque boîte, de façon à ce que, après l’enregistrement informatique de la vente, il y ait effacement de la boîte du marché et qu’elle ne puisse plus y revenir, de quelque manière que ce soit. Grâce à notre système, qui est un outil professionnel, nous aurons un contrôle des flux depuis l’usine jusqu’au consommateur, de sorte qu’il sera impossible qu’une boîte soit recyclée deux fois ou même qu’un contrefacteur imagine une boîte avec le même numéro. Cela se fera, bien entendu, quand les outils informatiques seront développés, mais cela peut aller relativement vite. Il y a quatre ou cinq ans, les industriels n’étaient pas très réceptifs au problème de la contrefaçon alors que, aujourd’hui, nous travaillons en grande harmonie avec eux et des grandes firmes informatiques, dont IBM, nous aident à construire des outils qui devraient nous permettre, demain, de saisir sans souci des milliards de boîtes. Les capacités des ordinateurs le permettent aujourd’hui.
Mme Catherine Lemorton, rapporteure : Je veux également saluer la capacité du réseau officinal à récupérer des lots rapidement quand l’AFSSAPS signale un problème.
M. Jean Parrot : Dans le DP, nous n’enregistrons que les consommations des quatre derniers mois mais, dans le cas d’un retrait de lot, il y a ce qu’on appelle le « bris de glace ». Dès qu’on aura saisi le lot – ce qui va se produire dans peu de temps puisqu’on va passer de treize chiffres de saisie à quinze, seize ou dix-huit –, on pourra redescendre par le serveur informatique jusqu’au pharmacien qui a vendu la boîte et repérer l’identité de la boîte et du patient. L’opération « bris de glace » fonctionnera jusqu’à la date de péremption de la boîte, puisque, après, on estime qu’on n’a plus à la suivre.
Mme Catherine Lemorton, rapporteure : Que pensez-vous de la délivrance exacte du nombre de comprimés nécessaires à un traitement comme dans les pays anglo-saxons, dans le cas, notamment, des pathologies aiguës ?
M. Jean Parrot : Cette question doit être réglée en amont, c’est-à-dire au niveau de l’autorisation de mise sur le marché (AMM), pour que le conditionnement soit adapté à l’optimisation du traitement. Si le traitement est de huit jours et si la posologie est de deux comprimés par jour, il faut faire des boîtes de seize. Si le traitement est plus court, il faut faire des boîtes encore plus petites. C’est ce que la commission d’AMM et les laboratoires ont progressivement mis au point. Aujourd’hui, les restes médicamenteux pour des traitements courts sont de plus en plus réduits.
Par ailleurs, il faut savoir que le prix d’un médicament n’est pas lié au nombre de comprimés qu’il y a dans la boîte. Il est souvent lié au coût de traitement journalier par rapport à une pathologie donnée ainsi qu’à la difficulté de production de la molécule et au nombre d’étapes de production de celle-ci. Il est plus difficile de fabriquer une antiprotéase que de l’aspirine. Les conditions de synthèse et de fabrication ne sont pas les mêmes. Un élément du prix est lié à la molécule elle-même ; un autre élément est lié à la fois au coût de traitement journalier et au nombre de patients à traiter. Quand un bassin de population de plusieurs millions de patients est à traiter, la négociation entre le Comité économique des produits de santé et les laboratoires tiennent compte de cet élément ainsi que de l’encombrement éventuel d’autres produits sur ce même bassin de population, dans la mesure où il peut y avoir des situations concurrentielles.
Cela étant, l’une des meilleures réponses que l’on peut donner à votre question en tant que pharmacien est la suivante : si on enlève la boîte et la notice et si on se met à déconditionner comme cela se faisait largement dans les pays anglo-saxons, le patient perd beaucoup de qualités liées à cette présentation dite « à la mode française », car beaucoup d’indications sont données sur la boîte et sur la notice, dont le numéro de lot n’est pas la moindre. Pour retrouver ce dernier et tous les éléments utiles de suivi du médicament, il faudrait faire des conditionnements individuels, c’est-à-dire indiquer sur chaque blister individuel tous les éléments d’identité du produit depuis sa fabrication.
Si l’on conditionne tous les médicaments en blisters individuels, ce sera très compliqué, surtout pour les patients qui souffrent de pathologies multiples et suivent des traitements lourds, et qui se retrouveront avec des quantités de petits blisters. Il leur sera difficile de les utiliser, cela demandera beaucoup de temps pour les reconditionner en officine et cela présentera un risque de mélange de spécialités, voire de non-reconnaissance exacte du produit ensuite par le patient. Il n’y a rien de pire que deux comprimés blancs, même s’il y en a un de taille différente par rapport à l’autre. Quand vous sortez deux blisters différents de boîtes de couleurs différentes, vous avez moins de risques de vous tromper.
Pour résumer, je ne suis pas très partisan des déconditionnements en général. Je trouve que c’est une prise de risques.
J’en profite pour vous dire qu’on a également eu une fausse bonne idée entre professionnels en demandant des prescriptions systématiques en dénomination commune internationale (DCI). Intellectuellement, c’est intéressant parce que cela permet aux pharmaciens ainsi qu’aux médecins d’avoir, grâce à la DCI, des références à l’identité de la molécule par rapport à la formule. En revanche le patient se retrouverait avec une ordonnance illisible pour lui. Il vaut mieux avoir des noms de fantaisie, même s’ils sont des noms commerciaux. Ce qui est le plus utile pour le patient, c’est la sécurité d’utilisation, même si, intellectuellement, il est contrariant de ne pas utiliser le nom réel du produit.
Il faut toujours faire attention aux fausses bonnes idées. J’ai, moi-même, milité à une époque pour la prescription en DCI et je me suis aperçu que son application au public n’était pas bonne.
M. Pierre Morange, coprésident : Vous nous avez dit que vous nous demanderez l’autorisation d’étendre le DP aux pharmaciens hospitaliers. Nous sommes ouverts à toutes suggestions pouvant être déclinées soit sous forme réglementaire, soit sous forme législative et participant de notre philosophie commune, à savoir l’intérêt des patients et la bonne gestion de l’argent public.
M. Jean Parrot : Je vous remercie. Pour le moment, le coût pour les pharmaciens du déploiement du dossier pharmaceutique est estimé à environ 20 millions d’euros sur cinq ans. Ce coût est financé par les cotisations des pharmaciens titulaires d’officines, lesquelles ont beaucoup augmenté. Nous avons demandé des possibilités de subventions de la part du Fonds d’intervention pour la qualité et la coordination des soins (FIQCS) puisque nous contribuons à l’amélioration de la qualité des soins. Mais, comme cela restera un outil professionnel, cela nous demandera quelques investissements.
Pour ce qui concerne les pharmaciens hospitaliers, je suis en train de faire réaliser une évaluation préalable afin d’être en mesure d’indiquer à la Fédération hospitalière de France le coût prévisionnel estimé et lui demander si elle pense pouvoir le supporter, compte tenu du nombre d’établissements à raccorder. Il faudrait que ce soit un prix par établissement. Je ne vois pas comment on peut faire autrement, à moins que l’État veuille prendre en charge ce coût.
Mme Catherine Lemorton, rapporteure : Nous avions bien d’autres questions à vous poser mais nous vous les communiquerons en vous demandant de bien vouloir y répondre par écrit.
M. Pierre Morange, coprésident : Nous vous remercions.
*
Audition de M. Philippe Gaertner, président de la Fédération des syndicats pharmaceutiques de France, accompagné de M. Jean-Pierre Lamothe, premier vice-président, président de la commission économie de l'officine, M. Claude Japhet, président de l'Union nationale des pharmaciens de France, et Mme Marie-Josée Augé-Caumon, membre du conseil d'administration de l'Union des syndicats de pharmaciens d'officine.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale. Je donne sans plus tarder la parole à notre rapporteure.
Mme Catherine Lemorton, rapporteure : Sans revenir sur le caractère juste ou injuste et sur l’efficacité économique de la franchise médicale de 50 centimes par boîte de médicaments, j’aimerais savoir ce que vous en pensez en termes de prescription et de consommation. La jugez-vous de nature à réduire la consommation des médicaments ?
Mme Marie-Josée Augé-Caumon : Il faut s’interroger sur la prise en charge de ces franchises. La décision n’est pas encore prise pour les complémentaires mais on entend dire qu’il se pourrait que le remboursement soit introduit dans les contrats responsables. Or il va de soi que si le reste à charge de l’assuré est remboursé, la disposition aura une portée moindre.
Il s’agit par ailleurs d’une mesure économique qui risque de pénaliser un certain nombre de patients et qui aura des effets induits. En premier lieu, ceux qui ont besoin de consommer beaucoup de médicaments, en particulier les patients en affection de longue durée (ALD), seront obligés de restreindre leur consommation au risque de ne pas se soigner comme il faut. Cela pourrait être néfaste à la santé des Français.
On risque en deuxième lieu d’assister à des transferts au sein des familles, le premier ayant atteint le plafond de 50 € cherchant à se faire prescrire la totalité des médicaments consommés ensuite par le reste de la famille.
Il me semble que l’on pourrait faire appel à d’autres outils, comme le dossier médical personnel (DMP) et l’observation de la redondance des actes, et faire confiance aux pharmaciens pour le suivi des patients en cas de trop forte consommation. Nous avons d’ailleurs signé une convention avec l’assurance maladie afin d’organiser le suivi des personnes âgées qui consomment beaucoup de médicaments.
M. Claude Japhet : Avant d’en venir aux franchises, je veux rappeler que le discours récurrent sur la surconsommation médicamenteuse dans notre pays traduit en fait une idée reçue. En effet, de 1994 à 2007, la consommation des médicaments remboursés n’a absolument pas varié. Plus exactement, elle a connu une faible augmentation jusqu’en 2004 et la tendance s’est inversée par la suite, ce qui fait que l’on n’a pas consommé davantage de boîtes cette année qu’il y a treize ans.
Qui plus est, la notion de surconsommation est assez floue. Il est en effet très difficile de faire des comparaisons internationales car le contenu des boîtes varie de 12 à 60 voire à 100 unités. En France, on raisonne en nombre de boîtes alors qu’en nombre d’unités, la consommation française a un peu moins progressé que celle des autres pays européens.
Nous demeurons toutefois largement au-dessus de nos voisins avec, en moyenne, quatre boîtes par personne et par mois. Pour disposer de comparaisons plus fines, il faudrait des méthodes de comptage similaires entre pays. En France, cela nous obligerait à remonter à la prescription et à la posologie, mais nous ne disposons pas d’outils suffisamment fins pour cela.
M. Jean Mallot, coprésident : S’il n’est pas possible de mener cette comparaison à grande échelle, on peut toutefois le faire sur un échantillon, ce qui permettrait d’avoir une idée.
Mme Marie-Josée Augé-Caumon : C’est ce qu’on a commencé à faire avec la DDD (Defined Daily Dose – dose journalière définie), mais on se heurte également à des différences de dosage, par exemple 200 mg en Angleterre et 400 mg en France.
M. Claude Japhet : J’en viens aux incidences que la franchise pourrait avoir sur la consommation et sur ses éventuels effets pervers.
Dès lors qu’il y a responsabilisation de l’assuré – et telle est la volonté du législateur – il y aurait autorégulation de la consommation si la mesure intervenait au moment même du paiement par l’assuré. Or tel ne sera pas le cas puisqu’elle sera appliquée par la suite par l’assurance maladie au moyen d’une retenue sur les remboursements qu’elle effectue. On peut donc penser qu’elle ne jouera pas comme un véritable frein à la consommation.
Nous pourrions même avoir un effet de seuil avec une accélération de sa consommation une fois que l’assuré aura atteint le plafond de la franchise. On a vu que le panier moyen de médicaments est de quatre boîtes par mois soit une cinquantaine de boîtes par an, ce qui correspond à peu près au plafond de la franchise. On assistera donc non seulement aux transferts intra familiaux que vient d’évoquer Mme Augé-Caumon, mais aussi à une surconsommation de fin d’année, une fois le plafond dépassée, afin d’anticiper le plafond de l’année suivante. On aura ainsi, au bout du compte, un effet inverse de celui que nous cherchons tous à atteindre, c’est-à-dire une régulation de la consommation de médicaments dans le temps.
Qui plus est, la fiabilité du dossier pharmaceutique personnel se trouvera affectée par les transferts de prescription, notamment au sein des familles, ce qui risque d’obliger les officines à un travail de recherche supplémentaire.
Mme Marie-Josée Augé-Caumon : Ces transferts devraient tout de même être limités par les médecins.
M. Claude Japhet : On voit déjà des anomalies flagrantes dans les prescriptions, par exemple avec un produit pour enfants sur une ordonnance destinée à un adulte. À nous de faire la part des choses, comme nous le faisons déjà.
M. Philippe Gaertner : Quand on parle de surconsommation, on a tendance à mélanger deux notions : la consommation en euros et la consommation en unités. Nous avons donc essayé d’y regarder de plus près et nous avons constaté qu’à partir d’une base 100 en 2001, le chiffre d’affaires industriel est passé à 130 en 2007, tandis que le nombre d’unités se situe entre 97 et 98, donc effectivement en légère diminution, et que la consommation en nombre de boîtes chute à 94. Les différents plans successifs ont donc bien eu un effet sur la quantité d’unités et de boîtes de médicaments consommées.
Je suis en légère divergence avec mes collègues en ce qui concerne la franchise.
J’observe pour ma part que les patients sont déjà fortement sensibilisés, qu’ils nous interrogent régulièrement à ce propos et qu’ils s’attendent même déjà à devoir la payer. Je pense donc que même si elle n’est pas prélevée en officine, elle aura bien un impact sur la consommation en ce qu’elle incitera à vider l’armoire à pharmacie. L’effet sera donc ponctuel mais réel.
Quant à l’effet de seuil, il est possible qu’il se manifeste à la fin de 2008, première année d’application du dispositif, mais plus les années suivantes car le phénomène de report anticipé ne jouera qu’une fois.
M. Jean-Pierre Lamothe : L’effet de la franchise va jouer sur la consommation, mais il s’estompera assez vite, en particulier parce qu’elle sera prélevée au moment du remboursement par l’assurance maladie.
La dérive que je crains est celle d’une demande accrue du patient sur le médecin afin d’avoir immédiatement des bons conditionnements, y compris en initiation de traitement. Cela pourrait entraîner un gâchis, source de surcoût pour l’assurance maladie. Nous commençons d’ailleurs déjà à ressentir ce phénomène au comptoir des officines.
J’exerce dans un quartier plutôt favorisé où les franchises ne sont pas très bien perçues parce que les patients cotisent beaucoup à l’assurance maladie, qu’ils ne sont pas très fréquemment malades et qu’ils n’arriveront pas forcément à obtenir un remboursement correct. Ils pourraient ainsi, à un moment, avoir une légitimité à demander à sortir du système solidaire pour aller vers un système assuranciel privé. Il y a donc là un réel danger pour notre système solidaire collectif.
Cela étant, le dispositif va se mettre en place et il serait malvenu en tant que professionnels de santé que, d’un côté nous demandions à ne pas être toujours la variable d’ajustement lorsqu’il y a un déficit de l’assurance maladie et que, de l’autre, nous refusions tout système responsabilisant le patient et procurant des recettes à l’assurance maladie. Simplement, il faudra veiller à ce que les dérives ne soient pas trop importantes et à ce que les grands conditionnements ne se développent pas à l’excès car cela pourrait être source de gâchis. S’il pouvait sembler logique que, pour un traitement de douze mois, douze boîtes soient remplacées par quatre, il ne faudrait pas que l’on arrive en fait à six ou sept boîtes car cela entraînerait un surcoût pour l’assurance maladie.
Mme Catherine Lemorton, rapporteure : À l’occasion d’un congrès, vous aviez, monsieur Gaertner, présenté un graphique qui montrait fort bien l’évolution de la consommation des médicaments en fonction des prescripteurs et des pharmaciens. Pouvez-vous en dire plus à ce propos ?
M. Philippe Gaertner : Je vous remettrai ce document. Il s’agit d’un graphique qui comporte des éléments purement économiques et d’autres éléments liés à la consommation : chiffre d’affaires industriel, marge des officines, nombre de consultations et de visites dont vos auditions précédentes ont montré qu’il était bien lié à la consommation. En effet, on constate que la courbe des consultations et celle de la consommation en unités sont parallèles.
M. Jean Mallot, coprésident : Comment analysez-vous les relations entre les pharmaciens et ce qui vient en amont, en particulier les efforts promotionnels des laboratoires ? Comment pourrait-on améliorer les choses ?
Mme Marie-Josée Augé-Caumon : On a coutume de dire que le médecin français est le plus innovant au monde, ce qui signifie que la pénétration des nouveaux médicaments, probablement induite par la visite médicale, réussit assez bien.
On constate toutefois que les choses changent dès lors que l’on introduit un autre intervenant dans le mécanisme, au lieu de laisser se dérouler un dialogue entre le médecin et l’industrie pharmaceutique. Ainsi, l’intervention du pharmacien est propice à une régulation : on a bien vu avec les génériques que les officines pouvaient agir sur la substitution.
Alors que jouent ce que l’on appelle les effets pervers du changement de prescription – c’est-à-dire que l’industrie s’efforce de maintenir un niveau élevé du prix du médicament – passer du duo médecin-industrie à un trio incluant le pharmacien pourrait bien avoir un effet sur l’économie du médicament.
M. Jean Mallot, coprésident : Il devrait même s’agir d’un quatuor car on ne saurait oublier l’assurance maladie…
Mme Marie-Josée Augé-Caumon : Les délégués de l’assurance maladie ont en effet un rôle efficace auprès des pharmaciens et des médecins, mais on n’en compte que moins d’un millier et leur poids est insuffisant au regard de celui des visiteurs médicaux.
M. Claude Japhet : Je ne me prononcerai pas sur la pertinence de la prescription médicale : les médecins prescrivent en toute liberté et nous n’avons pas à nous ingérer dans leurs relations avec l’industrie pharmaceutique.
Nous constatons néanmoins les effets sur les génériques de cette volonté de prescription favorisant plutôt l’innovation que ce qui existe déjà. Ainsi, on a vu la consommation des prazoles augmenter tandis que celle des produits de substitution demeurait stable. Le phénomène est encore plus frappant pour les statines dont le marché ne fait qu’augmenter jusqu’à ce que la substitution intervienne et que la consommation stagne avant de chuter. Le troisième exemple est celui des acides alendroniques pour lesquels les ventes de Fosamax ont progressé continûment jusqu’à ce que, quelques semaines avant que le brevet ne tombe, le nouveau médicament de contournement enregistre une forte progression tandis que l’ancien chutait brutalement, pratiquement au moment de la sortie du générique.
En disant cela, je ne me prononce nullement sur la pertinence d’aller vers le nouveau médicament. Simplement, il est surprenant qu’il y ait ce transfert de prescription au moment même où un produit est sur le point de tomber dans le domaine public, sachant que l’on n’est plus dans la même gamme de prix…
Ce n’est certes pas le cas pour tous les produits, mais on est quand même globalement dans un phénomène industriel et médical qui favorise de plus en plus l’innovation par rapport aux génériques. D’ailleurs, alors que le marché des génériques aurait dû enregistrer une progression constante, il est très loin des objectifs qui lui avaient été fixés. Cela montre que, quelle que soit la volonté de faire des économies, on ne parviendra jamais aux résultats escomptés au moment où on l’aurait voulu, tout simplement parce qu’il y a des transferts de prescription.
M. Jean Mallot, coprésident : Mais alors que faire ?
Mme Marie-Josée Augé-Caumon : Inciter à prescrire dans le répertoire des génériques.
M. Claude Japhet : Vous aviez raison de parler d’un partenariat à quatre : si l’industrie, les prescripteurs, les pharmaciens et l’assurance maladie ne se mettent pas d’accord sur des objectifs quantifiés, on ne parviendra pas à les atteindre.
M. Philippe Gaertner : À partir du moment où de nouvelles molécules innovantes sont mises sur le marché, nous sommes l’un des pays où l’accès à cette innovation est le plus rapide. Je ne porte pas de jugement de valeur sur ce phénomène, mais les citoyens, qui sont aussi les patients, attendent cet accès à l’innovation. Et c’est bien la mutualisation de la prise en charge qui le permet.
Je rappelle qu’au moment de l’arrivée des génériques, les officines ne demandaient rien. Cette arrivée a fait suite à une convention entre l’État et l’industrie pharmaceutique qui consistait à mettre en avant les génériques en contrepartie des progrès de l’innovation. C’est dans un deuxième temps, quand on a constaté la très lente montée en charge des génériques, que l’on s’est tourné vers le pharmacien en lui octroyant le droit de substitution, qui a été suivi par un certain nombre d’accords conventionnels. Toutes les conventions passées entre les pharmacies d’officine et l’assurance maladie ont été respectées et leurs objectifs ont même été dépassés.
Je ne reviens pas sur les déplacements de prescription que vient d’indiquer M. Claude Japhet, mais il est quand même quelque peu irritant, quand on a consenti au comptoir un effort important de substitution, de constater des effets de contournement liés à d’autres acteurs, médecins et industriels. Comme vous l’avez dit, il y a pourtant le quatrième partenaire, l’assurance maladie, ainsi qu’un objectif d’équilibre des dépenses.
S’agissant de la visite médicale, un autre phénomène provoque une forte irritation : celui des me too. Il pose un problème pour la pharmacie car on est bien cette fois sur une molécule de même DC (dénomination commune). Par le biais soit des dosages, soit de la forme galénique, on dévie la prescription.
Il ne s’agit pas de porter un jugement, mais simplement de décrire ce que ressentent les pharmaciens d’officine. Pour cela, je prendrai l’exemple d’un des derniers me too dans le domaine de la gynécologie.
Nous avions jusqu’à présent un dosage à 5 mg en boîte de 10 et nous voyons arriver à dosage à 3,75 mg en boîte de 14. Les études montrent bien entendu qu’en hormonothérapie ce dernier dosage a la même efficacité. On comprend fort bien que la femme ne continue pas à prendre 5 mg quand 3,75 sont suffisants et que le prescripteur tienne le même raisonnement. Il faut donc s’intéresser davantage à l’aspect économique pour comprendre l’irritation du pharmacien. Avec l’ancien princeps à 5 mg, la marge brute de la pharmacie était de 4,58 €. Quand on délivre le générique, en 2007, la marge est de 5,28 €, ce qui permet de compenser l’effort de substitution. Quand on passe au nouveau dosage, avec le princeps non substituable, la marge tombe à 3,20 €. La perte de marge est de 30,21 % par rapport au princeps initial et de 61 % par rapport au générique, pour un prix équivalent pour l’assurance maladie. Cela signifie que, dans une telle opération, la pharmacie supporte la totalité de la perte de marge, mais aussi que les pharmaciens ont l’impression que tout le travail qu’ils ont fait en faveur de la substitution est nié.
M. Jean-Pierre Lamothe : Ces dernières années, la consommation en nombre de boîtes vendues n’a pratiquement pas bougé et elle a même légèrement régressé. La visite médicale n’entraîne donc pas une surconsommation en unités, mais bien un changement dans la prescription. Il suffit, pour s’en convaincre, d’observer les contournements et les transferts de prescription liés au générique. L’influence essentielle de la visite est donc bien de dire que tel ou tel produit procure un meilleur bénéfice au patient et que c’est donc ce produit qu’il faut utiliser. Il reste toutefois à voir si cette utilisation est véritablement pertinente pour tous les patients. On peut se poser la question pour un certain nombre de produits…
S’agissant de la décision du médecin par rapport à une pathologie déterminée, les délégués de l’assurance maladie peuvent toutefois exercer une réelle influence. Ils peuvent en particulier rappeler qu’à qualité de traitement égal pour le patient il convient de prescrire dans le répertoire des génériques. En ce sens, on peut dire qu’il s’agit de l’ébauche d’un contre-pouvoir.
Par ailleurs, si l’on observe le marché du médicament au cours des quinze dernières années, on s’aperçoit que l’on est dans un modèle inflationniste pur puisque le nombre d’unités vendues ne bouge pas tandis que le chiffre d’affaires progresse. Autant il paraissait légitime de tendre vers une homogénéisation des prix industriels au niveau international, autant il faut être aujourd’hui vigilant quant aux produits de contournement. Observant un début de surchauffe sur les prix des spécialités pharmaceutiques, l’État avait mis en place le système des marges dégressives lissées, qui fait que, bon an mal an, les officines progressent deux fois moins que l’industrie.
S’il était logique dans ce contexte que la marge brute globale des officines soit limitée de la sorte, il faut toutefois faire attention car, avec ce dispositif, l’officine serait particulièrement sensible à un retournement du marché et à une déflation du prix du médicament. Il faudrait donc prévoir des taquets à la baisse. C’était d’ailleurs bien l’idée de l’égalité de marge entre le princeps et le générique. À défaut, la baisse du prix des médicaments aurait été répercutée directement sur la marge. On a vu avec le plan Médicament de 2006 que, alors que les revenus de l’industrie étaient stabilisés, les ressources des officines diminuaient.
On peut donc comprendre l’agacement dont a fait état M. Philippe Gaertner à propos de l’exemple précis d’un produit de contournement, tout simplement parce qu’il n’y a plus de mécanismes de régulation dans un contexte déflationniste. Or la plupart des États développés ont « sur-solvabilisé » la recherche et le développement industriel et l’on va, selon moi, vers une déflation inévitable et durable du prix des spécialités, même si, dans le contexte actuel de déremboursement et de développement des génériques, on assiste à une réallocation des ressources industrielles vers des produits plus onéreux. Les effets des plans médicaments successifs ne se feront sentir qu’à moyen ou à long terme, mais nous devons être vigilants pour que les réseaux ne soient pas déstructurés dans ce contexte déflationniste.
Enfin, s’il est légitime que des informations soient données dans le cadre de la visite médicale, il faut aussi que des contre informations puissent être délivrées.
M. Pierre Morange, coprésident : J’aimerais que nous abordions aussi le sujet des molécules onéreuses, car environ 30 % de l’augmentation des dépenses de médicaments en ville résultent du transfert de prescriptions hospitalières vers la médecine de ville.
Mme Marie-Josée Augé-Caumon : On a déjà tenu compte de l’arrivée des molécules onéreuses et de l’impact qu’aurait leur sortie de la réserve hospitalière quand on a renégocié la marge des pharmacies d’officine, il y a quelques années.
Aujourd’hui, nous sommes tributaires du transfert de l’hôpital vers la ville de molécules et de dispositifs médicaux que nous ne délivrions pas auparavant et ces consommations sont comptabilisées dans l’enveloppe ville et non dans l’enveloppe hôpital. Il y aura de plus en plus de molécules onéreuses supportées par l’enveloppe de ville, mais il y a aussi des traitements liés au suivi de l’hospitalisation, en particulier certains pansements, que l’on délivre désormais en ville, tout simplement parce que le malade reste souvent très peu de temps à l’hôpital.
M. Claude Japhet : Je rappelle que le prix moyen d’une boîte de médicament n’a pas évolué au cours des deux dernières années et qu’il reste autour de 10 €. En revanche, deux marchés se sont véritablement envolés ; celui des médicaments dont le prix public est supérieur à 500 €, – ils représentaient environ 1 % de la dépense de médicaments en ville en 2002 et atteignent 8 % aujourd’hui, ce que confirme le transfert de l’hôpital vers la ville – ; ainsi que celui des médicaments dont le prix public est entre 150 et 183 €, dont la part est passée de 7 % du total en 2004 à 14 % cette année.
Alors que l’on a fait des efforts pour abaisser les prix moyens ou faibles, l’impact des produits sortis de la réserve hospitalière a l’effet exactement inverse. Ce phénomène s’accélère en raison du raccourcissement des hospitalisations et il provoque une déstructuration économique, car un petit nombre seulement de patients est concerné.
Mme Marie-Josée Augé-Caumon : Cela n’a toutefois pas d’incidences sur le patient : le nombre de journées d’hospitalisation diminue, mais il n’y a pas d’incidents, ce qui signifie que les officines répondent bien à cette nouvelle demande.
M. Philippe Gaertner : En effet, les drames que l’on annonçait ne se sont pas produits.
Je vous remettrai également des documents concernant les médicaments sortis de la réserve hospitalière. Le premier montre que, jusqu’en 2005, ils représentaient 20 millions d’euros par mois dans le chiffre d’affaires des officines et qu’on en est aujourd’hui à 120 millions par mois. Ainsi, au mois de septembre dernier, les médicaments sortis de la réserve hospitalière ont représenté 6,3 % du chiffre d’affaires des officines et 3,5 % de la marge des pharmaciens sur les médicaments remboursables.
Il va falloir faire évoluer les modèles et s’intéresser à ce suivi, économiquement important, d’autant qu’un autre document indique que les officines profitent très inégalement de la sortie de la réserve hospitalière. Seules celles qui se trouvent à proximité d’un centre hospitalier universitaire (CHU) en bénéficiant vraiment.
Mme Catherine Lemorton, rapporteure : Je souhaite revenir sur les me too et sur les contournements de génériques.
Avez-vous remarqué, dans le territoire de votre officine, des variations de comportement des cabinets médicaux, par exemple pour une innovation thérapeutique donnée, après le passage d’un visiteur médical ?
Toujours dans ce territoire, disposez-vous de moyens pour dialoguer avec la profession médicale afin de leur expliquer les efforts que vous accomplissez en faveur des génériques et de leur demander pourquoi ils prescrivent une innovation thérapeutique dont on sait qu’elle n’en est pas vraiment une ?
J’ai ainsi le souvenir, il y a quelques années, d’un antibiotique qui n’apportait rien de plus mais qui était 30 % plus cher. Comment en parler avec les médecins ? Comment faire en sorte que la profession de pharmacien n’ait pas toujours l’impression d’être à la remorque du médecin au lieu d’être actrice ?
Mme Marie-Josée Augé-Caumon : Je ne suis pas sûre d’avoir compris si vous voulez nous faire dire que les visiteurs médicaux ciblent certains médecins ou qu’il y a des médecins plus sensibles que d’autres à l’influence de la visite…
Mme Catherine Lemorton, rapporteure : Je ne fais aucun procès d’intention aux médecins, mais je connais la difficulté qu’ils ont à se former, en particulier lorsqu’ils sont isolés, en milieu rural. Et je constate que les visiteurs médicaux, après s’être rendus chez les médecins, vont aussi dans les officines pour conseiller aux pharmaciens de se procurer un produit qui va être demandé très rapidement. Je me demande donc simplement si le pharmacien – qui a pu faire l’analyse de ce produit, en particulier grâce à la fiche de transparence – s’il constate qu’il s’agit d’un contournement de générique parce qu’il n’y a pas vraiment d’innovation thérapeutique, a la possibilité d’aller l’expliquer aux médecins, par exemple par l’intermédiaire des ordres professionnels.
Mme Marie-Josée Augé-Caumon : Nous avons toujours été d’une neutralité parfaite vis-à-vis de la prescription médicale. Quand on est au comptoir depuis des années, on sait très bien qu’il suffirait d’un haussement de sourcils, lorsqu’un patient présente une prescription, pour qu’il se détourne du traitement et que l’on pourrait donc ainsi inciter à la non-consommation. En fait nous agissons très peu, au moment de la dispensation, en direction du patient ou du médecin. En revanche, lorsque nous avons l’occasion de nous retrouver avec des médecins, nous ne nous privons pas de leur dire ce que nous pensons. Eux-mêmes nous ressortent souvent les arguments qu’ils ont entendus lors de la visite médicale, à savoir que l’on n’est pas au courant des nouvelles indications ou du RCP (résumé des caractéristiques du produit).
La seule possibilité d’être un tant soit peu efficace en la matière serait que médecins et pharmaciens reçoivent ensemble des formations et des communications, pour être au fait des nouveautés en même temps. Dans ces conditions, comme cela se fait ailleurs au sein de commissions médicales dans les hôpitaux, le pharmacien pourrait avoir une influence sur la prescription. Les réseaux de soins pourraient aussi nous permettre de nous rencontrer davantage.
M. Claude Japhet : Je me refuse à attaquer le choix thérapeutique du médecin, car il est de sa responsabilité de décider en toute connaissance du patient et de la pathologie, de la pertinence de sa prescription.
Par ailleurs, ce n’est pas parce qu’un nouveau produit sort que tous les médecins vont entrer dans le dispositif ; il est donc difficile d’apprécier l’impact de la visite en termes de pénétration des nouveaux produits.
J’ajoute que la relation entre médecins et pharmaciens dépend très largement de l’endroit où ils se situent : à la campagne quand, dans une zone déterminée, on a un ou deux pharmaciens pour deux ou trois médecins, il leur est assez facile de se rencontrer et de dialoguer. Des actions locales sont alors possibles parce qu’il y a de plus en plus de rencontres entre médecins et pharmaciens ainsi que des formations croisées, y compris avec l’hôpital. Les choses sont beaucoup plus difficiles en ville, où le nombre de médecins et la concurrence sont bien plus importants.
J’ajoute que je suis assez sceptique quant à la possibilité de nouer ce dialogue dans le cadre des syndicats ou des ordres professionnels.
Mme Catherine Lemorton, rapporteure : Je n’évoquais pas l’action du pharmacien au moment où il délivre les médicaments, mais j’envisageais bien une intervention en amont, dans un cadre collégial, autour par exemple des préconisations de la Haute Autorité de santé.
M. Pierre Morange, coprésident : L’objectif que nous partageons est celui de la santé de nos concitoyens et, dans ce cadre, chacun doit assumer ses responsabilités : le médecin prescrit, le pharmacien délivre. Il ne faudrait donc pas donner l’impression que l’on fait ici le procès des uns ou des autres, d’autant que le niveau des professionnels de santé français est remarquable et qu’ils recherchent systématiquement le travail en équipe.
Cela étant, la formation en pharmacologie des médecins est faible, en formation initiale comme en formation médicale continue, et il est évident que des progrès sont à faire.
M. Philippe Gaertner : J’ai la chance de travailler en milieu rural et de bien connaître les prescripteurs ; s’ils m’appellent fréquemment pour avoir un renseignement d’ordre pharmacologique, ils n’attendent absolument pas de moi que je les oriente dans la prescription.
S’agissant de l’influence de la visite médicale sur les me too, j’ai déjà dit que cela fonctionnait très bien et que le report de prescription était général et complet.
À propos de l’innovation, je vais prendre l’exemple des antiulcéreux.
Lors du passage de la cimétidine à la ranitidine, utilisées pour les traitements antiulcéreux, on s’est interrogé sur la nécessité d’aller vers un produit plus cher. On s’est ensuite posé la même question au moment de l’apparition des inhibiteurs de la pompe à protons. Aujourd’hui, compte tenu des effets positifs mais aussi des effets indésirables, aucun prescripteur n’envisage de revenir à la cimétidine. On a donc là un exemple manifeste d’innovation véritable, qui nous montre qu’il faut éviter de généraliser.
Enfin, je me suis occupé de formation pendant vingt-cinq ans. Si je suis convaincu de l’intérêt d’aller vers des formations communes, je mesure combien cela est difficile dans la pratique, car les pharmaciens y sont toujours beaucoup plus nombreux que les médecins et cela ne reflète pas l’équilibre normal entre les deux professions. Qui plus est, les contenus des formations nécessaires aux uns et aux autres sont différents.
Il me semble en revanche que les réseaux de soins peuvent être des lieux privilégiés d’échange des pratiques. Il faut donc s’efforcer de bâtir des outils qui soient transposables afin que chacun puisse les utiliser dans sa pratique quotidienne.
M. Pierre Morange, coprésident : Merci d’avoir répondu de façon aussi précise à nos questions.
*
Audition de MM. Christian Lajoux, président du LEEM – Les entreprises du médicament, et de Claude Bougé, directeur général adjoint.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale pour cette audition qui s’inscrit dans le travail qu’effectue notre mission sur la prescription, la consommation et la fiscalité du médicament.
Vous êtes des acteurs à la fois de la santé et de l’économie. Il nous paraît donc utile de savoir comment vous analysez la pertinence et l’efficacité de la fiscalité française du médicament.
M. Christian Lajoux : C’est au regard de la compétitivité de notre industrie du médicament que la fiscalité doit être considérée.
Auparavant, je veux rappeler qu’aujourd’hui les enjeux de la santé concernent l’industrie des sciences du vivant et du médicament. Quelques pays seulement continueront à participer à la compétition internationale en matière de recherche, car c’est bien là l’essentiel, l’innovation thérapeutique étant la finalité de l’industrie du médicament. Or la France, premier producteur et exportateur en Europe, est aujourd’hui confrontée à de nouvelles concurrences venant d’autres continents.
Dans ce contexte, il faut se demander si notre fiscalité nous pénalise, notamment au regard de l’attractivité de nos centres de recherche. Or, en dehors de ceux de l’industrie française, il y a en France très peu de centres de recherche de grands groupes internationaux. C’est un véritable handicap. De même, pour la production, la France est rarement choisie, les industriels préférant s’installer en Espagne, en Irlande ou en Hongrie.
C’est là qu’intervient notre fiscalité qui est extrêmement complexe, peu lisible, instable et plus lourde que celle de nos voisins. Il s’agit en fait d’une fiscalité d’exception, avec pas moins de onze taxes qui ont perdu leur caractère structurant pour devenir des taxes de rendement. Certaines sont des formes de débudgétisation de services publics, comme celle qui, en 2007, a financé à hauteur de 60 millions d’euros l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) et qui porte en particulier sur la recherche biomédicale. De même, les industriels du médicament contribuent chaque année à hauteur de 9 ou 10 millions d’euros au financement du nouveau Centre national de gestion des essais de produits de santé (CeNGEPS).
De façon plus directe, nous sommes peut-être la seule industrie à être taxée sur notre chiffre d’affaires. Nous sommes également taxés sur les ventes directes aux officines ainsi que sur la promotion.
Nous préférerions de beaucoup une fiscalité structurante, par exemple destinée à réduire la consommation de médicaments ou le déficit des comptes sociaux. Tel n’est pas le cas et, même si l’on note certaines évolutions récentes, par exemple en ce qui concerne le crédit d’impôt recherche, cette fiscalité n’est pas de nature à faire choisir la France par les groupes internationaux.
M. Pierre Morange, coprésident : Ce constat est partagé par la Cour des comptes qui a noté le manque de lisibilité et de stabilité de cette fiscalité, que vous-mêmes jugez également contreproductive. Quelle devrait être cette fiscalité « structurante » que vous appelez de vos vœux ?
M. Christian Lajoux : Elle pourrait prendre la forme d’une clause de sauvegarde : dans un cadre défini de croissance du médicament, un accord entre l’industrie et le Comité économique des produits de santé (CEPS) permettrait de taxer les dépassements de chiffre d’affaires.
M. Claude Bougé : Autre exemple : parce qu’il s’agit d’une taxe structurante destinée à relancer la recherche clinique, l’industrie n’a pas contesté il y a deux ans la création de la taxe spécifique destinée à organiser les recherches cliniques au niveau départemental.
En revanche, d’autres taxes ont perdu leur effet structurant. Tel est le cas de la taxe sur le chiffre d’affaires, qui demeure cette année « exceptionnellement » fixée à 1 % au lieu du taux normal de 0,6 % et qui est indéniablement une taxe de rendement. Tel est encore le cas de la taxe sur la publicité qui est devenue complexe, illisible, et qui fait l’objet d’innombrables contentieux.
Au total, nous faisons une nette différence entre les taxes structurantes et les taxes de rendement, illisibles internationalement, d’autant que la France est vue comme un pays où la fiscalité nationale et locale est plutôt élevée, ce qui nuit à son attractivité.
M. Christian Lajoux : Au cours de l’histoire de notre industrie, on a ajouté des taxes aux taxes et les objectifs d’un certain nombre d’entre elles ont été largement oubliés. On peut par exemple s’étonner qu’un récent rapport de l’Inspection générale des affaires sociales (IGAS) préconise de taxer la promotion des médicaments alors que cette dernière l’est déjà très lourdement. Peut-être cela fait-il tellement longtemps et l’effet structurant est-il tellement modeste qu’on l’a oublié !
À l’inverse, les accords conventionnels passés avec le CEPS dans les années quatre-vingt-dix ont permis de moderniser l’encadrement du chiffre d’affaires de l’industrie du médicament.
M. Jean Mallot, coprésident : S’agissant précisément de la promotion, je propose que nous en venions à la visite médicale. Comment s’assurer qu’elle joue son rôle sans qu’il y ait de dérive ? Que pensez-vous des propositions qui sont faites pour organiser une forme de contre-pouvoir ?
M. Christian Lajoux : Il y a bien un lien avec la fiscalité puisqu’une partie de la visite médicale est très fortement taxée…
La visite médicale est en complète reconstruction depuis un certain nombre d’années. Nous sommes d’ailleurs le seul pays d’Europe à avoir un tel encadrement de la qualité de la visite. Cela ne signifie bien sûr pas que les choses ne peuvent pas être encore améliorées et qu’il ne faut pas travailler sur un certain nombre de propositions, en particulier celles de l’IGAS.
À la différence des autres pays européens, en France les visiteurs médicaux doivent passer un diplôme reconnu par l’État. En 2003, les industriels du médicament ont élaboré leur propre charte de la visite de façon à promouvoir les bonnes pratiques. En 2004, dans le cadre de négociations avec le CEPS, a été élaborée une charte de la visite médicale qui n’existe que dans notre pays. À partir de cette charte, la Haute Autorité de santé (HAS) a proposé en 2005 que l’on aille vers un processus de certification, qui est en cours. À ce jour, 24 entreprises sont certifiées et 60 autres devraient l’être, au plus tard au début de l’année prochaine. Aucune des entreprises engagées dans ce processus n’a été recalée.
M. Claude Bougé : Les entreprises qui n’auront pas été certifiées en juillet 2008 ne pourront plus être conventionnées dans le cadre de l’accord avec l’État. Voilà qui devrait motiver la centaine d’entre elles qui ne se sont pas encore engagées.
M. Christian Lajoux : Cela montre que les autorités et les industriels ne sont pas restés inactifs et qu’ils ont pris à bras le corps les questions du bon usage, de l’objectivité de l’information et du contenu du métier.
Le récent rapport de l’IGAS ne fait état d’aucun dérapage de l’industrie du médicament par rapport à l’ensemble de ces principes, mais il pose clairement la question d’une nouvelle gouvernance de la santé à travers la répartition des rôles entre les différentes institutions.
M. Jean Mallot, coprésident : Il ne paraît pas anormal que les entreprises assurent la promotion de leurs produits auprès de ceux qui les prescrivent. Pour autant, un dispositif permettant de faire circuler des contre informations paraît utile et l’on parle souvent à ce propos des délégués de l’assurance maladie, les DAM. Comment serait-il possible de compléter le dispositif actuel afin que nous y trouvions collectivement notre compte ?
M. Christian Lajoux : Nous, industriels du médicament, considérons que nous avons des comptes à rendre et nous respectons cette exigence. Nous le faisons vis-à-vis de la HAS comme de l’AFSSAPS, et cette dernièure contrôle tous les documents promotionnels qui sont remis au corps médical. Les interdictions de publicité sont extrêmement rares puisqu’elles ne représentent qu’environ 1 % des documents que nous mettons en distribution et le nombre des mises en demeure est stable.
Cela ne signifie pas que le système ne peut pas évoluer. Des discussions sont en cours avec l’ensemble des autorités pour codifier davantage l’exercice de notre métier. Ainsi, fin 2006, le président du LEEM et le ministre de la santé ont conclu une convention instituant un code de bonnes pratiques des entreprises du médicament dans la formation médicale continue.
Les industriels ne contestent nullement le rôle des DAM, ils demandent simplement qu’ils répondent aux mêmes exigences, en particulier de diplôme, que les visiteurs médicaux. En effet, alors qu’ils assurent le contre-pouvoir des visiteurs médicaux ils n’ont ni la même qualification, ni la même certification par le biais de la HAS. Mais je crois savoir que le directeur de l’Union nationale des caisses d’assurance maladie, l’UNCAM, est plutôt favorable à cette avancée.
Pour notre part, nous ne refusons aucune évolution. Au contraire, nous avons montré depuis des années que nous acceptons le principe de la régulation et de l’encadrement, au service de la qualité et de l’objectivité de notre métier.
Néanmoins, je veux insister sur le fait que le débat sur la visite médicale ne saurait être l’enjeu majeur pour l’industrie du médicament. Le véritable enjeu est de savoir si l’on fera encore demain de la recherche dans notre pays et si nous serons présents dans la compétition et dans la production des biotechnologies.
Qui plus est, s’il faut la traiter, nous devons être conscients du fait que la question de la visite médicale relève désormais plutôt du passé : la montée en puissance des génériques va conduire des bataillons entiers de visiteurs médicaux à perdre leur emploi dans les mois et les années qui viennent.
M. Pierre Morange, coprésident : Pouvez-vous faire une projection qui tienne compte de l’entrée des blockbusters dans le domaine public, mouvement déjà largement engagé, et du développement des thérapeutiques issues du génie génétique et des biotechnologies ?
M. Christian Lajoux : C’est un exercice difficile. Cela étant, on voit bien que les nombreux visiteurs médicaux que nous utilisions pour promouvoir les blockbusters ne seront plus nécessaires dès lors que ces produits deviendront des génériques. Au cours de chacune des prochaines années, 800 millions d’euros de médicaments sont ainsi appelés à devenir des génériques. Or il s’agit précisément de médicaments pour lesquels s’exerçait une promotion assez intensive.
M. Pierre Morange, coprésident : Pouvez-vous donner une estimation chiffrée ?
M. Christian Lajoux : Il y a actuellement 23 000 visiteurs médicaux, dont 5 000 à 10 000 devraient disparaître dans les cinq prochaines années. Il s’agit donc d’un mouvement extrêmement important qui pose un problème, au-delà de la gestion des ressources humaines.
Aujourd’hui, lorsqu’un médicament est enregistré et que l’on débat de son remboursement, les autorités de santé font de plus en plus en sorte – et ce n’est pas contestable – de cibler la population à laquelle il s’adresse. Les médicaments innovants vont donc être destinés à des populations réduites. Cela s’ajoutant à la disparition des blockbusters tels que nous les connaissons aujourd’hui, il est évident que l’exercice d’information des visiteurs médicaux sera beaucoup plus ciblé et beaucoup moins lourd.
Il ne faut pas oublier par ailleurs que ce sont les visiteurs médicaux qui remontent les informations en matière de pharmacovigilance. Alors qu’il existe des structures régionales de pharmacovigilance, sur les 20 000 informations en la matière que recueille chaque année l’AFSSAPS, la moitié provient des visiteurs médicaux. Ne pensons donc pas que le visiteur médical se contente de brandir une pancarte pour que l’on prescrive un produit, son rôle est beaucoup plus complexe et le rapport de l’IGAS montre d’ailleurs les multiples facettes de ce métier.
Mme Catherine Lemorton, rapporteure : J’entends bien que vous êtes préoccupés de l’avenir de notre industrie du médicament et de sa présence future sur le territoire national. C’est bien évidemment aussi le souci du législateur, mais ce dernier ne saurait oublier que c’est également de santé publique qu’il s’agit. Si notre mission a choisi de s’intéresser au médicament, c’est tout simplement parce que nous nous demandons pourquoi, dans notre pays, 90 % des consultations débouchent sur une prescription de médicaments, alors que ce n’est le cas que pour 75 % au plus des visites en Allemagne et pour 45 % aux Pays-Bas.
On pourrait se dire que c’est tant mieux si cela profitait à la santé publique et si les comptes publics se portaient bien, mais tel n’est pas le cas. On ne saurait par exemple ignorer qu’il y a en France 130 000 accidents iatrogènes chaque année.
Dans la mesure où, grâce à vos exportations, vous êtes aussi présents dans les autres pays, avez-vous des explications aux différences que je viens de mentionner ?
M. Christian Lajoux : Nous sommes observateurs et acteurs du système de santé.
En tant qu’observateurs, nous constatons que la médecine préventive est insuffisamment développée dans notre pays.
Si nul ne conteste le chiffre que vous venez de donner, il faut aussi l’analyser au regard du niveau sanitaire de la France : nous sommes l’un des pays d’Europe où l’espérance de vie est la plus élevée, où le taux de mortalité infantile est le plus faible, où l’on vit le mieux lorsque l’on est très âgé. On ne peut donc pas prétendre que l’importance de la prescription qui fait suite à chaque consultation provoque un retard sanitaire. Cela a vraisemblablement un coût pour la collectivité, mais si ce coût permet que notre niveau sanitaire et que la qualité de vie de nos concitoyens soient meilleurs, on ne saurait le regretter ; il faut simplement voir comment assurer le financement du système.
S’agissant de la promotion, je puis vous dire, pour avoir exercé des responsabilités à l’échelle européenne, qu’elle ne présente en France aucune spécificité : nous exerçons notre métier de la même façon dans tous les grands pays européens. Il y a même en France beaucoup plus d’encadrement de l’action des industriels. Ainsi, il n’existe nulle part ailleurs l’équivalent de la commission publicité de l’AFSSAPS. Or celle-ci a précisément pour mission de vérifier la qualité des messages et des documents que nous adressons aux médecins.
Contrairement à ce que laisse entendre le rapport de l’IGAS, la France n’est pas le pays où la visite médicale exerce la plus forte pression. Au Royaume-Uni, le nombre des visites est plus important.
Il me semble donc que la prescription systématique trouve son origine dans les carences de notre système de prévention, dans la formation des médecins ainsi que dans la générosité de notre système de prise en charge.
Aujourd’hui, 96 % des demandes d’inscriptions en affection de longue durée – les ALD – sont acceptées ainsi que 99 % des demandes de renouvellement. C’est un système qui encourage la prescription de médicaments. Ainsi, pour la population hors ALD, la consommation de médicaments progresse de 1 %, tandis que pour les huit millions de patients en ALD, la croissance est de 8 ou 9 %
M. Claude Bougé : La Caisse nationale d’assurance maladie des travailleurs salariés (CNAMTS) vient de publier une étude d’une grande qualité sur la croissance des soins de ville au premier semestre 2007. Les dépenses de médicaments de la population en ALD ont augmenté de 6,5 %, tandis qu’elles diminuaient de 2,3 % pour le reste de la population.
L’entrée en ALD et la gestion des personnes qui y sont posent un vrai problème de santé et de sécurité sociale. Il faut donc s’intéresser à toute la politique menée en la matière.
M. Christian Lajoux : Cela étant, les choses semblent bouger et le projet de loi de financement de la sécurité sociale (PLFSS) pour 2008, comporte un certain nombre de dispositions, comme les contrats individuels entre les caisses d’assurance maladie et les médecins, qui permettront de traiter ce sujet. La HAS a également engagé un travail sur la rationalisation des ALD.
Mme Catherine Lemorton, rapporteure : Si la visite médicale est désormais mieux encadrée en France, cela répond peut-être à un besoin que l’on ressent d’ailleurs depuis longtemps, puisque c’est en 1993 qu’a été adoptée la loi « anti-cadeaux ».
M. Christian Lajoux : Une telle loi n’existe toujours pas dans la plupart des autres pays et la France était donc déjà en avance en 1993 quand, en concertation avec les industriels du médicament, on a adopté ce principe. Il existe désormais un code européen de bonne conduite, qui est largement appliqué en France puisque nous sommes beaucoup plus exigeants dans la plupart des domaines.
Mme Catherine Lemorton, rapporteure : Pourquoi 60 % des médecins ne sont-ils pas au courant du contenu de la charte de la visite médicale ? Certes, ce n’est pas à vous mais à l’UNCAM qu’il revient de la diffuser, mais, pour qu’elle soit appliquée, il faut bien qu’on la connaisse…
M. Christian Lajoux : Sa signature a fait l’objet d’une large publicité. Pour ma part, je retiens du récent rapport que 72 % des médecins se disent satisfaits de la façon dont s’exerce la visite médicale. Sur ce total, 30 % estiment tout de même qu’il est possible de l’améliorer, ce qui montre bien qu’ils ne sont pas nos « otages », comme on a pu le lire. Nous sommes face à des gens éduqués sur le plan thérapeutique et qui savent comment l’industrie du médicament fonctionne. Ils ont tout à fait la possibilité de fermer leur porte ; d’ailleurs une partie d’entre eux ne reçoit pas du tout à son cabinet, tandis que d’autres refusent de recevoir les visiteurs de telle ou telle entreprise, au motif que ses pratiques ne sont pas conformes à la charte.
On le voit, ce système est propice à l’amélioration de la qualité du service que la visite médicale apporte aux médecins en termes d’information.
M. Claude Bougé : J’ajoute que le système de sanctions fonctionne, soit par le biais de la commission de publicité, soit en application de la charte.
M. Pierre Morange, coprésident : En effet, les praticiens ne sont pas pris en otage puisqu’ils peuvent refuser de recevoir les visiteurs médicaux. Pour ma part, je leur demandais de se contenter de déposer les notices.
Pour en revenir à la recherche et développement, il convient en effet de faire en sorte que la France ne soit pas seulement un pays de consommateurs, mais aussi que l’on sache y créer et y produire des médicaments destinés à la population du monde entier. De ce point de vue, il faut prendre en considération d’une part la population solvable, qui se trouve désormais aussi dans certains pays émergents – qui d’ailleurs mènent eux-mêmes une politique active en la matière –, d’autre part les populations non solvables, qui sont les plus nombreuses. Il me semble que c’est un sujet que nous devrions aborder car ce n’est qu’en apparence qu’il sort du champ de notre mission, celle-ci étant vouée à s’assurer de la bonne utilisation de l’argent public au bénéfice du plus grand nombre.
M. Christian Lajoux : Il faut absolument éviter que les entreprises installées en France ne se délocalisent, mais il convient aussi de s’efforcer de faire venir chez nous les grandes entreprises internationales, dont les centres de recherche et les sites de production ne sont pour l’heure pas assez présents sur notre territoire.
Aujourd’hui, la concurrence n’est plus uniquement aux États-Unis, mais aussi en Angleterre et en Allemagne, pour la recherche sur les biotechnologies, et en Inde, en Chine et au Brésil pour la production. Cela est vrai en particulier pour les génériques que ces pays produisent moins à destination de leurs habitants que de ceux des pays développés où ils les vendent avec des marges colossales.
Certaines sociétés indiennes font de la rétro-ingénierie, c’est-à-dire qu’elles partent de la molécule finie et qu’elles redéfinissent des process de fabrication qui rendent les nôtres obsolètes. Nous aurions tort de considérer que cela ne concerne que la production : elles vont devenir demain de grandes sociétés de recherche. L’échiquier des pays qui vont jouer un rôle dans les sciences du vivant est ainsi en train d’être complètement redessiné. Et il ne faut pas négliger le fait que l’indépendance sanitaire d’un pays est un enjeu aussi stratégique que son indépendance militaire.
La France doit aussi se poser la question du rôle qu’elle veut jouer dans la diplomatie sanitaire. Celui-ci sera d’autant plus important qu’elle aura réalisé sur son propre territoire l’intégration totale des différents acteurs qui contribuent à bâtir un modèle de santé efficace, mais aussi qui permettent de réduire les tensions internationales, qui porteront de plus en plus non seulement sur l’accès à l’énergie mais aussi sur l’accès aux médicaments.
Il s’agit donc d’un sujet essentiel et c’est bien pour cela que j’insiste pour que l’on ne résume pas l’industrie du médicament à la simple question de la promotion. C’est un débat d’hier, qui n’a que peu d’incidence et pour lequel nous sommes tous disposés à corriger ce qui peut l’être. Le véritable enjeu, c’est celui de la recherche, celui de l’existence de la France dans les sciences du vivant, celui de son rôle d’acteur majeur pour le médicament.
M. Pierre Morange, coprésident : S’agissant de ces aspects géopolitiques, vous prêchez un converti : M. Jean-Pierre Raffarin, alors premier ministre, m’avait en effet confié, en 2004, la mission d’évaluer l’action de la France en faveur de la réalisation des Objectifs du Millénaire pour le développement dans le domaine de la santé. Je m’étais alors en particulier attaché à définir les moyens permettant au secteur français de la santé d’exercer dans le monde une forte influence, au-delà de la seule notion de diplomatie sanitaire.
Vous avez insisté sur la nécessité que les centres de recherche et de production des grands groupes internationaux soient présents sur le territoire français. Avez-vous des propositions concrètes à faire pour en renforcer l’attractivité ?
M. Christian Lajoux : Nous avons exprimé le souhait que cette nouvelle législature soit l’occasion de relancer le Conseil stratégique des industries de santé (CSIS) qui avait été institué par M. Jean-Pierre Raffarin, afin de rechercher avec les responsables des grands groupes comment la France pouvait être plus compétitive et plus attractive. Il ne s’était depuis lors réuni qu’une seule fois et il faut remercier M. Xavier Bertrand et M. François Loos d’avoir su organiser une deuxième réunion début 2007. Ce conseil sera véritablement relancé au début de l’année prochaine, sous la responsabilité du Premier ministre.
Pour notre part, nous formulons un certain nombre de propositions afin de renforcer l’attractivité de la recherche. Ainsi, nous considérons qu’il faut internationaliser les pôles de compétitivité, sur le modèle de Cambridge. Actuellement, en France, 8 pôles sur 71 sont consacrés à la santé, mais on a du mal à savoir quels sont véritablement les grands pôles internationaux.
Nous devons aussi nous efforcer de conserver nos excellents chercheurs sur le sol national, alors qu’ils sont aujourd’hui particulièrement nombreux à s’expatrier, un doctorant sur dix seulement trouvant un poste en France.
Il convient également que nous nous intéressions à la question des recherches cliniques. Cela concerne au premier chef le législateur, car il faudrait sans doute faire évoluer les lois bioéthiques pour que la France soit aussi compétitive que ses concurrents. Nous avons en particulier besoin de pouvoir suivre de grandes cohortes de population afin de mener des études épidémiologiques.
Ces propositions ont reçu un accueil extrêmement favorable dans les ministères concernés.
Il faut aussi s’intéresser de près à la mutation industrielle afin que nous ne perdions pas tous nos emplois sous la concurrence des pays du Sud et des pays leaders dans les nouvelles technologies.
Nous devrons aussi parler de la fiscalité, dont j’ai déjà dit qu’elle devait être structurante.
M. Pierre Morange, coprésident : Avez-vous des propositions précises afin de rendre cette fiscalité plus lisible, plus stable, plus favorable à la recherche-développement, plus profitable aux comptes sociaux ?
M. Christian Lajoux : Le crédit impôt recherche tel qu’il vient d’être redéfini va dans un sens qui convient aux industriels, en particulier parce qu’il est plus lisible.
La politique fiscale pourrait également être orientée de façon à ce que les investissements se dirigent davantage vers la recherche et vers les pôles de compétitivité. Des propositions ont déjà été faites en la matière, sur lesquelles les industriels du LEEM auront l’occasion de se prononcer. Dès que les ministères, les parlementaires, l’ensemble des institutions et les industriels se mettent à travailler ensemble, on voit bien qu’un certain nombre de pistes qui ont déjà été évoquées n’ont pas été suffisamment explorées. Nous souhaitons non pas que l’on ajoute des dispositifs les uns aux autres, mais que l’on se mette autour d’une table, par exemple au sein du CSIS, pour élaborer un véritable projet redéfinissant l’attractivité du pays.
M. Claude Bougé : Nous avons effectivement des propositions concrètes, par exemple pour que l’on admette dans l’assiette du crédit impôt recherche l’investissement en faveur des études épidémiologiques, ce qui nous permettrait de rattraper notre retard sur les pays très actifs dans le domaine des essais cliniques. S’il y a des études épidémiologiques en Grande-Bretagne, c’est aussi parce que le régime fiscal y est favorable. En France, on a fait le choix inverse et ces études sont insuffisamment développées.
Par ailleurs, quand un produit tombe dans le domaine public, le laboratoire est en fait taxé sur la promotion d’un médicament qui va donner lieu à substitution. Il faut que le laboratoire soit singulièrement vertueux pour continuer de le promouvoir et il y a quelque paradoxe de la part des pouvoirs publics à taxer ce qu’ils entendent développer.
M. Christian Lajoux : Jusqu’à ces derniers jours, il y avait une inégalité dans les conditions commerciales entre les fabricants de génériques et ceux de princeps, mais les règles du jeu semblent devoir être modifiées, ce qui va dans le bon sens.
M. Jean Mallot, coprésident : Nous avons beaucoup parlé de la promotion des médicaments dans le circuit habituel, mais un grand nombre de ventes se font désormais sur Internet. Comment contrôler le phénomène et s’assurer de la qualité des produits ?
M. Christian Lajoux : C’est une catastrophe, tous les industriels vous le diront, car cela permet tout et n’importe quoi.
M. Pierre Morange, coprésident : M. Jean Parrot, président du Conseil national de l’ordre des pharmaciens, nous a dit que 15 % des médicaments vendus sur le territoire américain étaient des contrefaçons.
M. Christian Lajoux : On considère que c’est le cas de 10 % des médicaments vendus dans le monde.
En France, on a beaucoup progressé ces dernières années, grâce à la collaboration entre les industriels, les services des douanes, la police et l’AFSSAPS. Nous étions, il est vrai, en retard puisque la directive européenne de 2004 vient seulement d’être transcrite en droit français. Il était également nécessaire de considérer qu’il ne s’agit pas seulement d’un délit mais d’un crime, car la contrefaçon d’un médicament a d’autres effets que celle d’un tee-shirt. Auparavant, les contrefacteurs étaient passibles de trois ans de prison, désormais celui qui contrefait un médicament encourt cinq ans. L’instruction va en outre être accélérée.
Il faut absolument maintenir notre système de distribution, qui est un des meilleurs du monde, notamment parce que son caractère monopolistique offre une véritable sécurité. Nous sommes sur ce point en accord total avec les pharmaciens d’officine. Ce système est certes onéreux, mais c’est le prix à payer pour la qualité et pour la sécurité. Ainsi, aujourd’hui en France pas un seul citoyen n’a intérêt à acheter ses médicaments sur Internet.
M. Pierre Morange, coprésident : Est-il vraiment possible d’agir à l’encontre des ventes sur Internet, dont on sait qu’elles sont le fait de sociétés extraterritoriales, contre lesquelles les recours judiciaires sont aléatoires ?
M. Christian Lajoux : Il est vrai que l’on n’est ainsi plus dans le cadre du territoire, donc des lois de la République.
Un certain nombre de sociétés ont mis en place des systèmes de détection qui leur permettent de remonter les filières. Il s’agit en fait d’un travail de police, fort long. On découvre ainsi qu’à travers une multitude de petits sites, on a affaire à des sociétés très organisées, qui relèvent de la grande criminalité.
M. Pierre Morange, coprésident : Il est en effet démontré qu’il s’agit pour les mafias d’une nouvelle branche d’activité, plus lucrative que le commerce de la drogue.
Il nous faut maintenant interrompre cette audition. Je vous remercie d’y avoir participé. Nous vous demanderons de bien vouloir répondre par écrit aux questions que nous n’avons pas pu vous poser ce matin.
*
Audition de Mme Marie-Noëlle Banzet, vice-présidente des laboratoires Servier, M. Éric Ducourneau, secrétaire général des laboratoires Pierre Fabre, et M. Christian Lajoux, président-directeur général de Sanofi Aventis France, accompagné de M. Philippe Cheng, directeur de la stratégie.
M. Jean Mallot, coprésident : Madame, messieurs, je vous prie de bien vouloir excuser l’absence de M. Pierre Morange, coprésident de la MECSS, mais aussi rapporteur du projet de loi relatif au pouvoir d’achat qui est inscrit aujourd’hui à l’ordre du jour de la séance publique.
La MECSS travaille sur la prescription, la consommation et la fiscalité des médicaments. De nombreux acteurs de ce dossier ont d’ores et déjà été auditionnés et, ce matin, c’est au tour des représentants de différents laboratoires. À l’issue de l’ensemble de ces entretiens, le rapport de Mme Catherine Lemorton fera état d’un certain nombre de préconisations afin d’améliorer aussi efficacement que possible la situation dans le domaine du médicament.
M. Christian Lajoux : Avant que ne commence l’audition, je souhaite savoir quelle est la logique qui a présidé au choix des participants aux tables rondes.
M. Jean Mallot, coprésident : Il y a bien une logique, mais toutes les règles comportent des exceptions, lesquelles sont susceptibles d’être interprétées différemment. Une audition implique nécessairement un regroupement des personnes afin de faciliter le bon déroulement de la séance.
M. Christian Lajoux : Il y a des regroupements d’entreprises et des regroupements de groupes d’entreprises.
Mme Catherine Lemorton, rapporteure : Nous avons tout d’abord souhaité entendre les laboratoires français, puis les laboratoires fabricants de médicaments génériques et, enfin, des laboratoires étrangers, notamment Ranbaxy, génériqueur indien.
Nous aurions souhaité également auditionner un représentant d’un laboratoire biotech, mais cela n’a pas été possible, ce que nous regrettons. J’ajoute que nous aurions voulu inclure dans votre groupe le représentant du laboratoire homéopathique Boiron, mais votre groupe, précisément, nous a fait savoir que ce laboratoire n’avait pas sa place parmi vous, ce qui m’a un peu surprise. Le représentant de ce laboratoire a néanmoins accepté d’être auditionné dans une autre table ronde.
M. Christian Lajoux : Je vous prie d’excuser mon insistance, mais il ne faut pas confondre les laboratoires et les groupes. Les représentants de GÉnériques Même MÉdicaments (GEMME) sont-ils invités en tant que laboratoire ou en tant que GEMME ?
Mme Catherine Lemorton, rapporteure : En tant que GEMME.
M. Jean Mallot, coprésident : Et nous posons quant à nous nos questions en tant que parlementaires.
Je vous laisse auparavant présenter de manière synthétique les entreprises que vous dirigez.
Mme Marie-Noëlle Banzet : Le groupe Servier est le second laboratoire français après Sanofi et le premier laboratoire français indépendant. Il réalise un chiffre d’affaires qui s’élève à 3,5 milliards d’euros. Ce groupe présente deux caractéristiques : un fort engagement dans la recherche puisque 23 % à 25 % du chiffre d’affaires y sont réinvestis ; ses activités sont en outre fortement internationalisées, tout en étant ancrées en France où nous réalisons le plus possible d’activité de production et de recherche. La totalité de la production chimique est réalisée en Normandie, 60 % de la production pharmaceutique dans l’Orléanais et 70 % à 75 % des activités de recherche en France.
Ce positionnement spécifique induit certains ratios un peu atypiques : 80 % de notre production est vendue à l’étranger dans 140 pays ; notre part de marché, s’agissant des princeps remboursables, s’élève à un peu moins de 3 % ; le ratio des dépenses de recherche et de développement (R&D) sur le chiffre d’affaires en France est de 77 % ; nous participons pour plus de 28 % à l’excédent de la balance commerciale du secteur. Par ailleurs, malgré un contexte économique difficile, nous continuons à créer des emplois : plus de 400, cette année, en France. Enfin, 75 % des impôts dus par le groupe sont payés en France.
Nous avons également une activité générique avec la filiale Biogaran qui représente 22 % de ce marché en France ; son chiffre d’affaires s’élève entre 400 et 420 millions ; sur les cinq dernières années, l’activité générique du groupe Servier a contribué pour plus de 700 millions aux économies de l’assurance maladie.
M. Christian Lajoux : Sanofi Aventis, fortement implanté en France, est le quatrième groupe international dans le domaine du médicament. M. Philippe Cheng va le présenter plus précisément.
M. Philippe Cheng : Sanofi est le premier groupe européen et français de médicament. Il comprend 100 000 collaborateurs, dont 29 000 en France, alors que 13 % seulement de son chiffre d’affaires mondial y sont réalisés. L’investissement en R&D représente 15 % du chiffre d’affaires mondial avec 4,4 milliards en 2006, soit, une progression de 9 %. Plus de la moitié des sites de R&D se situent en France ; on y dénombre 9 000 chercheurs sur les 18 000 que compte le groupe.
Des partenariats étroits sont noués entre les secteurs public et privé, notamment à travers les pôles de compétitivité. En France, 13 600 personnes travaillent à la production sur 25 sites, dont 10 de production chimique, 9 de production pharmaceutique et 6 de distribution. Les exportations s’élèvent à 9 milliards d’euros pour Sanofi Aventis France avec un solde positif de 4,7 milliards. Le solde des emplois est resté positif en 2006 avec 960 emplois créés dans la recherche et la production.
M. Christian Lajoux : J’ajoute que le groupe paie en France près de 1,5 milliard d’euros d’impôts sur les sociétés et de taxes spécifiques au secteur des médicaments.
M. Éric Ducournau : Les laboratoires Pierre Fabre ont été créés il y a quarante ans dans le sud-ouest de la France. Ils travaillent à la fois dans le domaine dermo-cosmétique avec un chiffre d’affaires qui s’élève à un peu plus de 700 millions et dans le domaine de la médication familiale où il est de 830 millions. L’entreprise emploie 8 500 personnes, dont les deux tiers en France. L’intégralité des activités de production – 4 sites –, de R&D – 7 sites – et de chimie fine – un site – sont situées en France. En 2007, nous avons investi 230 millions en R&D et 200 millions pour la partie médicament dont 100 millions en faveur de la seule oncologie. Nous effectuons des recherches dans les domaines cardio-vasculaire, urologique et sur le système nerveux central. L’an dernier, 200 emplois ont été créés.
Mme Catherine Lemorton, rapporteure : Que pensez-vous de la fiscalité française sur le médicament ?
M. Christian Lajoux : La multiplicité des taxes – il y en a onze – constitue un lourd handicap : contributions sur les dépenses de publicité, sur les grossistes-répartiteurs, sur le chiffre d’affaires, clause de sauvegarde – en cas de dépassement de l’objectif de chiffre d’affaires –, contributions à la charge des fabricants ou distributeurs de dispositifs médicaux, taxes qui rémunèrent des services rendus.
Certaines sont versées à l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) : taxe sur les spécialités bénéficiant d’une autorisation de mise sur le marché (AMM), redevance pour autorisation d’importation parallèle, redevance pour la publicité, taxe sur les essais cliniques ; d’autres sont versées à la Haute Autorité de santé (HAS) comme la redevance pour inscription sur la liste des médicaments remboursables. Ces taxes sont très nombreuses et peu lisibles. On peut en outre s’interroger sur l’efficacité de la taxe sur la promotion. Non seulement il est regrettable qu’une approche plus structurante ne soit pas développée, mais les taxes sur la publicité, sur le chiffre d’affaires, la clause de sauvegarde ou les taxes sur les ventes directes n’existent pas dans les autres pays.
M. Jean Mallot, coprésident : Vos collègues partagent-ils ce point de vue ? Qu’entendez-vous, par ailleurs, par une approche plus structurante de la fiscalité ?
Mme Marie-Noëlle Banzet : Je suis d’accord avec M. Christian Lajoux, ces taxes sont très pénalisantes. On peut citer la réforme du crédit d’impôt recherche comme exemple de fiscalité plus structurant qui va dans le bon sens.
M. Philippe Cheng : Encore peut-on souligner que cette réforme fait suite à celle qui prévoyait une majoration de l’abattement R&D qui devait s’appliquer en 2007, et que le projet de loi de finances pour 2008 prévoit de supprimer.
M. Éric Ducournau : Le taux d’imposition des laboratoires Pierre Fabre, en France, dépasse 34 % alors qu’il est de 27,76 % dans les pays de l’Union européenne – hors la France, donc – et de 31,7 % dans les pays hors Union Européenne. Cela représente un différentiel de 3 à 7 points.
M. Christian Lajoux : Outre que les entreprises se porteraient beaucoup mieux si la fiscalité était moindre, il est évident qu’à la taxe sur l’emploi, comme l’est celle sur la publicité, ou la taxe sur le chiffre d’affaires qui pénalise la performance, nous préférons des taxes relevant d’un processus conventionnel tel qu’il a été ébauché dans l’accord-cadre avec le Comité économique des produits de santé (CEPS).
Mme Catherine Lemorton, rapporteure : La taxe sur la publicité inclut-elle la visite médicale ?
M. Christian Lajoux : Oui. C’est pour cela que je la qualifie de taxe sur l’emploi.
Mme Catherine Lemorton, rapporteure : Pourquoi les Français consomment-ils globalement plus de médicaments qu’ailleurs ? Aujourd’hui, 90 % des consultations chez le médecin généraliste sont suivies d’une prescription de médicaments contre 72 % en Allemagne et 45 % aux Pays-Bas. Le passage des médicaments « over the counter » (OTC) – « au-delà du comptoir » ou médicaments conseil – en libre-service dans les pharmacies permettra-t-il de modifier le comportement des Français ?
M. Christian Lajoux : Sur quelles sources reposent de tels chiffres ?
Selon les données d’IMS Health, qui ne relèvent pas de l’étude d’opinion, les pourcentages de diagnostics débouchant sur une prescription sont les suivants : Portugal, 97 %, Italie, 93 %, Belgique et Espagne, 83 %, Slovaquie, 77 %, France, 76 %, Angleterre, 68 %, Pays-Bas, 60 %. Je tiens ces données à votre disposition, de même que des données sur la consommation de médicaments en France où l’on ne peut en l’occurrence pas parler de surconsommation. L’Union nationale des caisses d’assurance maladie (UNCAM) l’a d’ailleurs reconnu puisqu’en termes d’unités de prescription, la France se situe plutôt en deçà des autres pays européens.
J’ajoute que si, sur le plan européen, les protocoles thérapeutiques ne sont guère respectés, la France est encore le pays qui les respecte le plus. Cette consommation, enfin, doit être rapportée à l’état sanitaire du pays ; or celui de la France est plutôt meilleur que celui d’autres pays européens.
M. Jean Mallot, coprésident : Je vous prie de bien vouloir nous communiquer ces chiffres.
M. Christian Lajoux : Bien sûr.
M. Éric Ducournau : La question de la médication familiale se pose dans un contexte de changement de l’exercice libéral de la pharmacie. Le passage « devant le comptoir » de certains médicaments permettra de développer l’automédication à condition de respecter les conseils des pharmaciens et les règlements d’usage de certains produits dont, par exemple, les produits de sevrage tabagique. Il faut par ailleurs que les produits obtenant ce statut ne soient pas dénigrés, comme cela arrive parfois.
M. Christian Lajoux : S’agissant de l’automédication, la France est l’un des seuls pays européens où le principe de l’accès direct à une catégorie de médicaments – pour les « petits bobos » – n’est pas autorisé. Notre pays doit donc se mettre en conformité sur ce plan-là avec l’Europe, sachant que le monopole de la pharmacie est intangible et que le contrôle du pharmacien demeure essentiel. Enfin, s’il faut responsabiliser les patients, l’automédication ne doit en aucun cas être un alibi au déremboursement de médicaments.
M. Jean Mallot, coprésident : Je comprends, sans la partager, votre position sur la fiscalité, mais cela implique de ne pas attendre pour soi le bénéfice de l’impôt des autres. Il n’y aurait donc pas de surconsommation de médicaments en France ?
M. Christian Lajoux : Non seulement une consommation élevée n’est pas une surconsommation, mais de nombreuses maladies ne sont toujours pas prises en charge.
M. Jean Mallot, coprésident : Il ne faut pas mélanger, en l’occurrence, démarche globale de surconsommation et démarche ciblée sur telle ou telle pathologie.
Que pensez-vous des franchises médicales ? Quels effets auront-elles sur la consommation de médicaments ?
M. Christian Lajoux : Il est possible de donner un avis en tant que citoyen, mais je ne suis pas sûr que nous soyons légitimés à le faire en tant que représentants de l’industrie du médicament. Il s’agit pour nous d’encourager la responsabilisation des patients mais il n’est pas certain que l’établissement de franchises ira en ce sens. Quid, également, de la possibilité pour chaque patient d’accéder aux soins ?
Mme Marie-Noëlle Banzet : Je partage ce point de vue.
Mme Catherine Lemorton, rapporteure : Le schéma transmis par l’Union nationale des pharmacies de France (UNPF), que je viens de faire distribuer, montre un décrochage – par le haut ! – du chiffre d’affaires de l’industrie pharmaceutique à partir de 1999-2000 et par rapport au nombre d’unités de médicaments consommés par personne qui, lui, demeure constant. Comment l’expliquer ?
M. Christian Lajoux : L’UNCAM l’a dit : moins d’unités sont consommées et la valorisation des unités est plus importante. Si la croissance a été en effet forte jusqu’en 2005, le ralentissement a été ensuite sensible. Le taux de croissance de remboursement de médicaments durant le premier semestre de cette année est globalement de 2,6 %, mais il atteint 6,5 % pour les patients en affection de longue durée (ALD) et il est de moins 2,3 % pour les autres patients. Pour 45 millions de patients, le taux de croissance de consommation de médicaments n’est donc pas très élevé et pour 8 millions, en ALD, ce taux est de 9 %. La consommation de médicaments est de plus en plus concentrée sur les personnes âgées et les personnes en ALD.
En outre, 96 % des demandes d’inscription en ALD sont acceptées ainsi que 99 % des demandes de renouvellement. À cela s’ajoute que les patients en ALD seront sans doute de plus en plus nombreux, certains évoquant même le chiffre de 15 millions pour les années à venir. Leur prise en charge mériterait donc d’être réévaluée. La Haute Autorité de santé se penche actuellement sur ce sujet.
Mme Catherine Lemorton, rapporteure : Le décrochage augmentera donc encore selon vous ?
M. Christian Lajoux : Les médicaments de spécialités sont innovants : au nom de quoi ne pas les diffuser ? Ils visent à soigner des malades atteints du cancer, du SIDA ou encore de polyarthrite rhumatoïde !
Mme Catherine Lemorton, rapporteure : Ce que vous venez de dire est important. Cela devrait donc conduire à réévaluer l’objectif national des dépenses de l'assurance maladie (ONDAM) en conséquence.
M. Christian Lajoux : L’ONDAM ne tient en effet pas compte des besoins de santé du pays : ces dernières années, il était complètement irréaliste. Tous les observateurs s’accordent à dire que, compte tenu du vieillissement de la population et des progrès techniques, on ne peut maîtriser les dépenses de santé en dessous d’une croissance de 4 %. La part dévolue aux médicaments dans l’ONDAM est d’ailleurs faible alors que ce sont ces derniers qui permettent de faire des économies en évitant des récidives ou en raccourcissant des hospitalisations. Une bonne utilisation des médicaments amoindrit les dépenses de santé.
Mme Catherine Lemorton, rapporteure : La sortie de médicaments de la réserve hospitalière compte pour beaucoup dans cette augmentation de l’ONDAM.
M. Christian Lajoux : Aujourd’hui, 20 % des médicaments sortant de l’officine sont en effet des médicaments issus de spécialités hospitalières.
M. Jean Mallot, coprésident : Comment comptez-vous vous organiser pour contribuer à une bonne utilisation des médicaments ? Comment faire en sorte que la visite médicale serve aussi à cela ?
M. Christian Lajoux : J’insiste : les dépenses de santé ne sont pas aussi exponentielles que le prétendent certains responsables politiques. Il n’a par ailleurs jamais été question d’un « guichet ouvert » du médicament.
On ne peut à la fois mener une politique de santé publique, une politique de réduction des déficits des comptes sociaux et une politique industrielle du médicament, sachant que cette dernière est étroitement liée à la politique de santé publique. Si l’on oppose en permanence la contrainte du déficit des comptes sociaux – qui doit être en effet réduit – la France risque de perdre des places dans la compétition mondiale et cela nous coûtera cher.
Les industriels du médicament, notamment Sanofi, rappellent la nécessité du bon usage du médicament, car il n’est pas possible de construire un « marché » du médicament sur des gaspillages. Nous soutenons les mesures qui ont été mises en place, je pense en particulier au « Web médecin », géré par la Caisse nationale d’assurance maladie des travailleurs salariés (CNAMTS), qui permettra d’éviter des prescriptions redondantes. Le développement de l’automédication peut également permettre de réaliser des économies, de même que l’identification de la prescription à l’hôpital. Enfin, il faut s’emparer de la question des ALD car il est urgent de prendre des mesures radicales.
Mme Marie-Noëlle Banzet : Nous ne considérons pas que la visite médicale soit un gros mot, bien au contraire. Le bon usage du médicament passe aussi par une bonne utilisation de la visite médicale. Vous avez déjà entendu, lors de vos précédentes auditions, parler de la charte de la visite médicale. À ce propos, le groupe Servier est en cours de certification, et celle-ci sera sans doute acquise début 2008. De toute façon il ne s’agit que de la mise en forme d’une éthique de la visite médicale que nous respectons depuis très longtemps.
La grande spécificité du groupe Servier tient dans la longueur de la formation des visiteurs médicaux. Quand ils arrivent, munis de leur diplôme, nous leur assurons, la première année, une formation de l’ordre de dix semaines dans les domaines scientifique, professionnel, éthique. Ils subissent un contrôle très rigoureux prévu par la charte, mais que nous avions déjà mis en place auparavant.
La formation et les contrôles des visiteurs médicaux font partie du bon usage du médicament. Cela est indispensable et nous y sommes très attachés.
M. Christian Lajoux : À travers les différents rapports et la manière d’aborder le sujet, on observe une volonté de stigmatisation : on a l’impression que les visiteurs médicaux portent une pancarte et disent aux médecins de prescrire leurs produits. Ce n’est pas cela, et je vous invite à tourner avec certains de nos visiteurs médicaux. Vous constaterez qu’ils exercent un métier d’information, de formation et qu’ils sont ainsi parties prenantes au dialogue nécessaire pour faire évoluer les progrès thérapeutiques. On ne peut pas contester aujourd’hui que l’activité du visiteur médical dépasse, et de très loin, le champ de la promotion.
L’honnêteté oblige cependant à reconnaître que la prise en charge d’un certain nombre de médicaments très nouveaux et très sophistiqués par l’ensemble du corps médical, qui prescrit plutôt bien, passe, entre autres, par la qualité des visiteurs médicaux. Nous sommes le seul pays où existent, outre un diplôme de visiteur médical, une charte conventionnée avec le Comité économique des produits de santé, et un processus de certification avec la Haute Autorité de santé ; cela s’est fait dans les trois dernières années.
Au moment où nous nous engageons dans ce processus, en concertation avec les autorités de ce pays, selon les critères définis par la HAS et l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS), il y a une reconnaissance par les différentes autorités de l’État de l’utilité et de l’impact de la visite médicale, moyennant le respect d’un certain nombre de critères de qualité, même si des améliorations sont nécessaires. Sanofi Aventis fait partie des premières sociétés certifiées en termes de qualité. Je sais ce que cela implique en termes de travail et de contrôle interne, dans une société qui emploie 10 % des visiteurs médicaux employés en France alors qu’elle réalise 15 % du chiffre d’affaires du secteur.
La visite médicale participe à l’information des médecins. Selon le rapport de l’Inspection générale des affaires sociales (IGAS), 72 % des médecins sont satisfaits de la visite médicale, mais 25 % d’entre eux considèrent qu’on pourrait l’améliorer. Cela signifie bien qu’ils ne sont pas passifs vis-à-vis de la visite médicale. En tant que dirigeant d’entreprise, j’ai des contacts avec des médecins qui désirent parler de la façon dont se déroule la visite médicale. D’ailleurs, ce sont eux qui organisent les modalités de réception, qui demandent aux visiteurs médicaux d’aborder certains sujets. Dans notre pays, il y a un encadrement très spécifique et original de qualité, à caractère évolutif. Sanofi Aventis, pour sa part, ne refuse pas d’évoluer dans le cadre de la qualité, au service d’un meilleur usage du médicament.
M. Éric Ducournau : C’est une certification qui va en profondeur. Les visiteurs médicaux sont même interviewés un par un par la personne qui fait l’inspection. J’insisterai sur un autre élément : le rôle des visiteurs médicaux dans le domaine de la pharmacovigilance. Ils font remonter vers les laboratoires un certain nombre d’informations leur venant des médecins.
M. Christian Lajoux : Globalement, sur 20 000 remontées de pharmacovigilance, 10 000 sont assurées directement par les visiteurs médicaux.
M. Jean Mallot, coprésident : Iriez-vous jusqu’à en déduire que les délégués de l’assurance maladie (DAM) ne sont pas forcément indispensables ? Ils correspondent à une autre démarche, à d’autres critères et à un autre point de vue.
M. Christian Lajoux : Nous ne pouvons pas empêcher l’assurance maladie de pratiquer une forme de visite médicale. Toutefois, nous souhaiterions que les DAM suivent le même processus de formation et de certification que celui que l’on exige de l’ensemble de nos visiteurs médicaux. Il est tout de même paradoxal que les DAM ne rentrent ni dans le cadre de la charte de la visite médicale, ni dans celui de certification par la HAS.
Certes, il peut y avoir d’autres moyens de communication et d’information du corps médical et Sanofi Aventis admet leur multiplication, mais d’un côté on est sévère vis-à-vis de la visite médicale en en ignorant le contenu et la réalité de la pratique, de l’autre on laisse se multiplier les sites internet qui donnent des informations directes aux patients et aux médecins sans aucun contrôle parce qu’ils n’appartiennent pas à l’industrie du médicament ou n’ont aucune connexion avec elle.
La situation de notre pays est paradoxale : les industriels du médicament, qui sont des experts en termes de recherche et de production de leurs produits, n’ont pas la capacité de communiquer directement, en dehors du cadre que je viens d’évoquer, avec les médecins et aucunement avec les patients. En même temps, il y a une profusion d’acteurs qui communiquent sur le médicament sans avoir la même expertise que nous et sans être contrôlés sur les éléments d’information qu’ils diffusent.
Mme Catherine Lemorton, rapporteure : Internet n’est pas la panacée, en effet.
Parlons chiffres : quand vous trouvez une molécule, vous la mettez sur le marché français, une fois qu’elle a obtenu son autorisation de mise sur le marché (AMM). Quelle est en France la part de la visite médicale par rapport au prix du médicament, en comparaison avec la même entrée de ce médicament sur le marché des autres pays européens ? Avez-vous une idée ?
M. Christian Lajoux : La part de la visite médicale est de 10 % du chiffre d’affaires. Dans les autres pays, pour une firme comme Sanofi, c’est exactement pareil. Dans certains pays, c’est au-dessous, dans d’autres c’est au-dessus.
Dans le rapport de l’IGAS, on trouvait une information qui n’était pas exacte, à savoir qu’en Angleterre, il y avait moins de visiteurs médicaux que dans les autres pays. Nous avons repris les calculs et nous avons constaté que, dans ce pays, on trouve un visiteur médical pour trois médecins, ce qui n’est pas le cas en France.
Nos entreprises sont des entreprises internationales. Nos façons d’exercer dans les différents pays sont pratiquement les mêmes. À l’intérieur des entreprises, existent de plus en plus souvent des directions européennes dont la mission est de faire en sorte qu’il y ait une réalité de l’Europe, au moins en termes économiques et industriels. Je pourrai sans doute vous donner d’autres chiffres tout à l’heure.
Mme Catherine Lemorton, rapporteure : Je voudrais revenir sur les ALD.
Moins on permettra à des gens d’accéder au système de soins, et plus les personnes âgées qui sont grandes consommatrices de médicaments, auront tendance à maintenir leur ALD, parce qu’elles ne peuvent pas accéder à des assurances complémentaires. Je le dis par expérience de terrain. Lorsqu’on examinera les ALD, il faudra le prendre en compte.
M. Jean Mallot, coprésident : Nous avions encore mille questions, mais nous devons passer à l’audition suivante. Je vous remercie et je vous propose, dans les jours ou les semaines qui viennent, de compléter les réponses sur les points que vous n’auriez pas eu le temps d’exposer de façon exhaustive, ou de répondre à celles que nous n’avons pas posées, mais que nous aurions dû vous poser. Nous vous remercions.
*
Audition de Mme Anne Baille, présidente des Laboratoires Ranbaxy pharmacie génériques, Mme Marie-Josèphe Baud, présidente directrice générale de Sandoz, Mme Catherine Bourrienne-Bautista, déléguée générale de GEnériques Même MEdicaments (GEMME), M. Maurice Chagnaud, président-directeur général du Laboratoire Teva Classics, et M. Gilles Chaufferin, directeur général délégué adjoint des Laboratoires Boiron.
M. Jean Mallot, coprésident : Merci d’être venus. Pourriez-vous préalablement à nos échanges faire une présentation générale de chacune de vos entreprises, de votre démarche et de votre spécificité ?
Mme Anne Baille : Le laboratoire international Ranbaxy est d’origine indienne. Dans les années soixante, il a commencé avec la mise sur le marché et le développement d’un médicament, le chloramphénicol. Son activité industrielle s’est d’abord développée en Inde. Dans le milieu des années soixante-dix, il est entré en bourse sur les marchés indiens. Dans les années quatre-vingt-dix, il a connu deux étapes importantes : une expansion internationale, d’abord sur l’Asie et l’Afrique, donc dans des pays en voie de développement, ensuite dans des pays développés, les États-Unis et l’Europe. Son entrée sur le marché européen s’est faite dans le milieu des années quatre-vingt-dix en Angleterre. Autre étape importante pour notre groupe : le démarrage de l’investissement en recherche et développement (R&D) ; aujourd’hui, il compte 1 500 chercheurs, essentiellement en discovery.
L'une des particularités du groupe est de chercher de nouvelles entités chimiques, puis de les mettre à la disposition de partenaires industriels de l’industrie pharmaceutique et probablement, à terme, de les produire sous ses propres marques.
En ce qui concerne le marché français, Ranbaxy a fait l’acquisition en janvier 2004, de la filiale génériques des laboratoires Aventis, qui s’appelle toujours RPG. Depuis cette époque, nous développons en France notre gamme de médicaments génériques.
Mme Catherine Maurienne-Bautista : Je représente GEMME, l’association des laboratoires des médicaments génériques, qui regroupe les principaux acteurs actifs sur le marché français. Il a été créé il y a un peu plus de cinq ans pour accompagner les laboratoires dans le développement des médicaments génériques en France.
M. Jean Mallot, coprésident : Sous quelle forme ?
Mme Catherine Maurienne-Bautista : Nous sommes une association régie par la loi de 1901.
M. Maurice Chagnaud : Teva est aujourd’hui le leader mondial du médicament générique. C’est une entreprise israélienne, qui a connu un fort développement à partir des années quatre-vingt aux États-Unis. Elle s’est ensuite développée en Europe, essentiellement par acquisitions, dans un grand nombre de pays européens : Angleterre, Hollande, Hongrie, Italie et, en 2002, en faisant l’acquisition de la filiale génériques de Bayer, Bayer Classics, d’où notre nom de Teva Classics.
Ce laboratoire est à la fois le numéro 1 dans le médicament générique et le dix-septième acteur de la pharmacie mondiale. En effet, Teva ne fait pas que du générique. Il a une activité d’innovation, avec le Copaxone, promu d’ailleurs en France par Sanofi Aventis. Il travaille dans le domaine de la maladie de Parkinson, avec d’autres produits en cours d’enregistrement et de développement. Depuis la fin de 2005 et le début de 2006, il travaille dans le domaine du respiratoire, par le biais de l’acquisition, au niveau mondial, des activités de Aivax, qui intervenait dans le domaine du respiratoire et du générique.
Je suis le président de Teva Classics, de Teva Pharma et de Aivax Pharmaceutics en France. La volonté du groupe Teva est de continuer dans le même esprit, en développant une activité hybride : partir du générique, avec des activités de recherche et de développement suffisamment importantes pour faire de l’innovation.
M. Gilles Chaufferin : Boiron est un groupe familial indépendant, qui a soixante-quinze ans aujourd’hui. Ce groupe français fait un peu plus de 400 millions d’euros de chiffre d’affaires, réalisés pour 60 % en France et pour 40 % à l’international, au travers d’une vingtaine de filiales. Il emploie à peu près 4 000 personnes, dont 2 800 en France. Il assure aujourd’hui, depuis la France, plus de 90 % de sa production. Il est spécialisé dans un domaine particulier, le médicament homéopathique et en vend deux catégories principales : d’abord, des médicaments homéopathiques à nom commun, proches des médicaments génériques dans la mesure où ils sont vendus sous un nom commun, au même prix, et à un prix extrêmement bas, aujourd’hui remboursés à 35 % par l’assurance maladie ; ensuite, des spécialités de médication familiale, non remboursables, qui font aujourd’hui de Boiron le deuxième laboratoire de médicaments conseil en France.
Pour des raisons historiques et logistiques, Boiron assure la distribution de ses médicaments, ce qui représente une charge très importante. L’enjeu majeur de cette entreprise est aujourd’hui la recherche et le développement. Comme beaucoup de médicaments et de stratégies thérapeutiques traditionnelles et anciennes, ces médicaments ont été insuffisamment évalués jusque-là. L’enjeu de l’entreprise est de développer de façon encore plus active la R&D et de démontrer l’intérêt de santé publique de cette stratégie thérapeutique, traditionnelle et largement répandue dans notre pays.
Mme Marie-Josèphe Baud : Je suis présidente de Sandoz depuis le mois de mars 2007. C’est une filiale, spécialisée dans le domaine des génériques et des biosimilaires, du groupe Novartis. Sandoz est sur le marché français des génériques depuis 2001, essentiellement grâce à l’achat d’une société qui s’appelait GNR. En 2006, à l’issue d’une fusion mondiale avec le groupe Exal, Sandoz a acheté une société Exal qui exerçait en France sous le nom de Gegam dans le domaine du générique, et une autre activité, Biopharmaceutical, qui nous permet d’être le premier laboratoire à proposer non seulement des génériques, mais aussi le premier médicament biosimilaire sur le marché français, le similaire d’une hormone de croissance, qui est un nouveau champ sur le marché du médicament.
Mme Catherine Lemorton, rapporteure : Je tiens à apporter une information à la connaissance des personnes présentes.
Nous avions prévu d’entendre un représentant des laboratoires homéopathiques au cours de notre première table ronde, en l’occurrence celui des laboratoires Boiron, mais nous avons reçu des appels de la part de certains participants nous demandant de ne pas être auditionnés en même temps que les laboratoires Boiron. Je remercie donc les laboratoires du générique de ne pas avoir fait la même requête. Il fallait le dire, en toute transparence.
Ma question est assez simple. Vous êtes tous présents sur le même marché, avec les mêmes molécules. Comment faites-vous pour assurer la promotion de vos produits auprès des médecins et des pharmaciens ?
Mme Anne Baille : Nous devons résoudre au quotidien cette question, s’agissant du générique.
Quand je développe un médicament générique, je dois démontrer que je suis en train de développer un médicament qui sera la reproduction de celui qui existe déjà et qui a fait ses preuves. Ensuite, quand j’arrive devant un médecin ou un pharmacien, je dois lui expliquer que celui qui est fabriqué par les laboratoires Ranbaxy est celui qui lui convient le mieux, car nous sommes douze ou treize laboratoires en concurrence.
Nous avons tous nos stratégies, nos tactiques et nos particularités. Nous travaillons les uns et les autres sur nos gammes, qui ne sont pas exactement les mêmes. Ces particularités répondent à des besoins qui sont différents. Par ailleurs, nous développons tous des services particuliers, par l’intermédiaire de nos visiteurs pharmaceutiques et de nos clients pharmaciens.
Dans ce pays, le marché du médicament générique repose sur un pilier principal : le pharmacien d’officine, qui a le droit de substitution. Tout notre investissement promotionnel, pour développer ce marché, et nos entreprises au sein de ce marché, repose donc sur le pharmacien. Nos équipes vont visiter les pharmaciens d’officine et leur apportent des services. Ensuite, les pharmaciens d’officine prennent leur décision et achètent, ou non, nos produits.
M. Maurice Chagnaud : Le marché du générique est relativement jeune en France. Certains acteurs ont commencé en 1996-1997, mais les règles ne sont apparues évidentes qu’en 1999, avec le droit de substitution qui a été octroyé aux pharmaciens.
Entre 1996 et 1999, tous les laboratoires présents faisaient le choix du pharmacien, le choix du médecin, le choix d’une stratégie DCI, le choix d’une stratégie de générique « de fantaisie ». Plusieurs solutions étaient possibles. Il fut d’ailleurs très difficile de faire démarrer le médicament générique en France.
Certains acteurs avaient fait très tôt le choix du pharmacien, ce qui était logique. Cependant, on ne pouvait pas demander à un pharmacien d’avoir quinze fois le même produit. À l’époque, Mme Martine Aubry, alors ministre en charge de la santé, accorda un droit de substitution très large, allant jusqu’au médicament princeps, qui permettait au pharmacien de référencer la molécule originale, plus une ou deux molécules génériques, et d’être beaucoup plus efficace.
Nos entreprises sont jeunes, elles ont du mal à se structurer parce qu’elles doivent gérer plusieurs difficultés, à commencer par une croissance très rapide. Aujourd’hui, il y a plus de 300 produits sur le marché. Gérer 300 produits alors que vous démarrez ne se réalise pas du jour au lendemain. Il faut de la qualité, il faut des pharmaciens responsables, il faut libérer des lots, il faut des usines, des centres de conditionnement, etc. En outre, chaque année, en fonction de l’évolution de l’environnement économique, les règles sont un peu modifiées. Cela met l’entreprise en péril, car elle doit chaque année trouver des systèmes encore plus efficaces, pour être toujours aussi compétitive sur le marché.
M. Jean Mallot, coprésident : De quelles règles parlez-vous ?
M. Maurice Chagnaud : Prenez les baisses de prix systématiques sur le médicament générique. Le médicament générique a une logique économique, mais, à un moment donné, il faut laisser aux acteurs le temps d’être efficaces sur le marché. Or, on est allé très vite. On n’a pas attendu que le marché ait atteint 30 % en volume. À peine avait-il atteint 7 % que les baisses de prix ont commencé, voici deux ans. Gemme nous avait alertés, et nous avions communiqué à ce sujet. Aujourd’hui, nous nous retrouvons dans la même situation.
Nous avons de grands conditionnements à réaliser. Nous devons faire en sorte, et c’est une spécificité française, d’être au plus près du médicament éthique, ce qui nous oblige à avoir des conditionnements spécifiques, ce qui n’est pas le cas des autres pays européens ou anglo-saxons. En France, c’est une question de choix et de qualité, mais cela a un coût. Si l’on accepte de dire que la qualité et le packaging sont nécessaires, qu’il faut du blister et du grand conditionnement ; nous sommes prêts à suivre, mais il faut savoir que, à un moment donné, certains produits ne passeront plus en termes de prix de revient industriel.
Sur la base des derniers éléments connus concernant les nouvelles modifications qui s’appliqueront en 2008, 2009 et 2010, on sait, d’ores et déjà, qu’il faudra prendre des décisions et, éventuellement, arrêter certains produits. Dans le domaine hospitalier, c’est encore pire : au bout de trois ou quatre ans, les prix se sont tellement effondrés, que certains acteurs sont obligés de sortir du marché.
Il convient donc de faire très attention : nos entreprises sont jeunes, leur équilibre est extrêmement fragile, très lié à l’environnement et aux prises de décisions politiques, logiques avec l’environnement économique, mais sans qu’on ait toujours bien conscience des enjeux industriels que cela représente.
Mme Catherine Bourrienne-Bautista : L’ensemble de l’industrie des génériques ne génère pas de bénéfices depuis son arrivée sur le marché français. On a demandé aux laboratoires de lancer les produits, d’investir auprès des pharmaciens pour que le marché se développe. Ces investissements pèsent encore très lourd sur les entreprises. Quand interviennent des baisses de prix, quand l’environnement est chahuté très régulièrement, le risque est que certains acteurs et que certains produits disparaissent, notamment les produits anciens, les produits franco-français qui sont déjà à des prix très bas.
Mme Marie-Josèphe Baud : On peut aussi parler des rachats de sociétés : M. Maurice Chagnaud a évoqué le rachat de Aivax par Teva et nous pouvons rappeler celui de Gegam-Exal par Sandoz. Ces sociétés étaient sur le marché du générique et subissaient des pertes abyssales par rapport à leur taille.
Mme Anne Baille : S’agissant des prix, il y a, dans notre environnement économique, quelques mesures qui nous paraissent contreproductives. Je peux vous en citer trois.
La première est la taxe sur les ventes en gros, dont M. Christian Lajoux vous a parlé tout à l’heure et que nous subissons de plein fouet car elle n’est pas déductible.
En effet, nos laboratoires, de manière à développer le marché des médicaments génériques, ont appliqué des techniques de distribution, que nous appelons, dans notre jargon, des « ventes directes ». Celles-ci avaient initialement pour objectif de permettre aux pharmaciens d’avoir un stock suffisant leur permettant de substituer en cas de besoin. Comme il n’est pas possible à un pharmacien de dire à un patient de revenir plus tard pour chercher son médicament générique, on constitue ces stocks par l’intermédiaire de nos visiteurs qui travaillent avec les pharmaciens. Nous avons développé cette stratégie de vente directe, mais, chaque fois que nous développons le marché en utilisant cette tactique, nous payons un peu plus de taxe sur les ventes en gros. Nous sommes taxés exactement comme les grossistes répartiteurs.
La seconde mesure contreproductive est relative aux franchises.
En tant que citoyenne, j’ai mon avis sur les franchises, mais j’ai aussi mon point de vue de chef d’entreprise. Le prix fabricant moyen hors taxe d’un médicament générique est en France de 3 euros ; le prix public est de 5,70 euros. Imaginez ma mère, ma tante ou ma grand-mère : elle paiera 50 centimes pour une boîte qui coûte 5,70 euros ! Et à côté de cela, dans la plupart des départements français, on lui expliquera que, si elle n’accepte pas le générique, elle ne bénéficiera pas du tiers payant. D’un côté, on répète qu’il faut éduquer les patients, qui doivent prendre en charge leur santé, et de l’autre, on leur dit que cela leur coûtera 50 centimes, même s’ils prennent des génériques. Il y a quelque chose à faire, mais je n’ai pas la solution.
Le troisième point sur lequel je souhaite appeler votre attention est celui des grands conditionnements qui est un vrai problème industriel. Certes, le patient ne paiera qu’une fois ces 50 centimes, mais, sur nos séries de produits, nous en sommes à imaginer du conditionnement manuel. Nous faisons les boîtes une par une. Cela n’est pas compatible avec le prix du générique. Résultat : on hésite à entrer sur le marché ; on dit au pharmacien qu’il doit donner des conditionnements industriels, et l’offre générique n’est pas complète. Par rapport à la dynamique que nos autorités semblent vouloir donner à ce marché, c’est une mesure qui ne fonctionne pas.
M. Jean Mallot, coprésident : Si la franchise est forfaitaire, lorsqu’elle s’applique sur un produit peu cher, la proportion du médicament payée par le patient est plus élevée. Il peut ainsi avoir effectivement le sentiment que le générique est moins bien pris en charge.
S’agissant des médicaments homéopathiques, la problématique doit être voisine.
M. Gilles Chaufferin : La problématique est assez proche quant à l’impact de la franchise. Pour nous, le prix de fabrication hors taxe est de 0,90 euro. Avec un taux de remboursement de 25 %, la franchise aboutit quasiment à un déremboursement de l’ordonnance, d’autant que, malgré un coût de l’ordonnance beaucoup plus faible, le volume d’unités délivrées peut être plus élevé. Cela induit un coût supplémentaire par rapport à une ordonnance classique.
Par ailleurs, on nous encourage, ce qui n’est pas simple, à envisager d’autres conditionnements, mais les investissements industriels que cela implique sont très difficiles à supporter.
Mme Catherine Lemorton, rapporteure : Que pensez-vous de la remise en cause des marges arrière, dans le cadre de la politique de développement du générique ?
Pensez-vous que cette remise en cause mettra un frein, voire stoppera le développement du générique ? Car le but est de baisser encore le prix du générique.
M. Maurice Chagnaud : Dans tous les pays européens, on a tendance à se rapprocher de la pharmacie. Le pharmacien est à la fois un acteur de santé et un acteur économique. Il a sa propre entreprise. Si l’on fait un rapide calcul, compte tenu de la répartition entre les produits soumis au tarif forfaitaire de responsabilité (TFR) et les produits qui n’y sont pas soumis, avec la diminution des remises et des marges arrière de 25,74 % à 17 %, lesquelles seront désormais calculées sur des prix diminués en conséquence, le pharmacien va voir sa rémunération diminuer de 4,8 % et 5 % selon les volumes des médicaments vendus soumis au TFR. Au final, l’économie de l’officine en souffrira.
Les syndicats de pharmaciens d’officine ont accepté le principe de continuer à souffrir, mais il est bien évident que, pour nous, une compétition économique va se développer. Cette perspective nous amène à réfléchir à avoir une présence différente aux points de vente : comment faire la différence entre Sandoz, Ranbaxy, Teva, Biogaran et consorts sur le marché, quand vous avez le même produit, aux mêmes conditions économiques, au même moment, car toutes nos sociétés sont capables de lancer un même produit au même moment ?
Il faut donc faire preuve d’un savoir faire intéressant et différent, c’est-à-dire développer d’autres gammes de médicaments, apporter des services différents. Aujourd’hui, l’une des pistes de développement de Teva, suite au rachat d’Aivax, est d’aller très fortement dans l’asthme et dans le service auprès du pharmacien dans l’asthme. Nous allons donc, au cours de l’année 2008, dire aux pharmaciens : si vous le voulez, vous référencerez de manière un peu plus privilégiée la gamme Teva, parce que nous sommes capables de vous apporter un service additionnel – ce que tout le monde n’est pas en mesure de faire – et parce que nous proposons une gamme dans l’asthme, ce que tout le monde n’a pas.
Il nous faut, dans un marché compétitif, parce que nous sommes dans une démarche commerciale, avoir la capacité de faire la différence. Marges arrières ou pas, à un moment donné, il faudra mener une réflexion beaucoup plus globale sur la vente du médicament, notamment générique.
Je ne sais pas si le développement du générique sera freiné. Les marges arrière ont baissé d’une année sur l’autre, passant de quelque chose qui n’était pas structuré, à 20 %, puis à 15 %. Et quand vous regardez le taux de pénétration, au bout de trois mois, des groupes génériques lancés en 2006 et en 2007, vous constatez qu’ils sont inférieurs au taux de pénétration, au bout de trois mois de lancement, des groupes lancés en 2004, 2003 et en 2002. Avec l’oméprazole, on a rapidement atteint un taux de substitution de 75 %. Or, cette année, on n’a pas obtenu, au bout de trois mois, les mêmes résultats sur la modipine ou la terminafine. Il faut bien voir que la compétitivité économique n’est plus tout à fait la même.
Mme Anne Baille : La situation est désormais satisfaisante s’agissant des marges arrière sur les médicaments génériques, notamment grâce à la régulation introduite par la loi Dutreil. Non seulement tout travail mérite salaire, mais il n’est, en l’occurrence, pas facile d’opérer la substitution d’un produit, d’autant que nous lançons environ 40 à 50 nouvelles spécialités chaque année ce qui signifie que, une fois par semaine, les pharmaciens doivent s’informer sur la nature d’un nouveau médicament et repérer les patients qui peuvent être concernés. Cela justifie leur sur-rémunération.
Existe-t-il un risque d’atténuation de la dynamique du marché des génériques ? Compte tenu de la baisse des prix, nous ne pouvons pas investir plus alors qu’il aurait sans doute été possible de dégager des moyens pour nos entreprises, mais aussi, peut-être, pour les officines, afin d’en poursuivre le développement. Nos entreprises internationales, enfin, investissent à perte depuis quelques années et je ne suis pas certaine que l’on pourra convaincre longtemps nos commettants de continuer ainsi.
Mme Catherine Bourrienne-Bautista : Le pharmacien doit en effet conserver un intéressement à la délivrance de médicaments génériques, mais il convient sans doute de réfléchir à l’implication d’autres acteurs qui n’ont pas encore pris leur part dans le développement de ce marché, notamment, les médecins.
Mme Catherine Lemorton, rapporteure : Comment réagissez-vous en cas de contournement de médicaments génériques : disposez-vous d’un moyen d’action ou pouvez-vous, à tout le moins, donner un avis ?
M. Maurice Chagnaud : Il n’est pas possible de donner un avis, mais une telle pratique fait partie des règles du jeu. Si cela nous empêche de réaliser une économie, nous sommes également incités et contraints à œuvrer pour rattraper le chiffre d’affaires. À un certain stade, il n’y a de toute façon plus d’échappatoire. La durée de vie du médicament est un peu prolongée dans ce cas-là, mais c’est tout. Nous avons été challenger des brevets sur le plan européen, mais nous considérons que c’est à la Commission européenne de dire que ces produits ont le même service médical rendu (SMR) et qu’ils n’apportent pas d’amélioration du service médical rendu (ASMR).
Il avait été envisagé voilà deux ans, dans le cadre de l’examen du projet de loi de financement de la sécurité sociale (PLFSS), de considérer ces produits comme des génériques. Nous ne pouvons donc que constater la situation ou essayer de l’anticiper de manière à pouvoir « génériquer » ces produits le plus vite possible sur les marchés. Deux ou trois ans sont en général nécessaires.
Mme Marie-Josèphe Baud : Je suis d’accord avec M. Maurice Chagnaud : cela fait partie des règles du jeu et ce n’est pas nous qui pourrons mettre en place une régulation.
Mme Anne Baille : Notre éthique implique de ne pas contrefaire un brevet mais nous challengeons les produits s’ils sont brevetés et s’ils disposent d’une patente solide, ou s’ils sont protégés pour des raisons commerciales et non pour des raisons de propriété intellectuelle.
M. Jean Mallot, coprésident : Dans les années à venir, un grand nombre de médicaments arriveront dans le champ du « génériquable ». Qu’en est-il précisément ? Comment les laboratoires sont-ils organisés ?
M. Maurice Chagnaud : Chaque pays européen produit des brevets à peu près en même temps. Nous disposons en l’état de plus de 700 produits en développement pour être capables de lancer le brevet le jour J dans tous les pays d’Europe. Parce que cela demande des investissements très importants, nous essayons de mettre au point de nouveaux processus de fabrication plus économiques.
Par ailleurs le design des produits change : un grand nombre d’entre eux seront de plus en plus importants en valeur, mais ils auront une rotation très faible compte tenu de leur volume. Tous les laboratoires lanceront entre 40 et 70 lignes de produits par an, mais il ne faut pas s’attendre à de très grands taux de substitution, faute d’une grande visibilité. Les pharmaciens devront donc s’adapter.
Le marché du générique est aujourd’hui très instable parce que tous ses éléments constitutifs ne sont pas figés et que le cycle de vie et de mort du produit est plus rapide : à l’hôpital, des offres de produits disparaissent ! En termes d’échéance de brevets, un grand nombre de molécules de produits hospitaliers tomberont dans le domaine public entre 2008 et 2010.
Mme Anne Baille : La substitution dans le répertoire s’effectue aujourd’hui par groupes génériques ; il s’agit d’une définition réglementaire du médicament générique en référence à l’autorisation de mise sur le marché (AMM). Or les médicaments de demain seront de plus en plus complexes et relèveront du bio-similaire ou associeront dispositif médical et molécule. Il faudra donc travailler avec les autorités de santé pour trouver des cadres réglementaires permettant aux pays de bénéficier de l’arrivée de médicaments génériques – au sens commun du terme, et pas réglementaire –, mais aussi pour élargir ou créer un répertoire d’équivalents pharmaceutiques.
Mme Marie-Josèphe Baud : Si moins de grandes molécules tomberont dans le domaine public à partir de 2010, nombre de brevets de produits de biotechnologies expireront, ce qui représente des enjeux financiers très importants et implique une approche différente. Sandoz expérimente cela avec une hormone de croissance. Alors qu’avec les génériques nous allons voir les pharmaciens pour favoriser la substitution, nous devons en l’occurrence faire comprendre aux médecins que nous sommes en présence d’un produit dit « bio-similaire » qui coûte moins cher et qui est fabriqué à partir des technologies les plus performantes. Il faut donc créer un nouveau modèle.
Des procès sont en cours sur le droit d’utilisation des marques. Or, si nous ne pouvions plus définir nos produits comme génériques ou bio-similaires d’une marque, nous serions confrontés à un grave problème, puisque nous ne saurions pas comment en parler. Dans ce cas, le droit s’apposerait à nos AMM.
M. Maurice Chagnaud : Teva est également intéressée par les bio-similaires, mais l’investissement en R&D et le taux de succès en bio-équivalence n’ont en l’occurrence rien à voir avec l’investissement générique : les études de bio-équivalence dans le générique s’élèvent entre 600 000 et 2 millions d’euros quand, dans le bio-similaire, les sommes se situent entre 50 et 70 millions de dollars. À cela s’ajoute que, dans le cas d’espèce, les chances de réussite ne sont que de 50 %.
Mme Anne Baille : Les bio-similaires étant en outre des médicaments dont la matière première est biologique, il n’est possible d’optimiser les processus industriels que jusqu’à un certain point, lequel ne sera jamais comparable à celui qu’il est possible d’obtenir dans la chimie classique.
Mme Marie-Josèphe Baud : J’ajoute que, en tant que laboratoire de génériques, nous disposons de services de pharmaco-vigilance qui se montrent encore plus vigilants, bien entendu, avec ce type de produits.
Mme Catherine Lemorton, rapporteure : Pouvez-vous promouvoir les génériques auprès des médecins ?
Mme Anne Baille : Je souhaite que cela soit possible, mais je n’en ai pas les moyens. La « fuite de prescription hors du répertoire » n’est pas due à la mauvaise volonté des médecins et il faut bien par ailleurs que les laboratoires d’innovation promeuvent de nouvelles molécules. Il s’agit bien plutôt d’une responsabilité collectivité : que voulons-nous ?
En outre, la promotion du maintien de la prescription des standards thérapeutiques doit s’accompagner d’une réflexion sur leur nature que l’AFSSAPS et la HAS peuvent fort bien mener en indiquant ce que doit être, par exemple, l’utilisation de la première génération de statines par rapport à la deuxième. Enfin, à force de vouloir faire trop d’économies, il ne faut évidemment pas priver les patients des bénéfices thérapeutiques de l’innovation.
M. Maurice Chagnaud : Nous disposons de 300 produits et nous en aurons 600 en 2011 : il est inimaginable d’en faire la promotion auprès des médecins car cela coûterait une fortune. Une solution possible consisterait à utiliser les logiciels informatiques de manière à ce que l’ordonnance indique systématiquement les deux produits, sous le nom de marque et sous le nom de dénomination commune internationale (DCI), en laissant aux pharmaciens la possibilité de substitution.
Mme Catherine Bourrienne-Bautista : Les laboratoires ne peuvent certes pas aller voir tous les médecins pour les inciter à prescrire des médicaments génériques, mais ceux-ci disposent de plus en plus de logiciels d’aide à la prescription qui les informent des coûts de l’inscription d’un produit au répertoire et de sa possible substitution. De plus, les médecins peuvent être incités à prescrire dans le répertoire à travers des contrats individuels. Enfin, la mise en place d’un objectif de prescription dans le répertoire serait bienvenue, à l’instar du taux de substitution pour les pharmaciens.
Mme Catherine Lemorton, rapporteure : Comment les laboratoires Boiron procèdent-ils auprès des médecins ? Interviennent-ils uniquement auprès de ceux qui sont déjà homéopathes ? Les incitent-ils à se tourner vers l’homéopathie ?
M. Gilles Chaufferin : Notre action est relativement limitée, les médecins choisissant eux-mêmes de se former et d’embrasser cette discipline ; en outre, nous n’aurions pas les moyens de les solliciter : nous disposons de 80 visiteurs médicaux quand Sanofi en a 2 600 et que l’on en dénombre 23 000 en tout dans notre pays.
Nous sommes convaincus que le médicament homéopathique est une chance pour la médecine et pour la santé. La France a d’ailleurs le leadership mondial dans ce domaine. De plus, ces médicaments traditionnels continuent de se développer dans le contexte d’innovation tous azimuts que nous connaissons. L’utilisation de l’homéopathie croît également en automédication. Comment imaginer, dès lors, que des patients achèteraient des médicaments inefficaces ou toxiques ? Nous devons aujourd’hui trouver les moyens économiques permettant d’apporter la preuve scientifique de l’intérêt thérapeutique de l’homéopathie. Une révision de nos prix devrait nous y aider.
Mme Catherine Lemorton, rapporteure : Pensez-vous que l’épouvantail des excipients à effet notoire que certains brandissent contre les médicaments génériques – comme s’ils n’existaient pas déjà dans les médicaments princeps – peut toujours faire peur ?
M. Maurice Chagnaud : Comment pourrait-il en être autrement, puisque l’on a répété cela à 23 000 visiteurs médicaux ?
Mme Anne Baille : Les supermarchés sont remplis d’excipients à effet notoire avec le sucre, le pain etc. Un peu de bon sens devrait permettre de remédier à cette situation.
Mme Marie-Josèphe Baud : La suspicion sur les produits génériques touche aujourd’hui un traitement contre l’épilepsie. Il faudra donc insister encore longtemps sur leur bio-équivalence.
M. Jean Mallot, coprésident : Je vous remercie de vos interventions. Si vous avez des remarques ou des suggestions à faire, n’hésitez pas à nous les faire parvenir.
*
Audition de MM. Christophe Weber, président de Laboratoires internationaux de recherche (LIR), Jean-Christophe Tellier, président de Novartis Pharma, Louis Couillard, président directeur général de Pfizer France, et Mme Sabine Dandiguian, présidente directrice générale de Janssen-Cilag France.
M. Jean Mallot, coprésident : Je vous remercie de votre présence et je vous invite, dans un premier temps, à présenter brièvement vos entreprises.
M. Jean-Christophe Tellier : Novartis, troisième groupe pharmaceutique mondial, emploie 100 000 personnes, dont 3 000 en France. Il travaille à 96 % dans le secteur de la santé, en particulier sur les vaccins et diagnostics, les médicaments conseil, la pharma-innovation et les génériques, avec sa filiale Sandoz. Sa part de marché en France s’élève à environ 4 %. Le groupe est présent dans la médecine de ville et hospitalière et possède deux centres de recherche et deux centres de production, dont un centre de biotechnologies.
Mme Sabine Dandiguian : Janssen-Cilag est la partie pharmaceutique d’un grand groupe américain, Johnson & Johnson. Janssen est uniquement centré sur l’innovation et la recherche et ne comporte aucune filiale dévolue au médicament générique. Il possède en France un site industriel et un centre de recherche. À la différence de certains autres groupes, la première préoccupation est de maintenir l’emploi, non de créer des postes. Enfin, le groupe intervient dans le domaine de la santé mentale – schizophrénie – et, plus récemment, dans la recherche sur le VIH et l’onco-hématologie.
M. Christophe Weber : Je suis président de LIR et de GlaxoSmithKline France. Le premier groupe est constitué par une association de 15 laboratoires internationaux de recherches et vise à faire des propositions quant à l’accès aux progrès thérapeutiques en France. Le second comprend 6 600 collaborateurs, possède cinq centres de production, deux centres de recherches, une division pharmacie-recherche et une division santé-grand public.
M. Louis Couillard : Pfizer est le premier investisseur privé mondial en recherche bio-pharmaceutiques. Il est le deuxième en France et en Europe. Il possède trois sites manufacturiers en France, le siège étant situé Porte d’Orléans à Paris. Le groupe comprend plus de 3 500 collaborateurs en France et il est premier dans les domaines cardio-vasculaire et ophtalmologique, le troisième en oncologie.
Mme Catherine Lemorton, rapporteure : Un modèle fiscal vous semble-t-il préférable à celui de la France ? Quelles seraient d’après vous les améliorations à apporter au modèle français ?
Mme Sabine Dandiguian : Avec onze taxes, comme l’a rappelé M. Christian Lajoux, le système français est très complexe et insuffisamment lisible : ainsi, le triplement de la taxe sur le chiffre d’affaires décidé il y a quelques années lors de l’examen d’un projet de loi de financement de la sécurité sociale a impacté les entreprises dès le mois de janvier et une telle imprévisibilité peut remettre en cause des investissements. Il est très difficile de faire comprendre cela aux Américains.
La clause de sauvegarde est également interprétée comme la taxation du succès, de la performance et de l’innovation : on donne à l’État français le produit de notre croissance alors que l’on s’est battu pour obtenir des prix sur des médicaments innovants ! Pendant ce temps, la bagarre est rude et je dois me battre pour préserver nos sites en France.
M. Christophe Weber : La clause de sauvegarde nuit en effet fortement à l’image de la France. La Cour des comptes a estimé qu’elle n’avait pas de rendement, mais ce n’est pas exact, comme l’atteste le rapport du Comité économique des produits de santé (CEPS).
Mme Sabine Dandiguian : À cela s’ajoute que nous devons faire des projections sur l’évolution possible du marché pharmaceutique. Nous dépensons donc une énergie considérable pour essayer d’intégrer dans nos calculs le produit de la taxe.
M. Louis Couillard : Une décision d’investissement sur le territoire français ne se limite pas aux considérations fiscales. En ce qui nous concerne, nous travaillons à l’amélioration de la productivité de nos sites qui font de la recherche clinique, mais il est difficile de le faire en France lorsque l’on considère l’image que les médias en donnent : les patients inscrits dans ces études seraient ni plus ni moins que des cobayes. Dans ces conditions, il est difficile d’attirer des investissements.
À cela s’ajoute la rigidité du code du travail, les incertitudes pesant sur les investissements en raison de cycles de planification très longs. Il est très difficile d’adapter notre outil manufacturier sur le territoire français.
M. Christophe Weber : La France doit se rendre compte du niveau de compétition actuel pour attirer la recherche biomédicale. Récemment, nous souhaitions acheter un terrain pour agrandir un site, mais nous pensons désormais ne pas le faire, car les autorités locales nous ont demandé de nous engager à construire sur ce site dans les cinq à dix ans. Dans certains pays, en revanche, on vous attire en vous donnant le terrain et en construisant les bâtiments.
M. Jean-Christophe Tellier : La situation est d’autant plus complexe, en raison d’un manque de lisibilité. On a parfois le sentiment que la complexité des niveaux de taxation vise plus à identifier des poches potentielles de revenus possibles qu’à illustrer une politique générale.
Nous gagnerions à avoir un positionnement plus clair, plus lisible, plus prévisible pour que, dans nos centres de décision, chez nous, nous puissions positionner la France par rapport à d’autres pays sur certains avantages. On peut défendre une politique par rapport à une autre, mais on a très souvent le sentiment que la France utilise toutes les réserves de productivité sans avoir pour autant une politique générale. Elle se rend ainsi très obscure et très peu prévisible.
Par ailleurs, nous sommes sur des cycles longs. C’est particulièrement vrai pour la recherche, mais cela l’est aussi pour les autres investissements. En tout cas cela nécessite des règles du jeu claires et relativement pérennes. La perte de confiance quant à cette pérennité rend l’attractivité difficile.
M. Louis Couillard : Je veux revenir sur la cohérence de la politique fiscale.
Vous avez parlé de taxes sur le succès et sur la productivité des industries de recherche pharmaceutique. Financer l’impôt recherche en partie par une taxe sur la croissance du chiffre d’affaires générée par l’innovation pharmaceutique relevait d’une bonne intention. Pourtant, au niveau international, ce fut tout à fait contre-productif en raison du moyen utilisé pour financer cette politique.
M. Jean Mallot, coprésident : Comment faites-vous pour étudier, post AMM, un médicament sur les plans scientifique, médical et économique, afin d’orienter vos politiques de recherche, d’action économique, de prix, donc de promotion ?
M. Christophe Weber : Chaque organisation a sa propre méthodologie pour faire ses choix d’investissement recherche, mais ces choix se font dix ou quinze ans avant le lancement du médicament. Dans ces conditions, il y a énormément d’incertitudes : on ne sait pas quelle sera la thérapeutique, d’autres médicaments sont développés par d’autres laboratoires, et l’on sait que 85 % des essais échouent. Il faut donc prendre beaucoup de paris – le plus important est le progrès médical que l’on pense apporter avec ce médicament-là – d’autant que les systèmes de santé seront de plus en plus vigilants pour analyser le bénéfice thérapeutique des médicaments et définir des populations cibles.
C’est donc le progrès médical, ainsi que les probabilités de succès, qui guident les choix d’investissement. Cela relève de l’expertise du laboratoire. Bien sûr, le potentiel économique entre aussi en jeu, mais seulement en troisième position.
Pour confirmer ce progrès thérapeutique, par le biais des études post AMM, nous manquons cruellement, en France, de la possibilité de suivre les patients dans la vie réelle. Une base de données pourrait être mise en œuvre par la Caisse nationale d’assurance maladie des travailleurs salariés (CNAMTS) ou par d’autres. Cela nous permettrait de mesurer l’impact du médicament sur de larges populations, sur le système de santé et sur son évolution.
Aujourd’hui, pour faire des analyses en population réelle, nous devons nous tourner vers des bases de données existant aux États-Unis ou dans d’autres pays, parce qu’il est très difficile, en France, d’avoir cette capacité d’analyse. On n’y recueille pas suffisamment de données organisées.
Les études post AMM qu’on nous demande de faire sont beaucoup trop coûteuses, puisque nous devons carrément construire des cohortes pour suivre l’impact des médicaments. De nombreux débats ont eu lieu sur les études post AMM. Il semble qu’il y ait un blocage en France. Pourtant, tant que nous n’aurons pas une base de données solide, permettant de suivre des populations réelles dans le système de santé français, nous aurons énormément de mal à mesurer l’impact des médicaments sur la population traitée.
Mme Sabine Dandiguian : Il y a quelques années, on célébrait les décisions d’AMM. Maintenant, on célèbre les autorisations de remboursements. Aujourd’hui, le développement clinique doit intégrer la valeur ajoutée, d’abord pour le patient, pour le thérapeute, mais aussi pour le système de santé. La pression a changé. Aujourd’hui, quand on prévoit des études comparatives, on essaie de les faire par rapport au standard of care, à savoir le médicament qui est le plus prescrit et qui est reconnu comme une référence ; et l’on essaie de faire mieux que cette référence, d’où l’obligation de cibler les populations.
De plus en plus souvent, on proposera des médicaments qui apportent une valeur ajoutée sur une population restreinte. C’est ce que nous avons développé, s’agissant du sida. Aujourd’hui, les trithérapies existent, certes, mais le virus est très intelligent et continue à muter. Nous avons donc ciblé notre recherche sur tous les patients qui résistent à tout ce qui existe aujourd’hui. Ce n’est qu’ainsi que nous parvenons à avoir des remboursements à la hauteur des remboursements européens.
J’ai été très intéressée par certains propos de la précédente table ronde : pour transformer une molécule en médicament, il faut un milliard de dollars, sans être sûrs du succès. Ce sont des modèles économiques tout à fait différents.
Pour revenir sur ce que disait M. Christophe Weber, il est exact que je rêverais d’avoir accès, tout simplement, aux bases de données de la CNAMTS.
Pourquoi nous demande-t-on de faire des suivis post AMM ? Parce que les autorités de santé veulent voir ce que le médicament permettrait comme économies, en dehors de l’enveloppe médicaments : moins de jour de réhospitalisation, moins de rechutes, moins de durée d’hospitalisation. On ne peut pas se fonder sur de telles mesures aux États-Unis, parce que nous sommes dans un système de santé français. Par exemple, nous avons des médicaments contre la schizophrénie. Or la sectorisation psychiatrique est un modèle typiquement français. Il faut donc des mesures dans le système français. Je suis ainsi obligée de créer des cohortes spécifiques en accord avec les autorités, cohortes que je finance totalement – les dépenses ne sont pas déductibles, pour démontrer l’avantage perçu et consolider le remboursement. On pourrait peut-être simplifier, être plus transparents, travailler davantage ensemble en partenariat.
M. Jean-Christophe Tellier : Je pense que la segmentation entre le médical et l’économique est un élément essentiel. Comme le disait M. Christophe Weber, le point de départ est toujours médical. L’un des dangers dans la construction du système futur serait de vouloir trop tôt et trop complètement intriquer l’économique au médical. Selon moi, le point de départ de toute recherche publique biomédicale est de répondre à un besoin médical. La notion de temps est essentielle et le taux d’accrétion est de un pour mille, avec les coûts qui ont été évoqués.
Toutefois le point le plus important reste l’évaluation du médicament. Je prends un exemple : alors que, il y a dix ans, lorsque vous lanciez dans le monde un antihypertenseur, il était testé en gros sur 3 500 patients, nous avons déposé un dossier d’enregistrement sur une nouvelle classe thérapeutique, l’année dernière, avec 12 500 patients. Les exigences de développement se sont donc accrues et son coût devient important.
En outre, même si vous augmentez le nombre de patients et les sous populations sur lesquelles vous testez vos médicaments, notamment en pédiatrie et en gériatrie, vous ne serez jamais dans le contexte réel de prescription. Ce qui manque parfois en France par rapport aux autres pays, c’est une culture épidémiologique, qui fournirait des bases communes accessibles de données de santé publique, sur lesquelles nous pourrions, plus naturellement que nous ne le faisons aujourd’hui, inscrire nos plans de développement post AMM.
On nous demande des plans de gestion de risques, mais cela est relativement peu coordonné : la lisibilité et la clarté des demandes adressées à nos entreprises sur la gestion du risque une fois que nous avons une AMM peuvent être très différentes en fonction de nos interlocuteurs et le manque de coordination crée un délai additionnel. C’est ainsi que nous avons obtenu une AMM européenne, il y a quelque temps, et que nous attendons depuis deux mois la lettre écrite du plan de gestion des risques qui nous permettrait de commencer à travailler sur ce que nous mettrons en œuvre dans le suivi. Cependant, le problème principal tient au fait que les dépenses sont considérées par enveloppe. Il est très difficile de sortir un médicament de l’enveloppe médicament pur, et de démontrer à la collectivité l’avantage économique ou l’économie réalisée sur la prise en charge globale.
Il nous semblerait intéressant de développer dans l’avenir des réseaux épidémiologiques. Il faudrait pouvoir démontrer, plus qu’aujourd’hui, le gain économique que le médicament apporte dans la prise en charge thérapeutique globale. Si vous prenez, sur les cinquante dernières années, dans le domaine de la pathologie cardiovasculaire, le gain en morbimortalité que les traitements ont apportés, vous constatez que le taux de mortalité a été réduit de 80 % et le taux de morbidité de 50 %. Or vous n’avez aucun moyen de valoriser cela dans le système actuel.
M. Louis Couillard : Il faut sept à dix ans pour développer une molécule. La difficulté est d’anticiper le standard de prise en charge dans cette pathologie et les critères qui seront utilisés par les autorités pour évaluer cette amélioration. La prise en compte de ces bases de données nous permettrait d’améliorer l’observance thérapeutique et l’utilisation de nos produits. Chacun sait en effet que la principale cause d’échec thérapeutique est la non-observance dans l’utilisation de nos produits.
Mme Catherine Lemorton, rapporteure : Avez-vous pu comparer la vitesse de pénétration des nouvelles molécules dans les différents pays ? Avez-vous pu établir des tableaux comparatifs pour savoir quel temps met une molécule pour pénétrer de manière conséquente en France, en Italie, en Espagne, etc., dans les pays où vous intervenez ? Y a-t-il un rapport évident entre cette vitesse de pénétration et le système de prise en charge des pays, notamment celle des assurances de base ?
M. Christophe Weber : Le groupe LIR a réalisé une étude comparative des taux de pénétration. La conclusion qui en a été tirée est qu’il est quasiment impossible d’apporter une réponse ferme. En fait, il y a des spécificités très fortes en fonction des médicaments.
Par exemple, la France a un atout, l’ATU ou autorisation temporaire d’utilisation d’un médicament. Grâce à elle, des médicaments très innovants, souvent hospitaliers, peuvent être fournis aux patients qui en ont besoin. De cette manière, des patients sont traités plus tôt que dans d’autres pays. Sur ce plan, la France est très bien positionnée. En revanche, pour certaines molécules évaluées par la commission de la transparence, avec une ASMR IV ou V, les discussions qui s’instaurent sont très longues entre la commission de la transparence, le CEPS et le laboratoire. Dans ce cas, en moyenne, le médicament est mis à disposition en France un an et demi ou deux ans après des pays comme l’Angleterre, l’Allemagne, l’Italie ou l’Espagne. Cela dépend de la situation dans laquelle vous êtes.
J’ai pris deux exemples extrêmes : l’ATU qui permet un accès à l’innovation thérapeutique, et les difficultés dues au système existant en matière de négociation des prix, de remboursement, etc.
M. Jean-Christophe Tellier : Il est exact que les ATU et les procédures de dépôt de prix sur les ASMR I, II et III ont permis à la France de venir sur les niveaux de prix européens et sur des délais d’accès pour les patients à ces molécules assez proches de ce qui existe dans les autres pays.
Il s’agit en général de produits ou de pathologies pour lesquelles les conditions de consommation sont relativement bien alignées. On voit que, sur les AMM les plus récentes, sur les classes thérapeutiques les plus récentes, il y a moins d’écart entre le moment où les produits arrivent dans les différents pays. C’est probablement là où il y a le plus de convergence d’utilisation des produits dans les différents pays.
Mme Catherine Lemorton, rapporteure : Vous avez souligné le manque d’études épidémiologiques. Quel serait, selon vous, le modèle dont nous devrions nous rapprocher ?
M. Christophe Weber : On se rend compte qu’en France, il n’existe pas de gestionnaire de soins. La CNAMTS n’a pas un tel mandat, la HAS non plus. Pour qu’il y en ait un, il faut à la fois qu’il soit bien identifié et qu’il puisse couvrir l’ensemble des soins d’une pathologie donnée.
Les bases de données les plus robustes, actuellement, nous sont données par les Health maintenance organizations (HMO) américaines. Il s’agit d’organisations intégrées de management des soins au États-Unis, qui traitent les patients de la médecine libérale à l’hôpital et ont souvent des cohortes très puissantes pour analyser l’effet des médicaments.
M. Jean Mallot, coprésident : Ces bases sont-elles gérées de façon partenariale ?
M. Christophe Weber : Non, ce sont en règle générale des organisations privées. Néanmoins, l’important est que ce sont des gestionnaires de soins. Il faudra qu’un jour, en France, on crée un gestionnaire de soins.
Mme Sabine Dandiguian : L’Union nationale des caisses d’assurance maladie (UNCAM) possède beaucoup de données sur les affections de longue durée (ALD) et l’on peut faire le lien entre le diagnostic et la prescription. Cela est déjà très intéressant. Il faudrait pouvoir aller au-delà des seules données concernant le médicament, pour disposer de données concernant tous les frais pris en charge et obtenir un coût par patient. Le système américain est tellement axé sur la rentabilité qu’il intègre même la prévention. Toutefois si nous avions au moins accès aux données relatives aux ALD, qui sont assez claires parce qu’elles intègrent le diagnostic, le traitement et le nombre de consultations, ce serait intéressant. Or ces données existent en France.
M. Louis Couillard : La France dispose d’un outil phénoménal avec cette base de données de la CNAMTS, mais on ne sait pas l’exploiter. Pour des raisons qui semblent parfois dogmatiques, on ne se donne pas les moyens d’exploiter des gisements de productivité et d’efficacité.
Mme Sabine Dandiguian : Je ne crois pas qu’il s’agisse de raisons dogmatiques. Les données existent, mais encore faut-il organiser l’information, savoir qui y a accès et comment. Ce n’est pas facile techniquement. En tout cas s’il y avait un jour une vraie volonté politique d’y aller, au moins disposerions-nous immédiatement de données objectives existant sur le territoire national.
M. Jean-Christophe Tellier : Culturellement, on cherche dans l’analyse des données ce qu’on veut y trouver. En fonction de la position que vous avez sur l’échiquier, vous sortez les données qui vous intéressent et, à la fin de la discussion, si ces données vont dans le sens que vous êtes censé défendre, vous les prenez, sinon vous ne les utilisez pas !
C’est là où la France pêche culturellement. Elle a plutôt une histoire anatomo-clinique que statistique. La capacité à mettre en commun des informations, à les analyser et à en sortir des données est relativement récente.
Mme Catherine Lemorton, rapporteure : Cela ne vient-il pas aussi du cloisonnement des professionnels de santé ? Je pense notamment aux médecins, avec la médecine de ville et la médecine hospitalière. Ce cloisonnement n’existe peut-être pas dans les autres pays. Sans compter leur résistance à rendre des comptes sur l’efficacité d’un acte médical. Ne pensez-vous pas que cela participe aux lacunes de l’épidémiologie ?
Mme Sabine Dandiguian : La médecine est une vocation et il est très difficile aux médecins de rendre des comptes, surtout économiques, sur leurs prescriptions. N’oublions pas non plus que la médecine n’est pas une science exacte. S’il suffisait d’avoir des ordinateurs pour établir la prescription idéale, cela ne donnerait pas autant de soucis au corps médical. Il y a, en tout cas, une vraie résistance de la part de celui-ci, en France peut-être plus qu’ailleurs. Un médecin ne gagne pas plus selon que la prescription médicale porte sur deux ou quatre médicaments ; c’est l’intuition médicale qui lui a fait penser que son choix était le meilleur pour son patient.
Il y a également un manque de transparence entre le monde hospitalier et la ville. On a parlé du dossier médical. Pourquoi n’arriverait-on pas à avoir une vraie continuité des soins ? Aujourd’hui, le parcours de soins a redonné un certain sens et il a été finalement été bien accepté. Il faut aller plus loin.
Cela étant, je suis assez d’accord : les médecins n’acceptent pas forcément ce genre de discours.
M. Jean-Christophe Tellier : Une bonne partie de l’effet de croissance du médicament de ville est liée en fait à des transferts de l’hôpital vers la ville, sur des prescriptions de médicaments qui sont normalement hospitalières. À partir du moment où l’on joue sur les enveloppes pour contrôler les coûts d’un côté et mettre une croissance artificielle de l’autre, on perd la vision globale qui permet de prendre des décisions d’allocation de ressources ayant du sens par rapport à la santé publique.
Je veux également revenir sur la séparation du médical et de l’économique qui a été évoquée au début de cette table ronde.
Historiquement, la France a défendu depuis toujours la position suivante : indépendance médicale, responsabilité du médecin dans l’acte de prescription et de diagnostic. C’est un élément fondamental et l’arrivée de l’économie est souvent perçue par le monde médical comme une limitation et une contrainte à sa liberté de prescription.
Il ne faut pas trop rapprocher le médical et l’économique. Souvent, la contrainte ou le cadre médical n’est présenté que sous l’aspect économique. Le bon usage du médicament, la conservation de la liberté du médecin dans le cadre de ses prescriptions sont essentielles, mais cela n’est pas antinomique avec la volonté d’avoir un gestionnaire de soins qui donne des faits, qui partage ses informations et qui permet de progresser.
M. Louis Couillard : Une partie du problème provient de la question initiale posée. Les intervenants du domaine de la santé se positionnent différemment selon que l’objectif prioritaire est de réduire les coûts, ou bien de maintenir ou d’améliorer la qualité des soins.
Mme Catherine Lemorton, rapporteure : Que pensez-vous de la nouvelle compétence médico-économique que la loi de financement de la sécurité sociale pour 2008 donne à la HAS ?
M. Jean-Christophe Tellier : L’évaluation médico-économique est nécessaire, d’autant qu’elle s’inscrit dans le cadre réel de la prescription, plutôt post que pré AMM.
Par ailleurs, il est souhaitable que l’on développe l’épidémiologie. Pour ce faire, il faudra avoir des bases et des structures dont nous ne disposons pas forcément aujourd’hui.
Enfin, je reste un fervent adepte de la séparation des pouvoirs. Je pense qu’une AMM qui prend en compte le bénéfice et le risque sur le simple et unique fait médical résultant des études cliniques, une commission de la transparence qui définit un service médical rendu et un comité économique qui fixe un prix, garantissent que l’économique et le bénéfice collectif ne prendront pas le pas sur l’intérêt individuel du patient. Je serais moins en confiance si l’on mettait en place, demain, un système d’évaluation médico-économique en amont, qui justifierait des choix sur de seules bases économiques et ferait perdre des chances aux patients.
M. Jean Mallot, coprésident : Est-ce un point de vue partagé ?
Mme Sabine Dandiguian : Absolument ! Le système français, au niveau européen, est assez exemplaire grâce à la transparence des travaux du Comité économique des produits de santé, qui ne fonctionne pas mal : des experts médecins traitent de la valeur ajoutée médicale du médicament. La prise en compte de critères médico-économiques est normale dans le monde où nous vivons. Évaluer le vrai apport médico-économique et sociétal d’un médicament après sa mise sur le marché dans le système de santé tel qu’il est, est légitime.
Cependant j’ai des craintes quand je constate le pouvoir donné aux caisses d’organiser des arrangements médico-économiques individuels avec les médecins.
On voit ce qui se passe dans d’autres pays, en particulier en Angleterre, où l’on vient de vivre un cas dramatique de refus de remboursement de certains de nos médicaments. Lorsque le médico- économique prime, on en vient à parler de la valeur de la vie d’un patient âgé par rapport à celle de la vie d’un patient actif. En Angleterre, quand vous avez plus de soixante-quinze ans, vous avez du mal à être dialysé ; quand vous avez un cancer et que vous n’avez pas arrêté de fumer, vous n’avez plus la deuxième chimiothérapie : cela coûte trop cher à la société, vous n’avez pas consenti les efforts nécessaires. Un jugement moral est donc porté à cet égard.
Beaucoup de choses ont été mises en place par le législateur. Des précautions ont été prises, mais une brèche est ouverte. Je parle plutôt en tant que citoyenne. Vous êtes les élus du peuple et vous devez faire attention. Que les médecins en France ne soient pas obsédés par l’économique est un des points fort du système français, qui reste généreux. Quand on est très âgé, on a exactement les mêmes droits et le même accès aux médicaments. Nous avons des médicaments qui concernent les cancers de patients extrêmement âgés et nous rencontrons des difficultés en Angleterre parce que la vie d’un patient de plus de soixante-quinze ans n’a pas la même valeur que celle d’un patient actif. Ce sont des débats auxquels je me refuse absolument. Faites donc bien attention, en tant que législateurs, à mettre des barrières et à contrôler ce type de brèche.
Encore une fois, je parle en tant que citoyenne, parce que, en tant que laboratoire, des études médico-économiques nous sont demandées, et nous les ferons. En revanche, j’ai été assez inquiète de voir que des caisses locales de sécurité sociale commençaient à dire aux médecins que, pour avoir une prime à la fin de l’année, il ne fallait pas dépasser l’enveloppe fixée. Cela signifie qu’ils essaieront de limiter la prise en charge de patients atteints du VIH ou des patients très âgés en ALD.
M. Jean Mallot, coprésident : On a beaucoup parlé du circuit de mise au point, de la distribution du médicament, de sa surveillance et de son suivi. Cependant, il existe d’autres moyens de promotion et de commercialisation du médicament, via Internet notamment. Qu’en pensez-vous ? Comment réagissez-vous ? Il y a des risques bien connus : mauvaise utilisation, contrefaçon, perte de contrôle du système. Comment êtes-vous organisés pour lutter contre ces problèmes ?
M. Louis Couillard : Internet est une porte d’entrée à la contrefaçon et peut poser un problème important de santé publique. En tant que représentant de Pfizer, je peux vous dire que l’un de nos médicaments a probablement été le plus sujet à la contrefaçon et à la distribution dans les canaux alternatifs de distribution.
En France, nous n’avons pas rencontré ce problème de façon marquée parce que les réseaux de distribution sont bien hermétiques. En revanche nous avons eu des soucis importants en Angleterre au cours de la dernière année, avec plusieurs exemples de thérapies pour des problèmes chroniques, un fort volume de contrefaçon ayant été introduit dans les réseaux de distribution.
M. Jean Mallot, coprésident : Quelles solutions proposer pour répondre à de tels problèmes ?
M. Christophe Weber : Même aux États-Unis, la délivrance du médicament par Internet n’a pas pris une part très importante du marché, en tout cas moins importante qu’on ne l’avait pensé il y a dix ans : aujourd’hui, elle est de l’ordre de 20 %. Finalement, les patients aiment bien aller dans leurs officines pour chercher leurs médicaments. Néanmoins, Internet répond à une demande précise de certains patients ; Je pense donc que ce serait une erreur de fermer complètement cette option Internet. Il convient simplement de trouver un moyen pour la maîtriser. Ne pourrait-on pas autoriser les pharmacies à utiliser Internet pour envoyer des médicaments aux patients qu’ils connaissent, par exemple pour les renouvellements ?
Certes, Internet dématérialise la chaîne de production, ce qui présente des risques de contrefaçon. On peut donc interdire, mais on peut aussi décider de construire un système Internet maîtrisé, qui repose sur la chaîne française actuelle qui est malgré tout très solide par rapport à la contrefaçon.
Mme Sabine Dandiguian : S’agissant de la contrefaçon et de la distribution, nous sommes assez protégés. Parlons cependant de la communication aux patients.
Aujourd’hui, les patients vont sur Internet pour avoir des informations sur nos médicaments, informations qui ne sont absolument pas contrôlées. Je pose la question au législateur : comment encadrer l’industrie pharmaceutique pour l’autoriser à communiquer avec les patients, en particulier sur la problématique de l’observance ? Aujourd’hui, il y a des peurs et des stigmatisations. Je suis prête à prendre le pari de faire une expérimentation dans un domaine très particulier, celui de la santé mentale. Si l’on n’aide pas les patients et les familles à bien observer les traitements on n’en sortira pas, aussi géniales que puissent être nos molécules.
Aujourd’hui, sur Internet, il y a un accès non contrôlé et une communication sauvage sur les maladies et les médicaments. Comment pourrait-on faire pour que nous soyons un peu plus libres de communiquer ? On pourrait, ensemble, mettre en place des programmes, y compris sur Internet, pour améliorer l’observance des médicaments.
Actuellement, il n’y a aucune confiance entre nous, les acteurs, les autorités et les laboratoires pour mettre en place des programmes. Internet vit sa vie et je demande à mes équipes de ne pas trop s’en occuper, car c’est très compliqué. Clarifions la législation, menons des expérimentations sur certaines pathologies extrêmement graves – pour moi, ce serait la santé mentale – et voyons s’il est possible de ne pas utiliser cet outil. Le monde de demain se fera avec Internet, avec une information 24 heures sur 24 dans le monde entier. Le monde de demain devra intégrer complètement Internet parmi les outils de communication.
Je ne pense pas qu’en France on utilise au mieux ce qui pourrait être une opportunité, probablement pas dans la distribution, en ce qui me concerne, mais dans la communication vis-à-vis des patients et de leurs familles.
M. Louis Couillard : Je suis d’accord sur l’utilisation d’Internet en tant qu’instrument de communication, mais pas pour la distribution.
M. Christophe Weber : On est tous d’accord.
Mme Catherine Lemorton, rapporteure : Toutes vos interventions depuis une heure sont extrêmement pertinentes, mais j’ai entendu parler du « modèle » américain.
M. Christophe Weber : Il a bien des défauts.
Mme Catherine Lemorton, rapporteure : Néanmoins, il permet de réunir les conditions pour mieux suivre la vie réelle du médicament au jour le jour. Malgré tout cela, il reste quand même 50 millions d’Américains sans couverture sociale.
M. Christophe Weber : Nous sommes bien d’accord.
Mme Catherine Lemorton, rapporteure : Êtes-vous d’accord pour dire qu’il faut maintenir une prise en charge la plus large possible et que le législateur doit être très vigilant sur ce point ?
M. Christophe Weber : Le choix de solidarité de la France est très respectueux de l’intérêt du patient.
Mme Sabine Dandiguian : Le système américain n’est pas un système exemplaire, bien au contraire. Nous parlions du suivi épidémiologique, qui est une grande tradition dans la culture américaine. Le calcul du risque cardio-vasculaire par Framingham, par exemple, a beaucoup apporté. Cependant, en dehors de cela, je suis tout à fait d’accord avec vous. Le système français a une certaine générosité dont nous devons nous enorgueillir. Ce n’est pas un hasard si tant d’Anglais viennent se faire soigner en France.
Il reste encore beaucoup à faire en termes d’observance, car le médicament n’est qu’une réponse partielle à une maladie. J’ai l’impression qu’on n’accueille pas suffisamment les industriels du médicament autour de la table pour réfléchir à la mise en place de prises en charge globales des pathologies. On les exclurait plutôt. Or nous ne pourrons vaincre certains fléaux que tous ensemble.
Le médicament est très important, mais, je le répète ce n’est qu’une réponse partielle. Il doit être correctement accompagné. Pour notre part, nous parlons de schizophrénie, de patients qui rechutent et qui deviennent des serial killer ou des SDF. Comment faire si le médicament n’est pas pris correctement et que les familles ne sont pas associées à la prise en charge ? Mettons nous tous ensemble autour de la table et expérimentons.
M. Jean-Christophe Tellier : Souvent, on pense que la sévérité de la maladie est en corrélation directe avec l’observance. Mais ce n’est pas si évident. Nous avons eu le bonheur de produire un produit actif dans une maladie qui était 100 % mortelle, la leucémie myéloïde chronique. On pouvait imaginer qu’avec un tel médicament qui permettait aux patients de vivre, un produit qui n’a pas de problèmes particuliers et qui n’est pas contraignant, l’observance serait au maximum. Or, on s’est aperçu qu’on avait 60 à 80 % d’observance seulement.
J’ai pris cet exemple parce qu’il montre que laisser le patient trop en dehors de la prise en charge d’une maladie qui est la sienne conduit forcément, quel que soit le bien fondé de ce qui est fait autour, à des échecs au moins partiels. Quand vous savez que vous contrôlez de façon satisfaisante un diabétique sur deux et un hypertendu sur trois, vous mesurez les progrès qu’il reste à accomplir, même si les chiffres sont peut-être un peu gonflés.
Responsabiliser le patient passe forcément par un accès à l’information et par l’implication du patient et de son entourage dans la chaîne des soins. Nous laisser en dehors de cette problématique, c’est perdre une opportunité. Nous ne connaissons pas trop mal nos médicaments et l’on peut imaginer que notre intérêt est que les patients soient bien traités. Je n’ai aucun intérêt, de mon côté, à ce que des patients soient traités alors qu’ils n’en ont pas besoin, ou qu’ils ne prennent pas les médicaments dont ils ont besoin.
M. Jean Mallot, coprésident : Merci de votre participation fort intéressante et je renouvelle ma demande : si vous souhaitez compléter la séance de ce matin par des contributions, quelles qu’elles soient, n’hésitez pas à nous les faire parvenir.
*
Audition de Mme Annie Podeur, directrice de l'hospitalisation et de l'organisation des soins au ministère de la santé, de la jeunesse et des sports, M. Bernard Ortolan, président du conseil national de la formation médicale continue des médecins libéraux, M. Alain Beaupin, président du conseil national de la formation médicale continue des médecins salariés, et M. Dominique Bertrand, président du conseil national de la formation médicale continue des médecins hospitaliers.
M. Jean Mallot, coprésident : Madame, messieurs, je vous souhaite la bienvenue à l’Assemblée nationale. Peut-être pourriez-vous commencer par quelques observations générales sur la formation médicale continue et sur le bon usage du médicament.
Mme Annie Podeur : La direction de l’hospitalisation et de l'organisation des soins (DHOS) a deux missions essentielles qui sont ses deux lignes stratégiques. En tant qu’administration centrale elle mène une action de pilotage du système de santé qui porte sur l’ensemble de l’organisation des soins et pas seulement sur les soins hospitaliers. En ce moment, la DHOS s’investit particulièrement dans l’organisation des soins de premier recours, des soins primaires, des soins ambulatoires, notamment au travers des États généraux de l’organisation de la santé (EGOS) que la ministre m’a chargée de copiloter.
Toujours au sein du pilotage, le deuxième axe majeur concerne les professions médicales et paramédicales. Depuis septembre 2007, à la faveur d’une clarification des compétences entre la direction générale de la santé (DGS) et la DHOS, cette dernière est désormais en charge de l’ensemble du pilotage des professions c’est-à-dire de la vision démographique, de la formation initiale et continue, et non plus seulement de la gestion des personnels hospitaliers, dont une partie qui concerne les praticiens hospitaliers (PH) a été confiée au centre national de gestion. Il lui reste également, en coopération avec l’éducation nationale, l’enseignement supérieur et la recherche, la gestion des effectifs hospitalo-universitaires.
Notre deuxième grande orientation stratégique a trait à l’efficience des établissements de santé, qui passe, d’une part, par l’animation d’une politique de la fonction publique hospitalière, donc par une modernisation, d’autre part, par la gestion de la réforme du financement des établissements, notamment par la mise en œuvre de la fameuse tarification à l’activité (T2A). Je rappelle d’ailleurs que celle-ci n’est pas exclusive et que demeure à ses côtés un financement sous une forme forfaitaire notamment en ce qui concerne les médicaments, les molécules innovantes étant remboursées à l’euro/l’euro sur une liste dite « en sus ». La DHOS porte aussi un regard sur l’optimisation du fonctionnement interne des établissements, en termes d’efficience, ce qui signifie aussi qu’elle s’intéresse à la performance globale, c’est-à-dire à la qualité des soins. C’est bien là que l’on rejoint votre préoccupation quant à la politique du médicament.
En effet, l’amélioration de la prise en charge des patients et la recherche de l’efficience sont au cœur de notre politique, tout particulièrement en ce qui concerne le médicament. Je me propose donc d’aborder devant vous notre rôle dans la formation initiale et continue, de l’illustrer par l’exemple de la démarche intégrée formation initiale -formation continue - évaluation des pratiques professionnelles (EPP) en ce qui concerne les antibiotiques, enfin de vous dire un mot quant à notre politique en faveur du bon usage du médicament dans les établissements hospitaliers.
La formation des médecins est effectivement un élément déterminant de l’amélioration de l’usage du médicament dans le système de santé. Nos attributions en la matière sont récentes. Pour autant, dès ma prise de fonctions, nous avons commencé à travailler dans le champ de la formation médicale continue, avec les trois conseils nationaux qui sont ici représentés par leurs présidents.
C’est en revanche le ministère de la santé qui gère, en relation avec le ministère de l’enseignement supérieur et de la recherche, la formation initiale des médecins. Les maquettes de formation ont évolué et il est d’usage de souligner que la part dévolue à la formation en pharmacologie y a diminué. Mais le programme de la deuxième partie du deuxième cycle des études médicales comporte un certain nombre d’items qui permettent d’envisager de façon novatrice l’initiation à la politique du médicament et, surtout, de préparer le bon usage. On trouve ainsi un item sur la protection sociale, la consommation médicale et l’économie de la santé. Il est en effet indispensable que les jeunes médecins aient une vision des grands enjeux médico-économiques du système de santé, au-delà de la seule pharmacologie.
D’autres items touchent plus précisément à leur pratique quotidienne :
– les thérapeutiques antalgiques médicamenteuses et non médicamenteuses ;
– les thérapeutiques médicamenteuses et non médicamenteuses, vues dans le cadre réglementaire de la prescription, et les recommandations qui doivent être formulées par les praticiens ;
– la prescription et la surveillance des antibiotiques ;
– la prescription et la surveillance des anti-inflammatoires ;
– la prescription et la surveillance des anti-thrombotiques ;
– la prescription et la surveillance des psychotropes ;
– la iatrogénie qui est également une problématique importante, surtout lorsque la population vieillit et que les prescriptions s’alourdissent ;
– le diagnostic et la prévention car le métier du généraliste est bien le diagnostic et il faut qu’il puisse le poser sans avoir automatiquement recours à une panoplie de médicaments.
C’est pourquoi je juge essentiel d’associer théorie et pratique dans la formation médicale initiale, en développant un enseignement véritablement intégré du médicament. Il n’y a pas d’un côté des enseignements théoriques en pharmacologie, d’un deuxième côté l’apprentissage des grandes lignes des grands équilibres médico-économiques et de la place des différents acteurs, d’un troisième côté la pratique. De même que nous disposons à l’hôpital de lieux pour former les jeunes médecins, dès lors que l’on développe la filière universitaire en médecine générale, il est important que les maîtres de stage se préoccupent de la formation pratique à la bonne prescription du médicament.
M. Pierre Morange, coprésident : Nous avons bien compris qu’il ne convenait pas de se concentrer uniquement sur la pharmacologie et qu’il fallait avoir une approche transversale qui balaie les différents champs de la stratégie thérapeutique. Mais quel volume horaire d’enseignement jugeriez-vous utile d’y affecter en formation initiale comme en formation continue ?
Mme Annie Podeur : Je souhaite travailler avec la Haute Autorité de santé (HAS) et avec l’enseignement supérieur et la recherche sur les maquettes de formation, afin de préciser les volumes horaires et l’organisation des enseignements. Cependant, je n’ai pris mes fonctions qu’en septembre 2007 et tout cela n’est pas encore défini. Je puis toutefois vous indiquer que je souhaite qu’un accent tout particulier soit mis sur la formation du médecin généraliste car nous développons une filière universitaire et parce qu’il s’agit bien d’un enjeu majeur, les généralistes étant, au quotidien, les premiers prescripteurs auprès des patients.
S’agissant de la formation médicale continue, le dispositif qui a été prévu par l’article 59 de la loi du 4 mars 2002 relative aux droits des malades et à la qualité du système de santé sera effectif en 2008, les derniers arbitrages étant actuellement en cours. Nous souhaitons qu’il soit très lisible et très accessible pour les médecins. Les obligations de formation pour les médecins devront être respectées, dans un délai de cinq ans. À l’issue de cette période, la formation médicale continue devra comporter, notamment dans le champ du médicament, des objectifs dont on pourra vérifier qu’ils ont ou non été atteints. C’est tout l’intérêt de ce système.
La question de la place de l’industrie pharmaceutique dans la formation médicale continue a été fréquemment posée. Il faut aujourd’hui dépasser les questions de principe et s’assurer que, dès lors que des fonds sont alloués par l’industrie pharmaceutique, ils le sont en toute transparence et dans le respect de l’indépendance des professions médicales. C’est la raison pour laquelle la signature d’un code de bonnes pratiques entre le LEEM – les entreprises du médicament – et l’État va dans le bon sens, puisque le premier s’engage à garantir aux organismes de formation continue qu’il finance une indépendance scientifique et pédagogique. Certes, la Cour des comptes a relevé qu’il ne s’agissait pas d’un système contraignant, mais si l’on se donne les moyens de l’évaluation – et le rôle de la DHOS est bien de s’assurer que cette charte est scrupuleusement respectée – on aura déjà beaucoup avancé et un contenu aura été donné à la formation médicale continue.
Compte tenu du poids de la visite médicale, y compris à l’hôpital, je souhaite que ce dernier puisse également disposer d’une charte de la visite, sur le modèle de celle qui a été signée en juillet 2006, pour la médecine de ville, entre le LEEM et le CEPS, le comité économique des produits de santé. C’est un moyen d’aller vers une bonne prescription, économe des deniers publics.
Au vu du nombre croissant de malades chroniques et du vieillissement de la population, il est également souhaitable de prendre véritablement en compte la iatrogénie, à l’hôpital comme en ville. Cela signifie que la formation médicale continue est indissociable de l’évaluation des pratiques professionnelles. Nous devons d’ailleurs faire comprendre aux médecins généralistes que l’EPP s’inscrit bien dans l’obligation de formation et que l’on tiendra compte de leur capacité à s’engager dans une démarche d’évaluation de leurs pratiques. Cela devrait permettre des avancées considérables.
J’en viens à l’exemple des antibiotiques.
Un accord-cadre sur l’amélioration des pratiques hospitalières a été signé entre l’État, l’Union nationale des caisses d’assurance maladie, l’UNCAM, et les représentants des établissements afin d’améliorer la qualité des prescriptions en établissements de santé et de préserver leur efficacité. Parce qu’il peut être audité, l’effort qui a été demandé a été assorti d’une formule d’intéressement. Sur la base de cet accord-cadre national signé en 2006, on a signé 391 accords locaux, ce qui est loin d’être négligeable sur un total d’environ un millier de gros établissements.
On a aussi mobilisé les prescripteurs, avec le concours des sociétés savantes et des conférences médicales, qui ont largement contribué à la préparation de l’accord et qui se sont ensuite chargées de l’expliquer dans les établissements. Cela montre toute l’importance de s’appuyer sur les professionnels.
Nous avons également travaillé en étroite coopération avec la HAS afin que la démarche liée à la bonne utilisation et à la bonne prescription des antibiotiques puisse être une possibilité d’EPP. Le Conseil national de la formation médicale continue hospitalière a également été sensibilisé et, parce que nous avons toujours le souci de l’évaluation, nous avons diffusé un indicateur qui reflète l’usage des antibiotiques au sein des établissements. Il faut maintenant que les informations remontent pour que nous puissions en tirer des conclusions, mais nous souhaitons généraliser cet indicateur en tant qu’indicateur de performance hospitalière qui sera, à terme, rendu public.
M. Pierre Morange, coprésident : Avez-vous les moyens concrets de mettre en œuvre, sur le terrain, ces nouvelles missions et ces nouvelles ambitions ?
Mme Annie Podeur : Par moyens, on entend surtout moyens financiers, mais je crois que les ressorts sont avant tout managériaux et qu’il faut mettre l’accent sur la diffusion des démarches contractuelles.
Votre question me permet d’en venir à la politique contractuelle que mène la DHOS dans les établissements en ce qui concerne le médicament.
Dans tous les établissements de santé, une commission du médicament joue un rôle extrêmement important et réunit prescripteurs et pharmaciens. Regrouper de la sorte l’ensemble des acteurs autour des thèmes de la prescription et de l’usage du médicament est déjà un atout. Nous avons créé à l’échelon régional des commissions similaires, qui ont une vision générale sur le fonctionnement des commissions dans les établissements ainsi que sur la bonne utilisation des molécules onéreuses, qui représentent, en y incluant les dispositifs médicaux, un volume global de 3,6 milliards d’euros, pour lesquelles la progression de la consommation est très supérieure à l’évolution de l’Ondam, l’objectif national des dépenses de l'assurance maladie, et qui exigent donc un pilotage important.
Entre les deux commissions, le lien se fait sous la forme d’un contrat du bon usage du médicament. Cela permet d’aller plus loin que par le seul pilotage de la liste des molécules innovantes remboursées à l’euro/l’euro ; de travailler à l’articulation avec l’accord-cadre et avec le contrat local pour les antibiotiques ; de développer la prescription en dénomination commune internationale (DCI), ce qui est sans doute la clé d’une bonne utilisation des génériques là où ils présentent le plus grand intérêt, c’est-à-dire en ville ; de mener une réflexion globale sur la bonne prescription afin d’éviter les complications et la iatrogénie liées à un mauvais usage du médicament.
M. Pierre Morange, coprésident : Où en est-on de la contractualisation régionale entre les structures hospitalières et l’assurance maladie, qui avait été prévue par des projets de loi de financement de la sécurité sociale (PLFSS) antérieurs ?
Mme Annie Podeur : Pour les antibiotiques un accord-cadre est décliné en accords locaux. Le contrat de bon usage du médicament était également prévu dans le cadre de la réforme du financement. Les contrats pluriannuels d’objectifs et de moyens entre chaque établissement de santé et l’Agence régionale de l’hospitalisation (ARH) ont été généralisés. Ils sont pilotés par la DHOS, en liaison étroite avec l’UNCAM, et ils prévoient des objectifs et des revues annuelles systématiques. Il s’agit incontestablement d’un moyen de pilotage important.
Beaucoup d’ARH ont signé avant le 31 mars 2007 des contrats centrés sur les volumes d’activité et les grandes orientations stratégiques. Les directeurs ont jusqu’au 30 juin 2008 pour compléter le dispositif.
Mme Catherine Lemorton, rapporteure : Les présidents des conseils nationaux pourraient-ils nous expliquer pourquoi il a été prévu trois secteurs de formation continue alors que l’on peut penser que les médecins manipulent tous les mêmes médicaments ?
M. Dominique Bertrand : Nous sommes entrés en fonction en 2004 et le dispositif sera opérationnel cette année. Ce délai peut paraître long, mais un grand nombre de textes réglementaires étaient nécessaires et nous attendons d’ailleurs la publication du dernier décret pour que la formation médicale obligatoire soit véritablement lancée.
Si cette formation est obligatoire depuis toujours d’un point de vue déontologique, il y a longtemps que l’on essaie de la faire entrer dans les faits : des expériences avortées ont été menées en 1992-1993, ainsi qu’en 1996, avant qu’on ne lance le dispositif en 2002, puis qu’on ne le complète en 2003 et en 2004.
L’idée est de poser le principe que tout le monde peut et doit se former. L’exigence ne sera pas trop forte au début, mais l’objectif est bien que tous les médecins soient excellents et que tous soient aptes à la prescription et à la prise en charge efficace et exigeante des soins, tout en restant en concordance avec la science car, contrairement à une idée reçue, l’obsolescence des connaissances médicales est rapide. Il faut donc engager très rapidement tous les médecins dans ce processus : ils devront satisfaire à l’obligation de formation, dans le délai de cinq ans, à partir de 2008. Par la suite, le système évoluera pour chaque période de cinq ans, sans doute avec une fermeté plus grande, afin de répondre aux grandes dérives constatées et de remédier aux prescriptions inadéquates. C’est à ce titre qu’il y aura sans doute un jour un lien avec l’assurance maladie. Toutefois, pour l’instant, il est impératif de lancer sans retard le processus.
Bien que notre métier soit unique, ce sont les différences entre nos pratiques qui justifient l’existence des trois conseils. Dès l’origine, il a été prévu de procéder de la sorte. Pour autant, nous travaillons ensemble au sein d’une coordination que chacun d’entre nous préside à tour de rôle, pour un an. Notre entente est parfaite et notre conduite identique, d’autant que le praticien hospitalier peut devenir médecin libéral et qu’un médecin libéral comme un médecin salarié peuvent devenir praticien hospitalier.
Les différences entre les pratiques s’expriment par exemple dans le temps disponible pour la formation : les praticiens hospitaliers disposent de 15 jours sur leur temps de travail, ce qui n’est pas le cas des libéraux, tandis que, pour les salariés, les situations varient en fonction des conventions. Un de nos objectifs est donc d’uniformiser les règles.
M. Jean Mallot, coprésident : Existe-t-il des actions de formation qui mêlent deux catégories de médecins, voire les trois ?
M. Dominique Bertrand : C’est l’une de nos préoccupations essentielles et notre bonne entente nous a permis d’aller dans ce sens, ce qui n’était pas prévu à l’origine. Une de nos missions consiste à faire en sorte que l’on dispose d’organismes de formation agréés. Dans ce cadre, nous nous efforçons qu’il soit parfois possible de trouver dans la même session de formation des libéraux et des salariés. Nous veillons également à la représentation de la diversité des pratiques au sein des conseils d’administration des organismes.
M. Alain Beaupin : Depuis 2002, nous avons pour mission de fixer les orientations nationales de la formation médicale continue, d’agréer les organismes formateurs et de fixer les règles que doivent suivre les médecins.
Nous nous sommes bien fixé pour règle que les formations dispensées par les organismes agréés puissent s’adresser à des médecins de différentes catégories : avec un seul cahier des charges et un seul dossier, l’organisme peut postuler aux trois agréments et c’est assez fréquemment le cas.
On sait qu’aujourd’hui une part importante du financement de la formation médicale continue provient de l’industrie pharmaceutique. Or, à l’occasion de l’agrément, il nous est demandé de veiller à la transparence du financement de la formation.
M. Jean Mallot, coprésident : Qu’entendez-vous précisément par « une part importante » ?
M. Alain Beaupin : Nous sommes dans l’incapacité de vous donner des chiffres, mais le rapport de l’Inspection générale des affaires sociales (IGAS) fournit des ordres de grandeur. La campagne d’agrément ayant commencé début 2007 nous ne disposons pas encore des rapports des organismes que nous agréons.
Parmi les 159 établissements qui ont été agréés à ce jour, 20 % ont fait l’objet d’une observation en vue d’améliorer la qualité de leur financement ; 18 % ont reçu une observation au titre de leur politique de gestion des conflits d’intérêts ; 10 % ont eu une observation dans les deux registres. Près de la moitié des organismes ont donc fait l’objet d’une recommandation les incitant à améliorer encore leur attitude dans ce domaine. Quant aux 52 organismes ayant essuyé un refus, celui-ci a été motivé dans 73 % des cas par un problème de conflit d’intérêts ou de manque de transparence du financement.
Tout cela montre que nous nous sommes donné les moyens d’identifier les difficultés et de faire progresser la qualité des organismes.
S’il y a trois conseils, c’est bien sûr parce qu’il y a trois modes d’exercice. Pour ma part, je suis médecin généraliste salarié en centre de santé, mais je représente aussi les médecins de santé scolaire, les médecins du travail, les médecins de la protection maternelle et infantile et les médecins conseil. Or, il me semble que nous avons une voix à faire entendre dans la réflexion sur la consommation de médicaments. En effet, nous ne saurions oublier qu’il ne convient pas d’apporter à tout problème médical une réponse par un médicament et que, si l’on pense bien sûr à la prévention, il est également pertinent d’évoquer les conditions de travail et les conditions sociales.
M. Bernard Ortolan : La loi a effectivement prévu trois conseils. Le travail que nous effectuons ensemble tous les trois depuis quatre ans est, sans doute, exemplaire. Notre volonté sans faille de nous entendre sur un discours qui soit lisible par tous les médecins, quel que soit le statut de leur exercice, et qu’ils puissent s’approprier, traduit une écoute mutuelle et un décloisonnement entre les trois modes d’exercice qui n’a guère de précédent dans l’histoire de la médecine. Cela n’était pas évident et il serait préjudiciable pour la lisibilité du dispositif par l’ensemble des médecins de casser cette dynamique en créant un seul conseil. Il serait en outre hautement souhaitable que cette entente survive aux titulaires actuels des postes.
Mme Annie Podeur a parfaitement résumé les besoins que nous avons identifiés depuis longtemps : il y a maintenant un bon moment que dans la formation médicale continue nous travaillons à l’amélioration des pratiques, à la meilleure connaissance et à la diffusion des référentiels, à la vérification de l’impact des formations sur les comportements. Ce travail n’est pas axé sur le médicament et je dirais même que, la réflexion à ce propos étant polluée par la question du financement, on en parle le moins possible.
D’ailleurs, si l’objectif est de réduire les prescriptions, ce n’est à l’évidence pas seulement celui de la formation médicale continue. Cette dernière vise en effet à améliorer les pratiques, c’est-à-dire à faire en sorte que les médecins soient mieux au fait des connaissances scientifiques, qu’ils agissent en termes de prévention et de santé publique – ce qu’ils ne font pas naturellement dans le tissu libéral. Tout cela procède d’une nouvelle culture de réduction des risques et d’amélioration des comportements des malades, qui doit être apportée dès la formation médicale initiale et qui est relayée par la formation médicale continue.
Dans ce contexte, le choix des cinq thèmes prioritaires de formation médicale continue n’est pas anodin :
– rôle et place des praticiens en situation de crise sanitaire ;
– iatrogénèse ;
– prévention vaccinale ;
– prévention et dépistage des cancers ;
– prévention et réduction des risques environnementaux, comportementaux et professionnels.
Dans ce dernier domaine, il est évident qu’on demande au médecin, non plus de clôturer sa consultation par une liste de prescriptions sur une ordonnance mais, par de nouvelles attitudes et par de nouvelles relations avec le malade, de rechercher chez ce dernier les motivations et les ressources pour modifier ses comportements. Ainsi, on ne traite plus un diabétique avec des médicaments si l’on n’a pas provoqué la prise de conscience qu’il doit modifier lui-même ses comportements.
Si l’on souhaite agir davantage sur les accords de bon usage des soins, il convient sans doute de développer les relations avec les caisses d’assurance maladie. Cette année, ces dernières ont décidé de cibler leurs objectifs de formation médicale continue, donc leurs financements, sur ces accords conventionnels, sur la maîtrise médicalisée et sur la santé publique. Cela donnera sans doute des résultats intéressants en ville, qu’on ne pourra toutefois évaluer qu’ultérieurement, quand nous disposerons de notre système d’information sur le portail dont nous avons absolument besoin pour faire le lien entre la formation médicale continue et les modifications de comportement. Cependant on voit bien que, si tout le financement vient des caisses, c’est ce type d’objectifs qui sera privilégié.
Politiquement, deux objectifs semblent aujourd’hui particulièrement mis en avant : l’amélioration des comportements dans les établissements et les nouvelles formes d’exercice de la médecine générale, dans le cadre des états généraux de l’offre de soins. J’observe à ce propos que les trois conseils de la formation médicale continue ne sont pas invités à ces états généraux. C’est dommage car ils auraient sans doute des choses à dire sur l’évolution des métiers.
M. Pierre Morange, coprésident : On voit bien que l’on est dans un domaine où la multiplicité des acteurs et le manque de coordination posent problème. Or la MECSS a précisément la volonté d’aider à la clarification de l’ensemble du système de protection sanitaire et sociale, non parce qu’elle est obsédée par la simplification, mais parce qu’elle estime que cela concourt à l’optimisation des moyens.
Avez-vous en ce qui vous concerne des propositions concrètes pour passer de la théorie à la pratique ?
Mme Annie Podeur : Nous allons formaliser le dispositif, mais nous ne pouvons plus attendre parce que des médecins sont dans les starting-blocks et que certains ont même déjà pris le départ, par exemple au sein de groupes de pairs. Nous nous trouvons en effet au sein d’une sorte de coordination de très nombreux acteurs au niveau national et nous devons particulièrement veiller à ne pas casser les dynamiques qui sont en train d’apparaître sur le terrain.
M. Pierre Morange, coprésident : Si tout le monde est prêt, qu’est-ce qui vous fait aujourd’hui défaut pour accélérer le lancement du dispositif ? Est-il nécessaire de lever les blocages qui subsistent par des dispositions législatives ou réglementaires ? Comment notre mission peut-elle vous venir en aide ?
M. Dominique Bertrand : Le dispositif est prêt, il y a un barème et un certain nombre d’actions vont entrer en application dès maintenant avant d’être évaluées au bout de cinq ans. Ce qu’il faut désormais, c’est que les gens se lancent. Toutefois cela ne se fera pas forcément dans le domaine du médicament car l’activité des médecins est hétérogène. Pour autant, il est indispensable de parvenir à une homogénéité des pratiques vis-à-vis d’un même patient : celui-ci ne peut pas se voir proposer deux thérapeutiques différentes à deux endroits différents.
Le dernier décret doit paraître prochainement. Dès qu’il aura été publié et que le dispositif d’évaluation individuelle aura été arrêté, nous saurons comment chacun pourra valider sa formation continue et nous pourrons passer à l’étape suivante pour rendre le dispositif utile, efficace et efficient. Néanmoins la première étape est celle de la mise en route au cours de laquelle les exigences ne seront sans doute pas celles que vous appelez de vos vœux. Par la suite, nous disposerons sans doute d’informations en provenance des caisses d’assurance maladie qui nous permettront d’homogénéiser et d’améliorer les pratiques.
M. Bernard Ortolan : Nous avons pleine confiance dans les services de la DHOS qui s’efforcent d’aplanir les dernières difficultés. Il est certain que la mise en place d’un tel dispositif n’est pas chose aisée, mais bientôt douze ans se sont écoulés depuis les ordonnances Juppé. Cela semble long, d’autant que les médecins y avaient adhéré en 1996, qu’on leur en a parlé à nouveau en 2002 et qu’on continue à leur en parler, depuis quatre ans que nous sommes installés. Si le dispositif n’était pas opérationnel dans le courant de cette cinquième et dernière année, on pourrait véritablement parler d’échec.
Il ne manque donc qu’un décret, qui pourrait toutefois remettre en question un certain nombre de dispositions prévues par les textes antérieurs. Nous souhaitons que sa rédaction permette de lancer officiellement la première période quinquennale, au plus vite et si possible de manière rétroactive au 1er janvier 2008, sans pour cela installer obligatoirement les conseils régionaux de la formation médicale continue, dont on n’a pas besoin tout de suite et qui pourraient attendre la nouvelle loi sur la régionalisation de l’organisation du système de santé, annoncée par le premier ministre.
Nous voudrions que la volonté politique soit suffisamment forte pour que le dispositif de formation médicale continue soit opérationnel au cours du premier trimestre 2008 car, comme l’a dit Mme Annie Podeur, les médecins ont déjà commencé : plus de 8 000 cycles d’évaluation des pratiques professionnelles (EPP) ont été lancés ; 105 organismes ont été agréés par la Haute Autorité de santé pour délivrer les procédures d’EPP éligibles à l’obligation de formation ; nous-mêmes avons agréé plus de 150 organismes qui mènent des actions éligibles au crédit-formation et un certain nombre de médecins disposent de justificatifs qu’ils aimeraient faire valoir. Si tout cela ne fonctionne pas, le risque de découragement est grand.
M. Alain Beaupin : On a aujourd’hui quelques financements publics avec l’assurance maladie et la formation professionnelle conventionnelle. Je forme le vœu que des dispositifs permettant aux médecins de financer leur formation de façon plus indépendante voient le jour, par exemple sous la forme d’un crédit d’impôt leur permettant de suivre la formation médicale continue obligatoire.
M. Dominique Bertrand : Nous allons créer dans les six mois qui viennent, grâce au soutien de la DHOS, un portail qui permettra à chacun non seulement de présenter les actions qu’il mène mais aussi de les valider de façon quasi automatique, ce qui nous dispensera de la lecture fastidieuse d’un grand nombre de documents.
Mme Catherine Lemorton, rapporteure : Sait-on quel est l’investissement de l’industrie pharmaceutique dans son ensemble dans la formation continue des médecins libéraux ?
Le but de notre mission est de comprendre pourquoi trop de médicaments sont consommés en France. À cet égard, on peut penser que la mise en vente libre de certains médicaments dans les pharmacies, devant les comptoirs, conduira rapidement, au regard des 9 milliards de chiffres d’affaires qui sont en jeu pour l’industrie pharmaceutique, à ce que ces médicaments arrivent dans les rayons des supermarchés. Cette mesure ne risque-t-elle pas d’avoir un effet pervers au regard de tous les efforts que nous faisons, vous comme nous, pour réguler la consommation de médicaments dans notre pays ?
M. Bernard Ortolan : Le financement est la bouteille à l’encre de la formation médicale continue comme de l’évaluation des pratiques professionnelles. Le rapport de l’IGAS, de 2006, concernant la formation médicale continue, indiquait le montant de l’argent public mobilisé, soit 60 à 70 millions d’euros par an : 35 millions au titre de la formation professionnelle conventionnelle, 5 millions au titre de la cotisation obligatoire des médecins de 47 €, collectée par le Fonds d'assurance-formation des professions libérales (FAF-PL), auxquels il faut ajouter les sommes qu’injecte la direction générale de la santé à l’occasion de campagnes comme celle de 1990 sur le sida ou celles de 2006 et 2007 sur la grippe aviaire.
Le rapport de l’IGAS indiquait aussi que le besoin de financement était couvert dans une fourchette de 300 à 600 millions d’euros par l’industrie pharmaceutique. Pour notre part, nous n’avons pas les moyens d’effectuer de tels calculs, d’autant que nous ne savons pas ce qui se passe partout sur le terrain et que sont probablement incluses dans cette enveloppe toutes les petites soirées des laboratoires qui étaient organisées jusqu’ici et qui, désormais, ne seront pas interdites mais ne pourront plus être validées au titre de la formation médicale continue.
M. Pierre Morange, coprésident : Nous sommes tout prêts à vous apporter notre aide pour que le décret que vous attendez paraisse le plus rapidement possible.
S’agissant du besoin de financement, vous avez donné une fourchette très large. En tant que responsable de la formation médicale continue, avez-vous une idée plus précise des sommes qui seraient nécessaires à une formation de qualité ?
M. Bernard Ortolan : Si l’on exclut ce qui relevait jusqu’ici de la promotion et si l’on ne permet à l’industrie de financer que des programmes conformes à la charte du LEEM, qui laissent en particulier toute liberté pédagogique aux opérateurs agréés, je pense que 200 millions d’argent public supplémentaires permettraient aux 200 000 médecins en exercice de réaliser leur formation annuelle.
Le Fonds interprofessionnel de formation des professions libérales (FIF-PL) réfléchit actuellement à une augmentation de la cotisation des autres professionnels libéraux. Dans notre rapport 2005, nous avions suggéré que l’on augmente également la cotisation de 47 € perçue par le FAF-PL. On pourrait sans problème la doubler ou la tripler car elle est recueillie par les Urssaf et totalement indolore. Cela procurerait 10 millions d’argent public supplémentaires, ce qui n’est quand même pas négligeable pour garantir l’indépendance des médecins dans le financement de leur formation médicale continue.
Les choses sont plus compliquées en ce qui concerne le médicament. Les médecins n’avaient jusqu’ici pas été formés à la négociation avec les patients pour réduire les prescriptions. Désormais, on leur apprend à savoir dire non, on les forme à l’éducation des patients, en particulier à l’éducation thérapeutique et à l’éducation pour la santé. À eux ensuite de former leurs patients, mais tout cela prend du temps et n’est pas inclus dans le prix de la consultation.
Peut-être les jeunes générations pourront-elles se tourner davantage vers les recommandations que vers les prescriptions et assisterons-nous au développement des médicaments OTC (Over the counter), vendus devant le comptoir. Mettre les médicaments dans les grandes surfaces, comme en Angleterre, est une vraie question de santé publique, sur laquelle je ne veux pas me prononcer pour l’heure, même s’il est vrai que cela pourrait avoir des effets néfastes sur la santé.
M. Jean Mallot, coprésident : Il nous faut malheureusement interrompre cette audition. Je vous remercie d’y avoir participé. Nous vous demanderons de bien vouloir répondre par écrit aux questions que nous n’avons pas eu le temps de vous poser mais aussi de nous faire toutes les suggestions que vous jugerez utiles.
*
Audition de Mme Nathalie Tellier, chargée de mission à l’Union nationale des associations familiales et membre du bureau du Collectif interassociatif sur la santé, M. Jacques Mopin, administrateur de l’Union fédérale des consommateurs - Que choisir, accompagné par M. Christophe Le Guehennec, chargé de mission santé, et M. Thierry Saniez, délégué général de la Confédération de la consommation, du logement et du cadre de vie.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale. M. Christian Saout, président du bureau du Collectif interassociatif sur la santé (CISS), qui devait également intervenir au cours de cette audition, s’est excusé. Mme Nathalie Tellier s’exprimera en son nom.
Mme Catherine Lemorton, rapporteure : En médecine de ville, la prescription de médicaments est effectuée dans un colloque singulier entre médecin et patient, régi par une charte datant de 1928. D’une certaine manière, madame, messieurs, vous représentez les patients. Selon vous, pourquoi 90 % des consultations chez un médecin généraliste en France sont suivies d’une prescription de médicaments alors que ce taux est bien plus bas dans les autres pays européens ? Pensez-vous que le patient exerce une pression sur le médecin pour obtenir à tout prix un médicament, ou que la prescription est du seul ressort du médecin ?
M. Thierry Saniez : D’après les retours que nous avons, le problème est surtout d’ordre culturel et forme donc un tout. En France, le curatif l’emporte sur la prévention. Lorsque le patient va chez son médecin, plus il y a de médicaments prescrits, plus cela lui semble crédible.
Un chantier énorme reste ouvert en matière de prévention. S’agissant des médicaments, un travail de sensibilisation et même d’éducation est nécessaire tant auprès du personnel médical que de la population pour faire prendre conscience que le fait de prescrire plus de médicaments ne correspond pas forcément à une meilleure action curative.
Mme Nathalie Tellier : Il s’agit en effet d’un problème de société. Cependant, en tant que représentante tant de l’UNAF que du CISS, il m’est impossible de ne pas réagir aux propos tenus lors de l’audition qui a précédé la nôtre. On est dans la droite ligne d’une déclaration de la Confédération des syndicats médicaux français aux termes de laquelle « la prescription est l’issue d’une négociation avec le patient. Si, par exemple, une prescription comporte un antibiotique alors que le patient ne souffre que d’une maladie virale, c’est que le médecin a subit une énorme pression. » Pourtant, jusqu’à preuve du contraire, c’est le médecin qui détient le savoir et qui tient le stylo. La relation avec le patient est asymétrique. Certains patients peuvent demander telle ou telle prescription, mais c’est le médecin, in fine, qui prescrit. Certes, l’UNAF a observé que certaines crèches exigent une prescription d’antibiotiques pour reprendre un enfant malade, mais l’exemple reste marginal.
On ne saurait donc parler de pression du malade. Le problème tient à l’habitude qu’ont prise les médecins et les patients. Le fait que l’assurance maladie garantisse un remboursement correct est aussi un facteur non négligeable.
On ne fera évoluer les choses qu’à la condition d’une prise de conscience générale. La campagne menée sur les antibiotiques est de ce point de vue exemplaire car elle a délivré les mêmes informations aux patients et aux médecins et s’est traduite par une baisse de 20 % des prescriptions d’antibiotiques. C’est bien la preuve que le travail éducatif doit s’exercer en direction de tous les acteurs pour être efficace.
M. Thierry Saniez : Il arrive de plus en plus que des patients collectent des informations sur l’Internet, puis en fassent part au médecin pour exiger telle ou telle prescription. En outre, la médecine est soumise à une vision trop utilitariste : on prescrit pour résoudre le problème immédiat, sans s’attacher suffisamment à la résolution du problème de fond. L’état des personnes consommant des antidépresseurs est souvent dû à des modes de vie, à des conditions de travail, etc., qui en sont les vraies causes.
M. Jacques Mopin : L’UFC-Que choisir a réalisé des études qui démontrent clairement la responsabilité de l’industrie pharmaceutique dans la surconsommation de médicaments. Dans une enquête auprès de nos lecteurs, nous avons posé la question : « À qui accordez-vous votre confiance ? », en proposant de choisir parmi une soixantaine de professions. Le médecin généraliste, avec 90 % de confiance absolue, est arrivé juste après les sapeurs-pompiers. Que l’on n’aille donc pas parler de rapport équilibré ! La demande des patients, indéniable, est sans proportion avec la surconsommation actuelle de médicaments. Une étude de l’Institut de recherche et documentation en économie de la santé (IRDES), citée dans le rapport de l’IGAS sur l’information des médecins sur le médicament, établit formellement qu’à chaque campagne de promotion d’un médicament correspond un pic de prescription dudit médicament. Il n’est plus ici question de santé publique et d’intérêt du patient.
A contrario, lorsqu’une campagne aboutit à une réduction de 20 % des prescriptions d’antibiotiques, on peut en conclure qu’il existait auparavant 20 % de prescriptions erronées. Le ratio entre le nombre de visiteurs médicaux et le nombre de médecins est de un pour neuf en France. Il n’est inférieur qu’aux États-Unis. Aux Pays-Bas, il s’élève à un pour trente-quatre, et le patient sort de chez le médecin avec une ordonnance moins d’une fois sur deux. La corrélation est patente.
Pour arriver à enrayer ce qui est, à nos yeux, une catastrophe de santé publique assortie d’une catastrophe financière, il faut impérativement rééquilibrer l’information délivrée au médecin. Nous avons donc proposé de promouvoir l’information publique en créant un corps de visiteurs médicaux dépendant de la Haute Autorité de santé (HAS), tout en ramenant la part de l’activité des visiteurs médicaux de 10 % actuellement à 4 % du chiffre d’affaires des sociétés pharmaceutiques.
M. Thierry Saniez : Nous partageons l’analyse de l’UFC-Que choisir. Le problème est connu de tous et il faut œuvrer pour une information plus indépendante et assurée par le secteur public. Lors de l’audition qui vient de se dérouler, un intervenant a affirmé que l’on pourrait assurer la formation de 200 000 médecins une semaine par an avec 170 millions d’euros. Cela peut constituer une piste.
M. Pierre Morange, coprésident : La mise en place d’un corps de quelque 1 700 intervenants constitué de médecins pour un tiers et d’infirmières pour deux tiers entraînerait un transfert vers la Haute Autorité de santé des fonds affectés actuellement par l’assurance maladie à l’action des délégués de l’assurance maladie, les DAM. Il convient aussi de prendre en considération les incidences techniques et statutaires d’une telle réforme, à un moment où l’assurance maladie s’emploie à optimiser sa politique de ressources humaines. Est-il souhaitable de soustraire les DAM à l’assurance maladie alors que la vocation légitime de celle-ci est d’accompagner ce dispositif ? La volonté de s’en remettre à la HAS tient, certes, à ce que les interventions de l’assurance maladie sont parfois mal vécues par les praticiens, mais la multiplicité des structures ne risque-t-elle pas de retarder la mise en œuvre d’un dispositif efficace ?
M. Christophe Le Guehennec : Nos propositions sont précisément guidées par la volonté de faire avec ce qui existe. Les mesures pragmatiques que nous préconisons sont en grande partie reprises des rapports de la Cour des comptes et de l’IGAS. Elles sont conçues pour être appliquées le plus rapidement possible.
Ainsi, nous souhaitons que les délégués de l’assurance maladie deviennent des « visiteurs médicaux publics » dépendant de la Haute Autorité de santé. Il ne saurait être question de faire disparaître leur réseau pour en créer un autre.
M. Pierre Morange, coprésident : Cependant les DAM, quelles que soient par ailleurs leurs compétences, ne sont ni des médecins ni des infirmières.
M. Christophe Le Guehennec : Nous en sommes bien conscients, mais ils ont acquis une expérience de terrain dont il serait dommage de se priver. En ajoutant à cet effectif des médecins et des infirmiers, on pourra atteindre le chiffre de 1 700 visiteurs médicaux publics, qui nous semble adapté.
M. Pierre Morange, coprésident : Donc la proportion d’un tiers de médecins et de deux tiers d’infirmiers est un objectif de moyen ou de long terme.
M. Christophe Le Guehennec : En effet. Dans un premier temps, la Haute Autorité de santé pourrait porter un regard sur le réseau des DAM pour vérifier que l’action menée est conforme, du point de vue médical, aux avis de la commission de la transparence et aux recommandations de bonnes pratiques.
Mme Nathalie Tellier : Il est à la mode de parler de la « responsabilisation du patient », expression qui laisse entendre que celui-ci n’est pas responsable. Dans les années 1990, une commission du syndicat national de l’industrie pharmaceutique avait travaillé sur l’éducation des enfants au médicament au sein du système scolaire. L’initiative a été ensuite abandonnée, mais il s’agit là d’un bon moyen pour parvenir à une prise de conscience. Il faut persuader patients et médecins que l’on ne peut tout résoudre avec les médicaments et il serait bon d’associer de nouveau l’éducation nationale à des actions en direction des enfants.
Mme Catherine Lemorton, rapporteure : Les actions engagées comportaient-elles un volet de prévention ?
Mme Nathalie Tellier : Non, la démarche de prévention n’en était qu’à ses débuts alors qu’à l’heure actuelle c’est la principale perspective pour l’avenir. Le Collectif interassociatif sur la santé regroupe essentiellement des associations de malades qui mettent toutes l’accent sur la prévention et dont beaucoup développent des programmes en ce sens.
M. Pierre Morange, coprésident : Il n’est nullement dans notre intention de nous prononcer sur les responsabilités respectives des médecins et des patients. Le praticien, détenteur du savoir médical, est évidemment en position dominante. Inversement, la demande de la part du patient ne saurait être niée.
À côté de cela, beaucoup d’informations circulent sur l’Internet et échappent aux réseaux structurés qui garantissent la certification sanitaire des produits, notamment le réseau des pharmaciens. Ces derniers, de par l’obligation de conseil à laquelle ils sont soumis, participent à l’éducation sanitaire. Or, l’Internet permet la livraison de médicaments en dehors de tout contrôle ou propose des produits qui répondent à une logique purement mercantile. Le monde associatif prend-il des initiatives pour évaluer et combattre ces risques ?
M. Thierry Saniez : Notre expérience sur le terrain nous montre, d’une part, que les personnes sont demandeuses d’information, d’autre part, que tout un public n’est atteint ni par l’information institutionnelle classique ni par celle qui circule sur l’Internet. Cependant, lorsqu’un message est clair et lisible, les personnes se montrent prêtes à évoluer. Ainsi le programme national nutrition santé (PNNS) a-t-il réussi à faire passer son message « cinq fruits et légumes par jour » auprès d’une grande partie de la population. Concernant l’Internet, ne pourrait-on concevoir un grand portail public d’information qui constituerait une référence ?
M. Jacques Mopin : La commission de la qualité de l’information médicale de la Haute Autorité de santé, dont je fais partie, travaille sur ces sujets, puisque la loi dispose que la HAS devra certifier les sites Internet consacrés à la santé. S’il est impossible de certifier un site en particulier, il est au moins possible de valider des procédures permettant au patient de se faire une idée de l’endroit où il met les pieds : qui finance le site, qui délivre l’information, d’où l’information provient-elle, de quand date-t-elle ? La HAS s’oriente donc vers un dispositif de certification des procédures, afin de dissuader le patient de consulter le site si celui-ci ne répond pas à une série de critères. Le travail sur ce sujet touche à sa fin. Il conviendra d’assurer sa diffusion auprès du public car c’est une approche qui permettra de limiter les dégâts tout en se préservant de la tentation irréaliste de régenter le Web.
M. Christophe Le Guehennec : La Haute Autorité de santé a choisi un institut suisse, Health on the net (HON), pour être l’organe certificateur qui appliquera cette charte aux multiples sites consacrés à la santé. Sans être infaillible, tant s’en faut, la procédure permet une certaine lisibilité.
Du reste, la multiplicité des sources d’information renforce encore le rôle du médecin et du pharmacien, qui devront filtrer ce qui a été trouvé sur l’Internet. Il est d’autant plus important qu’ils disposent d’une information objective, émanant des autorités de santé et formatée pour répondre à leurs besoins quotidiens. Nous en sommes loin aujourd'hui.
M. Jacques Mopin : Si nous souhaitons instituer des visiteurs médicaux publics, c’est aussi parce que les médecins apprécient de recevoir des visiteurs. L’enquête de la HAS fait ressortir un plébiscite (environ 90 %) en faveur de ces visites, alors même que seuls 26 % des médecins interrogés considèrent comme objective l’information délivrée. Ils sont sensibles à ce qu’on leur apporte de l’information mais l’information objective publique emprunte à l’heure actuelle des canaux inadaptés à leurs besoins.
Mme Catherine Lemorton, rapporteure : Quels retours avez-vous des usagers du système de soins ? Estiment-ils que la consultation médicale leur apporte une bonne information en matière de prévention, notamment sur les sujets d’hygiène diététique ? Le message « Cinq fruits et légumes par jour », massivement diffusé, est en effet bien passé, mais qu’en est-il dans le colloque singulier entre le médecin et le patient ?
Puisque vous représentez les consommateurs – et que les patients sont devenus, pour une part, clients –, que pensez-vous de la mise en vente libre de médicaments devant les comptoirs des pharmacies, qui prélude peut-être à une commercialisation dans les supermarchés comme dans les pays anglo-saxons ? On sait que, aux États-Unis, l’iatrogénie figure parmi les dix premières causes de mortalité.
Mme Nathalie Tellier : La démarche de prévention est en train de se développer dans la pratique des médecins. Ceux-ci formulent de plus en plus de préconisation sur l’hygiène de vie. L’action des associations de patients est également importante, mais M. Christian Saout vous en aurait parlé mieux que moi.
En ce qui concerne l’automédication, il n’y a pas forcément d’opposition de la part des représentants des patients, à la condition que ce ne soit pas un objet économique. Aujourd'hui, on constate que les médicaments augmentent de 30 % à partir du moment où ils font l’objet d’un déremboursement. La mise en place de l’automédication suppose une information et une éducation. Le pharmacien doit jouer son rôle, par exemple grâce au dossier pharmaceutique que l’on est en train de mettre en place : le professionnel de santé pourra alors avoir connaissance de tous les médicaments, qu’ils soient prescrits ou non, achetés par le patient durant les quatre derniers mois, ce qui devrait permettre d’éviter les redondances et le risque d’iatrogénie. Par ailleurs, le « Web médecin » permet d’ores et déjà aux professionnels de santé, en attendant le dossier médical personnel, d’avoir accès à l’historique des remboursements dont a bénéficié le patient depuis un an.
Aujourd'hui, le prix des produits en vente libre dans une pharmacie n’est pas toujours clairement affiché. Or, à la suite d’un rapport du Conseil national de la consommation, un arrêté a été pris en 2003 pour obliger les pharmaciens à rendre publics et visibles les prix des médicaments non remboursables, qu’il y ait prescription obligatoire ou non, à tenir un répertoire de tous les médicaments à prix libre délivrés uniquement sur prescription médicale, et à indiquer sur une affichette que le prix des médicaments non remboursables est libre. En 2007, l’enquête menée par la DGCCRF – la direction générale de la concurrence, de la consommation et de la répression des fraudes – sur ce sujet dans 1 000 pharmacies s’est traduite par 638 rappels à l’ordre.
Si l’avenir est assurément à l’automédication, il faut cependant que chacun tienne son rôle. Les prix doivent rester raisonnables et le pharmacien, tout comme le médecin, doit donner l’information nécessaire au patient.
Thierry Saniez : Pour en revenir à la campagne du PNNS, la question qui se pose maintenant est de savoir ce que signifie exactement « manger cinq fruits et légumes ».
En matière de prévention, on constate certes des avancées sur le terrain mais la situation est loin d’être satisfaisante. Le vrai souci des patients actuellement est de savoir comment ils pourront continuer à payer les soins alors qu’ils doivent désormais acquitter des franchises. Avec 2 % des dépenses consacrées à la prévention, nous sommes de toute façon loin du compte.
Pour ce qui concerne l’automédication, notre confédération considère que, si un médicament ne nécessite pas de prescription médicale et se trouve en vente libre, il faut un assouplissement des conditions de sa commercialisation. De même, nous sommes favorables à un assouplissement du numerus clausus des officines.
L’an dernier, une étude que nous avons menée sur le paracétamol a révélé que les pharmaciens délivraient systématiquement les produits de marque, beaucoup plus chers que les « non-marques ». Dès lors que le produit n’est pas en accès direct et que le pharmacien est rémunéré au pourcentage du chiffre d’affaires, il est forcé que l’on ne propose pas le produit le moins cher, le générique, etc. C’est tout le système, fondé uniquement sur la vente, qui est à revoir à l’aune des objectifs poursuivis. Le chantier est considérable.
Une autre de nos études met en évidence notre retard en matière de génériques par rapport à des pays comme le Royaume-Uni ou l’Allemagne. Un important travail d’information reste à mener en ce domaine. Comment inciter le pharmacien à distribuer plus de médicaments génériques alors que sa rémunération est au pourcentage ? Nous nous sommes opposés aux franchises mais, dès lors qu’elles ont été instaurées, ne pourrait-on en dispenser les génériques ?
Mme Catherine Lemorton, rapporteure : Je précise que c’est la commission des affaires culturelles, familiales et sociales de l’Assemblée nationale qui a écarté l’idée d’exempter de franchise les médicaments génériques.
M. Jacques Mopin : La prévention se heurte à d’énormes obstacles économiques. Lors du lancement de notre campagne de lutte contre l’obésité infantile, nos associations locales ont mené des enquêtes sur le terrain et ont analysé les aliments pour enfants qui faisaient l’objet d’une démarche marketing dans la grande distribution. Il est apparu que ceux-ci, dans leur quasi-totalité, étaient extrêmement déséquilibrés : trop gras, trop sucrés, trop salés. Nous avons ensuite organisé une centaine de conférences de presse pour présenter ces résultats. Or, pas plus d’un média sur dix ne s’est déplacé, les plus honnêtes reconnaissant qu’ils ne venaient pas car ils ne voulaient pas être obligés de dire du mal de leurs annonceurs. Cela ne laisse pas d’inquiéter.
Tout notre système tend à vendre le plus de produits possible. On trouve normal que l’obésité fasse l’objet d’une prise en charge médicale, tandis que l’information relative à la prévention se heurte à des obstacles économiques, apparemment insurmontables. On ne parvient même pas à obtenir des grands distributeurs l’engagement de ne plus mettre ces aliments déséquilibrés aux caisses de sortie, là où ils sont à la portée des enfants. L’association des professionnels s’occupant des devants de caisse, dont j’ai découvert l’existence à cette occasion, n’est assurément pas prête à nous emboîter le pas en matière de prévention !
L’UFC-Que choisir est très réticente en ce qui concerne l’automédication. Nous partons du principe qu’un médicament est toujours, sous tel ou tel aspect, un poison. Le développement de l’automédication risque de provoquer des problèmes de santé publique. Lors des travaux préparatoires pour le rapport sur l’automédication présenté par M. Alain Coulomb, l’industrie pharmaceutique a argué de ses efforts en matière de prix des médicaments remboursés pour demander l’ouverture du marché de l’automédication. D’où nos réticences…
M. Thierry Saniez. La CLCV partage ces constats. Elle travaille actuellement au développement d’un grand portail d’information sur l’alimentation. En analysant plusieurs publicités télévisées dans ce domaine, nous avons établi que les règles du Bureau de vérification de la publicité (BVP) – aujourd'hui dénué de tout pouvoir de sanction – étaient systématiquement bafouées. Nous appelons de nos vœux un système comparable à celui en vigueur au Royaume-Uni, où les consommateurs peuvent saisir directement une autorité administrative pour édicter des règles claires et interdire telle ou telle publicité. On ne peut rechercher d’un côté à réduire les dépenses de santé et la consommation de médicaments, et faire tout, d’un autre côté, pour que les gens en arrivent à des situations où ils sont obligés d’aller voir le médecin et de consommer des médicaments.
M. Jean Mallot, coprésident : Je vous remercie pour vos contributions et vous invite à communiquer à la Mission les enquêtes ou les études dont elle n’aurait pas eu connaissance.
*
Audition de MM. Bertrand Garros, président de l’Institut national de prévention et d'éducation pour la santé, et Philippe Lamoureux, directeur général.
M. Pierre Morange, coprésident : Quelles préconisations l’INPES pourrait-il formuler dans le domaine de la prescription et de la consommation des médicaments afin d’améliorer la qualité de soins et l’efficience ?
M. Bertrand Garros : Tout d’abord, si vous le permettez, quelques mots de présentation générale.
L’INPES est un établissement public qui dispose d’un conseil d’administration réunissant des représentants de l’État, des organismes d’assurance maladie obligatoire et complémentaire, des associations de prévention ainsi que des usagers. La loi lui confie cinq missions principales : participation à la mise en œuvre des programmes de santé publique, expertise et conseil, développement de l’éducation pour la santé, participation à la gestion des situations de crise, établissement des programmes de formation à l’éducation pour la santé.
L’INPES dispose d’un budget de 120 millions et 140 personnes y travaillent. Il s’appuie sur les réseaux d’éducation pour la santé – comités régionaux et départementaux – et participe aux groupements régionaux de santé publique, les GRSP. Onze programmes prioritaires ont ainsi été mis en place, la plupart thématiques. L’INPES publie tous les cinq ans des « baromètres santé » permettant de mesurer l’évolution des comportements et développe également une activité importante dans le domaine de la communication – l’INPES étant le plus gros acheteur public d’espace publicitaire – à travers notamment un pôle de téléphonie sociale : Tabac info service, Sida infos service… Près de 70 millions de documents sont en outre distribués chaque année.
L’INPES développe aussi des sites Internet en fonction des différentes campagnes de prévention. Sa lettre électronique, enfin, est distribuée à 15 700 exemplaires.
L’INPES soutient par ailleurs les acteurs de terrain à travers des subventions ou des aides à la diffusion de matériels pédagogiques. Il a également signé un partenariat avec la Caisse nationale de solidarité pour l’autonomie (CNSA) afin de mieux prendre en compte les besoins de la population.
Les questions de la prévention et du médicament sont en effet fondamentales depuis longtemps, comme en atteste par exemple la vaccination. L’utilisation de médicaments – les statines, par exemple – en prévention primaire soulève également un certain nombre de problèmes. Certains experts considèrent d’ailleurs qu’il serait bienvenu de généraliser leur prise à condition qu’elles soient faiblement dosées.
Si l’on évoque souvent la prévention, la promotion de la santé est d’ordinaire moins mise en valeur alors qu’elle concerne toutes les actions visant à favoriser ce qui conditionne favorablement notre santé : bien-être, qualité de l’environnement, alimentation, sexualité. En relèvent par exemple la contraception orale, le traitement hormonal substitutif, les dysfonctionnements érectiles, la lutte contre le vieillissement ou la douleur.
Nous n’ignorons pas non plus la question de l’usage détourné des médicaments à des fins toxicomaniaques ou de dopage.
L’INPES s’est également efforcé de poser un certain nombre de questions transversales, dont l’intervention de la protection sociale obligatoire et complémentaire : jusqu’où faut-il aller dans la prise en charge de la prévention ou de la promotion de la santé ? Pourquoi certains produits sont-ils remboursés et d’autre pas ? Comment réduire les inégalités sociales et territoriales ? Quid des effets à long terme de médicaments préventifs destinés à un grand nombre ? Quelle est la place du médicament dans des stratégies de prévention ? Comment, par exemple, favoriser l’accès aux diététiciens ?
Je note, enfin, que la question de la réduction de la consommation des antibiotiques avait d’abord été abordée sous l’angle économique et que cela n’a pas donné les résultats escomptés. En revanche, lorsqu’elle l’a été sous l’angle médical en mettant en avant les problèmes liés aux résistances bactériologiques, le message est beaucoup mieux passé, et auprès des professionnels, et auprès des patients.
Mme Catherine Lemorton, rapporteure : L’impact de la dernière campagne de l’INPES consacrée à la dépression a-t-il été évalué auprès des médecins ?
M. Philippe Lamoureux : Avant de répondre à votre question, je tiens tout d’abord à préciser que les politiques de prévention visent à augmenter l’espérance de vie en bonne santé. Les relations entretenues avec le médicament sont dès lors ambivalentes puisque, dans certains domaines, les actions de prévention contribueront à en accroître la consommation. Il est en outre plus facile, parfois, de prescrire un médicament plutôt que de conseiller simplement une pratique sportive ; la lutte contre l’obésité en est un bon exemple.
Il est donc encore trop tôt pour évaluer l’impact de la campagne sur la dépression mais d’ores et déjà, 600 000 guides ont été diffusés dont 255 000 par commande spontanée des personnes ayant vu ou entendu les spots. Nous sommes de surcroît en relation avec l’assurance maladie de manière à pouvoir bénéficier d’un certain nombre d’indicateurs. Il faut noter que le dispositif d’information à destination du grand public est également couplé avec un dispositif à l’adresse des professionnels de santé. Nous avons considéré, enfin, qu’il fallait changer de regard sur la dépression en la considérant vraiment comme une maladie et non comme un trouble de l’humeur.
La logique des politiques de prévention repose sur le développement des aptitudes individuelles et sur la création d’environnements favorables à la santé. L’INPES s’époumone ainsi à organiser des campagnes de promotion pour le sport, mais 20 000 vélos en accès libre dans une ville permettent d’économiser le prix d’une campagne et les effets sur la santé sont bien réels. Il en va de même si l’on retire les distributeurs de produits gras et sucrés dans les écoles ou si l’on interdit de fumer dans les lieux publics.
Enfin, travailler sur la problématique de la prévention et du médicament, c’est également travailler sur la question de l’observance – prévention tertiaire – puisque 50 % des patients atteints de maladie chronique ne sont pas observants pour des raisons d’ailleurs très variables. Une approche globale est donc nécessaire. Nous venons à cette fin de publier un guide sur l’éducation thérapeutique du patient : c’est d’abord à partir de lui qu’il importe de raisonner.
Mme Catherine Lemorton, rapporteure : Travaillez-vous en partenariat avec l’éducation nationale ? Si oui, sur quels thèmes ?
M. Philippe Lamoureux : Ce partenariat est très étroit. Nous avons signé une convention cadre avec le ministère de l’éducation nationale, en particulier avec la direction générale de l’enseignement scolaire. Nous considérons les professionnels de l’éducation à l’instar des professionnels de santé. Une circulaire de 1998 et un décret de 2007 prévoient de développer l’éducation pour la santé en milieu scolaire en incluant cette discipline dans le socle des connaissances. Notre but est de travailler, dès la maternelle, sur le développement des aptitudes individuelles : estime de soi, capacité à gérer des conflits, esprit critique, lesquels constituent autant de facteurs de protection.
Nous avons en outre une chance historique, puisque 50 % des maîtres seront remplacés dans les sept années à venir et que l’on peut d’ores et déjà mieux former les étudiants en IUFM – institut universitaire de formation des maîtres – à l’éducation à la santé. Un partenariat avec les IUFM a d’ailleurs été conclu. Nous créons également des outils pédagogiques gratuits que nous mettons à la disposition des écoles ; cette année, 30 000 mallettes pédagogiques seront distribuées. Des difficultés demeurent néanmoins, puisque les projets dépendent sans doute trop étroitement de ceux qui les mettent en œuvre dans les établissements. Notre but est de faire en sorte que les projets d’éducation à la santé s’inscrivent dans le cahier des charges de chaque établissement scolaire.
M. Bertrand Garros : Les comités départementaux et régionaux d’éducation à la santé sont également très impliqués, notamment dans le cadre des schémas régionaux d’éducation à la santé.
Mme Catherine Lemorton, rapporteure : Considérez-vous que la médecine scolaire contribue à véhiculer les informations sur la prévention ? Le dispositif actuel est-il suffisant ?
Êtes-vous par ailleurs confrontés à des pressions provenant des groupes alimentaires par exemple ? De quels moyens d’action disposez-vous ?
M. Bertrand Garros : Non seulement la médecine scolaire manque malheureusement de moyens mais le binôme médecins - infirmières soulève parfois quelques problèmes hiérarchiques. En outre, les situations varient beaucoup en fonction des endroits. Il convient, enfin, de s’interroger sur l’articulation entre la médecine scolaire et la médecine ambulatoire.
L’INPES n’est pas quant à lui à l’abri du lobbying.
M. Philippe Lamoureux : Nous pensons que la politique de santé doit être portée par l’ensemble de la communauté éducative et non seulement par les médecins et les infirmières scolaires. Avant de se poser la question de savoir si les médecins scolaires sont assez nombreux, leur demande-t-on d’accomplir les actions les plus adéquates ? Ne serait-il pas possible, par exemple, de leur demander de faire un peu moins d’actions individuelles et un peu plus d’interventions collectives, notamment dans les classes ?
La logique du programme national nutrition santé (PNNS) est positive et il n’est pas question de montrer du doigt tel ou tel produit ou telle ou telle marque, même si nous avons parfois des discussions assez franches avec les représentants de l’industrie agroalimentaire. Nous souhaiterions en revanche que certaines marques fassent preuve d’un plus grand souci de vérité dans leur communication car il est toujours étonnant de lire des slogans tels que : « La première sucette sans gras » ou « Mangez de la confiture, vous aurez vos cinq fruits et légumes ». L’INPES n’a pas les moyens de s’opposer à ce type de publicités et le bureau de vérification de la publicité (BVP) étant un office professionnel et non une autorité administrative indépendante ou un établissement public, lui non plus ne peut intervenir sur un plan juridictionnel. Il est vrai, par ailleurs, que les principaux repères de consommation sont bien connus mais qu’il reste à agir.
M. Pierre Morange, coprésident : De quelle manière ?
M. Philippe Lamoureux : À travers un certain nombre de campagnes. Il faudra en outre se rapprocher des lieux de consommation en expliquant par exemple que les fruits et légumes peuvent être frais, surgelés ou en conserve. Il conviendra également de tenir compte du gradient social, la prévention profitant d’abord à ceux qui en ont le moins besoin. Comment travailler auprès des populations les plus concernées ? Cela passe sans doute davantage par des actions de terrain que par des campagnes nationales.
M. Bertrand Garros : À cela s’ajoute la question des alicaments, qui estompe la distinction entre médicament et aliment. La promotion de certains produits tels que des yaourts, par exemple, passe parfois par la mise en avant de leurs effets positifs sur la santé.
M. Jean Mallot, coprésident : Quels autres obstacles rencontrez-vous dans l’exercice de votre mission ?
M. Bertrand Garros : Sans doute faut-il revoir la formation des professionnels de santé afin qu’ils ne prescrivent pas systématiquement à chaque consultation, je pense notamment à une formation à l’écoute et à la gestion de la relation avec le patient.
Quid, par ailleurs, de la coopération interprofessionnelle ? Le médecin doit bénéficier des appuis qui s’imposent dans l’accompagnement de ses patients – que l’on songe par exemple aux maisons de santé. Une expérience est actuellement menée en Poitou-Charentes où des médecins généralistes travaillent avec des infirmières de santé publique. On peut également songer au rôle des pharmaciens. La réorganisation des pratiques permettra peut-être d’améliorer la situation, le bon usage des médicaments supposant une politique globale.
M. Philippe Lamoureux : Des études ont été réalisées afin de déterminer quels sont les freins à la prévention en médecine générale. Le facteur principal, bien compréhensible, est le manque de temps. On peut d’ailleurs s’interroger pour savoir si, telle qu’elle est actuellement conçue, la consultation est adaptée à la pratique de la prévention. Des évolutions positives ont tout de même lieu. Un programme d’accompagnement des patients diabétiques a par exemple été mis en place avec l’assurance maladie.
Autre problème fondamental : l’isolement des pratiques en médecine générale. Sans doute pourrait-on s’inspirer des exemples étrangers tels que les groupes de pairs.
Enfin, il faudrait remédier à la surabondance d’informations qui empêche les médecins de se repérer correctement.
M. Jean Mallot, coprésident : Considérez-vous que les visiteurs médicaux entravent votre action ?
M. Bertrand Garros : Il n’est pas choquant que des laboratoires veuillent être associés à des études sur l’observance, à condition de ne pas être directement intéressés au sort de leurs propres médicaments. Un travail commun est tout à fait envisageable. Sachant néanmoins que, dans les années à venir, le nombre de visiteurs médicaux diminuera de moitié, sans doute pourrait-on envisager des reconversions. Les médecins libéraux du Nord Pas-de-Calais utilisent ainsi les services de ces professionnels pour porter des messages généraux de santé publique.
M. Philippe Lamoureux : Les relations que l’on entretient avec la visite médicale et l’industrie pharmaceutique sont complexes. Les convergences d’intérêt peuvent être par exemple bien réelles quand il s’agit de promouvoir la lutte contre le tabagisme.
Par ailleurs, nous nous sommes interrogés sur la mise en place de visiteurs de santé publique qui interviendraient pour le compte de l’ensemble des agences sanitaires. Ce système, à ce jour, a été écarté en raison de sa complexité et de son coût, mais nous essayons de nous appuyer de plus en plus sur le réseau des délégués de l’assurance maladie, lesquels relaient les informations provenant de l’INPES. Cette collaboration, en l’état, est assez satisfaisante.
M. Jean Mallot, coprésident : Comment pouvez-vous évaluer votre propre action ?
M. Philippe Lamoureux : L’évaluation de la prévention, en dehors des études classiques « post-tests », est délicate en raison de la multiplicité des facteurs qui entrent en jeu. Une campagne sur l’arrêt du tabac sera d’autant plus efficace que, comme c’est le cas en ce moment, elle s’accompagne de diverses autres mesures : augmentation des taxes, avertissements inscrits sur les paquets de cigarettes etc. Il est donc plus facile de mesurer l’évolution des représentations que celle des comportements, même si l’on se dote peu à peu d’un certain nombre d’outils, dont les « baromètres santé » ou le croisement des indicateurs fournis avec des données macro-économiques comme par exemple les ventes de cigarettes, d’alcools ou de casques à vélo.
M. Pierre Morange, coprésident : Rapporteur de la mission d’information sur l’interdiction du tabac dans les lieux publics, j’ai pu me rendre compte de l’efficacité des mesures réglementaires préconisées.
M. Bertrand Garros : Nous nous heurtons également aux délais d’application de ces mesures, mais il est vrai que l’efficacité de la prévention est parfois spectaculaire, comme ce fut le cas dans la lutte contre la mort subite du nourrisson.
Mme la rapporteure a évoqué les médicaments vendus dans les pharmacies, devant le comptoir. Peut-être serait-il utile de procéder à des études de type « post-AMM » – autorisation de mise sur le marché – de manière à évaluer la iatrogénie et à éviter toute banalisation du médicament.
M. Philippe Lamoureux : Des résultats spectaculaires ont été enregistrés dans deux domaines : la lutte contre le tabac et les accidents de la route, où des mesures réglementaires très fortes ont en effet été prises. Sans doute, ces mesures ont-elles été efficaces parce que l’opinion publique était prête à les accepter et à entendre le message diffusé. Qu’en aurait-il été dans d’autres domaines ?
Mme Catherine Lemorton, rapporteure : Une campagne a eu lieu contre la consommation de cannabis.
M. Philippe Lamoureux : En effet et c’est un cas d’école intéressant car la situation est particulièrement complexe entre des centaines de milliers de consommateurs qui pensent consommer un produit bio et des millions de parents qui considèrent que le cannabis est la première étape vers la consommation d’héroïne. Nous avons dû mettre en place une campagne qui correspond à une voie moyenne en attendant de pouvoir mener une nouvelle campagne de prévention, peut-être beaucoup plus brutale, du type de celle qui a été menée contre le tabagisme, précisément parce que l’opinion est plus préparée à l’entendre. Il faut se donner du temps pour « installer le sujet » dans l’opinion. La France a tout de même été le premier pays d’Europe à faire des campagnes de prévention contre l’usage du cannabis.
M. Jean Mallot, coprésident : Je vous remercie pour vos interventions. Nous sommes évidemment prêts à prendre connaissance d’éventuelles contributions complémentaires.
*
Audition de MM. Pierre-Louis Bras, inspecteur général, et Aquilino Morelle, inspecteur, membres de l’Inspection générale des affaires sociales (IGAS).
M. Pierre Morange, coprésident : Je souhaite la bienvenue à M. Pierre-Louis Bras, coauteur du rapport de l’inspection générale des affaires sociales (IGAS), de septembre 2007, relatif à « l’information des médecins généralistes sur le médicament », et M. Aquilino Morelle, coauteur du rapport de l’IGAS, d’août 2007, relatif à « l’encadrement des programmes d’accompagnement des patients associés à un traitement médicamenteux, financés par les entreprises pharmaceutiques ».
Mme Catherine Lemorton, rapporteure : Quelles sont les principales conclusions de vos rapports respectifs ?
M. Pierre-Louis Bras : L’industrie pharmaceutique joue un rôle prééminent dans l’information des généralistes sur le médicament, notamment à travers la visite médicale : chaque médecin reçoit de 300 à 330 visites par an ; les laboratoires dépensent 25 000 euros par généraliste et par an. La visite médicale est évidemment aussi un instrument de promotion, avec trois défauts majeurs au regard des standards de l’objectivité : les visiteurs médicaux sont intéressés aux ventes ; ne sont promus que les médicaments générant une marge ; la visite médicale, qui dure huit minutes, ne permet pas de discuter en détail des produits et de les comparer entre eux. En outre, le coût de la visite médicale est élevé et les laboratoires le font payer à la collectivité.
L’action des pouvoirs publics reste timide. La Haute Autorité de santé, la HAS, vient juste de lancer une politique de communication. Quant aux délégués de l’assurance maladie, les DAM, ils font des contre-visites mais le système est lui aussi coûteux pour la collectivité.
Il conviendrait d’abord de conforter l’action de l’HAS en lui conférant un rôle de promotion du bon usage du médicament, de veille sur la qualité de l’information et d’intervention vis-à-vis des laboratoires.
Il faudrait ensuite exiger des résultats de la part des DAM.
On devrait enfin réduire le volume de la promotion pharmaceutique, information dispensée gratuitement et agréable pour les médecins mais moins objective que la consultation des sites des agences sanitaires, et recourir davantage à l’évaluation des pratiques professionnelles – l’EPP – ou la formation médicale continue.
M. Aquilino Morelle : Par rapport aux visites médicales, les programmes dits d’« observance » ou d’« apprentissage » constituent une figure nouvelle de l’intervention de l’industrie pharmaceutique dans le monde de la santé. Ils sont supposés encadrer les patients pour l’apprentissage technique de l’administration des médicaments et l’éducation thérapeutique en vue de réduire le défaut d’observance. Depuis l’apparition de ces programmes, en 2003, le nombre de dossiers présentés devant l’Agence française de sécurité sanitaire des produits de santé, l’AFSSAPS, a considérablement crû, surtout cette année. Dans le même temps, la France cherchait à introduire dans son droit des dispositions communautaires ouvrant la voie à ce développement, la procédure suivie au départ étant sui generis.
M. Pierre Morange, coprésident : Avez-vous des chiffres illustrant ce développement des programmes d’observance ?
M. Aquilino Morelle : De 2003 à 2008, dix-huit dossiers ont été présentés, quinze ont déjà été examinés, huit acceptés et sept refusés. Les chiffres peuvent paraître limités mais la croissance est soutenue.
M. Pierre Morange, coprésident : Qu’est-ce qu’un « dossier » ?
M. Aquilino Morelle : Il s’agit d’une demande d’autorisation déposée par un laboratoire pharmaceutique en vue d’organiser un programme d’observance, c’est-à-dire, une fois le médicament prescrit, un suivi du patient par téléphone à travers une plate-forme médicale. Le volume de patients est faible, de l’ordre de quelques centaines – les indications portent sur l’ostéoporose ou d’autres maladies nécessitant des traitements très lourds –, mais le mouvement est croissant.
La procédure n’entre dans aucune case juridique existante : ni dans le régime de la publicité grand public, ni dans celui de la publicité professionnelle, ni dans celui de l’information.
Nous n’avons pas prouvé l’existence d’un modèle économique expliquant l’insistance avec laquelle l’industrie pharmaceutique souhaite développer ces programmes. Il est certain, en revanche, qu’ils suscitent une très grande méfiance de la part des autorités de santé, des associations de patients et des acteurs importants du secteur, notamment la revue indépendante Prescrire et de nombreux parlementaires. Cette réticence est fondée sur la crainte que ces programmes ne visent en réalité à fidéliser les patients à un produit donné, avec des préoccupations plus mercantiles que de santé publique.
M. Jean Mallot, coprésident : Ces craintes s’appuient-elles sur l’expérience vécue dans d’autres pays ?
M. Aquilino Morelle : Ces programmes existent ailleurs, notamment aux États-Unis, sous une forme radicale : des publicités directes auprès du grand public pour tous les médicaments. Beaucoup de professionnels ont craint que la France ne s’engage dans cette voie au moment précis où les pays qui l’ont expérimentée tentent de revenir en arrière.
M. Pierre Morange, coprésident : Le caractère particulier du système assurantiel américain introduit lui-même un biais en matière d’observance des traitements. La comparaison est plus aisée avec les autres systèmes européens. Les tendances de fond qui y sont observées offrent-elles un éclairage pour la France ?
M. Aquilino Morelle : Pour ma part, je ne dispose pas d’éléments de comparaison.
M. Pierre-Louis Bras : L’observance est un problème réel mais l’intervention des laboratoires n’est pas la seule manière de la promouvoir. Aux États-Unis, tenant compte du conflit d’intérêt qui risque de toucher les laboratoires, les assureurs ont développé le disease management. En France, une première expérimentation de ce type vient d’être annoncée par la caisse nationale d’assurance maladie des travailleurs salariés, la CNAMTS, avec le service « sophia », qui concerne l’observance et la modification des comportements des patients chroniques. Au Royaume-Uni, le nouveau système de rémunération des cabinets à la performance est destiné à inciter ces derniers à adopter une attitude proactive vis-à-vis du comportement des patients.
L’observance est un vrai problème et les laboratoires, quels que soient leurs espoirs commerciaux, n’ont pas les moyens d’investir à la hauteur des besoins.
M. Aquilino Morelle : Notre première recommandation est d’interdire explicitement dans la loi française le contact direct ou indirect entre l’industrie pharmaceutique et le patient. Il serait toutefois possible de déroger à ce principe de portée générale dans les cas où les programmes seraient conduits au bénéfice exclusif ou principal du patient et non dans une logique condamnable de fidélisation. Nous préconisons de réserver cette possibilité de dérogation aux programmes d’apprentissage, c’est-à-dire portant sur des traitements pour lesquels une petite formation apparaît souhaitable. Pour les programmes d’observance, la plus grande rigueur nous semble requise.
Par ailleurs, pour clarifier la procédure, deux voies sont possibles : laisser l’AFSSAPS continuer d’instruire les dossiers ou en transférer la responsabilité à l’HAS. C’est un choix de pure opportunité qui incombe au ministre.
Il convient aussi d’encadrer le travail des plates-formes pour s’assurer que leur fonctionnement est le plus compatible possible avec l’intérêt des patients et les impératifs de santé publique.
Enfin, il nous paraît nécessaire de promouvoir la position française au sein de l’espace européen. La Commission européenne se montre assez allante sur ce sujet, contrairement à la Cour de justice des Communautés européennes, favorable à une définition la plus large possible de la publicité afin de protéger les populations du contact direct avec l’industrie pharmaceutique. Nous pensons qu’il convient de s’appuyer sur cette jurisprudence.
M. Pierre-Louis Bras : Dans les critères de bonne information qu’elle propose, la Commission européenne considère que l’identité de l’émetteur de l’information n’est pas un élément pour juger de la qualité de cette information, même s’il est animé par un intérêt matériel. Cette appréciation laisserait le champ libre à l’industrie pharmaceutique. Les laboratoires français se défendent d’être intéressés et déclarent que seuls leurs concurrents étrangers souhaitent voir étendues en Europe les pratiques ayant cours ailleurs.
Mme Catherine Lemorton, rapporteure : Le système des visites médicales est-il aussi prégnant, à l’étranger, notamment dans les autres pays européens ? La charte de la visite médicale a-t-elle vraiment modifié les comportements ? Les médecins sont-ils au courant de son existence ? Quand un visiteur médical se présente dans un cabinet de médecine libérale, la jolie plaquette de promotion du produit est-elle toujours accompagnée de sa fiche de transparence ?
M. Pierre-Louis Bras : Les entreprises du médicament – LEEM – et IMS Health ont refusé de nous communiquer des données mesurant l’intensité relative des visites médicales. Nous nous sommes donc appuyés sur des chiffres provenant du réseau international de la CNAMTS et sur ceux concernant les États-Unis, qui sont clairs. Nous avons calculé le rapport entre le nombre des visiteurs médicaux et celui des médecins. Le résultat doit être considéré avec prudence, ce n’est qu’une invitation à la réflexion, mais il en ressort que l’intensité de la visite médicale est plus forte en France que dans tous les autres pays européens – nous arrivons juste après les États-Unis. Si le LEEM, comme il le prétend, dispose d’autres chiffres, qu’il les produise. Le fait est que la rentabilité de la visite est plus élevée en France qu’ailleurs en Europe, ce qui incite les laboratoires à investir.
La charte de la visite médicale a permis d’améliorer la qualité des échantillons, impératif de santé publique. Pour le reste, je ne pense pas qu’elle ait entraîné des progrès significatifs, les laboratoires n’étant pas disposés à s’imposer des contraintes supplémentaires en matière de marketing et de vente. Le volet engagement quantitatif sur le nombre de visites mériterait des commentaires particuliers.
D’après l’observatoire du médicament mis en place par la revue Prescrire, un document est diffusé assez systématiquement : la fiche de transparence. De toute façon, au-delà des documents, l’essentiel, dans la visite, est le face-à-face personnel entre le visiteur et le médecin.
M. Pierre Morange, coprésident : Dans votre rapport, vous préconisez d’aller au bout de la logique qui avait conduit à la création de l’HAS. Parallèlement, vous insistez sur le rôle des DAM. Comment le travail de l’HAS doit-il s’articuler avec celui de la CNAMTS, sur les plans structurel, hiérarchique et budgétaire ?
M. Pierre-Louis Bras : Si nous étions devant une page blanche, il serait nettement préférable de créer un réseau public de visiteurs médicaux adossé à l’HAS plutôt qu’à la CNAMTS.
M. Pierre Morange, coprésident : L’UFC-Que choisir propose la constitution d’un corps de quelque 1 700 délégués, dont un tiers de médecins et deux tiers d’infirmières. Au regard des contraintes budgétaires et de la démographie médicale et paramédicale, cette idée est bien théorique.
M. Pierre-Louis Bras : Si le travail des DAM se médicalise – ce qui est souhaitable –, il faudra certifier leur réseau et exiger des formations équivalentes à celles dispensées par l’industrie pharmaceutique à travers les diplômes de visiteur médical. Des contacts informels existent déjà : la CNAMTS soumet certains documents à l’HAS et tient compte de ses avis. Il faut maintenant formaliser ces habitudes.
Par ailleurs, puisque nous proposons que l’HAS aille plus loin dans la politique de promotion du bon usage, il faut aussi qu’elle puisse recourir à l’outil le plus efficace pour faire passer un message auprès du médecin : la visite en face-à-face. Nous avons donc suggéré que l’HAS perçoive un budget afin de lancer des campagnes de visites médicales. Pour ne pas l’enfermer dans une relation avec la CNAMTS, nous recommandons qu’elle puisse passer des appels d’offres en direction de prestataires de l’industrie pharmaceutique. Si le réseau est subordonné à un double pilotage, cela ne marchera pas, tout le monde le sait.
M. Jean Mallot, coprésident : Comment seraient financés ces moyens supplémentaires dont l’HAS aurait besoin ?
M. Pierre-Louis Bras : Pour quelques campagnes, nous avons chiffré le besoin à un montant de 10 à 18 millions d’euros. Nous proposons par ailleurs d’accroître la taxe sur la promotion.
M. Pierre Morange, coprésident : Ces moyens proviendraient-ils d’un transfert budgétaire de la CNAMTS vers l’HAS ou d’une dotation d’État spécifique prévue dans la loi de financement de la sécurité sociale ?
M. Jean Mallot, coprésident : Si les moyens alloués sont faibles, l’impact sera faible.
M. Pierre-Louis Bras : La plupart des actions entreprises par les pouvoirs publics en direction des médecins visent à contrecarrer les messages envoyés par l’industrie pharmaceutique – essayer, par exemple, de promouvoir à nouveau les antibiotiques de première génération, extrêmement indiqués pour les affections ORL. Par souci des deniers publics, le premier enjeu consiste donc à faire baisser le niveau de la promotion. Ensuite, l’augmentation de la taxe sur la promotion pourrait tout à fait financer cette capacité de l’HAS à mobiliser un réseau de visites médicales.
Mme Catherine Lemorton, rapporteure : Quelle est la place des professionnels de santé dans les dispositifs d’observance ? Les compétences des réseaux existants – maintien à domicile, diabète, toxicomanie, etc. – ne sont-elles pas négligées ? Ne serait-il pas possible d’agir avec ces réseaux ?
M. Aquilino Morelle : Il faudrait non seulement agir avec eux mais aussi les développer. L’éducation thérapeutique est aujourd’hui le parent pauvre de la santé publique, elle-même dans un état peu glorieux. Céder le peu d’espace existant à l’industrie pharmaceutique est une mauvaise idée pour le patient comme pour le corps médical et tout le monde paramédical, notamment les pharmaciens. Cela revient en effet à vider de son sens la relation médecin-malade, rencontre d’une conscience et d’une confiance, et à dénaturer toute la chaîne médicale. Tous les professionnels, à commencer par l’Ordre des médecins et l’Ordre des pharmaciens, sont rétifs à l’idée de confier l’éducation thérapeutique à l’industrie pharmaceutique. Les réseaux de santé devraient être développés dans des conditions garantissant leur indépendance.
M. Pierre Morange, coprésident : Il faut rappeler que la CNAMTS vient de prendre des mesures d’éducation thérapeutique, concernant notamment la prise en charge du diabète et la prévention des amputations qui peuvent en résulter.
Mme Catherine Lemorton, rapporteure : Quels types de personnels la CNAMTS emploie-t-elle, ou compte-t-elle employer, sur ses plates-formes téléphoniques ? S’agit-il d’acteurs des réseaux ou de nouvelles recrues ?
M. Pierre-Louis Bras : Il vaudrait mieux poser la question à la CNAMTS mais j’ai cru comprendre qu’il s’agirait d’infirmières. Sur les plates-formes téléphoniques d’accompagnement des patients victimes de maladies chroniques, la personne qui répond doit avoir une capacité de jugement clinique. À l’étranger, ce sont au minimum des personnels paramédicaux. Je suppose que la CNAMTS va recruter des infirmières. Mais l’expérience est limitée puisqu’elle ne concernera que 136 000 patients.
M. Jean Mallot, coprésident : Les moyens contemporains de communication modifient les relations, d’une part, entre les laboratoires et les prescripteurs, d’autre part, entre les laboratoires et les patients. Quel est l’impact qualitatif, notamment du point de vue de la maîtrise ?
M. Pierre-Louis Bras : N’ayant pas travaillé sur le thème de l’automédication, je préfère m’abstenir de répondre.
M. Aquilino Morelle : La Cour de justice des Communautés européennes a explicitement reconnu le caractère de publicité aux informations sur les médicaments diffusées sur Internet. Je n’ai pas visité les sites un par un, j’ignore ce qui s’y passe en pratique, mais ils restent soumis à l’interdiction.
Mme Catherine Lemorton, rapporteure : Avez-vous entendu des médecins formuler des demandes précises en ce qui concerne leur information ?
M. Pierre-Louis Bras : Nous avons réalisé un sondage auprès des médecins sur la façon dont ils ressentent l’information. D’abord, 90 % d’entre eux se disent bien informés sur les médicaments. Leur première attente porte sur la comparaison entre médicaments, pour établir un bilan des mérites respectifs. D’autres demandes concernent les aspects pratiques – effets secondaires, posologies, prescriptions pédiatriques, etc.
Mais les besoins varient selon la typologie des médecins. Ceux qui travaillent intensément voient beaucoup de visiteurs médicaux et sont très satisfaits de leur information sur les médicaments. Ceux qui voient le moins de visiteurs médicaux sont les plus insatisfaits de leur information sur les médicaments. Attention, cela peut signifier que les médecins les plus critiques vis-à-vis de la visite médicale sont ceux qui s’estiment mal informés et qui se montrent les plus proactifs dans la recherche d’informations. Et puis, entre les deux extrémités du spectre, il existe une série d’attitudes intermédiaires. Il ne faut jamais généraliser.
Pour les médecins, la visite médicale est ambivalente : ils la voient comme un outil intéressant, adapté à leur pratique – les visiteurs sont de bons commerciaux, disponibles, agréables, qui patientent longtemps dans la salle d’attente –, mais ils ne sont que 27 % à la considérer objective. Les médecins pensent avoir une capacité de discernement vis-à-vis de ces démarches commerciales mais certaines études anglo-saxonnes montrent que tout le monde a tendance à s’illusionner sur sa capacité de résistance à un message commercial bien présenté.
En revanche, les médecins plébiscitent les autorités sanitaires pour leur objectivité, à hauteur de 90 % environ. Néanmoins, l’information qu’elles dispensent paraît moins adaptée.
La CNAMTS et les DAM sont jugées objectives à 46 %. Elles suscitent moins de suspicion que l’industrie pharmaceutique mais les médecins leur reprochent de dévaloriser les médicaments, pour des motifs économiques.
M. Pierre Morange, coprésident : Avez-vous programmé une évaluation des dispositifs de plates-formes téléphoniques ?
M. Pierre-Louis Bras : Ce n’est pas au programme de l’IGAS, mais la CNAMTS a prévu une évaluation de cette expérience à l’horizon 2010.
M. Pierre Morange, coprésident : C’est une idée que j’ai véhiculée car un regard extérieur ne serait pas inintéressant. Je crois aussi que la MECSS pourrait se saisir du sujet.
M. Pierre-Louis Bras : La CNAMTS pilote deux initiatives : Infosoins, qui concerne l’ensemble de la population, et un dispositif plus spécifique.
Mme Catherine Lemorton, rapporteure : Avez-vous perçu une différence de comportement entre les médecins installés depuis de nombreuses années et ceux qui sortent de leur formation hospitalo-universitaire ?
M. Pierre-Louis Bras : Dans la typologie, l’âge est effectivement un critère discriminant : plus le médecin est âgé, plus il rencontre de visiteurs médicaux et plus il leur fait confiance ; plus il est jeune, plus il a une démarche proactive pour rechercher de l’information. Mais je ne suis pas sûr que ce vecteur de différenciation soit déterminant.
M. Pierre Morange, coprésident : Nous vous remercions.
*
Audition de M. Philippe Brunet, directeur du cabinet du commissaire européen en charge de la santé.
M. Pierre Morange, coprésident : Je souhaite la bienvenue à M. Philippe Brunet, directeur du cabinet du commissaire européen en charge de la santé, M. Markos Kyprianou. Nous souhaitons recueillir son analyse sur la prescription, la consommation et la fiscalité des médicaments en Europe.
Mme Catherine Lemorton, rapporteure : Quelles sont les missions de la Commission européenne en matière de médicament ?
M. Philippe Brunet : Historiquement, la politique européenne du médicament a commencé avec la préoccupation d’instituer un marché commun du médicament. Le médicament, comme tout autre bien de consommation, a d’abord été pris en compte du point de vue de l’autorisation. L’harmonisation des critères et la mise en commun des résultats de l’évaluation n’ont pas suffi car les États membres arrivaient à des conclusions différentes sur le même médicament. Après plus de vingt ans, nous sommes parvenus à mettre en place des procédures communautaires centralisées, en vigueur depuis cinq ou six ans : une évaluation unique par l’Agence européenne des médicaments, située à Londres ; une décision d’autorisation de mise sur le marché – AMM – délivrée par la Commission, pour 90 % des médicaments nouveaux ; une évaluation par chacun des États membres pour les 10 % restants et une obligation de reconnaissance mutuelle des autorisations délivrées, avec, en cas de désaccord, une procédure communautaire.
Le début de l’harmonisation est intervenu en 1965, après l’affaire de la thalidomide. La Commission a maintenu les industries pharmaceutiques dans le portefeuille du commissaire à l’industrie, actuellement détenu par M. Günter Verheugen, vice-président de la Commission. Le commissaire à la santé est associé à la gestion de la législation sur les médicaments, notamment pour tout ce qui a trait aux conditions d’autorisation, aux mises à disposition de médicaments et à l’évaluation de la valeur thérapeutique ajoutée. Si le Traité de Lisbonne est ratifié et entre en vigueur, il y a de fortes chances que les portefeuilles soient réaménagés car la base juridique concernant la gestion des médicaments sera l’article 152, qui traite de la santé.
De 95 à 98 % de la législation relative aux AMM est totalement harmonisée : la marge de manœuvre des États membres est donc de très réduite à nulle.
La législation sur les essais cliniques n’est pas la plus efficace parce qu’elle ménage deux ou trois approches différentes, liées à des considérations culturelles et aux différences de puissance de l’industrie pharmaceutique d’un pays à l’autre. Le texte esquive soigneusement les questions d’éthique – les comités d’éthique nationaux conservent la totalité de leurs compétences – mais les États fédéraux et ceux qui ont régionalisé la gestion de ce dossier posent un problème particulier, car des approches totalement différentes peuvent prévaloir d’une partie à l’autre du pays. Ainsi, en Allemagne fédérale, les comités d’éthique des länder traditionnellement protestants et ceux des länder à dominante catholique émettent des avis différents.
L’objectif principal de la directive sur les essais cliniques était d’harmoniser plus ou moins complètement les critères scientifiques de manière à entraîner une économie d’échelle au niveau communautaire. De fait, la plupart des médicaments font maintenant l’objet d’essais cliniques multicentriques, impliquant de larges cohortes de patients, et nous devons tout faire pour les maintenir en Europe, ce qui n’est pas acquis, loin de là. De ce point de vue, la directive a échoué car elle n’a pas inversé la tendance à la délocalisation des essais.
M. Pierre Morange, coprésident : Avez-vous des chiffres précis ?
M. Philippe Brunet : Même l’industrie pharmaceutique ne les communique pas.
M. Pierre Morange, coprésident : Mais quel est votre sentiment ?
M. Philippe Brunet : Tout dépend des domaines. Dans les années soixante et soixante-dix, l’Europe était leader ; aujourd’hui, elle parvient péniblement à conserver sa seconde place, devant l’entité Japon-Australie.
M. Pierre Morange, coprésident : Les essais sont-ils délocalisés vers des plates-formes occidentales ou au profit de territoires aux systèmes de surveillance sanitaire rudimentaires ?
M. Philippe Brunet : Les deux. Ce ne sont pas les essais cliniques qui se délocalisent mais le promoteur. Une fois celui-ci parti aux États-Unis, c’est la Food and Drug Administration, la FDA, qui prend le contrôle en charge.
Dans le système communautaire centralisé, lorsqu’il présente les données résultant de ses essais cliniques, le demandeur doit justifier de ses méthodes, de son lieu et de ses modalités de travail, examinées en particulier sous l’angle de l’éthique. Si ces modalités ne sont pas validées, il risque toujours de se voir opposer un refus de prise en compte des résultats. La pression exercée sur le promoteur est énorme car ces essais cliniques coûtent excessivement cher et il court le risque de devoir les refaire voire, pire, de ne pas obtenir l’AMM ou une indication particulière.
La directive sur les essais cliniques a cherché à intensifier l’interpénétration entre recherche académique publique et recherche privée, qui constitue la clé des succès américain, australien et maintenant japonais dans le domaine du médicament. Hélas ! en dépit de nouveaux programmes communs de recherche, cette fécondation mutuelle entre le public et le privé n’est pas la règle au niveau européen, hormis dans certains États membres comme le Royaume-Uni.
M. Pierre Morange, coprésident : Pouvez-vous nous citer quelques exemples de délocalisations ?
M. Philippe Brunet : C’est très difficile car nous ignorons quels étaient les choix ex ante ! Certaines délocalisations sont motivées par des considérations scientifiques. C’est notamment le cas pour les recherches concernant les vaccins contre le sida. De très larges cohortes sont requises, dans des pays où la prévalence est extrêmement élevée, c’est-à-dire en Afrique noire ou en Thaïlande, pas en Europe. Par contre, pour d’autres médicaments, le promoteur recherche des facilités hospitalières qu’il trouvera très facilement aux États-Unis mais pas en Europe.
La Belgique, par exemple, dispose d’hôpitaux de bon niveau dans la plupart de ses grandes villes. Mais cet État fédéral, qui comporte trois régions et où l’on parle deux langues, a mis en place un système de comités d’éthiques locaux, par hôpital, mal coordonné au niveau national. Par conséquent, pour y conduire un essai multicentrique dans quatre établissements, il faut gérer quatre procédures, qui durent de deux à cinq ans. En Californie, État presque aussi grand que la France, l’avis est rendu dans les quatre mois, alors le promoteur n’hésite pas !
L’autre facteur déterminant est la qualité des infrastructures hospitalières mises à disposition, son potentiel d’expertise et l’excellence de son plateau technique.
M. Pierre Morange, coprésident : Venons-en à l’aval. Certaines stratégies thérapeutiques sont admises sur le continent européen et refusées outre-Atlantique, ou l’inverse. Qu’en pensez-vous ?
M. Philippe Brunet : Avant d’évoquer l’aval, je précise qu’il serait mal avisé de penser que l’AMM relève exclusivement de critères scientifiques. Lorsqu’un médicament est soumis à l’étude d’experts américains et européens en vue de sa validation sur la base des mêmes essais cliniques, les indications et les conditions de mise sur le marché peuvent être différentes. En effet, la valeur attachée à l’effet clinique final est différente entre l’Europe et les États-Unis. Les Américains ont une conception plus arithmétique, plus chiffrable de l’effet. Ils accorderont l’AMM à un médicament anticancéreux permettant une survie de quatre mois tandis que les Européens la refuseront ou le classeront en produit de deuxième ou de troisième ligne de défense. De même, à partir de quel taux de répondants l’AMM doit-elle être accordée ? 5 % ? 20 % ? La décision ne dépend pas de critères scientifiques.
Une fois l’AMM accordée, la question consiste à déterminer la place du nouveau médicament dans l’arsenal thérapeutique. Le choix a des conséquences importantes en termes de stratégie thérapeutique mais aussi de prix et de taux de remboursement. En France, on appelle cela l’amélioration du service médical rendu, en Europe, c’est la valeur thérapeutique ajoutée et, aux États-Unis, le health technology assessment.
Dans cette procédure, deux phases doivent être distinguées : d’une part la démarche purement scientifique, qui consiste à calibrer les thérapeutiques existantes, à y classer le nouveau médicament et à déterminer sa valeur thérapeutique intrinsèque ; d’autre part la conclusion économique, à savoir le choix du niveau de remboursement. Il me semble que la deuxième phase doit être laissée à l’appréciation des payeurs, c’est-à-dire des États membres ou des systèmes d’assurance, quels qu’ils soient. La première phase, en revanche, doit être gérée au niveau européen, sans quoi le patient n’y comprend rien et les conditions de l’AMM risquent d’être indirectement remises en cause. Hormis les antibiotiques, dont l’efficacité varie avec la géographie à cause des phénomènes de résistance, les effets des médicaments sont identiques partout. Après, la décision de rembourser doit incomber aux États membres ou aux systèmes d’assurance. Mais, si l’efficacité d’un médicament est reconnue dans vingt-six pays, son inefficacité ne saurait servir de prétexte à refuser son remboursement dans le vingt-septième. Cela accentuerait encore la mobilité des médicaments et des patients, déjà très forte dans les régions frontalières – entre la Catalogne espagnole et la Catalogne française, entre Liège ou Maastricht et Aix-la-Chapelle ou encore entre Strasbourg et la rive droite du Rhin. Au XXIe siècle, il est inconcevable qu’un médicament perde une indication en traversant la frontière alors que les structures hospitalières des deux pays fonctionnent en réseau, d’autant que les produits nouveaux ont fait l’objet d’une AMM communautaire et sont théoriquement accompagnés de la même notice partout. Bref, pour les médicaments du futur, la prochaine étape à franchir, au niveau communautaire, serait de créer un embryon de commission de transparence, tandis que la fixation des prix resterait une prérogative nationale.
Nous sommes préoccupés par le fait qu’aucun État membre n’est armé pour faire face aux nouvelles approches thérapeutiques. Dans les dix ans à venir, une branche va énormément se développer, en lien avec le décodage du génome humain : les médicaments personnalisés. Contrairement à une idée reçue, il ne s’agit pas forcément de nouveaux médicaments mais de produits très efficaces dans 20 % des cas, moyennement efficaces dans 20 % des cas et complètement inefficaces dans 60 % des cas, pour des raisons d’expression génétique. La prescription sera donc précédée d’une détermination de la présence du gène incriminé. Ce pilotage en amont de la prescription, en particulier pour les médicaments anticancéreux, est comparable à ce que fut l’antibiogramme dans les années cinquante. Mais il réduira à néant la pertinence de la notion d’ « amélioration du service médical rendu » ou de « valeur thérapeutique ajoutée ».
M. Pierre Morange, coprésident : Sans parler des modifications de méthodologie statistique des essais cliniques.
M. Philippe Brunet : Absolument. Il nous faudra par conséquent revoir les critères de sécurité, d’efficacité et de prescription des médicaments. Des produits aujourd’hui frappés d’une prétendue inefficacité notoire devront être remboursés si la population sur laquelle ils font effet peut être identifiée. Et cette méthodologie s’appliquera vraisemblablement aussi en psychiatrie.
La classification des médicaments sera également compliquée par le passage de certains produits de la catégorie curative ou palliative, à la catégorie préventive. Pour certaines maladies cardiaques – infarctus, lésions coronaires –, qui auraient dû apparaître après quarante, cinquante, soixante ou soixante-dix ans, la détermination de gènes dès l’adolescence et la prescription de bêtabloquants adaptés permettront de faire cesser le compte à rebours.
L’Union européenne, en particulier la Commission, est très souvent accusée de s’occuper de ce qui ne la regarde pas. Les essais cliniques et les AMM sont pris en compte au niveau communautaire. Les critères médicaux et techniques de l’aval devraient également l’être, par cohérence, pour des considérations économiques mais aussi parce que L’Europe, avec 480 millions de personnes, peut ambitionner un rôle de leader mondial si elle se dote d’un système intégré. Contrairement aux administrations nationales, qui rechignent à jouer le jeu de la coopération européenne, la communauté scientifique est prête.
Mme Catherine Lemorton, rapporteure : Dans quels pays de l’Union l’industrie pharmaceutique est-elle particulièrement puissante ?
M. Philippe Brunet : L’Angleterre, la France et l’Allemagne disposent d’une industrie pharmaceutique de pointe. Le reste est anecdotique. J’exclus la Suisse, où sont situées deux des plus grandes compagnies pharmaceutiques. Lorsque nous avons discuté des essais cliniques, le gouvernement anglais s’est montré très préoccupé de se prémunir contre une délocalisation de sa recherche. Les Français sont aussi vigilants. Les pays où sont menés très peu d’essais cliniques sont prêts à accepter n’importe quoi car ils ne se sentent pas concernés.
Outre la puissance de l’industrie pharmaceutique, le potentiel en nombre de patients et en infrastructures hospitalières joue également.
En outre, dans ce domaine comme dans les autres, l’absence de brevet communautaire est dramatique. À force de refuser de déposer des brevets en anglais, dans quinze ou vingt ans, nous serons obligés d’en déposer en coréen, en hindi ou en mandarin ! Personne ne traduira jamais un brevet en vingt-sept langues. L’Union a essayé de réduire le nombre de langues employées – anglais, français, allemand et espagnol – mais la directive transposant le brevet européen en brevet communautaire est en complète déshérence. La Commission actuelle et la précédente s’y sont cassé les dents. Si la prochaine Commission et le prochain Parlement n’accomplissent pas une percée, les conséquences à long terme seront désastreuses. En effet, quand les compagnies voudront un parapluie de propriété industrielle assez vaste, ils iront ailleurs, là où toute la production sera délocalisée.
M. Jean Mallot, coprésident : Certains insistent sur la singularité de la France en matière de prescription médicamenteuse, d’autres défendent des thèses moins affirmatives. Qu’en pensez-vous ? Disposez-vous d’éléments chiffrés permettant de trancher ?
M. Philippe Brunet : Il est injustifié de stigmatiser une singularité française. La quantité de prescriptions est peut-être plus élevée qu’à l’étranger mais les habitudes de prescription pourraient également être appréciées à l’aune de la qualité des soins. Les incidents indirects – par exemple la progression de la résistance aux antibiotiques pour les accidents thérapeutiques – sont beaucoup trop fréquents au regard du niveau de développement de l’Europe. Beaucoup de progrès sont à accomplir dans ce domaine.
Cela dit, les habitudes de prescription passent d’abord par l’attitude individuelle des médecins et les caractéristiques de l’environnement économique. Les données sont difficilement comparables d’un pays à l’autre. À Chypre, 50 % des citoyens sont couverts par une assurance volontaire. En Grande-Bretagne, les médecins sont sévèrement encadrés par les instances locales du National Health Service, le NHS, à travers un système décentralisé où la liste des médicaments prescriptibles varie selon le code postal. L’Espagne, la Grèce et, dans une moindre mesure, l’Italie sont régies par des systèmes analogues. Si elles veulent relever les défis actuels, elles devront changer de méthode. Il est tout de même incroyable que les Anglais traversent la Manche pour venir chercher leurs médicaments en France ou en Belgique.
En résumé, les habitudes de prescription dépendent du régime d’assurance dont le patient relève, de son médecin et de l’organisation du système de soins. Le biais scientifique est la seule façon d’infléchir la situation, par un dialogue avec les médecins, qui doit débuter à l’université. Malheureusement, dans les cours de médecine, les maladies sont décrites en détail tandis que les traitements sont traités en cinq lignes.
M. Pierre Morange, coprésident : Que pensez-vous de la qualité scientifique et de la certification sanitaire dans les pays où sont inexorablement délocalisés les centres de production de génériques, notamment la Chine et l’Inde ?
M. Philippe Brunet : En théorie, que le médicament soit générique ou princeps, les critères étudiés pour l’AMM sont identiques : le dossier doit décrire les méthodes de fabrication, la qualité des substances de base et les mécanismes de surveillance.
M. Pierre Morange, coprésident : En théorie ?
M. Philippe Brunet : Aux États-Unis, la FDA envoie systématiquement des inspecteurs chez les fabricants des pays tiers. L’Europe, quant à elle, s’est inspirée du système qui existait dans la plupart des États membres : nous attendons des signes d’appel pour envoyer une équipe de contrôle. Chaque système a ses avantages et ses défauts. En voulant tout contrôler, les Américains perçoivent une sorte de bruit de fond de problèmes qui les empêche d’identifier le problème le plus important. Le système européen semble finalement plus efficace.
M. Pierre Morange, coprésident : Avez-vous accès aux informations de la FDA ?
M. Philippe Brunet : Les agences nationales des pays européens et la FDA étaient liées par des accords bilatéraux. À la fin des années quatre-vingt-dix, un accord de coopération a été signé entre l’Agence européenne des médicaments et la FDA. Dès qu’un rapport d’inspection identifie un problème particulier, l’autorité l’envoie à son homologue.
Mme Catherine Lemorton, rapporteure : La France, et plus généralement l’Europe, font parfois preuve d’une grande inertie face à des médicaments au service médical rendu contestable. Le Canada, il y a six ou sept mois, a modifié les indications figurant sur la notice d’emploi de l’antibiotique Ketek, mais aucune modification n’est encore intervenue en Europe. Je pourrais donner d’autres exemples.
M. Philippe Brunet : Pourquoi les Canadiens ont-ils modifié la notice ?
Mme Catherine Lemorton, rapporteure : À cause des effets secondaires du médicament.
M. Philippe Brunet : Le milieu pharmaceutique reproche souvent à l’Europe d’être trop conservatrice sur les indications. Les décisions dépendent aussi des autres médicaments disponibles : la révision de la notice est plus facile s’il existe une alternative sur le marché. Par ailleurs, la révision est encadrée par le droit : si sa motivation est fragile, il arrive que la Cour de justice des Communautés européennes annule une décision.
Les systèmes européens de surveillance du marché et de pharmacovigilance sont, certes, largement perfectibles, mais leur efficacité est comparable à celle constatée aux États-Unis.
Je ne connais pas le cas que vous évoquez mais je répète que les antibiotiques présentent la particularité de ne pas être aussi efficaces dans toutes les régions du monde.
M. Pierre Morange, coprésident : J’ai été l’un des premiers à souligner le caractère problématique de l’application de la directive Bolkestein dans le secteur médico-social. Où en est le texte sur les services ?
M. Philippe Brunet : La question est opportune. Pour moi, ce sujet est un grand motif de frustration.
La directive proposée par le commissaire Bolkestein excluait les services de santé, non pas par refus de toute approche communautaire mais par volonté d’adopter une démarche spécifique pour ce secteur. Entre-temps, la Cour de justice des Communautés européennes a renforcé la jurisprudence précédente, confirmant que l’article 49 s’appliquait aux services de santé.
Il a donc été convenu de rédiger une directive relative aux services de santé mais prenant le contre-pied de la directive Bolkestein, d’une part, parce qu’elle était axée non pas sur les services mais sur la mobilité du patient et, d’autre part, parce qu’elle ne cherchait pas à modifier la détermination du droit applicable ou de la règle d’origine.
M. Pierre Morange, coprésident : Le sujet est hautement stratégique.
M. Philippe Brunet : Ce projet visait donc à codifier la jurisprudence et à élaborer un volet exclusivement santé afin d’y intégrer des aspects très prometteurs à moyen et long terme comme la création de centres thérapeutiques de référence ou l’adoption d’une approche en matière de technologies de l’information. La directive faisait pour la première fois référence au principe selon lequel sa transposition devait respecter les trois principes fixés par le conseil des ministres de la santé : universalité des soins, solidarité et équité. Enfin, elle prévoyait un mécanisme tendant à améliorer et à homogénéiser la qualité des soins en Europe – une femme victime d’un cancer du sein n’a pas la même espérance de vie en Lituanie et en France, même avec un traitement identique.
Il nous semblait important qu’une directive sur la mobilité des patients voie le jour. De fait, lors des négociations du traité de Lisbonne, il est apparu que plus aucun État ne contestait l’application de l’article 49 – c’est-à-dire des règles concernant la prestation de service – aux services de santé, conformément à la jurisprudence de la Cour de justice des communautés européennes, constante depuis trente ans. Par ailleurs, tout le monde admet dorénavant que des dispositions spécifiques sont nécessaires. Enfin, eu égard à la spécificité des services de santé, au fait que le payeur est souvent l’État, aux problèmes de planification des soins, à la difficulté d’équilibrer les budgets sociaux, aux différences d’organisation géographique selon les États membres, il a été décidé de ne pas transférer les compétences vers l’échelon européen mais de coordonner les systèmes nationaux. La Commission a suivi l’avis du Parlement européen, qui avait posé ces trois critères.
Le hic est que ce texte essentiel arrive pratiquement en fin de mandat de la Commission puisque la Commission Barroso quittera ses fonctions en novembre 2009. Nous sommes également en période de ratification, et force est de constater que l’Europe, dès qu’elle cherche à s’occuper des citoyens, est toujours bloquée par des échéances électorales. Enfin, les discussions n’aboutiront pas d’ici à la fin du mandat du Parlement européen, c’est-à-dire juin 2009, et même janvier 2009, mois à partir duquel l’accord institutionnel avec la Commission stipule que plus aucun texte législatif ne sera examiné.
Le texte est donc prêt. Le commissaire en charge de la santé y est favorable.
M. Pierre Morange, coprésident : Serait-il possible que nous disposions de ce texte, ou du moins de son exposé des motifs ?
M. Philippe Brunet : Il n’est pas encore formalisé.
Un formidable créneau d’opportunité s’offrait à nous car la France s’apprête pour la dernière fois à occuper la présidence tournante dans le cadre que nous connaissons.
La directive Bolkestein a été débarrassée de tous ses défauts et cela a permis de donner corps à la solidarité européenne dans le domaine sanitaire. Les Français, les Britanniques et les Allemands peinent à le comprendre mais certaines thérapeutiques et certains plateaux techniques ne seront jamais disponibles dans certains pays, même dans certains pays riches comme le Luxembourg. Nicosie ne possédera jamais l’arsenal de l’Assistance publique - Hôpitaux de Paris ; or les Chypriotes, faute d’accord communautaire, se font soigner au Liban plutôt que dans un autre État membre de l’Union.
Cela me fait penser à un autre problème : celui des technologies de l’information. Nous gérons les technologies du XXIe siècle avec des procédures datant du XIXe siècle. Les modèles bismarckien et beveridgien sont éminemment respectables mais complètement inadaptés aux puces électroniques. Le nec plus ultra de l’interprétation d’imageries de scanner se trouve parfois à Birmingham ou à Munich même si l’examen a été pratiqué à Paris. Or la coordination réalisée dans l’aviation, les transports, le secteur bancaire ou la finance n’existe toujours pas en matière de santé.
M. Pierre Morange, coprésident : Si je ne m’abuse, Bruxelles a expérimenté un système de compression numérique des images pour permettre leur transfert, et cela a donné lieu à des certifications.
M. Philippe Brunet : Il faut simplement régler le problème de l’interopérabilité. Puisque les opérateurs de GSM n’arriveront jamais à se mettre d’accord sur un standard, les décideurs politiques doivent trancher et choisir le système le plus adapté. Avec la directive, c’était possible.
Je pense que le texte est mûr mais, compte tenu du calendrier, je suis pessimiste quant à son adoption.
M. Pierre Morange, coprésident : La MECSS souhaite vivement que des documents précisant l’état de la réflexion lui soient communiqués. Il ne faudrait pas que l’Europe, après la directive Bolkestein, fonce à nouveau dans le mur. La présidence française constitue peut-être une opportunité pour se mettre en conformité avec l’histoire.
M. Philippe Brunet : J’ajoute que cette directive est le premier texte communautaire à prévoir la mise en place d’une structure d’évaluation des thérapeutiques.
M. Pierre Morange, coprésident : La directive traite-t-elle spécifiquement de la question du médicament ?
M. Philippe Brunet : Non. Elle porte sur tout l’arsenal réglementaire en vigueur et aborde pour la première fois la question de l’aval, c’est-à-dire les essais cliniques et les AMM, avec la mise sur pied d’un réseau de coordination autour d’une commission de transparence.
M. Pierre Morange, coprésident : Je vous remercie.
*
Audition de M. Bruno Toussaint, directeur de la rédaction de la revue Prescrire.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale.
Mme Catherine Lemorton, rapporteure : Nous nous intéressons aux prescriptions et à la consommation de médicaments et à la fiscalité du médicament en France.
Quel est votre avis sur la prégnance de la visite médicale auprès des médecins libéraux ? Que pensez-vous de l’arrivée de me-to sur le territoire ? Les médecins sont-ils suffisamment bien informés de manière indépendante ?
M. Bruno Toussaint : Je vous ai amené le numéro du journal Impact Médecine, du 24 janvier 2008. Dès la page 2, on peut lire une publicité pour Sanofi : « Nous vous accompagnons depuis les études jusqu’à la retraite. »
De fait, les firmes pharmaceutiques entourent les médecins prescripteurs depuis la faculté jusqu’à tard dans leur vie. Beaucoup d’enseignants sont des leaders d’opinion – parfois sous influence. Dans les staffs hospitaliers, les étudiants rencontrent déjà les visiteurs médicaux dans les couloirs. Certains s’étonnent que la salle de travail des externes soit parfois réquisitionnée pour recevoir les visiteurs médicaux. Ce sont d’ailleurs ces derniers qui financent le pot d’arrivée ou le pot de départ de tel ou tel. Il y a souvent, dans les hôpitaux, des dossiers de comptes rendus d’électromyogrammes, ou de je ne sais quel examen complémentaire, qui sont estampillés avec telle marque de tel produit de telle firme pharmaceutique.
Vous avez sans doute constaté, lors de vos auditions, que, dans nos pays, les pouvoirs publics sous-traitaient aux firmes pharmaceutiques beaucoup de choses qui ne sont pas de leur domaine. Ce sont elles qui financent totalement ou presque la recherche clinique, ou la formation, en tout cas dans certaines disciplines. Il existe des officines, subventionnées par les firmes pharmaceutiques, qui préparent les externes qui s’inscrivent aux concours de spécialistes.
Une fois installés, les médecins ont recours à la visite médicale. Vous avez examiné le rapport de l’IGAS sur ce sujet ; il est explicite. La revue Prescrire a animé, pendant une quinzaine d’années, un réseau d’observation de la visite médicale. Les données étaient à chaque fois convergentes. La visite médicale est agréable pour celui qui la reçoit, mais il s’agit, pour le visiteur médical, de vendre en augmentant les avantages du médicament et en diminuant ou en omettant ses effets indésirables, les contre-indications, les interactions médicamenteuses, bref tous les défauts. On ne peut pas s’attendre au contraire : la visite médicale est une force de vente et il est logique que les vendeurs mettent en avant les qualités du produit et pas ses défauts.
Intervient ensuite la formation médicale continue. Dans ce rapport, il apparaît que l’essentiel du financement vient des firmes pharmaceutiques. Encore une fois, en sous-traitant, on économise les deniers publics, mais cela a des effets sur les prescriptions et les dépenses de sécurité sociale, et probablement aussi sur la qualité des soins.
Dans notre système français et en Europe, aux États-Unis d’Amérique et au Japon, qui sont les trois grands marchés du médicament actuellement, la règle est à peu près la même. Pour que le médicament soit autorisé sur le marché, la firme doit obtenir l’AMM, mais il n’est pas exigé que celui-ci présente un progrès thérapeutique. À partir du moment où la balance bénéfice-risque paraît acceptable par rapport à un placebo, la nouveauté est autorisée. Souvent elle n’apporte pas de progrès. Cependant, il faut bien vendre ce médicament et l’on déploie pour cela beaucoup d’énergie : d’où les visiteurs médicaux, les congrès, la mobilisation des leaders d’opinion. Et cela fonctionne.
Un bon exemple est celui d’un médicament anti-cholestérol, la Provastatine, vendue en France sous la marque Tahor par Pfizer. Il est arrivé après d’autres médicaments de la famille, pour lesquels il existait des preuves scientifiques de bon niveau, une efficacité concrète puisqu’ils permettaient de réduire les accidents cardiovasculaires, voire la mortalité des patients. Cette nouveauté est arrivée sans apporter ses preuves dans son dossier, parce qu’elle était trop récente. Malgré tout, elle était très bien placée, voire au top de cette catégorie.
Mme Catherine Lemorton, rapporteure : Les professionnels se rendent tout de suite compte lorsque des contournements de génériques arrivent sur le marché. Ces contournements ont-ils la même prévalence dans les autres pays de l’Union européenne, l’Allemagne par exemple ? Est-ce que les autres pays, une fois que la politique du générique est décrétée, font en sorte que ces contournements ne s’imposent pas ?
M. Bruno Toussaint : Je ne connais pas précisément le marché des autres pays, mais la tactique de contournement des génériques n’est pas spécifique à la France. Le développement d’isomères, de métabolites, de nouveaux dosages, de nouvelles formulations est multinational. Il y a quelques années, le brevet de l’oméprazole Mopral, l’un des médicaments les plus efficaces contre l’acidité digestive, le reflux oesophagien, l’ulcère de l’estomac, etc., arrivait à échéance. La firme Astra a préparé le lancement d’un successeur, qui est un isomère, avec une partie de la substance active, que les patients absorbaient déjà. Il n’apportait aucun progrès thérapeutique. Pourtant il y eu un vrai lancement, au point que, sur la base d’accueil du site internet du Congrès américain, figurait un bandeau publicitaire prônant le nouveau médicament. Il faut dire qu’aux États-Unis, la publicité pour les médicaments est autorisée, même sur un tel site.
M. Pierre Morange, coprésident : Selon vous, quelles seraient les mesures à adopter pour que les dispositions concernant le service médical rendu (SMR) ou l’amélioration du service médical rendu (ASMR) prennent en compte le critère sanitaire, voire le critère médico-économique ?
M. Bruno Toussaint : Il y a sûrement quelque chose à faire. Une solution radicale serait de modifier les critères d’octroi de l’AMM. Si l’on exigeait des preuves de progrès thérapeutique, cela faciliterait beaucoup les choses pour la suite.
Il faudrait aussi sortir les firmes pharmaceutiques du conflit d’intérêts où les pouvoirs publics les placent. Encore une fois, on leur demande de développer les produits, de financer la recherche, de présenter leurs dossiers et de demander l’autorisation de vendre ces produits qui les font vivre. C’est tout de même préoccupant. Les dérapages sont inévitables.
Il faudrait enfin, à plus court terme, garantir véritablement l’indépendance des experts qui examinent les dossiers d’AMM et de transparence.
M. Pierre Morange, coprésident : Au-delà d’une déclaration générale sur l’amélioration des critères utilisés pour l’évaluation du SMR, quels seraient selon vous, et de manière plus précise, les éléments à mettre en œuvre ?
M. Bruno Toussaint : Actuellement, en France, l’appréciation de l’amélioration du service médical rendu par la commission de la transparence est plutôt indulgente, mais elle n’est pas caricaturale. La commission se prononce sur le dossier qu’on lui soumet pour l’agrément aux collectivités ou l’inscription sur les listes des spécialités remboursables. L’autorisation de mise sur le marché est la clé du problème. La transparence de la commission de la transparence est toute récente et elle n’est pas encore parfaite s’agissant de la transparence des experts ou de la diffusion des avis.
Les médecins quant à eux savent bien, plus ou moins confusément, que le progrès d’un médicament est nul ou minime, mais, si on les laisse sous l’influence des firmes qui leur serinent le contraire, on ne doit pas s’étonner qu’ils le prescrivent. Si le médicament n’avait pas obtenu d’AMM, cela changerait tout de même les choses.
M. Pierre Morange, coprésident : Quelles seraient donc les modifications à mettre en œuvre en matière d’AMM ?
M. Bruno Toussaint : Exiger des preuves de progrès thérapeutiques.
Prenez l’exemple de l’Epoxine, un antiinflammatoire : son intérêt, disait-on, était de soulager les douleurs et de ne pas faire de mal à l’estomac. Il n’y avait pas de preuves très solides de ces assertions. On s’en est contenté en France et dans d’autres pays. L’ASMR a été cotée au niveau III, c’est-à-dire modéré. Au fil des années, les données se sont complétées, l’ASMR est descendue et actuellement elle se trouve au niveau V. Cela veut dire qu’après quelques années, la commission de la transparence a constaté que ce médicament n’apportait rien par rapport aux autres. Si l’on avait été plus exigeant au départ, si ce médicament n’avait pas obtenu d’AMM, cela aurait limité les dépenses. Parce qu’il était vendu beaucoup plus cher que les autres.
Mme Catherine Lemorton, rapporteure : Savez-vous quelle est, en pourcentages, l’évolution de réelles évolutions thérapeutiques ces dix dernières années ?
M. Bruno Toussaint : Il y a un décalage entre l’avis de la commission de la transparence et celui de la revue Prescrire. Cela dit, le pourcentage de vrais progrès est tangible.
Chaque année, comme cela ressort du bilan de la revue, ce pourcentage est de l’ordre de 10 %. Il a tendance à diminuer depuis le début des années 2000. Les progrès concernent surtout des maladies rares ou des situations très précises, je pense aux maladies orphelines. Pour la plupart des problèmes de santé courants qui concernent beaucoup de gens, il n’y a pas de progrès. Pourtant, apparaissent beaucoup de me-to, d’isomères, etc. En revanche on trouve de plus en plus de médicaments qui sont des régressions thérapeutiques, dans la mesure où ils sont moins bons que ceux qui sont déjà sur le marché. Encore une fois, si on en arrive là, c’est parce qu’on n’exige pas de preuves de progrès.
M. Gérard Bapt : Je suis étonné. Vous avez dit que les progrès réalisés ces dernières années portaient surtout sur des maladies rares ou des maladies orphelines. Pourtant on a enregistré des progrès sur les cancers, notamment en matière de chimiothérapie.
M. Bruno Toussaint : Il y a des progrès médiatisés, annoncés, avec beaucoup d’éléments de spéculation, mais il y a aussi des progrès réels dans le domaine du cancer. La semaine dernière, la revue Prescrire a décerné des diplômes aux firmes qui avaient vraiment fait des efforts. Nous avons notamment décerné une récompense au trastuzumab Herceptin, en tant que traitement adjuvant du cancer du sein. Donné en complément de la chirurgie aux femmes atteintes d’un cancer du sein, et dont la tumeur surexprime des protéines, soit un quart d’entre elles, il augmente de 4 % en valeur absolue le taux de survie à quatre ans, et ce au prix d’une incidence de 3 ou 4 % d’insuffisance cardiaque. Il coûte cher, mais il a des effets intéressants et c’est l’un des meilleurs médicaments du lot. En matière de cancer, il y a beaucoup de nouveaux médicaments, vendus fort cher, mais on ne note pas vraiment de progrès radicaux.
M. Gérard Bapt : Jusqu’à maintenant, le discours a toujours été qu’on ne reculait pas devant le coût de thérapeutiques nouvelles s’agissant du cancer. La recherche clinique porte notamment sur des produits nouveaux dont on évalue l’efficacité et qui sont parfois très chers, pour des pourcentages de survie ou de guérison relativement modestes par rapport aux protocoles antérieurs. Néanmoins, ce n’est plus un problème de laboratoire pharmaceutique ; c’est un problème à la fois médico-économique et éthique. Il est vrai qu’on assiste à un développement exponentiel des médicaments à haut risque, parfois prescrits en dehors de la dotation hospitalière, ce qui pose des questions en matière d’évolution du coût de la santé. Toutefois, personne n’a, jusqu’à présent, franchi le pas de dire qu’il ne faut pas prescrire un médicament parce qu’il est trop cher.
M. Bruno Toussaint : Cela dit, il reste à examiner le bien-fondé de ces prix très élevés. Notre impression est que cela est le résultat du rapport de forces et de l’idée générale que vous avez évoquée, selon laquelle on ne discute pas et que l’on paie pour soigner le cancer, si menus soient les résultats. Ces derniers peuvent même être défavorables. En effet, beaucoup de nouveautés ont des effets indésirables impressionnants, parfois pires que les anciens médicaments.
On ne semble pas discuter du prix de ces médicaments-là. Pourtant, on sait bien que les 800 millions de dollars qui sont avancés comme coût de la mise au point d’un nouveau médicament sont très largement surévalués. Par exemple, la fabrication des produits par biotechnologie, c’est-à-dire par culture de colibacilles, une fois que l’on a mis au point la technique, ne coûte pas cher.
M. Jean Mallot, coprésident : Pouvez-vous nous en dire un peu plus sur les propositions que vous pourriez formuler pour améliorer le système ?
M. Bruno Toussaint : Il y a sûrement beaucoup de choses à faire.
J’ai évoqué les critères de l’AMM, car cela me semble important. Le financement de la recherche clinique est encore très largement sous-développé ; là aussi, il y a du travail. L’indépendance des experts et de l’expertise est également un vaste chantier à mettre en œuvre. Je pense à la transparence des dossiers, des délibérations et, surtout, des motifs de décision. On a beaucoup progressé en ce domaine, mais il faut prendre garde que cela continue. Il faudrait cantonner l’industrie pharmaceutique à son vrai métier, qui est de fabriquer de bons médicaments de bonne qualité.
Aujourd’hui, nous sommes le 31 janvier, date limite pour répondre au premier tour de consultation sur un projet de la Commission européenne relatif à la pharmacovigilance européenne. On constate que ce projet comprend toutes sortes de mesures qui aboutissent à confier la surveillance et le recueil des données en matière d’effets indésirables des médicaments aux firmes. Cela est mal venu et particulièrement grave. Ce n’est qu’un projet, mais il paraît extraordinaire qu’il ait été pensé par la Commission européenne. Nous espérons bien sûr qu’il va évoluer.
La collectivité a tout intérêt à ce que la formation des médecins soit indépendante et facile d’accès. Les visiteurs médiaux sont très agréables pour les prescripteurs : ce sont des gens bien policés, qui arrivent avec des solutions et pas de problèmes. On a expérimenté dans d’autres pays des systèmes efficaces de visite médicale, non plus au service des firmes, mais au service de la qualité des soins. Certes, c’est un effort pour la collectivité, mais ce serait utile et cela constituerait un progrès.
Il faut penser aussi à l’information du grand public. En France, la publicité grand public n’est pas autorisée pour les médicaments. C’est une bonne chose et il faut veiller à ce que cela continue. En effet, certaines actions de promotion contournent cette interdiction : elles sensibilisent le public à tel ou tel problème de santé, la solution proposée étant d’aller voir le médecin. Cependant comme les visiteurs médicaux travaillent le corps médical sur le même sujet, la promotion aboutit.
M. Jean Mallot, coprésident : Ce que vous indiquez paraît relativement simple et évident. Alors pourquoi ne le fait-on pas ? Quels sont les points de blocage ?
M. Bruno Toussaint : C’est vous qui êtes les représentants politiques. Je ne suis qu’un professionnel.
Ce qu’on peut constater, c’est que l’Agence européenne du médicament dépend de la direction générale « Entreprises ». Ainsi, la Commission européenne met la santé des firmes avant la santé publique. Si cela n’est pas clairement écrit, cela se lit entre les lignes et se voit dans les décisions. Il s’agit d’une attitude générale.
Il est extraordinaire de voir avec quelle lenteur la France retire du marché les médicaments dont le bénéfice risque est défavorable. Ainsi, cet été, l’Agréal a, enfin, été retiré du marché. Il s’agit d’un neuroleptique, de la famille des antipsychotiques, contre la schizophrénie. Ce médicament était proposé contre les bouffées de chaleur de la femme ménopausée, même si le rapport n’est pas direct. Depuis des années, on savait que ce médicament avait les effets indésirables de sa famille, notamment qu’il pouvait induire des syndromes parkinsoniens. Cela a pourtant duré longtemps. Un premier pays européen, l’Espagne, a décidé d’arrêter les dégâts, mais il a fallu plusieurs années pour que la France s’y résolve.
La lenteur de ces décisions est affligeante. On voit bien qu’actuellement les pouvoirs publics font passer la santé des firmes avant celle des patients. C’est un choix.
Mme Catherine Lemorton, rapporteure : Certaines campagnes témoignent d’une volonté d’information et d’éducation à la santé. Je pense à une des dernières campagnes de l’Institut national de prévention et d’éducation pour la santé (INPES) sur les états dépressifs. Est-ce à ce type de campagnes que vous faisiez référence ?
M. Bruno Toussaint : Je faisais plutôt référence à ce que l’on présente en prévention d’une maladie. Par exemple, on appelle l’attention de la population sur l’impuissance et les troubles de l’érection. Puis il y a un congrès sur ce thème, où l’on invite des journalistes. Le nombre des articles sur ce problème de santé augmente…
Le cas de la campagne de l’INPES sur la dépression est différent. Il ne lance pas de campagnes de ce genre très souvent, alors que, dans les magazines, on trouve beaucoup de choses sur de prétendus nouveaux problèmes de santé. À la base, un petit nombre de personnes est gêné par certains troubles. Il s’agit alors de faire croire que c’est préoccupant et que cela concerne beaucoup de gens. L’année dernière, on a parlé de l’insuffisance de désir sexuel féminin pour vendre de la testostérone. Pourtant, scientifiquement, on n’a pas de preuves véritables que cela change quelque chose. Reste qu’est apparu un site internet où l’on peut faire un test, et il est bien sûr très facile d’arriver à la conclusion qu’il faudrait en parler à son médecin.
La campagne de l’INPES est sans doute animée de bonnes intentions, mais, toute seule, elle ne saurait suffire. En effet, en France, les médecins libéraux sont rémunérés à l’acte. Or cela va beaucoup plus vite de conclure une consultation par une prescription de médicaments, qui sont par ailleurs remboursés, que d’inviter le patient à suivre une psychothérapie qui sera longue et qui n’apportera pas une rémunération supérieure au généraliste.
M. Jean Mallot, coprésident : Pensez-vous qu’il faille faire évoluer la rémunération des médecins ?
M. Bruno Toussaint : Vraisemblablement. Je ne peux que constater que le paiement à l’acte favorise la prescription de médicaments, parce que c’est la solution la plus rapide. De la même façon le pharmacien est rémunéré au volume de ses ventes, et pas au conseil et à la difficulté de l’ordonnance.
Mme Catherine Lemorton, rapporteure : Je précise que la question de la rémunération du médecin est abordée dans la loi de financement de la sécurité sociale pour 2008. L’article 44 de cette loi prévoit la possibilité d’expérimenter de nouveaux modes de rémunération.
M. Pierre Morange, coprésident : On ne peut pas accepter le propos réducteur selon lequel les pouvoirs publics financeraient la santé de l’industrie pharmaceutique au détriment de la santé des patients, ne serait-ce que sur la base des documents budgétaires.
Pour le reste, qu’il y ait, au travers de notre système actuel – qui s’est construit par sédimentation – des stratégies de contournement et de non-observance de ce qui est important – à savoir l’amélioration de la santé nos concitoyens, l’amélioration indispensable, en termes de coût-efficacité, de notre capacité à adapter les stratégies thérapeutiques en fonction de nos connaissances et l’amélioration des formations –, c’est incontestable. Tel est bien d’ailleurs l’objet de notre mission. Je tenais à recentrer le propos, eu égard à votre phrase, encore une fois bien réductrice.
M. Bruno Toussaint : Je n’ai pas dit cela : je parlais des priorités.
M. Pierre Morange, coprésident : Je pense néanmoins qu’il était important de clarifier les choses.
Mme Catherine Lemorton, rapporteure : Au début de nos auditions, en octobre, on avait évoqué le fait que le contrôlé finançait le contrôleur. En effet, les taxes sont collectées par l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) et la Haute Autorité de santé (HAS). On peut faire confiance à ceux qui participent à ces institutions, mais il est vrai qu’il peut toujours y avoir un doute. Auriez-vous des propositions à présenter à ce sujet ? Comment faire pour que l’AFSSAPS et l’HAS puissent fonctionner sans problème et sans qu’on puisse soupçonner l’existence de conflits d’intérêts ?
M. Bruno Toussaint : Aujourd’hui, l’AFSSAPS et les autres agences du médicament des pays européens ou des pays riches sont financées en grande majorité par des redevances, par les frais de dépôt de dossiers, de demandes d’AMM, etc. C’est dommage. Je ne suis pas spécialiste de financements publics, mais on peut tout de même imaginer qu’il serait assez facile d’organiser autrement, d’une part la collecte des frais et des redevances, et d’autre part le financement du fonctionnement des agences. D’ailleurs, il n’en a pas toujours été ainsi. Il est difficile de mesurer les conséquences de ce conflit d’intérêts, mais il ne doit pas être très difficile de régler le problème, si on le souhaite vraiment.
Mme Catherine Lemorton, rapporteure : Je vous remercie.
M. Pierre Morange, coprésident : Merci.
*
Audition de M. Jacques Sauret, directeur du Groupement d’intérêt public du dossier médical personnel (GIP-DMP).
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale pour cette audition qui s’inscrit dans le travail que mène notre mission sur la prescription, la consommation et la fiscalité des médicaments. La mission d’information sur le dossier médical personnel (DMP) conduite par M. Jean-Pierre Door, vient de rendre ses conclusions et elle a vous déjà auditionné. Il nous a paru néanmoins utile de vous entendre à nouveau sur le sujet du médicament.
Je donne sans plus tarder la parole à notre rapporteure.
Mme Catherine Lemorton, rapporteure : Pouvez-vous tout d’abord nous dire où en est le DMP ?
M. Jacques Sauret : Le DMP est à la fois dans une phase de travail sur le fond, puisque nous continuons à avancer au sein du GIP et avec les acteurs concernés sur les spécifications et la description du dispositif, et dans une phase d’attente de décisions politiques puisque, à l’issue de la revue de projet menée par l’Inspection générale des finances (IGF), l’Inspection générale des affaires sociales (IGAS) et le Conseil général des technologies de l’information (CGTI), qui a rendu ses conclusions en novembre dernier, et des travaux de la mission parlementaire, le gouvernement a constitué une mission de relance du DMP, qui sera suivie d’une concertation avec l’ensemble des acteurs. Présidée par M. Michel Gagneux, membre de l’IGAS, cette mission doit rendre ses conclusions avant la fin du mois de mars. Nous espérons qu’immédiatement après un certain nombre de décisions seront prises par le gouvernement afin de redonner une bonne visibilité aux différents acteurs, indépendamment de la concertation qui permettra pour sa part d’approfondir un certain nombre de sujets.
M. Jean Mallot, coprésident : Pouvez-vous préciser quelles pourraient être ces décisions politiques ainsi que les dispositions que vous souhaiteriez que le gouvernement prenne ?
M. Jacques Sauret : La principale décision consisterait à donner plus de visibilité aux acteurs, aussi bien aux industriels, qui sont prêts à investir des lors qu’ils sont assurés qu’il va se passer quelque chose, qu’aux professionnels de santé, que le GIP-DMP a aidés sur le terrain, grâce à un appel à projets, à progresser vers les spécifications et l’interopérabilité nécessaires au DMP. Or, faute de visibilité, les porteurs de projets locaux ont de plus en plus de mal à mobiliser les professionnels de santé de base, qui sont réticents à se lancer dans un projet dont ils ignorent s’il sera mené à bien.
Les décisions devraient porter par exemple sur l’hébergement des données partagées, car je distingue les données sur le poste de travail des professionnels de santé, que ce soit en officine, en établissement de santé ou chez un médecin libéral, et les données qui sont mises à disposition par un professionnel de santé pour que ses confrères puissent en prendre connaissance dans le cadre de la prise en charge sanitaire, dès lors que le patient en est d’accord. Nous nous demandons s’il convient d’aller vers un hébergement mutualisé, qui permettrait des économies d’échelle importantes, ou au contraire vers un hébergement totalement réparti, qui donnerait une meilleure visibilité aux acteurs régionaux. À ce stade, le GIP considère qu’il faudra sans doute aller vers un hébergement réparti, mais qu’il ne faut pas commencer par cela, sauf à prendre le risque de complexifier le projet. Mieux vaut faire simple et peu coûteux au début, tester pendant quelques années, ce qui permettra ensuite au gouvernement de décider en connaissance de cause d’aller vers plusieurs hébergeurs nationaux ou des hébergeurs territoriaux.
M. Pierre Morange, coprésident : Compte tenu de la montée en puissance progressive et de ce qui a déjà été fait, par exemple pour le dossier pharmaceutique (DP), quel est selon vous l’agenda crédible de mise en place du DMP et pour combien de personnes ?
M. Jacques Sauret : On peut imaginer une mise en œuvre opérationnelle, au départ pour quelques millions de personnes, dans un délai compris entre 24 et 36 mois, le temps que l’identifiant national de santé soit constitué, que la carte de professionnel de santé soit diffusée dans un nombre suffisant d’établissements, que les appels d’offres relatifs à l’hébergement et au portail soient lancés. Par la suite, la montée en puissance dépendra des premiers résultats observés et de la volonté politique.
Ce délai est nécessaire pour les infrastructures. Mais, comme la mission présidée par M. Jean-Pierre Door l’a préconisé, il ne faut pas attendre qu’elles soient en place pour accompagner le changement sur le terrain et pour mener des expérimentations. On pourrait ainsi tester une sorte de DMP régional avec le DP, un autre avec l’historique des remboursements de l’assurance maladie, mais en se plaçant déjà dans une logique d’industrialisation car les industriels sont réticents à investir dans des solutions qui s’écarteraient du dispositif final d’interopérabilité nationale.
Une fois que les infrastructures seront en place, on entrera dans une logique de montée en charge. Il faudra alors se demander si l’on commence par les affections de longue durée (ALD) ou par les maladies chroniques ou si, pour éviter toute stigmatisation, il vaut mieux commencer par des zones géographiques représentant une masse plus importante de population. Ne risque-t-on pas en effet de susciter un rejet de la part des médecins s’ils constatent que le délai de saisie est fort long pour les personnes en ALD, alors que ces dernières ne représentent qu’un peu plus de 10 % de la population ? Il faudra en discuter dans le cadre de la concertation afin de déterminer s’il y a lieu de privilégier les personnes qui en ont le plus besoin ou l’appropriation par les professionnels de santé.
M. Pierre Morange, coprésident : Si l’on prend l’hypothèse des ALD, qui représentent les dépenses les plus importantes de l’assurance maladie et dont les pluri pathologies aggravent le risque iatrogène, pensez-vous qu’au bout du délai de 24 à 36 mois il serait possible de mettre en place le DMP pour les 6 millions de personnes concernées ? Quel en serait le coût ?
M. Jacques Sauret : Il sera tout à fait possible de commencer au terme de ce délai et il faudra quelques mois pour couvrir l’ensemble de cette population : une fois que l’on disposera des infrastructures, la montée en charge pourra être très rapide.
S’agissant du coût, la revue de projet a considéré que nos évaluations étaient sous-estimées. Tout dépend du périmètre retenu. Si l’on intègre la carte Vitale, on fait exploser les coûts. Pour notre part, nous avons considéré que les coûts de l’évolution vers la carte Vitale 2 et de la diffusion de la carte de professionnel de santé (CPS) ne pouvaient être imputés au DMP. En effet, même si l’on renonçait aujourd’hui au DMP, le décret du 15 mai 2007 sur la sécurisation de la conservation et de la transmission des informations médicales par voie électronique entre professionnels serait appliqué et les établissements de santé seraient effectivement contraints à distribuer la CPS.
Le coût de la constitution du portail a été estimé par la Caisse des dépôts et consignations à une vingtaine de millions d’euros par an. La vérification par des audits externes a été interrompue dès lors que le gouvernement n’a pas souhaité, dans le projet de loi de financement de la sécurité sociale (PLFSS), habiliter explicitement la Caisse des dépôts pour le portail. Il s’agit d’une des décisions politiques que nous attendons. Si la Caisse des dépôts n’est pas retenue, il nous faudra lancer un appel d’offres mais l’ordre de grandeur devrait être le même pour un portail capable d’absorber l’ensemble de la population. Une montée en charge progressive réduirait légèrement les coûts.
Pour un hébergement extrêmement sécurisé et une disponibilité garantie, si l’on traite plus de 10 millions de dossiers et si l’on mutualise, le coût devrait être autour d’un euro par DMP et par an. S’il y avait plusieurs hébergeurs, le coût global serait plus élevé.
Mais l’essentiel des coûts tient à l’accompagnement du changement. Les éditeurs estiment que l’adaptation des logiciels des professionnels libéraux devrait revenir à 200 € par poste. L’assurance maladie dépensant déjà une centaine de millions d’euros chaque année pour les aides à la télétransmission, peut-être pourrait-on négocier une enveloppe globale annuelle de soutien à l’utilisation d’outils informatiques qui éviterait de prendre en charge la totalité de cette adaptation. Nous estimons à environ 90 millions d’euros le coût de mise à niveau des logiciels, d’autant qu’un certain nombre d’éditeurs facturent les mises à jour ultérieures ; que la quasi-totalité des pharmaciens sont informatisés avec des dispositifs parfois obsolètes, mais en général à jour ; que 80 % des médecins sont informatisés et qu’ils utilisent l’informatique à des fins médicales dans 60 % des cas, même s’ils se contentent parfois de relever l’état civil des patients et d’imprimer leurs ordonnances.
Il y aura aussi un coût d’appropriation de l’usage, que nous estimons à 20 millions d’euros par an pendant quatre ou cinq ans. Nous considérons en effet qu’il faudra une ou deux journées de formation, non pas à l’utilisation du DMP, qui devra être très simple et nécessiter un ou deux « clics », mais pour l’appropriation psychologique et l’évolution des pratiques liée à la possibilité d’un partage d’informations. Il reste à déterminer qui prendra en charge cette formation. Nous souhaitons donc lancer en 2008 une étude pour déterminer si cela doit relever des institutions de formation médicale continue ou des éditeurs de logiciels, car le DMP ne sera accessible aux professionnels que par l’intermédiaire de leur logiciel métier. Entre 300 et 600 millions d’euros sont consacrés chaque année à la formation médicale des médecins libéraux, dont 60 millions d’argent public. Tout le reste provient de fonds privés, essentiellement des laboratoires pharmaceutiques. Il faut donc trouver la façon de mobiliser l’intégralité des moyens, se demander à quoi doit être destiné en priorité l’argent public, s’interroger sur les synergies possibles avec l’argent privé afin d’amener les professionnels de santé libéraux à utiliser le DMP.
S’agissant de la formation des personnels hospitaliers, des crédits de formation continue existent, mais il est très difficile de les mobiliser car ils servent souvent à boucler les fins de mois des établissements… Les hospitaliers considèrent qu’il convient de prévoir également le financement d’un dispositif de formation des personnels qui seront le plus amenés à utiliser le DMP et le système de partage des informations.
Pour le support, c’est-à-dire la « hot line », le coût peut être extrêmement élevé ou très réduit. Il dépendra très fortement de la montée en charge. Il nous est apparu impossible de lancer un appel d’offre à ce stade car il risquerait d’être surdimensionné ou sous-dimensionné. Qui plus est, nous disposons de la plus grande plate-forme de services en Europe, celle de l’assurance maladie. Si cette dernière acceptait de consacrer un numéro de téléphone spécifique à une sorte de « SOS-DMP », on pourrait profiter de son adaptabilité pour mesurer au cours des premières années le nombre et le type d’appels par DMP ouvert et décider ensuite s’il convient de poursuivre avec elle ou de lancer un appel d’offres. L’idée est de préserver l’avenir, le temps que l’on y voie plus clair. Chaque praticien dispose déjà d’une « hot line », opérée par les éditeurs de logiciels et, d’une certaine façon, par l’assurance maladie. Nous pensons qu’il faudrait aller vers un point d’accès unique pour le praticien mais il convient de réfléchir avec les éditeurs et avec l’assurance maladie et de tester le mécanisme avant de se lancer dans un dispositif très onéreux. Le risque de saturation de la « hot line » paraît assez limité avec l’assurance maladie, qui peut lui dédier 2 500 téléopérateurs, ce dont aucun opérateur privé n’est capable. Il serait dommage de ne pas utiliser cette capacité.
Nous sommes toutefois confrontés à une difficulté qui devra être levée à l’occasion de la concertation : pour l’instant, la loi du 13 août 2004 ne permet qu’aux professionnels de santé délivrant des soins d’accéder au DMP. Jusqu’ici, nous avons considéré qu’un téléopérateur qui répond aux patients ne pouvait pas disposer de cet accès. Or, au moment des expérimentations conduites en 2006, nous avons constaté que les patients parlaient de leur santé, ce qui pouvait d’ailleurs poser des problèmes psychologiques aux téléopérateurs des hébergeurs, qui n’ont pas été formés à cela. Nous devrons donc nous demander s’il convient de modifier la loi, non pas afin que les téléopérateurs puissent apporter des réponses médicales, mais pour qu’ils puissent avoir un aperçu du contenu afin de savoir si un document a été effectivement déposé ou par qui le DMP a été consulté. Il conviendrait sans doute que l’opérateur puisse répondre au patient qui s’inquiète de savoir pourquoi tel professionnel a accédé à son dossier. Bien évidemment, on oppose à cette idée la protection de la liberté individuelle et du secret médical. Mais il me semble qu’une « hot line » qui ne pourrait répondre qu’à des questions techniques susciterait des frustrations voire des angoisses chez les patients.
S’agissant toujours du financement, certains syndicats de professionnels de santé considèrent que le DMP demandera un travail supplémentaire et que le système ne pourra fonctionner qu’avec une aide financière. Pour ma part, je juge cela inutile et contre-productif. La demande est de deux C par patient et par an pour un médecin, soit un coût total de 3 milliards d’euros. Or cela ne changerait rien au pouvoir d’achat des médecins libéraux : si le temps de consultation augmente de 10 % en raison du DMP, les deux C ne changeront pas grand-chose au pouvoir d’achat. À l’inverse, si le DMP apporte un véritable service, on n’aura pas besoin des deux C. Qui plus est, dès lors que le gouvernement ou l’assurance maladie céderaient sur ce point, les pharmaciens, les infirmiers, les kinésithérapeutes demanderaient à être traités de la même façon.
Il me paraît d’autant plus injustifiable de consacrer de telles sommes à ce service qu’il suffit d’attendre. Microsoft est en train de tester le dispositif dans deux États des États-Unis, en vue d’une généralisation en Amérique du Nord, voire en Europe, d’ici deux ans. Deux cents personnes se consacrent à plein-temps à élaborer les interfaces avec les plates-formes techniques de Microsoft, afin que les logiciels des professionnels de santé, en hôpital comme en ville, puissent envoyer très simplement, dès lors que le patient en est d’accord, les informations qu’ils produisent vers le dispositif Healthvault. Par conséquent, si on ne fait rien, le DMP existera, simplement, il sera stocké en Amérique du Nord, dans des conditions qui ne dépendront pas des lois françaises, même si France Télécom est en discussion avec Microsoft pour essayer d’être l’opérateur du dispositif en France et en Europe. Il y a donc là aussi une décision politique à prendre pour savoir s’il appartient à la France de décider elle-même des règles selon lesquelles les informations produites par les professionnels de santé sont partagées. En tout cas, le montant à mettre pour conserver un pouvoir de décision en la matière ne doit certainement pas être de plusieurs milliards d’euros. Nous estimons qu’une fois le dispositif installé tout au plus 300 millions d’euros seront nécessaires.
Mme Catherine Lemorton, rapporteure : Pouvez-vous préciser si on parle ici de données partagées ou de données personnelles mais aussi nous indiquer quels professionnels de santé auront accès au DMP et quelles informations y figureront ?
M. Jacques Sauret : Le débat entre données partagées et données personnelles est ancien. Un peu partout dans le monde on observe qu’il s’agit de données partagées mais de plus en plus accessibles aux patients. Il s’agit donc bien de données de nature professionnelle, fournies par les professionnels de santé à des fins de meilleure coordination des soins et pas d’un outil de vulgarisation de l’information médicale. La grande différence avec le projet anglais – qui est toutefois en train d’évoluer pour donner un accès aux patients via Internet – c’est qu’en France le législateur a dit dès 2004 qu’il voulait que le patient puisse avoir accès à ces informations et s’opposer au partage. En fait, cette possibilité d’opposition remonte à la loi du 4 mars 2002 relative aux droits des malades et à la qualité du système de santé. Quand on regarde attentivement les textes, on s’aperçoit que dès aujourd’hui, dans le code de la santé publique, un patient peut s’opposer à ce qu’une information soit transmise du professionnel de santé qui le prend en charge à tout autre professionnel. En fait, la seule différence entre le DMP et les dossiers de réseaux de soins ou tout autre échange, même par messagerie ou par courrier, c’est que le DMP donnera dès l’origine un accès extrêmement simple au patient. En créant la Commission d’accès aux documents administratifs (CADA), la loi du 17 juillet 1978 a rendu tous les documents accessibles, même si cela est parfois compliqué. Si toutes les administrations mettaient en ligne l’ensemble des documents administratifs, comme cela se fait en Suède, les modalités d’accès seraient extrêmement simplifiées. Pour moi, le DMP est une première étape pour rendre réellement et simplement applicable la loi de 2002 et il n’y a pas de différence de substances entre ce dossier et les autres partages et échanges d’informations de santé personnelles.
La mission dirigée par M. Michel Gagneux réfléchit aux modalités de ces échanges, les professionnels de santé ne souhaitant pas qu’on leur demande de poser de multiples questions au patient : êtes-vous d’accord pour ouvrir un DMP ? Êtes-vous d’accord pour ouvrir un DP ? Êtes-vous d’accord pour participer à un réseau de soins sur le diabète ? etc. L’une des pistes de réflexion est d’uniformiser les règles de consentement du patient, qui consentirait simplement au partage des informations de santé entre les professionnels qui le prennent en charge. Dans le cadre du DP, une fois cet accord explicite donné, par la suite l’accord est présumé implicite par tous les pharmaciens, sauf indication contraire du patient. Ne pourrait-on généraliser cette formule à l’ensemble des dossiers partagés, ce qui rendrait le dispositif plus compréhensible par le patient ? Il faut prendre garde à ce que la complexité n’exclue pas toute une partie de la population qui ne comprendrait pas ce qu’on lui demande.
Je pense qu’il est temps de réconcilier les notions de données personnelles et de données partagées en disant bien qu’il s’agit de données à destination des professionnels pour la coordination des soins mais accessibles au patient et sous son contrôle. Ce que fait ensuite le patient de ces données est un autre sujet : il peut télécharger son DMP sur son poste de travail et on ne peut lui interdire ni juridiquement ni techniquement d’en faire ce qu’il veut et de le montrer à qui il veut, par exemple en l’envoyant sur un site qui lui permet de vérifier les interactions médicamenteuses ou la monographie des médicaments. S’il apparaît dans la presse qu’une information du DMP a été rendue publique, nous devrons prouver que la « fuite » ne provient pas de la chaîne du DMP, d’où l’importance de la traçabilité pour savoir qui a accédé aux informations et à quel moment.
S’agissant de la liste des professionnels de santé, nous sommes confrontés à de vraies difficultés parce qu’en 2004 le législateur a visé l’ensemble des professionnels de santé et uniquement eux. Ainsi, les opticiens lunetiers, les orthophonistes, les orthoptistes ont accès au DMP, mais à quoi exactement au sein de ce dossier ? Chacune des professions qui prescrit considère que, dans un certain nombre de situations, elle a légitimement intérêt à avoir accès aux informations de santé. Certes, mais ces cas exceptionnels justifient-ils un accès aux DMP de toute la population ? Les antécédents gynécologiques sont importants pour beaucoup de professions, mais comment faire une sélection document par document ? Notre souhait est de commencer, dans la grille d’habilitation qui sera annexée au décret, à limiter très fortement, c’est-à-dire aux principales professions, là où il y a la plus forte valeur ajoutée en termes de coordination des soins : médecins, pharmaciens, infirmières, sages-femmes. Quelques informations devraient également être disponibles pour les dentistes et les kinésithérapeutes. Après quelques années d’utilisation, il faudra regarder comment les choses se passent avant d’envisager de légères modifications.
Il me semble absolument indispensable de gérer plus finement la question du masquage, qui a fait couler beaucoup d’encre. On ne saurait se placer dans le tout – chaque professionnel de santé a accès à toutes les informations – ou rien – je refuse que l’information soit dans mon DMP. Si l’information n’est pas mise dans le dossier par le professionnel de santé qui la génère, elle n’y figurera jamais. On constate par exemple que lorsqu’une personne atteinte du sida découvre sa séropositivité, elle est assez réticente à ce que cette information soit connue, mais au bout de quelques semaines ou de quelques mois, elle s’aperçoit que c’est une nécessité pour qu’elle soit correctement prise en charge. Si elle n’avait à l’origine pas d’autre choix que d’interdire que l’information figure dans le DMP, elle n’y serait jamais transcrite. Nous rejoignons donc le rapport présenté par M. Jean-Pierre Door sur l’idée qu’il faut se donner le temps de voir ce qui se passe concrètement sur le terrain, d’observer la réalité du masquage, de constater la réalité des besoins et des pratiques.
Mais on peut aussi considérer que la liste établie par le législateur est trop étroite en ce qu’elle est exclusivement limitée aux professionnels de santé. À la différence du code de la santé publique, elle ne prend pas en compte les professionnels qui travaillent sous l’autorité du professionnel de santé : secrétaires médicales ou aides pharmaciens. Or, les professionnels de santé ont besoin que ces personnes, couvertes par le secret professionnel, puissent continuer à les aider dans la prise en charge des patients. À défaut, ils ne bouleverseront pas leurs pratiques, qui leur ont permis de se concentrer sur la valeur ajoutée médicale, et ils n’utiliseront pas le DMP.
Dans le cadre des discussions internationales sur les normes, nous avons proposé de distinguer l’auteur de la donnée, l’émetteur – celui qui donne l’ordre et qui choisit de transférer – et le transmetteur. Mais nous avons bien besoin d’être assurés que le transmetteur est également habilité. Pour l’instant, si ce n’est pas le professionnel de santé qui transmet, nous rejetons la demande.
Ce thème est aussi l’objet d’une concertation qui pourrait conduire à demander au législateur d’étendre le droit d’émission. Là aussi, il conviendra de garantir la traçabilité, c’est-à-dire de savoir concrètement ce qui se passe sous l’autorité du professionnel de santé titulaire du cabinet ou de l’officine.
Mme Catherine Lemorton, rapporteure : Tous les médicaments en automédication ont été intégrés dans le DP actuellement en test dans six départements, ce qui paraît important au regard des interactions médicamenteuses. Le DMP intégrera-t-il également l’automédication ?
M. Jacques Sauret : L’accord que nous avons trouvé avec le Conseil national de l’Ordre des pharmaciens prévoit que le DP soit le volet médicament du DMP, c’est-à-dire que les éléments du dossier pharmaceutique soient transmis automatiquement dans le dossier médical, sauf opposition expresse du patient. Afin d’unifier les modalités de consentement du patient, nous souhaitons que les mêmes règles s’appliquent au DP et au DMP. Si tel est le cas, le transfert pourra être automatique, sauf opposition, et cela concernera l’intégralité des médicaments, qu’ils soient ou non remboursés.
M. Jean Mallot, coprésident : Même si vous n’êtes pas directement en charge de la prospective, je suppose que vous avez également travaillé sur les effets attendus du DMP, en particulier sur la prescription et la consommation de médicaments, et sur son impact économique. Pouvez-vous nous donner des éléments à ce propos ?
M. Jacques Sauret : Les gains attendus, possibles ou réels, pouvant ou non être imputés au DMP, ont fait l’objet de nombreux débats. Pour moi, peu importe si cela est dû au DMP, ce qui compte, c’est qu’il y ait un effet réel. Il est assez difficile en la matière de passer du micro ou macro-économique. Les études internationales montrent un effet réel avec une diminution de la dépense en médicaments liée au partage des informations.
En Andalousie, où la population représente environ 10 % de la population française et le nombre de professionnels de santé également 10 % des professionnels français, on a conditionné la délivrance du médicament par le pharmacien à la consultation du DMP, à la vérification de l’absence de iatrogénie et au respect d’un certain nombre de règles de bonnes pratiques, en particulier en matière de délivrance des génériques. Je ne dispose pas ici des éléments précis sur les résultats et sur les mécanismes et je vous les transmettrai ultérieurement, mais je puis vous dire que cela a effectivement débouché sur des baisses très importantes des coûts.
Cette expérience n’est toutefois pas directement transposable chez nous puisque le législateur français n’a pas prévu que le DMP puisse être un élément de contrainte de la délivrance par le pharmacien.
On constate à travers l’expérience menée sur le DP dans six départements, que dès qu’une interaction peut déboucher sur l’absence de délivrance, cela a un impact sur le chiffre d’affaires de l’officine, les économies s’effectuant sur la base des dépenses payées aux pharmaciens. La profession considère légitimement que l’effet sanitaire vertueux a pour elle des conséquences économiques dont il conviendrait de tenir compte, par exemple par une augmentation du taux de marge. De tels effets induits pourraient limiter les impacts effectifs du DMP comme du DP.
L’étude que nous avons réalisée évalue les gains de productivité liés au DMP, une fois qu’il sera monté en charge, à un milliard d’euros par an en effets directs et à un milliard d’euros également en effets indirects. Ces estimations ont suscité une certaine incrédulité. Pourtant, dans le monde industriel, qui est plus avancé dans l’utilisation des technologies de l’information pour réaliser des gains de productivité, un gain d’un pour cent est le minimum que l’on peut escompter d’une telle opération. Ici, il s’agit d’un projet qui a vocation à améliorer l’ensemble des systèmes d’information de santé ; aussi, si on le rapporte à l’ONDAM – objectif national des dépenses de l’assurance maladie –, un gain de productivité de un pour cent représenterait bien plus d’un milliard d’euros. La société qui a réalisé l’étude considère même que notre estimation est plutôt minorée car on est très loin d’avoir réalisé tous les gains de productivité possibles dans le monde de la santé…
Certains financeurs publics ont par ailleurs fait valoir qu’il s’agissait de gains de productivité et non d’économies. Il faut toutefois prendre en considération que l’on est sur une courbe de dépenses croissante et que, simplement en abaissant la pente de la courbe, les gains de productivité seront immédiatement traduits en économies. Cela ne signifie pas que les dépenses vont baisser mais qu’on va améliorer la productivité, c’est-à-dire la capacité à faire ce que l’on fait à moindre coût. Si ensuite, on décide de faire plus en raison de l’évolution démographique et du progrès technique, cela relève d’un choix politique.
Bien évidemment, ceux des financeurs qui s’opposent au DMP ne peuvent entendre cet argument car ils raisonnent en termes d’annualité budgétaire, alors qu’il faudra un certain nombre d’années pour que cette démarche produise des résultats importants.
M. Jean Mallot, coprésident : Les effets qualitatifs sont sans doute plus difficiles encore à étudier, je suppose que vous avez aussi des objectifs en la matière.
M. Jacques Sauret : La preuve est déjà faite par le DP. Après quatre ou cinq mois d’expérimentation et alors qu’un peu moins de 200 pharmacies testaient le dispositif, 70 000 dossiers avaient été ouverts et l’on avait recensé 70 alertes graves. Dans la littérature internationale, l’utilité de partager l’information apparaît comme une évidence que nul ne conteste.
Pour que cela soit utile il faut toutefois que cela soit utilisable, c’est-à-dire que l’accès à l’information se fasse à travers le logiciel métier. Le professionnel refuse en effet de perdre du temps à effectuer une démarche en plus de sa démarche professionnelle de prescription ou de délivrance du médicament. Cet aspect psychologique ne me paraît pas suffisamment pris en compte. L’accès Internet doit concerner uniquement le patient tandis que le professionnel doit pouvoir continuer à utiliser son interface utilisateur métier habituelle, par l’intermédiaire du logiciel dont il se sert à longueur de journée. C’est seulement si le DMP s’inscrit dans ce contexte qu’on peut en escompter les effets positifs que tout le monde lui reconnaît.
Mme Catherine Génisson : On ne peut pas fonder la mise en place du DMP sur l’analyse d’objectifs quantitatifs. On a donc bien commis une erreur en le présentant d’abord, en 2004, comme un gage d’économies substantielles. La priorité doit être donnée à l’exigence qualitative, l’augmentation moins forte des dépenses ou les économies suivront. C’est d’ailleurs cette logique qui devrait présider à toute politique de santé.
Alors que l’on est parti de l’idée que le DMP serait une coproduction entre le malade et le professionnel de santé, on a l’impression qu’il se transforme peu à peu en roman-feuilleton dans lequel tout le monde, par exemple la secrétaire médicale, peut ajouter sa petite histoire. Il faut donc revenir à une relation directe entre le patient et celui qui est le mieux à même de l’accompagner, c’est-à-dire le médecin traitant tel qu’il est défini par la loi de 2004. Sans doute conviendrait-il pour cela de commencer par faire simple, en indiquant les informations fondamentales pour une bonne connaissance du patient, avant d’en venir à des choses plus compliquées et à l’intervention d’autres personnes dans le dossier. Évitons une confusion peu propice à la mise en place rapide du DMP !
M. Pierre Morange, coprésident : Merci, Monsieur Sauret, de nous avoir permis de visiter à nouveau ce sujet, après le rapport de la mission d’information présidée par M. Jean-Pierre Door. On le sait, la pédagogie est dans la répétition.
*
Audition de M. Éric Woerth, ministre du budget, des comptes publics et de la fonction publique.
M. Pierre Morange, coprésident : Monsieur le ministre, je vous souhaite la bienvenue au sein de la mission d’évaluation et de contrôle des lois de financement de la sécurité sociale. Notre réflexion porte actuellement sur la prescription, la consommation et la fiscalité des médicaments. C’est bien entendu sur le troisième thème que nos questions se concentreront. La Cour des comptes a formulé des remarques sur le caractère instable et quelque peu illisible de la fiscalité afférente au médicament, ainsi que sur la spécificité des taxes qui concernent ce secteur.
Quels sont les produits générés par les taxes sur le médicament ? Une relance de l’activité du comité stratégique des industries de santé (CSIS) est-elle envisagée ? Les remarques de la Cour vous amèneront-elles à repenser la fiscalité du médicament, en prenant en compte non seulement le volet sanitaire, mais aussi le volet industriel, avec les questions de recherche et de développement et d’attractivité du territoire qui lui sont liées ?
M. Éric Woerth, ministre du budget, des comptes publics et de la fonction publique : Dans la nouvelle architecture gouvernementale, je suis responsable de l’équilibre des finances publiques, donc de celui des recettes et des dépenses de la sécurité sociale. Mon ministère s’intéresse au médicament sous deux angles : son impact sur les comptes de l’assurance maladie d’une part, la fiscalité qui lui est attachée d’autre part.
Le déficit prévisionnel de l’assurance maladie pour 2008 s’élève à 4,2 milliards d’euros, avec une augmentation de 2 milliards d’euros par an. Le non-respect de l’ONDAM est l’un des principaux aléas susceptibles de nous empêcher d’atteindre nos engagements en matière de finances publiques. Or le médicament contribue fortement au rythme élevé de croissance des dépenses d’assurance maladie. Entre 1995 et 2005, les dépenses de médicaments remboursables ont augmenté de plus de 5 % par an. L’année 2006 a constitué une sorte de palier, avec moins de 1 % de hausse, mais 2007 a été marquée par un redémarrage important, l’augmentation s’établissant à 3,6 %. Le dépassement de l’ONDAM pour 2007 est largement lié à ces dépenses de médicaments : l’écart entre le sous-objectif prévisionnel « produits de santé » et le chiffre réalisé est estimé à environ 2 milliards d’euros.
Cette situation s’explique par des phénomènes communs à tous les pays, comme la déformation du marché vers des médicaments de spécialité particulièrement onéreux – notamment les anticancéreux –, mais elle tient aussi aux caractéristiques du secteur du médicament en France : des prescriptions et une consommation excessives, la tendance des médecins français à prescrire en priorité des médicaments récents et coûteux sans que cela soit toujours justifié sur le plan médical, et enfin des coûts de distribution très élevés.
La politique menée ces dernières années a permis de ralentir l’accroissement des dépenses dans ce secteur. Le plan Médicaments, notamment, a généré 2,2 milliards d’euros d’économies. Ses effets sont cependant terminés et les dépenses repartent à la hausse. La politique du médicament est du ressort du ministère de la santé mais nous sommes étroitement associés à ses efforts pour maîtriser les prix des médicaments innovants et pour améliorer la qualité des prescriptions par une meilleure information des médecins et en contrôlant mieux les prescripteurs abusifs, comme les lois de financement de la sécurité sociale en donnent désormais la possibilité.
La fiscalité du médicament est très critiquée par l’industrie pharmaceutique, qui en dénonce régulièrement la lourdeur, la complexité et l’instabilité.
Pour ce qui est de l’attractivité du territoire pour les entreprises de ce secteur, on notera que la fiscalité n’est pas déterminante. Cela dit, il existe de réels problèmes et des possibilités d’amélioration. Nous sommes tous attachés au développement de l’industrie pharmaceutique en France, tant ce secteur est porteur de croissance et de progrès. Il nous faut donc donner plus de visibilité sur l’évolution de la fiscalité. Il n’est pas sain de faire varier tous les ans les taux de certaines taxes ou de créer des contributions « exceptionnelles » que l’on reconduit souvent d’année en année. Les règles doivent être clarifiées de manière à ce que les entreprises n’aient pas de doutes sur leur bonne application et ne recourent pas systématiquement au contentieux.
Nous avons commencé à traiter ces questions dans la loi de financement de la sécurité sociale pour 2008. Il faut maintenant aller plus loin. Je suis prêt à répondre à vos questions sur ce sujet.
Mme Catherine Lemorton, rapporteure : Les représentants de l’industrie pharmaceutique ont en effet souligné l’opacité de la fiscalité du médicament lors d’une audition devant la MECSS en décembre dernier. Ils appellent de leurs vœux une fiscalité structurante et souhaitent que l’on mette fin aux taxes imprévues, qu’ils ne peuvent budgétiser pour l’année suivante. Le « taux K » qui correspond à la clause de sauvegarde semble faire partie de ces taxes imprévues. Or, si l’on en croit la communication remise par la Cour des comptes à la MECSS au mois de mai 2007, le rendement de cette taxe est nul. Quel est votre commentaire sur ce point, monsieur le ministre ?
M. Éric Woerth : Si le taux K ne rapporte rien, c’est qu’il est fait pour cela : il s’agit d’une mesure dissuasive en vertu de laquelle, au-delà d’une certaine augmentation du chiffre d’affaires, une taxe s’applique. Ce taux, qui a d’ailleurs été élargi dans la loi de financement de la sécurité sociale pour 2008, ne s’applique pas car il existe des conventions signées entre chaque laboratoire et le Comité économique des produits de santé – CEPS –, au sein duquel est représentée l’assurance maladie, pour fixer à l’avance, de façon fine, l’augmentation du volume de vente des médicaments. Le taux K n’est utilisé qu’en l’absence de convention. C’est une sorte de filet de sécurité, destiné à obliger les industriels à se mettre autour de la table.
M. Catherine Génisson : Vous avez dénoncé à juste titre la surprescription médicamenteuse en France, monsieur le ministre, mais vous incluez dans ce phénomène la prescription de molécules destinées à la prise en charge de pathologies très lourdes. N’est-ce pas faire fausse route ? Autant je conviens que la prescription de « poudres de perlimpinpin » est excessive – et c’est plus l’éducation du patient qu’il convient alors d’améliorer, en montrant que l’exigence diagnostique et thérapeutique ne se traduit pas forcément par des ordonnances bien remplies –, autant je sais, de par mon expérience personnelle, qu’aucun médecin ne se mettra à prescrire à tort et à travers des antimitotiques ou d’autres molécules entrant dans des traitements très lourds. Au demeurant, ces prescriptions font l’objet de protocoles très rigoureux.
M. Pierre Morange, coprésident : Je crois que Mme Bachelot, que nous entendrons tout à l’heure, sera plus à même de vous répondre, ma chère collègue.
Mme Catherine Lemorton, rapporteure : La Haute Autorité de santé – HAS – et l’Agence française de sécurité sanitaire des produits de santé – AFSSAPS – sont chargées de collecter certaines taxes. La HAS est même financée à 90 % par ces revenus. Ces instances, qui surveillent les conditions de mise sur le marché des médicaments, sont les garantes de la santé de nos concitoyens. Le fait qu’elles tirent une part importante de leur financement de l’industrie du médicament ne risque-t-il pas de jeter parfois un doute sur la transparence du dispositif, sachant par exemple que des médicaments contournant les génériques sont mis sur le marché ? Ne serait-il pas préférable que la collecte de ces taxes soit assurée pas l’administration fiscale, l’ACOSS ou les URSSAF ?
M. Éric Woerth : Contrairement à la Cour des comptes, je ne suis pas choqué par le système actuel. Il s’agit en effet de taxes affectées, nullement de subventions. Les verser par un autre canal ne changerait rien : la procédure est de toute façon transparente et je ne crois pas qu’elle puisse réduire en quelque façon que ce soit l’autonomie de ces organismes par rapport aux industriels du médicament. En revanche, l’existence d’un florilège de taxe est en effet un problème.
M. Pierre Morange, coprésident : À ce sujet, monsieur le ministre, je me rappelle que le Gouvernement, sous la législature précédente, s’était engagé sur un pacte de stabilité, lequel n’a guère été respecté. Avez-vous des solutions concrètes pour répondre à ces problèmes ? Plus précisément, qu’en est-il de la relance du Comité stratégique des industries de santé ?
M. Éric Woerth : Personne ne peut être favorable à la multiplicité des taxes – onze actuellement – auxquelles est soumise l’industrie du médicament. Il faut cependant savoir que l’essentiel du produit est issu de deux d’entre elles : la taxe sur le chiffre d’affaires et la taxe sur la promotion, qui porte principalement sur la masse salariale des délégués médicaux.
M. Pierre Morange, coprésident : Le récent rapport de l’Inspection générale des affaires sociales (IGAS) sur l’information des médecins sur les médicaments propose de renforcer cette taxe sur la promotion, dans la mesure où le dispositif qu’elle vise est susceptible d’inciter à la consommation pharmaceutique. Que pensez-vous de cette recommandation ?
M. Éric Woerth : Le travail de simplification ne saurait se faire sans préserver la ressource. De plus, la stabilisation des règles devrait s’accompagner d’une meilleure acceptation, de la part des industries du médicament, de leurs rapports complexes avec les instances publiques qui sont aussi les payeurs.
S’agissant des dépenses de promotion, l’enjeu est assurément de limiter les visites médicales, qui poussent beaucoup à la prescription et à la consommation. Ces visites sont encadrées par une charte qu’il convient sans doute de renforcer. L’augmentation de la taxe peut aussi être envisagée.
Depuis le début de 2007, les prescriptions recommencent à augmenter fortement. Le système de promotion fait que le médecin a souvent tendance à prescrire un médicament nouveau, mais dont l’apport n’est pas évident, plutôt qu’un produit générique. Il y a donc là une vraie question.
M. Jean Mallot, coprésident : Vos services sont-ils à même de mesurer les effets que l’augmentation de cette taxe aurait sur la visite médicale et sur la prescription ?
M. Éric Woerth : À ma connaissance, non. Les choses ne sont d’ailleurs pas simples : la visite médicale est aussi un secteur d’emploi important. La taxe est certainement un outil pour limiter la promotion du médicament, mais ce n’est pas le seul. Nous préférons les systèmes conventionnels, tel celui dans lequel s’inscrit la charte de la visite médicale, que nous nous emploierons à privilégier.
Quant à la taxe sur le chiffre d’affaires, qui s’applique de façon indifférenciée à une donnée brute, elle ne permet pas un pilotage politique fin. Sans doute faudra-t-il inventer une modification du dispositif : je n’y suis pas opposé dès lors que l’on protège aussi la ressource.
Le Premier ministre réunira à nouveau le comité stratégique des industries de santé fin avril ou début mai afin de relancer son action. Les priorités qui lui seront indiquées sont l’indépendance sanitaire, les infrastructures propices au développement des activités de recherche et de production de médicaments sur notre territoire, ainsi que la recherche et l’innovation.
Sur ce dernier point, il faut noter que le crédit d’impôt recherche est presque taillé sur mesure pour l’industrie du médicament, qui en bénéficiera à hauteur de 500 ou 600 millions d’euros dans le dispositif mis en place à partir du 1er janvier 2008, contre 100 millions environ dans le dispositif précédent.
M. Guy Malherbe : On met sur le marché de nouveaux médicaments dont on sait que le service médical rendu est infime par rapport aux médicaments existants, mais dont on sait aussi qu’ils vont accroître le déficit de la sécurité sociale. Pourquoi, dans ces conditions, leur délivrer une autorisation de mise sur le marché alors que l’on pourrait tarir le déficit à la source ?
M. Pierre Morange, coprésident : Il me semble que cette question relève plutôt de la compétence de Mme Bachelot.
M. Éric Woerth : Je ne peux en effet me prononcer sur le service médical rendu. Plusieurs organismes examinent de près la question. Nous rencontrons régulièrement le CEPS et la HAS pour faire en sorte que les prix soient adaptés.
Mme Catherine Génisson : On a beaucoup parlé de la visite médicale, mais il faudra aussi que l’on se penche un jour de façon sérieuse sur la formation médicale continue, qui est au cœur du sujet.
M. Éric Woerth : S’il s’agit en effet d’un problème culturel, il n’est pas sans effets sur le plan comptable. On ne saurait trop insister sur l’importance de mécanismes de régulation. Pour nous, plus ils s’inscriront dans un cadre contractuel stabilisé dans le temps, meilleurs ils seront. Des efforts restent néanmoins à accomplir, tant du côté de l’État que de celui des industries pharmaceutiques.
Mme Catherine Lemorton, rapporteure : L’assiette de la taxe annuelle sur le chiffre d’affaires des spécialités pharmaceutiques est déterminée sur la base des déclarations des industriels. Ne serait-il pas opportun de contrôler ces déclarations ? Aujourd'hui, cela n’est apparemment pas fait.
M. Éric Woerth : Les industriels se plaignent plutôt d’être trop contrôlés !
Mme Catherine Lemorton, rapporteure : Ils se sont plaints en effet, lorsque nous les avons entendus, d’être maltraités pas le fisc. Des comparaisons ont-elles été menées au niveau européen ?
M. Éric Woerth : Pas vraiment. Le système économique du médicament doit être rendu plus transparent pour que l’on puisse y voir plus clair. Beaucoup d’éléments, comme les remises, ne sont pas très lisibles.
D’une manière générale, je n’ai pas le sentiment que les prélèvements obligatoires aient atteint un niveau insupportable pour cette industrie. En revanche, la fiscalité ne permet pas suffisamment le pilotage et les modifications brutales des règles pour remédier dans l’urgence aux dérapages sont mal supportées par les industriels, qui se voient soudainement réclamer des dizaines, voire des centaines de millions d’euros supplémentaires. Nous préférerions trouver avec eux des mécanismes de régulation forts, là où, aujourd'hui, ils ont tendance à contester la plupart des décisions sur la forme et à créer beaucoup de contentieux, ce qui est une source de crispations et de malentendus avec les organismes sociaux ou l’État.
Nous souhaitons naturellement leur développement, mais ils doivent reconnaître dans le même temps que c’est bien le système public français qui rembourse 20 milliards d’euros de consommation médicamenteuse en ville.
M. Pierre Morange, coprésident : Comme l’a suggéré Mme la rapporteure, il serait intéressant de mener une comparaison au niveau européen sur la fiscalité du médicament.
Vous évoquiez, monsieur le ministre, la nécessité d’affiner la taxe sur le chiffre d’affaires. Je note que plusieurs nations anglo-saxonnes modulent leurs taxes en fonction de l’importance du secteur recherche et développement et de l’implantation de sites industriels. Votre ministère examine-t-il une telle orientation ?
M. Éric Woerth : Il n’existe pas, à ma connaissance, d’étude comparative de la fiscalité. Au demeurant, une telle étude sera difficile à réaliser tant les politiques d’État sont différentes : la fixation du prix n’est pas la même dans tous les pays, il peut exister des mécanismes de régulation des prescriptions, des mécanismes de régulation des marges, etc., les circuits de distribution ne sont pas les mêmes, non plus que les dispositifs de remboursement… Il faudra donc prendre en compte une multitude de critères, qui de surcroît n’apparaissent pas tous au grand jour.
Je m’en tiens pour l’instant à des idées simples : nous consommons beaucoup, nous remboursons beaucoup, des médicaments innovants existent sur le marché. Il est prouvé que les systèmes de régulation peuvent donner des résultats mais ils doivent s’inscrire dans une plus grande concertation avec l’industrie pharmaceutique si l’on veut éviter les à-coups en cours d’année.
M. Jean Mallot, coprésident : Vous souhaitez donc réviser la fiscalité actuelle, dont on peut imaginer qu’elle a également peu d’impact sur la localisation des laboratoires. Quel sera le calendrier ?
M. le ministre. Un calendrier plutôt bref. Les discussions sont en cours pour les deux taxes principales. Nous devrions être prêts pour le projet de loi de financement de la sécurité sociale pour 2009, en veillant à rester en cohérence avec la révision générale des prélèvements obligatoires.
M. Pierre Morange, coprésident : Votre ministère mène-t-il une réflexion spécifique sur deux secteurs stratégiques dans la compétition mondiale, celui des génériques et celui des biotechnologies ?
M. Éric Woerth : Le sujet est plutôt du ressort du ministère de l’économie.
En ce qui concerne la consommation, si les génériques sont entrés dans les mœurs, il reste beaucoup de progrès à faire pour aller jusqu’au bout de la démarche. On ne peut laisser passer des médicaments qui ressemblent à des génériques mais qui n’en sont pas. Le système demeure un peu ambigu.
M. Jean Mallot, coprésident : Lors que nous avons auditionné les représentants des laboratoires spécialisés dans les génériques, ils ont souligné la relative fragilité économique de leurs entreprises : contrairement à leurs concurrents, ils ne peuvent se « rattraper » sur des médicaments nouveaux. Il serait paradoxal que la puissance publique ne se préoccupe pas de cette vulnérabilité au moment même où elle souhaite développer la production et l’usage des génériques.
M. Éric Woerth : Je partage votre analyse sur la fragilité de ces laboratoires, même si des entreprises plus importantes fabriquent à la fois des génériques et des non-génériques.
De plus, pour être incitatifs, les taux de remise consentis aux pharmaciens sont aussi élevés que sur les autres médicaments. Il existe donc un problème de composition de la marge pour les génériqueurs.
Pour le reste, je constate une progression satisfaisante de l’usage du générique, même si nous restons derrière la plupart de nos voisins.
M. Pierre Morange, coprésident : Les génériques représentent un marché mondial gigantesque. Si l’on ne réfléchit pas aux moyens de renforcer les entreprises implantées en France, la production, puis la conception, se verront transférées dans des pays tiers.
M. Éric Woerth : Le diagnostic est juste.
Mme Catherine Lemorton, rapporteure : Le LEEM, lors de son audition, a soutenu que le débat sur la visite médicale était un débat du passé et que la question était bien plutôt de savoir comment conserver sur le territoire la recherche et le développement, voire la production, de nos industries pharmaceutiques. Quel est votre avis sur le sujet ?
M. Éric Woerth : C’est un discours que l’on entend souvent dans la bouche des industriels. Fort heureusement, nous pouvons garder une industrie pharmaceutique sur notre territoire : le marché intérieur est très important et nous offrons des formations et des compétences adaptées. Les propos des industriels me semblent surtout dictés par la peur que le marché français ne se réduise, mais nous sommes en présence d’une industrie exportatrice. De plus, nous voulons tout au plus réduire l’augmentation du marché français.
Il faut également insister sur le caractère extrêmement incitatif des dispositifs de crédit d’impôt recherche votés cet automne. En la matière, nous sommes plus compétitifs que de nombreux pays voisins. Je ne crois vraiment pas qu’il existe un problème d’attractivité. Des laboratoires étrangers viennent s’implanter en France, y maintiennent et y développent des usines, parfois dans des territoires qui ne sont pas les mieux desservis par les infrastructures de transport.
L’industrie pharmaceutique en France connaît un développement que la régulation de la visite médicale ne saurait mettre en péril. Elle a en revanche intérêt à accepter les mécanismes de régulation car, si nous continuons à connaître des déficits de cette ampleur, les ajustements seront de plus en plus brutaux. Mieux vaut piloter ensemble un système offrant durablement des soins de meilleure qualité pour un meilleur coût. C’est vers cette culture que l’on doit se diriger, en cessant de se jeter à la tête des anathèmes ou des menaces de délocalisation. Le consommateur de médicament est privé, mais le remboursement est public.
Mme Catherine Lemorton, rapporteure : Dans les décennies à venir, les pouvoirs de l’Agence européenne du médicament en matière d’autorisations de mise sur le marché vont s’accroître. Peut-on évaluer le manque à gagner que cela représentera pour l’AFSSAPS ?
M. Éric Woerth : Les taxes qui s’appliquent lors de la mise sur le marché ne représentent pas un montant considérable. La taxation principale concerne la production. Je vous donnerai toutefois les chiffres exacts pour 2007.
M. Jean Mallot, coprésident : Vous avez appuyé à plusieurs reprises votre argumentation sur le fait que la prescription et la consommation sont excessives dans notre pays. Deux mesures ont été prises pour tenter de les réguler dans les articles 43 et 44 la loi de financement de la sécurité sociale pour 2008 : les contrats d’engagements individuels des médecins et l’évolution des modes de rémunération des professionnels de santé. Où en est-on dans l’élaboration des textes d’application que nécessitent ces articles ?
M. Éric Woerth : Il s’agit de bons outils, fondés sur la contractualisation et qui ont fait leurs preuves dans d’autres domaines. Les décrets d’application ne sont pas encore signés. Un contrat individualisé type sera proposé à la fin du premier trimestre. Mme Bachelot pourra certainement vous en dire plus.
On peut également imaginer des expérimentations sur les nouveaux modes de rémunération. Beaucoup de jeunes médecins, me semble-t-il, y sont prêts.
Mme Catherine Lemorton, rapporteure : Je reviens sur un autre type de promotion, celle qui concerne les médicaments en vente libre.
Ne conviendrait-il pas, dans un souci de santé publique, de taxer bien plus fortement la promotion télévisuelle ou médiatique qui banalise auprès du grand public des médicaments qui, s’ils ne sont pas ou plus remboursés, n’en restent pas moins des médicaments et présentent des risques d’interaction parfois importants ?
M. Éric Woerth : Il s’agit généralement de médicaments à faible service médical rendu.
Mme Catherine Lemorton, rapporteure : Pourtant, les contre-indications peuvent être importantes.
M. Éric Woerth : L’automédication, qui est une source d’économies non négligeables pour la sécurité sociale, semble bien fonctionner dans d’autres pays.
Les publicités dont vous parlez poussent à la consommation, mais en dehors de la sphère du remboursement. On notera que d’autres publicités appellent à ne pas consommer, notamment les antibiotiques.
Je conclurai en rappelant que la régulation comporte également la sanction. L’assurance maladie, qui dispose aujourd'hui des moyens de sanctionner les surprescriptions, ne doit pas hésiter à relever les excès, même si ceux-ci ne concernent qu’une infime minorité, et à mettre sous entente préalable des médecins qui abuseraient de certaines prescriptions. Il existe en outre des logiciels qui permettent, sans porter atteinte à l’exercice individuel, d’orienter les prescriptions.
M. Pierre Morange, coprésident : Monsieur le ministre, nous vous remercions de vous être prêté à cet exercice. Si vous disposez d’éléments de réflexion sur la façon dont le législateur pourrait participer à la rationalisation de la dépense publique dans ce domaine, nous vous saurions gré de nous les communiquer.
*
Audition de Mme Roselyne Bachelot, ministre de la santé, de la jeunesse et des sports.
M. Pierre Morange, coprésident : Je vous souhaite la bienvenue à l’Assemblée nationale pour cette audition qui s’inscrit dans le travail qu’effectue notre mission sur la prescription, la consommation et la fiscalité du médicament. Je donne, sans plus tarder, la parole à notre rapporteure.
Mme Catherine Lemorton, rapporteure : Même si l’industrie pharmaceutique dit parfois le contraire, les Français sont de gros consommateurs de médicaments par rapport à leurs voisins européens. Or, on sait que la visite médicale est une des raisons de cette hyperconsommation. Quels moyens avez-vous de réduire la visite médicale, tout en continuant à répondre au besoin d’information sur les innovations thérapeutiques ? En d’autres termes, comment faire que la visite médicale soit une source d’information et non de promotion ?
Mme Roselyne Bachelot, ministre de la santé, de la jeunesse et des sports : Il y a effectivement un problème de surconsommation de médicaments dans notre pays : 90 % des visites chez le médecin en France se terminent par une prescription alors que le taux est bien moindre dans d’autres pays.
Les actions de maîtrise médicalisée ont été renforcées au travers de nombreuses mesures que j’ai portées et qui ont été adoptées dans la loi de financement de la sécurité sociale pour 2008, notamment la mise sous entente préalable pour les gros prescripteurs. Il faut concilier la maîtrise médicalisée avec le principe, auquel nous sommes très attachés, de l’accès généralisé et équitable de la population à l’ensemble des médicaments.
La visite médicale n’est pas seule responsable de la surconsommation de médicaments. Cette dernière résulte également du fait que nous avons un champ de protection sociale très large et que nous gardons le taux de prise en charge socialisée des dépenses de santé parmi les plus élevés du monde. La France figure parmi les trois pays qui garantissent le taux de couverture socialisée le plus haut. Par ailleurs, la pratique généralisée du tiers payant crée par nature une demande plus importante, comparativement à nos voisins européens. En notre qualité de docteurs en pharmacie, madame la rapporteure, nous avons pu constater les dérives dues à la déresponsabilisation de la demande. Loin de moi l’envie de critiquer ce que je considère comme une avancée sociale, sur laquelle il ne s’agit pas de revenir, mais on peut quand même indiquer que nous payons celle-ci d’une certaine surconsommation.
L’impact direct ou indirect de la consommation des médicaments sur la morbidité et la mortalité doit être évaluée plus finement, en définissant des indicateurs par pathologie, région, mode de prise en charge, stratégies thérapeutiques, etc. pour mieux cibler les traitements. La faiblesse de l’épidémiologie et notamment de la pharmaco-épidémiologie dans notre pays rendent le suivi de ce type d’indicateurs complexe. J’ai souhaité que ce sujet soit identifié comme prioritaire dans le cadre des travaux du comité stratégique des industries de santé (CSIS).
J’ai également souhaité que la Haute Autorité de santé (HAS) soit investie d’une mission médico-économique, dont elle était privée jusque-là. Elle devra ainsi mieux analyser le rapport coût-bénéfices d’une stratégie de traitement.
Cela étant, il n’y a pas que des inconvénients à cette consommation de médicaments. Les résultats d’une étude académique récente du Commonwealth Fund placent la France à la première place de dix-neuf pays industrialisés pour la prise en charge de maladies médicalement curables. Il ne s’agit pas de clamer que nous sommes les meilleurs mais il faut veiller à ce que les mesures de régulation soient compatibles avec ces bons résultats.
Si j’ai fait ce préambule avant de répondre à votre question sur la visite médicale, c’est parce qu’il ne suffit pas de crier haro sur le baudet et de penser que la surconsommation est uniquement liée à celle-ci. Il faut faire, évidemment, des efforts dans ce domaine. La visite médicale pratiquée par les laboratoires pharmaceutiques n’est pas optimale : elle est trop chère, subjective, orientée vers les médicaments nouveaux, et donc, en général, les plus chers. Mais elle a le mérite d’apporter des connaissances aux prescripteurs : données cliniques, pharmacologiques. La visite médicale doit être améliorée. Les industriels du médicament s’y sont d’ailleurs engagés avec la signature de la charte de la visite médicale, en décembre 2004, et la mise en place d’une certification. La politique de « désarmement » prônée par l’Inspection générale des affaires sociales (IGAS) est une piste à étudier, même si ce désarmement a déjà commencé du fait du déclin de certaines classes « grand public » et par la montée des génériques.
Dans le cadre de la mise en place de la charte, le comité économique des produits de santé (CEPS) a demandé aux laboratoires, en accord avec le LEEM, de diminuer le nombre de contacts avec les visiteurs médicaux pour quatre classes thérapeutiques : les statines, les sartans, les fluoroquinolones et les médicaments de l’asthme. Sur les quatre classes, seules deux – les sartans et les fluoroquinolones – n’ont pas rempli le contrat alors que les deux autres ont vu leur nombre de contact diminuer significativement – autour de 25 %. Pour les deux classes n’ayant pas rempli le contrat, des sanctions de baisse de prix ont été prises.
Cet effort doit être poursuivi en proposant l’élargissement de ce suivi par le CEPS à d’autres classes thérapeutiques. C’est un sujet que je suis d’extrêmement près. Il faut toujours manier la carotte et le bâton.
Mme Catherine Lemorton, rapporteure : Est-il envisagé une alternative à la visite médicale des laboratoires afin d’offrir une information objective répondant au double souci de l’équilibre des comptes de l’assurance maladie et du fait qu’une innovation thérapeutique doit être promue en tant que telle en respectant les deux parties ?
Mme Roselyne Bachelot : La question de l’information des médecins est, effectivement, plus globale que celle de la visite médicale.
L’information des médecins doit d’abord, à mon sens, être assurée par les institutions publiques. Le système existant gagnerait en crédibilité, en efficacité et en « audibilité », si vous me permettez ce néologisme, à être plus cohérent et mieux coordonné et en prévoyant des mesures d’impact des actions d’information.
Les anciens médicaments qui gardent leur place dans la stratégie thérapeutique ne donnent plus lieu à une information promotionnelle. Or, dans certains domaines, ces médicaments restent le meilleur choix. Il faut donc contrebalancer l’information de la visite médicale qui ne porte que sur les nouveaux médicaments.
A côté des informations sur les effets indésirables dont l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) assure de mieux en mieux la diffusion, il est nécessaire d’informer et de former les prescripteurs sur la meilleure façon de gérer les risques médicamenteux.
Les efforts réalisés par les caisses d’assurance maladie – entretiens confraternels et délégués de l’assurance maladie, DAM – semblent utiles mais le double rôle de contrôleur et d’informateur peut paraître difficile à comprendre par certains prescripteurs. La visite médicale publique ne pouvant rivaliser en puissance avec la force de vente des laboratoires, elle doit être centrée sur des problèmes de santé publique particuliers et coordonnée avec la politique de santé publique.
Les informations de l’AFSSAPS concernent les médicaments alors que la Haute Autorité de santé s’adresse plus généralement à la prise en charge thérapeutique des patients, l’usage du médicament en étant un élément. Une étroite coordination doit se mettre en place entre les deux organismes, les champs d’intervention de ces deux émetteurs étant distincts et complémentaires.
L’ensemble des recommandations devrait figurer sur les sites des deux autorités de santé, in extenso ou sous la forme de liens bien identifiés. Un effort de lisibilité doit être fait et sera fait sur les sites Internet de l’AFSSAPS, et plus encore sur celui de la Haute Autorité de santé, afin de faciliter l’accès à l’information pour les professionnels de santé et le grand public.
Pour l’avenir, une réflexion est à mener pour mieux articuler les différents intervenants actuels – AFSSAPS, HAS, CNAMTS –, réflexion qui pourrait s’enrichir de l’expertise acquise par les associations de malades et les usagers du système de santé et des actions de communication menées par l’Institut national de prévention et d’éducation pour la santé (INPES) en direction de divers publics sur les enjeux de santé.
Pour ce qui concerne les données scientifiques et administratives, ces informations sont disponibles sur le site Internet de l’AFSSAPS. Cette agence construit une banque, déjà bien avancée, de données administratives et scientifiques sur les médicaments et les dispositifs médicaux.
La Haute Autorité de santé construit, quand à elle, une base de données indexée comportant les avis de transparence. En complément pour ce qui concerne la prise en charge, le site Internet de l’assurance maladie met à disposition les données sur les taux de remboursement selon les types de médicaments. Il serait souhaitable que les trois bases, AFSSAPS, HAS et assurance maladie, soient reliées entre elles dès qu’elles seront finalisées.
La Cour des comptes reconnaît que, si la base Thesorimed – ex base Thériaque – permet une mise à disposition gratuite de l’information, elle est difficilement compatible avec les logiciels d’aide à la prescription (LAP). En revanche, les bases de données privées sont payantes, mais elles sont facilement intégrables avec ces logiciels. Dans ce contexte, il conviendrait de conduire une réflexion en vue de développer une base de données publique unique, accessible à tous gratuitement pour répondre aux besoins de chacun des acteurs du système de santé : médecins, pharmaciens, établissements de santé, ministères, etc. Quand nous aurons bâti ce système, la visite médicale prendra toute sa place mais rien que sa place.
M. Pierre Morange, coprésident : Selon vous, à quel horizon peut-on espérer la constitution de cette base de données ?
Mme Roselyne Bachelot : Il faut compter à peu près deux ans. Si l’on pouvait en disposer pour la fin de 2009, ce serait bien.
M. Pierre Morange, coprésident : Cela vous semble techniquement et financièrement réalisable ?
Mme Roselyne Bachelot : Oui.
Mme Catherine Génisson : Vous avez souligné les interrogations des professionnels de la santé vis-à-vis des délégués de l’assurance maladie. Cette dernière s’immisce de plus en plus – et de manière heureuse – dans la politique de santé. Ne devons-nous pas, à partir de ce constat, avoir une réflexion plus globale sur l’avenir de la politique de santé ?
Mme Roselyne Bachelot : Pourriez-vous préciser votre question, madame Génisson ?
Porte-t-elle sur l’avenir de la politique de santé – ce qui déborde un peu du cadre de l’audition d’aujourd’hui – ou sur le rôle de la CNAMTS dans les procédures d’admission au remboursement et de validation ?
Mme Catherine Génisson : Je vous concède, madame la ministre, que ma question déborde un peu le cadre de l’audition de ce matin. J’avais une petite arrière-pensée en la posant dans la mesure où nous venons de prendre connaissance du rapport de la mission d’information, présidée par M. Yves Bur, sur les agences régionales de santé (ARS). Celui-ci contient une série de propositions qui m’ont fait penser qu’en parlant des délégués de l’assurance maladie, le cercle vertueux serait constitué…
Mme Roselyne Bachelot : J’ai eu l’occasion de venir devant cette mission pour décrire l’architecture que j’entends proposer à partir des agences régionales de santé et que le Parlement examinera lors de la grande loi sur la modernisation de l’organisation de la santé, aussi bien pour les soins de premier recours que pour la gouvernance de l’hôpital. Je souhaite que les agences régionales de santé aient un périmètre le plus large possible avec le médicosocial et le système de santé publique. Il est bien évident que, dans le cadre du rôle de territorialisation des politiques de santé publique assurés par les ARS, la bonne utilisation du médicament sera un élément important.
Votre question m’amène à parler du rôle de la caisse nationale d’assurance maladie des travailleurs salariés (CNAMTS). La Cour des comptes considère que la caisse nationale d’assurance maladie devrait avoir plus de place dans la procédure d’admission au remboursement. Même si je salue le rôle de la CNAMTS, je trouve que la vision de la Cour des comptes est un peu idyllique et je vais donc vous donner mon sentiment à ce sujet.
Mme Catherine Génisson : La vision de la Cour des comptes non seulement est idyllique mais encore pose des problèmes de fond.
Mme Roselyne Bachelot : Je vous remercie de le faire remarquer.
M. Pierre Morange, coprésident : L’état d’esprit à la MECSS, madame la ministre, est particulièrement consensuel !
Mme Roselyne Bachelot : Ce que je constate, c’est que les résultats des actions de maîtrise médicalisée de la CNAMTS sont mitigés – pour employer un terme diplomatique. Sur la base des données du Groupement pour l’élaboration et la réalisation de statistiques (GERS) et des données de remboursements du régime général, les économies dues à la maîtrise médicalisée en 2006 – les derniers chiffres à notre disposition – sont évaluées à environ 460 millions d’euros, ce qui correspond à un taux d’atteinte des objectifs initiaux de l’ordre de 58 %. Il n’y a pas de quoi s’esbaudir !
Depuis plusieurs années, les caisses d’assurance maladie cherchent un rôle plus actif dans la gestion des soins. On pense, par exemple, au plan stratégique proposé en 1999 par M. Gilles Johannet.
M. Pierre Morange, coprésident : Cela nous permet de prendre un peu de distance par rapport à l’histoire de notre système sanitaire !
Mme Roselyne Bachelot : Le rôle théorique d’un « assureur » – terme que la caisse nationale d’assurance maladie utilise volontiers à son profit – impliquerait la possibilité de sélection des patients en fonction du risque, voire celle de modifier le niveau des primes d’assurance ou de déterminer le contenu des biens et des services pris en charge. Aucun de ces éléments n’est réalisable ni surtout souhaitable – pour répondre directement à la question de Mme Génisson – pour les caisses d’assurance maladie en France. Il y a une confusion sémantique qui relève d’une confusion conceptuelle.
Les caisses interviennent actuellement dans le processus de remboursement et de fixation des prix en étant représentées auprès de la Commission de la transparence et au sein du Comité économique des produits de santé (CEPS). Elles ne sont donc pas exclues de la réflexion sur ce sujet. Elles y participent à leur place. Elles interviennent également dans le bon usage du médicament à travers des actions influençant la pratique de prescription des praticiens. Le conventionnement des médecins et l’application des accords de bon usage (AcBUS), l’information par des moyens comme les lettres aux médecins, les entretiens confraternels et les délégués de l’assurance maladie (DAM) en constituent des exemples.
Je le répète de façon solennelle, en France, c’est à l’État de garantir la santé de la population en assurant l’équité d’accès aux soins et en tenant compte de l’efficience. Il n’est donc pas envisageable que les caisses puissent s’affirmer davantage dans le rôle d’assureur dans ces domaines.
L’image de la CNAMTS bon gestionnaire des deniers collectifs, bon père de famille avisé est un peu idyllique. Certaines actions ont porté leurs fruits, mais les effets globaux sont insuffisants, en tout cas pas meilleurs que ceux assurés par d’autres acteurs.
Je ne propose pas un remplacement mais, plus exactement, une meilleure coordination avec la politique du Gouvernement, notamment en mettant à disposition les bases de données et en contribuant à rendre cette politique plus efficiente. Les objectifs sont partagés. Pour moi, une querelle de chapelle serait contre-productive.
Je crois que nous avons, madame Génisson, des philosophies très proches sur ce sujet.
M. Jean Mallot, coprésident : Du fait de la grande consommation de médicaments par les Français, le médicament prend un part non négligeable dans le déficit de l’assurance maladie. Parmi les mesures prises récemment, la franchise médicale a été abordée avec plusieurs de nos interlocuteurs au cours de nos auditions. Aucun n’a soutenu ce dispositif et plusieurs ont tiré à boulets rouges dessus. Avez-vous déjà quelques éléments d’information sur la mise en place de cette mesure et, notamment sur son impact ?
Mme Roselyne Bachelot : Permettez-moi de rappeler plusieurs points concernant les franchises.
Elles ont été créées pour permettre le financement de nouveaux besoins de santé : maladie d’Alzheimer, cancer, soins palliatifs. J’ai donné, devant la représentation nationale les chiffres très précis et je rendrai compte de l’utilisation de ces sommes. Elles gardent un niveau de prise en charge socialisée des dépenses de santé parmi les plus élevées du monde. Celle-ci reste, même avec les franchises, à près de 80 %, ce qui est considérable. Il faut garder cela à l’esprit.
Un Français sur quatre ne paie pas la franchise : les bénéficiaires de la CMUC, les femmes enceintes et les enfants. Par ailleurs, la franchise est plafonnée à 50 euros. Elle représente donc une dépense de 4 euros par mois en moyenne.
Il est vrai que c’est un effort pour certaines personnes. En cas de très grandes difficultés, j’ai indiqué les mécanismes qui permettaient de faire appel au fonds dédié de l’assurance maladie, sur lequel une somme de 224 millions d’euros a été provisionnée pour 2008. Elle permet de financer aussi bien des actions collectives – pour 30 millions d’euros – d’associations qui s’occupent de personnes particulièrement précaires que des dossiers individuels qui peuvent être présentés soit directement par les personnes, soit par des associations ou des systèmes sociaux, comme les centres communaux d’action sociale.
J’ai choisi, pour supporter la franchise, les secteurs qui, dans notre système de santé, sont, non pas les plus dérapants, mais les plus dérivants par rapport à d’autres pays ayant des performances identiques et même supérieures à celles de la France. Les médicaments en font partie. Tout le monde reconnaît que la consommation de médicaments par les Français n’est pas en corrélation avec nos performances en matière de morbidité et de mortalité. J’ai donc fait porter sur les médicaments une franchise de 50 centimes par boîte.
Il est encore trop tôt pour faire une évaluation. Pour certaines maladies importantes qui nécessitent des médicaments prescrits en fonction de l’autorité du médecin prescripteur, je n’ai pas pensé que cela diminuerait les prescriptions, même si on peut parfois se poser la question de l’adéquation de celles-ci avec les pathologies qu’elles sont destinées à soigner, même dans le cas des affections de longue durée (ALD). On a vu, pour le traitement de l’hypertension ou du diabète, des délivrances de médicaments parfois erratiques.
Les franchises n’ont été appliquées qu’il y a un mois. Les statistiques ne sont pas encore remontées. Mais, dès que j’aurai des évaluations à ce sujet, je vous les communiquerai.
Mme Catherine Génisson : Je vous remercie, madame la ministre, pour la réponse que vous avez donnée à ma question qui débordait de notre ordre du jour.
Vous avez évoqué la question de l’information et de la formation des médecins. J’aimerais que vous parliez de la formation médicale continue, sujet qui est un peu un serpent de mer. Même si des dispositions ont été prises, elles sont encore trop peu appliquées.
De par l’affectation de leurs recettes, les franchises sont un peu la gabelle moderne. Et il est de la responsabilité de l’État de définir comment sont orientées les priorités à partir des recettes ou des éventuelles économies réalisées.
Il faut examiner les conséquences des franchises médicales sur la santé publique. Empêchent-elles ou non l’accès aux soins primaires, y compris pour les pathologies mineures, pour les personnes dont la moyenne des revenus se situe juste au-dessus de celle donnant droit à l’exonération des franchises ? Les études et analyses doivent être orientés sur ce sujet car on peut craindre des problèmes.
Mme Roselyne Bachelot : Vous posez le problème de l’accès aux soins pour cette zone grise de personnes qui ont trop de revenus pour être prises en charge par les systèmes socialisés et pas assez pour vivre. Le travailleur pauvre en fait partie mais cela touche aussi d’autres catégories.
Les difficultés d’accès aux soins peuvent résulter de plusieurs facteurs. Le premier est l’éloignement géographique, non seulement dans les zones rurales, mais également dans des quartiers populaires. L’application du ticket modérateur n’est pas vraiment en cause. Mais le problème majeur est les dépassements d’honoraires, qui ne représentent pas loin de 2,5 milliards d’euros, sur 18 milliards d’honoraires. J’ai inscrit comme priorité de mon action de les maîtriser. Certains sont justifiés, en particulier pour garder des praticiens de niveau international dans les CHU. Mais il faut combattre de façon déterminée les dépassements d’honoraires illégaux : les dessous de table et les pratiques condamnables.
Il faut également revisiter la notion d’acte. J’ai pris dans la loi de financement de la sécurité sociale un certain nombre de dispositions et je travaille actuellement à un décret sur le montant qui oblige à présenter un devis.
La question que vous posez, madame Génisson, ouvre le débat du bouclier sanitaire. Le pacte de 1945, dont on parle beaucoup même si ceux qui en parlent ignorent généralement quelles étaient les modalités de prise en charge par la protection sociale en 1945, a introduit le principe : « Chacun paie selon ses moyens et reçoit selon ses besoins. » La question a été soulevée par des experts et des philosophes de tous les horizons politiques de savoir si, devant la difficulté d’accès aux soins pour cette classe intermédiaire, il ne convenait pas d’instaurer un bouclier sanitaire. J’en appelle à la représentation nationale pour que le débat ait lieu au Parlement.
Mme Catherine Génisson : Le débat est actuellement au sein du Gouvernement. La personne qui vous a précédée dans les auditions de ce matin n’y est pas favorable.
M. Pierre Morange, coprésident : Revenons à l’objet de la mission d’évaluation et de contrôle. Peut-être pouvez-vous dire quelques mots de la formation des médecins, madame la ministre, avant que nous vous posions d’autres questions.
Mme Roselyne Bachelot : La Cour s’est également intéressée à la formation des médecins.
Le ministère de la santé ne gère pas seul la formation initiale des médecins. Il le fait avec le ministère de l’enseignement supérieur et de la recherche. Je travaille actuellement avec Mme Valérie Pécresse sur la première année de médecine.
Il faut donner toute sa place à la thématique médicament du fait des enjeux majeurs qu’elle représente tant en termes de santé publique qu’en termes d’économie de la santé.
L’évaluation des pratiques professionnelles (EPP) constitue un levier fort, d’autant que la validation de l’EPP permet une validation d’une partie du barème de la formation médicale continue (FMC).
Les critères médico-économiques ne sauraient être occultés. Il faut sensibiliser, dès leur formation, les futurs médecins à la nécessité d’une prise en charge qui conjuguera qualité et optimisation des coûts, dans le respect de l’information et des attentes du malade. L’étudiant ne peut pas vivre toutes ses études dans un monde éthéré où la question du rapport qualité-coût ne serait jamais évoquée. Ce souci d’efficience ne peut pas être limité aux quelques heures consacrées à l’économie de santé et doit être présent dans l’esprit de chacun, si nous voulons préserver la qualité de notre protection sociale. Nous y travaillons avec nos collègues du ministère de l’enseignement supérieur et de la recherche, en lien étroit avec la HAS.
A la demande de la direction générale de la santé, a été créé, il y a sept ans, un enseignement de la iatrogénie pour les étudiants en médecine de sixième année – cet enseignement fait l’objet d’un séminaire obligatoire – et en pharmacie. Par ailleurs, la direction générale de la santé a transmis l’an dernier au ministère de la recherche et de l’enseignement supérieur des propositions visant à améliorer la formation des étudiants en médecine et en pharmacie tout au long de leur cursus.
Les réflexions en cours sur le LMD (licence, master, doctorat) devraient notamment porter sur la nécessité de bien prendre en compte la thématique du médicament.
Pour ce qui concerne la formation médicale continue, le dispositif professionnel de FMC sera installé en 2008. L’ensemble des dispositifs doit être très lisible pour les médecins : il faut qu’ils puissent s’approprier ces démarches et qu’elles puissent, en particulier pour l’EPP, faire partie intégrante de leur pratique.
Je ne reviens pas sur la visite médicale et la place de l’industrie pharmaceutique car j’ai déjà longuement répondu sur ces deux points.
Mme Catherine Lemorton, rapporteure : Je souhaite revenir sur la question de l’octroi des autorisations de mise sur le marché (AMM) par l’AFSSAPS. Des médicaments sont autorisés à être mis sur le marché alors qu’ils sont des contournements de génériques, ce qui oblige ensuite les pouvoirs publics, via le CEPS, à revoir les prix à la baisse. S’ils n’avaient pas reçu d’AMM au départ, on aurait évité les effets de contrôles en cascade, qui mettent parfois à mal la politique du générique sur certaines molécules.
Mme Roselyne Bachelot : Il y a eu, certes, des dérives, mais elles sont tout à fait limitées. M. Noël Renaudin, président du CEPS, qui a été interpellé sur cette affaire, a indiqué qu’un produit et son dérivé commercial, le Mopral et l’Inexium, avaient échappé à son rayon laser mais qu’en fait le contournement de génériques était extrêmement difficile. Les produits peuvent être admis au remboursement mais la simple mise à disposition sous une autre forme galénique, qui pouvait d’ailleurs ne pas être sans intérêt, n’entraîne pas une majoration du prix.
Mme Catherine Lemorton, rapporteure : La grève du générique des pharmaciens a quand même eu des répercussions sur les comptes de l’assurance maladie. Ces produits ne sont pas anodins. Ils sont chers.
Mme Roselyne Bachelot : C’est effectivement un vrai problème, mais il ne faut pas, à partir d’un ou deux exemples malencontreux, qui ont échappé aux radars, et qui ont été rectifiés par la suite, dire que c’est une pratique généralisée.
M. Jean Mallot, coprésident : Ces cas ne posent-ils pas la question de l’attribution de l’AMM en général ? Ne peut-on imaginer d’ajouter aux critères d’attribution celui de progrès thérapeutique ?
Mme Roselyne Bachelot : C’est la base. Si la molécule est la même, il n’y a pas de raison qu’un laboratoire n’ait pas l’AMM. C’est ensuite au niveau du remboursement qu’il y a des différences. S’il n’y a pas d’amélioration du service médical rendu, le produit est moins remboursé.
Je vais faire un point sur l’admission au remboursement. La Cour estime, en effet, que le circuit de mise sur le marché et d’admission au remboursement est insuffisamment sélectif. Je veux donc en dire quelques mots.
La commission d’AMM évalue le profil d’efficacité et de sécurité individuel des médicaments. La commission de la transparence doit évaluer l’impact par rapport aux alternatives thérapeutiques disponibles, établir la place dans la stratégie thérapeutique et estimer la population-cible des médicaments. La commission d’AMM a une vision individuelle et la commission de la transparence une vision populationnelle, collective, donc de santé publique.
L’intérêt de santé publique (ISP) est un sous-critère du service médical rendu (SMR). Il évalue l’apport d’un médicament dans une population. C’est donc un critère collectif. Aujourd’hui, l’évaluation du médicament repose sur le service rendu par un médicament à l’individu plus qu’à la collectivité, comme le prouve le nombre de SMR importants attribués.
L’ISP introduit dans l’évaluation la dimension de l’accessibilité aux soins. La politique des études post-AMM découle de l’importance donnée à ce critère. C’est un sujet qui intéresse beaucoup Mme Lemorton qui m’avait interrogée sur les études post-AMM. Les médicaments doivent être réévalués sur la base des résultats des études post-AMM et au vu de l’évaluation de l’ISP réellement rendu. Les conséquences doivent en être tirées en termes de population prise en charge.
Nous réfléchissons à l’opportunité d’une réforme de la transparence, pour mieux définir les critères appréciés par la commission de la transparence afin de conduire à une plus grande sélectivité dans le choix des médicaments remboursés par la collectivité.
Toutefois, il faut être prudent avant de changer à nouveau les textes et de bouleverser les règles du jeu : il est nécessaire que la commission de la transparence se concentre sur son rôle propre, qui est de définir la valeur thérapeutique ajoutée d’un médicament, sans doublonner le travail de la commission d’AMM. Il faut qu’elle ose attribuer un SMR insuffisant si son intérêt ne justifie pas sa prise en charge par la collectivité, sans considérer que cela remet en cause l’AMM.
Il est à signaler que l’avis du Haut conseil pour l’avenir de l’assurance maladie du 29 juin 2006 indiquait que le système français de fixation des prix du médicament était plutôt efficace. Les principes y sont considérés comme sains et conduisent à des niveaux de prix cohérents. Le rapport de l’Office of Fair Trading sur les prix pharmaceutiques anglais de février 2007 cite à plusieurs reprises le modèle français en exemple – ou plutôt un modèle qui est le modèle français mais sans dire qu’il est français.
La mise en œuvre d’essais comparatifs est effectivement un sujet fondamental et doit monter en puissance. Mais il ne faut pas perdre de vue que nous sommes aujourd’hui dans un contexte européen et qu’être les seuls à exiger des essais cliniques supplémentaires pourrait conduire à créer des freins majeurs à l’accès aux innovations dans notre pays. Cette question doit être introduite progressivement, par exemple au moment de la réévaluation du SMR et de l’ASMR des médicaments ayant obtenu un prix élevé. Ce sera un des enjeux de la mission médico-économique confiée à la HAS.
C’est une particularité des Français d’anticiper un certain nombre de réglementations européennes avant même qu’elles ne soient mises en œuvre, puis, quand elles sont appliquées au niveau européen, de montrer l’Europe du doigt et de s’offusquer de ce que l’Europe nous impose des législations alors que nous les avons inventées nous-mêmes.
Mme Catherine Lemorton, rapporteure : S’il y a eu un « bug » dans le système concernant la molécule qui a été citée, cela signifie qu’il y a une faille dans ce système.
Mme Roselyne Bachelot : Dans tous les systèmes humains, il y a des « bugs » !
Mme Catherine Lemorton, rapporteure : Quand on voit les problèmes d’épidémiologie qui existent sur certains traitements, dont l’affaire est actuellement en justice, je ne voudrais pas qu’il y ait d’autres failles.
Du fait de la pyramide des compétences sanitaires, il est parfois difficile de discerner le contour de celles-ci. La loi de financement de la sécurité sociale pour 2008 octroie une compétence médico-économique à la HAS. Quelle est la différence entre la compétence économique de la HAS et celle du CEPS ?
Mme Roselyne Bachelot : La Cour a relevé un besoin d’analyse médico-économique permettant de rapporter l’efficacité des médicaments à leur coût. Elle a cité, à cet égard, l’exemple de la Grande-Bretagne et de l’Allemagne.
L’une des mesures de la loi de financement de la sécurité sociale pour 2008 que j’ai portée permet, comme vous l’avez rappelé, à la HAS de compléter son approche des stratégies thérapeutiques par la prise en compte de considérations médico-économiques, lui permettant ainsi de privilégier les parcours de soins les plus efficients.
Cette approche médico-économique est développée avec les homologues britanniques – National Institute of Clinical Excellence (NICE) – et allemand – Institut pour la qualité et l’efficience en santé (IQWIG) – de la HAS, avec lesquels celle-ci développe des contacts réguliers.
Le développement de cette nouvelle approche permettra aux décideurs de bénéficier d’une expertise globale en contribuant à la promotion des stratégies de soins les plus efficaces, à une grande sélectivité dans la prise en charge, à une allocation efficiente des moyens et à une information opérationnelle adaptée aux besoins des médecins.
Les deux premiers champs sur lesquels la médico-économie pourrait intervenir sont, d’une part, la prise en charge médicamenteuse de l’hypertension artérielle, pour laquelle plusieurs types de médicaments sont en concurrence, les plus coûteux devant être réservés aux patients pour lesquels ils apportent véritablement un bénéfice supérieur aux alternatives moins coûteuses et, d’autre part, le traitement de l’obésité sévère des patients atteints de diabète, les traitements médicamenteux ne devant être prescrits qu’en deuxième recours, si les patients ne voient pas leur indice de masse corporelle diminuer après une diète et une activité physique adaptées.
Les prescripteurs, en tant qu’ordonnateurs de soins, doivent également être sensibilisés aux aspects médico-économiques de leurs prescriptions. La diffusion par la HAS de recommandations de bonne pratique et de stratégie thérapeutique prenant en compte les aspects médico-économiques devrait y contribuer. Une autre mesure de la loi de financement de la sécurité sociale pour 2008 consiste à intégrer dans les logiciels d’aide à la prescription certifiés par la HAS l’affichage des prix au moment de la prescription. C’est une mini-révolution, car les prescripteurs ne connaissent pas aujourd’hui le prix des médicaments qu’ils prescrivent.
En résumé, et pour répondre à la question, le CEPS négocie les prix. La HAS indique à ses interlocuteurs une stratégie qui intègre la notion médico-économique.
M. Jean Mallot, coprésident : Nous vivons à l’époque d’Internet. C’est un moyen important pour l’information des usagers et à la fois l’information et la formation des professionnels de santé. Cet outil est également à la disposition des laboratoires pour faire la promotion de médicaments. Comment la puissance publique peut-elle garder la maîtrise de ces mécanismes et en assurer la moralisation ? Ne peut-on envisager qu’elle mette en place des contre-feux pour contrebalancer l’information fournie aux consommateurs ?
M. Pierre Morange, coprésident : Sur Internet, se pose également le problème des médicaments contrefaits, qui échappent à tout contrôle sanitaire.
Mme Roselyne Bachelot : La question d’Internet est évoquée dans bien d’autres enceintes.
Pour ce qui concerne l’information donnée aux médecins, les logiciels d’aide à la prescription sont certifiés par la HAS, et j’engage les médecins à n’utiliser que ceux-ci qui leur donnent une totale garantie.
En ce qui concerne l’information accessible à la population, un groupe de travail a été constitué pour réfléchir sur les contre-feux à appliquer. C’est extrêmement difficile à mettre en œuvre. Mais ce n’est pas pour cela que nous ne faisons rien.
Pour ce qui est des contrefaçons et la mise à disposition d’un certain nombre de médicament plus ou moins fiables sur des sites dédiés à certains types de produits qui font florès sur Internet, nous sommes totalement dénués de moyens. Nous ne pouvons faire que de l’information aval, en incitant les personnes à ne pas se procurer ces médicaments sur Internet, certains produits ne respectant ni les présentations galéniques efficientes, ni le dosage en principes actifs. Surtout, comme ce sont des médicaments qui ne sont pas dépourvus d’effets secondaires ni de dangers, ils doivent être prescrits selon les règles de l’art.
Les risques de ces produits sont connus. Ils relèvent donc d’une information grand public dispensée par l’ensemble de la chaîne de l’information : les pouvoirs publics, les ministères et les médecins. Il est impossible d’empêcher la vente de ces produits sur Internet. On ne peut qu’inciter les citoyens à suivre le réseau pharmaceutique et le réseau médical, car ce sont eux qui leur donnent la meilleure information et la meilleure protection. Le médicament n’est pas un produit banal.
M. Pierre Morange, coprésident : L’industrie de la contrefaçon sévissant à l’échelle planétaire, le taux de pénétration de ces produits est très élevé.
Mme Roselyne Bachelot : Il faut reconnaître, à ce sujet, que grâce à la structuration de son système de santé et de délivrance pharmaceutique, notre pays est protégé, comparé à d’autres pays, notamment aux pays en voie de développement où la contrefaçon est dramatique : on vend sur les marchés de telle ou telle capitale d’Afrique des produits totalement inefficaces et même nocifs.
M. Jean Mallot, coprésident : Où en est l’application des articles 43 et 44 de la loi de financement de la sécurité sociale pour 2008 concernant le contrat d’engagements individuel et l’expérimentation de nouveaux modes de rémunération des médecins ? Des textes restent certainement à produire pour que ces mesures soient effectives et des concertations avec les acteurs du système doivent être engagées pour qu’elles prennent effet.
Mme Roselyne Bachelot : Je pourrai revenir devant vous pour l’évaluation de ces dispositions à la fin du premier trimestre 2008.
J’ai indiqué aux directeurs des missons régionales de santé que je souhaitais que l’on soit très allant sur ces nouvelles dispositions. J’ai d’ailleurs été très sensible au fait que la Cour des comptes ait salué la loi de financement de la sécurité sociale pour 2008 comme ayant pris en compte certaines de ses préconisations, même si je ne partage pas toutes les analyses de la Cour.
J’ai ouvert la voie à de nouveaux modes de rémunération et à de nouvelles dispositions dans la loi de financement de la sécurité sociale pour 2008. Ces innovations doivent être considérées aussi bien par l’assurance maladie que par les médecins et les professionnels de santé comme quelque chose qui ne vient pas combattre la logique conventionnelle, mais s’y ajouter et l’améliorer. Pour des médecins qui souhaitent aller plus loin, en particulier en matière de santé publique, je considère que c’est une opportunité intéressante.
Au cours des états généraux de l’organisation de la santé, j’ai été frappée de voir, dans les discours tenus par les jeunes médecins, à quel point ils étaient réceptifs à ces nouveaux modes de rémunération. Pour prendre en charge du dépistage et des politiques de santé publique, c’est une voie extrêmement prometteuse.
M. Pierre Morange, coprésident : Nous vous remercions, madame la ministre.
*
AcBUS : Accord de bon usage des soins
ACOSS : Agence centrale des organismes de sécurité sociale
AFSSAPS : Agence française de sécurité sanitaire des produits de santé
AGF : Assurances générales de France
ALD : Affection de longue durée
AMC : Assurance maladie complémentaire
AMM : Autorisation de mise sur le marché
AMO : Assurance maladie obligatoire
ANAES : Agence nationale d’accréditation et d’évaluation en santé
ARCHIMED : Archives médicales de la Mutualité sociale agricole
ARH : Agence régionale de l’hospitalisation
ARS : Agence régionale de santé
ASMR : Amélioration du service médical rendu (I : majeure, II : importante, III : modérée, IV : mineure)
ATU : Autorisation temporaire d’utilisation
BVP : Bureau de vérification de la publicité
CADA : Commission d’accès aux documents administratifs
CDES : Comité départemental d’éducation à la santé
CeNGEPS : Centre national de gestion des essais de produits de santé
CEPS : Comité économique des produits de santé
CGTI : Conseil général des technologies de l’information
CHMP : Comité des médicaments à usage humain
CHU : Centre hospitalier universitaire
CIP : Code identifiant de présentation
CISS : Collectif interassociatif sur la santé
CLCV : Confédération de la consommation, du logement et du cadre de vie
CMU : Couverture maladie universelle
CMUC : Couverture maladie universelle complémentaire
CNAF : Caisse nationale des allocations familiales
CNAMTS : Caisse nationale d’assurance maladie des travailleurs salariés
CNAVTS : Caisse nationale d’assurance vieillesse des travailleurs salariés
CNFMC : Conseils nationaux de la formation médicale continue (des médecins salariés, des médecins libéraux, des médecins hospitaliers)
CNHIM : Centre national hospitalier d’information sur le médicament
CNIL : Commission nationale de l’informatique et des libertés
CNIS : Conseil national de l’information statistique
CNSA : Caisse nationale de solidarité pour l’autonomie
COFRAC : Comité français d’accréditation
CPAM : Caisse primaire d’assurance maladie
CPS : Carte de professionnel de santé
CREDES : Centre de recherche, d’étude et de documentation en économie de la santé (devenu IRDES)
CRES : Comité régional d’éducation à la santé
CRPV : Centre régional de pharmacovigilance
CSIS : Conseil stratégique des industries de santé
CSMF : Confédération des syndicats médicaux français
CSP : Code de la santé publique
DAM : Délégué de l’assurance maladie
DC : Dénomination commune
DCI : Dénomination commune internationale
DDASS : Direction départementale des affaires sanitaires et sociales
DDD : Defined daily dosis (dose journalière définie)
DGCCRF : Direction générale de la concurrence, de la consommation et de la répression des fraudes
DGI : Direction générale des impôts
DGS : Direction générale de la santé
DHOS : Direction de l’hospitalisation et de l’organisation des soins
Desease management : Soutien à la prise en charge thérapeutique des patients atteints de maladies chroniques
DMP : Dossier médical personnel
DP : Dossier pharmaceutique
DPI : Déclaration publique d’intérêts
DRASS : Direction régionale des affaires sanitaires et sociales
DREES : Direction de la recherche, des études, de l’évaluation et des statistiques
DSS : Direction de la sécurité sociale
EGOS : États généraux de l’organisation de la santé
EMEA : European agency for the evaluation of medicinal products (agence européenne du médicament)
EPAR : Rapport européen public d’évaluation
EPP : Évaluation des pratiques professionnelles
EuroMedStat : Statistics on Medicines in Europe
FAF-PL : Fonds d’assurance-formation des professions libérales
FDA : Federal drug administration
FDA : Food and Drug Admistration
FFSA : Fédération française des sociétés d’assurances
FIF-PL : Fonds interprofessionnel de formation des professions libérales
FIQCS : Fonds d’intervention pour la qualité et la coordination des soins
FMC : Formation médicale continue
FOPIM : Fonds pour la promotion de l’information médicale
FSPF : Fédération des syndicats pharmaceutiques de France
GEMME : GEnériques Même MEdicaments
GERS : Groupement pour l’élaboration et la réalisation de statistiques
GIP-DMP : Groupement d’intérêt public du dossier médical personnel
GIS : Groupement d’intérêt scientifique
GRSP : Groupement régional de santé publique
GTCPD : Groupe de travail sur les conditions de prescription et de délivrance
HAS : Haute Autorité de santé
HCAAM : Haut conseil pour l’avenir de l’assurance maladie
HMO : Health maintenance organization (organisation intégrée de management des soins aux États-Unis)
HON Health on the net foundation (fondation suisse choisie par la Haute Autorité de santé pour la certification des sites de santé)
IEC : Inhibiteurs de l’enzyme de conversion
IGAS : Inspection générale des affaires sociales
IGF : Inspection générale des finances
IMS : Intercontinental Marketing Services
INca : Institut national du cancer
INPES : Institut national de prévention et d’éducation pour la santé
IPP : Inhibiteur de la pompe à protons
IQWIG : Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen – Institut pour la qualité et la rentabilité dans la santé publique (homologue allemand de la Haute Autorité de santé)
IRDES : Institut de recherche et documentation en économie de la santé
ISP : Intérêt de santé publique
IUFM : Institut universitaire de formation des maîtres
IVG : Interruption volontaire de grossesse
LAP : Logiciel d’aide à la prescription
LEEM : Les entreprises du médicament
LFSS : Loi de financement de la sécurité sociale
LGO : Logiciel de gestion d’officine
LIR : Laboratoires internationaux de recherche
LMD : Licence, master, doctorat
LPP : Liste des produits et prestations
Médicaments conseil : Médicaments pouvant être délivrés sans ordonnance par le pharmacien, faisant l’objet d’une AMM mais non remboursés
MECSS : Mission d’évaluation et de contrôle des lois de financement de la sécurité sociale
MSA : Mutualité sociale agricole
NHS : National Health Service
NICE : National Institute of Clinical Excellence – Institut national d’évaluation clinique (homologue britannique de la Haute Autorité de santé)
NR : Non remboursable
OMS : Organisation mondiale de la santé
ONDAM : Objectif national de dépenses d’assurances maladie
OTC : Over the counter (« au-delà du comptoir » ou médicaments conseil)
PGR : Plan de gestion des risques
PH : Praticiens hospitaliers
PLFSS : Projet de loi de financement de la sécurité sociale
PNNS Programme national nutrition santé
PSUR : Periodic Safety Update Reports (rapport périodique actualisé de pharmacovigilance)
PTT : Protocole temporaire de traitement
R&D : Recherche et développement
RAPDI : Rapport annuel des déclarations d’intérêt
RAPPE : Rapport d’évaluation d’AMM
RCP : Résumé des caractéristiques du produit
RSI : Régime social des indépendants
Sartans : Antagonistes des récepteurs AT1 de l’angiotensine
SERC : Service rendu à la collectivité
SIPS : Système d’information sur les produits de santé
SML : Syndicat des médecins libéraux
SMR : Service médical rendu
SMRI : Service médical rendu insuffisant
T2A : Tarification à l’activité
TDR : Test de diagnostic rapide
THS : Traitement hormonal substitutif
TFR : Tarif forfaitaire de responsabilité
UFC : Union fédérale des consommateurs
UNAF : Union nationale des associations familiales
UNCAM : Union nationale des caisses d’assurance maladie
UNOCAM : Union nationale des organismes d’assurance maladie complémentaire
UNPF : Union nationale des pharmacies de France
URCAM : Union régionale des caisses d’assurance maladie
USPO : Union des syndicats de pharmaciens d’officine
VM : Visite médicale
*
ANNEXE 5 : COMMUNICATION DE LA COUR DES COMPTES CONCERNANT LES TAXES SUR LE MĖDICAMENT HUMAIN

COMMUNICATION À LA COMMISSION DES AFFAIRES CULTURELLES, FAMILIALES ET SOCIALES
DE L’ASSEMBLÉE NATIONALE
MISSION D’ÉVALUATION ET DE CONTRÔLE DES LOIS
DE FINANCEMENT DE LA SÉCURITÉ SOCIALE
Article LO 132-3-1 du code des juridictions financières
LES TAXES SUR LE MÉDICAMENT HUMAIN
Mai 2007
![]()
Sommaire
PARTIE I : UN DISPOSITIF COMPLEXE ET INSTABLE 3
I. - Les taxes rémunérant des services rendus 3
A. La taxe perçue par la Haute Autorité en santé 3
B. Les taxes perçues par l’Agence française de sécurité sanitaire des produits de santé 4
II. - Les taxes affectées à l’assurance maladie 8
A. Les taxes destinées à maîtriser la dépense de médicaments remboursés par l’assurance maladie 10
B. Les taxes destinées à procurer des recettes à l’assurance maladie 12
C. La TVA brute collectée par les commerçants en gros en produits pharmaceutiques 15
Partie II : Les problèmes juridiques posés par certaines taxes 16
I. - Les contentieux portant sur l’assiette 16
A. Une complexité d’ensemble 16
B. Deux taxes présentent des difficultés particulières 17
II. - Les difficultés liées aux contrôles 19
A. L’existence d’un plan de contrôle 19
B. La contestation des contrôles 20
Partie III : le rendement des principales taxes 21
A. Les taxes affectées à l’assurance maladie 21
B. L’évolution des montants de TVA 24
C. Les échanges d’informations entre administrations et organismes percevant des taxes 27
![]()
Les entreprises qui produisent ou distribuent des médicaments sont redevables de taxes spécifiques dont le nombre a été croissant au cours du temps. On en compte actuellement onze d’importance très inégale. En 2006, le montant total de ces taxes a atteint 1 021 M€ contre 742 M€ en 2005, soit une progression de 37,8 %.
Cette diversité des taxes se justifie au moins en partie par les finalités différentes qui sont poursuivies : prix à payer ou contrepartie de services rendus pour les unes, régulation économique ou encadrement sanitaire pour d’autres ou encore recettes destinées au financement de l’assurance maladie.
Ces taxes ont représenté 3,1 % du chiffre d’affaires (France) des industries pharmaceutiques en 2005 et environ 4 % en 2006 et les recettes affectées à l’assurance maladie 2,9 % et 3,8 % des dépenses de médicaments.
PARTIE I : UN DISPOSITIF COMPLEXE ET INSTABLE
I. – LES TAXES RÉMUNÉRANT DES SERVICES RENDUS
A. La taxe perçue par la Haute Autorité en Santé
1. Son objet
En vue de l’inscription d'un médicament sur la liste des spécialités pharmaceutiques remboursables ou sur la liste des médicaments agréés à l’usage des collectivités publiques, du renouvellement ou de la modification de cette inscription, les entreprises pharmaceutiques paient une taxe (article L. 5123-5 du code de la santé publique). Son objet est de couvrir le coût de la prestation d’évaluation du service médical rendu. Les montants forfaitaires de la taxe sont fixés par arrêté et varient en fonction du type de demande (demande initiale, modification, renouvellement). La taxe est due par dossier présenté.
Cette contribution a été perçue jusqu’en 2004 par l’AFSSAPS puis à compter du 1er janvier 2005 par la HAS conformément aux dispositions de la loi du 13 août 2004 qui a transféré à cette dernière les activités de la commission de la transparence et celles de la commission d’évaluation des produits et prestations (CEPP).
2. L’évolution de son rendement
Les recettes de cette taxe ont représenté respectivement en 2005 et 2006, 4,1 m € soit 6,43 % du montant total des recettes enregistrées par la Haute Autorité et 4,2 m € soit 6,89 % de ce même montant. Cette évolution correspond à une augmentation de 3,7 % des montants encaissés. Ces recettes couvrent environ 90 % du coût de fonctionnement de la commission de transparence.
![]()
3. Les conditions de calcul, de recouvrement et de contrôle
S’agissant du calcul des taxes, les services de la Haute Autorité n’enregistrent aucun contentieux. Si des problèmes surviennent, ils ont pour origine une erreur matérielle qui peut être facilement corrigée.
Le recouvrement, de la responsabilité de l’agent comptable, pose essentiellement des difficultés liées aux modalités de règlement ; quand celui-ci est opéré par virement, et non par chèque, la difficulté consiste à rattacher le versement à la spécialité pharmaceutique concernée.
Quand un montant de taxe supérieur à celui qui était dû, au regard du type de demande faite, a été encaissé, un remboursement a lieu : là encore la difficulté consiste pour l’agent comptable à rattacher le versement opéré au médicament pour lequel la demande a été faite. En 2006, 11 500 € ont été remboursés ce qui représente moins de 0,3 % du montant total perçu.
b. Les taxes perçues par l’Agence française de sécurité sanitaire des produits de santé
1. La taxe annuelle sur les spécialités pharmaceutiques bénéficiant d’une autorisation de mise sur le marché
L’AFSSAPS perçoit chaque année une taxe sur chaque spécialité pharmaceutique bénéficiant d’une autorisation de mise sur le marché (AMM) dite taxe sur le chiffre d’affaires des spécialités pharmaceutiques. Le montant de cette taxe est fonction du montant des ventes hors taxes de la dite spécialité au cours de l’année civile précédente, à l’exclusion des ventes à l’exportation (article L 5121-17 du code la santé publique).
Elle est due par le bénéficiaire de l’autorisation. La loi a prévu un barème comportant un minimum de cinq tranches ; il en comporte actuellement neuf.
Les spécialités pharmaceutiques bénéficiant d’une autorisation d’importation parallèle font dans les mêmes conditions l’objet d’une taxe annuelle (article L. 2124-17-2 du Code de la Santé Publique).
Cette taxe peut s’apparenter au prix à payer par les laboratoires pharmaceutiques pour les spécialités bénéficiant d’une AMM « vivante » en vue de contribuer à l’ensemble des activités réalisées par l’Agence au titre de sa mission générale de sécurité sanitaire tant en termes d’évaluation des spécialités, de contrôle des produits, d’inspection des sites, de veille et de surveillance des effets inattendus ou indésirables des spécialités pharmaceutiques ou encore de bon usage des dits médicaments.
Le montant de la taxe perçue par l’AFSSAPS était de 17,7 M€ en 2004, de 18,6 M€ en 2005 et de 19,3 M€ en 2006. Sur la période il a augmenté de 8,9 %. Il a représenté en 2006, 21 % du montant des ressources de l’AFSSAPS. Le nombre d’entreprises concernées est passé de 370 en 2004 à 397 en 2006.
2. Les autres taxes perçues par l’AFSSAPS
![]()
L’agence française de sécurité sanitaire des produits de santé perçoit quatre autres taxes se rapportant directement aux médicaments ;
La loi a institué un droit dit « progressif » payé par les laboratoires pharmaceutiques pour toute demande et examen de dossier d’autorisation de mise sur le marché de médicaments (nouveaux principes actifs ou nouvelles indications), ou de modification ou encore de renouvellement de cette autorisation (article L. 5121-16 du code la santé publique). Son montant est fixé par décret dans une limite actuellement de 25 400 euros définie par la loi. Le droit payé varie en fonction du type d’évaluation à réaliser (nouveau principe actif, nouvelle indication pour un produit disposant déjà d’une AMM, etc.…). Le barème actuellement applicable est celui qui résulte de l’article D. 5121-64 du code de la santé publique.
La loi a aussi prévu un droit dit « progressif » à payer pour l’enregistrement des médicaments homéopathiques (article L.5121-15 du code la santé publique). Le montant en est fixé par décret dans une limite édictée par la loi, actuellement de 7 600 euros. Le droit payé varie en fonction du type d’évaluation à réaliser. Le barème applicable est celui qui résulte de l’article D. 5121-65 du code de la santé publique.
L’agence reçoit aussi une taxe pour toute demande d’importation parallèle (article L.5124-17-1 du code la santé publique).
Enfin, elle perçoit une taxe pour toute demande de visa ou de renouvellement de visa de publicité (article L.5122-5 du code la santé publique).
Le barème du droit progressif en matière d’AMM a été réévalué en 2004 ; il était inchangé depuis 1996 (décret n° 96-653 du 16 juillet 1996 modifié par l’ordonnance n° 2000-916 du 19 septembre 2000 relatif à la transposition en euros des taux). Cette réévaluation avait pour objectif de tenir compte des coûts supportés par l’agence. Le montant forfaitaire du droit payé est de 25 400 euros pour une demande d’examen de dossier au titre d’un nouveau principe actif, de 16 790 euros pour une demande de dossier faite au titre d’une nouvelle indication, de 10 110 euros pour une demande de dossier faite au titre d’un produit similaire, de 1 011 euros pour une demande de dossier faite au titre d’une modification, enfin de 674 euros pour une demande de renouvellement quinquennal.
Le rendement des droits progressifs payés par les laboratoires est directement lié au nombre de dossiers de demande, de modification ou de renouvellement d’AMM déposés.
3. Une évolution des rendements liée aux choix de stratégie industrielle et commerciale des entreprises
Le tableau qui suit récapitule l’évolution des montants des taxes sur les médicaments perçues par l’AFSSAPS entre 2004 et 2006.
![]()
L’augmentation du rendement des taxes a été relativement forte, de 12,6 % entre 2004 et 2006. Elle est pour une large part due à l’augmentation du nombre de dossiers examinés en vue de renouvellements quinquennaux et de modifications d’AMM.
Tableau n° 1 : Évolution du rendement des taxes sur le médicament perçues par l’AFSSAPS
M€ |
2004 |
2005 |
2006 |
Evol 2004-2006 |
Taxes sur le CA des spécialités pharmaceutiques disposant d'une AMM |
17,7 |
18,6 |
19,3 |
8,9 % |
Droits pour l'examen des dossiers d'AMM |
||||
Nouveaux principes actifs |
1,7 |
0,8 |
0,9 |
-43,6 % |
Nouvelles indications |
2,7 |
1,3 |
1,5 |
-43,8 % |
Produits similaires |
8,5 |
11,4 |
9,2 |
8,1 % |
Modifications |
12,2 |
15,1 |
16,6 |
36,0 % |
Renouvellement quinquennal |
1,3 |
1,7 |
2,1 |
64,2 % |
Enregistrements homéopathiques |
0,2 |
0,3 |
0,1 |
-46,9 % |
Droits pour examen des dossiers de demande d'importation parallèle |
0,009 |
0,06 |
||
Visa publicité |
4,8 |
5,2 |
5,6 |
14,0 % |
Total |
49,3 |
54,5 |
55,5 |
12,6 % |
Budget total |
92,7 |
98,8 |
100,0 |
|
Part des taxes dans Budget en % |
53 % |
55 % |
55 % |
Cour des Comptes d’après données AFSSAPS
Le tableau suivant figure l’évolution du nombre de demandes par types de dossiers.
Tableau n° 2 : Évolution du nombre de demandes par types de dossiers examinés par l’AFSSAPS
2004 |
2005 |
2006 |
Evol 2004-2006 | |
Droits pour l'examen des dossiers d'AMM |
Nbre de dossiers |
Nbre de dossiers |
Nbre de dossiers |
|
Nouveaux principes actifs |
77 |
36 |
48 |
-37,7 % |
Nouvelles indications |
174 |
90 |
103 |
-40,8 % |
Produits similaires |
863 |
1 129 |
915 |
6,0 % |
Modifications |
12 437 |
15 022 |
16 783 |
34,9 % |
Renouvellement quinquennal |
2 027 |
2 518 |
3 168 |
56,3 % |
Enregistrements homéopathiques |
341 |
415 |
181 |
-46,9 % |
Droits sur examen de demande d'importation parallèle |
1 |
7 |
||
Visa publicité |
10 035 |
10 151 |
10 897 |
8,6 % |
Cour des Comptes d’après données AFSSAPS
![]()
Entre 2004 et 2006, le nombre de dossiers sur les nouveaux principes et les nouvelles indications a baissé. Cette évolution peut s’expliquer à la fois par des choix de stratégie industrielle et commerciale des entreprises du secteur et par un recours plus important aux procédures européennes.
Si le montant des droits progressifs encaissés au titre des demandes d’AMM sur de nouveaux principes actifs et de nouvelles indications diminue du fait d’un probable transfert des demandes vers l’EMEA (european agency for the evaluation of medicinal products ou agence européenne des produits de santé), il y a lieu de noter que les recettes provenant des expertises rendues pour le compte l’EMEA, lorsque la France est rapporteur sur des demandes d’AMM, enregistrent une augmentation. Ces recettes (non comptabilisées avec les taxes) sont passées de 2 M€ en 2004 à 3 M€ ce qui représente une augmentation de 50 %.
Les recettes de l’agence comme son activité sont ainsi dépendantes du partage qui s’opère dans les demandes de mise sur le marché entre le niveau national et communautaire.
Hormis la taxe sur le chiffre d’affaires qui progresse de façon assez linéaire, les montants des autres taxes peuvent connaître des évolutions importantes d’une année sur l’autre. L’exemple le plus significatif est celle du droit progressif sur les demandes d'A.M.M. « similaire » dont le rendement est très instable depuis 2004 en raison de la politique industrielle et commerciale des laboratoires : le nombre de dossiers examinés a été respectivement en 2004, 2005 et 2006 de 863, 1 129 et 923 et les rendements correspondants de 8,5 M€, 11,4 M€, et 9,2 M€.
Concernant les rendements prévisionnels sur les années à venir, une étude devrait être lancée en 2007 couvrant l’évolution possible des taxes pour la période 2008 – 2010.
4. La liquidation et le recouvrement des taxes perçues
S’agissant de l’assiette de la taxe annuelle sur le chiffre d’affaires des spécialités pharmaceutiques bénéficiant d’une autorisation de mise sur le marché, les informations relatives aux chiffres de vente de chacune des spécialités concernées sont transmises par les laboratoires pharmaceutiques. Le système est donc exclusivement déclaratif. L’agence n’est pas en mesure de vérifier la véracité et l’exhaustivité des informations relatives aux chiffres d’affaires ainsi transmises.
Nonobstant ce fait majeur, l’agence ne fait pas état d’autre problème particulier lié à l’établissement de la taxe. Les calculs réalisés à partir des éléments d’assiette ainsi transmis, n’ont pas donné lieu à contentieux.
Dans l’hypothèse où l’Agence reçoit un montant de taxe ou de droit supérieur à celui qui est dû, elle émet, soit une réduction de titre de recette (partielle ou totale) si la demande intervient dans le même exercice budgétaire que le titre de recette initial, soit un mandat d’annulation du titre de recette si la demande n’intervient pas dans le même exercice budgétaire.
En matière de recouvrement, les éventuelles difficultés peuvent provenir de l’utilisation massive du virement bancaire en lieu et place du recours au chèque, ce qui génère des difficultés d’identification du paiement et de rapprochement entre les montants encaissés, les dossiers reçus et les titres de recettes correspondants. Pour y remédier, l’agence fait état du lancement prochain d’un projet de télédéclaration et de télépaiement.
![]()
Compte tenu des risques de conflit d’intérêt entre l’activité de l’AFSSAPS et son financement par une taxe perçue auprès des professionnels, la Cour considère qu’il serait préférable que sa collecte ne soit pas assurée par l’agence mais plutôt par la direction générale des impôts en même temps que la TVA.
II. – LES TAXES AFFECTÉES À L’ASSURANCE MALADIE
Depuis plusieurs années, le législateur mène une politique de maîtrise des dépenses de médicament avec plusieurs objectifs : limiter la part des remboursements de médicaments dans les dépenses d’assurance maladie afin de contribuer au respect de l’ONDAM, contenir le déficit de l’assurance maladie et rationaliser les prescriptions médicamenteuses elles-mêmes. Plusieurs taxes ont été créées pour répondre à ces objectifs (v. tableau n° 3).
![]()
En outre, depuis 2006, la sécurité sociale dont, principalement l’assurance maladie, bénéficie, au titre de la compensation des exonérations de cotisations sociales sur les bas salaires, du reversement du produit de la TVA brute sur les médicaments collectée par les grossistes répartiteurs.
Tableau n° 3 : Tableau des taxes affectées à l’assurance maladie
Nom |
Texte institutif |
Débiteur |
Bénéficiaire |
Organisme de recouvrement |
Contribution des entreprises assurant l'exploitation en France, au sens de l'article L. 596 du code de la santé publique, d'une ou plusieurs spécialités pharmaceutiques donnant lieu à remboursement par les caisses d'assurance maladie (art L245-1 du CSS) dite "taxe sur les dépenses de promotion" |
Loi n° 83-25 du 19 janvier 1983 et loi n° 97-1164 du 19 décembre 1997 |
Entreprises assurant l'exploitation en France des dites spécialités |
caisse nationale de l'assurance maladie des travailleurs salariés |
ACOSS |
Contribution à la charge des entreprises assurant l'exploitation d'une ou plusieurs spécialités pharmaceutiques dite "contribution de la clause de sauvegarde de l’ONDAM" (Article L138-10 du CSS) |
Loi nº 98-1194 du 23 décembre 1998 |
Entreprises assurant l'exploitation d'une ou plusieurs spécialités pharmaceutiques |
assurance-maladie |
ACOSS |
Contribution assise sur le chiffre d'affaires hors taxes réalisé en France au titre des spécialités pharmaceutiques due par les entreprises de vente en gros dite "taxe sur les grossistes répartiteurs"(L138-1 du CSS) |
Loi n° 91-738 du 31 juillet 1991 ; Loi n° 96-1160 du 27 décembre 1996 |
Entreprises de vente en gros de spécialités pharmaceutiques |
assurance maladie |
ACOSS |
Contribution des entreprises assurant l'exploitation en France, au sens de l'article L. 5124-1 du code de la santé publique, d'une ou plusieurs spécialités pharmaceutiques donnant lieu à remboursement par les caisses d'assurance maladie assise sur le chiffre d'affaires hors taxes dite "taxe sur le chiffre d'affaires" (art L245-6 du CSS) |
Loi du 2003-1199 du 18 décembre 2003 de financement de la sécurité sociale pour 2004 et loi nº 2004-810 du 13 août 2004 |
Entreprises assurant l'exploitation en France des dites spécialités |
caisse nationale de l'assurance maladie des travailleurs salariés |
ACOSS |
Contribution exceptionnelle de régulation, assise sur le chiffre d'affaires hors taxes pour l'année civile 2006 réalisé en France au titre des spécialités inscrites due par les entreprises de vente en gros de spécialités pharmaceutiques |
Loi n° 2006-1640 du 21 décembre 2006 |
Entreprises de vente en gros de spécialités pharmaceutiques |
assurance-maladie |
ACOSS |
Source Cour des comptes
CSS : code de la sécurité sociale
![]()
CSP : code de la santé publique
A. Les taxes destinées à maîtriser la dépense de médicaments remboursés par l’assurance maladie
1. La taxe sur les dépenses de promotion des médicaments
Cette taxe est due par les entreprises assurant l’exploitation en France d’une ou de plusieurs spécialités pharmaceutiques inscrites sur la liste des médicaments remboursables aux assurés sociaux (article L.162-17 du code de la sécurité sociale) ou sur la liste des médicaments agréés à l’usage des collectivités (article L.5123-2 du code de la santé publique) dont le chiffre d’affaires hors taxes réalisé en France métropolitaine et / ou dans les DOM a égalé ou excédé 15 millions d’euros au cours de l’exercice considéré au titre des dites spécialités pharmaceutiques (article L.245-1 du code de la sécurité sociale). La définition précise de l’assiette s’est avérée délicate et fait l’objet de nombreux contentieux exposés plus loin (cf. partie II).
Le montant de la contribution se calcule à partir d’un système de 4 tranches d’assiette auxquelles correspondent 4 taux de contribution différents. Ces quatre taux étaient respectivement en 2004 de 16 %, 21 %, 27 % et 32 %, en 2005 et 2006 de 19 %, 29 %, 36 % et 39 %.
2. La contribution de la clause de sauvegarde de l’ONDAM
a. Son objet et sa finalité
L'article 31 I, II 2ºde la loi du 23 décembre 1998 de financement de la sécurité sociale pour 1999 a institué une contribution due par les entreprises assurant l’exploitation d’une ou de plusieurs spécialités pharmaceutiques remboursables en fonction de l'évolution du chiffre d'affaires de l'industrie pharmaceutique au regard de la progression de l'ONDAM et répartie entre laboratoires selon trois éléments : leur chiffre d'affaires, la progression de celui-ci et leurs dépenses promotionnelles (article l38-10 du code de la sécurité sociale).
Seules sont soumises à cette contribution les entreprises pharmaceutiques qui ne souhaitent pas passer convention avec le Comité économique des produits de santé (CEPS) portant sur l’ensemble des chiffres d’affaires de leurs médicaments remboursables en ville et garantissant le respect des engagements conventionnels.
![]()
Lors de son adoption, le législateur mentionnait que mis à part le dispositif de taxation assis sur la distribution des médicaments et les dispositions prévues dans les conventions passées entre le CEPS et les laboratoires pharmaceutiques sur certains produits, aucun mécanisme ayant pour objectif de compenser le dépassement constaté du sous-objectif des prescriptions de l’ONDAM n’existait encore. Entre autres objectifs, le législateur entendait, avec une politique de conventionnement, contribuer à la diminution des dépenses promotionnelles des laboratoires.
b. Le mode de calcul de la taxe
Cette taxe dite « de la clause de sauvegarde » est versée par les entreprises concernées lorsque l’évolution de leur chiffre d'affaires hors taxe réalisé en France en spécialités remboursables, à l’exception des médicaments orphelins, entre l’année civile N-1 et l’année N, est supérieure à un pourcentage excédant un taux K fixé chaque année par la loi de financement de la sécurité sociale.
L'article 29 de la loi du 29 décembre 1999 de financement de la sécurité sociale pour 2000 a fixé, pour la première fois, un seuil de déclenchement de la contribution à 2 %, sans lien direct avec l'ONDAM. Cette modification visait à prendre en compte le nouveau mode de calcul de l’ONDAM défini en année N sur la base de la prévision d'exécution et non plus sur l'objectif initial de l'année passée.
L'article 49 de la loi de la loi du 23 décembre 2000 de financement de la sécurité sociale pour 2001 a porté ce taux à 3 %. Il a été maintenu à ce niveau par la loi du 21 décembre 2001 de financement de la sécurité sociale pour 2002 (article 23).
Pour 2003, le taux K a été fixé à 4 % (par l’article 17 de la loi du 20 décembre 2002 de financement de la sécurité sociale) puis à nouveau à 3 % pour 2004 (par l’article 16 de la loi du 18 décembre 2003 de financement de la sécurité sociale). Enfin l’article 74 de la loi du 13 août 2004 relative à l'assurance maladie a réduit ce taux à 1 % pour 2005, 2006 et 2007.
La loi de financement de la sécurité sociale pour 2001 a prévu un mécanisme de récupération linéaire en fixant un barème de taxation dont chaque taux (50 %, 60 % et 70 %) s'applique successivement à une tranche déterminée de supplément de chiffre d'affaires par rapport au taux de l'objectif K (respectivement inférieur à 0,5 %, compris entre 0,5 et 1 % et supérieur à 1 %).
Le montant de la contribution due est calculé globalement puis ensuite réparti entre les entreprises redevables selon trois critères : le niveau brut du chiffre d'affaires, pour 30 % ; la progression du chiffre d'affaires pour 40 % ; les frais de promotion pour 30 % (article L138-11 du code de la sécurité sociale).
Chaque entreprise redevable acquitte une fraction de chacune des parts ainsi fixées. Chaque fraction est calculée selon une formule prévue par l’article L138-12 du code de la sécurité sociale.
L’exposé technique des modalités de calcul de cette contribution met en évidence sa complexité ; celle-ci tient aussi au déclenchement de la taxation à un niveau collectif avant toute valorisation individuelle et au renvoi pour son calcul individualisé, pour partie, à la taxe sur les dépenses de promotion versée par l’entreprise concernée.
![]()
Théoriquement cette taxation, sous conditions, du chiffre d’affaires global des entreprises du secteur peut avoir pour résultat qu’une entreprise non conventionnée dont le chiffre d’affaires progresse de façon importante ou supérieure au taux de progression global retenu comme seuil de déclenchement échappe à toute taxation.
c. Une taxe au rendement nul
En dépit du fait que le taux K a été abaissé à un niveau bas, aucune entreprise susceptible d’y être soumise ne fait l’objet d’une taxation. La complexité du dispositif rend difficile l’appréciation du bien fondé de ce résultat. Si cette taxe devait être maintenue dans son principe, il conviendrait d’en simplifier le dispositif de façon à s’assurer de son application effective.
Le mécanisme qui a perdu tout objectif de rendement ne constitue plus qu’un dispositif d’incitation à la contractualisation avec le CEPS. Chaque année, seules deux ou trois entreprises exploitant une spécialité pharmaceutique ne disposent pas de la convention nécessaire. En revanche, les entreprises conventionnées versent aux termes de leurs engagements des « remises conventionnelles » qui ont représenté 190 millions d’euros en 2003, 350 millions d’euros en 2004, 309 millions d’euros en 2005 et 507 millions d’euros en 2006.
d. L’application de cette taxe aux spécialités pharmaceutiques faisant l’objet d’une rétrocession hospitalière
La loi de financement de la sécurité sociale pour 2006 a étendu le champ d’application de la taxe.
L’article 138-10 relatif à la contribution dite de sauvegarde fait désormais référence dans son paragraphe II aux spécialités pharmaceutiques visées à L. 5126-4 du code de la santé publique c'est-à-dire aux médicaments que certains établissements de santé, disposant d'une pharmacie à usage intérieur, sont autorisés à vendre au public et au détail.
Cette disposition n’a pas encore fait l’objet d’une application. La circulaire visant à préciser les questions d’interprétation du droit existant, notamment en ce qui concerne l’assiette et les modalités de recouvrement de la dite contribution est en cours d’examen pour validation au ministère de la santé et des solidarités. Une fois ces conditions précisées, un arrêté ministériel déterminera le modèle à partir duquel les entreprises feront leur déclaration aux URSSAF (article R138-10 du code de la sécurité sociale). Il conviendrait que le dispositif permettant l’entrée en vigueur de cette contribution soit précisé et appliqué.
B. Les taxes destinées à procurer des recettes à l’assurance maladie
1. La taxe sur les grossistes répartiteurs
La contribution sur les ventes directes a été créée par la loi du 12 avril 1996 portant diverses dispositions d'ordre économique et financier. Elle était initialement assise sur le chiffre d'affaires hors taxes réalisé par l'ensemble des établissements assujettis, comportait six tranches d’imposition et était recouvrée trimestriellement.
![]()
La loi du 18 décembre 2003 de financement de la sécurité sociale pour 2004 en a modifié l’économie en l’individualisant par entreprise.
Elle est due actuellement par les entreprises de vente en gros de spécialités pharmaceutiques (grossistes - répartiteurs) et par les entreprises assurant l’exploitation d’une ou de plusieurs spécialités pharmaceutiques lorsqu’elles vendent en gros des spécialités pharmaceutiques (article L 138-1 du Code de la Sécurité Sociale).
La contribution est assise sur le chiffre d’affaires hors taxes réalisé en France au titre des spécialités pharmaceutiques remboursables à l’exclusion des médicaments orphelins. L’assiette est composée de deux parts : une première constituée par le chiffre d’affaires réalisée par l’entreprise au cours de l’année civile, une seconde constituée par la différence entre le chiffre d’affaires hors taxes réalisé au cours de l’année civile et celui réalisé l’année précédente.
Le montant de la contribution est calculé par application d’un taux de 1,9 % à la première part et un taux de 2,25 % à la seconde. Ces taux sont restés inchangés en 2004, 2005 et 2006. Le montant de la contribution ne peut excéder 2,7 % ni être inférieur à 1,4 % du chiffre d'affaires hors taxes réalisé par l'entreprise. Elle est due annuellement avec paiement d’une provision en septembre de l’année N et régularisation en février de N+1. Selon l’ACOSS, la réforme intervenue en 2004 en a simplifié la gestion.
Il convient, ici, de préciser que la loi de financement de la sécurité sociale pour 2007 (art. 5) a créé une « contribution exceptionnelle de régulation », assise sur le chiffre d'affaires hors taxes pour l'année civile 2006 réalisé en France auprès des pharmacies d'officine, des pharmacies mutualistes et des pharmacies de sociétés de secours minières au titre des spécialités et due par les entreprises de vente en gros de spécialités pharmaceutiques ainsi que par les entreprises assurant l'exploitation d'une ou plusieurs spécialités pharmaceutiques et les vendant directement aux pharmacies. Son assiette est composée de deux parts. Une première part est constituée par le chiffre d'affaires hors taxes réalisé par l'entreprise au cours de l'année 2006 ; une seconde part est constituée par la différence entre le chiffre d'affaires hors taxes réalisé au cours de l'année 2006 et celui réalisé au cours de l'année 2005. Le montant de la contribution est calculé en appliquant un taux de 0,21 % à la première part et un taux de 1,5 % à la seconde part.
Lors du dépôt du projet de loi devant le Parlement la contribution devait rapporter 50 millions d’euros. Après discussion de la loi, le taux applicable à la première tranche est passé de 0,28 % à 0,21 %. En conséquence de quoi le rendement de la contribution devrait être de 37 millions d’euros en 2007.
2. La taxe sur le chiffre d’affaires
La loi 18 décembre 2003 de financement de la sécurité sociale pour 2004 a créé une contribution exceptionnelle de l'industrie pharmaceutique aux ressources de la branche maladie assise sur le chiffre d’affaires des laboratoires.
L’institution de cette taxe avait pour objectif de compenser la baisse des recettes qui résultait de la modification par la même loi des tranches du barème de taxation des dépenses de promotion des médicaments. Son taux était de 0,525 %.
![]()
La loi du 13 août 2004 relative à l’avenir de l’assurance-maladie a pérennisé cette nouvelle contribution sur le chiffre d'affaires des entreprises pharmaceutiques (article L.245-6 du code de la sécurité sociale). En outre, elle a étendu le champ des
entreprises redevables (des seules entreprises titulaires d'une autorisation de mise sur le marché des médicaments à toutes les entreprises assurant l'exploitation de médicaments), a exclu du chiffre d'affaires pris en compte celui réalisé au titre des spécialités génériques, sauf si elles sont remboursées sur la base d'un tarif forfaitaire de responsabilité, et précisé le mode de versement de la taxe. Ces modifications se sont appliquées pour la première fois au titre de la contribution due en 2005. Son taux était fixé à 0,6 %.
a. Son principe
Toutes les entreprises exploitant un médicament à usage humain et donnant lieu à remboursement doivent l'acquitter. Son assiette est constituée du chiffre d'affaires hors taxe réalisé sur le territoire national sur les médicaments bénéficiant d'une autorisation de mise sur le marché, à l'exclusion des spécialités génériques (ne donnant pas lieu à remboursement sur la base d’un tarif forfaitaire de responsabilité) et des médicaments orphelins sous réserve que le chiffre d’affaires de ces médicaments ne soit pas supérieur à 20 millions d’euros.
Une contribution provisionnelle est versée au 15 avril. Celle-ci est égale au produit du chiffre d’affaires de l’année N-1 par le taux applicable. La régularisation de l’année précédente se fait sur le montant du chiffre d’affaires de l’année passée une fois connu et est due à la même date que la contribution provisionnelle.
b. La question de sa compatibilité avec le droit européen
La Cour s’est déjà interrogée sur la compatibilité de cette taxe avec le droit européen (1).
En effet l’article 33 de la Sixième directive 77/388/CEE du Conseil, du 17 mai 1977, prise « en matière d'harmonisation des législations des États membres relatives aux taxes sur le chiffre d'affaires - système commun de taxe sur la valeur ajoutée assiette uniforme » dispose « sans préjudice d'autres dispositions communautaires, les dispositions de la présente directive ne font pas obstacle au maintien ou à l'introduction par un État membre de taxes sur les contrats d'assurance, sur les jeux et paris, d'accises, de droits d'enregistrement, et, plus généralement, de tous impôts, droits et taxes n'ayant pas le caractère de taxes sur le chiffre d'affaires ».
![]()
Cette disposition a été interprétée par la Cour de justice des Communautés européennes comme interdisant aux États membres de maintenir ou d’introduire toute taxe ayant les caractéristiques suivantes : s’appliquant de manière générale aux transactions ayant pour objet des biens ou services, étant proportionnelle au prix de ces biens et de ces services, perçue à chaque stade du processus de production et de distribution enfin s’appliquant à la
valeur des biens et des services, la taxe due lors d’une transaction étant calculée après déduction de celle qui a été payée lors de la transaction précédente (2).
Selon l’avis donné par la direction de la législation fiscale dans le cas présent, « trois de ces caractéristiques ne sont pas remplies : la contribution ne s’applique pas de manière générale aux transactions car elle a pour assiette le seul chiffre d’affaires constitué par les ventes de certaines entreprises exploitantes de médicaments, elle est due à un stade unique du circuit économique, elle n’est pas applicable aux ventes ultérieures à celle de l’entreprise exploitante et s’applique au chiffre d’affaires et non à la valeur ajoutée, et, enfin, les entreprises redevables ne bénéficient d’aucune déduction ou remboursement » (3).
c. Une stabilisation souhaitable
Le taux initial de la contribution applicable en 2005 était de 0,6 %. Il a significativement augmenté, passant à 1,76 % pour 2006. L’article 22 de la loi du 21 décembre 2006 de financement de la sécurité sociale pour 2007 en a fixé le taux « à titre exceptionnel, à 1 % »
La Cour estime nécessaire d'offrir plus de stabilité à la taxe sur le chiffre d'affaires, considérée par l'industrie comme donnant l'image d'une régulation aléatoire par rapport à ce qui se passe dans les autres États européens.
c. La TVA brute collectée par les commerçants en gros en produits pharmaceutiques
Dans le cadre des nouvelles modalités de financement des exonérations de cotisations sociales, la loi de finances pour 2006 a prévu d’affecter au financement de régimes de base de la sécurité sociale un panier de taxes dont le produit de la TVA brute collectée par les commerçants de gros en produits pharmaceutiques. L’assurance maladie en reçoit l’essentiel.
Un arrêté du 6 février 2006 (JO 10/02/2006) a fixé des quotes-parts provisoires de répartition du « panier fiscal » entre les régimes ; et un arrêté fixant les quotes-parts définitives de répartition à compter de 2006 est en cours de signature.
Parallèlement, une convention a été conclue entre l’État et l’ACOSS le 28 décembre 2005, renouvelée en décembre 2006. Elle définit le rythme et les montants des versements opérés par l’État en matière de TVA brute collectée par les commerçants de gros en produits pharmaceutiques. Les versements opérés le 20 de chaque mois constituent un à valoir sur la TVA brute collectée déclarée au cours de ce même mois. La régularisation se fait le mois suivant ou, si cela n’est pas possible, en mois M+2.
![]()
PARTIE II : LES PROBLÈMES JURIDIQUES POSÉS PAR CERTAINES TAXES
I. – LES CONTENTIEUX PORTANT SUR L’ASSIETTE
La loi du 20 décembre 2002 portant loi de financement de la sécurité sociale pour 2003 avait prévu, à son article 65, une compétence de recouvrement direct par l’ACOSS de certaines contributions ; parmi elles, figuraient la taxe sur les grossistes répartiteurs, la contribution de la clause de sauvegarde, enfin la taxe sur les dépenses de promotion. La loi prévoyait notamment l’assistance de l’ACOSS par des inspecteurs du recouvrement habilités. La loi prévoyait qu’un décret en Conseil d'État fixerait, en tant que de besoin, les modalités d'application de ces dispositions.
Ce décret n’ayant pas été pris et des difficultés contentieuses visant le recouvrement des contributions existant, le législateur par la loi du 21 décembre 2004 portant loi de financement de la sécurité sociale pour 2005 a transféré la compétence du recouvrement des taxes pharmaceutiques exercée jusque-là par l’ACOSS à certaines URSSAF désignées par l’ACOSS. Depuis 1er janvier 2005, le recouvrement et le contrôle des taxes dues par les laboratoires pharmaceutiques sont de la compétence des URSSAF de Paris et Lyon.
Ce transfert a répondu à la fois à la préoccupation de recentrer l’ACOSS sur son rôle de tête de réseau et à la volonté de sécuriser les opérations en faisant entrer les règles de recouvrement et de contrôle relatives aux dites taxes et contributions dans le droit commun des opérations menées par les URSSAF.
A. Une complexité d’ensemble
Le recouvrement de ces taxes est jugé par l’ACOSS, si ce n’est difficile, du moins présentant une certaine complexité due à la nature de l’ensemble du dispositif et du secteur économique visé et d’autre part à la spécificité même des dites contributions.
Les règles de calcul ne sont pas les mêmes d’une taxe à l’autre alors qu’elles visent a priori les mêmes entreprises. L’ACOSS doit donc gérer de façon concomitante des règles différentes et complexes s’appliquant aux mêmes assujettis.
La nature même du secteur économique où la structure des entreprises peut évoluer rapidement du fait d’opérations de fusion, de cession, ou d’absorption implique un suivi et une actualisation permanente des fichiers, ce qui est un facteur supplémentaire de complexité de la gestion. À cela s’est ajoutée l’évolution rapide des textes définissant les assiettes et les taux et l’insuffisance de doctrine permettant de les interpréter.
![]()
Cette situation a amené l’ACOSS et les URSSAF concernées à développer des outils de suivi. Les informations des fichiers d’assujettis des deux URSSAF font l’objet d’opérations régulières de consolidation par recoupement de leurs données avec d’autres sources comme les informations légales ou les déclarations faites à l’AFSSAPS. Les
entreprises du secteur font l’objet d’une veille particulière par l’exploitation de la presse, des relations directes avec les entreprises ou les organisations représentatives de celle-ci comme le LEEM. Des réunions entre les deux URSSAF spécialisées ont été instituées et une base documentaire partagée entre elles a été mise en place. La Cour estime souhaitable de renforcer les outils de suivi et de veille du secteur.
B. Deux taxes présentent des difficultés particulières.
1. La contribution de la clause de sauvegarde de l’ONDAM
Le périmètre des entreprises concernées s’avère délicat à établir. Les entreprises entrant dans le champ d’application de la taxe sont celles qui ne bénéficient pas d’une convention avec le CEPS à la date du 31 décembre de l’année précédant celle de l’établissement des déclarations. La liste des entreprises ayant conclu une convention est publiée au Journal officiel. Les entreprises sont donc celles ne figurant pas sur cette liste. Pour les identifier, il convient donc de comparer la liste parue et le fichier général des entreprises du secteur. De là, découlent plusieurs questions pratiques qui, même si elles ne sont pas extrêmement difficiles à résoudre, nécessitent une organisation et des vérifications importantes.
D’une part, les conventions du CEPS sont passées avec des groupes alors que l’assujettissement se fait à partir de l’enregistrement des entreprises sur la base de leur immatriculation SIREN. Le délai entre la date de publication de la liste, généralement fin janvier, et la date limite d’envoi des déclarations fixée au 15 février, est assez bref. Cette situation a conduit l’ACOSS à initier une collaboration avec le CEPS pour identifier facilement les différents groupes et entreprises qui ne sont pas conventionnés. Toutefois, certaines des informations transmises par le CEPS sont apparues insuffisamment fiables à l’ACOSS qui a procédé aux opérations de calcul mais n’a pas recouvré.
D’autre part, l’ACOSS et les URSSAF déplorent la complexité des opérations pratiques et notamment le fait que le calcul de la contribution doit tenir compte du montant versé au titre de la taxe sur les dépenses de promotion laquelle peut faire l’objet d’un versement provisionnel en année n et d’une régularisation en année n+1. Les calculs opérés peuvent donc être remis en cause après régularisation des montants effectivement dus. Enfin toute rectification même matérielle du chiffre d’affaires est de nature à remettre en cause bon nombre des opérations déjà réalisées.
La Cour relève la complexité des opérations à mener et les difficultés rencontrées par les URSSAF et l’ACOSS.
2. La taxe sur les dépenses de promotion des médicaments
![]()
Les difficultés constatées en matière d’établissement de la taxe ne tiennent pas, ici, à la difficulté de déterminer le périmètre des entreprises redevables mais à un défaut de précision quant à la définition des éléments constituant l’assiette.
a. Les difficultés subsistantes
Normalement, sont prises en compte les charges comptabilisées au titre des frais de publication et des achats d’espaces publicitaires, dès lors qu’une spécialité pharmaceutique remboursable ou agréée auprès des collectivités y est mentionnée ; tous les frais engagés au titre d’une publication, quelle qu’en soit la forme, sont intégrés dans l’assiette hors les frais d’acheminement.
Ne sont pas prises en compte les dépenses d’information relatives à l’emballage, aux effets indésirables, à la santé humaine et aux maladies humaines sans référence avec un médicament, au bon usage ou à la pharmacovigilance, faites dans la presse médicale professionnelle ou dans les dictionnaires professionnels.
Des difficultés subsistent cependant. On citera par exemple l’intégration dans le champ des dépenses des frais professionnels des visiteurs, selon qu’ils sont remboursés ou pris en charge directement par les laboratoires, celle des dépenses de fabrication des objets promotionnels remis aux prescripteurs ou encore de publication de certains documents obligatoires remis aux prescripteurs.
Dans le premier cas, l’imprimé déclaratif homologué visait en 2003 « les seuls frais remboursés aux visiteurs » alors que le guide de déclaration établi en 2006 accompagnant le même imprimé vise aujourd’hui globalement « les frais des visiteurs médicaux ». En l’absence d’un texte officiel précisant le changement de doctrine du ministère, il est à craindre que les contrôles débouchant sur une réintégration de charges soient contestés par les entreprises.
Dans le second cas, l’ACOSS et les entreprises pharmaceutiques s’opposent sur la notion d’achat d’espaces publicitaires ; l ‘ACOSS considère que la fabrication et la remise d’objets sur lesquels figure le nom d’une spécialité médicale remboursable sont une dépense de promotion de cette spécialité ; les laboratoires estiment que la notion d’achats d’espaces publicitaires ne vise pas cette catégorie d’objets.
Dans le dernier cas, les laboratoires font valoir que la tolérance accordée par le ministère en ce qui concerne l’exclusion de l’assiette des frais de publication des résumés des caractéristiques des médicaments et de l’avis de la commission de transparence accordée au titre de l’exercice 2004 s’applique rétroactivement aux dépenses faites au titre des deux précédents exercices.
La circulaire ministérielle ayant pour objet de préciser l’esprit, le cadre et les éléments à prendre ou non en compte n’a toujours pas été publiée, malgré une annonce de publication pour décembre 2006.
b. La nécessité de préciser et de sécuriser ses conditions d’application
Pour ce qui est de la taxe sur les dépenses de promotion des médicaments, les entreprises du secteur contestent toujours, notamment au plan contentieux, le bien fondé du périmètre des charges en constituant l’assiette.
![]()
C’est la finalité explicite de cette contribution, de limiter les dépenses favorisant, hors d’un besoin objectif, la consommation des spécialités pharmaceutiques, qui est mise en cause par les entreprises du secteur.
En assimilant les spécialités pharmaceutiques à des produits dont la prescription peut relever de motivations qui ne soient pas strictement rationnelles, cette taxe suscite une forte opposition de ceux qui financent la promotion.
La Cour juge nécessaire de faire disparaître rapidement toutes les incertitudes relatives à l’application de la taxe sur les dépenses de promotion des médicaments et pour parachever le dispositif d’adopter la circulaire précisant le champ des charges et dépenses à prendre en compte.
Dans le même ordre d’idée et de façon complémentaire, la circulaire relative à l’application de la contribution sur les dépenses de promotion des dispositifs médicaux devrait aussi être publiée.
La Cour relève la nécessité pour le ministère de préciser, si nécessaire, les textes applicables et à tout le moins de publier les circulaires attendues dans un but de sécurisation des opérations de déclaration, de calcul, de contrôle et de bon recouvrement des taxes.
Par ailleurs, se pose la question de l’articulation de cette taxe avec d’autres dispositifs de maîtrise de la promotion des médicaments (v. infra, partie III, A, 2).
II. – LES DIFFICULTÉS LIÉES AUX CONTRÔLES
Depuis 1er janvier 2005, le recouvrement et le contrôle des taxes dues par les laboratoires pharmaceutiques sont de la compétence des URSSAF de Paris et Lyon. La répartition entre les deux URSSAF s’est faite en tenant compte de la localisation du siège social de l’entreprise : l’URSSAF de Paris est compétente pour les entreprises dont le siège social est situé en région parisienne et dans les départements d’Outre-mer et l’URSSAF de Lyon est compétente pour les entreprises dont le siège est situé en province et à l’étranger.
En 2005, le coût estimé des opérations de recouvrement et de contrôle des taxes sur les médicaments (y compris la contribution sur les dépenses de promotion les dispositifs médicaux – article L.245-1 du code de la sécurité sociale – dont les URSSAF sont chargées) était de 324 928 € comprenant les coûts de gestion des comptes et de contentieux y compris les charges de personnel mais hors coût des fonctions support.
a. L’existence d’un plan de contrôle
De 2002 à 2004 l’ACOSS puis à partir de 2005 les URSSAF ont eu la responsabilité du recouvrement direct des contributions. Pour la période 2002 à 2005 l’ACOSS avait établi un plan initial de contrôle.
Au 31 décembre 2004, sur 296 entreprises assujetties soit à la taxe sur les « grossistes-répartiteurs » soit à la taxe sur les dépenses de promotion, 101 avaient vu leurs dossiers contrôlés. Le contrôle de 93 autres entreprises a priori non redevables mais dont l’assujettissement ne pouvait être exclu a aussi été réalisé.
![]()
Au 31 octobre 2005, 259 laboratoires sur les 296 assujettis avaient été contrôlés.
Le montant total des redressements notifiés était de 165,8 m€ au 31 décembre 2005. Selon l’ACOSS, les montants notifiés concernent pour une très large part la taxe sur les dépenses de promotion et leur importance s’explique par le fait qu’il s’agissait d’une taxe nouvelle dont le principe et les modalités faisaient l’objet d’interprétations ou d’hésitations de la part des entreprises concernées.
L’ACOSS n’a pas été en mesure de communiquer le détail des sommes effectivement recouvrées à la suite de ces notifications.
Pour 2006, les URSSAF de Paris et de Lyon ont établi un plan de contrôle des entreprises redevables des taxes sur les médicaments ; ont été sélectionnées les entreprises présentant un enjeu financier substantiel et n’ayant pas encore fait l’objet d’un contrôle. Pour 31 entreprises contrôlées, le montant des redressements s’est élevé à 126 000 €.
b. La contestation des contrôles
Les opérations de contrôle menées par les URSSAF ont fait l’objet de recours par les laboratoires pharmaceutiques devant les tribunaux judiciaires.
Les actions contentieuses qui étaient de nature à fragiliser le dispositif dans son ensemble ont conduit le législateur à valider par l’article 73 de la loi du 18 décembre 2003 de financement de la sécurité sociale pour 2004 les opérations de contrôle et de redressement qui feraient l’objet de contestation au moyen de l’irrégularité des agréments des inspecteurs ayant mené les contrôles et redressements.
Devant les juridictions judiciaires, ces dispositions ont été contestées par des laboratoires pharmaceutiques ayant fait l’objet de redressements. Le moyen en était que si le législateur peut adopter, en matière civile, des dispositions rétroactives, le principe de prééminence du droit et la notion de procès équitable consacré par l'article 6 de la Convention européenne de sauvegarde des droits de l'homme et des libertés fondamentales s'opposent, sauf pour des motifs d'intérêt général, à l'ingérence du pouvoir législatif dans l'administration de la justice, afin d'influer sur le dénouement judiciaire des litiges.
La Cour de cassation dans un arrêt du 8 novembre 2006 a jugé qu'obéit « à d'impérieux motifs d'intérêt général l'intervention du législateur, qui, sans régler le fond du litige ni priver le débiteur de la contribution du droit de contester le bien-fondé du redressement, est destinée à éviter le développement d'un contentieux de nature à mettre en péril le recouvrement des cotisations de sécurité sociale et par suite la pérennité du système de protection sociale » et a fait prévaloir les arguments défendus par les organismes de sécurité sociale.
![]()
Les appels formés conjointement par le ministère et les inspecteurs à l'encontre de décisions rendues par le Tribunal administratif de Paris le 9 janvier 2006, lesquelles ont annulé les agréments de 6 inspecteurs de l'URSSAF de Paris, ne sont pas encore jugés par la Cour administrative d'appel saisie. La décision de la Cour de cassation vient en appui des arguments pour demander l’annulation des jugements de première instance.
PARTIE III : LE RENDEMENT DES PRINCIPALES TAXES
Seront examinés, ici, le rendement des principales taxes perçues à savoir les quatre taxes recouvrées par l’ACOSS ainsi que les produits de la TVA applicable aux entreprises du secteur.
A. Les taxes affectées à l’assurance maladie
1. Le rendement global des taxes
Le rendement des taxes et contributions recouvrées par l’ACOSS puis par les URSSAF, tel qu’il a évolué entre 2004 et 2006, est présenté dans le tableau récapitulatif ci après :
Tableau n° 4 : Rendement des taxes sur le médicament perçues par l’ACOSS
En M € |
2004 |
2005 |
Evolution 2004-2005 |
2006 |
Evolution 2005-2006 |
Taxe sur le chiffre d'affaires |
103,4 |
119,0 |
15,1 % |
375,3 |
215,3 % |
Taxe sur les grossistes répartiteurs |
377,6 |
350,5 |
-7,2 % |
374,7 |
6,9 % |
Contribution clause de sauvegarde "ONDAM" |
0,0 |
0,0 |
0 |
||
Taxe sur les dépenses de promotion |
197,3 |
214,3 |
8,6 % |
211,8 |
-1,1 % |
Total |
678,3 |
683,8 |
0,8 % |
961,8 |
40,6 % |
Source ACOSS
N.B. Le rendement de la clause de sauvegarde a été de 0,042 M€ en 2001 et de 0,487 M€ en 2002.
Le rendement total de ces contributions s’élevait en 2004 à 678,3 millions d’euros, en 2005 à 683,8 millions d’euros, en 2006 à 961 millions d’euros et une prévision de rendement a été établie par la direction de la sécurité sociale pour 2007 à hauteur de 770 millions d’euros (dont 210 millions d’euros pour la taxe sur le chiffre d’affaires).
Pour s’en tenir à la période la plus récente, le montant de la taxe sur le chiffre d’affaires a connu en 2006 une hausse significative, après augmentation du taux applicable de 0,6 à 1,76 %, son produit étant de la sorte multiplié par 3. Pour 2007, la diminution du taux ramené à 1 % par l’article 22 de la loi de financement de la sécurité sociale de financement devrait entraîner une baisse du rendement.
![]()
La progression de plus de 40 % du rendement total de ces taxes sur la période 2004 à 2006 est due, pour l’essentiel, à l’évolution significative et positive de la taxe sur le chiffre d’affaires. L’augmentation de son rendement a, à lui seul, contribué pour 92 % à l’augmentation des sommes recouvrées en 2006. La taxe sur le chiffre d’affaires a représenté en 2006 près de 40 % des recettes ainsi recouvrées.
2. Le cas particulier de la taxe sur les dépenses de promotion
Avec un montant de 214 M€ en 2005, la taxe sur les dépenses de promotion a représenté 0,9 % du chiffre d’affaires (France) de l’industrie pharmaceutique. Malgré l’ancienneté de cette taxe (instaurée en 1983), aucune étude n’a cherché à évaluer son impact sur les dépenses de promotion des laboratoires. Il est vraisemblable que son effet régulateur est faible. Toutefois, la taxation n’est pas le seul instrument d’encadrement des dépenses de promotion dont disposent les pouvoirs publics, notamment en matière de transparence et de neutralité des informations mises à la disposition des praticiens.
Ainsi le législateur a encadré par la loi du 13 août 2004 l’activité de visite médicale des laboratoires pharmaceutiques dont l’objectif est d’assurer directement la promotion des médicaments auprès des prescripteurs. L’article L. 162-17-8 du code de la sécurité sociale prévoit qu’une charte de qualité des pratiques professionnelles des personnes chargées de la promotion des spécialités pharmaceutiques par prospection ou démarchage est conclue entre le comité économique des produits de santé et un ou plusieurs syndicats représentatifs des entreprises du médicament qui vise, notamment, à mieux encadrer les pratiques commerciales et promotionnelles qui pourraient nuire à la qualité des soins.
L'article L. 162-17-4 du même code dispose que les entreprises signataires doivent s'engager à respecter la dite charte et, selon une procédure établie par la HAS, à faire évaluer et certifier par des organismes accrédités la qualité et la conformité à cette charte de la visite médicale qu'elles organisent ou qu'elles commanditent. Cette certification vise à examiner dans quelle mesure les laboratoires ont développé une démarche « qualité » nécessaire pour répondre aux engagements de la charte signée par le CEPS et le LEEM le 22 décembre 2004. Les principaux mécanismes prévus par la certification sont opérationnels depuis la fin 2006 :
– Un référentiel de certification a été établi. Il comprend quatre exigences auxquelles correspondent plusieurs critères. Ces quatre exigences visent les connaissances et les compétences des visiteurs médicaux, les documents mis à leur disposition, les moyens mobilisés pour le respect des règles déontologiques et les moyens utilisés par les laboratoires pour s’assurer de la qualité des visites. Pour chacun des critères, des éléments de réponse et des preuves devront être apportées par les entreprises dans le cadre du processus de certification ;
– La certification des entreprises sera réalisée par des organismes accrédités. À ce jour, cinq organismes certificateurs ont fait une demande d’accréditation auprès du comité français d'accréditation (COFRAC). L’audit de ces organismes et la décision d’accréditation devraient intervenir pendant l’année.
D’ores et déjà, plusieurs laboratoires pharmaceutiques ont contacté ces organismes certificateurs, soit pour conduire des audits à blanc (début février, on compte 11 audits à blanc), soit pour mener des audits réels, devant déboucher sur une certification (début février : 3 audits réels).
![]()
La HAS a prévu une mise en œuvre de la certification des entreprises pharmaceutiques pour 2007. Celle-ci débouchera sur l’attribution de certificats aux
entreprises. La mise en œuvre effective de la certification permettra de relever les difficultés d’application et de procéder à leur analyse. Elle dépend cependant de la détermination par le CEPS du délai au terme duquel l’ensemble des entreprises devra être certifié. L’hypothèse de la fin 2007 est envisagée avec possibilité d’un délai supplémentaire pour l’obtention du certificat.
Toutefois, malgré l’existence d’une charte de la visite médicale signée en 2004 et d’obligations légales préexistantes, la qualité de l’information ne semble pas avoir significativement progressé. Dans un contexte de concurrence assez vive entre entreprises du médicament, les progrès attendus risquent d’être encore longs à se matérialiser. Il serait utile que plusieurs laboratoires obtiennent rapidement la certification, ce qui en démontrerait la faisabilité du dispositif et entraînerait un intérêt pour son développement.
![]()
Il conviendra d’examiner le bon niveau de complémentarité entre la taxe et la charte de qualité de la visite médicale.
B. L’évolution des montants de TVA
1. L’évolution de la TVA nette déclarée
Si on agrège l’ensemble des montants perçus entre 2004 et 2006 sur toutes les entreprises du médicament, à l’exception des autres produits de santé (4), l’évolution du rendement est la suivante :
Tableau n° 5 : Montant de TVA nette déclarée par les entreprises du médicament
Montant global de TVA nette déclarée |
2004 |
2005 |
2006 |
Evolution 2004-2006 |
Activité cumulée des secteurs 244 A, C, D, 514 N, 523 A |
1 034 |
1 175 |
1 142 |
10 % |
Source Ministère de l’Économie, des Finances et de l’Industrie –
Direction Générale des Impôts(DGI)
Ainsi en 2006, le montant de TVA nette déclarée par les entreprises du secteur a été de 1,1 milliard d’euros en augmentation de 10 % par rapport à 2004.
Le tableau qui suit présente l’évolution du rendement des montants annuels agrégés de TVA nette déclarée en millions d’euros, par code d’activité NAF, pour 2004, 2005 et 2006 (année de dépôt de la déclaration).
Tableau n° 6 : Evolution des montants annuels agrégés de TVA nette déclarée en M€, par code d’activité NAF
Année |
Fabrication de produits pharmaceutiques de base |
Fabrication de médicaments |
Fabrication d'autres produits pharmaceutiques |
Commerce de gros de produits pharmaceutiques |
Commerce de détail de produits pharmaceutiques |
2004 |
79 |
150 |
58 |
547 |
200 |
2005 |
126 * |
170 |
46 |
604 |
229 |
2006 |
41 |
173 |
45 |
654 |
229 |
Evolution 2004-2006 |
-48,1 % |
15,3 % |
-22,4 % |
19,6 % |
14,5 % |
* une entreprise du secteur a déposé une déclaration de TVA au titre de juin 2005 faisant figurer un montant net à payer exceptionnel de 55 M€.
![]()
Source Ministère de l’Économie, des Finances et de l’Industrie – direction générale des impôts
2. L’évolution du montant de TVA brute collectée par les entreprises du secteur
L’évolution du montant de TVA brute collectée par les entreprises appartenant aux catégories d’activités liées au secteur du médicament a augmenté de 10,8 % entre 2004 et 2006.
Tableau n° 7 : montant de TVA brute collectée par les entreprises du secteur du médicament
Le Montant global de TVA brute collectée (M €) |
2004 |
2005 |
2006 |
Évolution 2004-2006 |
Activité cumulée des secteurs 244 A, C, D, 514 N, 523 A |
6 842 |
7 303 |
7 580 |
10,8 % |
Source : DGI
Au 31 décembre 2006, l’ensemble des versements opérés au profit de l’ACOSS par le ministère de l’économie au titre de cette mesure représentait la somme de 2,9 milliards d’euros de TVA brute collectée.
3. Une connaissance partielle des effets liés aux décisions de déremboursement
S’agissant des décisions de déremboursement intervenues depuis 2005 et ayant eu pour conséquence pour un certain nombre de spécialités pharmaceutiques d’augmenter mécaniquement le taux de TVA applicable de 2,1 % à 5,5 %, leur impact n’est ni mesuré ni connu de façon précise.
Au ministère de l’économie, des finances et de l’industrie, l’effet des décisions de déremboursement sur le rendement de la TVA sur le médicament n’a pu être mesuré.
De façon classique deux éléments jouent : les prix et les volumes.
Les prix de vente des médicaments déremboursés tendent à augmenter sensiblement, ce qui pourrait avoir pour conséquence d’augmenter le montant de la TVA collectée. Néanmoins, c’est sur le volume des ventes que les opérations de déremboursement ont en général un impact significatif.
![]()
En effet selon la direction de la sécurité sociale, les opérations de déremboursement déjà effectuées auraient en général eu pour effet de réduire le chiffre d’affaires des médicaments concernés de près de 70 %. Cette réduction inclut les hausses de prix.
Ces observations sont corroborées par l’analyse faite de l’évolution des ventes de médicaments en officines et aux hôpitaux en France. Les chiffres établis montrent que dans les officines, entre 2000 et 2005 (date des derniers chiffres disponibles), la part relative des ventes de spécialités non remboursables diminue régulièrement (5). Pour que le taux de croissance des spécialités non remboursables dépasse celui des spécialités remboursables, de nombreux déremboursements de médicaments réalisant des chiffres d’affaires très significatifs devraient être réalisés et simultanément des spécialités à forte croissance devraient accéder au marché sans être admises au remboursement.
En l’état, le déremboursement de spécialités pharmaceutiques conduit à une diminution des dépenses d’assurance maladie et met fin au versement par les entreprises des taxes dues au titre de l’exploitation de ces spécialités, sans que l’on puisse en établir un bilan.
4. L’efficacité des opérations de vérification menées
Les entreprises du secteur du médicament humain qui ont fait l'objet d'une vérification de comptabilité par les services de la DGI n'ont pas été programmées en contrôle sur la base d'éléments ou de critères spécifiques différents de ceux habituellement utilisés par les services fiscaux.
Le tableau ci après figure le ratio du nombre de contrôles avec rappel TVA / nombre d’entreprises moyen sur les 3 années et pour chacun des types d’entreprises selon la nature de leur activité (codes NAF) :
Tableau n° 8 : Ratio du nombre de contrôles avec rappel TVA / nombre d’entreprises
Activité |
244A |
244C |
244D |
514N |
523A |
Ratio |
Fabrication de produits pharmaceutiques de base |
Fabrication de médicaments |
Fabrication d'autres produits pharmaceutiques |
Commerce de gros de produits pharmaceutiques |
Commerce de détail de produits pharmaceutiques |
taux |
2,80 % |
5,30 % |
3,70 % |
2,60 % |
0,20 % |
Le ratio est plus faible concernant le commerce de détail de produits pharmaceutiques, s’agissant d’une très large population (environ 60 contrôles pour 25 000 déclarants TVA).
![]()
Du point de vue des enjeux budgétaires, ce sont évidemment les fabricants de médicaments et les commerces de gros qui font apparaître les redressements intervenus après vérifications les plus importants.
Le tableau suivant montre sur la période 2004 à 2006 l’évolution du nombre de contrôles opérés sur les entreprises du médicament (hors commerce de détail des produits médicaux et orthopédiques) et l’évolution des redressements opérés en euros.
Tableau n° 9 : Évolution du nombre de contrôles opérés sur les entreprises du médicament et des montants de redressements intervenus
2004 |
2005 |
2006 |
Évol 2004-2006 | |
Redressement (M€) |
11,6 |
30,9 |
53,3 |
360 % |
Nbre de contrôles |
135 |
175 |
156 |
16 % |
Source DGI
Il apparaît clairement que les contrôles opérés ont été productifs, leur augmentation sensible (+ 16 %) faisant passer les sommes recouvrées en plus d’un peu moins de 12 millions d’euros à plus de 50 millions d’euros soit une multiplication par plus de 4.
La Cour souligne la nécessité de poursuivre la politique de contrôles et de vérifications engagée.
C. Les échanges d’informations entre administrations et organismes percevant des taxes
De façon générale, il n’existe pas beaucoup de relations formalisées, régulières ou même ciblées entre les différentes administrations ou organismes en charge de percevoir des taxes sur les spécialités pharmaceutiques et les entreprises du médicament.
Les informations relatives à leurs activités, produits, volumes de ventes, ou chiffres d’affaires ne sont pas systématiquement partagées. Deux raisons peuvent être évoquées.
D’une part, les éventuels échanges d’informations sont régis par les textes d’application générale. Ainsi, la direction générale des impôts, quand elle a besoin d’informations, exerce son droit à communication en application de l'article L.83 du livre des procédures fiscales pour demander aux établissements ou organismes soumis au contrôle de l'autorité administrative les documents de service dont elle a besoin. C’est notamment le cas des demandes qu’elle formule auprès de l'agence française de sécurité sanitaire des produits de santé.
D’autre part, la pratique n’en est pas établie. Si l’ACOSS a des relations régulières et fréquentes avec le comité économique des produits de santé dans le cadre de la procédure de recouvrement de la contribution de la clause de sauvegarde de l’ONDAM, l’essentiel des échanges d’informations qui ont lieu s’effectue avec le ministère de tutelle (direction de la sécurité sociale).
![]()
Une mutualisation, même partielle, des informations transmises pourrait s’avérer utile tant pour valider les informations recueillies que pour réaliser une veille à finalité prospective des stratégies ou hypothèses de rendement des différentes taxes concernées.
*
* *
Le rendement de l’ensemble des contributions existantes apparaît globalement stable, le surcroît de recettes en 2006 étant directement lié à l’augmentation exceptionnelle du taux de la contribution sur le chiffre d’affaires.
Il convient cependant de s’interroger sur l’efficacité des différentes taxes. L’architecture d’ensemble paraît davantage résulter d’une sédimentation progressive ou coïncidente des diverses contributions que d’une analyse visant à remettre à plat, dans le sens d’une simplification et d’une stabilisation, la fiscalité existante.
De façon plus générale est posée la question de la cohérence du dispositif au regard de la poursuite de deux objectifs de l’action publique distincts, pour ne pas dire divergents : d’une part, celui de la taxation des entreprises sur leur chiffre d’affaires avec la préoccupation d’un rendement au profit de l’assurance maladie, d’autre part celui de la limitation des dépenses de consommation de médicaments, particulièrement importantes dans notre pays, et dont les conditions de prescription comme d’usage font l’objet d’interrogations sur leur bien fondé. Les considérations qui motivent les mesures fiscales et financières prises apparaissent disjointes voire indépendantes.
Au total, l’ensemble du dispositif que constituent les taxes sur le médicament humain apparaît complexe : les modifications nombreuses et successives entraînent une instabilité et une hésitation sur les objectifs. Pour renforcer sa fiabilité et sa crédibilité, il serait utile de le simplifier.
![]()
ANNEXE 6 : COMMUNICATION DE LA COUR DES COMPTES CONCERNANT LA CONSOMMATION ET
LA PRESCRIPTION DES MĖDICAMENTS
COMMUNICATION À LA COMMISSION DES AFFAIRES CULTURELLES, FAMILIALES ET SOCIALES
DE L’ASSEMBLÉE NATIONALE
MISSION D’ÉVALUATION ET DE CONTRÔLE DES LOIS
DE FINANCEMENT DE LA SÉCURITÉ SOCIALE
Article LO 132-3-1 du code des juridictions financières
LA CONSOMMATION ET LA
PRESCRIPTION DE MÉDICAMENTS
juillet 2007
Sommaire
I. ETAT DES LIEUX ET ENJEUX 1
A. LA CONSOMMATION DE MÉDICAMENTS EN FRANCE 1
1. Les ventes de médicaments en France 2
2. Une consommation de médicaments supérieure à celle des autres pays européens 3
B. LA PRESCRIPTION 5
1. Les caractéristiques générales de la prescription 5
2. La qualité des prescriptions 6
C. LES ENJEUX 7
1. Les enjeux de santé publique 7
2. Les enjeux en termes de maîtrise des dépenses 7
II. DE LA MISE SUR LE MARCHÉ À L’ADMISSION AU REMBOURSEMENT 8
A. LA MISE SUR LE MARCHÉ DU MÉDICAMENT 8
1. La procédure d’AMM 8
2. Les médicaments sans AMM et les prescriptions hors AMM 10
B. L’ADMISSION AU REMBOURSEMENT 13
1. Les critères de l’évaluation initiale 13
2. L’absence d’analyse médico-économique 15
C. LE SUIVI DES MÉDICAMENTS EN PRATIQUE MÉDICALE RÉELLE 17
1. Pharmacovigilance et suivi post-AMM : une réévaluation timide de la balance
bénéfice-risque 17
2. Le suivi des médicaments après l’admission au remboursement 21
III. DE LA PRESCRIPTION À LA CONSOMMATION 23
A. L’INFORMATION SUR LE MÉDICAMENT 23
1. La transparence des travaux d’évaluation des médicaments 23
2. Les bases de données sur le médicament 26
B. LES DETERMINANTS DE LA PRESCRIPTION DE MÉDICAMENTS 30
1. La formation médicale 30
2. L’information des médecins 37
3. Les actions de l’assurance-maladie pour maîtriser les prescriptions 43
4. Les accords de bon usage des soins 57
C. LES ACTIONS SUR LES COMPORTEMENTS DES PATIENTS 58
1. L’information grand public sur le médicament 58
2. Les programmes d’aide à l’observance 60
D. LA PROMOTION DES GÉNÉRIQUES 63
1. Le retard français 63
2. La mise sur le marché des génériques 64
3. Les actions pour accroître la délivrance des génériques 66
INTRODUCTION
La France se caractérise par un niveau de prescription et de consommation de médicaments supérieur à celui de ses voisins européens sans que cela se justifie par des indicateurs de morbidité ou de mortalité différents.
Les motifs de cette forte consommation sont à rechercher tant dans les modalités de prescription et de consommation (poids des facteurs culturels et organisationnels, information médicale…) qu’en amont, dans le circuit de mise sur le marché et d’admission au remboursement du médicament.
Le présent rapport6 a pour objectif d’analyser les facteurs susceptibles d’expliquer la surconsommation de médicaments en France : pour les raisons mentionnées supra, seront successivement analysés, après un état des lieux et des enjeux, le circuit de la mise sur le marché et de l’admission au remboursement, puis les facteurs intervenant directement sur les comportements de prescription et de consommation : information sur le médicament, formation médicale, actions sur les comportements des patients…
Le champ du rapport se limite à la médecine de ville. Sont ainsi exclues du périmètre de l’enquête les dépenses liées aux médicaments délivrés à l’hôpital. Les prescriptions faites à l’hôpital et délivrées en ville seront incluses dans les montants cités, mais ne font pas l’objet d’une analyse spécifique. Sont également écartées du périmètre de l’enquête les dépenses liées à l’automédication, qui ne représentent qu’une part très faible dans la consommation (6 %).
A. La consommation de médicaments en France
Il n’existe pas d’indicateur précis de la consommation de médicaments en raison de l’impossibilité de distinguer entre ce qui est acheté ou remboursé par l’assurance maladie et ce qui est réellement consommé par le patient.
En dehors d’études ponctuelles sur les prescriptions ou sur la consommation de médicaments, les chiffres qui sont utilisés proviennent principalement de deux sources : les ventes de médicaments et les dépenses remboursées.
![]()
Ces indicateurs placent la France au premier rang en Europe pour le niveau de médicaments vendus ou prescrits.
1. Les ventes de médicaments en France
Les ventes de médicaments en ville atteignaient 20,4 milliards d’euros en 2006. Elles ont cru un peu moins vite en 2006 (+ 3,55 %) qu’en 2005 (+ 5,82 %).
Tableau n° 1 : Marché du médicament vendu en ville
2004 |
2005 |
2006 | |
Valeur (en M€) |
18 623 |
19 706 |
20 405 |
Taux de croissance annuelle |
NC |
+ 5,82 % |
+ 3,55 % |
Volumes (nombre de boîtes) |
3 092 520 935 |
3 165 092 958 |
3 152 631 197 |
Taux de croissance annuelle |
NC |
+ 2,35 % |
- 0,39 % |
Source : IMS Health - mars 2007
Le marché français est concentré. En 2006, les dix médicaments7 les plus vendus représentaient 11,6 % du total des ventes (2,36 milliards d’euros).
Tableau n° 2 : Les 10 médicaments les plus vendus en ville
Produit |
Classe |
Valeur en € |
Valeur en € |
Valeur en € |
PLAVIX |
Anti-agrégant plaquettaire |
382 250 784 |
450 257 664 |
510 773 056 |
TAHOR |
Hypolipidémiant |
363 126 316 |
403 925 779 |
401 703 991 |
SERETIDE |
Antiasthmatique |
276 866 142 |
294 935 687 |
309 225 794 |
INEXIUM |
Antiulcéreux |
153 096 822 |
173 978 314 |
220 966 498 |
ARANESP |
Antianémique |
--- |
82 384 074 |
182 860 641 |
SYMBICORT |
Antiasthmatique |
125 643 292 |
141 422 348 |
154 168 227 |
GLIVEC |
Antinéoplasique |
112 599 561 |
133 676 296 |
151 904 628 |
ELISOR |
Hypolipidémiant |
228 020 984 |
216 344 040 |
149 955 143 |
ENBREL |
Traitement polyarthrite rhumatoïde |
77 550 800 |
113 185 548 |
142 764 920 |
LOVENOX |
Anticoagulant |
121 936 586 |
128 660 387 |
137 176 761 |
Total |
1 841 091 285 (9,9 %) |
2 138 770 137 (10,9 %) |
2 361 499 659 (11,6 %) | |
Total marché ville |
18 622 610 040 |
19 705 859 042 |
20 404 771 853 |
![]()
Source : IMS Health - mars 2007
2. Une consommation de médicaments supérieure à celle des autres pays européens
a. Analyse globale
La France se caractérise par un niveau de consommation de médicaments supérieur à celui de ses voisins européens sans que cela se justifie par des indicateurs de morbidité ou de mortalité différents.
Ainsi, une enquête réalisée par Ipsos Santé pour le compte de la CNAMTS8 auprès de 4 000 patients et 1 000 médecins européens9 fait apparaître un niveau de consultations proche entre les pays étudiés, mais un niveau de médicaments consommés et prescrits très supérieur en France :
France |
Allemagne |
Espagne |
Pays-Bas | |
Consultations par an |
4,9 |
5,2 |
4,8 |
3,2 |
Médicaments pris dans les 7 derniers jours |
1,9 |
1,6 |
1,4 |
1,3 |
Médicaments prescrits par le médecin |
1,6 |
1,2 |
1,2 |
0,9 |
L’analyse des non-consommants vient renforcer ces différences de comportements : si 38 % des Français n’ont pris aucun médicament au cours de la semaine écoulée au moment de l’enquête, la proportion de cette population s’élève en Allemagne et en Espagne pour atteindre 52 % aux Pays-Bas.
Selon une étude de la DREES10 portant sur cinq pays européens (France, Allemagne, Royaume-Uni, Italie, Espagne), la France arrive en 2004 largement en tête pour les quantités vendues, avec une consommation de l’ordre de 1 500 unités par habitant, soit 40 % environ de plus que la moyenne. De ce fait, la France enregistre en 2004 les ventes de médicaments par habitant les plus élevées (284 € par an), devant l’Allemagne (244 €), le Royaume-Uni, l’Italie (202 €) et l’Espagne (193 €), et ce, bien que le prix moyen par unité soit inférieur de 20 % par rapport à la moyenne (notamment par rapport à l’Allemagne et l’Italie).
La dépense de médicaments est très concentrée : 10 % des assurés consomment 47 % de médicaments remboursés et 5 % représentent 30 % de la consommation totale (232 boîtes de médicaments par an). Il s’agit de personnes en affection de longue durée (ALD) et de personnes âgées, ces deux catégories se recoupant largement.
![]()
Les médicaments consommés sont pour l’essentiel des médicaments prescrits : la place de l’automédication en France est faible11 (6 % des achats de médicaments en
pharmacie) et l’essentiel de ce qui est consommé est constitué par des produits à prescription médicale obligatoire.
b. Analyse par classe thérapeutique
Ce constat global est également valable pour certaines classes thérapeutiques.
Ainsi, la France demeure l’un des pays d’Europe où l’on consomme le plus d’antibiotiques. Si la consommation de ces médicaments a baissé de 13 % entre 2002 et 2005, la France continue de se caractériser par une consommation d’antibiotiques nettement plus élevée que celle de ses voisins européens : elle représentait en 2005 plus de 30 doses journalières pour 1000 personnes en France en moyenne annuelle, soit deux fois plus que la consommation observée en 2003 en Allemagne et au Royaume-Uni et trois fois plus qu’aux Pays-Bas12. Par ailleurs, certaines familles d’antibiotiques, pour lesquels les experts recommandent de limiter la prescription afin de préserver leur efficacité et d’éviter l’émergence de nouvelles antibio-résistances, sont plus fréquemment consommées en France que dans les autres pays européens. C’est notamment le cas des céphalosporines de troisième génération, dont la consommation est en France quatre fois plus importante qu’en Allemagne et dix fois plus importante qu’au Royaume-Uni, au Luxembourg ou aux Pays-Bas.
S’agissant des psychotropes, la France se situe depuis de nombreuses années parmi les plus grands consommateurs de tranquillisants et d’hypnotiques.
S’agissant de la classe des statines, une étude du CREDES montrait dès 1992 que la France se situait parmi les plus grands consommateurs d’hypolipémiants, alors que le risque de maladies cardio-vasculaires y est plus faible que dans les pays nordiques. Une étude publiée en 2004 sur des données de 200013 montre que la consommation quotidienne moyenne varie de 1 à 4 pour 14 pays européens, sans que les différences épidémiologiques le justifient. Les auteurs de l’article estiment que la prédominance sur le marché français de molécules n’ayant pas fait la preuve de leur efficacité en termes de prévention de la mortalité plaide pour une influence non négligeable du marketing des firmes pharmaceutiques. Les statines sont encore à l’heure actuelle la classe thérapeutique la plus coûteuse pour l’assurance maladie (plus d’1 Md € en 2006), même si une décroissance est amorcée par rapport à 2005 en raison de la générication de la simvastatine (mai 2005) et de la pravastatine (juillet 2006).
![]()
Les inhibiteurs de la pompe à protons (médicaments de la classe des anti-ulcéreux) figurent également parmi les médicaments les plus remboursés en France. Si la croissance du nombre de boîtes remboursées (47 millions en 2005 contre 25 millions en 2000) peut s’expliquer en partie par l’élargissement des indications thérapeutiques14, la CNAMTS souligne cependant que la consommation de ces médicaments est particulièrement forte en
France (19 comprimés par personne par an contre 12 en Allemagne) sans raison médicale apparente15.
Le niveau de prescription de veinotoniques et de vasodilatateurs, en dépit de leur classement parmi les médicaments à service médical rendu insuffisant, demeure enfin une singularité française. Selon une étude internationale16, les médecins français prescrivent 14 lignes de veinotoniques par habitant et par an, contre 12 au Royaume-Uni, 11 en Allemagne, et 8 au Canada ; seule l’Espagne se situe au même niveau que la France.
1. Les caractéristiques générales de la prescription
Le niveau de prescription des médecins est élevé en France par rapport aux autres pays européens et ce constat n’a pas évolué depuis vingt ans. Ainsi, selon l’étude IPSOS précitée, 90 % des consultations de généralistes donnent lieu à prescription d’un médicament en France, contre 83 % en Espagne, 72 % en Allemagne et 43 % aux Pays Bas. Cette forte prescription n’est pas le fait de quelques médecins mais d’une pratique d’ensemble : les experts parlent d’« un modèle français de prescription17 ».
Les médicaments prescrits sont plutôt des spécialités récentes. Les médecins français ont en effet la réputation d’être très sensibles à la mise sur le marché de nouveaux médicaments : ainsi que le rappelle une étude internationale citée par l’IRDES18, pour les médicaments remboursables, les spécialités de moins de dix ans représentaient en 2001 38 % du chiffre d’affaires de l’industrie pharmaceutique et les spécialités de moins de cinq ans 22 % ; en Allemagne, pour la même année, la part de marché en valeur des médicaments de moins de 10 ans n’était que de 26 %.
À l’inverse, le marché des génériques est longtemps resté insuffisamment développé, et progresse désormais davantage par le droit de substitution exercé par les pharmaciens que par la prescription des médecins (cf. infra).
![]()
Par ailleurs, selon une étude internationale19, les médecins français prescrivent beaucoup plus de médicaments à service médical rendu insuffisant (SMRI – cf. infra) que les médecins des autres pays étudiés : 2,2 lignes de prescription par habitant et par an sont consacrées en France aux médicaments à SMRI (sur un total de 14 lignes de prescription tous médicaments confondus), contre 1 ligne de prescription sur 11 en Allemagne, 0,8 sur 14 en Espagne et 0 sur 12 au Royaume-Uni et 0 sur 8 au Canada. C’est
également la France qui réalise la plus grosse part des ventes en volume de médicaments à SMRI prescrits et non prescrits : plus de 17 % des ventes totales de médicaments contre moins de 3 % au Royaume-Uni. En termes de ventes par habitant, un patient français consomme 8 fois plus de boîtes de médicaments à SMRI qu’un patient canadien ou anglais.
Le comportement des médecins en France est enfin très variable d’un département à l’autre. Des analyses faites par la CNAMTS en 200420 font ainsi apparaître qu’à clientèle identique, le nombre de boîtes d’antibiotiques prescrites par un médecin par consultation peut varier de 0,23 à 0,41, soit un rapport de 1,8.
Des disparités ont également été mises en évidences entre les catégories de médecins. À titre d’exemple, le niveau de prescription des médicaments anti-cholestérol (statines), premier poste en montant des médicaments remboursés varie par département de 1 à 10 chez les cardiologues et de 1 à 2 chez les généralistes21.
2. La qualité des prescriptions
Outre le niveau excessif de prescription qui vient d’être rappelé, la qualité des prescriptions est régulièrement mise en cause par l’assurance-maladie, même si, ainsi que l’avait relevé la Cour dans son rapport sur la sécurité sociale de 2004, ces études restent dispersées et sans cohérence d’ensemble. Les études suivantes peuvent être citées à titre d’exemple :
– une étude menée en 2004 en Lorraine et Champagne-Ardenne auprès d’un échantillon de patients souffrant de dépression montre que seuls 4 traitements sur 10 sont conformes aux recommandations scientifiques ;
– une étude menée par l’URCAM du Nord-Pas-de-Calais sur un échantillon de 35 000 patients ayant reçu une ou plusieurs prescriptions de coxibs montre que 30 % d’entre eux sont traités par coxib en dehors des indications prévues par l’AMM22 ;
– une enquête de la CNAMTS menée en 2002 visant à mesurer la conformité des pratiques d’instauration des traitements hypolipémiants aux recommandations de l’ANAES et de l’AFSSAPS a montré que dans 30 % des cas, la prescription d’un traitement de ce type n’avait pas été précédée d’une analyse du taux de LDL cholestérol et que, même dans un tel cas, la moitié des prescriptions concernait des patients dont le taux était inférieur au seuil préconisé pour l’instauration d’un traitement hypolipémiant ;
– l’analyse de la prescription de Plavix publiée en 2002 par la CNAMTS a montré que, dans 40 % des cas, les motifs de prescription n’étaient pas conformes à l’indication stricte de l’AMM ;
– ![]()
une enquête de la CNAMTS sur la consommation de trois benzodiazépines (Tranxène, Nordaz et Rohypnol) publiée en 2001 a montré que les posologies usuelles maximales recommandées par l’AMM n’étaient pas respectées et que
les durées de traitement étaient dépassées dans près de la moitié des cas pour deux de ces médicaments.
1. Les enjeux de santé publique
Une prescription et une consommation excessives de médicaments peuvent avoir des effets négatifs en termes de santé publique. L’expression de « mauvais usage du médicament » regroupe ainsi trois types de problèmes : le niveau excessif de prescription et de consommation de médicaments ; la mauvaise qualité de la prescription ; la non-observance par le patient du traitement ou des conseils du médecin.
Ces trois phénomènes peuvent se traduire par des risques de iatrogénie médicamenteuse (effets indésirables liés à la prise simultanée de plusieurs produits) pouvant aller jusqu’à l’association contre-indiquée de médicaments. Ils peuvent également conduire au développement de résistances comme dans le cas des antibiotiques.
S’agissant tout particulièrement de la iatrogénie médicamenteuse, les pouvoirs publics en ont fait un enjeu prioritaire de santé publique. On estime en effet qu’environ 130 000 personnes sont hospitalisées en France chaque année en raison d’un accident lié à la prise de médicaments. Or 40 à 60 % de ces accidents seraient évitables. Ce sont bien sûr les personnes âgées qui sont les plus vulnérables : selon les estimations du régime général, 1,5 million de personnes de plus de 65 ans consomment régulièrement plus de 7 médicaments de classes thérapeutiques différentes.
2. Les enjeux en termes de maîtrise des dépenses
Le total du marché de ville du médicament s’élève à 20,4 Mds € en 2006 (+3,5 % par rapport à l’année précédente), ce qui représente plus de trois milliards de boîtes vendues.
Les dépenses de médicaments remboursables délivrés en ville ont évolué très rapidement depuis plus de 10 ans. Elles représentent un tiers des dépenses de médecine de ville et « tirent » la croissance des soins de ville : elles ont contribué à plus de 50,7 % à la croissance de ces dépenses entre 2004 et 2005.
Toutefois, après une progression forte ces dernières années (+ 6,4 % en 2004, + 4,6 % en 2003, + 8,1% en 2001), elles évoluent moins vite : + 4,8 % en 2005, +1 % seulement en 2006, les premiers mois de l’année 2007 manifestant cependant une nouvelle accélération des dépenses de médicaments, qui sont en hausse de 3,3 % en février 2007 par rapport à février 200623.
![]()
La croissance des dépenses de médicaments remboursables est due à plusieurs facteurs :
– le coût particulièrement élevé de nouveaux médicaments mis sur le marché (les médicaments coûtant plus de 15 € ne représentent que 16 % des unités vendues mais 65 % des dépenses en 2005 contre 49 % en 200024). Ainsi, des médicaments nouveaux et chers se substituent régulièrement à des médicaments anciens et peu onéreux. C’est ce qu’on appelle l’effet structure25.
– le poids des médicaments traitant des pathologies lourdes (cancer, hépatite, sida, sclérose en plaques, leucémie), dont les prix sont élevés26, ne cesse de croître.
– enfin, le taux de prise en charge des médicaments augmente : en 2005, près de 43 % des médicaments sont pris en charge à 100 % contre 36 % en 2000.
II. – DE LA MISE SUR LE MARCHÉ À L’ADMISSION AU REMBOURSEMENT
a. La mise sur le marché du médicament
Un médicament ne peut être commercialisé sur le marché français que s’il dispose d’une autorisation de mise sur le marché (AMM) délivrée soit par l’agence française de sécurité sanitaire des produits de santé (AFSSAPS), soit par son homologue européenne, l’agence européenne du médicament (EMEA).
Les procédures communautaires de mise sur le marché des médicaments
L’enregistrement d’un médicament susceptible d’être commercialisé dans plusieurs pays de l’Union européenne doit obligatoirement passer par l’une des deux procédures suivantes :
- ![]()
la procédure centralisée est une procédure unique aboutissant à une autorisation de mise sur le marché (AMM) unique, valable dans tous les pays de l'Union européenne. L’évaluation du médicament est alors réalisée par l’EMEA par l'intermédiaire de son comité des médicaments à usage humain (CHMP). Lorsque le CHMP rend un avis favorable, un rapport européen public d'évaluation (EPAR) est réalisé et une recommandation est présentée à la Commission européenne en faveur de l'octroi d'une autorisation de mise sur
- le marché pour ce produit. La procédure centralisée est obligatoire pour les produits issus des biotechnologies et, depuis novembre 2005, pour les nouvelles substances destinées à traiter le sida, le cancer, les maladies neurodégénératives, le diabète, ainsi que pour les médicaments orphelins. Elle s’appliquera aussi, à partir du 20 mai 2008, aux médicaments des maladies auto-immunes et virales ;
- la procédure de reconnaissance mutuelle : le demandeur dépose son dossier dans l’un des États membres dit « État membre de référence ». Si l’autorisation est accordée, elle peut être étendue aux autres États membres par une procédure de reconnaissance mutuelle. Ce système est ouvert à tout médicament non soumis obligatoirement à la procédure centralisée destinée aux marchés de plus d’un État membre.
Tableau n° 3 : Evolution du nombre d’AMM en procédure centralisée
2003 |
2004 |
2005 |
2006 | ||
Evaluation initiale |
Demandes d’AMM27 |
39 |
48 |
41 |
65 |
Avis favorable |
24 |
34 |
24 |
51 | |
Avis défavorable |
4 |
0 |
1 |
4 | |
Demandes retirées |
2 |
7 |
15 |
8 | |
Modifications d’AMM |
Demandes |
996 |
1 101 |
1 213 |
1 588 |
Source : Rapports annuels de l’EMEA
En raison de l’extension du champ de la procédure centralisée, la procédure nationale tend à se limiter aux génériques, aux « me-too »28 et à certains médicaments dont la commercialisation se trouve limitée au marché d’un seul Etat membre. Ainsi, en 2006, sur 890 nouvelles AMM délivrées par l’AFSSAPS, 718 concernaient des génériques.
Tableau n° 4 : Evolution de la procédure nationale d’AMM
1999 |
2000 |
2001 |
2002 |
2003 |
2004 |
2005 |
2006 | |
Octrois d’AMM |
782 |
683 |
632 |
559 |
644 |
596 |
828 |
890 |
Refus d’AMM |
279 |
238 |
235 |
175 |
270 |
302 |
82 |
117 |
Modifications d’AMM |
8 480 |
8 590 |
8 749 |
10 386 |
12 416 |
15 810 |
15 702 |
12 630 |
Renouvellement quinquennal |
1 364 |
1 927 |
2 411 |
3 129 |
2 121 |
1 752 |
1 965 |
1 693 |
![]()
Source : AFSSAPS
L’attribution de l’AMM, qu’elle soit française ou européenne, est fondée sur trois critères : l’efficacité, l’innocuité et la qualité du médicament29. Le médicament, qui doit être jugé « sur ses mérites propres », obtient une AMM dès lors que son rapport bénéfices/risques a été jugé positif. Il en résulte que l’AMM n’a pas pour objet de limiter le nombre de médicaments admis sur le marché, ni de comparer les médicaments entre eux.
En particulier, peuvent être admis sur le marché des médicaments n’apportant pas de réelle valeur ajoutée par rapport aux médicaments existants. L’AFSSAPS indique que « les procédures d’AMM pour des principes actifs réellement innovants se font plus rares, en tout cas en procédure nationale ». La cotation des nouveaux médicaments examinés en 2006 par la revue Prescrire fait apparaître que plus de 85 % des dossiers examinés « n’apportent rien de nouveau ».
Cette comparaison est censée intervenir au stade suivant, où la commission de la transparence formule, à la demande du laboratoire concerné, un avis sur l’admission au remboursement du produit.
2. Les médicaments sans AMM et les prescriptions hors AMM
Les médicaments ne disposant pas d’une AMM peuvent exceptionnellement être commercialisés en obtenant une autorisation temporaire d’utilisation (ATU). Par ailleurs, les médicaments disposant d’une AMM peuvent être prescrits par les médecins en dehors des indications de l’AMM.
Ces deux pratiques, qui tendent à se développer, vont dans le sens d’un contournement de l’AMM.
a. L’autorisation temporaire d’utilisation
À titre exceptionnel, les médicaments nouveaux peuvent être prescrits avant même l’obtention d’une AMM dans le cadre d’autorisations temporaires d’utilisation (ATU), délivrées par l’AFSSAPS dans le cas de pathologies graves ou rares lorsqu’il n’existe pas de traitement approprié (article L. 5121-12 du CSP).
Les médicaments bénéficiant d’ATU ne peuvent être délivrés qu’à l’hôpital, soit parce qu’ils sont réservés à l’usage hospitalier, soit dans le cadre de la rétrocession.
En 2006, l’AFSSAPS a ainsi autorisé 219 spécialités dans le cadre d’ATU dites « nominatives », délivrées pour un seul malade désigné, à la demande et sous la responsabilité du médecin prescripteur. Les conditions de délivrance des ATU individuelles ont été étendues par la loi du 26 février 2007. Les médicaments disposant d’une ATU « nominative » sont réputés rétrocédables.
![]()
L’AFSSAPS délivre également des ATU dites « de cohorte », qui concernent un groupe de patients, et qui sont délivrées à la demande du titulaire des droits d’exploitation, lequel s’engage à déposer une demande d’AMM dans un délai fixé. Les médicaments
bénéficiant d’une ATU de cohorte doivent être inscrits sur la liste officielle des médicaments rétrocédés.
Ces ATU de cohorte sont parfois utilisées par les firmes pour commercialiser un produit non finalisé avant son AMM. Le passage des ATU vers l’AMM est particulièrement long (7 ans pour certaines molécules). Pour limiter cette dérive, il conviendrait que l’AFSSAPS publie les avis défavorables qu’elle prononce en matière d’ATU de cohorte. En 2005, l’AFSSAPS indiquait qu’un projet en ce sens a été proposé au LEEM et que « la publication des avis défavorables en matière d’ATU de cohorte est actée et sera réalisée dans le courant du premier semestre 2006 ». Cette publication n’est cependant toujours pas effective.
Par ailleurs, le prix des médicaments disposant d’une ATU est libre. Jusqu’en 2006, le titulaire d’une ATU avait pour unique obligation d’adresser périodiquement au ministre chargé de la santé des informations sur le coût pour l'assurance maladie du médicament bénéficiant de l'autorisation octroyée. La LFSS pour 2007 a inséré dans le code de la santé publique un article L. 162-16-5-1 ainsi rédigé : « Le laboratoire titulaire des droits d'exploitation d'un médicament bénéficiant d'une autorisation temporaire d'utilisation prévue à l'article L. 5121-12 du code de la santé publique déclare au comité économique des produits de santé (CEPS) le montant de l'indemnité maximale qu'il réclame aux établissements de santé pour le produit. En l'absence de laboratoire exploitant, toute pharmacie à usage intérieur intéressée à l'achat de ce médicament déclare au comité le montant de l'indemnité qui lui est réclamée pour acquérir le produit si cette indemnité n'a pas déjà fait l'objet d'une déclaration au comité. Le comité rend publiques ces déclarations. Le laboratoire exploitant la spécialité ou, à défaut, les pharmacies à usage intérieur qui se sont procuré ce produit informent annuellement le comité économique du chiffre d'affaires correspondant à ces spécialités ainsi que du nombre d'unités fournies ou reçues. Si le prix ou le tarif de remboursement fixé ultérieurement par le comité économique des produits de santé pour le médicament lors de son inscription au remboursement au titre d'une autorisation de mise sur le marché est inférieur au montant de l'indemnité déclarée au comité, ce dernier demande au laboratoire de reverser à l'agence centrale des organismes de sécurité sociale, sous forme de remise, tout ou partie de la différence entre le chiffre d'affaires facturé aux établissements de santé sur la base de l'indemnité et celui qui aurait résulté de la valorisation des unités vendues au prix ou au tarif de remboursement fixé par le comité. Le produit de cette remise est affecté aux régimes d'assurance maladie selon les règles prévues à l'article L. 138-8. »
b. Les prescriptions hors AMM
La prescription hors AMM d’un médicament se définit comme son utilisation en dehors des mentions légales figurant dans le résumé des caractéristiques du produit (RCP). L’utilisation hors AMM peut concerner la posologie, les modalités d’administration, la durée de traitement, les indications du médicament.
![]()
Si l’on écarte les erreurs de prescription, l’utilisation des médicaments en dehors des indications de l’AMM peut être due, soit à un décalage entre les données scientifiques et les délais administratifs de modification de l’AMM, soit au refus de modification de l’AMM par la firme pharmaceutique : en raison du coût des essais, les laboratoires peuvent ne pas
prévoir l’ensemble des indications thérapeutiques pour lesquelles la spécialité serait susceptible d’être prescrite30.
La prescription hors AMM pourrait représenter entre 15 et 20 % du total des prescriptions, mais elle reste très mal évaluée. En théorie, la prescription hors AMM n’est pas admise au remboursement, mais cette obligation ne fait qu’exceptionnellement l’objet d’un contrôle.
À l’hôpital, la prescription hors AMM a été officiellement admise avec la mise en place de la tarification à l’activité. L’utilisation hors AMM des médicaments innovants et coûteux31 doit être conforme à des "protocoles thérapeutiques" définis par l'AFSSAPS, la Haute Autorité de Santé (HAS), ou l'Institut national du cancer (INCa)32 pour être admise au remboursement. Ainsi, en application de la mesure 46 du plan Cancer qui vise à permettre aux patients une égalité d'accès aux médicaments et dispositifs onéreux et innovants, l’AFSSAPS et l’Inca ont signé en octobre 2005 un protocole temporaire de traitement (PTT) pour HERCEPTIN®, valable jusqu’à obtention de l’AMM. Ce protocole devait être révisé en septembre 2006, dans le cas où l’AMM n’aurait pas été obtenue.
![]()
En médecine de ville, la pratique hors-AMM, son impact financier et son impact en termes de santé publique restent largement méconnus. Il serait nécessaire de mieux évaluer et d’encadrer cette pratique. L’article 56 de la loi de la LFSS 2007, qui autorise à titre dérogatoire et pour une durée limitée le remboursement de médicaments prescrits hors AMM lorsque les assurés sont en affection de longue durée ou atteints d’une maladie rare, à condition qu’il n’existe pas d’alternative appropriée et que les médicaments concernés figurent dans un avis ou une recommandation de la HAS pris après consultation de l’AFSSAPS33, ne constitue à cet égard qu’une première étape dans la recherche d’un encadrement de ces prescriptions. D’une part, il conviendra de s’assurer que les laboratoires concernés par ces autorisations dérogatoires n’utilisent pas ce dispositif pour contourner l’AMM34. D’autre part, cette évolution législative ne tranche pas la question des prescriptions hors AMM ne donnant pas lieu aux dérogations données ci-dessus.
b. L’admission au remboursement
La commission de la transparence, transférée de l’AFSSAPS à la HAS par la loi du 13 août 2004, est chargée d’évaluer, indication par indication, le service médical rendu par un médicament (SMR), ainsi que l’amélioration du service médical rendu par le même médicament (ASMR)35, avec pour objectif de conseiller le ministre en vue de l’admission au remboursement et d’éclairer le CEPS dans la fixation des prix. L‘évaluation de la commission de la transparence intervient à la fois au stade de la demande de première inscription d’un médicament et au stade de la demande d’extension d’indication.
1. Les critères de l’évaluation initiale
a. Le SMR
L’appréciation du SMR prend en compte six critères, fixés par le décret du 27 octobre 1999 (art. R. 163-3-I. du CSP) : l'efficacité, les effets indésirables du médicament, sa place dans la stratégie thérapeutique, la gravité de l'affection à laquelle il est destiné, le caractère préventif, curatif ou symptomatique du traitement médicamenteux et enfin son intérêt pour la santé publique (ISP).
Le SMR est mesuré sur une échelle comprenant 4 niveaux. Le niveau du SMR détermine le taux de remboursement36 : les médicaments ayant un SMR important sont remboursés à 65 %, ceux ayant un SMR modéré ou faible à 35 %, tandis que les médicaments présentant un SMR insuffisant ne peuvent être inscrits sur la liste des médicaments remboursables.
Le SMR, tel qu’il est aujourd’hui appliqué, ne permet pas une grande sélectivité dans l’admission des spécialités au remboursement. En 2006, seules 16 spécialités ont été considérées comme ayant un « SMR insuffisant » au stade de la première inscription (soit 3,5 % de l’ensemble des SMR attribués). La même proportion se retrouve au stade des extensions d’indications. De manière symétrique, le taux d’attribution de « SMR important » est très élevé (87 % en 2006). Ainsi, la quasi-totalité des médicaments ayant obtenu une AMM sont admis au remboursement.
![]()
Cette situation est en partie due au fait que le SMR est principalement évalué à partir de l’efficacité et des effets indésirables du médicament, pondéré par la gravité de la pathologie. Le critère d’intérêt de santé publique, qui fait certes l’objet d’une définition approfondie dans un document élaboré par la HAS37, est peu utilisé en pratique. En particulier, les trois dimensions de l’impact de santé publique (l’impact du médicament sur l’état de santé de la population, la réponse apportée à un besoin de santé publique, l’impact du médicament sur le système de santé) ne sont pas toujours distinguées dans les avis de la
transparence. Il en est de même pour la pondération respective qui est attribuée à chacune de ces trois dimensions pour aboutir à la cotation de l’ISP. Par ailleurs, la question du poids respectif de l’ISP et des autres critères du SMR demeure entière. La DGS estime dans sa réponse que la prise en compte d’une dimension collective à travers ce critère pourrait ouvrir la voie à une plus grande sélectivité.
Il en résulte une confusion entre les notions de « SMR insuffisant » et d’inefficacité du produit, alors que le terme de « SMR insuffisant » devrait signifier plutôt « insuffisant pour justifier une prise en charge par la solidarité nationale », soit parce qu’il présente un intérêt clinique limité, soit parce qu’il n’est pas considéré comme prioritaire.
La DGS souligne dans sa réponse que la réforme, envisagée depuis 2004, n’a pu aboutir pour l’instant en raison des enjeux tant politiques qu’économiques qui l’entourent. Deux précisions peuvent toutefois être apportées à ce stade. La première est d’ordre terminologique : conformément aux dispositions de la loi de 2004 sur l’assurance-maladie, le service médical sera qualifié d’ « attendu » lors de l’évaluation initiale et de « rendu » uniquement lors de l’évaluation a posteriori. Par ailleurs, il est prévu d’abandonner toute référence à la notion de service médical « insuffisant » et de retenir trois niveaux de cotation : important, modéré et mineur, le dernier niveau emportant systématiquement la non-inscription.
Une autre difficulté réside dans la possibilité pour certains médicaments de se voir attribuer des indications remboursables et d’autres non remboursables, puisque la commission de la transparence se prononce indication par indication. Aux termes de l’article L. 162-4 du code de la sécurité sociale, le médecin est censé faire figurer sur l’ordonnance la mention « NR » (non remboursable) lorsqu’il prescrit un médicament dans une indication non remboursable. Toutefois, outre la difficulté pour le médecin de faire le partage entre les indications remboursables et non remboursables d’un même produit, les contrôles que peuvent réaliser les caisses d’assurance maladie sur ce phénomène demeurent très théoriques, puisqu’elles ne peuvent remonter à la pathologie. Cette situation se complique encore davantage dans les cas où il existe des dosages différents pour un même principe actif et une même indication, et où seuls certains dosages sont admis au remboursement38.
b. L’ASMR
![]()
L’avis de la commission de la transparence comporte également, de manière concomitante, l’appréciation, pour chacune des indications thérapeutiques, de l’amélioration du service médical rendu (ASMR) apportée par le médicament. Ce critère suppose donc une comparaison du médicament avec les alternatives thérapeutiques existantes, qui peut s’effectuer soit directement (en comparant un produit A au produit B), soit indirectement en comparant les deux produits à un placebo.
L’ASMR détermine également le niveau de prix (ASMR I, II et III = avantage de prix, IV = léger avantage de prix possible, V = prix plus bas que celui des médicaments comparables).
Aux termes de l’article R. 163-5 du code de la sécurité sociale, les médicaments qui n’apportent pas d’amélioration du service médial rendu (ASMR V) ne peuvent être inscrits sur la liste des médicaments remboursables que s’ils apportent « une économie dans le coût du traitement médicamenteux ». Il en résulte que l’admission au remboursement dépend à la fois du SMR et de l’ASMR.
L’analyse des décisions rendues par la commission de la transparence fait apparaître une majorité de médicaments ayant une ASMR V, c'est-à-dire n’apportant aucune amélioration39, ce qui montre que la réalité de l’innovation, mesurée par la commission de la transparence, est assez limitée.
Le caractère non obligatoire et donc non systématique des essais cliniques contre comparateurs est problématique. La commission de la transparence ne dispose pas du pourcentage exact de dossiers présentant des essais cliniques contre comparateurs, mais une estimation réalisée à partir d’un échantillon laisse à penser que moins de la moitié des dossiers dispose de ces données. Or les évaluations indirectes contre placebo n’apportent pas le même niveau de preuve.
Enfin, ainsi que l’avait déjà souligné la Cour dans son rapport sur la sécurité sociale de 2004, on peut regretter qu’il n’existe pas à ce jour de liste des médicaments classés par niveau d’ASMR. Le bilan d’activité de la commission de la transparence n’est pas détaillé et la base Medicam de la CNAMTS n’intègre pas non plus le critère du niveau d’ASMR. Il est donc impossible, à ce jour, de savoir quelle est la contribution à l’accroissement des remboursements de chaque catégorie, notamment de celle des médicaments sans amélioration du service médical rendu40.
2. L’absence d’analyse médico-économique
Le constat dressé par la Cour dans son rapport sur la sécurité sociale de 2004 sur l’absence d’expertise médico-économique est toujours vrai : « ni la commission de la transparence recentrée sur sa mission d’expertise médicale, ni le comité économique des produits de santé (CEPS) dont la mission est de réguler les prix, n’assument actuellement la mission transversale d’analyse médico-économique ».
Les comparaisons internationales montrent que d’autres pays, sans recourir à la notion de SMR, définissent des priorités et restreignent plus énergiquement l’accès au remboursement de certains médicaments.
![]()
Ainsi en Allemagne, selon une étude de l’IRDES41, une réforme conduite en 2004 a redéfini les contours du panier de médicaments pris en charge, en excluant de nombreux produits. Il en résulte que :
– les médicaments de prescription non obligatoire ne sont plus pris en charge, même s’ils sont prescrits, sauf lorsqu’ils sont utilisés dans le traitement standard de maladies graves et/ou pour les enfants de moins de 12 ans.
– certains médicaments de prescription obligatoire ne sont pas couverts pour les plus de 18 ans, en particulier ceux utilisés pour les problèmes de santé mineurs (états grippaux, rhumes).
– ne sont pas pris en charge les « lifestyle drugs », c'est-à-dire les médicaments dont l’indication ne répond pas à la définition d’une maladie (ex : chute des cheveux) ou les médicaments destinés à traiter une maladie résultant des choix de vie d’une personne (obésité, sevrage tabagique, etc.).
En Angleterre, selon la même étude, la décision de prise en charge d’un médicament appartient au ministre de la santé, qui établit deux listes après consultation des différentes parties (industrie, médecins, pharmaciens, patients et Institut national d’évaluation clinique -NICE) : une liste négative qui recense les médicaments que les médecins n’ont pas le droit de prescrire dans le cadre du National Health Service (liste noire) ; une liste restrictive qui recense les molécules dont la prescription est prise en charge seulement pour des indications et des catégories d’individus précises (liste grise).
Plusieurs groupes de produits sont ainsi exclus de la prescription, notamment :
– des produits exclus sur la base d’arguments médico-économiques. Pour une liste de 17 médicaments, seules les versions les moins chères sont prises en charge (antalgiques, laxatifs, benzodiazépines, etc.) ;
– les médicaments à risque de mésusage ;
– les produits dont les coûts ne sont pas justifiés au regard des priorités du NHS ;
– les produits administrés par un mécanisme d’injection prérempli si une alternative plus économique est disponible.
Le rapport coût-efficacité d’un médicament est ainsi un critère essentiel dans les décisions de prise en charge en Angleterre.
![]()
En France, la réforme de l’assurance-maladie par la loi du 13 août 2004 n’a pas permis de combler cette lacune. L’UNCAM s’est certes vu transférer la compétence de fixer le taux du remboursement mais avec une limite importante, puisque l’UNCAM n’a aucune compétence sur les prix auxquels s'applique ce taux.
C. Le suivi des médicaments en pratique médicale réelle
1. Pharmacovigilance et suivi post-AMM : une réévaluation timide de la balance bénéfice-risque
a. Pharmacovigilance : une réactivité parfois insuffisante, une information à améliorer
La surveillance du risque et du bon usage des produits de santé constitue une mission essentielle de l’AFSSAPS. La possibilité introduite par la loi du 26 février 2007 de renouveler l’AMM sans limitation de durée42 va encore accroître les exigences en matière de pharmacovigilance. En effet, l’article L. 5121-8 du code de la santé publique dispose désormais que l’AMM peut être renouvelée « le cas échéant, sans limitation de durée dans des conditions fixées par un décret en Conseil d’État, sauf si l’AFSSAPS décide, pour des raisons justifiées ayant trait à la pharmacovigilance, de procéder à un renouvellement supplémentaire, sur la base d’une réévaluation des effets thérapeutiques positifs du médicament ou produit au regard des risques (…). »
Le directeur général de l’AFSSAPS a la possibilité de suspendre ou de retirer une autorisation de mise sur le marché notamment lorsque la balance bénéfice-risque n’est pas considérée comme favorable dans les conditions normales d’emploi ou lorsque l’effet thérapeutique annoncé fait défaut43.
Dans les faits, les décisions de retrait ou de suspension d’AMM interviennent parfois tardivement. Ainsi, plusieurs anorexigènes amphétaminiques, qui avaient fait l’objet d’une suspension d’AMM depuis le 15 octobre 1999, n’ont vu leur AMM retirée qu’en 2006. En ce qui concerne les immunostimulants, retirés du marché en 2005, le centre régional de pharmacovigilance de Saint-Etienne avait, en 2001, rapporté les résultats d’une enquête nationale de pharmacovigilance rassemblant les effets indésirables notifiés en France depuis la commercialisation de ces produits jusqu’en 1998 : 315 effets indésirables, dont 121 graves, avaient été identifiés. Mais alors que l’efficacité de ces médicaments n’était pas avérée, leur RCP a simplement été modifié en 2003, et ce n’est qu’en 2005, soit 4 ans après les résultats de l’enquête, qu’ils ont été retirés du marché.
Certains médicaments, dont la balance bénéfices-risques est contestée depuis plusieurs années et qui ont été retirés du marché dans des pays voisins, restent commercialisés en France (cf. encadré).
![]()
Certes, la réévaluation de la balance bénéfice-risque d’un médicament est un processus complexe et la pharmacovigilance ne se limite pas aux retraits et suspensions d’AMM. Par ailleurs, les médicaments précités avaient fait l’objet de modification de leur résumé des caractéristiques du produit (RCP) et/ou de l’envoi de lettre aux prescripteurs. Il n’en demeure pas moins que l’introduction de restrictions d’indications dans les RCP n’a évidemment pas le même effet qu’une mesure de suspension ou de retrait d’AMM, ne serait-ce que parce que les médecins ne sont pas nécessairement informés de ces restrictions
d’indications et que le respect de ces indications dans les prescriptions ne fait l’objet d’aucune vérification. De même, l’efficacité des lettres aux prescripteurs n’est pas mesurée.
Exemple 1
Un dérivé amphétaminique titulaire d’une AMM française, indiqué comme traitement adjuvant des hypertriglycéridémies et du diabète avec surcharge pondérale, est également utilisé hors AMM comme médicament amaigrissant. Alors que ce médicament a été retiré du marché espagnol en 2003 et que les anorexigènes amphétaminiques ont été retirés du marché en raison d’une balance bénéfice-risque défavorable (risque grave d’hypertension artérielle et valvulopathies), il est resté commercialisé en France, où il réalise l’essentiel de ses ventes.
Lors d’un comité technique de pharmacovigilance de fin 2004, plusieurs notifications d’effets indésirables pouvant évoquer un effet de type amphétaminique ont été rapportées. Une actualisation des données relatives aux troubles neuro-psychiatriques observés avec cette spécialité pharmaceutique a alors été décidée. Par la suite, du fait d’une notification d’un cas d’hypertension artérielle pulmonaire associée à la prise de ce médicament, cette enquête a été étendue aux hypertensions artérielles pulmonaires. Pourtant, au vu de ces enquêtes, la commission de la pharmacovigilance ne s’est prononcée, fin 2005, que pour une réévaluation de la balance bénéfice-risque de ce médicament et pour des études complémentaires.
Au vu de ces nouvelles données, certains membres de la commission nationale de pharmacovigilance, en mars 2007, se sont prononcés pour un rapport bénéfice/risque défavorable de ce médicament. De son côté, la commission d’AMM, sur la base de l’ensemble de l’évaluation des données, a émis en avril 2007 un avis défavorable au maintien de l’indication comme adjuvant au régime adapté dans les hypertriglycéridémies et a donc jugé le risque/bénéfice du produit défavorable dans cette indication. Seule l’indication adjuvant au régime adapté chez les diabétiques avec surcharge pondérale a été maintenue, en l’attente de données complémentaires et des résultats de l’inspection de la seule étude clinique retenue supportant cette indication, étude qui présentait des lacunes méthodologiques selon plusieurs membres de la commission d’AMM. Début juillet 2007, l’AFSSAPS n’a toujours pas communiqué sur cet avis de la commission d’AMM ni modifié l’AMM de ce médicament.
Il faut souligner que de son côté, la commission de la transparence avait, au printemps 2006, conclu que le SMR de cette spécialité était insuffisant dans l’indication adjuvant du régime adapté des hypertriglycéridémies mais a refusé de se prononcer sur le SMR apporté par cette spécialité dans l’indication diabète avant l’avis de la commission d’AMM. Cet avis n’a pas encore été pris en compte et ce médicament est toujours remboursé à 65 %.
Exemple 2
Un vasodilatateur périphérique est commercialisé en France depuis plus de trente ans soit sous forme orale, soit sous forme injectable. Deux enquêtes de pharmacovigilance et de toxicovigilance ont été réalisées en 1998 et 2005 à la suite de notifications de cas graves d’atteintes neurologiques et cardiovasculaires.
La commission de pharmacovigilance, en juillet 2005, avait conclu que « compte tenu des risques importants du produit, encore largement méconnus par les professionnels de santé, pour un bénéfice jugé insuffisant, [ses] membres ont voté à l’unanimité pour une réévaluation du bénéfice-risque [de ce médicament] par une commission commune (commission nationale de pharmacovigilance et commission d’AMM) intervenant au dernier semestre 2005, et une limitation de la publicité (…). » Des modifications de l’AMM avec des mises en garde ont été introduites suite aux résultats des enquêtes de pharmacovigilance de 1998 et 2005.
À la suite de la réévaluation, le groupe de travail thérapeutique de cardiologie aurait confirmé l’intérêt du produit pour sa forme orale. En dépit des conclusions du groupe de cardiologie, la commission nationale de pharmacovigilance, en juin 2006, s’est prononcée majoritairement pour une balance bénéfice-risque défavorable s’agissant de l’ensemble des formes orales, quel que soit leur dosage.
![]()
Mais en juin 2006, la commission d’AMM s’est prononcée pour un retrait de l’AMM de la forme comprimé pour le seul dosage 300 mg, et pour le renforcement du RCP de la forme orale (150 mg) et des formes injectables. En octobre 2006, le directeur général de l’AFSSAPS a décidé le retrait du marché de ces spécialités à compter de fin novembre 2006. Les autres formes et dosages restent commercialisés et remboursables à 35 %.
De son côté, la commission de la transparence, dans ses avis de 1999 et 2004, a jugé que le service médical rendu était insuffisant dans l’ensemble des indications thérapeutiques pour justifier une prise en charge. Chargée de réexaminer le SMR de ces spécialités par le ministre de la santé, la commission de la transparence, dans son avis rendu en juin 2006, a conclu que le service médical rendu par ces spécialités est faible dans l’indication « Traitement symptomatique de la claudication intermittente des artériopathies chroniques oblitérantes des membres inférieurs (au stade II) » et insuffisant dans l’ensemble des autres indications de l’AMM.
L’AFSSAPS a tardé à communiquer les conclusions de la commission de la pharmacovigilance et de la commission d’AMM. Le compte rendu de la réunion de la commission de la pharmacovigilance ne mentionnait pas les conclusions de la commission sur ce dossier. Il se contentait d’indiquer : « ce sujet donnera lieu à une information auprès des professionnels de santé à la mi-novembre 2006. La publication complète du compte rendu se fera la semaine suivant la diffusion de la lettre d’information. » De même, le compte rendu de la commission d’AMM de juin 2006 ne mentionne pas ce médicament. Ce n’est que fin novembre 2006 que ce retrait du marché a été annoncé, avec l’envoi d’un message d’information destiné aux professionnels de santé et la mise en ligne d’un complément au compte rendu de la commission nationale de la pharmacovigilance de juin 2006.
Exemple 3
Un neuroleptique prescrit dans les « bouffées de chaleur » a reçu une AMM française il y a une trentaine d’années. Susceptible de provoquer des syndromes parkinsoniens, il a été retiré du marché espagnol en mai 2005, mais continue d’être commercialisé sur le marché français. L’AFSSAPS indique qu’une enquête menée en 2005 a établi le caractère favorable de la balance bénéfice/risque de ce médicament sous réserve d’une modification du RCP. La commission d’AMM a donné en juillet 2005 un avis favorable au maintien de l’AMM sous réserve d’une série de modifications du RCP (limitation à 3 mois de la durée de traitement, contre-indication de l’association aux neuroleptiques, antipsychotiques et anti-émétiques, mises en garde renforcées sur les risques de dyskinésies et de troubles parkinsoniens exigeant l’arrêt du traitement, mention de la possible survenue de troubles de l’humeur et d’anxiété). Selon le directeur de l’AFSSAPS, un retrait du médicament n’a pas été envisagé compte tenu du positionnement de ce médicament par rapport aux traitements hormonaux : ce traitement est le seul pour les indications visées à ne pas être un traitement hormonal dans un contexte de publications sur les risques du traitement hormonal de la ménopause. La modification du RCP est intervenue en février 2006. Toutefois, ce n’est qu’en avril 2006 que l’AFSSAPS a mis en ligne une lettre du laboratoire faisant état d’une modification du RCP.
Le 13 juillet 2006, à la suite de plaintes de la part de patients concernant la survenue d’effets indésirables graves neuro-psychiques et de questions concernant la différence de statut de ce produit dans les pays européens considérés, la Commission Européenne a saisi l’EMEA afin de statuer sur le rapport bénéfice-risque de ce médicament. Un arbitrage a ainsi été notifié le 19 septembre 2006, la France étant le pays rapporteur et l’Espagne le co-rapporteur. Ce dossier devait être discuté à l’EMEA en février 2007. L’arbitrage en cours au niveau européen devra déterminer si les différences dans l’analyse du profil de sécurité d’emploi et de la balance risque/bénéfice de ce médicament peuvent être expliquées par un usage différent entre les pays et justifient le maintien (ou non) de l’AMM au niveau européen.
L’information relative à la pharmacovigilance pourrait être améliorée : les rapports d’enquête de pharmacovigilance ne sont pas publiés, ni, sauf exception, les études de pharmaco-épidémiologie ayant conduit à des modifications de RCP. Le signalement par l’AFSSAPS des modifications de RCP pour raisons de pharmacovigilance n’est pas systématique. Les comptes rendus de la commission de pharmacovigilance sont mis en ligne avec un délai de 3 ou 4 mois (cf. infra) et sont parfois incomplets (cf. encadré).
b. Plans de gestion des risques et études post-AMM
![]()
La surveillance des médicaments après leur commercialisation est longtemps restée insuffisante faute d’études ou d’essais post-AMM réalisés sous l’égide des pouvoirs publics.
Or, ces études sont indispensables pour poursuivre l’évaluation de la balance bénéfices-risques des médicaments en situation réelle, identifier les risques non étudiés ou tardifs qui pourraient apparaître lors de l’utilisation d’un médicament et étudier l’impact sur le système de soins de l’utilisation de ce médicament.
Ces études peuvent être réalisées par les laboratoires pharmaceutiques à la demande de la commission de la transparence, du CEPS ou de l’AFSSAPS, ou bien être financées ou réalisées par les pouvoirs publics.
Les études demandées aux industriels dans le cadre des plans de gestion du risque ou des études post-inscription
La réglementation communautaire impose depuis novembre 2005 aux laboratoires pharmaceutiques d’intégrer des plans de gestion des risques dans le dossier d’AMM pour certains produits (nouveaux médicaments, médicaments génériques lorsqu’un problème de sécurité a déjà été identifié avec le médicament princeps, médicaments déjà commercialisés mais dont la demande d’extension de l’autorisation de mise sur le marché entraîne des changements significatifs des conditions d’emploi du produit). Ces plans peuvent prévoir des études post-AMM et/ou un plan de minimisation des risques. Ils peuvent également être déposés en post-AMM suite à la mise en évidence d’un signal de pharmacovigilance. En 2006, 16 études post-AMM ont ainsi été soumises à l’AFSSAPS ainsi que 9 études observationnelles.
À ces études post-AMM demandées par l’AFSSAPS s’ajoutent des études post-inscription à la demande de la commission de la transparence. Toutefois, en dépit de l’accord-cadre44 conclu en juin 2003 entre le CEPS et le LEEM, supposé élargir le champ des études post-AMM et prévoir leur financement, le nombre d’études post-AMM effectivement menées à terme est extrêmement réduit : un bilan réalisé en mai 2006 sur les 105 études demandées depuis 1997 par le CEPS et/ou la commission de la transparence a montré que seulement 7 % des études avaient été menées à terme. Pour 30 % d’entre elles, aucun document n’avait été reçu, et pour 24 %, le protocole était en cours de validation. Ces chiffres ont légèrement évolué depuis : selon la DGS, à fin mai 2007, sur 131 études demandées depuis 1997, 12 % sont terminées, 27 % ont leur protocole en cours de validation tandis que pour 18 % d’entre elles (principalement des études demandées dans les six derniers mois), aucun document n’a été reçu.
Il reste qu’aucun dispositif explicite de sanction n’est prévu à l’encontre des laboratoires qui ne mettraient pas en œuvre ces études ou les mettraient en œuvre avec retard. L’avenant du 29 janvier 2007 à l’accord cadre du 13 juin 2003 se contente d’indiquer que « les délais dans lesquels les études doivent être entreprises (…) sont définis par l’avenant à la convention, qui peut prévoir également les conséquences à tirer du non-respect de ces délais ».
Les études commanditées et financées par les pouvoirs publics
De son côté, l’intervention publique dans le domaine des études post-AMM reste trop réduite.
![]()
L’AFSSAPS n’a financé que 6 études post-AMM de 1999 à 2004. Le retrait de plusieurs médicaments du marché entre 2001 et 2004 a cependant conduit à développer
davantage les études post-AMM. Depuis 2005, l’AFSSAPS a mis en place un programme d’études dont les grands axes sont approuvés par le conseil scientifique et doté d’un budget d’environ 800 000 €. Le nombre d’études de pharmaco-épidémiologie financées par l’agence a ainsi progressé : 19 études ont été financées de 2005 à 2006. Toutefois, ces études ne sont pas rendues publiques. L’AFSSAPS indique que dans la mesure où les études commanditées par elle font l’objet d’une restitution en conseil scientifique, il peut être envisagé à l’avenir de publier une synthèse des résultats de ces études.
Par ailleurs, le groupement d’intérêt scientifique « évaluation épidémiologique des produits de santé » créé en août 2004 entre le ministère de la santé, la CNAMTS et l’INSERM afin de développer l’évaluation post-AMM n’a eu qu’une activité limitée : seules trois études (Cadeus, Copeprev, Bloixa) ont débuté sur les 5 initialement prévues. Selon la CNAMTS, l’étude Emes n'a pas été réalisée par manque de prise en compte des contraintes techniques (absence de codage) par l'équipe de recherche. Quant à l’étude Eulam (sur Keppra®), elle n'a pas été lancée car le protocole prévoyait un retour aux dossiers, ce qui, selon la CNAMTS, pouvait permettre d’identifier les patients. La CNAMTS demandait à être associée au protocole, ce qui n'a pas été le cas. Il est à noter que cette étude avait reçu l’accord de la CNIL. Au-delà, la CNAMTS fait valoir que ces travaux ont mobilisé assez lourdement ses équipes et, pour certains d’entre eux, l’ensemble du réseau de l’assurance maladie (notamment pour les études faisant appel à des données nominatives...). Pour faciliter le recours aux informations de l’assurance maladie dans les cas où les études ne nécessitent que des données anonymes, la possibilité d’exploiter l’échantillon de bénéficiaires au 1/100e a été ouverte il y a quelques mois pour un ensemble d’institutions publiques, dont l’AFSSAPS fait partie. Il n’en reste pas moins que l’activité du GIS devrait être relancée.
2. Le suivi des médicaments après l’admission au remboursement
Pas plus qu’au stade de la première inscription et de l’admission au remboursement, la France n’a développé de système réellement sélectif au stade de la réévaluation des médicaments en cours de commercialisation.
En application de l’article R. 161-71 du code de la sécurité sociale, la HAS « formule des recommandations sur le bien-fondé et les conditions de remboursement d’un ensemble de soins ou catégories de produits et prestations ». Ces recommandations sont émises à l'initiative de la Haute Autorité ou à la demande du ministre chargé de la santé ou du ministre chargé de la sécurité sociale, du CEPS ou de l’UNCAM. La décision de dérembourser ou non appartient au ministre de la santé et de la sécurité sociale.
Entre 1999 et 2001, l’ensemble des médicaments remboursables par l’assurance-maladie a fait l’objet d’une réévaluation à la demande conjointe des ministres chargés de la santé et de la sécurité sociale. La commission de la transparence avait alors évalué le SMR de 4 490 spécialités. Elle a conclu que pour 835 d’entre elles, le SMR était insuffisant pour justifier leur remboursement par la sécurité sociale.
![]()
Mais dans un premier temps, ces spécialités n’ont pas été déremboursées : elles n’ont fait l’objet que d’une baisse de taux de remboursement et d’une baisse de prix. Dans un deuxième temps, les pouvoirs publics ont décidé de procéder à une actualisation de leur réévaluation en trois vagues.
La première vague a porté sur 60 médicaments (84 spécialités) n’ayant pas de place dans la stratégie thérapeutique, d’autres moyens de prise en charge leur étant préférables. Une douzaine de spécialités se sont vues retirer leur AMM et les autres ont été déremboursées par arrêté du 24 septembre 2003. L’économie qui en a résulté représente, selon la DSS, 43 millions en année pleine.
La deuxième vague a concerné 245 médicaments (403 spécialités) de prescription médicale facultative. La HAS a recommandé, le 14 septembre 2005, la radiation de la liste des médicaments remboursables de la très grande majorité de ceux-ci. Par arrêté du 17 janvier 2006, le ministre de la santé et des solidarités a décidé le retrait du remboursement de 152 de ces 245 médicaments à compter du 1er mars 2006 et le maintien temporaire de 61 veinotoniques à un taux de remboursement de 15 %45 (jusqu’au 1er janvier 2008). Toutefois, les veinotoniques ne peuvent plus être pris en charge à 100 % dans le cadre des ALD (mesure dite du « 15 à 15 »)46, ce qui a permis une économie de 70 M€ selon la DSS. La deuxième vague de réévaluation a permis au total 460 M€ d’économies en année pleine (345 M€ pour l’année 2006).
La troisième vague, qui a rapporté 34 M€ d’économies selon la DSS, concernait 133 médicaments (238 spécialités), principalement à prescription médicale obligatoire. La HAS a jugé le SMR insuffisant pour 89 d’entre eux. Le ministre n’a pas suivi l’avis de la HAS. Pour 48 vasodilatateurs, il a décidé de conserver leur prise en charge à hauteur de 35 %, tout en leur appliquant une baisse de prix pouvant aller jusqu’à 20 %. Les 41 médicaments restants (essentiellement des anti-diarrhéiques et des produits utilisés en ORL et en pneumologie) seront remboursés à 15 % au lieu de 35 % par l’assurance-maladie, avant d’être complètement déremboursés au 1er janvier 2008. Aucune mesure équivalente à celle du « 15 à 15 » prise lors de la deuxième vague de déremboursement n’a été décidée cette fois-ci.
Ces trois vagues de réévaluation appellent plusieurs remarques.
D’une part, les décisions prises lors des deux dernières vagues ne vont pas dans le sens d’une gestion active de la liste des produits remboursables, qui supposerait d’affecter les financements collectifs en priorité à la prise en charge des traitements les plus performants. Même si elles ne sont que temporaires, elles témoignent bien de la difficulté des pouvoirs publics à réviser la liste des spécialités remboursables.
![]()
D’autre part, parmi l’ensemble des scénarios possibles, c’est l’un de ceux qui rapportait le moins d’économies pour l’assurance-maladie qui a été retenu pour la troisième
vague : celle-ci n’a rapporté que 34 M€ d’économies, alors que le déremboursement total des spécialités à SMR insuffisant se serait traduit par 147 M€ d’économies pour l’année 2007.
III. DE LA PRESCRIPTION À LA CONSOMMATION
Les spécificités françaises en la matière peuvent s’expliquer par un certain nombre de facteurs, examinés ci-dessous, au premier rang desquels figurent les lacunes dans le domaine de la formation des médecins et dans celui de l’information mise à leur disposition.
A. L’information sur le médicament
1. La transparence des travaux d’évaluation des médicaments
Pour assurer leurs missions d’évaluation, l’AFSSAPS et la HAS s’appuient largement sur des experts externes. La crédibilité de cette expertise aux yeux du grand public comme des professionnels de santé suppose la transparence des procédures d’évaluation et la gestion des conflits d’intérêt. Or, celle-ci reste insuffisante.
La directive communautaire 2004/27 CE, qui a accru les obligations des agences en matière de transparence, a été transposée avec la loi du 26 février 2007 mais était applicable dès le 30 octobre 2005. Dans l’intervalle, l’AFSSAPS a commencé à mettre en œuvre ces dispositions, mais de manière partielle, alors même que la directive reprenait plusieurs obligations déjà en vigueur en droit français.
a. Les déclarations d’intérêts des agents et des experts
La déclaration d’intérêts des experts travaillant pour l’AFSSAPS constitue une obligation légale depuis la loi du 1er juillet 1998 sur le renforcement de la sécurité sanitaire. Depuis 2005, l’AFSSAPS a pris des mesures pour améliorer la détection et la gestion des conflits d’intérêts (déclarations électroniques, détermination des niveaux de conflits, exclusion de l’expert concerné par un conflit d’intérêt majeur). Toutefois, début juillet 2007, seul le rapport annuel des déclarations d’intérêt (RAPDI) 2005 était mis en ligne. Ce rapport montre que 135 déclarations d’intérêts ne sont pas parvenues et qu’environ 40 % des déclarations datent de plus d’un an. Selon le directeur de l’AFSSAPS, le rapport 2006 des déclarations d’intérêts, en cours d’édition fin juin 2007, montre une amélioration du taux de déclaration et de la part des déclarations de l’année.
La déclaration d’intérêts des agents de l’AFSSAPS n’a commencé à être mise en œuvre qu’au premier semestre 2006.
La loi du 26 février 2007, qui a retenu le principe d’une déclaration annuelle des experts et des agents, devrait améliorer le dispositif. Toutefois, la question du délai de publication de ces déclarations et de la gestion effective des conflits reste posée.
![]()
En ce qui concerne la HAS, les déclarations d’intérêts des rapporteurs extérieurs devant la commission de la transparence, pourtant prévues par l’article R. 163-17 du code de
la sécurité sociale, ne sont ni systématiques, ni actualisées. Selon la HAS, il est systématique de demander sa déclaration publique d’intérêts (DPI) à tout expert extérieur sollicité. Concernant leur actualisation, il appartient aux experts d’informer la HAS des éventuels nouveaux intérêts survenus depuis la remise de leur DPI. De plus, la HAS précise qu’elle demande systématiquement une nouvelle DPI si celle qui est disponible date d’un an ou plus. Toutefois, l’examen des DPI des rapporteurs de la commission de la transparence figurant sur le site de la HAS montre que 12 experts n’ont pas fait parvenir de déclaration d’intérêt. Par ailleurs, les déclarations d’intérêts datent souvent de plusieurs années.
b. La publication des rapports d’évaluation d’AMM (RAPPE)
Le nombre de RAPPE publiés
La publication des rapports d’évaluation d’AMM (RAPPE), prévue par la directive47 et figurant déjà à l’article 6 de loi de 1998 sur la sécurité sanitaire (art. L.793-1, devenu L.5311-1 du CSP), n’a débuté qu’en juin 2004.
Dans un premier temps, l’AFSSAPS a choisi de publier les RAPPE pour les seules AMM correspondant à de nouvelles entités chimiques ou biologiques ainsi que pour les extensions d’indications majeures.
Ce n’est que dans un second temps que la publication des RAPPE sera étendue à l’ensemble des nouvelles demandes d’AMM. L’AFSSAPS estime pouvoir atteindre cet objectif d’ici fin 2007, ce qui représenterait une cinquantaine de RAPPE.
Cette évolution constitue un progrès : il faut toutefois souligner qu’en raison du retard pris dans la publication des RAPPE, les médicaments ayant obtenu une AMM avant juin 2004 ne devraient pas faire l’objet de RAPPE.
Les délais de publication
Les délais de publication des RAPPE sont parfois excessifs : pour les cinq RAPPE mis en ligne entre janvier et juin 2007, le délai écoulé depuis l’octroi de l’AMM allait de 9 mois à un an et demi. Selon le directeur de l’AFSSAPS, une procédure interne mise en place en 2006 prévoit un délai de publication du RAPPE, après notification finale auprès du titulaire de l’AMM, d’environ 10 semaines. Ce délai inclut une procédure contradictoire pour les firmes de 4 semaines afin qu’elles puissent vérifier l’absence de données confidentielles. Il reste qu’il conviendrait également de prévoir un délai maximal entre la date délivrance de l’AMM et la notification finale du RAPPE auprès du titulaire de l’AMM.
La mise à jour des RAPPE
![]()
Les modalités d’actualisation des RAPPE risquent de susciter des difficultés. D’une part, la loi du 26 février 2007 n’a pas étendu l’obligation de publication d’un RAPPE aux
évaluations relatives au renouvellement de l’AMM. D’autre part, si après la mise sur le marché, de nouveaux éléments, notamment concernant le profil de sécurité du produit, sont notifiés à l’agence, cette information ne sera pas systématiquement intégrée dans le RAPPE : ce n’est qu’en cas de modification notable de l’indication thérapeutique du médicament ou d’une extension d’indication, que l’information contenue dans le RAPPE sera mise à jour.
À minima, il conviendrait de préciser dans le RAPPE ou dans un document qui lui serait attaché les éventuelles réévaluations en cours.
Un exemple récent illustre cette nécessité. Pour les antithrombines [anticoagulants] Melagatran Astrazeneca® ou Exanta® (mélagatran, ximélagatran), l'AMM européenne date du 23 décembre 2003, mais le RAPPE n’a été publié que le 2 décembre 2005, soit avec près de deux ans de retard. Cette publication très tardive du RAPPE serait due au fait que, suite au refus de la Food and Drug Administration (FDA) d’octroyer une AMM à ces spécialités, une réévaluation a été engagée par l’AFSSAPS en septembre 2004. Le RAPPE n’a été publié qu’après qu’a été rendu l’avis européen. Le médicament a finalement été retiré du marché en février 2006.
Ainsi, dans ce cas, la découverte de risques sanitaires sérieux peu après la délivrance de l’AMM a bloqué la publication du RAPPE, alors que c’est précisément dans de tels cas que la transparence des données aurait été la plus utile. Il aurait sans doute été concevable, au lieu de reporter la publication du RAPPE, de publier ce dernier en indiquant, dans le RAPPE ou dans un document attaché, qu’une réévaluation était en cours.
c. La publication des ordres du jour, des comptes rendus et du règlement intérieur des commissions
La disposition la plus novatrice de la directive communautaire concerne la publication du règlement intérieur, des ordres du jour, et des comptes rendus, assortis des décisions prises et du détail des votes et des explications de vote, y compris les opinions minoritaires des commissions en charge de l’évaluation des médicaments (commission d’AMM, commission de la pharmacovigilance, commission chargée du contrôle de la publicité, commission de la transparence).
![]()
Le site Internet de l’AFSSAPS ne comporte actuellement qu’une partie des informations requises par la directive communautaire de 2004, contrairement aux engagements pris par l’AFSSAPS dans des communiqués de presse de mars 2006. Début juillet 2007, les ordres du jour des commissions ne sont toujours pas rendus publics. En moyenne, les comptes rendus des commissions d’AMM sont mis en ligne avec sept mois de retard, et ceux de la commission de pharmacovigilance avec un délai de 3-4 mois. S’agissant des sept autres commissions48, seule leur composition figure sur le site de l’AFSSAPS, à l’exception de la commission chargée du contrôle de la publicité, dont le règlement intérieur a été mis en ligne en 2007.
Des améliorations sont toutefois attendues. La mise en ligne du règlement intérieur de la commission nationale de la pharmacopée et de la commission nationale des stupéfiants et psychotropes est projetée pour la fin de l’année 2007. S’agissant les délais de publications des comptes rendus, l’objectif de l’AFSSAPS est de les réduire à 10 semaines à compter de l’été 2007. Ces engagements vont dans le sens des orientations souhaitées par la Cour.
S’agissant de la commission de la transparence, la publication des comptes rendus, des ordres du jour n’est pas encore effective.
d. Les progrès attendus
Au regard de la situation actuelle, les dispositions de la loi du 26 février 2007 constituent un net progrès dans la recherche d’une transparence accrue. Néanmoins, la loi n’impose aucun délai de publication des comptes rendus des réunions des commissions et des RAPPE. Or, si l’on peut comprendre que la mise en ligne de ces documents ne puisse être immédiate, il n’en demeure pas moins que le délai actuel pour les comptes rendus de la commission d’AMM n’est pas satisfaisant. De même, les délais de publications des RAPPE sont parfois excessifs.
D’autre part, la question de la transparence du fonctionnement des groupes de travail dépendant de l’AFSSAPS et travaillant en amont des commissions reste entière. Ces 51 groupes de travail effectuent l’essentiel du travail d’analyse des évaluations réalisées par les experts internes ou externes, les commissions n’examinant au fond que les rares dossiers qui ne font pas l’objet d’un consensus. Or, le site internet de l’AFSSAPS ne mentionne ni l’existence ni la composition de ces groupes d’experts. L’obligation de transmettre une déclaration d’intérêt est insuffisamment respectée dans ces groupes. S’agissant du groupe de travail sur les conditions de prescription et de délivrance (GTCPD), qui joue un rôle crucial pour la taille du marché du futur médicament puisqu’il peut restreindre ou non les conditions de prescription et de délivrance des médicaments, seuls 16 de ses 24 membres figuraient dans le RAPDI 2005, dont seulement 14 avaient fourni une déclaration d’intérêt.
2. Les bases de données sur le médicament
Une base de données médicamenteuse est une banque de données électroniques intégrant les informations produites par les autorités officielles sur les produits de santé, des informations scientifiques produites par les sociétés savantes et toute autre information sur le médicament.
Afin d’être utilisable par les prescripteurs, une base de données sur le médicament devrait présenter plusieurs caractéristiques :
– regrouper l’ensemble des données administratives et médicales des médicaments sur le marché, qui sont actuellement dispersées (AMM, dénomination commune internationale, SMR, ASMR, date éventuelle de commercialisation, taux de remboursement et prix) ;
– permettre la prescription en dénomination commune internationale (DCI) ;
– ![]()
être codifiée, structurée et intégrable dans les logiciels d’aide à la prescription (LAP).
Les bases actuelles (base AMM et bases utilisées par les prescripteurs) ne répondent pas à l’ensemble de ces critères.
a. La base AMM
La LFSS pour 2001 prévoyait dans son article 47 que « d’ici au 1er janvier 2003, l’AFSSAPS mettra en œuvre une banque de données administratives et scientifiques sur les médicaments et les dispositifs médicaux destinée à servir de référence pour l'information des professionnels de santé et des administrations compétentes en matière de produits de santé. Cette base sera rendue accessible au public dans des conditions fixées par décret ». Le décret d’application de cette disposition n’a été pris que le 25 mars 2007.
Le contenu de cette banque de données se limite aux données régulatoires de base : le décret précise en effet que la banque de données comprend les informations contenues dans le répertoire des spécialités pharmaceutiques et dans le répertoire des groupes génériques ainsi que les RCP et les notices des médicaments pour lesquels l’AMM est en cours de validité.
La base AMM, qui contient l’information source, à savoir la totalité des RCP et notices des spécialités titulaires d’une AMM, n’est toujours pas complète : elle ne contient pour l’instant que les résumés caractéristiques des produits des AMM délivrées après le 1er janvier 2002. La reprise du stock des 13 500 AMM antérieures à cette date n’a pu, faute de moyens suffisants, être engagée qu’en 2006. Selon l’AFSSAPS, elle devrait s’achever fin 2008.
b. Les bases utilisées par les médecins
Il existe aujourd'hui trois bases codifiées et structurées utilisées par les médecins. Deux bases sont privées (Vidal et Claude Bernard) tandis que la troisième est publique (Thériaque).
Thériaque
La base Thériaque a été créée il y a près de vingt ans par le centre national hospitalier d’information sur le médicament (CNHIM), association de loi 1901. Depuis janvier 2004, Thériaque est gérée et financée dans le cadre du système d’information sur les produits de santé (SIPS), groupement d’intérêt économique (GIE) associant le CNHIM (40 % des voix), la CNAMTS (40 %), la MSA (10 %) et la CANAM (10 %). Le projet initial consistait à transformer ce GIE en GIP associant l’AFSSAPS mais cette évolution n’a jamais eu lieu.
![]()
Le préambule de la charte constitutive du GIE précise que celui-ci a « pour but de traiter et d’exploiter tous supports d’informations scientifiques, réglementaires et tarifaires sur les produits de santé et d'élaborer en particulier une base de données sur les produits de santé. Cette base constitue la base de référence pour les besoins propres de l’assurance maladie obligatoire. Elle sera mise à disposition de l'ensemble des partenaires de santé et principalement des usagers, des professionnels de santé, des établissements de santé et du secteur médico-social, et des réseaux d’information intervenant dans le domaine de la santé. Les membres du GIE considèrent que cette base de données a pour vocation de contribuer à l’amélioration de la transparence de l’information sur les produits de santé ainsi qu'au développement de l’informatisation du système de soins ». Cette base devait être accessible aux professionnels de santé via Internet et être intégrée dans des logiciels d’aide à la prescription ou de dispensation (cela a bien été le cas, mais uniquement pour les logiciels hospitaliers).
Thériaque est la seule base de données indépendante sur tous les médicaments disponibles en France, en ville et à l'hôpital : génériques, à prescription restreinte, d'exception, agréés aux collectivités ou non, remboursés ou non. Cette base est constituée à partir de plusieurs sources d’informations, la très grande majorité étant des sources officielles et réglementaires (RCP, JO et recommandations AFSSAPS, INCA et HAS), d’autres bibliographiques. Pour tous les médicaments disponibles en France ayant une AMM et une ATU de cohorte, pour certains ayant une ATU nominative, Thériaque présente en détail différentes rubriques : composition et voie d'administration, renseignements administratifs et techniques, génériques, interactions médicamenteuses, contre-indications, précautions d'emploi et mises en garde, posologies et mode d'administration, indications, propriétés pharmacothérapeutiques, effets indésirables, grossesse et allaitement, (in)compatibilités physicochimiques, fiches de transparence de l’AFSSAPS, fiches d'information thérapeutique pour les médicaments d'exception, avis de la commission de transparence (ASMR, SMR) depuis 1995.
Aucune de ces trois bases ne répond totalement aux critères mentionnés ci-dessus, ce qui avait conduit à confier au FOPIM49, créé par la LFSS de 2001, la création d’une base de connaissances publique sur le médicament. Mais la disparition du FOPIM début 2004 et le transfert de ses missions à la HAS semblent s’être accompagnés de la renonciation au projet de base de données publique sur le médicament.
En effet, la HAS a commandé une étude afin d’évaluer les trois principales bases présentes sur le marché (Thériaque, Claude Bernard et Vidal) notamment dans la perspective de la certification des logiciels d’aide à la prescription. Cette étude a été réalisée par Cap Gemini en avril 200650. Trois conclusions principales s’en dégagent :
– en premier lieu, selon cette étude, la création d’une base publique ex nihilo n’est pas souhaitable vu le niveau de qualité des bases existantes.
– en second lieu, les trois bases répondent aux enjeux de la certification des logiciels d’aide à la prescription (cf. infra).
– en troisième lieu, l’étude, constatant que les bases actuelles présentent des lacunes dans le domaine de la prescription en dénomination commune internationale (DCI), préconise de confier à Thériaque la production d’une information publique complémentaire destinée à être utilisée par les bases de données existantes. Cette mission serait double : réalisation d’une nomenclature normalisée en DCI (ie d’un arbre d’équivalence entre le nom de la molécule et les différents noms de marque correspondants) et réalisation de RCP « virtuels » (ie de RCP pour les noms de molécule, qui feraient la synthèse des différents RCP pour les noms de marque).
![]()
![]()
La HAS endosse les deux premières conclusions de l’étude, mais rejette la troisième, portant sur la nécessité d’une production d’information publique en amont des bases de données existantes. La HAS estime en effet que des travaux en cours à l’AFSSAPS, postérieurs à l’étude Cap Gemini, rendent inutile la recommandation du consultant. Ces travaux, qui doivent déboucher en septembre 2007, devraient en effet permettre de fournir une table officielle des correspondances entre médicaments en DCI et spécialités pharmaceutiques. La Cour souligne qu’il ne s’agit toutefois que du premier des deux volets
identifiés par Cap Gemini comme nécessaires à la prescription en DCI : l’arbre de correspondance entre molécule et spécialité. Le second volet (la constitution de RCP virtuels), n’est pas abordé. À cet égard, la Cour ne peut que regretter que l’AFSSAPS n’ait jamais rejoint le GIE SIPS qui héberge Thériaque. Une telle configuration aurait eu tout son sens dans le schéma proposé par Cap Gemini et aurait permis à l’AFSSAPS d’exercer un contrôle sur le travail d’auteur conduit par Thériaque en matière de « RCP virtuel ».
Quelles que soient les motivations et considérations qui peuvent justifier la position de la HAS, elles ne devraient pas remettre en cause le diagnostic posé depuis de longues années sur la nécessité d’une base publique d’information sur le médicament qui semblait d’ailleurs partagé par de nombreux acteurs, au premier rang desquels les médecins. La CNAMTS, dans sa réponse, indique elle aussi être convaincue de l’intérêt d’une base de données sur les produits de santé, qui soit à la fois publique et indépendante. Elle ajoute que cette base de données pourrait être Thériaque puisque tel était l’objet du GIE-SIPS, que devait rejoindre l’AFSSAPS.
Dernier élément qui illustre la nécessité d’une base publique sur le médicament, l’IRDES est en train de constituer sa propre base, faute d’autre base existante, à partir des fichiers de base que Thériaque lui a transmis à titre gratuit. Cette base de données publique associerait, pour chaque spécialité, son code CIP, la DDD (defined daily dosis), le SMR, l’ASMR, la voie d’administration et le prix. Tout l’intérêt de cette base est de pouvoir opérer des tris et des croisements que les bases actuelles ne permettent pas d’effectuer (ex : trier tous les médicaments par niveau d’ASMR, etc.).
Dans ce contexte, Thériaque apparaît supérieure dans des domaines essentiels pour une base de données publique sur le médicament : indépendance à l’égard de l’industrie pharmaceutique51, exhaustivité (Thériaque recense tous les médicaments disponibles en France, AMM ville et hôpital, les ATU de cohorte, les ATU nominatives et les préparations hospitalières de l’AP-HP), contenu (elle est la seule à faire figurer le SMR et l’ASMR du médicament) et gratuité d’accès.
Il faut certes préciser que cette supériorité de Thériaque dans le domaine du travail d’auteur est contrebalancée par une faiblesse de cette base en matière informatique, et notamment de capacité d’interfaçage avec les LAP. Il semble toutefois à la Cour que ces manques peuvent être aisément comblés par une politique d’investissements appropriée, plus aisément sans doute que les lacunes des bases privées en matière d’exhaustivité des sources et de travail d’auteur.
La HAS estime pour sa part que le travail qu’elle conduit actuellement d’élaboration d’une charte de qualité des bases de médicaments permet de mettre sur le même plan les trois bases en ce qui concerne leur exhaustivité. Cette charte devra en effet être signée par les organismes gérant les bases de données sur lesquelles s’appuient les logiciels d’aide à la prescription pour que ces derniers puissent être certifiés. Dès lors, la HAS semble considérer que la garantie offerte par la charte qualité des BDM retire tout intérêt à l’existence d’une base publique, puisque la certification de bases privées permettrait à ces dernières d’atteindre le niveau souhaité.
![]()
Ce raisonnement ne satisfait cependant pas pleinement la Cour pour deux raisons :
– d’une part car les modalités de certification et de contrôle des BDM ne sont pas pas connues à ce stade ;
– et d’autre part, parce que ce raisonnement implique que la gratuité de l’information mise à disposition des prescripteurs, que seule permet une base publique comme Thériaque, ne constitue pas un critère pris en compte par la HAS.
c. L’avenir de Thériaque
Dans le même temps, l’avenir de Thériaque semble menacé puisqu’un confit a éclaté fin 2006 entre les partenaires du GIE.
En effet, fin novembre 2006, l’assurance maladie a indiqué vouloir réduire le financement du GIE (1 M€), améliorer sa gestion financière (recrutement d’opérateurs de saisie chargés de travailler sous la responsabilité de pharmaciens…) et réorienter le contenu de la base afin de satisfaire ses besoins propres. La CNAMTS précise dans sa réponse que la masse salariale représente 75 % du budget de fonctionnement du GIE-SIPS, tandis que les autres dépenses sont structurelles et peu compressibles. Il est donc légitime à ses yeux de rationaliser en priorité ce poste de dépenses (en remplaçant des pharmaciens par des opérateurs de saisie).
Le CNHIM a fait part de son désaccord : « les items seront réduits en conséquence (possiblement des 3/5), les données seront saisies par de simples opérateurs de saisie donc le travail d'auteur et de structuration/codification des données effectué jusqu’alors par des pharmaciens ne pourra donc plus être réalisé, l'intégration dans les logiciels de prescription/dispensation sera de facto remise en cause, la compétence hospitalière dans le développement de la base est totalement déniée par l'assurance maladie et les trois praticiens hospitaliers seront réintégrés dans leur corps d'origine (la directrice actuelle l’étant dès février 2007), enfin, aucune garantie sérieuse ne peut être apportée quant à la fiabilité future de la base Thériaque au travers du refus de test et de validation de la nouvelle méthode d'élaboration et de saisie des données par les représentants de l’assurance maladie. »
Les négociations entre le CNHIM et l’assurance-maladie semblent ne pas avoir permis de trouver un accord sur ces bases avec le CNHIM, qui envisage de quitter le GIP au 3 août 2007. La CNAMTS précise toutefois que ni l’existence de la base, ni son développement, ni la recherche d’une meilleure productivité du GIE ne sont remis en cause. La base du GIE sera rebaptisée (puisque le CNHIM est propriétaire du nom de marque Thériaque) mais restera selon la CNAMTS fidèle à la philosophie de base publique et indépendante.
B. Les déterminants de la prescription de médicaments
A. La formation initiale
![]()
La formation initiale des médecins n’a fait l’objet que d’une investigation réduite de la Cour. En effet, ce sujet aurait nécessité une enquête spécifique, en raison de la technicité
des problématiques qui le sous-tendent. Les développements qui suivent se réfèrent donc à un récent rapport du Sénat52 et à des travaux complémentaires de la Cour.
Le rapport du Sénat de juin 2006 avait souligné le caractère lacunaire de la formation médicale initiale, essentiellement orientée vers l’apprentissage clinique.
D’une part, cette formation ne laisse pas suffisamment de place aux questions thérapeutiques. L’enseignement de la pharmacologie est peu développé : avec un volume horaire d’environ 80 heures, il est devenu le temps d’apprentissage le plus court d’Europe. Des catégories entières de médicaments (sérums, vaccins, désinfectants, antidotes et antiparasitaires) sont passées sous silence et seules trois heures de formation sont consacrées aux antibiotiques. Les futurs praticiens ne sont pas non plus informés de l’inefficacité de certains produits, dont la France détient le record de prescriptions (vasodilatateurs, veinotoniques…). La commission des affaires sociales du Sénat considère en outre que cet enseignement est dispensé trop tôt dans le cursus universitaire, à un moment où les étudiants n’ont pas encore été en contact avec des patients, ce qui le rend très théorique. Une amélioration importante concerne toutefois les futurs médecins généralistes, qui doivent désormais effectuer un stage pratique d’un semestre auprès d’un médecin libéral.
En second lieu, l’enseignement en économie de la santé est largement insuffisant : les futurs médecins ne sont pas informés du coût des thérapeutiques prescrites et des moyens de financement de la solidarité nationale.
Par ailleurs, les étudiants en médecine sont soumis très tôt à l’influence des laboratoires tant dans le cadre de l’hôpital (fiches posologiques largement diffusées, présélection de médicaments par la pharmacie de l’établissement qui connaît souvent des pressions commerciales fortes), que par le rôle des professeurs d’université leaders d’opinion.
![]()
Le ministère chargé de la santé a indiqué vouloir s’impliquer davantage dans le cursus des médecins. Traditionnellement, bien que les textes relatifs aux études médicales soient cosignés par le ministère de l’éducation nationale et le ministère de la santé53, ce dernier a été pratiquement absent des réformes. Bien que membre de droit de la commission pédagogique nationale des études médicales54, chargée de donner son avis sur l'élaboration et la révision des programmes des enseignements des 1er et 2e cycles, la DGS ne participe aux
réunions de cette commission que depuis quelques années. Pour le moment, ses interventions sont toutefois restées limitées. À terme, le ministère chargé de la santé souhaiterait que les problématiques de santé publique soient mieux prises en compte dans l’élaboration des programmes de formation initiale des études médicales.
b. La formation continue et l’évaluation des pratiques professionnelles
L’évolution rapide des techniques et des connaissances médicales nécessite une formation médicale continue et une évaluation des pratiques professionnelles de qualité. Inscrite au code de déontologie depuis le décret du 6 septembre 1995, l’obligation de formation continue des médecins libéraux s’est vue conférer un caractère législatif par les ordonnances de 1996, puis par la loi du 4 mars 2002. La mise en œuvre de ce dispositif n’a véritablement commencé qu’à partir de février 2004, lors de l’installation par le ministre chargé de la Santé des conseils nationaux de formation médicale continue. Par ailleurs, la loi du 9 août 2004 a instauré une obligation d’évaluation de la pratique professionnelle (EPP). Mais le décret du 2 juillet 2006 a reporté l’entrée en vigueur de cette obligation à 2007, année de création des conseils régionaux de la formation médicale continue.
La situation actuelle : une obligation de formation peu respectée, des formations essentiellement financées par les firmes pharmaceutiques
Les données sur les pratiques et les financements de la formation médicale continue sont très lacunaires. D’après l’IGAS55, seul un médecin libéral sur cinq participe à des actions organisées par des organismes de formation.
L’essentiel de la formation médicale continue (entre 300 et 600 M€ par an selon l’IGAS) est financé par les firmes pharmaceutiques. Les actions de formation financées par l’industrie sont en principe soumises à l’ordre des médecins qui donne son avis sur leur compatibilité avec l’art. L. 4113-6 du Code de la santé publique (dit loi « anti-cadeaux »), mais il existe beaucoup de sous-déclarations.
Les financements institutionnels publics et privés sont très modestes :
– les financements conventionnels (CNAMTS) s’élevaient à 70 M€ en 2005, dont la moitié a servi à indemniser les médecins. La gestion de la FMC mise en place par la CNAMTS s’inspire des principes paritaires, avec un comité paritaire conventionnel (CPC) rassemblant les représentants des syndicats signataires de la convention et les représentants de la CNAMTS ;
– le fonds d’assurance formation des professions médicales (FAF PM), créé dans le cadre de la formation professionnelle de droit commun, est dirigé par un conseil d’administration où siègent uniquement les organisations syndicales de médecins. Ses dépenses étaient de 5 M€ en 2005.
![]()
Les dépenses de formation assumées directement par la profession (dépenses d’abonnement à des revues, acquisitions de supports pédagogiques et cotisations à des associations de formation) ne sont pas connues. Il est à noter que depuis 2005, les dépenses
de formation ouvrent droit à un crédit d’impôt dans la limite de 320 € par an et par médecin
La nouvelle organisation : un dispositif complexe et redondant
En dépit de leur caractère complémentaire, la formation médicale continue et l’évaluation des pratiques professionnelles sont régies par deux organisations distinctes.
La loi du 4 mars 2002 a confié la gestion de la FMC à la profession par l’intermédiaire de trois conseils nationaux de la formation professionnelle continue (CNFMC) compétents respectivement pour les médecins libéraux, les médecins hospitaliers, et les autres médecins salariés. Ces conseils, chargés d’agréer les organismes de formation et d’établir des barèmes nationaux pour s’assurer du respect de l’obligation de FMC, ont été installés en février 2004. Les conseils régionaux de la formation médicale continue (CRFMC), qui doivent être installés en 2007, sont chargés de vérifier le respect de l’obligation individuelle.
De son côté, l’évaluation des pratiques professionnelles est partagée entre la Haute autorité de santé et la profession. La HAS est chargée de définir les méthodes d’évaluation des pratiques professionnelles. Pour les médecins libéraux, les prestataires d’EPP peuvent être soit des médecins habilités par les URML et formés par la HAS, soit des organismes agréés par la HAS.
L’éclatement de la gestion de l’EPP et de la FMC est source de complexité. Les organismes de formation, qui sont souvent en même temps compétents en matière d’évaluation des pratiques professionnelles, doivent ainsi obtenir deux agréments (HAS, CNFMC), remplir deux dossiers de demandes d’agrément différents reposant sur deux cahiers des charges distincts. Afin d’assurer un minimum de cohérence entre FMC et EPP, le barème de la FMC prévoit que sur un ensemble de 250 crédits nécessaires pour satisfaire à l’obligation de formation continue, un forfait de 100 crédits est attribué à chaque médecin ayant satisfait à l’obligation d’évaluation dans les conditions définies par la HAS. Par ailleurs, le décret du 14 avril 2005 prévoit que le CNFMC donne un avis à la HAS pour l’agrément des organismes d’EPP. Mais la réciproque n’est pas vraie : les textes ne prévoient pas que la HAS donne son avis sur l’agrément des organismes de FMC.
La complexité du dispositif est accrue par le fait qu’il existe 3 CNFMC pour les médecins. Un comité de coordination de la formation médicale continue, comprenant 12 représentants des 3 CNFMC, 3 représentants du ministère de la santé et 1 représentant du ministère de l’enseignement supérieur, a dû être créé (art. R. 4133-12 à 14) pour tenter d’harmoniser le fonctionnement et les procédures des trois CNFMC56. Comme l’a souhaité le ministère chargé de la santé, un barème unique de FMC a été élaboré par les trois conseils nationaux de FMC, ce qui constitue un progrès notable. Toutefois, l’existence d’un barème unique et d’un cahier des charges unique pour l’agrément des organismes de formation souligne la redondance des structures mises en place.
![]()
![]()
Cette organisation complexe génère également des surcoûts (locaux, site internet spécifique à la FMC…). Les CNFMC, qui n’ont pas de personnalité juridique, ont obtenu
des moyens de fonctionnement dans le cadre d’une convention passée entre le ministère de la santé et le conseil national de l’ordre des médecins (art. L. 4133-5 du CSP). Le budget 2007 du ministère consacré à la FMC est de 4,465 M€ pour toutes les professions. Comme les textes permettant la mise en place de la FMC ne concernent pour le moment que trois professions (médecins, pharmaciens et dentistes), cette somme est répartie selon la clé de répartition suivante : 60 % pour les médecins (2,679 M€), 24 % pour les pharmaciens et 16 % pour les dentistes. Cette dotation devrait également permettre de financer les CRFMC.
L’IGAS, en janvier 2006, avait recommandé de modifier la loi afin d’adosser les CNFMC à la HAS. La Cour constate que cette mesure aurait permis de réaliser des économies et des synergies (guichet unique, mutualisation des ressources, site Internet unique) et d’harmoniser les doctrines en matière d’EPP et de FMC.
Une organisation qui ne permet pas d’éviter les conflits d’intérêts.
Aucun financement public nouveau n’étant consacré à la FMC, celle-ci continue à être essentiellement financée par les firmes pharmaceutiques. Initialement, il avait été envisagé d’interdire de délivrer un agrément aux organismes de formation financés à plus de 40 % par l’industrie pharmaceutique. Ce projet a été abandonné au motif de l’importance des financements de l’industrie, faute desquels la plupart des organismes de formation se trouveraient en grave difficulté financière, et de la possibilité de contrôler la qualité des formations via les procédures d’agrément. Le code de bonnes pratiques passé entre le LEEM et les CNFMC et signé par le ministre de la santé le 22 novembre 2006 a reconnu la possibilité du financement des organismes de formation par les firmes pharmaceutiques. La qualité et l’indépendance des formations sont censées être garanties par les nouvelles procédures d’agrément. Or, ces procédures ne permettent pas d’éviter les conflits d’intérêt.
Le code de bonnes pratiques passé entre le LEEM et les CNFMC est supposé encadrer les pratiques des entreprises dans le domaine de la FMC (qualité scientifique, transparence de financements, évaluation de la formation par les participants). Mais les engagements sont peu précis et le code est dépourvu de tout caractère contraignant57.
![]()
Le décret du 2 juin 2006 a précisé que l’agrément des organismes de formation est accordé sur la base d’un cahier des charges prenant en compte la qualité scientifique et pédagogique des programmes proposés, la conformité aux référentiels et aux bonnes pratiques de la profession dans tous les thèmes abordés, la transparence des financements ; l’engagement relatif à l'absence de toute promotion en faveur d'un produit de santé et à l'utilisation de la dénomination commune des médicaments, le respect des orientations nationales définies par le conseil national, l’acceptation du principe d'une évaluation externe du fonctionnement de l'organisme de formation et de la qualité des formations. Mais le cahier des charges figurant dans le dossier de demande d’agrément se limite à énoncer les informations devant être communiquées par les organismes de formation (déclaration d’intention, description de l’organisme, modalités de recrutement des experts et de gestion des conflits d’intérêt, description de la gestion financière…) sans préciser dans quelle mesure ces éléments sont susceptibles ou non de conduire à un refus de l’agrément.
Il serait donc indispensable de préciser le cahier des charges servant de base à l’agrément des organismes de formation afin d’en faire un véritable référentiel de qualité. Ainsi que le recommandait l’IGAS en 2006, le respect de ce référentiel pourrait être confié à des organismes agréés par la HAS. Des sanctions doivent être prévues en cas de non-respect de ce référentiel (requalification des dépenses de formation en dépense de promotion entrant dans l’assiette de la taxe et retrait de l’agrément).
La composition même des CNFMC est porteuse de risques pour l’indépendance des décisions d’agrément des organismes de formation. Les CNFMC, composés de façon quadripartite (organisations syndicales représentatives, organismes de formation, UFR, représentants de l’ordre), peuvent être tentés de délivrer facilement un agrément aux organismes de formation représentés en leur sein ou aux organismes de formation créés par les organisations syndicales. C’est particulièrement le cas du CNFMC des médecins libéraux dont plus de la moitié des membres sont des représentants des syndicats et des organismes de formation (art. R. 4133-7 du CSP). L’article R. 4133-10 du CSP prévoit que les membres des conseils nationaux doivent rédiger une déclaration d'intérêt selon un modèle établi par arrêté du ministre chargé de la santé et s’engagent à déclarer tout changement de leur situation. Cet arrêté n’a pas été pris et aucune déclaration d’intérêt n’a été réalisée. Au-delà, les procédures supposées éviter les conflits d’intérêt ne sont pas suffisantes. En effet, la publication des déclarations d'intérêt n’est pas obligatoire. L’article R. 4133-10 se contente d’indiquer que ces déclarations d’intérêt sont mises à la disposition du ministre chargé de la santé sur sa demande. L’IGAS, dans son rapport de janvier 2006, avait indiqué que la déclaration des conflits d’intérêt potentiels n’était pas suffisante et recommandait de confier l’instruction des demandes d’agrément auprès des CNFMC à des rapporteurs indépendants choisis sur une liste fixée par le ministre de la santé après avis des CNFMC. L’instruction par des rapporteurs indépendants n’a finalement été retenue que pour une durée de cinq mois. Selon la DGS, la direction du budget se serait opposée à un dispositif pérenne d’instruction par des rapporteurs indépendants en raison du coût de cette mesure.
S’agissant du barème à satisfaire pour respecter l’obligation de formation, l’IGAS avait recommandé d’exclure les études de phase IV (considérées comme des supports de promotion des firmes pharmaceutiques) des actions de recherche susceptibles d’être valorisées pour la FMC et l’EPP. Le barème défini par l’arrêté du 13 juillet 2006 a pris en compte cette recommandation. L’IGAS avait également recommandé de renforcer la sélectivité des critères de labellisation des revues médicales dont l’abonnement donne lieu à attribution de crédits de FMC, de manière à éviter que l’abonnement à des revues financées par des entreprises pharmaceutiques soit pris en compte au titre de la formation continue. Cette recommandation n’a été que partiellement suivie. Le barème prévoit en effet de limiter à 10 crédits sur 5 ans (sur un total de 250 requis) le nombre de crédits résultant d’un abonnement à un périodique médical ou à un ouvrage médical, cette valeur étant portée à 20 crédits sur cinq ans pour un abonnement à un périodique de formation répondant à des critères de qualité définis conjointement par les CNFMC.
![]()
En ce qui concerne l’EPP, le dispositif d’agrément, confié à la HAS, présente davantage de garanties. Toutefois, le cahier des charges utilisé pour l’agrément n’est pas plus précis que celui utilisé pour l’agrément des organismes de FMC. Il est actuellement en cours de refonte, ainsi que la procédure d’agrément. Surtout, les URML, qui sont chargées d’informer les médecins sur l’EPP et les différentes possibilités d’évaluation qui leur sont offertes, peuvent avoir intérêt à privilégier le recours à un médecin habilité ou le recours à un
organisme de formation se situant dans leur mouvance syndicale. S’agissant des risques de conflits d’intérêt avec l’industrie pharmaceutique, la HAS indique que certains organismes se sont vus refuser ou suspendre leur agrément pour ces motifs et qu’elle souhaite accompagner les démarches permettant de financer les actions d’EPP dans un cadre permettant de garantir leur indépendance par l’absence de financement provenant des industries de santé.
Les modalités de pilotage
Ni le ministère ni l’assurance maladie n’ont véritablement les moyens de définir des priorités en matière de FMC et d’EPP, ce qui empêche notamment d’en faire un moyen d’action sur les prescriptions.
En ce qui concerne la FMC, la DGS ne dispose que d’une voix consultative au sein des CNFMC. L’assurance maladie n’est pas représentée au sein des conseils. Les CNFMC doivent définir pour cinq ans des thèmes prioritaires de formation prenant en compte les objectifs de la politique de santé publique, les plans d'action de santé publique ainsi que les programmes de santé. Toutefois, ces thèmes, bien qu’ils fassent l’objet d'une bonification de 20 % de crédits de formation, ne sont pas obligatoires et les CNFMC n’auront pas les moyens de peser véritablement sur les choix de FMC, qui appartiendront en premier lieu aux financeurs. Sur les cinq priorités définies en 2006 pour les cinq prochaines années, et qui ont été élaborées en concertation avec le directeur général de la santé, une seule concerne le médicament (la iatrogénie)58.
En ce qui concerne l’EPP, ni la loi du 13 août 2004 ni le décret du 14 avril 2005 ne prévoient la possibilité pour la HAS de définir des thèmes prioritaires. La CNAMTS, qui participe au financement du dispositif via le FAQSV, n’est pas davantage fondée à demander un ciblage des crédits.
L’absence de sanction en cas de non-respect de l’obligation de formation médicale continue
Aux termes de l’article R. 4133-16 du CSP, le praticien dépose tous les cinq ans auprès du conseil régional de la formation médicale continue son dossier regroupant les justificatifs des formations suivies. Le conseil régional vérifie, au vu du dossier, le respect de l'obligation de formation continue dans les conditions prévues à l'article L. 4133-1 du code de la santé publique. Le conseil régional de la formation médicale continue valide le respect de l'obligation de formation continue en délivrant au praticien une attestation et en informe le conseil régional de l'ordre dont dépend le praticien au titre de son activité principale.
![]()
Un projet de décret modificatif allège ce dispositif, puisqu’il prévoit que le praticien transmet au CRFMC une déclaration attestant qu’il a satisfait à l’obligation de formation continue, le conseil régional ayant la possibilité de lui demander des justificatifs. Le contrôle se ferait de manière aléatoire, selon des modalités qui ne sont pas précisées.
Par ailleurs, aucune sanction n’est prévue en cas de non-respect de l’obligation de FMC. L’article R. 4133-1659 se contente de disposer : « Si, au terme de ces cinq ans, le praticien n'a pas envoyé son dossier au conseil régional de la formation médicale continue, celui-ci le met en demeure de produire tous justificatifs. En cas d'absence de production des justificatifs demandés dans un délai de six mois, le conseil régional de la formation médicale continue en informe le conseil régional de l'ordre dont dépend le praticien au titre de son activité principale. Lorsqu'au vu du dossier présenté, le conseil régional de la formation médicale continue estime que le praticien n'a pas respecté son obligation de formation continue, il arrête, de concert avec ce dernier, un plan permettant de compenser le retard pris dans le suivi des formations éligibles à la formation médicale continue. En cas de refus du praticien de s'engager à mettre ce plan en œuvre, le conseil régional de la formation médicale continue en informe le conseil régional de l'ordre dont dépend le praticien au titre de son activité principale. »
L’accès aux données de FMC
Un portail de la FMC est en cours de réalisation, qui permettra notamment aux médecins de transmettre par voie électronique leur déclaration attestant du respect de l’obligation de formation continue aux CRFMC. Un prestataire a été sélectionné et un groupe de pilotage réunissant des représentants du ministère chargé de la santé, des CNFMC et des ordres concernés (médecins, pharmaciens et chirurgiens-dentistes) se réunit régulièrement. Le coût total du projet est estimé à 1,2 M€. Les travaux en cours font apparaître deux difficultés.
D’une part, la HAS n’a pas été associée aux travaux sur le portail, qui n’intègre pas l’EPP. Les informations relatives à l’EPP sont mises en ligne sur le site de la HAS, et il n’existe pas de complémentarité entre les deux sites.
D’autre part, les modalités d’accès de la DREES aux données du portail en vue d’une exploitation statistique des dossiers individuels de formation des médecins n’ont pas été précisées. La DREES n’a pas été associée aux réunions du comité de pilotage. Or, la DREES devrait participer aux travaux sur le portail car il est essentiel qu’elle ait accès à ces données.
La Cour avait recommandé dans son rapport sur la sécurité sociale de 2004 de définir une stratégie cohérente de communication publique sur le médicament. Malgré des améliorations certaines depuis 2004, cette recommandation n’a pas été réellement mise en œuvre.
![]()
Il en résulte que la principale source d’information des médecins sur le médicament reste la visite médicale.
a. La visite médicale et son encadrement
L’influence de la visite médicale sur les prescriptions
La visite médicale constitue le premier moyen d’information des médecins sur le médicament. C’est également le premier outil de promotion pour l’industrie pharmaceutique, qui y consacre, selon le rapport du Sénat précité, 80 % de ses dépenses de marketing, soit l’équivalent de 8 500 € par médecin. On compte environ 24 000 visiteurs médicaux, ce qui place la France dans la moyenne européenne pour le rapport entre le nombre de visiteurs médicaux et le nombre de praticiens.
Toutefois, en France, la visite médicale influence considérablement les habitudes de prescription. Les médecins français ont ainsi la réputation d’être très sensibles à l’arrivée de nouvelles spécialités sur le marché pharmaceutique et de les intégrer très rapidement à leur pratique.
L’IRDES a tenté d’analyser les facteurs qui influencent la prescription de médicaments innovants. L’étude a été réalisée en comparant les prescriptions des médecins (collectées par l’enquête EPPM d’IMS Health) sur la période 1992-1998 à l’investissement promotionnel global des firmes ainsi qu’à l’investissement pour la visite médicale. Deux classes de médicaments ont été retenues : les macrolides (une catégorie d’antibiotiques) et les anti-dépresseurs. Les résultats de l’étude ont montré que, quel que soit le médicament étudié, le nombre de lignes prescrites par trimestre a été fortement corrélé à l’investissement promotionnel des firmes. Ainsi selon l’IRDES, à chaque pic marquant une action promotionnelle, correspond un pic du nombre de prescriptions des médecins.
C’est pourquoi différentes actions ont été entreprises récemment pour tenter d’encadrer la visite médicale (nouvelle mission incombant à la HAS) ou d’en contrebalancer les effets (création des délégués de l’assurance-maladie, cf. plus loin).
L’encadrement de la visite médicale
Afin d’encadrer la visite médicale, la loi du 13 août 2004 a confié à la HAS une mission de certification de la visite médicale des firmes60, afin d’en garantir la conformité à la charte de la visite médicale signée le 22 décembre 2004 entre le LEEM et le CEPS. La HAS a élaboré son référentiel de certification à partir du contenu de la charte et la certification est opérationnelle depuis novembre 2006.
Selon le préambule de la charte, la visite médicale a « pour objectif principal d’assurer la promotion des médicaments auprès du corps médical et de contribuer au développement des entreprises du médicament. Elle doit à cette occasion favoriser la qualité du traitement médical dans le souci d’éviter le mésusage du médicament, de ne pas occasionner de dépenses inutiles et de participer à l’information des médecins » : il s’agit là d’une formulation quelque peu contradictoire et qui ne laisse guère de doute sur le fait que la priorité est donnée à la première proposition.
![]()
Les objectifs de la charte manquent de précision : la charte prévoit ainsi que « l’entreprise privilégie le contenu de la visite médicale par rapport à la fréquence des visites afin que l’information délivrée soit la plus complète et objective possible et qu’en particulier le temps nécessaire à l’information du prescripteur sur le bon usage du médicament soit
disponible ». Ces indications sont particulièrement floues, sans parler de l’absence de possibilités de vérification par les organismes certificateurs. De la même façon, la charte prévoit que « l’entreprise veille à ce que l’activité de visite, tous réseaux confondus, relative à une même spécialité, ne revête pas un caractère abusif », sans préciser la définition de la notion d’abus ni la manière de le mesurer et les sanctions encourues.
Un avenant du 21 juillet 2005 prévoit toutefois que le CEPS arrête chaque année la liste des classes thérapeutiques pour lesquelles il estime qu’une réduction de la visite médicale est nécessaire, en fonction de considérations relatives au bon usage du médicament ou aux dépenses de l’assurance-maladie. En cas de non-respect du taux d’évolution du nombre de visites décidé par le CEPS, ce dernier peut procéder à une baisse temporaire ou définitive du prix des spécialités concernées. Il s’agit là de l’aspect le plus contraignant de la charte mais tout dépendra de l’application qui en sera faite61.
S’il est trop tôt pour apprécier les premiers résultats obtenus, on peut toutefois porter un jugement sur le principe même de la charte. Si ce dispositif constitue la première tentative d’encadrement de la visite médicale, sa principale limite réside dans les possibilités de vérification des principaux engagements pris par les firmes, qu’il s’agisse de la fréquence des visites, de la publicité comparative, du contenu de la présentation orale, des documents distribués au médecin, etc. Une note de la HAS indique d’ailleurs que la certification de la visite médicale n’est pas « un contrôle de l’activité des délégués médicaux dans le cabinet du médecin », ni « une validation des documents promotionnels », ni « un suivi quantitatif de la visite médicale » mais « une incitation à l’amélioration continue de la qualité fondée sur l’engagement des acteurs de l’entreprise » et « une certification évolutive grâce aux enseignements issus de la mise en œuvre ».
Enfin, on peut craindre que la certification par la HAS donne une caution à la visite médicale dont celle-ci ne disposait pas jusqu’à présent.
b. L’information publique destinée aux médecins
Comparée à l’information dispensée par l’industrie pharmaceutique, l’information publique apparaît souvent dispersée, sous-utilisée et peu adaptée aux pratiques médicales.
Les productions scientifiques de la HAS
![]()
La loi du 13 août 2004 assigne un rôle primordial à la HAS en matière d’information médicale. Son portefeuille comporte à ce jour 22 types de productions scientifiques. Ce chiffre apparaît excessif, surtout si l’on ajoute les produits des autres institutions. Il serait souhaitable à tout le moins de stabiliser le vocabulaire utilisé par la HAS, afin de favoriser une bonne appropriation des différents documents par les médecins. En quelques années, se sont en effet succédé les fiches de transparence, les fiches de stratégie thérapeutique, les
fiches-produit, les fiches de bon usage du médicament, les fiches Transpa-Flash, sans que les spécificités de chacune soient forcément évidentes pour les lecteurs.
Parmi les innovations intéressantes de la HAS, figurent les fiches de bon usage du médicament, consacrées à un médicament susceptible de faire l’objet d’un mauvais usage. À ce jour, cinq fiches de bon usage ont été mises en ligne62 ; il est prévu la publication d’une dizaine de fiches par an. Différentes actions de diffusion de ces fiches ont été réalisées (mise en ligne des fiches téléchargeables, envoi des fiches par courrier aux médecins généralistes, aux spécialistes concernés, et aux pharmaciens d’officine, organisation de petits-déjeuners avec la presse professionnelle, etc.) et des études d'impact de ces actions de diffusion sont en cours.
Par ailleurs, la HAS a décidé de reprendre la publication des fiches de transparence anciennement réalisées par l’AFSSAPS. Ces fiches, consacrées à une classe thérapeutique, soulignent les équivalences thérapeutiques entre produits d’une même classe. On peut regretter qu’une seule fiche soit disponible à ce jour.
La HAS a concentré ses efforts en 2005 sur la simplification des messages et l’amélioration de leur lisibilité (charte graphique). En 2006, les efforts ont porté sur la perception de ces documents à tous les niveaux : études pré-publication pour ajuster la communication, pré-tests de compréhension de nouveaux documents avant diffusion, post-tests d’évaluation de la perception des documents diffusés. L’ensemble de ces efforts, ainsi que leur caractère novateur, doivent être salués.
Les recommandations professionnelles de la HAS et de l’AFSSAPS
La HAS et l’AFSSAPS produisent toutes les deux des recommandations professionnelles.
Les recommandations de bonne pratique (RBP) de l’AFSSAPS jouent d’ailleurs un rôle croissant dans le contexte d’européanisation de l’AMM. En effet, le libellé des AMM européennes par reconnaissance mutuelle, de plus en plus fréquentes, est relativement général, pour fournir un dénominateur commun entre les différents pays. Il renvoie souvent à des recommandations nationales le soin de compléter l’AMM.
L’élaboration des RBP de l’AFSSAPS est longue (un an en moyenne, parfois trois ans) et la méthodologie peu adaptée aux exigences d’une communication réactive. Depuis 2002, l’AFSSAPS a mis en place des « mises au point », dont l’élaboration plus souple et plus rapide permet de communiquer plus facilement sur des sujets d’actualité (éléments d’information sur la sécurité d’emploi des médicaments, compléments sur la stratégie thérapeutique).
![]()
![]()
En l’état, les recommandations de bonne pratique de l’AFSSAPS souffrent d’un certain nombre de défauts. Le contenu des RBP est d’un accès parfois difficile et l’AFSSAPS n’a toujours pas engagé, contrairement à ce qu’elle avait affirmé à la Cour en 2006, d’enquête de satisfaction et d’étude d’impact auprès des prescripteurs concernant la qualité de ses productions et notamment des RBP. Par ailleurs, l’arrivée d’une nouvelle classe thérapeutique ne conduit pas nécessairement à l’élaboration d’une recommandation de bon usage d’un médicament : l’agence estime en effet qu’un temps suffisant d’utilisation du
produit est nécessaire pour recueillir toutes les données issues de la littérature et de la pratique. Or, c’est précisément pour les nouvelles classes de médicaments que les RBP seraient très utiles, afin d’aider les prescripteurs et les patients à bien employer ces nouvelles molécules, dont tous les effets ne sont pas encore connus. L’agence indique toutefois dans sa réponse utiliser plus volontiers le support des mises au point lorsqu’une nouvelle classe ou spécialité thérapeutique apparaît sur le marché et qu’il y a lieu de la positionner au sein d’une stratégie thérapeutique avec des messages clairement identifiés sur le risque63. S’agissant des critères de choix des thèmes des RBP, des progrès ont été enregistrés dans les années récentes : l’agence axe ses RBP sur les gammes thérapeutiques faisant l’objet d’un nombre important de prescriptions (statines, antibiotiques, antidépresseurs, antidiabétiques, antisécrétoires gastriques, traitement de l’ostéoporose).
Tableau n° 5 : RBP et mises au point publiées par l’AFSSAPS en 2005 et 2006
2005 |
2006 | |
RBP |
Prise en charge thérapeutique du patient dyslipidémique (actualisation) |
Traitements médicamenteux de l’ostéoporose post-ménoposique (actualisation) |
Traitement de l’anémie au cours de l’insuffisance rénale chronique de l’adulte |
Bon usage des médicaments antidépresseurs dans le traitement des troubles dépressifs et des troubles anxieux de l’adulte | |
Les traitements médicamenteux de l’endométriose génitale |
Traitements médicamenteux du diabète de type 2 | |
Mises au point |
Prise en charge de la fièvre chez l’enfant |
Antidépresseurs chez l’enfant |
Anti-dépresseurs chez l’adulte |
Bon usage de la desmopressine | |
THS (actualisation) |
Bon usage de Macugen | |
Sécurité d’emploi des coxibs |
TRALI (Transfusion Related Lung Injury) | |
Iatrogénèse chez le sujet âgé |
||
Prévention et prise en charge des tuberculoses survenant sous anti-TNFs |
||
Prise en charge thérapeutique de l’éradication de Helicobacter pylori chez l’adulte et l’enfant |
||
Traitement antibiotique probabiliste des urétrites et cervicites non compliquées |
||
Médicament et conduite automobile |
Les recommandations professionnelles élaborées par la HAS (RPC, conférences de consensus, consensus formalisé, audition publique) témoignent d’un plus grand souci d’accessibilité, avec notamment l’existence d’une synthèse.
![]()
La ligne de partage entre la HAS et l’AFSSAPS n’est toutefois pas toujours clairement définie. On peut regretter qu’il n’existe pas de portail Internet commun pour les recommandations professionnelles des deux agences.
Les outils de l’assurance-maladie
Enfin, l’assurance-maladie déploie de réels efforts pour diffuser aux professionnels de santé une information synthétique et facilement utilisable, sous différentes formes (lettre aux médecins, lettre aux pharmaciens, supports mémos), qui lui permet de mettre l’accent sur les axes de la maîtrise médicalisée. Ces outils, qui participent de l’information publique destinée aux médecins, seront analysés en détail plus loin.
c. Les outils d’aide à la prescription
La deuxième mission impartie à la HAS par la loi du 13 août 2004 est la certification des logiciels d’aide à la prescription (LAP), dont l’objectif est d’améliorer la sécurité et la qualité de la prescription, de faciliter le travail du prescripteur et de diminuer le coût du traitement à quantité égale.
Le cadre juridique
L’article L. 161-38 du CSS prévoit ainsi que « la HAS est chargée d’établir une procédure de certification des sites informatiques dédiés à la santé et des logiciels d’aide à la prescription médicale ayant respecté un ensemble de règles de bonnes pratiques. À compter du 1er janvier 2006, cette certification est mise en œuvre et délivrée par un organisme accrédité attestant du respect des règles de bonne pratique édictées par la HAS ». La certification concernera dans un premier temps les logiciels de ville. Une seconde version sera ensuite élaborée pour les LAP hospitaliers.
Le contenu
Le référentiel de certification est élaboré, il est actuellement en cours de relecture et fait l’objet d’un test de faisabilité. Les critères du référentiel sont organisés selon les chapitres suivants : information sur le médicament, information sur le patient (identification, antécédents et pathologies, histoire médicamenteuse), affichage des médicaments, rédaction de la prescription, alertes et messages d’information du prescripteur, finalisation de l’ordonnance, traçabilité, retour du médecin sur sa pratique de prescription, sécurité des données et confidentialité
Parmi les critères de certification figure la possibilité de prescrire en dénomination commune internationale (DCI), malgré l’opposition initiale des éditeurs de logiciels64. L’intérêt de la prescription en DCI est double : augmenter la sécurité des patients en diminuant le risque d’absorber deux fois le même médicament ayant deux noms de marque différents et accroître la substitution par les pharmaciens.
Le mode de certification est en cours de définition. Les organismes certificateurs des LAP seront accrédités par le comité français d’accréditation (COFRAC). La certification ne devrait pas démarrer avant la fin 2007.
Les difficultés
![]()
![]()
Comme précédemment, cette seconde mission de la HAS a le mérite de tenter d’encadrer des pratiques préexistantes. Toutefois, quelques difficultés peuvent d’ores et déjà
être soulignées. Ainsi, si la plupart des critères du référentiel doivent bien faire l’objet de « contrôles » de la part de l’organisme certificateur, certains, pourtant importants, ne font l’objet que d’« engagements » des éditeurs de LAP, ce qui affaiblit la portée réelle de la certification65. Le critère n° 9 du référentiel soulève pour sa part une difficulté spécifique. Il prévoit en effet que « le LAP met toujours à disposition l’information sur les médicaments qui provient des bases de données signataires de la charte de qualité. Cette information est différenciée de celle ayant une autre origine ». Cette formulation laisse entendre que l’information contenue par des logiciels certifiés peut provenir d’une autre origine que des bases de données signataires de la charte de qualité, ce qui affaiblit considérablement le degré de fiabilité du logiciel. Toutefois, la mise à disposition d’une information ayant une autre source que les bases de données signataires est sans doute inévitable66, même si la HAS reconnaît elle-même les risques qui en résultent en termes de neutralité.
Par ailleurs, une difficulté consiste dans la base de données à laquelle est adossé le LAP. En effet, la constitution des LAP met en jeu les performances de la base médicaments utilisée : exhaustivité de l’information, complétude, neutralité, sécurité. Si l’on souhaite que les LAP mettent à disposition des informations sur le SMR et l’ASMR d’un médicament, cela impose que soit enfin achevée la base de données médicaments offrant ces informations (cf. supra). Dès lors, la HAS considère que le préalable à la certification des LAP est l’adhésion de la base de données qu’ils utilisent à une « charte de qualité » qu’elle a elle-même définie67. Le rapport Cap Gemini précité indique que l’état actuel des bases de données médicaments « ne fait pas obstacle à la certification des LAP, sauf sur quelques points précis bien identifiés et qui peuvent être résolus68 ».
3. Les actions de l’assurance-maladie pour maîtriser les prescriptions
a. L’analyse des prescriptions par l’assurance-maladie
Il s’agit de la phase préalable, qui permet de déterminer où se situent les risques de dérives, aussi bien en termes de bon usage du médicament que de coûts.
Les limites du système d’information de la CNAMTS
Le mauvais usage du médicament peut être de nature quantitative (dans le cas d’une prescription excessive) ou qualitative (dans le cas d’une prescription non conforme aux référentiels ou à l’AMM).
![]()
Sur le plan purement quantitatif, la CNAMTS peut procéder à des contrôles automatiques en identifiant des volumes anormaux prescrits par un seul médecin ou
consommés par un seul assuré. Ainsi, la CNAMTS indique que les contrôles de « méga-consommants » ont permis de repérer 250 professionnels de santé ayant des pratiques douteuses.
Sur le plan qualitatif, le système d’information de la CNAMTS ne permet que des contrôles limités, puisque la CNAMTS n’a accès qu’à l’historique des remboursements des prescriptions d’un praticien, mais pas à l’indication ou à la pathologie. Elle ne peut donc rapprocher automatiquement la prescription de la pathologie (ce qui lui permet d’identifier des cas de mauvais usage du médicament) que lorsque la prescription est spécifique de la pathologie ou bien, depuis peu, en matière d’affections de longue durée (ALD).
En effet, l’avis CNIL du 14 juin 2005 autorise la CNAMTS à exploiter les données anonymisées du fichier des ALD. L’appariement des bases du service médical et de celles comportant les données de remboursement a été mis en œuvre dès le deuxième semestre 2005 et a d’ores et déjà produit des résultats. Outre des études conduites sur des patients diabétiques ou des pathologies cardio-vasculaires, la CNAMTS a notamment mis en place des outils de contrôle des prescriptions en rapprochant les consommations de soins des pathologies des patients en ALD, en particulier pour des prestations (médicaments, actes de kinésithérapie, LPP) sans rapport manifeste avec une ALD et pourtant prescrits dans la partie haute de l’ordonnancier bizone.
La CNAMTS indique toutefois que la généralisation de ce dispositif aux autres pathologies que les ALD n’est pas envisagée actuellement, estimant que les comparaisons internationales qu’elle a menées ne lui permettent pas d’établir le bénéfice d’une telle généralisation : aucun des dix pays européens étudiés n’aurait mis en place un tel dispositif de codage systématique.
Dans les cas autres que les ALD, la CNAMTS recourt à des enquêtes manuelles.
Le dispositif mis en œuvre par la MSA
La MSA expérimente pour sa part depuis septembre 2004 un système d’information médicalisée, sous la forme du logiciel ARCHIMED (analyse, recherche, informations médicalisées), généralisé à l’ensemble des services du contrôle médical en janvier 2005.
Ce logiciel, qui permet précisément de croiser les données médicales et administratives, intègre des fonctions avancées d’analyse sur la pratique des professionnels de santé et sur la consommation des assurés. Il intègre également une fonction « observatoire du médicament » et un module « gestion du risque », facilitant l’imputation sur le bon risque.
ARCHIMED permet au médecin conseil d’analyser les ordonnances au regard des pathologies et ainsi de repérer des anomalies dans la prescription. Lorsqu’une anomalie est repérée, un courrier est envoyé au médecin, afin de l’en informer. Si ce dernier le souhaite, un entretien confraternel peut avoir lieu. Cette dernière possibilité est rarement mise en œuvre, mais, selon la MSA, l’envoi du courrier suffit généralement à modifier les habitudes de prescription des médecins. Pour la MSA, la clé de la réussite de ce dispositif est d’offrir aux médecins une évaluation de leur pratique, peu fréquente en libéral, notamment en milieu rural.
![]()
Dans le cadre du plan de maîtrise médicalisée prévue par la convention du 12 janvier 2005, le logiciel ARCHIMED a été enrichi de modules spécifiques automatisés concernant le plan médicament (polymédication des personnes âgées, associations médicamenteuses formellement contre-indiquées, prescription des benzodiazépines après 70 ans, prescription
des statines après 80 ans, génériques) et le plan ALD (respect de l’ordonnancier bizone et des référentiels).
b. Les engagements de maîtrise médicalisée
La maîtrise médicalisée a pour objet d’inciter les médecins à réduire leurs prescriptions sur certains postes jugés prioritaires.
Les objectifs
Dans le cadre de la convention médicale du 12 janvier 2005, les médecins libéraux et l’assurance-maladie se sont ainsi engagés pour 2005 sur des objectifs de maîtrise médicalisée dans 5 domaines69, dont 3 concernent directement les médicaments : antibiotiques, psychotropes, statines. L’avenant n° 12 à la convention médicale (arrêté du 23 mars 2006) a ajouté deux nouveaux thèmes d’engagement dans le domaine du médicament70 : la réduction des dépenses d’inhibiteurs de la pompe à protons (IPP) dès 2005 et les hypertenseurs (IEC-Sartans) à partir de 2007.
Tableau n° 6 : Objectifs de maîtrise médicalisée dans le domaine du médicament en 2005, 2006 et 2007
Thème |
Objectif 2005 |
Objectif 2006 |
Objectif 2007 |
Antibiotiques |
Baisse de 10 % des montants tendanciels 2005 des prescriptions |
Baisse de 10 % des montants des prescriptions par rapport à 2005 |
Baisse de 5 % des montants des prescriptions par rapport à 2006 |
Statines |
Baisse de 12,5 % des montants tendanciels des remboursements |
Stabilisation en montant des prescriptions |
Stabilisation en montant des prescriptions |
Hypnotiques et anxiolytiques |
Baisse de 10 % des montants tendanciels 2005 des prescriptions |
Baisse de 5 % des montants de prescription |
Baisse de 5 % des montants de prescription par rapport à 2006 |
Génériques |
Augmentation de la prescription dans le répertoire générique sur l’ensemble des classes |
Augmentation de la prescription dans le répertoire générique, notamment sur la classe thérapeutique des statines, des IPP et des IEC-sartans |
Prescription dans le répertoire générique |
IPP |
- |
Baisse de 3 % des dépenses par rapport à l’évolution tendancielle |
Baisse de 3 % des dépenses par rapport à l’évolution tendancielle |
IEC Sartans |
- |
- |
Limitation à 6 % de la croissance des dépenses par rapport à 2006 |
![]()
Les objectifs retenus pour la maîtrise médicalisée appellent plusieurs remarques :
– tout d’abord, les objectifs sont définis en termes de montants et non en volumes (nombre de boîtes71). Ils intègrent par conséquent l’effet des baisses de prix décidées par le CEPS (effet prix) ainsi que l’économie générée par l’arrivée sur le marché de nouveaux génériques (effet structure). Or, un engagement de maîtrise médicalisée n’a de sens que s’il porte sur la modification des pratiques de prescription des médecins (volumes).
– par ailleurs, s’agissant des génériques, il n’existe pas de moyen de vérifier que les économies générées sur ce poste sont dues aux médecins. Elles peuvent en effet être dues à une substitution accrue opérée par les pharmaciens ou à une extension du répertoire des génériques.
– enfin, certains objectifs étaient ambigus. Pour les statines en particulier, il n’était pas précisé dans quelle mesure l’objectif d’infléchissement des prescriptions devait être atteint par le développement des génériques, par la limitation des prescriptions et/ou par un meilleur ciblage des prescriptions. Cette ambiguïté a rendu difficile la détermination des actions prioritaires de l’assurance maladie sur cette thématique (cf. infra).
Le bilan 2005 : des économies attendues surestimées, un impact sur les prescriptions très faible
L’ONDAM 2005 a été construit avec un objectif d’économies de maîtrise médicalisée de 1,2 milliard d’euros (hors indemnités journalières), sans que la part des économies liées aux produits de santé soit précisée. Les économies attendues de la maîtrise médicalisée ont été revues à la baisse (moins de 1 milliard d’euros) lors de la signature de la convention médicale. S’agissant des médicaments, la CNAMTS a évalué les économies susceptibles d’être générées par la maîtrise médicalisée à 285 M€, auxquels s’ajoutent 55 M€ d’économies sur les génériques.
Les hypothèses retenues pour chiffrer ces économies étaient contestables. Pour les statines et les hypnotiques/anxiolytiques, la CNAMTS a calculé ses prévisions de tendance à partir de la moyenne des années 2002, 2003 et 2004, alors que la croissance des remboursements était en diminution en 2004. Par exemple, pour les anxiolytiques et les hypnotiques, les calculs d’économies attendues ont été réalisés sur la base d’une hypothèse d’une tendance de 0 %, alors que les remboursements en 2004 pour ces classes étaient en décroissance de 15,8 % par rapport à 2003.
![]()
S’agissant des antibiotiques, la CNAMTS a utilisé une méthode de calcul de la tendance différente : au lieu de faire la moyenne des trois dernières années, comme pour les statines et les psychotropes, elle a considéré que l’évolution en 2005 prolongerait l’atténuation de la baisse des remboursements d’antibiotiques observée entre 2003 (-7,3 %) et 2004 (-3,8 %)72.
Au total, la CNAMTS a systématiquement utilisé la méthode de calcul de la tendance qui lui était la plus favorable, en prenant la moyenne 2002-2004 pour les médicaments pour lesquels les montants de remboursement commençaient à diminuer en 2004 et en prolongeant la tendance pour les antibiotiques, pour lesquels la baisse des remboursements se faisait moins forte en 2004 qu’en 2003.
Compte tenu des hypothèses retenues par la CNAMTS, les économies attendues ont été surestimées, comme le soulignait, avant même la signature de la convention médicale, une note de la DSS dressant un premier bilan financier du protocole d’étape du 15 décembre 2004. La DSS, faisant l’hypothèse que la tendance pour 2005 serait celle observée en 2004, en concluait que l’économie attendue était de 29 M€ (au lieu des 33 M€ annoncés) pour ces deux classes, de 80 M€ (au lieu de 90 M€) pour les antibiotiques, et de 140 M€ (au lieu de 150 M€) pour les statines.
En dépit de ce mode de calcul facilitant le respect des objectifs conventionnels, ces derniers n’ont pas été atteints. Selon la CNAMTS, les économies générées par la maîtrise médicalisée dans le domaine du médicament se sont élevées à 201 M€ en 2005, au lieu des 285 M€ attendues.
Tableau n° 7 : Bilan des économies réalisées selon la CNAMTS
Objectifs 2005 |
Réalisations 2005 | |||
Taux d’évolution des dépenses attendues |
Economies attendues |
Taux d’évolution des dépenses constaté |
Economies réalisées | |
Antibiotiques |
- 10 % |
91 |
- 3,8 % |
35 |
Statines |
- 1,5 % |
161 |
+ 1,9 % |
122 |
Psychotropes |
- 10 % |
33 |
-3 % |
11 |
Génériques |
55 |
33 | ||
Total médicament |
340 |
201 | ||
Total maîtrise médicalisée |
998 |
722 | ||
Source : CNAMTS
![]()
Les économies obtenues par la maîtrise médicalisée sont encore plus réduites si l’on neutralise les baisses de prix et la générication pour s’intéresser aux seuls volumes. En effet, l’année 2005 a été marquée par la baisse de prix de 88 présentations pharmaceutiques, parmi lesquelles des statines et des médicaments génériqués (dont deux statines). Au total, selon la DSS73, une économie de 155 M€ a été réalisée grâce aux baisses de prix ciblées, dont 47 M€ sur les statines. Au final, seuls 17 M€ d’économies auraient été générées par l’infléchissement des prescriptions sur les trois classes visées par la convention médicale.
Tableau n° 8 : Économies nettes des effets prix et générication
Tendance (1) |
Dépenses 2005 tendancielles |
Croissance 2005 constatée |
Dépenses 2005 constatées |
Economies nettes des effets prix et générication | |
Antibiotiques |
- 0,2 % |
845 |
4,3 % |
851 |
6 |
Statines |
+ 13 % |
1359 |
4,6 % |
1265 |
9 |
Psychotropes |
- 4 % |
313 |
- 2,7 % |
317 |
2 |
Total |
17 |
(1) : La période de régression retenue pour calculer la tendance est celle de janvier 2002 à décembre 2004 pour les statines et les antibiotiques, et de janvier 2001 à décembre 2004 pour les psychotropes.
Source : DSS/6B- données GERS
En faisant l’hypothèse, peu probable, que l’intégralité de la pénétration des génériques constatée en 2005 sur les médicaments visés par la convention médicale a été le fait des médecins, les économies de maîtrise médicalisée se situeraient à environ 70 M€ (50 M€ venant en déduction des économies sur les génériques).
Ainsi, l’impact de la maîtrise médicalisée sur les volumes de médicaments remboursés a été faible en 2005. Il n’en demeure pas moins qu’après plusieurs années de forte hausse, l’infléchissement des volumes observé en 2005, bien que très inférieur aux attentes, a constitué un progrès. Pour les statines, une baisse du nombre d’instaurations de traitements a ainsi été observée à compter d’avril 2005, ce qui marque une rupture par rapport à la tendance antérieure : sur les 4 derniers mois de 2005, la baisse atteint -28 % comparativement à la même période de 2004, ce qui s’est traduit par une baisse de 13 % sur l’ensemble de l’année. La CNAMTS indique que depuis la fin de l’année 2005, le nombre d’instaurations de traitements par statines est stable alors que l’effectif des malades à risque cardiovasculaire augmente d’environ 4 à 5 % par an (patients hypertendus et/ou diabétiques).
Tableau n° 9 : Évolutions des volumes (boites) et des montants remboursés en rapport avec des statines seules ou associées.
2003-2002 |
2004-2003 |
2005-2004 |
2006-2005* | |
Volumes (boîtes) |
12,3 % |
12,0 % |
8,7 % |
4,7 % |
Montants remboursés |
16,3 % |
13,8 % |
3,7 % |
-4,4 % |
* en incluant les données des SLM, pour les prescripteurs libéraux, ces évolutions étaient, respectivement de +6,0 % pour les volumes et de -3,7 % pour les montants remboursés.
Source : CNAMTS- Données brutes Medic’am du régime général, hors sections locales mutualistes (SLM), France métropolitaine. Médicaments délivrés en officine de ville.
Le bilan 2006 : un infléchissement plus marqué des volumes
![]()
Pour les trois classes thérapeutiques faisant l’objet d’un engagement de maîtrise médicalisée depuis 2005, les objectifs 2006 ont été définis de manière moins ambitieuse qu’en 2005. L’ensemble de ces quatre postes ainsi que l’augmentation de la prescription dans le répertoire des génériques devait dégager 222 M€ d’économies.
La CNAMTS estime que le montant des économies réalisées en 2006 sur le poste médicament a finalement atteint 239 M€ : 46 M€ pour les antibiotiques, 135 M€ pour les statines, 20 M€ pour les psychotropes, 13 M€ pour les IPP, 25 M€ pour les génériques.
Ainsi que cela a été vu supra, ce chiffrage a l’inconvénient de ne pas tenir compte des baisses de prix de certains médicaments, lesquels relèvent du plan médicament et non de la maîtrise médicalisée.
Selon les estimations de la DSS, réalisées sur la base des données GERS à fin octobre 2006, les économies nettes des effets prix et générication réalisées en 2006 sur les trois classes thérapeutiques seraient de 120 M€, contre 197 M€ attendus74.
Tableau n° 10 : Bilan des économies réalisées selon la DSS (en M€)
Objectifs 2006 |
Réalisations 2006 | ||
Taux d’évolution des dépenses attendues |
Economies attendues |
Economies nettes des effets prix et générication | |
Antibiotiques |
- 10 % |
63 |
21 |
Statines |
0 % |
93 |
62 |
Psychotropes |
- 5 % |
13 |
9 |
IPP |
- 2 % |
28 |
28 |
Génériques (1) |
25 |
- | |
Total médicament |
222 |
120 | |
Total maîtrise médicalisée |
791 |
460 | |
1) En ce qui concerne les génériques, la DSS considère que les économies y afférant sont prises en compte dans le volet génériques du plan médicament. Ce point est contesté par la CNAMTS qui indique que seule une partie des économies liées aux génériques (25 M€ sur 150 M€) ont été imputées aux médecins (celles correspondant à la prescription dans le répertoire). Il n’y donc pas de double compte avec le plan médicaments.
Source : DSS, données GERS (séries corrigées des effets de stockage des génériques et des effets épidémiologiques)
![]()
Le tableau ci-dessous, qui analyse la décomposition de la croissance du chiffre d’affaires hors taxes des laboratoires en ville, confirme que la maîtrise médicalisée a sans doute contribué, depuis 2006 à modifier les prescriptions des médecins, mais dans des proportions moindres que ce qui était initialement attendu.
Tableau n° 11 : Décomposition de la croissance du chiffre d’affaires hors taxes des laboratoires en ville sur les médicaments remboursables (source DSS)
Effet prix |
Effet boîtes |
Effet structure |
Croissance du CAHT | |||||
2005 |
2006 |
2005 |
2006 |
2005 |
2006 |
2005 |
2006 | |
Antibiotiques |
- 0,4 % |
- 5,2 % |
6,3 % |
- 6,5 % |
- 1,7 % |
- 1,3 % |
4,0 % |
-12,5 % |
Statines |
- 3,8 % |
- 6,0 % |
11,8 % |
6,0 % |
- 3,3 % |
- 6,0 % |
4,0 % |
- 6,3 % |
Psychotropes |
- 0,2 % |
- 7,2 % |
- 0,6 % |
-1,0 % |
- 1,8 % |
- 1,2 % |
- 2,6 % |
-9,2 % |
IPP |
- 6,1 % |
- 7,4 % |
8,0 % |
9,1 % |
- 4,1 % |
- 1,5 % |
- 2,8 % |
- 0,5 % |
Ensemble des médicaments remboursables |
- 1,0 % |
- 3,9 % |
3,4 % |
- 5,7 % |
4,4 % |
11,1 % |
6,8 % |
0,7 % |
Source : DSS, données GERS et données de remboursements du régime général, non corrigées des épidémies et des effets de stockage des génériques.
En ce qui concerne les médicaments figurant à l’avenant 12, le nombre de boîtes vendues diminue pour les antibiotiques (- 6,5 % en 200675 contre + 6,3 % en 2005) et les psychotropes (-1,0 % contre -0,6 %). Pour les statines, le nombre de boîtes continue à croître, mais moins vite (6,0 % contre 11,8 %). La diminution du CAHT des statines est toutefois essentiellement due aux baisses de prix du répertoire intervenues en février 2006 ainsi qu’à la générication de la simvastatine (mai 2005) et de la pravastatine (juillet 2006).
En revanche, les IPP connaissent une croissance en volume plus forte (+ 9,1 %) qu’en 2005 (+8%).
Quant au ralentissement de la croissance du CAHT pour l’ensemble de médicaments remboursables (0,7 % en 2006 contre 6,8 % en 2005 et 6,2 % en 2004), il est essentiellement aux baisses de prix et à la diminution des volumes de ventes, largement imputables aux déremboursements. En neutralisant l’effet des déremboursements, la décomposition de la croissance conduit à un effet prix de - 3,9 %, un effet boîtes de 1,1 % et un effet structure de 5,7 %.
Objectifs pour 2007
Les objectifs de maîtrise médicalisée pour 2007 et 2008 sont les mêmes qu’en 2006, sauf pour les antibiotiques pour lesquels l’objectif de diminution des montants de prescription est revu à la baisse. L’action de l’assurance maladie dans le domaine du médicament concernera également la lutte contre la iatrogénie médicamenteuse chez les personnes âgées76 (cf. infra).
![]()
La principale évolution apportée dans l’avenant 23 à la convention médicale est la déclinaison individuelle des objectifs nationaux concernant les génériques, les IPP, les antibiotiques et le respect de l’ordonnance bizone.
Cette déclinaison est susceptible d’améliorer le retour d’information sur les pratiques individuelles des médecins. Toutefois, ces objectifs ne seront pas opposables, et le texte ne précise pas comment sera vérifiée l’atteinte des objectifs individuels.
c. Les supports de communication de l’assurance maladie
Les campagnes de communication thématiques
La Cour avait déjà analysé dans son rapport sur la sécurité sociale de 2005 le succès de la campagne antibiotiques conduite par l’assurance-maladie entre 2002 et 2005 et avait regretté qu’une seule campagne de ce type ait été réalisée.
Ciblée à la fois sur les prescripteurs et sur les patients, cette campagne de communication avait contribué à la baisse de 13 % de la consommation d’antibiotiques observée entre 2002 et 2005. Sur la même période, l’exposition aux antibiotiques est passée de 36,6 DDD à 31 DDD pour 1000 personnes et par jour, soit une baisse de l’ordre de 15 %, même si, ainsi qu’on l’a vu plus haut, la France continue de se caractériser par une consommation d’antibiotiques nettement plus élevée que celle de ses voisins européens.
À ce jour, une seule autre campagne grand public est envisagée : elle concerne la lutte contre la iatrogénie médicamenteuse, également inscrite parmi les objectifs de maîtrise médicalisée pour 2008.
La iatrogénie médicamenteuse est un problème croissant de santé publique. On estime qu’environ 130 000 personnes sont hospitalisées en France chaque année en raison d’un accident lié à la prise de médicaments, et que 40 à 60 % de ces accidents sont évitables.
La CNAMTS s’est engagée à accompagner les médecins traitants dans une démarche de prévention en les encourageant à réviser périodiquement les traitements de leurs patients à risque. Environ 15 400 entretiens confraternels portant sur le thème de la iatrogénie ont été réalisés au cours du second semestre 2006. Ces entretiens doivent être poursuivis en 2007. Les actions d’accompagnement de l’assurance-maladie s’appuient largement sur les travaux des autorités sanitaires. Elles ciblent en priorité quelques facteurs de risque :
– les benzodiazépines à demi-vie longue (médicaments largement utilisés et susceptibles d’accumulation),
– les psychotropes en général, afin d’éviter la prescription concomitante de plus de deux de ces molécules
– la classe des vaso-dilatateurs, dont le SMR a été jugé insuffisant, et qui ne figurent donc pas parmi les priorités lorsqu’il s’agit de hiérarchiser les prescriptions.
![]()
Il est bien sûr trop tôt pour tirer le bilan de cette campagne mais on peut d’ores et déjà noter un effort de convergence entre les différentes institutions (comme pour les antibiotiques), nécessaire au succès de la démarche. La CNAMTS s’appuie en effet sur les recommandations de l’AFSSAPS de juin 2005 sur la prévention de la iatrogénèse chez le
sujet âgé et renvoie également aux outils d’auto-analyse que la HAS propose sur son site Internet pour réévaluer les traitements des patients à risque iatrogène accru77.
La lettre aux médecins et la lettre aux pharmaciens
La CNAMTS dispose également depuis 2004 d’une publication mensuelle au format 4 pages, destinée aux prescripteurs, la « lettre aux médecins ». Sur 21 numéros déjà parus, 10 portent, directement ou indirectement, sur le médicament. Les sujets abordés sont les suivants : les génériques (à plusieurs reprises), les anxiolytiques et les hypnotiques, les statines, la maîtrise médicalisée (à plusieurs reprises également), les antibiotiques ou encore la iatrogénie.
Cette publication, claire et synthétique, constitue un bon relais des plans d’action mis en œuvre par la CNAMTS.
Une « lettre aux pharmaciens » a également été créée en octobre 2006 sur le modèle de la lettre aux médecins, dans la foulée de la signature de la convention nationale entre l’assurance-maladie et les syndicats de pharmaciens en mars 2006.
Les supports « mémos »
Il s’agit de supports cartonnés, qui sont, selon les cas, soit distribués aux médecins par les délégués de l’assurance-maladie ou lors des entretiens confraternels, soit présentés dans une des publications de la CNAMTS destinées aux professionnels de santé (lettre aux médecins, lettre aux pharmaciens).
Sur 18 mémos réalisés au cours de l’année 2006, 11 portent sur le médicament. On retrouve les principaux thèmes d’action de l’assurance-maladie : inhibiteurs de la pompe à protons, statines, génériques, antibiotiques et benzodiazépines. Huit d’entre eux ont une visée médico-économique et trois ont une visée de bonnes pratiques.
Certains mémos constituent ainsi un résumé de recommandations professionnelles et portent d’ailleurs le double timbre CNAMTS/AFSSAPS. Il en va ainsi des mémos « angine aiguë – rappel des recommandations » ou « rhinopharyngite - rappel des recommandations » qui constituent un rappel des recommandations sur l’usage des antibiotiques et du test de diagnostic rapide (TDR).
D’autres ont un objectif clairement affiché en termes de réduction des dépenses. Ainsi, le mémo « statines » ou le mémo « angine aiguë – coût chez l’adulte »78 font apparaître les différences – significatives – de coût de traitement mensuel (dans le premier cas) ou total (dans le second) entre les différentes molécules. S’agissant des statines, la CNAMTS évalue le coût de traitement mensuel minimal au 1er mars 2006 à 8,28 € dans le cas de la molécule la moins chère (générique du Zocor et du Lodales), à 11,72 € pour le princeps le moins cher (Zocor ou Lodales) et à 25,36 € pour la molécule la plus chère (atorvastatine – Tahor, qui constitue d’ailleurs le deuxième médicament le plus remboursé par l’assurance-maladie avec 130 M€ en 2005).
![]()
Il existe enfin différents mémos « génériques », qui s’inscrivent bien sûr dans l’objectif de maîtrise des coûts. Ils ont pour objectif de faciliter la prescription de génériques
par les médecins, en établissant une correspondance entre le nom de marque et le nom de la molécule, en rappelant les molécules génériquées au cours du semestre concerné ou encore en confrontant les objectifs de pénétration des génériques dans le cadre de la maîtrise médicalisée au pourcentage atteint pour l’instant.
Ces mémos semblent constituer un bon outil de communication car ils sont manifestement conçus pour les prescripteurs et adaptés aux contraintes de la prise de décision du médecin (brefs, clairs et synthétiques), ce qui n’est pas toujours le cas des autres outils de communication disponibles.
d. Les actions individuelles sur les médecins
Aux côtés des actions collectives vis-à-vis des médecins (engagements conventionnels, supports de communication thématiques), la CNAMTS s’est engagée dans une série d’actions individuelles, la plus récente étant constituée par les délégués de l’assurance-maladie qui visent à contrebalancer la visite médicale de l’industrie pharmaceutique.
Les délégués de l’assurance-maladie (DAM) et les entretiens confraternels
Pour accompagner la réforme de l’assurance-maladie par la loi du 13 août 2004, la CNAMTS s’est dotée de compétences à vocation pédagogique en mettant en place les délégués de l’assurance-maladie (DAM). Suite à la convention médicale du 12 janvier 2005, les DAM ont pour mission de présenter aux professionnels de santé les priorités de la maîtrise médicalisée définies chaque année par l’assurance-maladie et les syndicats professionnels. Pour chacun des cinq thèmes évoqués plus haut, le délégué présente au médecin les objectifs de l’année en cours et lui remet des données chiffrées sur ses prescriptions qui lui permettent de se comparer à ses confrères.
L’objectif est de contrebalancer la pression exercée par les visiteurs médicaux des firmes pharmaceutiques. Les recherches menées au plan international dans ce domaine montrent en effet que le contact direct avec les professionnels79 et le retour d’informations sur leurs pratiques sont parmi les stratégies les plus efficaces pour agir sur les comportements individuels.
L’objectif est d’atteindre 1 200 DAM en 2009 sur l’ensemble du territoire.
Pour 2007, la cible prévisionnelle est de 300 000 contacts (avec des médecins ou des pharmaciens), avec une médicalisation plus importante sur trois thèmes : antibiotiques, statines et IPP. Cette action s’adressera à des professionnels ciblés en raison du potentiel important d’optimisation de leurs prescriptions.
Aucun bilan chiffré de l’action des DAM n’est disponible. Il est toutefois vraisemblable qu’elle ait un impact sur les prescriptions. La caisse primaire de Carcassonne fait ainsi figure de modèle : elle a obtenu une diminution de la consommation d’antibiotiques de 20 % contre 5 % en moyenne sur le territoire national, grâce à l’action de ses DAM qui rencontrent les médecins généralistes tous les mois.
![]()
Les entretiens confraternels existent pour leur part depuis 1999 pour accompagner des programmes d’actions sur le diabète et l’hypertension artérielle. Les modalités
d’intervention sont analogues à celles des DAM, mais les messages portent davantage sur les pratiques médicales individuelles.
En 2006, près de 25 000 échanges ont été réalisés auprès des médecins généralistes (majoritairement sur le thème de la iatrogénie médicamenteuse) et des cardiologues (statines). En 2007, les échanges confraternels porteront sur les thèmes suivants : statines, inhibiteurs de la pompe à protons, iatrogénie médicamenteuse, antiagrégants plaquettaires et IEC Sartans.
Les profils personnalisés
Le profil personnalisé a été mis en place dans le cadre de la maîtrise médicalisée à la suite de la signature de la convention nationale des médecins généralistes et libéraux en février 2005. Il concerne tous les médecins généralistes et les spécialistes rencontrés sur les thèmes médicaments de la maîtrise médicalisée (par exemple, les cardiologues pour les statines, les rhumatologues pour IPP). Les données individuelles sont comparées aux données départementales pour les généralistes et aux données régionales pour les spécialistes.
Les profils sont remis et commentés par les DAM ou les médecins conseils. À partir de 2007, les visites et donc les profils individuels seront ciblés sur les médecins ayant un montant total et un montant moyen par patient remboursés de la classe thérapeutique concernée (antibiotiques, statines, IPP) les plus importants. Un fort potentiel d’économies en est donc attendu.
Au total, tous les médecins généralistes (50 000) ont eu un retour d’information au moins deux fois au cours de l’année 2006 (sous forme de visites de DAM ou d’échanges confraternels). La totalité des médecins cardiologues80 ont eu un retour d’information sur leurs prescriptions de statines : type de molécules, pourcentage de génériques prescrits, coût total et par patient.
e. Les moyens mis en œuvre par la MSA
Cette section regroupe l’ensemble des actions mises en œuvre par la MSA au titre ou en accompagnement de la maîtrise médicalisée.
Le plan d’actions
Afin de mettre en œuvre les objectifs conventionnels, la MSA a défini un plan d’actions en 7 axes. Le plan d’actions pour l’année 2005 a été diffusé aux caisses le 2 mai 2005. Celui pour 2006 a été diffusé aux caisses le 2 juillet 2006, mais les actions et les méthodologies devant être mises en œuvre, notamment dans le domaine du médicament, n’a été détaillé qu’en décembre 2006. Il a par conséquent été décidé de le poursuivre en 2007.
Plan d’actions de la MSA pour 2006 et 2007
- Axe 1 : campagne de communication sur les thèmes de maîtrise médicalisée et le parcours de soins ;
- ![]()
Axe 2 : dialogue médical de groupe, afin que les médecins puissent collectivement échanger sur leurs pratiques avec les médecins-conseils de la MSA. Les modalités concrètes de mises en œuvre de cette action en 2006 devaient être définies au vu des résultats de l’évaluation de cette action en 2005 (évaluation attendue pour juillet 2006) ;
- Axe 3 : accompagnement des patients en ALD et contrôle de l’ordonnancier bizone ;
- Axe 4 : médicaments ;
- Axe 5 : contrôle des arrêts de travail ;
- Axe 6 : transports ;
- Axe 7 : dépistage du cancer du sein.
S’agissant de l’axe 4, il est à noter que les actions mises en œuvre par la MSA ne reprennent qu’en partie les objectifs conventionnels. Ainsi, en 2006, la MSA n’a réalisé aucune action concernant le champ des IPP, des statines81 et des antibiotiques. Concernant les antibiotiques, elle semble avoir considéré que les retombées de la campagne nationale financée par la CNAMTS étaient suffisantes. S’agissant des statines et des IPP, elle a privilégié la promotion des génériques, et donc une démarche non spécifique à ces deux classes thérapeutiques.
Inversement, elle a défini des actions (polymédication des personnes âgées, associations formellement contre-indiquées) ne figurant pas dans les objectifs conventionnels, mais répondant aux caractéristiques de ses assurés (population âgée, habitant souvent en milieu rural, 20 % d’assurés en ALD).
Les objectifs généraux des plans d’actions et les actions permettant d’atteindre ces objectifs (nombre de lettres par médecins, nombre d’assurés contactés…) ont été déclinés par régions, mais les caisses n’ont pas eu d’objectif chiffré d’économies, ni même d’objectifs en terme de prescriptions.
L’évaluation de ces plans d’actions de maîtrise médicalisée s’est limitée à analyser le nombre d’actions effectivement réalisées par les caisses et à évaluer qualitativement, essentiellement dans le cadre des entretiens confraternels, l’efficacité des actions réalisées. Par ailleurs, une demi-douzaine de caisses n’a réalisé aucune action en 2006.
Pour 2007, la MSA a décidé de passer d’une logique d’obligation de moyens à une logique d’obligation de résultats en fixant un montant national d’économies à réaliser sur les objectifs conventionnels déclinés par régions.
Les actions mises en œuvre en 2005 et 2006 :
– Statines
En dehors de la promotion des génériques, la MSA a réalisé deux actions spécifiques aux statines en 2005, mais qui ont dû être abandonnées en 2006, faute de référentiels scientifiques sur lesquels s’appuyer :
![]()
* CRESTOR : cette statine (dosée à 10 mg ou 20 mg), n’étant remboursable qu’en seconde indication, la MSA avait procédé au blocage du remboursement de cette statine lorsqu’elle était prescrite en première intention. Mais l’arrivée sur le marché d’un nouveau dosage (5 mg), qui a obtenu une indication en première intention, a conduit la MSA à abandonner cette action en 2006. En effet, il était difficile de bloquer les remboursements de CRESTOR 10 mg et 20 mg prescrits hors AMM, alors même que CRESTOR 5 mg était
remboursable sans restriction. Il est à noter que la CNAMTS a été confrontée à la même difficulté, qui illustre la nécessité, pour l’Etat, de mieux contrer les stratégies de contournement développées par les laboratoires ;
* la réduction de la prescription de statine en prévention primaire chez les personnes âgées de plus de 80 ans ne présentant pas de co-morbidité cardiovasculaire. Ce type de prescription, qui n’était pas recommandée par l’AFSSAPS, représentait 0,35 % des prescriptions dans cette classe d’âge. Ces prescriptions, détectées par ARCHIMED, ont donné lieu à l’envoi de 1 610 courriers de signalement, accompagnés d’une fiche sur le bon usage des statines chez la personne âgée. Cette action n’a pas été reconduite en 2006. En effet, des études récentes auraient montré l’intérêt des prescriptions de statines, quel que soit l’âge du patient (cf. supra).
– Benzodiazépines-hypnotiques
En 2005, l’objectif de la MSA était de réduire la consommation de benzodiazépines chez les plus de 70 ans. En effet, les benzodiazépines, qui peuvent nuire à la mémoire, aux fonctions cognitives et à l’équilibre, sont responsables de nombreuses chutes chez les personnes âgées, d’hospitalisations et de décès82.
Un courrier personnalisé et une fiche d’information sur les benzodiazépines ont été adressés aux médecins ayant prescrit des benzodiazépines à des patients de plus 70 ans. Au total, sur les 1,5 million d’ordonnances concernant des personnes de plus de 70 ans qui ont été analysées par ARCHIMED, 6,1 % comportaient une benzodiazépine. 5 500 courriers d’alerte ont été envoyés aux médecins.
Cette action a été abandonnée en 2006, l’évaluation de l’action réalisée en 2005 ayant montré qu’elle avait été inefficace, les patients, habitués depuis de nombreuses années à consommer ces médicaments, faisant pression sur les médecins.
En 2006, la MSA a donc réorienté son action vers les primo-prescriptions. L’objectif était de prévenir la survenance d’une dépendance aux benzodiazépines et de rappeler aux prescripteurs la règle d’une prescription courte ne devant pas dépasser 4 à 12 semaines selon la molécule. La méthodologie, définie en décembre, prévoyait l’envoi d’un courrier aux médecins et assurés concernés et au moins deux entretiens téléphoniques avec le prescripteur par mois et par médecin conseil.
– Associations formellement contre-indiquées
Cette action, réalisée en 2005 et poursuivie en 2006, avait pour objectif de diminuer le nombre d’interactions médicamenteuses.
L’action a consisté à envoyer un courrier d’information au prescripteur et au pharmacien d’une interaction médicamenteuse sur la prescription rédigée et délivrée. Entre juillet et février 2006, 1 100 alertes ont ainsi été envoyées aux médecins.
– Polymédication chez les personnes de plus de 70 ans
![]()
Le logiciel ARCHIMED a permis d’analyser les ordonnances comprenant plus de 10 médicaments, lesquelles étaient ensuite analysées par les médecins conseils afin de
détecter les anomalies. L’objectif fixé aux caisses était l’envoi de 5 courriers par mois et par ETP médecin conseil. Suite à leur analyse par un médecin conseil, 21 % ont été détectées comme comportant une anomalie. Un quart d’entre elles ont fait l’envoi d’un courrier signalant les anomalies au médecin prescripteur, accompagné le cas échéant de fiches sur la prescription des vaso-dilateurs et de benzodiazépines.
Selon la MSA, le taux de modification de l’ordonnance après envoi du courrier est très important. Cela serait dû au fait qu’il permet aux médecins d’évaluer leur pratique, ce qu’ils n’ont pas souvent l’occasion de faire en libéral, notamment en milieu rural.
Ce dispositif, expérimenté dans trois régions en 2004, a été généralisé en 2005 et poursuivi en 2006.
4. Les accords de bon usage des soins
L’assurance maladie dispose, depuis la loi de financement de la sécurité sociale pour 2000, d’un outil d’action supplémentaire : les accords de bon usage des soins (AcBUS). Ceux-ci peuvent être signés au niveau national ou régional, avec un syndicat représentatif d’une profession de santé ; le ministre peut, dans certaines conditions, en prendre l’initiative et ils sont soumis à son approbation.
Ainsi que le soulignait déjà la Cour dans son rapport sur la sécurité sociale de 2004, l’ampleur des progrès souhaitables dans le bon usage des prescriptions de médicaments pouvait conduire à penser que celui-ci constituerait un champ privilégié pour la conclusion de tels accords. Cependant, sept ans après l’ouverture de cette possibilité nouvelle, seuls deux accords nationaux ont été conclus dans le domaine du médicament.
– l’AcBUS relatif au test de dépistage rapide de l’angine vise à rationaliser le recours aux antibiotiques pour cette pathologie. Il engage les médecins généralistes à réduire leurs prescriptions d’antibiotiques en contrepartie d’une formation sur l’utilisation du test et de sa mise à disposition gratuite. Les actions prévues se sont effectivement déroulées et cet accord a certainement eu un impact positif, mais il est difficile d’évaluer ce qui, dans la réduction constatée de la prescription d’antibiotiques à partir de l’hiver 2002-2003, lui est imputable et ce qui résulte des campagnes d’information du grand public à partir d’octobre 2002, du plan de lutte contre l’antibio-résistance lancé par l’Etat en 2001, ou encore des 13 000 échanges confraternels menés par le service du contrôle médical avec des médecins libéraux.
– l’AcBUS relatif à l’usage des antiagrégants plaquettaires dans la prévention secondaire des accidents thrombotiques a été signé le 29 septembre 2005 mais il n’a pas été publié au journal officiel. Une nouvelle version de cet AcBUS a finalement été publiée au journal officiel du 5 janvier 2007.
![]()
Ce retard est dû aux difficultés suscitées par l’AMM de l’aspirine. En effet, cet ACBUS vise à privilégier, dans la classe des antiagrégants plaquettaires, la prescription de l’aspirine, qui est 24 fois moins chère que le clopidogrel (Plavix®), commercialisé par Sanofi-Aventis. Le Plavix® est le premier médicament remboursé par l’assurance-maladie (avec 350 M€ en 2005). Initialement, l’AcBUS indiquait que l'aspirine constituait le "choix préférentiel" dans l'artérite oblitérante des membres inférieurs. Or, le laboratoire Sanofi-
Aventis a rappelé qu'en France l'aspirine ne possède pas cette indication, contrairement au clopidogrel.
Il a donc fallu attendre que la HAS publie en mai 2006 une recommandation professionnelle sur la prise en charge de l’artériopathie chronique oblitérante athéroscléreuse des membres inférieurs pour valider l’AcBUS.
Ce cas illustre les difficultés posées par la procédure d’AMM dans le cas de médicaments anciens et la nécessité, à l’avenir, d’une plus grande coordination entre la CNAMTS, la HAS et l’AFSSAPS.
C. Les actions sur les comportements des patients
Si les comportements de prescription des médecins jouent un rôle déterminant pour expliquer la forte consommation de médicaments en France, les patients peuvent parfois influencer le comportement de prescription des médecins. Une étude récente indique que 46 % des médecins français déclarent faire l’objet d’une « pression à la prescription », contre 36 % des médecins allemands ou espagnols et seulement 20 % des médecins néerlandais. Il semble toutefois exister un malentendu entre médecins et patients en France, puisque 58 % des médecins français déclarent ressentir une attente de prescription pour les rhumes alors que seuls 24 % des patients disent l’estimer nécessaire.
Au-delà, les patients sont directement responsables de la correcte observance des traitements prescrits.
L’information grand public sur le médicament et les actions d’éducation thérapeutique constituent deux vecteurs pour infléchir les comportements des patients en matière de consommation de médicaments. Or, l’intervention publique dans ces deux domaines reste insuffisante.
1. L’information grand public sur le médicament
L’information sur le médicament à destination du grand public souffre, comme l’information à destination des prescripteurs (cf. infra), d’un trop grand éparpillement et de la place excessive laissée à l’information privée.
L’information publique a trois sources principales : l’assurance-maladie, l’AFSSAPS, la HAS.
a. L’assurance-maladie
Outre ses campagnes de communication thématiques, évoquées plus haut et qui comportent également un volet à destination des patients, l’assurance-maladie développe depuis peu de nouvelles méthodes de communication directe avec les assurés, notamment dans le domaine des génériques, qui semblent prometteuses.
![]()
La CNAMTS a ainsi mené une expérimentation en 2004 pour inciter les personnes prenant régulièrement le même médicament de marque à choisir un médicament générique aussi efficace mais moins cher. Cinq caisses (Angers, Béziers, Le Havre, Puy-de-Dôme,
Marne) ont ainsi adressé un courrier d’information personnalisé, ciblé sur les assurés qui consomment peu de génériques. Par ailleurs, la caisse des Vosges procède de façon différente en contactant ses assurés directement par téléphone pour les informer de l’existence d’un médicament générique correspondant au traitement qu’ils suivent.
Dans les deux cas, la CNAMTS a évalué à 49 % le taux de changement de comportement au bout de six mois. Forte de ces résultats concluants, l’assurance-maladie a donc décidé d’étendre à la France entière l’envoi de courriers personnalisés. Elle estime que cette opération pourrait lui permettre d’économiser annuellement entre 12 et 17 M€ supplémentaires grâce au développement des génériques. Début 2006, le taux de changement de comportement au niveau national était estimé à 40 %.
L’action de la MSA a également consisté à envoyer un courrier aux assurés pour lesquels il était apparu que d’importantes économies auraient pu être réalisées si des génériques leur avaient été prescrits. Ce courrier précisait le montant des économies qui auraient pu être réalisées. L’objectif assigné aux caisses était l’envoi par chaque médecin-conseil de 50 courriers par trimestre. Cet objectif était relativement limité par rapport aux possibilités offertes par ARCHIMED, qui permet de détecter automatiquement les assurés concernés et de générer automatiquement un courrier. La raison avancée est que la situation des assurés devait être vérifiée par les médecins conseils (par exemple, pour éviter d’envoyer des courriers aux personnes en fin de vie).
À noter que, contrairement à la CNAMTS, aucune action n’est destinée aux prescripteurs. La MSA semble considérer que le problème des génériques ne relève pas de leur niveau, puisque les pharmaciens ont le droit de substitution et sont encouragés à le faire.
Par ailleurs, l’action de la MSA à destination des assurés semble être appelée à disparaître, puisque les caisses d’assurance maladie subordonnent de plus en plus le tiers-payant à l’acceptation de la substitution.
b. L’AFSSAPS
L’AFSSAPS dispose également d’une gamme d’outils d’information à destination du grand public83. Ceux-ci sont principalement mis à disposition du public sur son site internet, d’un accès particulièrement difficile jusqu’à présent. La refonte de ce site, prévue en 2007, devrait toutefois permettre de structurer les informations aussi bien par type de public que par type de produit et par thème de santé. Il s’agira là d’une amélioration de taille.
L’AFSSAPS s’est par ailleurs employée à renforcer sa communication directe vers le grand public au-delà de la seule information disponible sur le site Internet. Une première initiative a ainsi été engagée en 2005 avec l’élaboration d’un dépliant grand public sur « médicaments et conduite automobile ». En 2007, trois campagnes d’information ont été organisées : médicament et contrefaçon (mai), bien lire la notice des médicaments (juin), attention aux tatouages éphémères au henné noir (juillet).
![]()
L’AFSSAPS cherche également à développer le partenariat avec les associations de patients. Elle a ainsi conçu un projet de « lettre aux associations » qui devrait voir le jour en
2007. Par ailleurs, l’AFSSAPS a constitué en 2006 un groupe référent d’associations chargé de la relecture des documents produits par l’agence. Si ce dispositif paraît être une bonne idée, encore faut-il s’assurer de l’indépendance des associations consultées. Le Sénat avait recommandé dans son rapport précité d’ « obliger les laboratoires à rendre publique la liste des associations de patients qu’ils subventionnent ». Cette disposition a été reprise à l’article 31 de la loi du 26 février 2007 (art. L. 1114-1 du CSP).
Enfin, l’AFSSAPS compte poursuivre le renforcement de l’information en direction des usagers via des partenariats complémentaires avec d’autres organismes intervenant dans ce champ, tels que la CNAMTS ou l’INPES.
c. La HAS
La HAS communique avec le grand public essentiellement par l’intermédiaire de son site Internet, qui lui, est déjà structuré par type de public. En 2006, le nombre de visites est en hausse de 34 % par rapport à l’année précédente.
Tout ceci constitue toutefois un ensemble assez hétérogène, qui suppose une coordination des interventions des différents acteurs et qui ne couvre pas l’ensemble des questions que se posent les assurés. Une place importante est donc laissée à l’information « privée » sur le médicament. C’est précisément pour tenter de la contrôler que la loi du 13 août 2004 confie à la HAS la mission « d’établir une procédure de certification des sites informatiques dédiés à la santé ». La HAS a pour l’instant élaboré une recommandation pour aider les internautes à chercher des informations sur la santé et à évaluer la qualité des sites Web84. Par ailleurs, un message de sensibilisation a été rédigé à destination des médecins afin qu’ils connaissent mieux l’utilisation d’Internet par les patients et soient incités à dialoguer avec eux sur le sujet. Mais il ne s’agit pas de certification à proprement parler. Sur ce point, la HAS n’a pour l’instant réalisé qu’un état des lieux sur les systèmes d’évaluation de la qualité des sites et envisage de sous-traiter à un organisme indépendant la certification des sites français.
2. Les programmes d’aide à l’observance
Depuis quelques années, les firmes pharmaceutiques proposent des programmes « d’aide à l’observance » ou « d’accompagnement des patients » qui peuvent combiner plusieurs types d’actions : formation et information des professionnels médicaux, envoi au patient de brochures d’information, relances téléphoniques et/ou visites visant à améliorer l’observance du traitement.
![]()
Ces programmes, qui visent à améliorer l’observance des traitements médicamenteux85, peuvent avoir des effets pervers lorsqu’ils s’apparentent à de la publicité et permettent de contourner l’interdiction de publicité directe auprès du public pour les médicaments de prescription.
Initialement développés aux Etats-Unis, ces programmes arrivent depuis peu en France : l’AFSSAPS, via la commission de contrôle de la publicité, a été saisie depuis 2001 de 15 programmes de ce type, dont 6 durant la seule année 2006. En l’état actuel de la réglementation, il n’existe pas de cadre juridique spécifique permettant d’encadrer de telles initiatives. La commission de contrôle de la publicité aurait exprimé des préoccupations tournant autour, d’une part, de l’absence de cadre juridique spécifique permettant d’encadrer de telles initiatives, d’autre part, de la crainte que des données recueillies au cours de la mise en œuvre de ces programmes puissent être utilisées à des fins étrangères aux nécessités du traitement des patients, notamment quand le prestataire désigné par le laboratoire se trouve faire partie d’un groupe d’assurance.
En l’absence de cadre juridique clair, les dossiers sont acceptés par l’AFSSAPS au cas par cas. Pour l’instant, huit programmes ont été autorisés. Selon l’AFSSAPS, ces programmes ne sont pas des programmes d’aides à l’observance, mais plutôt des programmes d’éducation des patients (modalités d’administration d’un produit, contexte de prise en charge globale d’une pathologie, etc.…). Toutefois, elle n’exclut pas que des programmes d’aide à l’observance aient été mis en place sans qu’elle soit consultée.
Au niveau européen, une annexe d’une recommandation de l’agence européenne du médicament du 14 novembre 2005, dépourvue de portée réglementaire, indique que les plans de gestion des risques (PGR) désormais requis pour certains médicaments peuvent comprendre des programmes d’aide à l’observance réalisés par les entreprises pharmaceutiques.
Dans une note du 23 janvier 2007 adressée à la DGS, l’AFSSAPS indique que « peu de véritables programmes d’accompagnement des patients » sont actuellement mis en place ou prévus par l’AMM. À ce jour, il s'agit surtout de programmes d'éducation des patients inscrits dans les plans de minimisation des risques des PGR européens pour des spécialités particulières nécessitant soit des procédures d'administration strictes (exemple Macugen® (Pfizer), injections intra-vitréennes), soit des mesures de surveillance active et des recommandations à destination des patients avec carnets de surveillance et brochures sur le médicament et ses risques (exemple : Tysabri® - natalizumab).
Seuls les PGR des spécialités Acomplia® et Intrinsa® prévoient en Europe un « programme d’accompagnement des patients ».
– ![]()
s’agissant d’Acomplia®, médicament anti-obésité commercialisé en France depuis mars 200786, « Sanofi Aventis va mettre en place un « programme d'accompagnement » de la prescription pour les patients via une société de service externe dès la commercialisation. Ce programme comprend un ensemble de services visant à aider les patients à mettre en œuvre les changements de comportement (régime, activité physique) complémentaires à la mise sous traitement. Les patients, sous réserve de leur inscription, bénéficieront d'outils d'auto-formation (DVD sur la pathologie, recommandations sur l’activité physique et le régime) et recevront des courriers réguliers de la société de service pour
entretenir la motivation et la compliance au traitement. Ils auront également la possibilité d'appeler un centre d'appel pour toutes questions relatives à la pathologie, la nutrition, l'activité physique.
– s’agissant d’Intrinsa®, commercialisé depuis février 2007, l’AFSSAPS indiquait que la filiale française des laboratoires Procter & Gamble Pharmaceuticals ne semble pas vouloir créer un site web destiné aux patients, comme cela était envisagé dans le PGR européen, mais devrait se contenter d’éditer une brochure destinée aux prescripteurs et qui a été validée par le département de la surveillance des risques et de bon usage des médicaments de l’AFSSAPS.
Pour ces deux spécialités, l’AFSSAPS indique que dans un premier temps, seule une action d’information/formation des professionnels de santé a été retenue en France. Un bilan à 6 mois est prévu qui réexaminera la question de la mise en place d’un plan d’accompagnement.
Le gouvernement avait introduit dans le texte initial de la loi du 26 février 2007 un article (art. 29-10) l’habilitant à mettre en place par voie d’ordonnance un régime spécifique d’encadrement de ces programmes. Le projet d’ordonnance prévoyait notamment que les firmes pourront, par l’intermédiaire des médecins, mettre en place des « dispositifs individualisés (relance téléphonique, numéro vert, éducation personnalisée pour les patients, envoi d’infirmiers à domicile, etc.…) ». Cette disposition, introduite sans débat parlementaire et très controversée87, a finalement été retirée, mais une proposition de loi est en cours de préparation. Une mission IGAS a également été diligentée
Le développement des initiatives des firmes pharmaceutiques dans ce domaine nécessite incontestablement une clarification du cadre juridique. Les travaux en cours devront encadrer strictement ces programmes, qui ne peuvent être autorisés qu’à la condition d’apporter une réelle valeur ajoutée, de respecter les droits des patients, et de concerner des médicaments apportant une véritable amélioration du service médical rendu. L’examen de ces programmes devra être réalisé collégialement par la commission de contrôle de la publicité de manière transparente.
La Cour considère en effet qu’il appartient en priorité aux pouvoirs publics de répondre au besoin, bien réel, d’accompagnement des patients et que celui-ci ne doit pas être abandonné aux firmes pharmaceutiques.
La HAS, dans un courrier du 20 décembre 2006 adressé au directeur général de la santé, mettait en avant la nécessité de ne pas limiter l’éducation thérapeutique aux actions sur l’observance et notait que le fait d’identifier un seul intervenant, à savoir l’industrie pharmaceutique, fut-ce à des fins d’encadrement présente des risques d’incompréhension quant au rôle dévolu par les pouvoirs publics aux autres acteurs, notamment les associations de patients ou les réseaux d’éducation thérapeutique.
![]()
Ce point de vue est pleinement partagé par la Cour. Il n’en reste pas moins que les interventions publiques dans le domaine de l’éducation thérapeutique devraient être
renforcées et rationalisées, notamment en recherchant une plus grande complémentarité entre les acteurs (assurance maladie, AFSSAPS, HAS, associations, INPES, etc.).
d. La promotion des génériques
Un médicament générique est une copie d’un médicament de marque (dit princeps) dont le brevet, qui lui assure une exclusivité, a expiré après une dizaine d’années de commercialisation. Il doit posséder la même composition qualitative (principe actif) et quantitative (dosage), la même forme d’administration ainsi que la bioéquivalence. Ces médicaments sont fabriqués par des laboratoires qui n’ont pas de frais de recherche et de développement. Un médicament générique est environ 30 % moins cher qu’un princeps.
Les génériques ne jouent bien sûr que sur un des deux volets de la prescription et de la consommation de médicaments : celui de la maîtrise des dépenses de santé, mais pas celui de la santé publique. S’agissant de ce premier volet, l’impact de la délivrance de génériques est d’autant plus crucial qu’entre 2004 et 2007, de nombreux brevets de molécules phares sont tombés dans le domaine public : les molécules concernées représenteraient une économie potentielle de plus de 3,8 milliards d’euros pour l’assurance maladie.
Longtemps en retard par rapport à ses principaux voisins européens88, la France a vu sa consommation de génériques progresser de façon considérable au cours de ces dernières années. La part des génériques dans les médicaments remboursables est ainsi passée de 5,4 % en 2000 à 15,6 % en 2005 et les économies permises par les génériques augmentent chaque année (561 M€ en 2005 contre 380 M€ en 2004)89. Toutefois, comme le souligne la CNAMTS90, ces résultats restent encore en retrait par rapport à des pays comme l’Allemagne ou le Royaume-Uni, où la part des ventes dans l’ensemble du marché était déjà supérieure à 20 % en 1996 ou 1997.
La France a désormais atteint le niveau international, avec un taux de pénétration dans le répertoire des génériques91 qui est passé de 63 % à 75 % entre décembre 2005 et mai 2007.
![]()
Dans son avis sur le médicament de juin 2006, le haut conseil notait que « la modestie relative du marché français des génériques est liée, à titre principal, à l’étroitesse persistante du répertoire français et à titre secondaire, à l’insuffisance du taux de délivrance de génériques dans le répertoire ». La CNAMTS confirme cette analyse : le problème aujourd’hui n’est plus la croissance de la part des génériques dans le répertoire, mais plutôt
la diminution de la part du répertoire dans l’ensemble des médicaments. Dans l’avenant 23 à la convention nationale des médecins libéraux, figure ainsi un objectif de prescription des IPP dans le répertoire.
2. La mise sur le marché des génériques
Le marché des génériques représente 10 % du marché mondial du médicament et il est appelé à se développer rapidement du fait des prochaines expirations de brevets de molécules phares. De nouvelles stratégies se développent et la concurrence devient vive entre les génériqueurs et les laboratoires détenant le princeps. L’AMM devient donc une étape importante puisqu’elle constitue le passeport pour la vente des futurs produits : les deux tiers des demandes d’AMM déposées auprès de l’AFSSAPS concernent des génériques.
L’IRDES a analysé les stratégies de contournement des génériques développées par les firmes pharmaceutiques92. Les deux principales consistent à défendre leur brevet par des actions en justice contre les génériques sur la date d’expiration du brevet et à accroître la durée de protection de leurs brevets en étendant les indications du médicament ou en diversifiant leur gamme de produits (nouveaux dosages, association de molécules, nouvelle présentation).
a. Les extensions d’indication et la diversification des médicaments
Cette stratégie semble être particulièrement développée en France. Elle permet parfois le report d’une partie des ventes de princeps aux dépens du générique, le générique du princeps concerné subissant la concurrence du nouveau princeps (très proche de l’ancien) développé par le laboratoire. Elle est facilitée par le fait que l’AMM ne repose pas sur l’exigence d’un progrès thérapeutique.
La transposition en droit interne de la directive européenne 2004/27 CE sur le médicament93 a toutefois introduit plusieurs dispositions favorables au développement des génériques, visant notamment à contrer ces stratégies commerciales.
![]()
Ainsi, la notion d’AMM globale94 rend désormais impossible une protection supplémentaire des données pour les extensions de gamme des princeps (nouveau dosage, forme pharmaceutique ou méthode d’administration). Par ailleurs, la définition du médicament générique a été étendue de manière à pouvoir considérer les différents sels, esters, éthers ou isomères d’un principe actif comme un même principe actif : autrement dit, quand un principe actif est substituable par un médicament générique, il en va de même pour l’ensemble de ses dérivés. Enfin, l’article 4 de la loi du 27 février 2007 a étendu le champ du répertoire des groupes génériques : une extension de gamme d’une spécialité de référence déjà inscrite au répertoire des groupes génériques, de même composition qualitative et
quantitative en substance active et de même forme pharmaceutique et dont la bioéquivalence avec cette dernière aura été démontrée par des études de biodisponibilité appropriées, pourra, sur demande du ministre de la santé, intégrer le groupe générique déjà constitué en tant que spécialité de référence (art. L5121-1 5° du CSP). Elle sera donc substituable elle aussi.
Cependant, le droit actuel ne permet pas de limiter les associations de médicaments. Or, ces associations, qui permettent de prolonger la période de protection des données, ne constituent pas une réelle innovation thérapeutique et accroissent le risque de surmédication. On peut à cet égard citer l’exemple de l’Inégy®, qui associe les principes actifs de Tahor® (simvastatine) et d’Ezétrol® (ézétimibe). Ce médicament, qui n’apporte aucune amélioration du service médical rendu (il a d’ailleurs obtenu une ASMR V selon l’avis de la transparence du 21 septembre 2005), a obtenu l’AMM le 28 juillet 2005, soit peu de temps avant la générication de la simvastatine.
b. La période de protection des princeps
La transposition de la directive 2004/27 CE par la loi du 27 février 2007 a introduit une procédure allégée pour les génériques et les produits bio-similaires : les demandes d’AMM pour ces médicaments peuvent désormais être déposées à partir de la fin de la huitième année suivant la délivrance de l’AMM à la spécialité de référence, c’est-à-dire 2 ans avant la fin de la période de protection de cette spécialité. Les spécialités génériques pourront donc être commercialisées dès la fin de la période de protection des spécialités de référence.
Toutefois, la période de protection des princeps est allongée d’un an (11 ans au lieu de 10) si pendant les huit premières années suivant l’autorisation de mise sur le marché de la spécialité de référence, le titulaire de celle-ci obtient une autorisation pour une ou plusieurs indications thérapeutiques nouvelles considérées comme apportant un avantage clinique important par rapport aux thérapies existantes. Cette disposition apparaît contradictoire avec le principe de base énoncé plus haut selon lequel l’AMM ne constitue pas une évaluation comparative d’un médicament.
La principale difficulté à laquelle se heurtent les génériqueurs est le manque d’information concernant la date d’expiration des brevets. Les obligations respectives des génériqueurs et des entreprises exploitant des princeps sont à cet égard asymétriques.
En effet, la loi du 27 février 2007 (4e alinéa de l’article L.5121-10 du CSP) oblige le titulaire de l’AMM d’une spécialité générique à informer le directeur de l’AFSSAPS des indications, formes pharmaceutiques et dosages de la spécialité de référence pour lesquels les droits de la propriété intellectuelle n’ont pas expiré au moment de la commercialisation du générique. Cette disposition, présentée comme un moyen d’éviter les contentieux, introduit une contrainte supplémentaire pour les fabricants de génériques et risque de retarder la commercialisation de ces produits.
![]()
De son côté, le deuxième avenant à l’accord-cadre conclu avec le LEEM prévoit que les entreprises exploitant des spécialités pour lesquelles elles détiennent des brevets ont la possibilité, mais non l’obligation, de déclarer au CEPS les titres considérés et leurs dates d’échéance.
3. Les actions pour accroître la délivrance des génériques
a. Les actions sur les prescripteurs
Le médecin a trois possibilités pour prescrire un générique :
– soit il prescrit directement un générique ;
– soit il prescrit dans le répertoire des génériques un princeps qui sera substituable ensuite par le pharmacien. C’est ce qu’on appelle la prescription dans le répertoire, par opposition à la prescription « hors répertoire », où le pharmacien n’a pas la possibilité de substituer un générique au princeps prescrit par le médecin.
– soit il utilise la dénomination commune internationale (DCI).
Le développement de la prescription des génériques par les médecins repose essentiellement sur l’action des pouvoirs publics, car l’industrie reporte ses efforts de promotion sur les médicaments les plus récents et encore protégés. Depuis 2002, l’assurance-maladie a ainsi mené des campagnes d’information auprès des médecins pour les convaincre de prescrire des génériques.
Les accords conventionnels
L’accord du 5 juin 2002, en contrepartie d’une revalorisation de la consultation (20 €), demandait aux médecins de rédiger 25 % en moyenne nationale de leurs lignes de prescription en DCI ou en génériques directement. Les médecins s’engageaient en outre à porter leur taux de prescription dans le répertoire des génériques à au moins 12,5 % des lignes de prescription médicamenteuses en moyenne nationale.
La dénomination commune internationale (DCI) des médicaments désigne la substance active ou la molécule qu’il contient (ex : l’oméprazole à la place du Mopral ou de l’Inexium ; le paracétamol à la place du Doliprane ou de l’Efferalgan). La DCI a été mise en place par l’OMS pour servir de langage commun à l’ensemble des professionnels de santé et des patients. En effet, les médicaments sont aujourd’hui commercialisés sous un nom de marque, qui ne fournit aucune indication thérapeutique et qui varie selon les pays.
La prescription en DCI diminue le risque d’interactions médicamenteuses et/ou de mésusage en permettant de repérer les médicaments qui contiennent la même molécule. Par ailleurs, la prescription en DCI facilite la substitution des génériques en officine : si la DCI est connue des pharmaciens, tel n’est pas forcément le cas des autres personnes travaillant dans les officines.
![]()
L’objectif de prescription en DCI n’a pas été atteint. Si la prescription en DCI a légèrement augmenté, elle reste très inférieure à l’objectif de l’accord. Chez les généralistes, la prescription en DCI a progressé, passant de 4,2 % en juin 2002 à 8,4 % en novembre 2005 et 11,2 en novembre 2006. Elle reste beaucoup moins fréquente chez les médecins spécialistes (4,1 % en novembre 2006). Sur la période allant de décembre 2004 à novembre 2005, 42 % des généralistes prescrivaient plus de 10 % de lignes en DCI, contre seulement 13 % des spécialistes. Inversement, 6 % des généralistes prescrivaient moins de 1 % de lignes en DCI, contre 40 % des spécialistes95.
En revanche, le second objectif de l’accord (prescription dans le répertoire des génériques) a été largement atteint, comme le souligne le HCAAM dans son avis de juin 2006 : « bien qu’il n’ait pas permis d’atteindre ses objectifs sur les prescriptions en DCI, l’accord de 2002 a contribué au développement des génériques, comme en témoigne la marche d’escalier de près de 8 points observée après juin 2002 ».
En mars 2006, les syndicats nationaux de médecins et de pharmaciens ont signé avec l’UNCAM un accord tripartite en vue d’améliorer la pénétration des génériques. Cet accord permet de coordonner les actions en faveur de la consommation de génériques. Les médecins se sont à nouveau engagés à prescrire, en priorité, des médicaments figurant au répertoire des génériques.
La maîtrise médicalisée
Le développement des génériques constituent un des axes des engagements de maîtrise médicalisée des dépenses des médecins libéraux dans la convention du 12 janvier 2005. Les DAM ont notamment pour mission d’informer les médecins sur leur niveau de prescription par rapport à leurs confrères dans le cadre du retour personnalisé d’information.
Les actions pour promouvoir la DCI
Plusieurs facteurs expliquent la faiblesse des prescriptions en DCI : formation insuffisante, habitudes des prescripteurs et des patients, publicité des firmes en faveur des noms de marque, impossibilité de prescrire en DCI pour une minorité de médicaments.
La CNAMTS a mobilisé plusieurs outils, déjà examinés plus haut, pour inciter les médecins à la prescription de génériques. Elle diffuse ainsi des mémos qui indiquent les principales molécules génériquées en les associant à leur nom de marque (notamment pour les statines) et s’appuie sur les lettres d’information des médecins.
Par ailleurs, la loi de février 2007 a fait de la prescription en DCI un critère de la certification des logiciels d’aide à la prescription.
Au-delà, il serait nécessaire de mieux prendre en compte la DCI dans le cadre de la formation initiale et continue des médecins.
b. Les pharmaciens
Le droit de substitution
Les pharmaciens ont obtenu en 1999 le droit de substituer un générique à un médicament original de marque. Toutefois, le pharmacien ne peut exercer son droit de substitution qu’au sein des médicaments inscrits dans un répertoire élaboré par l’AFSSAPS.
![]()
Tardive à se mettre en place, la mise en œuvre de ce droit de substitution s’est progressivement renforcée. C’est à ce droit de substitution exercé par les pharmaciens que l’on doit principalement les progrès enregistrés en matière de délivrance des génériques.
Les accords conventionnels
L’UNCAM et les trois organisations syndicales représentatives des pharmaciens (FSPF, USPO, UNPF) ont conclu le 6 janvier 2006 un accord pluriannuel (2006-2008) d’objectifs de délivrance de spécialités génériques. Cet accord fixe l’objectif de pénétration des médicaments génériques à 70 % du répertoire pour la fin 2006, ce qui représente une économie de 100 M€. Cet objectif a été décliné en objectifs individuels pour chaque officine : plus le taux initial de délivrance de médicaments génériques est faible, plus l’effort demandé est important. Une liste de 20 molécules96, dont l’augmentation du taux de pénétration est essentielle pour le succès du développement du générique, a également été définie pour l’année 2006. Cette liste permet d’organiser le suivi des objectifs au niveau national et local.
Les délégués de l’assurance-maladie ont été mobilisés pour rendre visite aux pharmaciens dans les officines ayant un taux de prescription de génériques nettement inférieur à la moyenne nationale (grandes villes d’Ile-de-France, région PACA).
Un premier bilan de l’accord publié par la CNAMTS en août 2006 montre que la délivrance des génériques a sensiblement progressé : fin avril 2006, le taux national de délivrance des génériques a atteint 67 %, dépassant l’objectif de 66 % prévu pour fin juin. Toutefois, cette progression n’a pas été également répartie entre toutes les pharmacies. En particulier, les pharmacies de petite taille et celles situées dans les grandes villes ont plus de difficultés à délivrer des génériques.
c. La promotion des génériques auprès des assurés
L’assurance-maladie s’oriente vers une politique vis-à-vis des assurés de plus en plus incitative en matière de génériques.
Les campagnes d’information
Des campagnes d’information ont tout d’abord été organisées en direction des patients pour les convaincre d’accepter les génériques. La dernière remonte à septembre 2005.
Elles ont été suivies par des campagnes de promotion directe des génériques auprès des assurés, plus incitatives puisqu’elles passent par un contact individuel avec les patients. Il ne s’agit plus de faire connaître ce que sont les médicaments génériques, mais de faire évoluer les comportements de ceux qui les utilisent insuffisamment. Ces actions, déjà évoquées plus haut, sont menées tant par la MSA que par la CNAMTS.
La suppression du tiers-payant
![]()
En 2006, l’assurance maladie a décidé de supprimer la dispense d'avance de frais (tiers-payant) lorsque les assurés sociaux refusent la délivrance d’un médicament générique par le pharmacien. Cette mesure, qui dans un premier temps a été limitée à huit départements ayant un faible taux de délivrance, ne pouvait s’appliquer aux bénéficiaires de la couverture maladie universelle (CMU).
La LFSS pour 2007 a étendu le principe « tiers-payant contre génériques » à l’ensemble des assurés et aux bénéficiaires de la CMU (art. L. 162-16-7 du code de la sécurité sociale).
Toutefois, cette disposition n’a pas été généralisée à l’ensemble du territoire. La loi prévoit en effet que l’accord national passé avec les pharmaciens peut décider de maintenir la dispense d'avance de frais dans les zones géographiques pour lesquelles les niveaux de substitution sont supérieurs aux objectifs fixés par cet accord.
Début 2007, cette mesure a été étendue aux 16 départements qui n'ont pas atteint l'objectif de 70 % de consommation de génériques dans le répertoire prévu pour l’année 2006 (Bouches-du-Rhône, Corse du Sud, Haute-Corse, Haute-Vienne, Val de Marne, Rhône, Var, Eure, Essonne, Val d'Oise, Creuse, Isère, Haute-Loire, Guadeloupe, Martinique et Guyane).
Dans le plan d’économies élaboré en juin 2007, la CNAMTS a proposé de généraliser cette mesure à l’ensemble du territoire. La mise en œuvre de cette mesure suppose une modification législative.
* *
*
Le circuit de la mise sur le marché et de l’admission au remboursement ne permet pas de jouer un rôle de filtre efficace dans l’introduction de médicaments, ni au stade de la réévaluation en cours de vie des produits.
Par ailleurs, en dépit de l’évolution apportée par la maîtrise médicalisée, les prescriptions des médecins restent très peu encadrées en France.
Il en résulte que la formation et l’information en matière de médicament seraient essentielles pour influencer le niveau et la qualité des prescriptions. Or celles-ci demeurent très insuffisantes, malgré certains efforts récents.
Ces éléments, ajoutés à l’insuffisance de l’accompagnement des patients, se conjuguent pour expliquer les spécificités françaises en matière de prescription et de consommation de médicaments.
![]()
1 () Lettre du Président de la 6ème chambre à la directrice de la législation fiscale en date du 19 juin 2006.
2 () Arrêt CJCE, 31 mars 1992, aff.200/90 Plén ;, dansk Denkavit et P.Poulsen Trading ; arrêt CJCE, 17 septembre 1997, aff.28/96,5è ch.,Fricarnes.
3 () Réponse de la directrice de la législation fiscale au Président de la 6ème chambre en date du 25 juillet 2006.
4 () Il s’agit des entreprises du secteur telles qu’identifiées par leur code d’activité selon la nomenclature d’activité françaises (NAF): 244A : Fabrication de produits pharmaceutiques de base ; 244C : Fabrication de médicaments ; 244D : Fabrication d’autres produits pharmaceutiques ; 514N : Commerce de gros de produits pharmaceutiques ; 523A : Commerce de détail de produits pharmaceutiques.
5 () En ce sens voir les rapports « Analyse des ventes de médicaments aux officines et hôpitaux, 1994-2004 », AFSSAPS, mai 2006 et « Ventes de médicaments aux officines et hôpitaux ; chiffre clef 2005 », AFSSAPS, octobre 2006
6 () La Cour s’étant intéressée régulièrement dans le passé à la question du médicament (cf. rapports sur la sécurité sociale de 2001 pp. 85-110, 2002, pp 368-382, 2003 pp. 213-216 et 2004 pp. 305-355), le présent rapport permet également de faire le suivi des recommandations qui y étaient énoncées.
7 () Ce classement porte sur les médicaments ayant un nom de marque : il intègre tous les médicaments, quels que soient leur forme, leur dosage ou leur taille de conditionnement, vendus sous un même nom de marque ainsi que les génériques qui ne sont pas commercialisés sous le nom de la dénomination commune internationale (DCI).
8 () Enquête intitulée « Les Européens, les médicaments et le rapport à l’ordonnance », Février 2005.
9 () En France, Allemagne, Espagne, Pays-Bas.
10 () DREES, « Le marché du médicament dans cinq pays européens : structure et évolution en 2004 », Etudes et résultats, n°502, juillet 2006.
11 () Source : Les entreprises du médicament (LEEM) : site Internet.
12 () CNAMTS, “La consommation d’antibiotiques : situation en France au regard des autres pays européens”, Points de repère, n°6, novembre 2006.
13 () « Variations and increase in use of statins across Europe », EuroMedStat 2004.
14 () Depuis leur mise sur le marché en 1998, ces médicaments ont bénéficié d’indications thérapeutiques élargies pour le reflux gastro-oesophagien, et, dans certaines conditions, pour la prévention des troubles gastriques due à la prise d’anti-inflammatoires.
15 () CNAMTS, Point d’information mensuel du 14 novembre 2006
16 () Fédération nationale de la mutualité française, direction de la santé, département politique du médicament, « Prescription et consommation de médicaments à SMR insuffisant : une comparaison internationale », 2005.
17 () Source : rapport du haut conseil pour l’avenir de l’assurance-maladie.
18 () « La diffusion de l’innovation pharmaceutique en médecine libérale : revue de la littérature et premiers résultats français », Questions d’économie de la santé n°73, novembre 2003.
19 () Fédération nationale de la mutualité française, direction de la santé, département politique du médicament, « Prescription et consommation de médicaments à SMR insuffisant : une comparaison internationale », 2005.
20 () CNAMTS, « Prescriptions médicales : disparités géographiques », novembre 2004.
21 () Source : id.
22 () Consommation étudiée sur un an entre le 1er mai 2002 et le 30 avril 2003.
23 () Selon la CNAMTS, une partie de ces évolutions s’explique par le phénomène épidémique qui a commencé fin décembre et s’est poursuivi en janvier.
24 () Source : CNAMTS, « Médicaments remboursables : analyse des principales évolutions de l’année 2005 ».
25 () A titre d'exemple, le Mopral, premier médicament consommé au niveau mondial, a été substitué il y a quelques années à un anti-ulcéreux classique, le Ranitilide Sinitilide, qui était meilleur marché. Ce dernier médicament avait lui-même remplacé des pansements gastriques de plus faible coût.
26 () Il est vrai cependant que ces médicaments remplacent parfois des traitements encore plus onéreux (hospitalisations, etc.).
27 () Le nombre de demandes d’AMM n’est pas égal à la somme des avis rendus par le CHMP et des demandes retirées en raison du délai de traitement des demandes d’évaluation.
28 () On appelle « me-too » un produit concurrent d’un produit innovant doté d’une structure chimique voisine, mettant en avant un effet au moins équivalent et qui cherche à prendre des parts d’un marché rentable.
29 () La législation en vigueur a été modifiée par la loi du 26 février 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicamentqui a transposé la directive communautaire 2004/27/CE.
30 () Ainsi, beaucoup de spécialités n’ont pas d’AMM pour l’utilisation chez l’enfant ou pour les pathologies rares.
31 () Ces médicaments ne sont pas pris en charge dans le cadre d’un groupe homogène de soins (GHS) mais doivent faire l’objet d’accords passés entre les établissements hospitaliers et les Agences Régionales de l’Hospitalisation (ARH) dont ils relèvent. Ces accords seront des contrats de bon usage décrivant, pour chaque médicament quelles sont les pratiques dont l’application ne met pas en cause le principe du remboursement. (Article L.162-22-7 du code de la sécurité sociale).
32 () Décret n°2005-1023 du 24/08/05 relatif aux contrats de bon usage des médicaments et des produits et prestations
33 () Article L. 162-17-2-1 du code de la sécurité sociale.
34 () L’arrêté d’inscription des produits peut fixer des conditions de prise en charge et comporter une obligation pour le laboratoire de déposer dans un certain délai une demande d’AMM ; si cet engagement n’est pas respecté, le CEPS peut prévoir une pénalité financière allant jusqu’à 10% du chiffre d’affaires réalisé en France par l’entreprise au titre du dernier exercice clos pour le produit considéré.
35 () L‘évaluation de la commission de la transparence intervient au stade de la demande de première inscription d’un médicament et au stade de la demande d’extension d’indication.
36 () La fixation du taux de remboursement appartient au directeur de l’UNCAM, mais la décision de celui-ci est liée par le SMR déterminé par la commission de la transparence.
37 () Rapport du groupe de travail de la commission de la transparence sur l’impact de santé publique des médicaments : « Intérêt de santé publique des médicaments : principes et méthode d’évaluation », Février 2006.
38 () A titre d’exemple, plusieurs spécialités de la classe des statines sont concernées par ce phénomène.
39 () 58 % en 2005, 54 % en 2006.
40 () Cette analyse est réalisée en Allemagne (cf. Arzneiverordnungsreport).
41 (). Source : IRDES, questions d’économie de la santé n°99 : « les politiques de prise en charge des médicaments en Allemagne, Angleterre, France », octobre 2005.
42 () Jusque-là, l’AMM devait être renouvelée tous les cinq ans.
43 () Article L.5121-9 du CSP issu de la loi du 26 février 2007 transposant la directive européenne 2004/27/CE sur le médicament.
44 () Article 6 « Suivi des nouveaux médicaments en pratique médicale réelle - Perspectives de santé publique ».
45 () Cette décision a nécessité une évolution du droit en vigueur pour autoriser le maintien temporaire de la prise en charge de médicaments ayant un SMR insuffisant. L’article 41 de la LFSS pour 2006 a modifié l’article R. 163-3 du code de la sécurité sociale, qui prévoit désormais que « les ministres chargés de la santé et de la sécurité sociale peuvent décider le maintien temporaire de la prise en charge de certaines spécialités (…) dont le service médical rendu est insuffisant ». L’alinéa 2 du même article prévoit un taux de remboursement à 15% pour ces spécialités.
46 () L’expression « 15 à 15 » signifie « 15 % remboursé 15 % ». En effet, sans cette modification de la réglementation, les médicaments dont le taux de prise en charge est abaissé à 15 % continueraient à être remboursés à 100 % dans le cadre des ALD. Cette possibilité d’exonération a été supprimée pour tous les veinotoniques, dont on est sûr qu’ils n’ont pas de rapport direct avec une ALD. Cela n’a en revanche pas été le cas pour les autres médicaments dont le taux de remboursement a été abaissé à 15 % (vasodilatateurs) et qui continuent donc à être remboursés à 100 % pour les patients en ALD.
47 () L’article 27 de la loi du 26 février 2007 a transposé les dispositions de la directive. L’article L. 5311-1 du code de la santé publique dispose désormais que l’AFSSAPS « rend publics un rapport de synthèse de l’évaluation effectuée pour tout nouveau médicament dans des conditions déterminées par voie réglementaire, ainsi que les décisions d’octroi, de suspension et de retrait de l’autorisation de mise sur le marché. »
48 () Commission chargée du contrôle de la publicité et de la diffusion des recommandations sur le bon usage du médicament ; commission nationale de matériovigilance ; commission nationale de biovigilance ; commission nationale de matériovigilance, commission nationale des stupéfiants et psychotropes ; commission chargée du contrôle de la publicité en faveur des objets, appareils et méthodes ; commission nationale de la pharmacopée ; commission de la cosmétologie.
49 () Fonds pour la promotion de l’information médicale.
50 () Etude de faisabilité portant sur le développement d’une base de connaissance publique pour la prescription et la dispensation de tous les médicaments répondant aux objectifs de la certification des logiciels d’aide à la prescription.
51 () Elle est en effet la seule base à n’avoir aucun lien, direct ou indirect, avec l’industrie pharmaceutique. A l’inverse par exemple, la base de données Claude Bernard fait partie du groupe Cégédim, qui compte parmi ses clients l’industrie pharmaceutique.
52 () Rapport d’information de la commission des affaires sociales du Sénat du 14 juin 2006 sur les conditions de mise sur le marché et de suivi des médicaments.
53 () L’article L. 632-1 du code de l’éducation dispose que le régime des études médicales et post-universitaires ainsi que l’organisation de la recherche est fixé par arrêtés du ministre chargé de l’enseignement supérieur et du ministre chargé de la santé.
54 () Le rôle et la composition de la CNEM sont définis dans le décret n°91-136 du 31 janvier 1991. Aux termes de l’article 11 de ce décret, les membres de droit de cette commission sont le directeur général de la santé, le directeur des hôpitaux, le directeur des enseignements supérieurs au ministère chargé de l’éducation nationale ou son représentant, un représentant du ministre chargé de la recherche, e directeur de la sécurité sociale au ministère chargé des affaires sociales ou son représentant, et un représentant du ministre chargé de la défense. La présidence de la commission est assurée pour un an, alternativement, par le directeur général de la santé au ministère chargé de la santé ou son représentant et par le directeur des enseignements supérieurs au ministère chargé de l’éducation nationale ou son représentant.
55 () IGAS, Rapport n°2006-002 « Mission relative à l’organisation juridique, administrative et financière de la formation continue des professions médicales et paramédicales, janvier 2006.
56 () Ce comité de coordination a pour mission de formuler à l'attention des conseils nationaux tous avis et propositions susceptibles d'améliorer l'efficacité des actions menées, d'harmoniser leur fonctionnement ainsi que la cohérence des procédures et des critères d'agrément et de procéder aux études et travaux que les conseils nationaux décident de lui confier.
57 () Il est simplement indiqué que le code fera l’objet d’une évaluation par les parties signataires à l’issue d’un délai de deux ans puis tous les deux ans. Au vu de cette évaluation, « le cas échéant, le présent code pourra faire l’objet de modifications. »
58 () Les autres sont le rôle et la place des praticiens en cas de crise sanitaire, la prévention vaccinale, la prévention et dépistage des cancers, la prévention des risques environnementaux, comportementaux et professionnels.
59 () Sur ce point, le projet de décret modificatif n’apporte que des corrections de forme à ces dispositions.
60 () Uniquement pour la médecine de ville.
61 () Fin mars 2007, le CEPS avait pris la décision de limiter la fréquence de la visite médicale pour quatre classes : les statines, les médicaments de l’asthme, les sartans et les antibiotiques fluoroquinolones. Pour ces quatre classes, l’objectif est une baisse étalée sur trois ans (-6%, -10%, -12%, avec 2005 comme année de référence). Le CEPS devrait être amené à prononcer « incessamment » des baisses de prix pour les produits pour lesquels l’objectif n’a pas été atteint (la décision n’était pas encore confirmée à la fin mars 2007, mais il s’agissait sans doute de certains produits de la classe des sartans).
62 () Ropinirole dans le traitement des jambes sans repos ; candésartan dans l’insuffisance cardiaque ; rimonabant dans l’obésité ou le surpoids ; l’ézétimibe dans l’hypercholestérolémie ; l’association amlodipine - atorvastatine chez les patients hypertendus.
63 () Exemple : Macugen et Tysabri.
64 () Un article a été introduit en ce sens par les parlementaires dans la loi du 27 février 2007 (article 17).
65 () Il en va ainsi par exemple du critère 26 sur l’absence de considérations promotionnelles dans la sélection, l’ordre et la présentation des médicaments ou du critère 62 sur l’absence d’affichage de publicité.
66 () La HAS précise en effet dans sa réponse que d’autres bases de données concernant le médicament peuvent aussi être utilisées dans les LAP, mais pour d’autres fonctions. Elles peuvent porter sur des produits non concernés par la certification (homéopathie, cosmétiques, etc.).
67 () Cette charte de qualité reprend les huit critères du référentiel de certification qui mettent en jeu la base médicaments.
68 () Il s’agit pour l’essentiel des SMR et des ASMR, absents de deux des trois bases examinées (Claude Bernard et Vidal).
69 () Les autres domaines prioritaires sont les arrêts de travail et les prescriptions remboursées à 100 % par l’assurance-maladie obligatoire (ALD).
70 () Le deuxième thème d’engagement porte sur les indemnités journalières.
71 () En toute rigueur, il faudrait tenir compte de la taille des conditionnements voire du dosage des médicaments.
72 () Source CNAMTS, en nombre de prescriptions d'antibiotiques pour 1000 habitants, corrigé des pics épidémiques (calculs Institut Pasteur).
73 () Note au Ministre du 15 mai 2006
74 () Ces évaluations sont réalisées en retenant comme tendance la croissance annuelle moyenne entre 2002 et 2005, en neutralisant l’effet des déremboursements pour les psychotropes (les autres classes visées par l’avenant 12 n’ayant pas fait l’objet de déremboursements) et en corrigeant des effets de stockage des génériques (statines, IPP, antibiotiques) et de l’épidémie de grippe (antibiotiques).
75 () Pour les antibiotiques, l’effet boites, une fois pris en compte l’épidémie de grippe de 2005 et le stockage des génériques en décembre 2005, est ramené à - 3,9%.
76 () Des objectifs chiffrés sont définis pour 2008 : « diminution de 10% du nombre de personnes ayant une prescription de benzodiazépine à demi-vie longue ; diminution de 10% du nombre de personnes ayant eu une prescription de vasodilatateur. »
77 () Mise au point sur la prévention de la iatrogénie médicamenteuse chez les personnes âgées, AFSSAPS, juin 2005 ; programme d’évaluation des pratiques professionnelles « prescription médicamenteuse chez le sujet âgé », HAS, octobre 2005.
78 () Tous deux datés de 2005.
79 () Efficacité des méthodes de mise en œuvre des recommandations médicales, ANAES janvier 2000.
80 () à l’exception des médecins à faible activité (3 500 à 4 000).
81 () La MSA a mis en place une action sur les statines en 2005 qui a dû être arrêtée.
82 () Santé Canada, « Vieillissement et aînés : Prévention des blessures non intentionnelles chez les aînés », Ottawa, ministre des Travaux publics et des Services gouvernementaux du Canada, 2002.
83 () Il s’agit de documents visant à répondre aux interrogations du grand public, intitulés « Vous et votre traitement » ou « Questions/réponses ».
84 () Intitulée : « Internet santé, faites les bons choix ».
85 () Selon Prescrire, certaines études montreraient que fidéliser un patient serait six fois moins coûteux que d’en trouver un nouveau. Les entreprises pharmaceutiques perdraient chaque année 30 milliards de dollars de ventes (sur 600 milliards) parce que les patients interrompent leur traitement.
86 () Il est à noter qu’un groupe d’experts de la Federal Drug Administration (FDA), équivalent américain de l’AFSSAPS, a refusé la mise sur le marché d’Acomplia en juin 2007, ce qui a conduit le laboratoire à retirer son dossier d’introduction de ce médicament aux Etats-Unis. De son côté, l'Agence européenne du médicament (EMEA) a initié un réexamen de ce produit qu'elle avait autorisé à la commercialisation dans l'Union européenne en juin 2006. Elle a annoncé qu'elle rendrait son avis sur le médicament à l'issue de la réunion de son comité d'experts qui se tiendra en juillet 2007.
87 () Le Collectif Europe et Médicament a notamment demandé l’interdiction des programmes d’aide à l’observance, comme toute publicité directe auprès du public pour les médicaments de prescription (art. L. 5122-6 du CSP), insistant sur le fait que l’information des patients doit être assurée par les professionnels de santé et les services sanitaires et sociaux, à partir de données solides, comparatives et émanant de sources indépendantes des firmes.
88 () A titre d’exemple, la promotion des génériques a été lancée en France en 1996, alors que l’Allemagne avait commencé dès les années 1980.
89 () Source : point d’information mensuel du 6 juin 2006.
90 () Points de repère n°2, août 2006.
91 () Le répertoire des génériques se définit comme la liste des spécialités princeps généricables et des génériques qui leur sont attachés. C’est au sein de ce répertoire que s’exerce le droit de substitution des pharmaciens.
92 () IRDES, « Les laboratoires pharmaceutiques face à l’arrivée des génériques : quelles stratégies pour quels effets ? », Questions d’économie de la santé, n°84, octobre 2004.
93 () Cette directive vient de finir d’être transposée par la loi n°2007-248 du 26 février 2007 portant diverses dispositions d’adaptation au droit communautaire dans le domaine du médicament.
94 () Cette notion avait déjà été transposée en droit français par le décret n°2005-156 du 18 février 2005.
95 () Source : IMS Health, EPPM
96 () Il s'agit essentiellement des statines, d'IPP (inhibiteurs de la pompe à protons), d'hypnotiques et d'anxiolytiques, des molécules chères et encore peu substituées.
© Assemblée nationale