

|
|
|
N° 3208
--
ASSEMBLÉE NATIONALE
CONSTITUTION DU 4 OCTOBRE 1958
ONZIÈME LÉGISLATURE
Enregistré à la Présidence de l'Assemblée nationale le 27 juin 2001
RAPPORT D'INFORMATION
DÉPOSÉ
en application de l'article 145 du Règlement
PAR LA MISSION D'INFORMATION COMMUNE PRÉPARATOIRE
AU PROJET DE LOI DE RÉVISION DES « LOIS BIOÉTHIQUES » DE JUILLET 1994 (1)
Président
M. BERNARD CHARLES,
Rapporteur
M. ALAIN CLAEYS
Députés.
--
TOME I
Rapport
(1) La composition de cette mission figure au verso de la présente page.
Bioéthique.
La Mission d'information commune préparatoire au projet de loi de révision des lois « bioéthique » de juillet 1994 est composée de : M. Bernard Charles, président ; M. Jean-Michel Dubernard, M. Roger Meï, vice-présidents ; Mme Yvette Benayoun-Nakache, Mme Christine Boutin, secrétaires ; M. Bernard Accoyer, M. Pierre Albertini, M. André Aschieri, M. Philippe Auberger, Mme Martine Aurillac, Mme Roselyne Bachelot-Narquin, M. Jean-Paul Bacquet, M. Serge Blisko, M. Bruno Bourg-Broc, M. Jean-Paul Bret, M. Yves Bur, M. Jérôme Cahuzac, M. Alain Calmat, M. Christophe Caresche, Mme Nicole Catala, M. Jean-Pierre Delalande, M. Patrick Delnatte, M. Jean-Jacques Denis, M. Bernard Derosier, M. Dominique Dord, M. René Dosière, M. Renaud Dutreil, M. Michel Etiévant, M. Claude Evin, M. Jacques Floch, M. Jean-Pierre Foucher, Mme Jacqueline Fraysse, M. Hervé Gaymard, M. André Gerin, Mme Odette Grzegrzulka, M. Jean-Claude Guibal, Mme Catherine Génisson, M. Pierre Hellier, Mme Bernadette Isaac-Sibille, Mme Muguette Jacquaint, M. Armand Jung, M. Jérôme Lambert, M. Jean-Yves Le Déaut, M. Jean-Marie Le Guen, Mme Jacqueline Mathieu-Obadia, M. Jean François Mattei, M. Jean-Pierre Michel, M. Philippe Nauche, M. Robert Pandraud, M. Bernard Perrut, M. Jean-Luc Préel, M. Didier Quentin, Mme Yvette Roudy, Mme Odette Trupin, M. Alain Veyret, M. Alain Vidalies.
SOMMAIRE
-
Pages
INTRODUCTION 11
PREMIÈRE PARTIE : LES LOIS DE BIOÉTHIQUE EN QUÊTE D'UN NOUVEL ÉQUILIBRE 15
I.- LA PERMANENCE DES DEVOIRS DU PARLEMENT EN MATIÈRE BIOÉTHIQUE : ASSURER LE RESPECT DES DROITS DE L'HOMME 15
A.- L'ÉTHIQUE DE RESPONSABILITÉ DU LÉGISLATEUR 15
1.- Le principe de responsabilité 15
2.- Le principe de précaution revisité 16
B.- UNE MISSION D'INFORMATION SOUCIEUSE D'ÉCLAIRER LES CHOIX POSSIBLES 17
1.- L'inscription dans un débat public et transparent 17
2.- La mise en lumière des enjeux 17
C.- COMMENT RÉPONDRE À DES ASPIRATIONS CONTRADICTOIRES ? 19
1.- Quelles sont ces aspirations ? 19
2.- Faire des choix en édictant des normes 20
II.- LE DROIT, LA SCIENCE, L'ÉCONOMIE : QUELLES NOUVELLES CONTRAINTES POUR LE LÉGISLATEUR ? 21
A.- L'ÈRE DES RÉVOLUTIONS BIOTECHNOLOGIQUES : LA LOI FACE À LA SCIENCE EN MOUVEMENT 21
1.- Le clonage humain 21
a) Le règne de la confusion 21
b) Une distinction indispensable 23
c) Le clonage reproductif humain : une perspective inacceptable 24
d) Sa très nette condamnation 25
e) Le débat outre-Atlantique 25
f) L'ouverture du débat sur le « clonage dit thérapeutique » 26
2.- La révolution génétique : du décryptage du génome aux thérapies cellulaires 29
a) Le décryptage du génome humain 29
b) Quel horizon pour les perspectives thérapeutiques ? 31
B.- L'HYDRE JURIDIQUE : LA LOI CONFRONTÉE À UN DROIT INTERNATIONAL EN CONSTRUCTION 32
1.- Les sources internationales du droit de la bioéthique : de la « soft law » à un droit relativement contraignant 32
a) Le rôle précurseur de la « soft law » 32
b) Le cadre plus contraignant des conventions : la Déclaration universelle de l'Unesco sur le génome humain et les droits de l'homme et la convention d'Oviedo de 1997 34
2.- Le droit national face aux sources externes du droit de la bioéthique 40
a) La reconnaissance de la valeur du droit international en droit interne 40
b) La difficulté d'assurer la conformité de la loi française à des textes internationaux ambivalents 42
c) Un chemin étroit pour assurer la cohérence des normes internes et externes 44
C.- LES LOIS BIOÉTHIQUES FACE AUX ENJEUX ÉCONOMIQUES 46
1.- Le développement du marché des biotechnologies 46
a) Le poids de ce secteur 46
b) La structure et l'évolution du secteur des biotechnologies 48
2.- L'avance indéniable des États-Unis 49
a) La dynamique conjointe de la science et de l'économie aux États-Unis 49
b) La ruée vers l'Amérique ? 52
DEUXIÈME PARTIE : L'ASSISTANCE MÉDICALE À LA PROCRÉATION (AMP) : UN PARCOURS DIFFICILE QUI NÉCESSITE DES AMÉLIORATIONS 53
I.- LA RÉALITÉ CHIFFRÉE DE L'AMP 54
II.- LES CONDITIONS DE RECOURS À L'AMP ET L'OPPORTUNITÉ DE LES RÉVISER 59
A.- LES CONDITIONS MÉDICALES SONT GLOBALEMENT SATISFAISANTES 59
B.- LES CONDITIONS SOCIALES FONT L'OBJET DE DEMANDES D'ÉLARGISSEMENT QUI SEMBLENT POUR CERTAINES SOUHAITABLES 61
1.- Les conditions tenant à la situation des futurs parents et des donneurs de gamètes doivent être adaptées 61
2.- Le transfert post mortem d'embryon doit être autorisé sous réserve du consentement du père de son vivant 62
3.- Les nouvelles demandes exprimées par les personnes homosexuelles ou transsexuelles semblent difficiles à satisfaire 65
III.- L'APPLICATION PRATIQUE DES TECHNIQUES D'AMP 67
A.- L'ÉVALUATION DES NOUVELLES TECHNIQUES D'AMP FAIT GRAVEMENT DÉFAUT 67
1.- Le cas de l'ICSI 67
2.- Les leçons pour l'avenir 71
B.- LES IMPERFECTIONS DE LA TECHNIQUE ONT CRÉÉ DES PROBLÈMES MÉDICAUX OU ÉTHIQUES 74
1.- Le recours excessif aux traitements de stimulation ovarienne 74
2.- Les difficultés liées à la conception d'embryons au-delà des besoins réels 76
3.- L'aide psychologique apportée aux couples est insuffisante 82
C.- LES PROBLÈMES SPÉCIFIQUES LIÉS AU RECOURS À UN TIERS DONNEUR 83
1.- La gestion du don de gamètes 83
a) Une situation de pénurie inquiétante 83
b) Une information et une sensibilisation insuffisantes en faveur du don 86
c) Les problèmes spécifiques liés au don d'ovocytes 86
d) L'alternative possible pour répondre au problème de l'insuffisance des dons de sperme 89
2.- La question de l'anonymat et du secret 90
IV.- LE TROP FAIBLE CONTRÔLE DES CENTRES D'AMP 94
A.- LE RÔLE CONFIÉ À LA COMMISSION NATIONALE DE MÉDECINE ET DE BIOLOGIE DE LA REPRODUCTION ET DU DIAGNOSTIC PRÉNATAL (CNMBRDP) 95
B.- LES FAIBLESSES ET LACUNES DE LA CNMBRDP 100
C.- LA DEMANDE QUASI-UNANIME DE CRÉATION D'UNE AGENCE DOTÉE DE RÉELS MOYENS POUR SUPPLÉER LA CNMBRDP 103
D.- L'EXPÉRIENCE CONCLUANTE DE LA HUMAN FERTILIZATION AND EMBRYOLOGY AUTHORITY (HFEA) BRITANNIQUE 107
E.- LES AMÉNAGEMENTS MINEURS NÉCESSAIRES AU DISPOSITIF D'ENCADREMENT DES CENTRES D'AMP 111
TROISIÈME PARTIE : LA RECHERCHE SUR L'EMBRYON 113
I.- L'INADÉQUATION DE LA LÉGISLATION DE 1994 114
II.- DES PERSPECTIVES DE RECHERCHE IMMENSES 118
A.- DES PISTES DE RECHERCHES NOMBREUSES 121
1.- Le pouvoir des cellules souches 122
2.- Les cellules souches embryonnaires 124
3.- Les cellules souches adultes 126
4.- Les cellules souches f_tales 129
5.- Le « clonage thérapeutique » 130
B.- EN L'ÉTAT DES CONNAISSANCES SCIENTIFIQUES ACTUELLES, IL REVIENT AU PARLEMENT DE DÉTERMINER LE CHAMP DE LA RECHERCHE AUTORISÉE 133
C.- DES ESPOIRS THÉRAPEUTIQUES CONSIDÉRABLES 138
1.- Des expérimentations réussies sur l'animal 139
2.- Des essais d'application chez l'homme. 140
a) Le succès de la thérapie sur les « bébés bulles » 140
b) Une récente réussite dans l'utilisation de cellules f_tales 141
c) Le pari des scientifiques pour l'avenir 142
III.- LES CONDITIONS INDISPENSABLES D'ENCADREMENT DE LA RECHERCHE SUR LES CELLULES SOUCHES 145
A.- DES INTERDITS À FIXER 146
1.- L'interdiction d'implanter un embryon ayant fait l'objet de recherches 146
2.- L'interdiction de toute thérapie germinale 146
B.- DES LIMITES ET DES CONDITIONS CLAIRES À POSER 148
1.- Le consentement des parents au don de leurs embryons surnuméraires à la recherche 148
2.- Le stade de développement de l'embryon sur lequel la recherche serait autorisée doit être précis et limité 149
3.- La nécessité absolue de protéger les femmes qui donneraient leurs ovocytes à la recherche 151
4.- La transparence des recherches doit être assurée 152
5.- La finalité précise de la recherche et l'absence d'alternative à l'utilisation de l'embryon ou de ses cellules doivent être prouvées 152
C.- GARANTIR LE RESPECT DE CES INTERDITS ET DE CES LIMITES PAR UN ENCADREMENT RIGOUREUX 153
QUATRIÈME PARTIE : DON ET UTILISATION DES ÉLÉMENTS DU CORPS HUMAIN 155
I.- LES LOIS DE 1994 ONT ÉTÉ ADOPTÉES DANS UN CONTEXTE DÉFAVORABLE À L'ACTIVITÉ DE TRANSPLANTATION 155
A.- UN CLIMAT DE CRISE 155
B.- UNE CONFIANCE ENTAMÉE 157
1.- Le cas du coma dépassé d'Amiens 157
2.- Le cas des multi-prélèvements 157
3.- La rumeur des voleurs d'organes 158
II.- LA RÉPONSE DU LÉGISLATEUR : DONNER UN STATUT AU CORPS HUMAIN 160
A.- L'INSCRIPTION DU RESPECT DU CORPS HUMAIN DANS LE CODE CIVIL : DES PRINCIPES GÉNÉRAUX 160
B.- L'APPLICATION DE CES PRINCIPES AU DON ET À L'UTILISATION DES ÉLÉMENTS ET PRODUITS DU CORPS HUMAIN. 161
III.- LA SITUATION ACTUELLE 161
A.- UNE AMÉLIORATION GLOBALE DE LA SITUATION 161
1.- L'activité de prélèvement et de greffe a retrouvé son niveau de départ 161
2.- Apaiser l'inquiétude 162
B.- LA PERSISTANCE D'UNE PÉNURIE D'ORGANES 164
1.- La pénurie : une inadéquation entre des besoins et des ressources 164
2.- Les besoins 164
3.- L'offre 166
4.- Le recours aux donneurs vivants 171
C.- L'ORGANISATION DE L'ACTIVITÉ DE PRÉLÈVEMENT PEUT ÊTRE AMÉLIORÉE 172
D.- LA TRANSPARENCE EST ASSURÉE PAR L'ÉTABLISSEMENT FRANÇAIS DES GREFFES 175
1.- La liste des patients en attente d'une transplantation ou d'une greffe 175
2.- La répartition et l'attribution des greffons 175
3.- Un accompagnement de sécurité sanitaire 176
4.- Les autres missions de l'EFG 177
IV.- LA QUESTION DU CONSENTEMENT, CONDITION DE L'AMÉLIORATION DU DISPOSITIF ? 178
A.- LA PLACE CROISSANTE DU CONSENTEMENT EN MATIÈRE MÉDICALE 178
B.- LE CONSENTEMENT AUX SOINS 179
1.- Qui s'exprime ? 180
2.- Pour quelle expression ? 182
3.- Les autres cas d'exigence du consentement 182
4.- Le consentement au prélèvement d'organe : cas du donneur vivant 182
a) L'élargissement de la catégorie des donneurs vivants et la notion de proche 183
b) Les comités d'experts 184
c) La notion d'urgence 188
5.- Le consentement au prélèvement d'organe : cas du donneur décédé 189
a) Consentement présumé ou autorisation de la famille ? 189
b) Le Registre des refus : un bilan prématuré 190
6.- Les tissus, cellules et produits d'origine humaine 192
a) Sécurité sanitaire et vigilance 192
b) La moelle osseuse 193
c) Le statut des déchets opératoires 194
d) Les tissus f_taux 195
V.- FAVORISER LE DON D'ORGANES 197
A.- COMMUNIQUER EN FAVEUR DU DON D'ORGANES : TENIR COMPTE DES RÉALITÉS HUMAINES 197
1.- Le prélèvement et la greffe : une dimension symbolique très forte 198
2.- Les circonstances du refus : un moment traumatisant 198
B.- L'INFORMATION DU PUBLIC : ASSOCIER LE PLUS GRAND NOMBRE D'ACTEURS 200
1.- Une responsabilité des pouvoirs publics 200
2.- Associer l'Éducation nationale 202
3.- Les associations participent à l'information 203
C.- LA FINALITÉ DE L'INFORMATION : DIMINUER LE NOMBRE DE REFUS 204
1.- À qui s'adresse le message ? 205
2.- Le contenu du message 205
D.- LA RECONNAISSANCE DE LA SOCIÉTÉ : LE DONNÉ ET LE RENDU 207
VI.- FINALITÉ SCIENTIFIQUE, MÉDICALE OU THÉRAPEUTIQUE 210
A.- UNE NÉCESSAIRE CLARIFICATION 211
1.- Finalité thérapeutique, finalité médicale 211
2.- Finalité scientifique 211
B.- L'AUTOPSIE, UN RÉVÉLATEUR DE CETTE CONFUSION 212
1.- L'utilité de l'autopsie 212
2.- L'application de la loi de 1994 serait la cause de son déclin 213
3.- La finalité thérapeutique ne doit pas devenir un alibi éthique 215
4.- La relation malade-médecin 217
CINQUIÈME PARTIE : LA MÉDECINE PRÉDICTIVE 219
I.- LA MÉDECINE PRÉDICTIVE AU CARREFOUR DES CRAINTES ET DES ESPOIRS 220
A.- DE LA SÉLECTION À LA DISCRIMINATION 220
1.- Vers un nouvel eugénisme ? 220
2.- La crainte et la condamnation de la discrimination génétique 224
B.- LES ESPOIRS LIÉS À LA DÉTECTION DES MALADIES D'ORIGINE GÉNÉTIQUE 225
1.- Les avancées récentes 225
2.- Relativiser la portée prédictive des examens génétiques 226
3.- Une réflexion nécessaire sur l'accompagnement des diagnostics 227
II.- LA PRATIQUE RASSURANTE DU DIAGNOSTIC PRÉIMPLANTATOIRE ET PRÉNATAL 230
A.- UN DÉBAT RÉCURRENT AUTOUR DE LA QUESTION DES FINALITÉS ET DES CONSÉQUENCES DU DIAGNOSTIC PRÉIMPLANTATOIRE 230
B.- L'ACTIVITÉ DES CENTRES DE DIAGNOSTIC PRÉIMPLANTATOIRE 232
C.- L'EFFICACITÉ CONSTATÉE DE LA LÉGISLATION EN VIGUEUR CONTRE TOUTE DÉRIVE EUGÉNIQUE 234
III.- LES EXAMENS GÉNÉTIQUES ET LES ASSURANCES 236
A.- DES INTÉRÊTS CONTRADICTOIRES 236
1.- Les contraintes propres au marché de l'assurance 236
2.- Le face-à-face inégal entre assureurs et souscripteurs 237
B.- LA PRATIQUE ACTUELLE DES ASSUREURS 238
1.- Le moratoire de 1999 238
2.- La mise en avant par les assureurs du modèle britannique 238
C.- LE DÉBAT AUX ÉTATS-UNIS 239
1.- Une question âprement débattue 239
2.- Vers une législation fédérale ? 240
D.- L'INTERVENTION DU LÉGISLATEUR 240
1.- Les préconisations insatisfaisantes du Conseil d'État 240
2.- La nécessité de légiférer 242
IV.- LES EXAMENS GÉNÉTIQUES ET LES EMPLOYEURS 243
A.- L'ÉVALUATION DU RISQUE DE DISCRIMINATION 243
1.- Les tests génétiques au service des employeurs ? 243
2.- La position du Conseil d'État : relativiser le risque de discrimination 244
B.- LA NÉCESSITÉ D'INTERDIRE DANS LA LOI TOUTE DISCRIMINATION À L'EMBAUCHE FONDÉE SUR LE PATRIMOINE GÉNÉTIQUE 246
1.- Une position du Conseil d'État peu convaincante 246
2.- Affirmer le principe de non-discrimination 246
SIXIÈME PARTIE : LA BREVETABILITÉ DU GÉNOME HUMAIN : DES ORIENTATIONS POUR TRAITER D'UN SUJET GRAVE 247
I.- LE DROIT DES BREVETS FACE AU VIVANT 248
A.- LE DROIT DES BREVETS : UN PROCÉDÉ CLASSIQUE POUR PROTÉGER LES INVENTEURS 248
1.- L'objet paradoxal du droit des brevets : protéger les inventeurs en rendant publiques les inventions 248
a) La fonction du droit des brevets 248
b) La construction de ce droit 249
2.- La protection européenne des brevets 250
a) L'Office européen des brevets 250
b) Les missions de l'Office 250
3.- Les brevets aux États-Unis 251
a) L'United States Patent and Trademark Office : une agence fédérale puissante et respectée 251
b) La règle du premier inventeur, une spécificité du droit des brevets américain 252
B.- LA PROBLÉMATIQUE ANCIENNE DE LA BREVETABILITÉ DU VIVANT 252
1.- Des précédents historiques et des initiatives américaines dans les années 80 252
2.- Les critères de brevetabilité du vivant 253
II.- LE DROIT DES BREVETS FACE À L'HUMAIN 255
A.- LA PROBLÉMATIQUE NOUVELLE DU GÉNOME HUMAIN : DE LA DÉMARCHE SCIENTIFIQUE À LA LOGIQUE ÉCONOMIQUE 255
1.- La logique de valorisation des découvertes par les entreprises américaines 255
2.- Le dépôt massif de demandes de brevets 256
B.- LE DÉBAT EUROPÉEN AUTOUR DE LA DIRECTIVE 98/44/CE 257
1.- La genèse mouvementée de la directive 257
2.- Les ambiguïtés de la rédaction 258
3.- La pratique contestable de l'Office européen des brevets 260
4.- Le débat en France 261
a) L'appel contre la brevetabilité des gènes humains 261
b) La position du Comité consultatif national d'éthique 262
c) La position du ministre de la recherche 263
5.- Le débat en Allemagne 265
a) Le dépôt d'un projet de loi par le Gouvernement fédéral 265
b) La position du Bundesrat 266
c) La position de la commission « droit et éthique de la médecine moderne » au Bundestag 266
C.- LE DÉBAT AUX ÉTATS-UNIS 268
1.- La nature spécifique de ce débat 268
2.- La pratique juridique plus stricte de l'USPTO 269
III.- QUATRE ORIENTATIONS POUR LE RÉGLEMENT DE CETTE QUESTION 271
A.- INTRODUIRE DES LIMITES ÉTHIQUES DANS LE DROIT DES BREVETS 271
1.- Réaffirmer les règles actuelles en matière d'ordre public et de bonnes m_urs 271
2.- Introduire des préoccupations éthiques dans le cadre de la procédure d'attribution des brevets 272
B.- PRÉSERVER LE LIBRE ACCÈS À LA CONNAISSANCE 272
1.- La déclaration Clinton-Blair 272
2.- Vers un domaine public en génomique ? 273
C.- S'OPPOSER À LA CONSTITUTION DE MONOPOLES 275
1.- La pratique des licences et des brevets dépendants 275
2.- Les moyens de s'opposer à la constitution des monopoles 275
D.- REMETTRE EN CAUSE LES BREVETS DE PRODUITS SUR LE VIVANT PAR UNE DÉMARCHE EUROPÉENNE ET INTERNATIONALE 276
1.- Remettre en cause les brevets de produits sur le vivant 276
2.- Engager une nouvelle politique à l'échelon européen et international 277
EXAMEN DU RAPPORT 279
ANNEXES 289
MESDAMES, MESSIEURS,
Une femme de 62 ans accouche en France après avoir bénéficié d'un don d'ovule aux États-Unis et avoir été inséminée par le sperme de son frère, lui-même ayant sollicité, de son côté, une mère porteuse pour donner naissance à un autre enfant. Le gourou français d'une secte établie au Canada témoigne devant le Congrès des États-Unis sur son projet de créer de toutes pièces un clone humain.
Ces deux événements récents montrent combien il est difficile aujourd'hui d'aborder les questions bioéthiques dans la sérénité. La crainte de voir l'homme dépassé par ses propres découvertes n'est pas récente mais elle prend désormais des proportions sans précédent.
Or, la recherche est un facteur de progrès. Trop souvent encore, les avancées de la science engendrent des réactions de méfiance, de crainte, voire de rejet. Le temps n'est certes plus celui du procès de Galilée. L'État, les autorités morales n'exercent plus la direction des esprits, en s'opposant au progrès de la science et de la connaissance. Ces phénomènes sont sans doute plus diffus, moins explicites. La médiatisation de certaines découvertes suscite à la fois de formidables attentes mais aussi des peurs, face à des innovations, à l'évidence, sans précédent.
Pourtant, les découvertes récentes en matière de médecine et de biologie laissent libre cours à une grande espérance : la maladie et la souffrance perdent chaque jour du terrain. Peut-on rejeter ce mouvement bénéfique ? Assurément non. Pour autant, il nous faut également répondre aux craintes qui peuvent apparaître ça et là.
Pour le Parlement, le meilleur moyen de relever ce défi est d'appliquer un principe de précaution qui n'est nullement le synonyme d'une mise sous le boisseau de la science. Au contraire, la meilleure des précautions est encore de disposer de connaissances précises pour que le politique puisse prendre les décisions nécessaires dans la plus grande clarté. L'application de ce principe ainsi revisité comme une règle d'action suppose la mise en _uvre de son corollaire nécessaire, l'évaluation.
Il appartient alors au politique de tracer le cadre dans lequel la recherche et les pratiques médicales peuvent être menées. La définition de ce cadre implique que le Parlement fixe des interdits, des limites et mette en place des mécanismes en vue d'assurer le respect des règles ainsi édictées.
Mais, ces questions ne peuvent se résoudre à la simple application de règlements et au suivi de procédures prédéterminées. Derrière ces normes, ces protocoles, apparaissent des femmes et des hommes, des médecins et des patients, qui ont à résoudre quotidiennement ces tensions qui caractérisent la bioéthique. Le parcours d'un couple s'engageant dans un processus d'assistance médicale à la procréation est difficile, physiquement et psychologiquement. Comment le soutenir et l'aider à faire les choix nécessaires tout au long de cette démarche ? Comment réagir devant les attentes des personnes malades, devant leurs souffrances, leurs espoirs ? Peut-on leur opposer des règles impersonnelles qui ne tiendraient pas compte de leurs difficultés quotidiennes ? Un effort doit être engagé vers eux en ce domaine, pour les accompagner dans les meilleures conditions possibles.
Emprunter cette voie étroite est pour le législateur un défi qu'il ne peut relever seul. La révision des lois bioéthiques ne peut être une démarche dans laquelle le Parlement se tiendrait éloigné des aspirations de la société. Le législateur doit susciter une discussion publique et la réflexion de chacun sur ces sujets. Il est nécessaire que le Parlement puisse inscrire son action dans la transparence et dans un débat largement ouvert aux citoyens.
C'est conscient de ces devoirs que le Parlement a décidé d'adopter le principe, salué par tous, d'une révision régulière des lois bioéthiques. L'article 21 de la loi n° 94-654 du 29 juillet 1994 a ainsi prévu que cette loi ferait l'objet, après une évaluation de son application par l'Office parlementaire d'évaluation des choix scientifiques et technologiques, d'un nouvel examen par le Parlement, dans un délai maximal de cinq ans après son entrée en vigueur.
Cette évaluation a eu lieu dans les délais requis. L'Office parlementaire d'évaluation des choix scientifiques et technologiques a remis deux rapports, l'un portant sur le clonage, la thérapie cellulaire et l'utilisation thérapeutique des cellules embryonnaires (1), l'autre sur l'application de la loi n° 94-654 du 29 juillet 1994 relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal (2).
Parallèlement, aux fins de préparer le projet de loi de révision des textes de 1994, le Premier ministre, M. Lionel Jospin, a saisi le Conseil d'État d'une étude sur ce sujet. La Haute juridiction a rendu son rapport en novembre 1999 (3). Le Conseil d'État s'est ici strictement conformé au champ défini par la lettre de mission du Premier ministre. Ainsi, selon ses termes, ce rapport ne pouvait se limiter à la seule loi n° 94-654 du 29 juillet 1994 relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal. La loi n° 94-653 relative au respect du corps humain devait entrer dans le cadre de cette étude.
Le rapport étant rendu, formulant une série de propositions sujettes à débat, le Parlement se devait de préparer l'examen probable du projet de loi révisant les lois de 1994.
C'est dans cette perspective que fut créée en mai 2000 une Mission d'information chargée de préparer la révision de ces lois. Cette Mission a été l'une des plus actives de la législature. Pour preuve, elle a procédé pendant un an à l'audition de près de quatre-vingt-dix personnalités, pendant un total de plus de cinquante heures.
Le présent rapport rend compte des travaux de cette Mission menée en France mais aussi à l'étranger, que ce soit en Allemagne ou aux États-Unis. Car cette année passée à préparer l'examen du projet de loi portant révision des lois de 1994 a permis de saisir combien ces questions ne peuvent être traitées du seul point de vue national. La science mais aussi les comportements sociaux et individuels tout autant que l'évolution de la règle de droit font éclater ce cadre trop restreint, pour ouvrir le champ des problématiques à l'échelle mondiale.
Face à ces évolutions rapides et difficiles à appréhender, quel doit être le rôle du législateur ? Peut-il encore adopter une loi qui puisse, dans son contenu et son application, apparaître crédible ? Cette année d'auditions nous rend enclin à répondre par l'affirmative à cette légitime interrogation, pourvu que le législateur ait à l'esprit trois préoccupations.
Il lui appartient, en premier lieu, de mesurer son rôle et sa responsabilité face au débat éthique. Le Parlement doit adopter des textes qui créent un équilibre entre plusieurs valeurs et aspirations.
Parallèlement, sous peine de ne pas être entendu, le législateur se doit de prendre en compte trois séries d'évolution dans les domaines scientifiques, socio-économiques et juridiques.
Enfin, il lui incombe de mettre en _uvre ses deux missions essentielles : l'organisation d'un débat institutionnel qui permette à la société de voir émerger les enjeux relatifs à la bioéthique ; établir des normes, résultats de cette discussion publique, au plus près des enjeux et dans l'équilibre des valeurs.
Dans le cadre de ce rapport, il sera utile d'analyser les conditions dans lesquelles le législateur peut assumer ces trois préoccupations. Dans un second temps, il conviendra de procéder à l'évaluation des lois de 1994, en se fondant sur les travaux menés par la Mission d'information depuis maintenant plus d'un an.
PREMIÈRE PARTIE :
LES LOIS DE BIOÉTHIQUE EN QUÊTE D'UN NOUVEL ÉQUILIBRE
I.- LA PERMANENCE DES DEVOIRS DU PARLEMENT EN MATIÈRE BIOÉTHIQUE : ASSURER LE RESPECT DES DROITS DE L'HOMME
Le législateur, et plus largement le politique, doit concilier deux impératifs : demeurer fidèle à certains principes, tout en étant attentif aux conséquences qu'une telle attitude peut entraîner. Les questions bioéthiques rendent encore plus aiguë cette tension permanente. La résolution de cette équation complexe passe par une ligne de conduite claire : le respect des droits de l'homme dont le Parlement a la charge, en vertu de la Déclaration de 1789, elle-même. C'est à la loi d'assurer la conciliation de principes contradictoires. C'est dans cet exercice que la politique révèle son essence même, qui peut se résumer en un mot : choisir.
A.- L'ÉTHIQUE DE RESPONSABILITÉ DU LÉGISLATEUR
1.- Le principe de responsabilité
Aujourd'hui, en réponse aux préventions de Rabelais, la science et la conscience semblent intimement liées. Les réflexions éthiques menées par nombre de savants en témoignent. Cette préoccupation constante n'est pas toujours perçue à sa juste mesure. Pourtant, la définition et le respect des principes éthiques ne peuvent être le fait de la seule communauté scientifique. Ces questions relèvent de l'ensemble du corps social et c'est dans un cadre démocratique que les éventuelles limites apportées à l'activité scientifique doivent être posées.
On peut s'interroger sur les moyens dont la société dispose pour maîtriser l'activité scientifique, lorsque celle-ci pourrait aller au-delà de l'acceptable. Le droit est, à l'évidence, l'instrument privilégié pour mener à bien cette mission. Son rôle est à la fois de fixer des interdits et les sanctions applicables aux contrevenants, mais aussi de réguler des pratiques, en instituant des modalités d'encadrement.
La définition des règles de droit, dans ce domaine, ne peut être fondée sur des a priori, sans lien avec la réalité sociale. La norme doit être l'expression de ce que la société tolère mais aussi de ses aspirations. Or les demandes sociales ne sont pas toujours cohérentes. Des contradictions et des tensions apparaissent.
Il appartient alors au politique d'évaluer ces forces divergentes et de tenter de trouver un équilibre, qui se traduira par l'adoption d'une norme, censée exprimer, à un moment donné et dans un lieu déterminé, la résultante de ces forces contraires. Quelle méthode doit alors présider à l'action du politique ?
Dans deux conférences données en 1919, le sociologue allemand Max Weber nous apporte des éléments de réponse (4). Il s'y demande comment peuvent s'articuler les fonctions scientifiques et politiques. Sont-elles compatibles ou diffèrent-elles intrinsèquement ? Parallèlement, Weber traite des conditions dans lesquelles les activités humaines, et plus particulièrement politiques, peuvent s'orienter selon l'éthique. Les réponses apportées par le sociologue, dans ce texte célèbre, sont aujourd'hui encore éclairantes. Cherchant à dégager l'éthique propre à chacune de ces activités, Weber en distingue deux formes : celle de la conviction et celle de la responsabilité.
Si, pour Weber, le tenant de l'éthique de conviction semble être un pur idéaliste, à l'inverse, le partisan de l'éthique de responsabilité agit en fonction des résultats qui seront attachés à ses actions.
Le politique est, pour l'essentiel, mû par une éthique de la responsabilité. Ainsi, dans sa fonction législative, le politique se doit de mesurer les conséquences juridiques et sociales des lois qu'il adopte. Cette prescription vaut dans tous les domaines investis par le politique.
La bioéthique met le politique aux prises avec des questions de principe essentielles. La recherche sur l'embryon, le clonage, la médecine prédictive, tous les sujets abordés dans le cadre des lois bioéthiques conduisent chacun d'entre nous à interroger sa conscience. Chaque parlementaire aborde ces problèmes selon ses convictions personnelles. Mais, conformément au principe de l'intérêt général, le législateur se doit d'aller au-delà et de rechercher, dans le cadre d'un débat ouvert, le point d'équilibre qui rende compte de l'état de la société vis-à-vis de ces questions.
Le législateur doit alors suivre un principe d'action clair qui lui permette d'exercer pleinement sa responsabilité.
2.- Le principe de précaution revisité
Un principe et une méthode doivent présider à la définition de ces choix. Le principe est celui de précaution ; la méthode celle de l'évaluation. Le principe de précaution, auquel il est souvent fait référence désormais, est parfois l'objet de confusion ou de malentendu. Il ne saurait être perçu comme synonyme d'un repli sur soi et d'un refus de décider. Le principe de précaution n'est pas la consécration juridique et politique d'une méfiance généralisée à l'égard de la science. La précaution ne consiste pas à interdire toute activité scientifique, dès lors qu'un doute pourrait apparaître dans les esprits. Elle est, au contraire, un principe d'action. Elle place le politique devant sa responsabilité, celle de choisir entre plusieurs solutions. Pour ce faire, une méthode doit être suivie, celle de l'évaluation, qui permet au législateur et au citoyen d'agir en connaissance de cause, trouvant un équilibre fécond entre le progrès scientifique et le respect des principes essentiels, comme la dignité humaine.
Cependant, face aux incertitudes de la science, le politique est parfois démuni. Le meilleur moyen d'agir selon le principe de précaution est de permettre à la science d'avancer, de lever le voile obscur qui empêche toute décision.
Cette règle d'action ne peut fonctionner que si les décisions prises sont réversibles. Une fois que la science a démontré que la voie choisie n'est pas la bonne, qu'elle soulève des difficultés et se heurte à des questions de principe, le politique doit pouvoir revenir sur sa décision et interdire, le cas échéant, une pratique qu'il avait autorisée auparavant. C'est aussi pour cette raison que la révision régulière des lois bioéthiques est une nécessité.
Par ailleurs, le Parlement doit légiférer en considérant la demande sociale face aux avancées de la science. C'est en tenant compte de cet impératif que la Mission d'information a organisé ses travaux.
B.- UNE MISSION D'INFORMATION SOUCIEUSE D'ÉCLAIRER
LES CHOIX POSSIBLES
1.- L'inscription dans un débat public et transparent
La mise en _uvre de la révision des lois bioéthiques de 1994 a pu sembler prendre trop de temps pour certains. Mais le temps pris à préparer le projet de loi qui vient d'être adopté en Conseil des ministres a été celui du débat. Le Gouvernement a, en effet, pris soin de procéder à toutes les consultations nécessaires afin de mener cette discussion dans la plus grande transparence. Dans un domaine aussi sensible, le caractère public de la discussion est souhaité par tous.
Comme on l'a indiqué, le Gouvernement a demandé au Conseil d'État de présenter un rapport sur la révision des lois de 1994. Puis, il a saisi le Comité consultatif national d'éthique ainsi que la Commission nationale consultative des droits de l'homme. À chaque étape de cette procédure, les positions du Gouvernement ont été exprimées clairement et publiquement. Les avis rendus par ces différentes instances ont été, pour leur part, également rendus publics afin que le débat puisse avoir lieu.
La Mission est donc intervenue aux fins de tirer un premier bilan de toutes ces consultations et de préparer l'examen par une commission spéciale puis par l'Assemblée, dans son entier, du projet de loi. Le souci de faire de cette Mission un lieu transparent a été constant durant cette année consacrée aux questions bioéthiques.
2.- La mise en lumière des enjeux
Les auditions auxquelles la Mission a procédé se sont articulées autour de plusieurs objectifs. Il s'est agi, en premier lieu, d'entendre les acteurs institutionnels. La Mission a auditionné ainsi le sénateur Claude Huriet, co-rapporteur de l'Office parlementaire d'évaluation des choix scientifiques et technologiques, mais également les représentants du Conseil d'État, de la Commission consultative nationale des droits de l'homme et du Comité consultatif national d'éthique.
Puis la Mission a souhaité entendre des personnalités reconnues dans le débat public comme scientifiques ou comme médiateurs de ce débat. Ainsi le Professeur Jacques Testart et le Docteur Axel Kahn mais aussi des journalistes spécialistes des questions médicales ont été auditionnés.
Puis la Mission a organisé des auditions thématiques autour de quatre questions : l'embryon, l'assistance médicale à la procréation, les dons et l'utilisation des produits du corps humain, la médecine prédictive. Elle a également entamé une série de consultations autour de la loi n° 88-1188 du 20 décembre 1988 relative à la protection des personnes qui se prêtent à des recherches biomédicales. L'adoption récente d'une directive européenne en date du 26 février 2001 sur ce sujet ouvre le débat autour de la révision de cette loi de 1988. En l'état de ses travaux, la Mission ne peut conclure sur ce sujet. Il appartiendra à la commission spéciale qui prendra la suite de la Mission d'approfondir ce sujet, le Sénat procédant à un travail analogue (5).
L'aspect international de ces problèmes n'a pas été négligé comme en témoignent les deux journées consacrées à la HFEA (Human Fertilization and Embryology Authority) et aux droits européen et international. Les déplacements de la Mission à Berlin, lors d'une rencontre avec des parlementaires allemands, et de votre Rapporteur aux États-Unis, allaient également dans ce sens, ainsi que l'audition de Mme Noëlle Lenoir, présidente du Groupe européen d'éthique des sciences et des nouvelles technologies auprès de la Commission européenne. Votre Rapporteur a également participé à la réunion, organisée par la commission temporaire sur la génétique humaine du Parlement européen, avec les parlements nationaux des États membres et des pays candidats.
Comme il est maintenant d'usage, la Mission a également souhaité entendre les représentants des principaux courants de pensée, qu'ils soient religieux ou philosophiques. Il appartient, en effet, au Parlement d'essayer de saisir les différentes sensibilités qui peuvent s'exprimer sur ces questions, dans les diverses familles de pensée présentes en France.
La Mission a aussi recueilli, lors de ses travaux, le témoignage des représentants d'associations, comme l'Union nationale des associations familiales ou l'Association française de lutte contre les myopathies, ou d'organismes professionnels tels France Biotech ou le Mouvement des entreprises de France (MEDEF). Votre Rapporteur a également pu entendre des témoignages individuels de personnes ayant, par exemple, suivi le parcours d'une assistance médicale à la procréation.
Les travaux de la Mission sont donc l'expression de plusieurs préoccupations. Ses membres ont souhaité être informés des avancées scientifiques accomplies depuis le vote des lois de 1994. Puis, il apparaissait naturel de mieux appréhender les demandes sociales, exercice souvent délicat compte tenu de la multiplicité des aspirations de nos concitoyens. Il importait enfin de déterminer le cadre normatif international qui s'impose au législateur national.
C.- COMMENT RÉPONDRE À DES ASPIRATIONS CONTRADICTOIRES ?
1.- Quelles sont ces aspirations ?
Le législateur doit tenir compte des aspirations de nos concitoyens. Mais comment les connaître, en évaluer l'importance, l'intensité ? De plus, au-delà de ces aspirations, le Parlement doit aussi maintenir un cadre social fondé sur certains principes structurants. Pourrait-on, face à une demande sociale explicite, faire immédiatement fi de la dignité humaine ? Il est du devoir du Parlement de ne pas nécessairement céder à des mouvements d'opinions brusques sur ces sujets délicats.
Car, souvent les avancées scientifiques sont porteuses, simultanément, d'espoirs considérables mais aussi de grandes inquiétudes. Des fronts diffus apparaissent et s'opposent. Les personnes atteintes de maladies jusqu'alors incurables attendent les bienfaits de la médecine. À l'inverse, d'autres personnes en appellent aux principes et interpellent la science et ses débordements.
Des questions auxquelles il est difficile d'apporter une réponse font alors jour. Doit-on abandonner certaines pistes de recherche qui pourraient permettre de développer des thérapies parce qu'elles apparaissent sujettes à caution sur le plan des principes ? Soigner des personnes malades n'est-il pas là un principe éthique également de premier ordre ? Doit-on encourir le risque de laisser la science explorer toutes les méthodes même les plus contestables, au risque d'altérer gravement l'essence même de l'humanité ?
L'audition que la Mission a organisée autour de journalistes a montré la difficulté d'entendre, sur ces sujets, une réponse claire de la part de la population.
Selon Mmes Martine Allain-Régnault, Hélène Cardin et Marianne Gomez, respectivement journalistes à France 2, France Inter et au journal La Croix, le terme d'éthique, et plus encore de bioéthique, n'a pas de sens précis pour nos concitoyens. En dépit d'une connaissance souvent solide des découvertes médicales, les Français ne semblent pas véritablement concernés par les questions éthiques. Selon les termes de Mme Allain-Régnault, ils attendent les progrès bénéfiques de la médecine mais n'en veulent pas la rançon désagréable. Ils craignent le commerce, mais sont prêts à payer un enfant s'ils n'en ont pas, à acheter un organe ou à aller chercher à l'étranger des médicaments fabriqués à partir de prélèvements humains rémunérés si on les leur refuse en France. Selon elle, l'opinion semble faire naturellement confiance aux chercheurs mais également aux élus pour limiter les débordements de la science.
Néanmoins, les craintes essentielles des Français portent plus sur la sécurité alimentaire que sur les problèmes bioéthiques. Ainsi Mme Allain-Régnault constatait lors de son audition : « Mais savoir s'il faut ou non couper en deux un embryon, dans la mesure où ce n'est pas le leur, cela n'intéresse pas les Français. Quant aux barrières difficilement franchissables, je ne suis pas sûre que les Français en voient, mais ils trouvent bien qu'on avance doucement. Ils aiment la prudence ».
En dehors de la détresse des familles confrontées aux maladies génétiques - l'un des points saillants évoqués lors de l'audition de ces trois journalistes - il apparaît que la principale préoccupation de nos concitoyens est, avant tout, d'avoir des enfants « normaux » et en bonne santé, sans se poser réellement la question de l'eugénisme, qui ne semble pas avoir grand sens pour eux. Les personnes qui ne peuvent pas avoir « naturellement » d'enfants sont prêtes à tous les sacrifices et à beaucoup de concessions pour arriver à leurs fins. Il s'agit là souvent d'un état quasi-obsessionnel, comme l'a confirmé devant la Mission, en particulier, le Docteur Salomon Kessous, médecin généraliste exerçant à Paris.
Au total, nos concitoyens apparaissent prêts à accepter de grandes ouvertures dans le domaine de la bioéthique. Cette tolérance à l'égard de la recherche est le fruit d'un refus de plus en plus marqué de la fatalité et une volonté forte de maîtrise des événements.
Mme Marianne Gomez, journaliste à La Croix, a également observé, en France, selon ses termes, une évolution à l'anglo-saxonne, vers plus de pragmatisme et d'utilitarisme. Pour elle, une partie de la société est de moins en moins réticente à l'idée que l'on puisse subordonner les moyens aux fins poursuivies, l'essentiel étant la lutte contre la souffrance.
Face à cette aspiration, Mme Marianne Gomez a jugé que, pour l'essentiel, les croyants, qui ne sont pas d'ailleurs unanimes sur ces questions, faisaient exception et mettaient encore en avant la préservation de certains principes. Elle a noté, toutefois, que ces préoccupations étaient proches de celles exprimées par le Docteur Axel Kahn, qui, s'appuyant sur la morale kantienne, affirme que l'homme doit toujours être traité comme une fin, jamais comme un moyen (6).
2.- Faire des choix en édictant des normes
Le législateur doit tenir compte de ces aspirations. Quelle légitimité aurait-il à s'y opposer ? Mais, comme le soulignait M. Claude Evin, lors des travaux de la Mission, il doit également expliquer la complexité des décisions que doivent prendre les pouvoirs publics. Il faut aussi savoir refuser de suivre l'opinion lorsque des raisons sérieuses le justifient. Cet exercice est celui d'un véritable funambule, soumis sans cesse à des vents nouveaux.
Ces choix s'expriment par l'adoption de lois dont l'objet est double : la définition des interdits et la mise en place de procédures et de structures qui permettent le respect de ces interdits.
Le législateur n'a pas pour fonction d'arbitrer entre différentes conceptions spirituelles ou philosophiques. Il a pour mission d'établir le droit, subtile combinaison entre des exigences immédiates et des principes plus constants. Pour ce faire, il doit tenir compte de l'évolution du monde qui l'entoure, sans rester enfermé dans ses certitudes.
II.- LE DROIT, LA SCIENCE, L'ÉCONOMIE : QUELLES NOUVELLES CONTRAINTES POUR LE LÉGISLATEUR ?
Le législateur ayant pour tâche de réviser les lois bioéthiques de 1994 doit faire face à de nouveaux enjeux. La science a considérablement évolué depuis six ans ; des problématiques nouvelles ont émergé laissant place à des enjeux inédits. On a également assisté à une explosion du secteur des biotechnologies qui impose d'évaluer ces questions en ayant en tête les rapports de force entre les différentes économies, même si la question bioéthique ne saurait se résoudre dans la simple équation des intérêts financiers et industriels. Ne pas tenir compte en amont des réalités économiques risque de porter atteinte à l'efficacité même des lois bioéthiques. Enfin, le cadre juridique international, et plus précisément européen, actuellement en construction, conduit à s'interroger sur les marges de man_uvres dont le législateur national dispose désormais.
A.- L'ÈRE DES RÉVOLUTIONS BIOTECHNOLOGIQUES : LA LOI FACE À LA SCIENCE EN MOUVEMENT
Notre intention n'est pas ici de présenter chacune des innovations - elles furent nombreuses - qui ont marqué le monde de la médecine et de la biologie depuis 1994. Il nous appartiendra d'aborder ces questions dans la partie suivante du rapport, consacrée à l'évaluation du dispositif législatif de 1994. À ce stade de la réflexion, on tentera simplement de mettre en lumière les deux principaux événements qui ont marqué l'opinion publique pendant cette courte période : le clonage et la révolution génétique. Chacun a, selon ses propres enjeux, modifié profondément notre horizon de pensée et nous devons désormais vivre avec ces nouvelles réalités que la science nous a livrées.
La question du clonage apparaît comme très caractéristique de la difficulté à prévoir le rythme et l'ampleur des avancées scientifiques. Alors qu'en 1994, le clonage n'avait pas suscité de réel débat, il est apparu comme l'une des interrogations majeures dans le domaine de la bioéthique, avec la naissance à l'Institut Roslin en Écosse de la brebis Dolly, premier animal issu de cette technique en 1997. À partir de cette naissance, qui a été suivie de plusieurs autres, se sont multipliées les prises de position autour d'une question, qu'il convient de traiter avec sérénité.
En effet, rarement une technique biomédicale aura ouvert la voie à tant de fantasmes, de craintes et d'attentes (7). La possibilité pour chacun de voir son double créé à partir de ses propres cellules laisse libre cours aux spéculations les plus folles. L'imagination est sans limite et la science semble, en apparence, lui donner raison. Car le clonage permet d'envisager des pratiques qui auraient semblé relever de la science fiction, il y a quelques années encore. Elles apparaissent aujourd'hui - à tort ou à raison - à portée de main.
Pourquoi ne pourrait-on créer un clone pour disposer le cas échéant de ses organes en cas d'accident ou de maladie ? À partir de quelques cellules ne pourrait-on pas donner naissance au clone d'un enfant prématurément disparu pour mettre fin au chagrin de ses parents ? Par le clonage de son propre être ne pourrait-on pas atteindre une forme d'immortalité ? On touche ici du doigt des interrogations qui ne sont pas seulement scientifiques. Notre rapport à autrui est fondamentalement éprouvé dans la question du clonage humain, qui renvoie à une tentation narcissique qui, pour le moins, nous installe dans une sensation de malaise.
Ces découvertes et les questions qu'elles engendrent, relayées par les médias, n'ont pas manqué de marquer l'opinion publique, au point d'engendrer des poussées d'inquiétude collective, comme le souligne le rapport du Conseil d'État de 1999.
Pourtant, le clonage ne doit pas être perçu comme la seule faculté - hypothétique aujourd'hui pour l'homme - de créer le double d'un être à partir d'un élément de son propre corps (8). Pour le dire de manière simple, il est avant tout le moyen de former des cellules à partir d'autres cellules. On est loin ici de la création de toutes pièces d'un être composé.
Il faut bien avouer que des initiatives scandaleuses ont contribué à alimenter ces confusions. Ainsi, un mouvement sectaire a créé aux États-Unis une société qui prétend pouvoir donner naissance au premier clone humain, à partir des cellules d'un enfant mort à l'âge de dix mois, moyennant la somme de 200.000 dollars. Ces rodomontades ont été relayées par les médias, mais aussi, à leur corps défendant, par des institutions qui, de facto, leur donnent un certain écho. Le fait qu'une commission du Congrès des États-Unis ait auditionné la responsable de ladite société fondée par cette secte l'a propulsée sur le devant de la scène. Cette secte a ainsi fait la couverture au printemps dernier de grands journaux comme le New York Times Magazine ou Time. Une telle initiative institutionnelle contribue à accréditer l'idée que le clonage d'une personne humaine est aujourd'hui possible, ce qui n'est pas le cas. Elle souligne, au passage, la difficulté pour nos démocraties de faire face à de tels mouvements, dont il faut parler pour appeler nos concitoyens à la méfiance et à l'exercice de leur jugement critique, avec le risque cependant d'offrir à ces sectes une audience inespérée.
Mais, avant même que cette secte ne s'empare de la question, des scientifiques parfois considérés comme sérieux par leurs pairs se lancèrent dans la surenchère médiatique. Le 6 janvier 1998, le physicien américain Richard Seed a annoncé son intention de pratiquer le clonage reproductif humain. En décembre 1998, un chercheur sud-coréen, le Professeur Lee Bo-Yon, s'attire les foudres de la communauté scientifique en annonçant avoir créé un embryon humain par clonage. L'opération, qui se serait interrompue juste avant l'implantation de l'embryon dans l'utérus d'une mère porteuse, n'a pas été confirmée par une revue scientifique. En 2001, c'est au tour de deux scientifiques, italien et grec, les Docteurs Severino Antinori et Panyiotis Zavos, de faire connaître leur volonté de se livrer à une telle expérience. Ces spécialistes de la procréation s'étaient déjà illustrés dans des entreprises douteuses. Le Docteur Antinori s'est ainsi fait connaître en développant une technique permettant à des femmes sexagénaires de donner naissance à un enfant. Le Docteur Zavos est, quant à lui, connu pour avoir mis en place une activité commerciale autour de la stérilité masculine, avec la vente de kits d'analyse de sperme, ou la proposition de services de congélation de cellules... Leur annonce a été jugée révoltante par les milieux scientifiques et politiques. Le Docteur Zavos, scientifique par ailleurs reconnu, semble-t-il, a été entendu sur ce sujet par la commission de la Chambre des représentants.
La presse médicale a pu également accréditer l'idée que le clonage reproductif humain était en passe d'être réalisé. L'hebdomadaire britannique de renommée internationale, The Lancet, a ainsi consacré un éditorial en janvier 1999 à cette question, jugeant qu'il était urgent de s'interroger dès aujourd'hui sur les droits des futurs clones, la réalisation de telles expériences sur l'humain étant inéluctable.
b) Une distinction indispensable
Ces initiatives médiatiques n'ont pas toujours permis de distinguer clairement les différentes réalités auxquelles la notion générique de clonage renvoie dans les faits. Comme le souligne à bon droit le Conseil d'État dans son rapport de 1999, « le clonage peut être défini comme une technique consistant à reproduire des organismes vivants génétiquement identiques. Il peut donc concerner de simples cellules (clonage cellulaire) ou des êtres humains, des animaux, ou des végétaux (clonage reproductif). En ce qui concerne le clonage reproductif, il convient également de distinguer entre les différentes techniques existantes : le clivage d'un embryon de quelques cellules en deux, afin de créer deux individus distincts génétiquement identiques ; le clonage par transfert dans des ovocytes énucléés des noyaux de cellules d'un même embryon ou le clonage par transfert de noyaux provenant d'un organisme adulte comme dans le cas de la brebis Dolly ou en mars 1998 de la génisse Marguerite à l'INRA (9) ».
Le clonage cellulaire évoqué par le Conseil d'État est également qualifié de « clonage thérapeutique » lorsque telle est sa vocation. Son objet n'est en aucun cas de créer un être semblable à un autre par voie de clonage. Le « clonage thérapeutique » permettrait de préparer des populations de cellules pluripotentes, grâce auxquelles on pourrait corriger, chez l'adulte, des maladies telles que des dégénérescences nerveuses, le diabète, des leucémies ou des brûlures graves (10). Dans son avis n° 9 du 28 mai 1997, le Groupe de conseillers pour l'éthique de la biotechnologie auprès de la Commission européenne a également appelé à la distinction entre le clonage reproductif et les autres formes de clonage. L'utilisation du terme « clonage thérapeutique » peut, à cet égard, être sujette à discussion. Car, même si la technique scientifique présente des similitudes avec le clonage reproductif, la finalité des deux pratiques est distincte. Le rapport de l'Office parlementaire d'évaluation des choix scientifiques et technologiques le rappelait : « L'objectif est donc, dans cette hypothèse, non de parvenir au développement d'un être humain mais d'obtenir, à partir des cellules somatiques d'un patient, les cellules souches dont la différenciation contrôlée permettrait de traiter l'affection dont il est porteur sans provoquer de phénomène de rejet. La technique est bien la même dans les deux hypothèses : reproduction non sexuée d'entités génétiquement identiques. La finalité diffère : création, dans le premier cas, d'un organisme complet, dans le second, de lignées cellulaires, le passage obligé étant, en l'état actuel de la science, le développement préalable d'un embryon in vitro » (11).
Sans doute faudrait-il trouver un terme mieux adapté et facteur d'une moindre confusion pour qualifier ce que l'on appelle le « clonage thérapeutique ». Pour autant, puisque cette expression, sans doute impropre, est consacrée par le débat public, votre Rapporteur s'y référera par convention et moyennant les précautions d'usage qui viennent d'être énoncées.
c) Le clonage reproductif humain : une perspective inacceptable
Pour couper court à tous les fantasmes, il convient de préciser que le clonage reproductif animal ne doit pas être perçu aujourd'hui comme une pratique courante et simple. Cette technique n'est pas sans danger, tant s'en faut. Le Professeur Jean-Paul Renard notait ainsi, en 1999, que, selon les expériences, entre 40 et 74 % des clones d'animaux adultes mouraient prématurément (12). En dehors même de la position de principe qui justifie l'interdiction du clonage reproductif sur l'homme, on imagine ce qu'auraient de scandaleux de telles pratiques sur des êtres humains.
Par ailleurs, les conséquences du clonage reproductif conservent encore leur part d'ombre. Ainsi en mai 1999, il est apparu que le clonage de la brebis Dolly, deux ans après sa naissance, soulevait des problèmes scientifiques, pour l'heure, insolubles. La presse s'est faite l'écho des interrogations des chercheurs sur l'âge de l'animal, après qu'ils eurent constaté, en examinant les télomères des chromosomes de Dolly - c'est-à-dire leurs extrémités - que ceux-ci avaient prématurément vieilli. Alors que la brebis avait moins de trois ans, ses chromosomes correspondaient à ceux d'un animal de neuf ans (13). Un être cloné accumulerait-il alors les effets de son âge mais aussi de celui dont il est issu ? La question n'est pas mince et elle met en lumière les incertitudes qui existent encore dans le domaine du clonage.
Le Groupe de conseillers pour l'éthique de la biotechnologie auprès de la Commission européenne avait également relevé ces interrogations dans son avis du 28 mai 1997 : « Les risques liés à l'utilisation éventuelle de cellules adultes comme cellules donneuses de noyaux sont largement inconnus. Les individus clonés auraient-ils une espérance de vie plus brève ? Auraient-ils un risque accru de développer un cancer ? Seraient-ils, ou non, féconds et s'ils sont féconds, leur descendance présenterait-elle un taux anormalement élevé d'anomalies génétiques ? ».
Même si la science ne semble pas aujourd'hui en mesure de donner corps à ce fantasme du clonage reproductif humain, le droit et la politique se sont emparés du sujet pour le condamner avec une fermeté exemplaire.
Le clonage reproductif d'une personne humaine a été unanimement condamné, tant au plan national qu'international. L'Organisation mondiale de la santé (OMS) a adopté une résolution, le 14 mai 1997, allant dans ce sens. L'article 11 de la Déclaration de l'Unesco, du 11 novembre 1997, sur le génome humain et les droits de l'homme a, quant à lui, clairement indiqué que « des pratiques qui sont contraires à la dignité humaine, telles que le clonage à des fins de reproduction d'êtres humains ne doivent pas être permises ».
Saisi de l'avant-projet de loi portant révision des lois de bioéthique, le Comité consultatif national d'éthique s'est déclaré opposé, à l'unanimité, au clonage reproductif dans son avis n° 67 du 27 janvier 2001. Ce faisant, il ne faisait que confirmer son avis n° 54 du 22 avril 1997, rendu suite à une saisine du Président de la République.
On observe que les réactions à la naissance de Dolly ont été vives. L'Europe n'est pas restée à l'écart de ce mouvement de réprobation. Ainsi le Parlement européen a adopté une résolution le 12 mars 1997, condamnant le clonage reproductif humain. Le Conseil de l'Europe lui emboîta le pas un an après, par la signature, en 1998, d'un protocole additionnel à la Convention du 4 avril 1997 sur les droits de l'homme et la biomédecine, signée à Oviedo.
Les politiques se sont également émus de cette perspective contraire à la dignité humaine. Le président Clinton a ainsi interdit en mars 1997, quelques jours après l'annonce de la naissance de Dolly, le financement sur fonds fédéraux de toutes les recherches consacrées au clonage humain.
Le débat ne semble pas clos sur cette question aux États-Unis. En effet, après l'annonce du président Clinton, le Congrès a refusé de voter une loi interdisant ces recherches, que ce soit dans le secteur public ou privé. L'adoption d'une telle loi ne pourrait avoir lieu, semble-t-il, dans le consensus. On notera cependant que des États fédérés américains, comme la Californie, le Michigan, la Louisiane et le Rhodes Island ont voté des lois interdisant le clonage. Lors de son déplacement outre-Atlantique, votre Rapporteur a pu constater que le Parlement américain était encore saisi de cette question. Comme on l'a évoqué précédemment, des auditions venaient, en effet, d'avoir lieu, en mars 2001, devant la commission chargée de ce dossier à la Chambre des représentants.
Cette absence de loi fédérale interdisant le clonage humain reproductif ne signifie nullement qu'une telle opération pourrait être menée à bien, si elle était techniquement possible, ce qui n'est pas aujourd'hui le cas. En effet, la puissante Food and Drug Administration (FDA) délivre les autorisations concernant les recherches menées sur l'homme. Selon les représentants de cette agence, que votre Rapporteur a rencontrés lors de son déplacement aux États-Unis, une telle autorisation serait requise pour toute recherche concernant le clonage humain. Or d'après la FDA, dont les seules préoccupations sont réglementaires et sanitaires, et non éthiques, de telles recherches ne pourraient être autorisées, en l'état actuel de la réglementation, compte tenu des risques sanitaires qu'elles représenteraient. En 1998, le Président Clinton avait préconisé cette solution, faute de pouvoir dégager une majorité au Congrès sur ce sujet. Le non-respect des prescriptions de la FDA peut exposer le contrevenant à une lourde peine d'amende voire de prison.
Il est vrai qu'une tentative de clonage reproductif humain poserait des problèmes sanitaires graves. Sur la base des travaux menés à l'Institut Roslin par le Professeur Wilmut pour donner naissance à Dolly, le journal Libération estimait ainsi en février 1998, après l'annonce médiatique du Professeur Seed, qu'il faudrait pas moins de 1 000 ovocytes pour espérer réussir un clonage reproductif humain. Sachant qu'une opération de stimulation ovarienne permet d'obtenir dix ovocytes chez une femme, il faudrait convaincre cent femmes d'accepter de se prêter à cette expérience, consistant à prélever chaque ovocyte sous anesthésie générale. Le même article estimait également à trente le nombre de mères porteuses nécessaire pour espérer mener à terme une seule et unique grossesse, sachant que les autres n'aboutiraient pas (14).
La FDA a conscience que le contrôle de ce type de recherches n'est pas toujours aisé. D'après le Docteur Kathryn Zoon, directrice du Center for Biologics Evaluation and Research, ce contrôle s'exerce par des vérifications des sites Internet qui pourraient être impliqués dans de telles recherches. Pour autant, la crédibilité des pouvoirs publics passe par cette capacité de faire respecter les règles juridiques et éthiques adoptées par la société.
f) L'ouverture du débat sur le clonage dit « thérapeutique »
La question du recours possible au « clonage thérapeutique » fait, quant à elle, l'objet d'un débat en Europe comme aux États-Unis d'ailleurs. La discussion avait été relancée au plan européen, quelque temps plus tôt, par l'annonce du Royaume-Uni en août 2000, d'autoriser, le « clonage thérapeutique ». Cette décision a suscité des critiques, la ministre allemande de la santé, Mme Andrea Fischer, estimant, par exemple, que faire des embryons un matériel pour les scientifiques était dangereux. Le Royaume-Uni a cependant annoncé sa volonté d'interdire par la loi le clonage reproductif, au mois d'avril dernier. La décision de ce pays d'autoriser le « clonage thérapeutique » a suivi une vaste consultation publique organisée par la HFEA (Human Fertilization and Embryology Authority).
En dehors de la question de principe, le débat sur le « clonage thérapeutique » renvoie à celui sur les dons d'ovocytes. En effet, le clonage d'une cellule par transfert de son noyau dans un ovocyte énucléé impose que soit pratiqué un tel don. Or ce type de don n'est pas une opération simple, à la différence du don de gamète mâle. Il suppose une stimulation ovarienne et une anesthésie générale pour les donneuses. Cette contrainte inquiète les différentes instances qui se sont prononcées sur ce type de clonage.
Le débat sur le « clonage thérapeutique » humain se déroule actuellement en France, les positions anglaises ayant contribué à le relancer. Si une unanimité s'est faite jour pour condamner le clonage reproductif, la question du clonage cellulaire à vocation thérapeutique apparaît moins tranchée.
Le Premier ministre, M. Lionel Jospin, s'est exprimé en faveur d'avancées en ce domaine, lors de son allocution devant le Comité consultatif national d'éthique, le 28 novembre 2000. Évoquant les espoirs nés de ces nouvelles techniques et s'interrogeant sur la possibilité de répondre ou non à la demande des personnes souffrant de maladies jusqu'à aujourd'hui incurables, le Premier ministre a estimé envisageable qu'une perspective soit ouverte en autorisant la constitution de cellules par transfert de noyaux de cellules somatiques, selon des protocoles strictement définis et encadrés. Dès lors, comme c'est l'usage dans une démocratie, le débat s'est ouvert, diverses autorités prenant position sur ce sujet.
Deux instances ont exprimé, plus ou moins explicitement, leur position sur la possibilité de recourir au « clonage thérapeutique » humain. Le 27 janvier 2001, le Comité consultatif national d'éthique s'est ainsi interrogé sur l'opportunité d'autoriser cette pratique dans son avis n° 67 sur l'avant-projet de loi portant révision des lois de bioéthique. La réponse à cette question a été très discutée au sein de cette instance, deux positions étant alors dégagées. Pour les opposants au « clonage thérapeutique », le risque apparaît d'une réification de l'embryon. Par ailleurs, l'accent a été porté sur la pression psychologique dont pourraient faire l'objet les femmes aux fins de donner des ovocytes, indispensables au « clonage thérapeutique ». La crainte de la création des conditions d'un véritable marché a également été soulevée. On notera que ce souci est partagé par le Groupe européen d'éthique, dans son avis du 15 novembre 2000.
Au sein du Comité consultatif national d'éthique, les partisans du « clonage thérapeutique » ont fait valoir que les perspectives thérapeutiques associées à cette technique justifient, à elles seules, que celle-ci soit autorisée. Pour eux, « le devoir de solidarité avec les personnes qui souffrent de maladies interdit en ce domaine d'entraver la recherche, au risque de pénaliser irrémédiablement les malades ». Il leur apparaît également souhaitable d'agir dans ce sens dans la mesure où la mondialisation de la recherche, la sévérité de la compétition scientifique internationale et des intérêts économiques sont en jeu. Pour les partisans de l'autorisation du « clonage thérapeutique », la France risquerait de dépendre, à terme, des recherches réalisées à l'étranger. Notre pays serait alors placé dans cette situation paradoxale selon laquelle nous ne manquerions pas de recourir aux thérapies ainsi développées hors de nos frontières, sans avoir pu exercer un quelconque contrôle sur le respect des règles éthiques auxquelles nous sommes attachés, voire en pleine contradiction avec ces règles.
Au vu de cette divergence d'opinions en son sein, le Comité consultatif national d'éthique a émis deux recommandations, constatant qu'une majorité de ses membres était cependant favorable à l'autorisation du « clonage thérapeutique » :
- combler le vide juridique qui entoure la question des dons d'ovocytes, afin de protéger les femmes ;
- introduire des modalités plus souples de révision des lois de bioéthique, « le temps de l'évolution des références éthiques ne [s'accordant] pas avec celui de la construction des normes juridiques, ni avec celui de l'avancée des connaissances scientifiques ».
La position du Comité consultatif national d'éthique illustre la difficulté de résoudre simplement cette question, les principes éthiques en jeu étant éminemment contradictoires.
En dépit d'un débat interne, à l'occasion duquel ses membres se sont, semble-t-il, partagés, la Commission nationale consultative des droits de l'homme (CNCDH) s'est, quant à elle, exprimée contre le « clonage thérapeutique », à l'occasion de la remise de son avis sur l'avant-projet de loi, le 31 janvier dernier. En cela, la CNCDH a repris les conclusions du Groupe européen d'éthique.
Ce groupe a rendu un avis sur ce sujet, le 15 novembre 2000. Il a estimé que « pour l'heure, la création d'embryons par transfert de noyaux de cellules somatiques (« clonage thérapeutique ») pour les besoins de la recherche sur la thérapie cellulaire serait prématurée, étant donné qu'il existe un vaste champ de recherches à explorer à l'aide d'autres sources de cellules souches humaines : à partir de d'embryons surnuméraires, de tissu f_tal ou de cellules souches adultes ».
Le Président de la République s'est également exprimé sur la question du « clonage thérapeutique », en février dernier, à Lyon. Il a fait connaître son opposition à l'ouverture de cette voie de recherche, qui rend, à ses yeux, le clonage reproductif possible et risque de conduire à des trafics d'ovocytes. Il a été rejoint sur cette ligne par le Conseil d'État, saisi de l'avant-projet de loi, et qui s'est prononcé également contre cette perspective.
Le débat sur la question du « clonage thérapeutique » est aujourd'hui ouvert devant le Parlement. Aucune unanimité n'apparaît sur cette question tant au plan national qu'européen. Les divers intérêts en balance rendent ce choix sans doute difficile. Il ne peut être issu que d'une discussion démocratique, après que les instances expertes et consultatives ont été entendues. Le Parlement aura donc à examiner ce sujet qu'il ne peut passer sous silence. Dans le même temps, il conviendra sans doute d'expliciter dans notre loi la condamnation formelle du clonage reproductif que la loi du 29 juillet 1994 ne prohibe pas expressément, comme l'a souligné le Conseil d'État dans son rapport de novembre 1999.
2.- La révolution génétique : du décryptage du génome aux thérapies cellulaires
a) Le décryptage du génome humain
L'entreprise de décryptage du génome humain est l'une des aventures scientifiques les plus impressionnantes que le siècle ait connue. Le défi dans lequel les chercheurs se sont lancés est colossal et la concurrence entre les principaux acteurs de cette « saga » est à la hauteur de l'enjeu.
Le décryptage du génome consiste à identifier l'ordre dans lequel s'enchaînent les trois milliards d'éléments qui constituent la molécule d'ADN. Ces éléments sont des bases appelées adénine, guanine, thymine et cytosine. Ces bases s'organisent en gènes qui donnent à la cellule une fonction, consistant en la fabrication d'une protéine particulière. Le séquençage du génome permet donc de repérer chaque gène, de connaître sa séquence et de découvrir sa fonction dans la cellule.
En 1990, les États-Unis ont lancé une initiative en invitant à la constitution d'un consortium aux fins de décrypter le génome dans un délai de quinze ans. Ce groupe intitulé « Programme génome humain » regroupe l'Allemagne, la Chine, les États-Unis, la France, la Grande-Bretagne et le Japon. Les National Institutes of Health (NIH), le département de l'énergie américain et le Wellcome Trust britannique figurent parmi les principaux bailleurs de fonds du projet.
Le travail semblait d'une ampleur incommensurable. Le consortium constitué autour de l'initiative des États-Unis a choisi alors une méthode : décrypter l'ADN humain chromosome après chromosome. Stimulé par la concurrence d'un acteur privé, le Programme génome humain va multiplier les efforts pour aboutir avant la date fixée initialement.
Comme le soulignait la presse française, ce qui a été présenté comme le « Programme Apollo des sciences de la vie » au début des années 90 est apparu comme un affrontement peu amène entre deux équipes de chercheurs, deux visions de la recherche, deux méthodes scientifiques (15).
En 1992, un généticien, Craig Venter, quitte le NIH pour créer Tigr, un institut à but non lucratif, qui s'associe à Human Genome Sciences, une société cotée en bourse et dirigée par William Haseltine. Cette société a pour objet de commercialiser les découvertes de Tigr. L'alliance entre les deux partenaires est cependant rompue en juin 1997, après que Human Genome Sciences eut souhaité empêcher Tigr de rendre public le génome d'une bactérie que cet institut avait décrypté.
Libéré de ses liens avec le secteur concurrentiel, Tigr décroche des contrats de recherche avec des organismes publics dont les NIH eux-mêmes, des fondations caritatives ou le ministère de l'environnement américain qui lui a confié son programme sur les génomes microbiens. Ces commandes assurent à cette structure un budget confortable de 30 millions de dollars par an, qui lui permet d'organiser son activité et sa stratégie.
En mai 1998, Craig Venter lance un véritable défi au Programme génome humain. Désormais, à la tête de la société Celera Genomics, filiale du fabricant d'automates Perkin Elmer, il fait le pari de séquencer le génome humain pour 2001, grâce à une technique originale et éprouvée sur les génomes de plusieurs bactéries : le whole genome shotgun. La méthode repose sur le séquençage de millions de fragments du génome humain éclaté. La difficulté majeure est de les assembler ensuite dans l'ordre pour reconstituer tout le message génétique sans erreur.
Le défi lancé par Craig Venter a conduit les responsables du consortium international à renforcer les moyens mis à la disposition de celui-ci. L'aventure du décryptage du génome humain s'est donc accélérée pour aboutir finalement en 2001 par la publication de la presque totalité de ce décryptage.
Le résultat de cette entreprise a été publié de manière simultanée le 11 février 2001 par les deux groupes de recherche : Celera Genomics dans la revue américaine Science et le Programme génome humain dans la revue britannique Nature.
Il n'a pas manqué d'étonner dans la mesure où l'on s'est aperçu que le génome humain comptait seulement 30.000 gènes. Ce nombre doit être rapproché de celui du nématode, un ver qui possède 19 000 gènes, de la drosophile, une mouche, qui en compte 14.000, et surtout du grain de riz composé de 50.000 gènes. Au début du projet de séquençage, l'estimation allait de 100.000 à 150.000 gènes, alors qu'en 1995, Craig Venter la fixait à 70.000. On mesure la surprise que fut celle de l'annonce de ce résultat.
De ce faible nombre de gènes, on a tiré quelques conclusions. L'idée selon laquelle à un gène seul correspondrait une seule fonction semble battue en brèche. Il faut désormais raisonner en termes d'interactions entre gènes, de combinaison de leurs fonctions, d'interférences entre l'ADN et l'environnement cellulaire. La conception d'une détermination génétique de l'homme sort également très affaiblie de cette découverte. Le défi pour les généticiens va être désormais de définir la fonction de chacun des 30.000 gènes, sachant que celle-ci peut varier selon les signaux émis par d'autres gènes, d'autres molécules cellulaires, d'autres cellules.
Le décryptage du génome a également dévoilé une autre découverte surprenante : l'extrême hétérogénéité de l'ADN humain. Des parties entières de l'ADN sont dénuées de fonction codante, alors que d'autres contiennent de nombreuses informations. Si le chromosome 19 est riche en gènes de régulation des fonctions nerveuses, le chromosome Y apparaît lui comme très pauvre en gènes.
On a pu aussi constater que les segments de l'ADN varient d'un individu à l'autre. La plupart des différences individuelles tiennent au changement d'une base dans un gène. À partir de la connaissance de ces variations, baptisées SNP's (single nucleotide polymorphisms), il serait possible de créer des médicaments ciblés. Alors que jusqu'à présent 800.000 SNP's avaient été précisément repérés sur le génome humain, Celera Genomics en a identifié 2,3 millions.
Une polémique est intervenue sur les résultats présentés par Celera Genomics. Certains ont contesté que le décryptage effectué par la firme privée soit complet, alors que le consortium international indiquait qu'il ne serait en mesure de proposer un décryptage exhaustif qu'en 2003. « L'article de Science est une imposture, Venter n'a pas terminé le séquençage. Un tiers des gènes est incomplet, c'est une séquence à 15.000 trous », a ainsi déclaré Jean Weissenbach, directeur du Centre national de séquençage à Evry dans un quotidien. L'un des principaux reproches fait à Celera Genomics est d'avoir, en fait, tiré profit du travail du consortium international, en se l'appropriant scientifiquement et médiatiquement.
La nouvelle du décryptage est apparue comme l'entrée dans une nouvelle ère scientifique et médicale. La perspective de guérir des maladies se profile de manière plus nette encore, suscitant des espoirs que la recherche tentera de combler.
b) Quel horizon pour les perspectives thérapeutiques ?
Comme le déclarait le Professeur Arnold Munnich, chef du service de génétique médicale de l'Hôpital Necker-Enfants Malades et directeur de l'unité de recherche sur les handicaps génétiques de l'enfant de l'INSERM, dans un entretien à Libération, le 26 juin 2000 : « Il y a d'énormes malentendus entre les scientifiques et le public. Le séquençage du génome en est un exemple. A-t-on attendu le séquençage exhaustif du génome pour localiser et identifier les gènes de maladies génétiques? Évidemment non. Les années 1980-2000 ont été nos vingt glorieuses, au cours desquelles on a découvert les gènes de plus de mille maladies: myopathie, mucoviscidose, hémophilie, amyotrophie spinale, presque tous les handicaps visuels et auditifs, et la majorité des malformations congénitales. Tout cela avant le séquençage exhaustif du génome ». Cela étant, le Professeur Arnold Munnich a estimé, dans cet entretien, que le décryptage permettrait évidemment d'accélérer le travail de localisations des gènes responsables de maladies génétiques.
La course au génome a duré dix ans. Reste maintenant à tirer profit des résultats obtenus pour mettre au point des médicaments. Le délai pour aboutir à ces résultats attendus risque d'être plus long que celui qui fut nécessaire pour décrypter le génome. « C'est la fin du début », soulignait Francis Collins, directeur du Programme génome humain pour les NIH, à l'occasion de l'annonce du décryptage. Les espoirs nés de cette découverte ne seront pas comblés dans l'immédiat. Espérons que cette attente ne sera pas à l'origine de trop grandes frustrations.
La « course au génome » a montré tout l'intérêt d'une coopération internationale et les effets de la concurrence. Elle montre aussi tout l'intérêt d'une action juridique dépassant les frontières nationales.
B.- L'HYDRE JURIDIQUE : LA LOI CONFRONTÉE À UN DROIT INTERNATIONAL EN CONSTRUCTION
Le fait que les pays connaissent des législations différentes peut conduire certaines personnes à rechercher ailleurs la possibilité d'obtenir un traitement, de bénéficier d'une pratique médicale, interdits dans leur pays d'origine. La question se posait déjà il y a trente ans lorsque la France n'autorisait pas l'interruption volontaire de grossesse, contrairement à certains de ses voisins. Le désir de donner naissance à tout prix à un enfant peut entraîner ce que certains qualifient, sans doute un peu rapidement, de « tourisme médical ». L'exemple récent de cette mère de 62 ans ayant bénéficié d'un don d'ovule et d'une fécondation à partir du sperme de son frère aux États-Unis - pratiques interdites en France - pour ensuite donner naissance à l'enfant dans notre pays illustre parfaitement cette situation. La mobilité des individus, la diffusion des informations via Internet rendent nos législations plus sensibles aux règles applicables dans les autres pays. C'est pourquoi la définition d'un droit international dans le domaine de la bioéthique apparaît comme une nécessité absolue.
La bioéthique est régie par des normes ressortissant à des sources multiples et en voie d'élaboration. Face à une activité scientifique dépassant le strict cadre national et des enjeux économiques qui transcendent les frontières, les États ont cherché à organiser un cadre juridique, plutôt souple, qui peut s'imposer aux droits nationaux.
Cette démarche n'est pas illégitime puisque la bioéthique renvoie aux fondements même de l'humanité, en s'articulant essentiellement autour de l'idée de dignité humaine. Elle a donc vocation à transcender les clivages nationaux et culturels.
Afin de mesurer l'impact du droit européen et international en ce domaine, la Mission d'information a organisé une journée d'auditions consacrée à ce sujet. Ce débat avec des personnalités européennes s'est déroulé autour des questions suivantes : quelles sont les règles supranationales qui s'imposent aux lois internes ? En conséquence, quelles sont les marges de man_uvre dont disposent les Parlements ?
Avant de présenter les réponses apportées à ces questions, il convient de revenir sur les caractéristiques de ce droit international de la bioéthique en devenir.
1.- Les sources internationales du droit de la bioéthique : de la « soft law » à un droit relativement contraignant
a) Le rôle précurseur de la « soft law »
Comme le soulignent justement Mme Noëlle Lenoir et M. Bertrand Mathieu (16), en tant que droit international, le droit de la bioéthique est exemplaire. C'est, en effet, « un droit qui se veut prospectif en tentant d'apporter une réponse aux immenses et nouveaux défis que la génétique adresse à l'humanité ». Pour ce qui concerne ses sources, ce droit est précurseur « car il annonce la diversification croissante des modes d'expression juridique au plan international (17)». Les normes de la bioéthique découlent d'actes unilatéraux d'organisations internationales ayant force obligatoire - c'est le cas des règlements et des directives communautaires -, mais aussi et surtout d'actes conventionnels, de traités ou de déclarations solennelles, à valeur indicative et incitative.
Mais ce qui confère au droit de la bioéthique son originalité la plus notable, c'est le rôle des textes publiés par des organismes non gouvernementaux qui, petit à petit, influencent le droit positif en l'enrichissant ou en l'interprétant. C'est le cas, par exemple, des avis rendus par des comités d'éthique internationaux existant dans le cadre de l'Union européenne et de l'Unesco.
Ainsi, à côté de normes qui s'imposent aux États, selon des modalités sur lesquelles nous reviendrons, existe ce que l'on qualifie en droit international de « soft law » (18), c'est-à-dire, d'une part des normes, contenues dans des résolutions, des recommandations ou des déclarations d'organisations internationales, qui ont une valeur exclusivement proclamatoire ou programmatoire (19), et, d'autre part, des textes émanant d'organisations non gouvernementales, sans portée juridique directe. On assiste ainsi, selon l'expression du Professeur Bertrand Mathieu, à l'émergence d'un droit souple, évolutif, parfois incertain, et finalement peu contraignant.
Dans ce cadre, les déclarations des organisations non gouvernementales (ONG) ont une influence particulière. L'Association médicale mondiale (AMM) a joué un rôle notable en ce domaine. La Déclaration d'Helsinki de 1964, élaborée sous son égide, peut être considérée comme le premier texte international en matière bioéthique. Modifiée à plusieurs reprises - en 1975, 1983 et 1989 - cette déclaration a fixé les orientations que doivent suivre les médecins lors d'expérimentations médicales sur l'homme. Des principes importants y sont énoncés : la conduite de la recherche doit être menée par une personne compétente ; celle-ci doit respecter un principe de précaution ; tout protocole expérimental doit être soumis, au préalable, à un comité indépendant ; le consentement de l'individu doit être libre et éclairé, etc.
Ces principes ne s'imposent pas formellement aux États ou aux praticiens. Mais, par la force de ces déclarations, ils connaissent une aura qui emporte leur respect par les différents acteurs.
Les comités d'éthique jouent également le rôle de producteurs de normes. Le développement de ces comités est issu des préconisations de la déclaration d'Helsinki de l'AMM telle que modifiée en 1975. Cette idée a été reprise et précisée par la déclaration de Manille de 1981, approuvée par l'Organisation mondiale de la santé (OMS) et le Conseil des organisations internationales des sciences médicales (CIOMS). Aux termes de cette déclaration, ces comités d'éthique doivent réunir des chercheurs et des non-spécialistes « pour représenter les valeurs culturelles et morales de la communauté ».
Il existe plusieurs comités d'éthique internationaux, parmi lesquels le Comité international de bioéthique (CIB) de l'Unesco, créé en 1983, dont les 55 membres issus d'une quarantaine de pays sont désignés par le directeur général de l'organisation. Comme le notent Mme Noëlle Lenoir et M. Bertrand Mathieu, la tâche majeure du CIB est de nature juridique. En effet, en 1993, un mandat a été confié à cet organe par la Conférence générale des États membres de l'Unesco : préparer un « instrument international » énonçant les principes éthiques applicables à la génétique humaine. Le CIB a décidé de recourir à la technique de la déclaration, plus facile à adopter mais moins contraignante pour les États (20). Ainsi, a été votée par l'Unesco, le 11 novembre 1997, la Déclaration universelle sur le génome humain (21).
L'Europe s'est également dotée d'un organe consultatif chargé des questions éthiques. En 1991, la Commission a décidé la constitution du Groupe de conseillers pour l'éthique de la biotechnologie (GCEB). Devenu le Groupe européen d'éthique (GEE) en 1997, son rôle est comparable à celui des comités d'éthiques nationaux. Composé de scientifiques, de juristes, de philosophes et de théologiens, le GEE a rendu de nombreux avis au cours de son premier mandat, de 1998 à 2001, sur des sujets aussi divers que la recherche sur les embryons humains, les banques de tissus humains, l'informatisation des données de santé ou l'utilisation des cellules souches humaines. Le 24 avril dernier, la Commission européenne a nommé les douze membres du GEE pour son second mandat (2001-2004), la présidence restant confiée à Mme Noëlle Lenoir.
Les avis du GEE ne s'imposent pas aux États. Mais, là encore, ils peuvent les influencer, soit par les inflexions auxquelles ils peuvent aboutir lors du processus d'élaboration des normes communautaires, soit en dégageant des principes que le législateur national jugera pertinents et reprendra à son compte.
Mais cette « soft law », ce droit déclaratoire, ne constitue pas la principale contrainte s'imposant aux législateurs nationaux. Quelques normes conventionnelles ont été édictées depuis une quinzaine d'années, s'appliquant en droit interne, avec plus ou moins d'intensité.
b) Le cadre plus contraignant des conventions : la Déclaration universelle de l'Unesco sur le génome humain et les droits de l'homme
et la convention d'Oviedo de 1997
Un certain nombre de conventions émanant de l'ONU sont intervenues, traitant le plus souvent indirectement des questions relevant de la bioéthique.
La Convention contre la torture et les autres peines ou traitements cruels inhumains ou dégradants, du 10 décembre 1984, est ainsi susceptible de s'appliquer aux expériences sur l'être humain contraires à l'éthique, dans la mesure où ce texte se fonde sur le principe de dignité inhérente à la personne humaine. Il en est de même pour des traités plus anciens comme la Convention du 9 décembre 1948 sur la prévention et la répression du crime de génocide et la Convention du 21 décembre 1965 sur l'élimination des discriminations raciales qui pourraient servir de fondement à l'interdiction de pratiques eugéniques ou de discriminations fondées sur le patrimoine génétique.
La Convention relative aux droits de l'enfant, du 26 janvier 1990, pourrait également être invoquée dans le cadre de la bioéthique, sur des sujets comme l'assistance médicale à la procréation. En effet, l'article 8 de cette convention reconnaît à l'enfant le droit à l'identité et aux relations familiales. De là, peut être posée la question du droit de l'enfant à connaître ses origines et s'ouvre alors le débat autour de l'accès de l'enfant, issu d'une AMP, à sa parenté biologique.
En dehors de diverses résolutions comme celle de 1991 relative à la protection des personnes atteintes de maladie mentale (22) et de celles adoptées par l'OMS (23), le texte onusien présentant le caractère le plus pertinent dans le domaine qui nous intéresse est la Déclaration universelle sur le génome humain et les droits de l'homme, adoptée par l'Unesco le 11 novembre 1997.
Comme on l'a souligné, la Déclaration de l'Unesco a été adoptée sous une forme déclarative plutôt que conventionnelle pour tenir compte du décalage existant entre les différents pays membres de l'Unesco. Conformément à la pratique de l'ONU, l'adoption de cette déclaration peut apparaître comme un premier pas vers la signature d'un traité de portée plus contraignante.
La Déclaration de 1997 ressortit, selon l'expression consacrée, à un droit de finalité. Elle se veut plus un texte de référence, servant de repère aux législateurs nationaux, qu'un cadre normatif contraignant. Au-delà d'un aspect symbolique évident, du fait de son caractère universel, elle peut contribuer utilement à un rapprochement des législations.
La Déclaration de 1997 s'articule autour d'un préambule et d'un dispositif. Le préambule rappelle les textes de références par rapport auxquels la Déclaration s'inscrit. Il s'agit, entre autres, de la Déclaration universelle des droits de l'homme de 1948 et des Pactes internationaux des Nations Unies relatifs aux droits économiques, sociaux et culturels, et aux droits civils et politiques du 16 décembre 1966, mais aussi des textes relatifs à la propriété intellectuelle, la dimension économique des recherches en matière biotechnologique étant prise en compte. Le préambule vise également la Convention de 1971 sur l'interdiction des armes bactériologiques, ce qui traduit le désir d'inciter les États à proscrire la fabrication d'armes constituées d'organismes modifiés génétiquement (24).
Le dispositif de la Déclaration s'organise autour de trois principales orientations : l'affirmation de la primauté de la dignité de l'individu ; la liberté de la recherche, qui doit être conciliée avec le principe précédent ; le rappel des droits de solidarité entre les États. L'article premier de la Déclaration proclame ainsi que « le génome humain sous-tend l'unité fondamentale de tous les membres de la famille humaine, ainsi que la reconnaissance de leur dignité intrinsèque et de leur diversité. Dans un sens symbolique, il est le patrimoine de l'humanité ».
Le respect de la dignité humaine passe par l'absence de discrimination en fonction du patrimoine génétique. L'article 2 de la Déclaration l'énonce clairement :
« a) Chaque individu a droit au respect de sa dignité et de ses droits, quelles que soient ses caractéristiques génétiques.
b) Cette dignité impose de ne pas réduire les individus à leurs caractéristiques génétiques et de respecter le caractère unique de chacun et leur diversité ».
L'article 6 le stipule avec plus de netteté encore : « Nul ne doit faire l'objet de discriminations fondées sur ses caractéristiques génétiques, qui auraient pour objet ou pour effet de porter atteinte à ses droits individuels et à ses libertés fondamentales et à la reconnaissance de sa dignité ».
L'article 3 de ce texte limite, pour sa part, la possibilité d'exploiter économiquement le génome humain dans une formule qui reste cependant sybilline, selon laquelle le génome « en son état naturel » ne peut donner lieu à des gains pécuniaires. Bien qu'elle ne soit pas compétente en matière de propriété intellectuelle, l'Unesco semble vouloir ici empêcher que la simple découverte d'un gène - c'est-à-dire sa localisation - sans apport inventif ne puisse conduire à l'obtention d'un brevet.
La Déclaration du 11 novembre 1997 arrête les principes qui doivent présider à toute recherche, traitement ou diagnostic portant sur le génome d'un individu. Une évaluation préalable et rigoureuse des risques et avantages potentiels est nécessaire (article 5a). Dans tous les cas, le consentement préalable, libre et éclairé de la personne intéressée doit être recueilli. Si cela n'est pas possible, le consentement ou l'autorisation est alors obtenu dans des conditions fixées par la loi et doit être guidé par l'intérêt supérieur de cette personne (article 5b). Les recherches menées, sans avoir pu recueillir le consentement de la personne, ne peuvent l'être qu'au bénéfice de la santé de celle-ci, dans le cadre d'une protection légale et dans le respect de la protection des droits individuels (article 5e). L'encadrement est donc ici particulièrement strict. La personne concernée a, par ailleurs, le droit d'accepter ou de refuser d'être informée des résultats obtenus (article 5c).
Sont également affirmés les principes de confidentialité des données génétiques et de réparation équitable du dommage subi à l'occasion d'une intervention portant sur le génome (articles 7 et 8).
Concernant, plus largement, la recherche sur le génome humain, la Déclaration du 11 novembre 1997 stipule qu'aucune recherche ne peut prévaloir sur le respect des droits de l'homme, des libertés fondamentales et de la dignité humaine des individus ou, le cas échéant, des groupes d'individus (article 10). Ainsi est explicitement déclaré contraire à cette dignité le clonage mené à des fins de reproduction d'êtres humains. La Déclaration va plus loin en invitant les États et les organisations internationales compétentes à coopérer afin d'identifier de telles pratiques et de prendre, au niveau national ou international, les mesures qui s'imposent (article 11).
Parallèlement, le principe de la liberté de recherche est reconnu ainsi que celui de libre accès aux progrès de la biologie, de la génétique et de la médecine (article 12).
La Déclaration de 1997 arrête aussi les principes qui doivent régir les conditions d'exercice de l'activité scientifique (articles 13 à 16). Elle définit également les devoirs de solidarité des États vis-à-vis des individus, des familles ou des populations particulièrement vulnérables aux maladies ou handicaps de nature génétique. Enfin, elle encourage les États à coopérer dans le domaine de la recherche sur le génome humain (articles 17 à 19).
Au total, la Déclaration de l'Unesco de 1997 constitue, avant tout, un cap vers lequel les législations nationales doivent tendre. S'il ne s'impose pas à notre loi, ce texte déclaratif pourrait cependant se voir reconnaître une certaine valeur juridique par les juridictions françaises. On verra plus loin dans quelles conditions une telle évolution serait possible.
Par rapport à la Déclaration de l'Unesco de 1997, la Convention sur les droits de l'homme et de la biomédecine, élaborée sous l'égide du Conseil de l'Europe et signée le 4 avril 1997, possède une véritable force contraignante. Cette convention, dite d'Oviedo, est l'aboutissement d'un travail mené de longue date par le Conseil de l'Europe à travers ses organes représentatifs (25). Avant d'aborder le contenu de ce traité, on présentera deux observations. Le Conseil de l'Europe s'est toujours montré plus actif que la Communauté européenne sur ces questions, qui relèvent de ses compétences alors qu'elles n'entrent pas dans le champ communautaire. On remarquera aussi que la Convention européenne de sauvegarde des droits de l'homme et des libertés fondamentales de 1950 peut servir de fondement à des revendications en matière bioéthique, y compris devant la Cour européenne des droits de l'homme de Strasbourg. En effet, la Convention proclame le droit à la vie (article 2), l'interdiction des traitements inhumains ou dégradants (article 3), le respect de la vie privée et familiale (article 8) ainsi que la liberté de pensée qui sert de fondement à la liberté de recherche (article 9). Il s'agit là d'autant de principes susceptibles d'être sollicités en matière d'éthique biomédicale.
Mais la Convention d'Oviedo est plus précisément consacrée à la bioéthique. Adopté par les États du Conseil de l'Europe en novembre 1996, ce texte est susceptible d'être complété par des protocoles additionnels consacrés à des questions spécifiques : l'embryon, la recherche sur les personnes, les transplantations d'organes, le génome humain.
Selon Mme Noëlle Lenoir et M. Bertrand Mathieu, ce texte reflète « le compromis obtenu entre diverses conceptions ayant cours en Europe. Certains des principes de la Convention, en effet, traduisent la valeur spirituelle attachée à la vie et à la personne humaine, tandis que d'autres se fondent sur l'affirmation de la liberté, liberté de l'individu comme liberté de la recherche » (26). Il est clair que les textes internationaux en général, et européens en particulier, doivent tenir compte d'approches et d'histoires diverses. Dans le cadre européen, il est patent que les positions britanniques et allemandes apparaissent peu compatibles.
La Convention d'Oviedo a été signée par vingt-neuf États membres du Conseil de l'Europe et ratifiée par sept d'entre eux. Elle est donc déjà entrée en vigueur dans ces sept pays. La France n'a pas encore ratifié la convention.
Dans son préambule, la Convention présente les principales préoccupations qui ont conduit à l'élaboration du texte. Les signataires déclarent avoir pris acte des rapides développements de la biologie et de la médecine. Ils sont convaincus de la nécessité de respecter l'être humain dans sa dignité, ayant conscience des actes qui pourraient mettre en danger la dignité humaine par un usage impropre de la biologie et de la médecine. Les signataires entendent promouvoir un débat public et soulignent « la nécessité d'une coopération internationale pour que l'humanité tout entière bénéficie de l'apport de la biologie et de la médecine ».
Dans le cadre de son dispositif, la Convention d'Oviedo fixe des règles, pour l'essentiel, peu éloignées de celles dont la France s'est dotée par les lois de 1994. L'objet de la Convention est défini à son article 1er :
« Les parties à la présente Convention protègent l'être humain dans sa dignité et son identité et garantissent à toute personne, sans discrimination, le respect de son intégrité et de ses autres droits et libertés fondamentales à l'égard des applications de la biologie et de la médecine.
Chaque partie prend dans son droit interne les mesures nécessaires pour donner effet aux dispositions de la présente Convention ».
L'article 2 de la Convention proclame la primauté de l'être humain, son intérêt et son bien devant prévaloir sur le seul intérêt de la société ou de la science. Comme la Déclaration de l'Unesco de 1997, la Convention d'Oviedo fixe le principe du consentement libre et éclairé de la personne devant subir une intervention dans le domaine de la santé. Ce consentement doit être le résultat d'une information adéquate. Il peut être retiré à tout moment (article 5). La question de la protection des personnes incapables de consentir est réglée de manière proche de ce que prévoit la Déclaration de l'Unesco, mais sans doute plus précisément (articles 6 à 9).
Par ailleurs, est proclamé le droit de chacun au respect de sa vie privée s'agissant des informations relatives à sa santé. En outre, il convient également de respecter la volonté exprimée par la personne de connaître ou non une information recueillie sur sa santé.
La première partie de la Convention d'Oviedo consiste ainsi en une codification du droit médical moderne. À ce titre, elle joue un rôle primordial dans la mise à niveau de la législation de certains États, en particulier ceux d'Europe centrale et orientale.
La Convention d'Oviedo consacre également plusieurs articles au génome humain. Fort opportunément, son article 11 interdit toute forme de discrimination à l'encontre d'une personne en raison de son patrimoine génétique. Cette stipulation présente un intérêt essentiel dans le débat autour de la médecine prédictive et l'accès à l'emploi et aux assurances. L'article 12 traite d'ailleurs des tests génétiques prédictifs. De tels examens, destinés à détecter une maladie génétique, à identifier le sujet comme porteur d'un gène responsable d'une maladie ou à établir une prédisposition ou une susceptibilité génétique à une maladie, ne pourront être effectués qu'à des fins médicales ou de recherche médicale, et sous réserve d'un conseil génétique approprié. De la sorte est interdit tout test dont l'objet serait, par exemple, de connaître le patrimoine génétique d'un individu aux fins de le juger apte ou non à occuper un emploi.
L'article 13 de la Convention d'Oviedo limite les cas où une intervention modifiant le génome humain peut être opérée. Seules des raisons préventives, diagnostiques ou thérapeutiques peuvent la justifier, pour autant qu'elle n'ait pas pour but d'introduire une modification dans le génome de la descendance de la personne concernée. Enfin, l'article 14 du traité interdit l'utilisation des techniques de l'AMP pour choisir le sexe de l'enfant à naître, hormis le cas où l'on souhaite éviter une maladie héréditaire grave liée au sexe.
Un chapitre entier de la Convention d'Oviedo est consacré, en outre, à la recherche scientifique. Un principe général de liberté est proclamé en ce domaine, à l'article 15, sous réserve des limitations contenues dans le traité et des autres dispositions juridiques assurant la protection de l'être humain. La Convention assure la protection des personnes se prêtant à une recherche, qu'elles aient ou non la capacité de consentir à celle-ci. Les conditions strictes ainsi requises sont proches de celles contenues dans la législation française depuis la loi n° 88-1138 du 20 décembre 1988, dite Huriet-Sérusclat, relative à la protection des personnes se prêtant à des recherches biomédicales. C'est le constat qu'a fait M. Carlos de Sola devant la Mission.
Selon lui, une légère nuance apparaît cependant s'agissant des personnes incapables, cette question ayant fait l'objet de controverses, en particulier en Allemagne. Si la loi française, quant à elle, précise que l'on peut pratiquer des recherches qui ne présentent pas un bénéfice individuel direct pour les personnes, à condition qu'il n'existe pas de risque majeur pour celle-ci, la Convention d'Oviedo est plus stricte : elle exige que les risques et la contrainte soient minimales. M. Carlos de Sola a estimé qu'il fallait faire preuve d'une rigueur particulière à l'égard, par exemple, des recherches menées sur des personnes plongées dans le coma, ne pouvant pas s'exprimer, alors que ces recherches ne leur apportaient aucun bénéfice direct.
L'article 18 de la Convention d'Oviedo revêt, quant à lui, une importance spécifique dans l'un des débats dont la Mission a été saisie. Il stipule que :
« Lorsque la recherche sur les embryons in vitro est admise par la loi, celle-ci assure une protection adéquate de l'embryon.
La constitution d'embryons humains aux fins de recherche est interdite ».
Si le premier alinéa de cet article laisse aux États une large marge de man_uvre pour adapter leur législation, le second fixe une interdiction beaucoup plus nette. On verra plus tard que l'interprétation de cet alinéa prête à discussion.
S'agissant des embryons surnuméraires, la Convention reste muette, les points de vue des États étant divergents sur cette question. Comme l'a montré M. Carlos de Sola, un État qui autorise la recherche sur des embryons surnuméraires peut ratifier la Convention, au même titre qu'un État qui interdit toute recherche conduisant à la destruction de ces embryons.
Enfin, la Convention du 4 avril 1997 fixe les conditions dans lesquelles peuvent être réalisés les prélèvements d'organes et de tissus sur des donneurs vivants à des fins de transplantation (articles 19 et 20). Elle interdit également, par son article 21, que le corps humain ou ses parties soient des sources de profit, arrêtant, par ailleurs, les règles d'utilisation des parties de corps humain prélevées au cours d'une intervention.
La Convention d'Oviedo a été complétée par un protocole additionnel en date du 12 janvier 1998, portant interdiction du clonage humain. Il prend acte des progrès que certaines techniques de clonage peuvent apporter à la connaissance scientifique ainsi qu'à ses applications médicales. Pour autant, il y est clairement établi que « l'instrumentalisation de l'être humain par la création délibérée d'êtres humains génétiquement identiques est contraire à la dignité de l'homme et constitue un usage impropre de la biologie et de la médecine » . En conséquence, l'article 1er du protocole additionnel interdit « toute intervention ayant pour but de créer un être humain génétiquement identique à un autre être humain vivant ou mort ». Le second alinéa de cet article précise que « l'expression être humain « génétiquement identique » à un autre être humain signifie un être humain ayant en commun avec un autre l'ensemble des gènes nucléaires » .
On constate que la Convention d'Oviedo fixe donc des règles qui s'imposent aux États signataires dans des conditions sur lesquelles il convient de s'arrêter.
2.- Le droit national face aux sources externes du droit de la bioéthique
a) La reconnaissance de la valeur du droit international en droit interne
La Constitution du 4 octobre 1958 a explicitement reconnu une valeur en droit interne des normes internationales. Son Préambule indique que « la République française, fidèle à ses traditions, se conforme aux règles du droit public international ». Plus explicite, l'article 55 de la Constitution dispose que « les traités ou accords régulièrement notifiés ou approuvés ont, dès leur publication, une autorité supérieure à celle des lois, sous réserve, pour chaque accord ou traité, de son application par l'autre partie ».
Sans entrer dans le détail d'une jurisprudence qui fut longtemps fluctuante sur ce sujet (27), on rappellera néanmoins les principes qui président à l'application du droit international en droit interne.
Les normes internationales ont une valeur supra législative, en vertu de l'article 55 de la Constitution. Depuis 1975, pour la Cour de cassation, et 1989, pour le Conseil d'État, les juridictions appliquent strictement cette règle (28). Désormais les cours font prévaloir les traités ainsi que les actes communautaires (29) sur les lois contraires, même postérieures.
Parmi les textes les plus directement applicables en droit français figurent ceux émanant de la Communauté européenne. On citera ainsi la directive européenne 98/44/CE du 6 juillet 1998 relative à la protection juridique des inventions biotechnologiques qui a un effet direct en droit français. Devant être transposée avant juillet 2000 - ce qui n'a pas été le cas pour des raisons que l'on abordera ensuite - cette directive peut venir à l'appui d'un recours contre des actes administratifs. Le juge national doit écarter l'application de toute norme nationale susceptible d'être contraire aux objectifs fixés par cette directive.
La règle d'application des traités ayant une force contraignante à l'égard des États est dénuée d'ambiguïté. C'est le cas pour la Convention d'Oviedo du 4 avril 1997. Encore faut-il que son contenu soit clair et tel n'est pas toujours le cas. On le constatera.
Cette règle d'application est, en revanche, moins nette pour les textes internationaux de portée plus indicative comme la Déclaration de l'Unesco de 1997 sur le génome humain. Comme l'a souligné le Professeur Bertrand Mathieu, lors de son audition par la Mission, il existe ainsi un paradoxe selon lequel la prégnance des normes internationales dans le droit de la bioéthique ne se traduit pas par un système normatif très fortement contraignant pour les droits nationaux. Concrètement, le droit international propre à la bioéthique ne représente pas pour le législateur national une réelle contrainte juridique.
Il n'en reste pas moins que le législateur national peut s'estimer légitimement tenu par des principes fixés dans des textes à vocation internationale, à l'élaboration desquels la France a souvent largement participé. Comme le constate le Professeur Bertrand Mathieu, les législateurs nationaux ne peuvent de la sorte ignorer ces textes internationaux, quelles que soient leur force ou leur faiblesse juridique qui les affectent.
D'un autre point de vue, il est clair que les normes non strictement juridiques jouent un rôle essentiel, non seulement dans la formation du droit, mais aussi dans la régulation des pratiques. Cet aspect a été très nettement illustré par les propos des scientifiques entendus lors de la journée d'auditions consacrée aux questions internationales, le 20 septembre 2000. En dépit de certains comportements que l'on peut qualifier de déviants, la communauté scientifique apparaît sensible à l'évolution des déclarations internationales, à l'élaboration desquelles elle contribue d'ailleurs fortement.
Ainsi les principes énoncés dans les déclarations, comme celle de l'Unesco de 1997, ne s'imposent pas directement aux États. Ceux-ci ont cependant une forme d'obligation politique - presque morale pour autant que ce terme ait un sens dans les relations inter-étatiques - de les respecter. Cette contrainte, qui n'est pas établie juridiquement au regard du droit international, apparaît donc plus diffuse. Comme on l'a remarqué, l'article 22 de la Déclaration de l'Unesco stipule que : « Les États devraient s'efforcer de promouvoir les principes énoncés dans la présente Déclaration et, par toutes mesures appropriées, favoriser leur mise en _uvre ». On mesure le caractère peu contraignant de ce dispositif.
À l'inverse, la Convention d'Oviedo impose de véritables obligations aux États signataires. Son article 23 stipule que : « Les Parties assurent une protection juridictionnelle appropriée afin d'empêcher ou faire cesser à bref délai une atteinte illicite aux droits et principes reconnus dans la présente convention ». De plus, l'article 25 prévoit que les Parties mettent en place des sanctions appropriées dans le cas de manquement aux dispositions de la Convention.
En dépit d'un dispositif d'application plus incitatif que contraignant, la Déclaration de l'Unesco de 1997 pourrait cependant se voir reconnaître une certaine force juridique sur le plan interne et, de manière indirecte, par les juridictions. En effet, les juges nationaux, notamment le Conseil constitutionnel, utilisent implicitement des principes issus du droit international pour interpréter la portée des textes nationaux, en particulier la Constitution. C'est par ce mécanisme assez discret que le juge constitutionnel a accueilli au sein du bloc de constitutionnalité des principes initialement formulés par le droit international ou par d'autres droits nationaux, comme celui de dignité de la personne humaine et celui du respect de la vie privée. Sans doute, pourrait-il tirer argument de la Déclaration de 1997, pour exciper d'autres principes applicables en droit interne.
b) La difficulté d'assurer la conformité de la loi française à des textes internationaux ambivalents
La législation française de 1994 est marquée par son caractère très protecteur des droits des individus et de la dignité humaine. Les décisions du Conseil constitutionnel en date du 27 juillet 1994, rendues sur ces lois prouvent que, de ce point de vue, celles-ci ne peuvent susciter aucun réel reproche. S'appuyant sur le Préambule de 1946 mais aussi implicitement sur les principes du droit international, le Conseil a affirmé que la sauvegarde de la dignité de la personne humaine contre toute forme d'asservissement et de dégradation était un principe à valeur constitutionnelle.
Les lois françaises ne présentent aucune contradiction majeure avec les textes internationaux qui, globalement, introduisent des protections, dont notre pays s'est déjà doté. Les lois de 1994 ont trouvé un équilibre qui paraît correspondre à celui que les normes internationales semblent faire émerger. En cela la position française est médiane au regard de celles du Royaume-Uni ou de l'Allemagne. Pourtant les traités, fruits de compromis, présentent souvent des ambiguïtés qui rendent difficiles les interprétations pouvant être données de leurs stipulations. De là naît une difficulté, pour les législateurs nationaux, de s'inscrire dans un cadre juridique international qui n'est pas toujours clairement défini. Cette souplesse peut cependant être aussi perçue comme une facilité appréciable laissée aux États.
À l'occasion de son intervention devant la Mission, le Professeur Bertrand Mathieu a ainsi tenu, tout d'abord, à souligner la relative faiblesse de la conceptualisation et de l'effectivité du droit international en matière bioéthique. Prenant l'exemple de la convention d'Oviedo, il a estimé qu'elle avait l'avantage de laisser au législateur national la faculté de faire ce qu'il entend, lui laissant dans les faits tout le poids de la décision.
À première vue, le texte de ce traité est, en effet, clair, mais on s'aperçoit que la notion d'être humain n'y est finalement pas définie, même si une protection de cet être y est affirmée. Il s'agit d'une souplesse considérable qui conduit à vider d'une partie de leur substance les dispositions de la Convention.
Ainsi son article 18 admet que certaines législations nationales permettent des recherches sur l'embryon in vitro, à condition qu'elles assurent une protection adéquate de l'embryon. Selon le Professeur Bertrand Mathieu, une interprétation littérale de ce texte devrait conduire à considérer que toute recherche induisant la destruction d'un embryon ne peut être jugée comme assurant une protection adéquate de ce dernier.
La notion de protection adéquate doit être interprétée au regard des stipulations de l'article 1er de la Convention. Celui-ci prévoit la protection de l'être humain dans sa dignité et son identité. Or, selon le Professeur Bertrand Mathieu, il est incontestable que l'article 18 n'est véritablement protecteur de l'embryon humain que dans l'hypothèse où l'on inclurait cet embryon dans la catégorie des êtres humains.
Mais l'interprétation de ce texte est moins claire qu'il n'y paraît. Car le rapport explicatif à la Convention de 1997 du Conseil de l'Europe introduit des ambiguïtés qu'il convient de relever. Ce rapport, connexe à la Convention, se définit lui-même comme n'étant pas un instrument d'interprétation authentique. Il apporte néanmoins un éclairage précieux sur les intentions des parties signataires. Il renvoie ainsi au droit interne des États le soin de préciser la signification des termes « être humain », tout en constatant, en même temps, que le respect, dès le commencement de la vie, de la dignité de l'être humain et de l'identité de la personne humaine est un principe généralement admis. Il existe donc bien un paradoxe à voir, d'une part, l'article 1er de la Convention affirmer le principe de dignité de l'être humain - distinct de la notion de « personne » et qui semble inclure l'embryon - et l'article 18 imposer une protection adéquate de l'embryon en cas de recherche et, d'autre part, laisser le soin, implicitement, aux États signataires de donner leur définition de l'être humain, y compris en excluant l'embryon de cette notion.
Cette réflexion subtile menée par le Professeur Bertrand Mathieu sur la notion d'être humain au sens de la Convention d'Oviedo et sur la protection qui en découlerait pour l'embryon humain, montre la difficulté de s'appuyer sur les textes internationaux pour fonder une législation nationale.
Au regard de ces textes, le rapport du Conseil d'État de novembre 1999 n'a mis en évidence qu'une seule réelle contrainte pour le législateur français. Elle concerne la création d'embryons à des fins de recherche.
La conception d'embryons à de telles fins est actuellement explicitement interdite par l'article L. 2141-8 du code de la santé publique. Or, la question de l'ouverture de cette possibilité a été au c_ur des débats tenus devant la Mission et, plus largement, devant le public, ces derniers mois. Comme la loi française, l'article 18 de la Convention d'Oviedo l'interdit expressément. Cette prohibition s'imposerait donc au Gouvernement et au Parlement français.
La stipulation interdisant la création d'embryons à des fins de recherche a été adoptée, non à l'unanimité, mais à une majorité supérieure aux deux tiers. Certains États ont voté contre, tel le Royaume-Uni. Du fait de ce vote à la seule majorité, les États peuvent donc émettre des réserves. Le Royaume-Uni pourra ainsi ratifier la convention en faisant une réserve sur ce point. Selon M. Carlos de Sola, le Conseil de l'Europe a accepté cette situation, estimant que le domaine d'application de la Convention d'Oviedo était suffisamment large pour qu'un point, si important soit-il, ne puisse pas empêcher un État d'accéder à l'ensemble du corpus commun européen dans le domaine de la biomédecine.
Au total, le caractère ambivalent des normes internationales laissent les États en première ligne sur des sujets sensibles. Il leur appartient de prendre les mesures les plus conformes à leur sensibilité propre, née de leur histoire et de leur culture.
S'oriente-t-on cependant vers une homogénéisation, à terme, des règles nationales ? La question a été posée aux intervenants entendus par la Mission lors de la journée consacrée aux enjeux internationaux le 20 septembre 2000.
c) Un chemin étroit pour assurer la cohérence des normes internes et externes
M. Carlos de Sola, chargé des questions de bioéthique au Conseil de l'Europe, a estimé, lors de son audition, que l'on se dirige vers une certaine homogénéisation des droits, dans la mesure où les sociétés deviennent de plus en plus semblables. Il a néanmoins insisté sur le fait que des traditions nationales demeuraient prégnantes, avec de nettes différences, en particulier entre les pays d'Europe continentale et le Royaume-Uni.
Le Professeur Alain Pompidou a exprimé son accord avec M. Carlos de Sola sur un mouvement tendant à un rapprochement des normes entre les pays occidentaux. Mais abordant dans une plus large vision, la question internationale, il a souligné la difficulté d'aboutir à un tel mouvement unitaire dans le cadre mondial. Pour lui, les pays asiatiques s'inscrivent dans une optique très différente de celle des pays de l'OCDE.
Dans le cadre strictement européen, la recherche d'un équilibre entre les traditions nationales est un souci constant pour le Groupe européen d'éthique. Mme Christiane Bardoux, secrétaire de cet organe, l'a montré fort clairement lors de son intervention devant la Mission. Selon elle, deux éléments essentiels guident l'approche communautaire de la bioéthique : respecter à la fois les droits fondamentaux tout en restant attentif à la diversité des États. La recherche permanente d'un équilibre sage entre une exigence commune et le souci de respecter la diversité historique, culturelle ou religieuse des États est ainsi l'une des exigences essentielles du débat bioéthique dans la Communauté européenne.
Si la Communauté européenne ne dispose pas de la compétence en matière de bioéthique, elle peut cependant jouer un rôle important dans le discussion qui se noue désormais au plan international sur ces sujets. N'ayant pas de compétences pour légiférer, la Communauté européenne n'est pas dans l'obligation de faire un choix crucial dans ces domaines. Elle peut donc permettre à des opinions diver-gentes de s'exprimer, dans le cadre de forums, en dehors de toute dramatisation des enjeux. Selon la secrétaire du GEE, c'est ici que réside l'apport constructif du débat communautaire.
Le caractère paradoxal des normes internationales de la bioéthique apparaît en pleine lumière. D'un côté, une aspiration à l'émergence de règles universelles est très nette. L'internationalisation des recherches, la mobilité des scientifiques mais aussi des patients, la diffusion des connaissances font éclater les frontières dans des domaines qui touchent l'humanité en son entier. De l'autre, demeurent des traditions qui conduisent à des approches différentes de ces sujets. La tentative d'ajustement de ces deux tendances conduit à l'adoption de textes internationaux qui constituent toujours des avancées.
Le législateur doit donc tenir compte des engagements pris par la France dans le cadre international. C'est pourquoi il apparaît nécessaire à votre Rapporteur que le Parlement puisse ratifier la Convention d'Oviedo parallèlement à la révision des lois bioéthiques de 1994. Il s'agirait là d'un signe fort adressé par le législateur quant à sa volonté d'insérer son action dans la perspective de l'émergence d'un droit international de la bioéthique. Le maintien de la cohérence entre le droit interne et les traités engageant notre pays doit être l'un des principaux soucis du Parlement. Encore faut-il que le politique prenne la mesure du développement d'un droit international de la bioéthique en construction, fondé sur des textes de portée plus ou moins contraignante et universelle, et qui recherche sans cesse à établir un équilibre entre des principes parfois contradictoires.
C.- LES LOIS BIOÉTHIQUES FACE AUX ENJEUX ÉCONOMIQUES
Le Parlement ne peut adopter la loi sans tenir compte de la réalité économique, aujourd'hui étroitement liée aux activités de recherches et aux pratiques médicales. Le secteur des biotechnologies tient désormais une place qui apparaît plus solide que celle de la nouvelle économie fondée sur l'Internet dont les résultats récents ont déçu. La première phase, celle des découvertes, est aujourd'hui relayée par celle des inventions et de la mise sur le marché prochaine de nouveaux produits. Face à cela, le législateur doit tenir compte de la réalité, sans surtout abdiquer ses prérogatives.
1.- Le développement du marché des biotechnologies
Les biotechnologies comprennent toutes les méthodes et techniques utilisant les capacités génétiques et physiologiques du vivant pour mieux conduire ou contrôler des processus naturels, ou mieux produire et purifier des substances issues de la transformation biologique de substrats naturels. Le champ des biotechnologies ne concerne donc pas seulement les sujets qui nous intéressent dans le cadre de la révision des lois de bioéthique. Il couvre aussi, par exemple, les OGM et les questions agricoles, l'environnement.
Le secteur des biotechnologies, et celui des biotechnologies médicales en particulier, connaît une croissance très forte depuis les années 80. Ce mouvement s'est notablement accéléré depuis le milieu des années 90, avec le projet de décryptage des génomes.
En 1998, le chiffre d'affaires mondial des biotechnologies atteignait 17 milliards de dollars ce qui apparaît assez peu au regard de celui de l'industrie pharmaceutique, de l'ordre de 307 milliards. Mais cette comparaison ne rend pas compte de la dynamique qui accompagne le développement des biotechnologies. Nous sommes aujourd'hui dans une phase d'investissement, matériel et surtout intellectuel, qui, dans ce secteur, peut s'avérer très longue. Des potentialités importantes apparaissent pourtant. D'ici quelques années, les résultats des recherches menées actuellement auront un impact économique et financier beaucoup plus massif. Les projections montrent, en effet, que ce chiffre d'affaires global pourrait atteindre 28 milliards de dollars en 2005 et 50 milliards en 2010, avec 35 milliards attendus pour les produits relatifs à la santé humaine, un peu plus de 5 milliards de dollars pour le diagnostic médical et autant pour les produits agricoles (30).
En termes d'effectifs travaillant dans ce domaine, en Europe, le secteur des biotechnologies représentait 46.000 emplois en 1998 contre 16.000 en 1994. Cette progression de 285 % montre la dynamique qui anime ce secteur d'activité. En France, il employait environ 5.000 personnes en 1998 alors qu'aux États-Unis 153.000 personnes y travaillaient (31).
Comme on l'a mentionné, les biotechnologies concernent des secteurs aussi essentiels que la santé, l'agriculture et l'environnement. Dans le secteur médical, on distingue principalement trois groupes de produits issus des biotechnologies :
- Les protéines dites recombinantes comme l'érythropoiétine (EPO), utilisée en cas d'anémies, ou comme l'interféron, qui permet de lutter contre des cancers et l'hépatite. Ces substances représentent actuellement un marché de 15 milliards de dollars par an.
- Les anticorps monoclonaux dont le champ d'application thérapeutique est extrêmement large : cancers, maladies auto-immunes, affections cardiaques, rejets de greffes. Ce marché s'avère très porteur puisque aujourd'hui 10 anticorps monoclonaux sont commercialisés et 150 sont en développement.
- Une nouvelle génération de produits biologiques comme les vaccins et les thérapies géniques visant des cibles mises à jour par la génétique. On connaît maintenant environ 500 de ces cibles. Après le déchiffrage du génome humain, on en disposera de plusieurs milliers. La perspective est tracée pour le développement de produits.
L'an passé, presque 80 médicaments issus des biotechnologies étaient commercialisés. Près de 1.500 étaient en développement dont 369 en développement clinique, ciblant plus de 200 maladies différentes. Au cours des années, ce nombre a crû régulièrement. Il était de 81 en 1988, de 143 en 1993, de 284 en 1996 et donc de 369 en 2000 (32).
Pour mémoire, on rappellera que, dans le secteur agronomique, les biotechnologies ouvrent aussi de nouvelles perspectives en matière d'amélioration des plantes en favorisant, entre autres : la résistance des végétaux aux parasites et aux insectes nuisibles ; la tolérance aux herbicides ; la résistance à la sécheresse ou aux basses températures ; la réduction des dépenses énergétiques. Enfin, dans le secteur de l'environnement, les biotechnologies ouvrent des perspectives dans les domaines de l'épuration par des méthodes biologiques ou du traitement de la biomasse.
Le secteur des biotechnologies se caractérise donc par un mouvement perpétuel de découvertes et d'inventions. Il est également marqué par une évolution de ses structures industrielles et financières.
b) La structure et l'évolution du secteur des biotechnologies
Le secteur des biotechnologies comprend essentiellement des sociétés de petite dimension, qui tentent de se regrouper depuis quelques temps aux fins de résister à l'appétit des grands groupes pharmaceutiques. La révolution génomique semble, en effet, bienvenue pour l'industrie pharmaceutique qui réalise aujourd'hui ses meilleures ventes avec des médicaments qui tomberont dans le domaine public vers 2005. On considère que la génomique permettrait à cette industrie de passer de 337 milliards de dollars de chiffre d'affaires en 2000 à plus de 500 milliards en 2004.
Ce mouvement de concentration des start-up est également mû par le souci, pour ces entreprises, de se crédibiliser sur le marché afin, notamment, de drainer des financements plus importants. Ces jeunes entreprises tentent aussi de sortir de leur activité de base fondée sur la recherche pour développer de véritables produits. Le fait que les deux entreprises françaises les plus en vue dans ce domaine - Genset et Transgene - aient confié récemment leur gestion à des personnes issues du monde de l'industrie pharmaceutique a été interprété comme une volonté de réorienter leur activité dans ce sens.
Le secteur des biotechnologies reste très dépendant de la capitalisation boursière, faute de retombées industrielles immédiates. Il a bénéficié en 2000 d'un transfert important de capitaux boursiers provenant des mauvais résultats des sociétés spécialisées dans le commerce électronique. Les investisseurs ont préféré se porter vers les biotechnologies qui ont profité de l'annonce du décryptage du génome humain. Ainsi entre le 15 juillet et le 15 août 2000, 28 sociétés de biotechnologies biomédicales sont entrées en bourse aux États-Unis, levant 2,2 milliards de dollars. Le NASDAQ Biotechnology a gagné près de 20 % entre le 1er janvier et la fin novembre 2000 (33).
La flambée des cours est fonction moins de résultats concrets que de publications d'articles scientifiques établissant l'avancée des recherches menées par les entreprises de ce secteur. Cette situation a profité aux sociétés qui se sont consacrées au décryptage du génome et qui peuvent donc présenter des résultats tangibles. Il s'agit, en particulier, de Celera Genomics ou encore de Myriad Genetics, cette entreprise de l'Utah qui, grâce à la généalogie et au code génétique d'un million de mormons, a pu identifier et breveter l'un des gènes responsables du cancer du sein.
Lors de son déplacement aux États-Unis, votre Rapporteur a pu mesurer aussi le poids de la bio-informatique qui apparaît comme une seconde étape dans le domaine de la génomique. Les entreprises qui interviennent dans ce nouveau secteur mettent en _uvre des programmes informatiques très puissants afin de traiter les milliards de données liées au génome. Des grands groupes informatiques, comme IBM ou Compaq, se sont associés à des sociétés pharmaceutiques ou de biotechnologies comme Celera Genomics, pour développer de nouveaux ordinateurs. Cette coopération constitue l'une des principales évolutions en la matière.
Sur le plan économique, la croissance globale du secteur devrait continuer à être soutenue grâce à la conjonction de plusieurs tendances favorables : l'arrivée à maturité de nouveaux médicaments produits par les biotechnologies, ainsi que la diversité des recherches et des secteurs d'application. Dans ce cadre, l'avance des États-Unis est patente.
2.- L'avance indéniable des États-Unis
a) La dynamique conjointe de la science et de l'économie aux États-Unis
Les États-Unis occupent une place prépondérante dans ce secteur d'activité (34). La France, et plus largement l'Europe, accusent un retard important, malgré un certain nombre d'atouts, notamment la qualité de leur recherche fondamentale en biologie.
Comme le notait le quotidien Les Echos, le 8 janvier dernier, la dernière décennie a pu apparaître comme merveilleuse pour les États-Unis, tant ce pays a renforcé son potentiel technologique et scientifique, en prenant une nette avance dans les techniques de l'information et dans les biotechnologies. Ce succès a semblé d'autant plus frappant que les États-Unis sortaient d'une période beaucoup moins brillante, les grands secteurs comme l'automobile et l'électronique étant concurrencés, lors de la décennie précédente, par le Japon et la recherche semblant en panne outre-Atlantique.
Ainsi que le souligne M. Pierre Papon, président de l'Observatoire des sciences et des techniques, l'écart s'est nettement creusé entre les États-Unis et l'Europe dans les années 90. Dans le secteur des sciences de la vie, les Américains auraient même pris une avance décisive (35).
Cette avance est due à un investissement massif dans la recherche à partir de 1994. Les budgets européens consacrés à la connaissance - la recherche et le développement, la formation et l'éducation - stagnent, alors que les États-Unis engagent des fonds importants dans ce domaine. En 1999, les États-Unis ont investi 75 milliards d'euros de plus que l'Europe dans la recherche, avec un produit national brut pourtant inférieur.
La prépondérance des États-Unis dans le domaine de la recherche est aussi fondée sur une très efficace complémentarité des pôles public et privé. Les universités et les laboratoires de recherches passent des accords avec des entreprises, à qui elles délivrent des licences d'exploitation. La mobilisation des structures scientifiques, notamment universitaires, aux fins d'obtenir des fonds de la part de l'État, est beaucoup plus intense qu'en Europe. Ainsi, il y a six ans a été créée la Science coalition, association qui regroupe des chercheurs publics et privés, des professeurs ainsi que des associations et des industriels. Cette structure s'est donné comme mission de sensibiliser le monde politique américain aux questions de recherche, avec, semble-t-il, un réel succès.
Le nombre de sociétés de biotechnologies aux États-Unis était de 1 238 en 1998, contre 140 en France et 1 038 dans le reste de l'Europe. A première vue, la comparaison ne laisse donc pas apparaître un si net retard de notre continent (36). Mais le poids des sociétés en question, notamment en termes de brevets déposés, est à l'avantage des États-Unis.
Comme le soulignait, lors de son audition, M. Gilles de Poncins, directeur général de GenOway, une entreprise française de biotechnologie, les sociétés américaines intervenant dans ce domaine peuvent s'appuyer sur une industrie pharmaceutique très puissante. La perspective de voir les États-Unis disposer d'une position totalement dominante dans ce secteur est réelle. Les recherches menées dans ce pays sur les cellules souches seraient, selon M. Gilles de Poncins, l'une des voies empruntées pour asseoir cette primauté.
M. Pascal Brandys, alors président-directeur général de Genset, a noté devant la Mission que l'industrie française est naissante, par rapport à celle des États-Unis. La majorité des sociétés de biotechnologie, dans notre pays, ont moins de dix ans d'existence et comptent seulement quelques dizaines de salariés. La doyenne d'âge est Transgènes, créée il y a vingt ans. Genset, née il y a onze ans, est actuellement la plus grande entreprise de biotechnologie en France, avec ses six cents salariés.
Dans ces entreprises de première génération, les capitaux sont réduits, à la différence des sociétés américaines qui ont déjà, dans la plupart des cas, des produits commercialisés issus de la recherche. À l'inverse, la majorité des entreprises françaises en sont au premier stade du développement de technologies ou d'essais cliniques. Selon M. Pascal Brandys, aucune d'entre elles ne commercialise véritablement de produits issus de sa recherche.
Comme le souligne le rapport du Conseil économique et social de 1999, les conséquences du succès américain sont de deux ordres. En premier lieu, le développement d'une nouvelle branche industrielle qui est déjà fortement créatrice de richesses et d'emplois. Si l'on reprend les données présentées précédemment, on constate qu'en 1998, les États-Unis comptaient plus de 1 200 sociétés de biotechnologies représentant 153 000 emplois et un chiffre d'affaires de 16 milliards d'euros contre seulement 46 000 emplois et 3,7 milliards d'euros pour l'Europe avec un nombre sensiblement équivalent de sociétés de biotechnologies.
En second lieu, une prééminence vis-à-vis du reste du monde qui s'exprime clairement dans les résultats commercialisés de l'innovation : près de la moitié des molécules pharmaceutiques nouvelles faisant l'objet d'une commercialisation mondiale est d'origine américaine. Il en va de même, dans le domaine agricole, pour la plupart des organismes génétiquement modifiés mis aujourd'hui sur le marché.
La France reste bien placée au plan européen. Traditionnellement, en deuxième position après le Royaume-Uni, elle est cependant récemment passée derrière l'Allemagne, qui a produit de très substantiels efforts dans ce secteur depuis quelque temps.
Pour M. Pascal Brandys, l'industrie française dispose néanmoins d'atouts : elle s'appuie sur une recherche, essentiellement publique, de très grande qualité dans le domaine des sciences du vivant. Aujourd'hui, les domaines d'excellence de la France se retrouvent dans l'industrie des biotechnologies, que ce soit la génomique, la thérapie génique ou l'immunologie.
Pour lui, l'environnement s'est également amélioré pour ce qui est des financements, grâce à l'ouverture des nouveaux marchés financiers. Cela a permis à plusieurs sociétés françaises d'accéder à la cotation en bourse et d'avoir ainsi accès aux moyens de se développer.
Selon les responsables d'entreprises de biotechnologies françaises, que la Mission a entendus, un travail important reste à accomplir. Contrairement aux États-Unis, les organismes de valorisation des laboratoires de recherche publics sont souvent sous-équipés. De plus, la direction de ces instituts ne se fixe pas de véritables objectifs, en termes de créations d'entreprises ou de développement de l'activité de licences.
Des initiatives comme le Génopole d'Evry montrent que l'on peut mettre en place des synergies efficaces. Créé en juillet 1998, le Génopole a pour mission de favoriser le développement de la biologie à grande échelle dans la recherche publique et privée comme dans le domaine industriel des biotechnologies. Son objectif est dynamique : positionner la France dans la compétition scientifique et industrielle engagée dans la génomique, la post-génomique et plus largement dans les sciences de la vie. Sa mission consiste aussi à animer le réseau national des génopoles de Lille, Strasbourg, Lyon-Grenoble, Aix-Marseille, Montpellier, Toulouse et Bordeaux. Le Génopole accueille sur son site des laboratoires comme le centre national de séquençage, le centre national de génotypage ou le laboratoire de thérapie génique de l'Association française contre les myopathies. Par ailleurs, est présent à Évry un campus universitaire multidisciplinaire. Il devrait rassembler en 2002 près de 30 laboratoires ou équipes de grands établissements publics de recherche et des universités. Le Génopole entend aussi faciliter la valorisation maximale de ces activités de recherche en transformant les découvertes en innovations brevetables puis en applications industrielles et commerciales. L'accent est donc mis sur ce qui est généralement considéré comme l'un des points faibles du secteur dans notre pays. Comme le souligne son directeur général, M. Pierre Tambourin, le Genopole se situe à l'interface du monde privé et public, ce qui constitue l'un de ses principaux atouts.
C'est sans doute dans cette direction que des efforts devront être accomplis dans notre pays si l'on souhaite que l'écart avec les États-Unis ne se creuse pas trop cruellement. Car cette domination américaine soulève le problème de la délocalisation d'une partie de la recherche vers les centres d'excellence américains.
Pour M. Gilles de Poncins, directeur général de GenOway, l'enjeu est actuellement de savoir si une entreprise européenne doit ou non partir s'installer outre-Atlantique si elle souhaite se développer. Les sollicitations sont nombreuses et, semble-t-il, attractives financièrement.
Les sociétés américaines déploient effectivement des stratégies de conquêtes efficaces. Ainsi, lorsqu'une entreprise européenne dépose une demande de brevet qui n'a pas encore été acceptée, les sociétés d'outre-Atlantique sollicitent l'entreprise pour le lui acheter, avant même que le brevet ait été accordé. Mais plus encore, ce sont les entreprises elles-mêmes que convoitent ces sociétés implantées aux États-Unis.
Le succès des États-Unis se manifeste aussi par l'afflux de post-doctorants venant de tous les pays, et plus spécialement d'Europe. Votre Rapporteur, qui a eu l'occasion de rencontrer certains d'entre eux lors de son déplacement à Washington, a pu mesurer l'attrait que les États-Unis exercent sur les jeunes scientifiques français, dont la qualité semble très appréciée par les responsables de laboratoires américains. Les moyens matériels qui sont mis à leur disposition dès leur arrivée sont sans commune mesure avec ce qu'ils peuvent espérer sur le vieux continent. On notera cependant que ces jeunes chercheurs souhaitent souvent revenir en France après quelques années passées aux États-Unis, leur séjour dans ce pays et l'expérience ainsi acquise pouvant bénéficier à nos centres de recherche.
Ces éléments ne sont pas mineurs dans la réflexion que nous devons mener sur l'évolution de notre législation en matière de bioéthique. La perspective de voir la recherche se développer ailleurs qu'en Europe ne doit pas nous conduire, dans un aveuglement total, à méconnaître les principes auxquels nous sommes attachés et à autoriser toutes les pratiques. Mais elle nous montre que tout effort mené pour trouver un équilibre propre à la bioéthique doit être porté sur la scène internationale pour avoir l'assurance que ces principes accèdent à une forme d'universalité.
Le droit international de la bioéthique est encore trop faible. Il ne permet pas de maîtriser pleinement la recherche et les pratiques médicales. Il ne constitue pas non plus une barrière suffisamment solide pour faire face aux éventuelles dérives, pour maîtriser les logiques de marchés pour lesquelles la bioéthique compte peu.
La révision des lois de 1994 doit donc s'apprécier au regard de ces avancées mais aussi de ces obstacles. C'est dans cet esprit empreint de réalisme que la Mission d'information a mené ses travaux, abordant, à l'instar du Conseil d'État, cinq grands champs de réflexion : l'assistance médicale à la procréation, la recherche sur l'embryon, le don et l'utilisation des éléments du corps humain, la médecine prédictive et la brevetabilité du génome humain.
DEUXIÈME PARTIE :
L'ASSISTANCE MÉDICALE À LA PROCRÉATION (AMP) :
UN PARCOURS DIFFICILE QUI NÉCESSITE DES AMÉLIORATIONS
L'assistance médicale à la procréation (AMP) permet de remédier à certains problèmes d'infertilité en améliorant le processus naturel de fécondation ou en reproduisant ce processus en laboratoire, c'est-à-dire in vitro. Sous ce thème sont ainsi regroupées des pratiques très différentes allant du simple traitement hormonal au transfert d'un embryon conçu in vitro, éventuellement avec don de gamète, spermatozoïde ou ovocyte. Pour reprendre la classification du Professeur Jacques Montagut, on pourrait distinguer :
- les techniques visant à optimiser la fécondation de gamètes in vivo, parmi lesquelles la stimulation ovarienne et l'ensemble des pratiques d'insémination artificielle de spermatozoïdes ;
- les techniques ayant pour but de féconder in vitro les gamètes et de transférer ultérieurement dans la cavité utérine de la mère l'_uf fécondé, improprement appelé « embryon » en dépit de son stade de développement ;
- et enfin les techniques de cryoconservation, c'est-à-dire de conservation par le froid des gamètes - actuellement possible pour les seuls spermatozoïdes - et des embryons.
Depuis la naissance d'Amandine en février 1982, premier enfant issu d'une fécondation in vitro (FIV) en France, grâce à l'équipe dirigée par le Professeur René Frydman, qui a suivi de près Louise Brown, premier enfant conçu grâce à cette technique, né en juillet 1978 en Grande-Bretagne, les techniques n'ont cessé d'être perfectionnées par l'amélioration de leur efficacité, ou l'extension de leurs indications à de nouvelles causes d'infertilité. Elles ont ainsi contribué à accroître la demande en ce domaine, mais aussi entretenu un peu plus l'idée que la science est toujours apte à satisfaire le désir d'enfant. Pourtant, le parcours dans lequel s'engagent les couples recourant à l'AMP est long, difficile, douloureux à plus d'un titre et surtout hypothéqué dans ses chances de succès, en dépit des progrès enregistrés ces dernières années.
Il convient à cet égard de rappeler la réalité humaine qui se cache derrière ces pratiques médicales. Mme Chantale Ramogida, fondatrice de l'association « Pauline et Adrien », dans son audition devant la Mission, l'exprimait ainsi : « L'homme et la femme sont obligés d'exposer leur vie privée devant le corps médical et, maintenant, devant des juges au tribunal de grande instance ou de simples greffiers. (...) Certains couples divorcent. (...) Mais les enfants sont là, ils ont un père et une mère, bien que séparés, et un certain équilibre demeure. Ce sont des enfants tellement désirés qu'il n'y a pas de demi-mesure. Si les techniques de procréation ne débouchent pas sur la grossesse, le couple divorce dans de nombreux cas, malheureusement trop souvent. Mais parfois, même en cas de naissance, le couple est si altéré par tout le parcours que l'homme et la femme ne savent plus trop pourquoi ils sont ensemble. L'association compte huit mille quatre cents adhérents dans toute la France et reçoit quatre-vingts appels par jour et cent cinquante lettres par semaine. Souvent ce sont des hommes qui nous appellent pour nous demander de parler à leur conjointe car il n'y a plus de vie de couple. Tout se focalise sur cette idée : l'enfant ».
I.- LA RÉALITÉ CHIFFRÉE DE L'AMP
Deux sources d'information renseignent sur l'évolution des activités d'AMP dans notre pays : celle de la Commission nationale de médecine et de biologie de la reproduction et du diagnostic prénatal (CNMBRDP), chargée par la loi du suivi des centres agréés pour l'AMP, et celle de la FIVNAT, association qui regroupe une grande partie de ces centres (37) sur la base du volontariat. Ces deux sources sont complémentaires pour comprendre et analyser la situation et son évolution. En effet, la FIVNAT regroupe les données individuelles relatives aux patients qui recourent aux techniques d'AMP, tandis que la CNMBRDP reçoit et synthétise l'ensemble des bilans d'activité que chaque centre agréé est tenu de lui transmettre chaque année.
S'il est difficile d'établir des comparaisons justes se basant sur les mêmes éléments statistiques, plusieurs spécialistes estiment que la France occupe le troisième rang mondial, après Israël et les pays nordiques, en termes de recours à l'AMP par rapport à la population, bien avant les États-Unis.
Cette place prééminente n'est pas étonnante, non pas en raison du caractère plus élevé de l'infertilité ou de la stérilité de nos compatriotes mais bien en raison, d'une part, du niveau atteint par ces techniques dans notre pays, et, d'autre part, du régime particulièrement favorable du remboursement de ces pratiques par la sécurité sociale. La France est en effet l'un des rares pays où la prise en charge des AMP s'effectue à 100 % pour quatre tentatives maximum dans le cas d'une FIV ou d'une fécondation par micro-injection intracytoplasmique (technique dite de l'ICSI qui signifie Intra-Cytoplasmic Sperm Injection en anglais). Le coût de ces pratiques est loin d'être négligeable puisque l'on peut chiffrer à près de 3 000 francs une insémination artificielle intra-conjugale (IAC) et à environ 20 000 francs une FIV ou une ICSI. De plus, une récente décision du Conseil d'État, dans l'affaire Mme Laguette et autres du 27 novembre 2000, a considéré comme illégale cette limite de quatre tentatives ainsi que celle liée à l'âge de la femme qui recoure à l'AMP (38), en considérant que les ministres n'étaient pas compétents pour édicter de telles limitations par le moyen des nomenclatures (39). Si l'on compare cette situation à celle du Royaume-Uni, où aucune pratique d'AMP ne fait l'objet d'une prise en charge, même partielle, on ne peut que se féliciter du souci de permettre un égal accès à l'AMP de tous les patients dans notre pays.
En nombre de naissances, l'AMP se chiffrait en 1998 à 13.453 enfants, soit 18 % du total des naissances dans notre pays. Ce chiffre est en amélioration régulière grâce à la conjonction de plusieurs facteurs : le renforcement de la technicité des centres, qui parviennent à améliorer leurs résultats de manière significative, le recours à la technique de l'ICSI, qui améliore sensiblement le taux de réussite dans certains cas d'infécondité masculine, une plus grande sélection des patients qui entrent dans une procédure d'AMP et, enfin, un effet positif en termes d'efficacité des traitements de stimulation ovarienne.
1998 |
1997 |
Hausse de | |
Nombre de naissances dans la population française |
740.300 |
726.300 |
+1,9% |
Nombre de naissances en PMA |
13.453 |
12.132 |
+9,8% |
Nombre de naissances en PMA pour 1.000 naissances |
18 |
17 |
Source : M. de Mouzon, Unité 292 de l'INSERM, sur la base des données fournies par la FIVNAT en 2000.
S'agissant de la répartition des techniques d'AMP, on note une place croissante du recours à l'ICSI, qui représentait, selon la FIVNAT, 46,6 % des ponctions en 1999, auxquelles il faut ajouter 2,4 % de cycles où une ICSI a été pratiquée en même temps qu'une FIV. Toutefois, cette place croissante de l'ICSI a tendance à stagner dans la mesure où les indications médicales pour l'utiliser sont limitées. Comme le montre la partie suivante du présent rapport, le rendement de l'ICSI est relativement élevé mais il convient d'atténuer ce constat par celui de l'âge moyen des femmes concernées. En effet, s'agissant en l'espèce d'infertilité masculine, les patientes sont en moyenne plus jeunes ; les traitements de stimulation ovarienne pratiqués sur elles donnent ainsi de meilleurs résultats, d'où une explication parmi d'autres du taux de succès élevé de cette technique.
RÉPARTITION DES TECHNIQUES UTILISÉES
Inséminationavec conjoint |
Insémination avec donneur |
FIV&ICSI (avec conjoint) |
Transfert d'embryons (avec conjoint) |
FIV&ICSI (avec donneur) |
Transfert d'embryons (avec don) |
Total | |
Nombre de tentatives |
44.661 |
8.117 |
38.461 |
7.117 |
1.296 |
175 |
99.827 |
Distribution des tentatives |
44,74% |
8,13% |
38,53% |
7,13% |
1,30% |
0,18% |
Source : M. de Mouzon, Unité 292 de l'INSERM, sur la base des données fournies par la FIVNAT en 2000.
Le tableau suivant présente les résultats obtenus pour chaque technique d'AMP en nombre de grossesses cliniques, en pourcentage par rapport aux nombres de cycles, en accouchements, en distinguant les grossesses simples et multiples, et en nombre d'enfants vivants.
Si l'on retient le nombre de ponctions opérées sur les patientes, on note, pour l'année 1999, une stabilisation du nombre d'ovocytes totaux inséminés ou injectés. En revanche, le taux de fécondation, c'est-à-dire le nombre d'embryons obtenus par ovocytes fécondés s'est amélioré.
Lorsqu'il y a eu congélation d'embryons, le nombre d'embryons congelés était en moyenne pour l'année 1999, de 3,9 en FIV et de 3,6 en ICSI. S'agissant du transfert des embryons in utero, il faut se féliciter de la baisse continue du nombre d'embryons transférés, qui est actuellement de 2,3 en FIV et de 2,4 en ICSI. Seulement 6 à 7 % des transferts réalisés l'ont été avec plus de trois embryons. Cette nouvelle pratique diminue considérablement le nombre de « réductions embryonnaires » qu'il était nécessaire parfois de pratiquer en cas d'implantation réussie d'un nombre trop élevé d'embryons. Les équipes françaises ont en effet choisi désormais, de transférer, dans 40 % des cas, deux embryons. Le taux de grossesses associé a été particulièrement amélioré puisqu'il est aujourd'hui de 29 %.
Cette évolution de la pratique vers une baisse notable du nombre d'embryons transférés est le résultat d'une prise de conscience par les équipes des risques que présentent les grossesses multiples pour la mère et les enfants. Les inconvénients et handicaps associés à la prématurité, le risque de fausses couches ou le recours, toujours douloureux et dangereux, à la réduction embryonnaire, ont en effet milité pour cette nouvelle pratique. Ce choix n'est pas partagé par d'autres pays. Ainsi, aux États-Unis, c'est la recherche d'une ou de plusieurs naissances « à tout prix » qui prime et qui conduit à transférer en moyenne quatre embryons in utero, avec des cas extrêmes de naissances multiples fortement contestables. En Suède, il est même envisagé de ne transférer qu'un seul embryon, ce qui réduit considérablement le taux de grossesses réussies.
BILAN DES RÉSULTATS DE L'AMP EN FONCTION DES TECHNIQUES UTILISÉES
1998 |
1997 |
1998 |
1997 |
1998 |
1997 |
1998 |
1997 |
1998 |
1997 | ||
Insémina-tion avec conjoint |
Insémina-tion avec conjoint |
FIV avec conjoint |
FIV avec conjoint |
FIV avec donneur |
FIV avec donneur |
Don de sperme |
Don de sperme |
Total |
Total | ||
Unité temps (Cycles / Ponctions / Transferts) |
n |
44.661 |
42.843 |
45.578 |
43.675 |
1.471 |
1.858 |
8.117 |
8.805 |
99.827 |
97.181 |
Grossesses cliniques |
n |
4.279 |
4.378 |
9.614 |
9.306 |
380 |
432 |
984 |
954 |
15.257 |
15.070 |
%/unité temps |
9,58% |
10,22% |
21,09% |
21,31% |
25,83% |
23,25% |
12,12% |
10,83% |
15,28% |
15,51% | |
Réductions embryonnaires |
n |
54 |
331 |
114 |
147 |
7 |
4 |
11 |
5 |
186 |
487 |
%/ gross. |
1,26% |
7,56% |
1,19% |
1,58% |
1,84% |
0,93% |
1,12% |
0,52% |
1,22% |
3,23% | |
Accouchements |
n |
3.067 |
2.724 |
7.188 |
6.617 |
307 |
318 |
769 |
698 |
11.331 |
10.357 |
%/gross. |
71,68% |
62,22% |
74,77% |
71,10% |
80,79% |
73,61% |
78,15% |
73,17% |
74,27% |
68,73% | |
%/unité temps |
6,87% |
6,36% |
15,77% |
15,15% |
20,87% |
17,12% |
9,47% |
7,93% |
11,35% |
10,66% | |
Gémellaires |
n |
312 |
276 |
1.643 |
1.456 |
92 |
83 |
60 |
65 |
2.107 |
1.880 |
%/accouch. |
10,17% |
10,13% |
22,86% |
22,00% |
29,97% |
26,10% |
7,80% |
9,31% |
18,60% |
18,15% | |
Triples |
n |
12 |
14 |
107 |
103 |
11 |
6 |
4 |
1 |
134 |
124 |
%/accouch. |
0,39% |
0,51% |
1,49% |
1,56% |
3,58% |
1,89% |
0,52% |
0,14% |
1,18% |
1,20% | |
Quadruples et plus |
n |
1 |
2 |
1 |
0 |
0 |
0 |
0 |
0 |
2 |
2 |
%/accouch. |
0,03% |
0,07% |
0,01% |
0,00% |
0,00% |
0,00% |
0,00% |
0,00% |
0,02% |
0,02% | |
Nombre d'enfants vivants |
n |
3.259 |
2.806 |
8.971 |
8.243 |
409 |
385 |
814 |
698 |
13.453 |
12.132 |
Source : Unité 292 de l'INSERM de M. de Mouzon, sur la base des données fournies par la FIVNAT en 2000.
Cette évolution positive de la pratique en France a cependant été possible grâce à l'amélioration des techniques associée à une politique de transfert plus appropriée en fonction des patientes.
Le tableau ci-après indique l'issue des grossesses, une fois la FIV ou l'ICSI pratiquée.
ISSUE DES GROSSESSES - FIV et ICSI (1995 - 1998)
FIV CLASSIQUE |
ICSI | ||
Nombre de grossesses |
8.512 |
5.803 | |
Avortements spontanés (%) |
19,4 |
18,0 | |
Grossesses extra-utérines (%) |
3,4 |
1,6 | |
Interruptions médicales de grossesse (%) |
0,6 |
0,6 | |
Accouchements : |
% |
76,8 |
79,7 |
N |
6.540 |
4.626 | |
Simples (%) |
71,9 |
73,5 | |
Multiples: |
Jumeaux (%) |
26,1 |
24,2 |
Triples (%) |
1,5 |
1,6 | |
Quadruples et + |
n=1 |
n=1 | |
Réductions Embryonnaires |
2,8 |
2,3 | |
Source : FIVNAT 2000.
À propos du profil des couples qui recourent à une FIV, il faut retenir la hausse continue de l'âge moyen des femmes comme des hommes, respectivement de 34,3 et de 36,3 ans en 1999, ce qui correspond à une entrée plus tardive dans le processus d'AMP, parallèle avec le retard, pour l'ensemble de la population, de la naissance du premier enfant, constatée depuis plusieurs années par les démographes. Ce facteur temps est un inconvénient à prendre en compte dans la mesure où le taux de succès de l'AMP diminue avec l'âge des patientes.
Une étude socio-démographique de l'INSERM (40) présentée en septembre 1999 et menée sur un échantillon de 5 000 patients pendant cinq ans dans plus de quarante centres d'AMP, montre que l'accès aux soins liés aux techniques d'AMP est relati-vement bien assuré dans notre pays. Parmi les patients qui y recourent, on retrouve en effet toutes les classes socio-économiques de la société française dans une proportion qui permet de conclure que l'AMP n'est pas réservée à une classe privilégiée.
II.- LES CONDITIONS DE RECOURS À L'AMP ET L'OPPORTUNITÉ
DE LES RÉVISER
A.- LES CONDITIONS MÉDICALES SONT GLOBALEMENT SATISFAISANTES
Nul ne remet en cause fondamentalement les conditions médicales de recours aux techniques d'AMP posées par la loi n° 94-654 du 29 juillet 1994. Celui-ci, rappelons-le, est limité à l'existence d'un intérêt thérapeutique, soit pour remédier à l'infertilité d'un couple, dont le caractère pathologique a été médicalement diagnostiqué, soit pour éviter la transmission à l'enfant d'une maladie « d'une particulière gravité ».
Mme Nicole Questiaux, rapporteure de la Commission nationale consultative des droits de l'homme (CNCDH) auditionnée par la Mission le 24 mai 2000, déclarait ainsi l'attachement de la Commission à la finalité thérapeutique de l'AMP : « on ne changera pas un équilibre bâti sur une vision un peu thérapeutique de cette intervention, une intervention qui n'est pas de convenance, par conséquent où la liberté et l'autonomie de chaque membre du couple sont fortement encadrées. Je ne crois pas qu'un problème nouveau se pose, en termes de droits de l'homme, en ce qui concerne cet encadrement et cette philosophie de l'assistance à la procréation ».
Le législateur de 1994 a donc clairement affirmé le caractère strictement médical du recours à l'AMP. Sans remettre en cause ce choix qui paraît indiscutable, l'assouplissement de plusieurs règles est souhaitable.
Il s'agit en premier lieu de celle relative à l'infertilité diagnostiquée du couple. Il apparaît nécessaire de permettre expressément l'autoconservation des gamètes ou des tissus germinaux de personnes qui subissent des traitements ou des opérations chirurgicales stérilisants.
Ainsi que l'a affirmé devant la Mission, le Professeur Pierre Jouannet, Président de la Fédération française des Centres d'études et de conservation des _ufs et du sperme humains (CECOS), le 12 juillet 2000, cette activité connaît depuis quelques années un développement extrêmement important bien qu'elle n'ait pas été prévue par le législateur de 1994. La loi, en effet, limite le recueil et la conservation de gamètes au seul bénéfice des couples engagés dans une procédure d'AMP. Les CECOS ont cependant accepté d'étendre leur intervention au bénéfice d'hommes qui subissent un traitement ou une intervention chirurgicale pouvant les rendre stériles à l'exemple des chimiothérapies ou des radiothérapies dans le cadre des traitements anticancéreux. Il peut aussi s'agir d'hommes qui vont subir une stérilisation volontaire (vasectomie).
Pour l'année 1999, les CECOS ont ainsi accepté de recueillir et de conserver le sperme de 3 803 hommes, soit près de 99 000 paillettes qui se sont ajoutées à celles recueillies, dans les années précédentes, dans les mêmes circonstances. Or, ainsi que le soulignait le Professeur Pierre Jouannet, ces hommes ne vivent pas forcément en couple. Ils peuvent être célibataires, jeunes, voire adolescents. La réglementation en vigueur et le régime d'autorisation des centres, qui ne prévoit pas d'autorisation spécifique pour ce type d'activité, sont donc tout à fait inadaptés.
Il convient de modifier la loi afin d'autoriser le prélèvement et la conservation des gamètes pour l'usage personnel, futur ou hypothétique, d'individus qui doivent subir des traitements stérilisants ou risquant de l'être. Cette modification est d'autant plus urgente que certains CECOS ont commencé à congeler des fragments ovariens de jeunes femmes, parfois même de jeunes filles, atteintes de tumeur et suivant des traitements stérilisants, parfois à la demande de leur cancérologue ou hématologue, alors même que les techniques d'utilisation de ces tissus, qui contiennent des follicules primordiaux, précurseurs des ovules, ne sont pas encore au point. Le Président de la Fédération des CECOS indiquait ainsi que « des recherches se font chez l'animal, la souris, la brebis et on pense que les techniques seront au point dans les années qui viennent. Nos collègues cancérologues nous disent que même si nous ne sommes pas capables d'utiliser aujourd'hui ces fragments de tissus ovariens pour aider à concevoir un enfant, quand ces petites filles ou ces jeunes filles auront grandi, nous pourrons sans doute les aider à devenir mères ».
Une modification de la règle posée par l'article L. 2141-6 du code de la santé publique, selon laquelle l'AMP avec tiers donneur ne peut être pratiquée que comme « ultime indication », c'est-à-dire lorsque l'assistance médicale à la procréation à l'intérieur du couple (AMP endogène) ne peut aboutir, doit également être revue à la lumière de la réalité des situations vécues par certains couples. Si l'on peut comprendre, en effet, le souhait du législateur de 1994 de limiter autant qu'il est possible le recours au don de gamètes, on ne peut que regretter que l'application stricte de cette règle puisse conduire, dans certains cas, à des pratiques proches de « l'acharnement procréatif » alors que l'AMP intraconjugale n'a aucune chance d'aboutir ou très peu. Il faut aussi considérer le cas de couples qui ont déjà eu un enfant grâce à un don et qui doivent recourir à l'ICSI (41)qui permet de remédier désormais à certaines causes sévères d'infertilité masculine, s'ils souhaitent un autre enfant, ce qui les place dans une situation psychologique difficile devant la possibilité d'avoir des enfants de statuts biologiques différents. La loi devrait donc favoriser l'AMP intraconjugale et non plus l'imposer tout en permettant de rechercher un équilibre entre l'appréciation des indications d'AMP par les praticiens et la demande des couples. Un tel changement permettrait, en outre, de valoriser l'AMP hétérologue qui fait figure, compte tenu des termes actuels de la loi, de « pis-aller » à forte connotation d'échec.
S'agissant de la seconde condition médicale de recours à l'AMP posée par la loi, qui concerne la non-transmission d'une maladie grave à l'enfant, on peut se réjouir qu'à la suite des protocoles de recherche appliqués dans certains CECOS, l'arrêté du 10 mai 2001 modifiant l'arrêté du 12 janvier 1999 relatif aux règles de bonne pratique cliniques et biologiques en AMP, ait permis la prise en charge des patients à risque viral, hommes ou femmes atteints notamment du virus du sida (HIV), de l'hépatite B ou C (VHB et VHC).
Aux exigences particulières posées quant à la composition des équipes prenant en charge les couples concernés, la sélection, les obligations et le suivi médical de ces derniers, s'ajoutent les techniques spécifiques de préparation et de lavage du sperme ou du liquide ovocytaire, d'évaluation de la charge virale des gamètes et éventuellement de leur traitement. On peut donc se réjouir de ces nouvelles règles permettant de garantir l'accès de ces couples à l'AMP, alors que près d'un tiers des praticiens interrogés en 1997 et 1998 par le Centre de recherche juridique de l'ouest (CRJO) déclaraient refuser de prendre en charge les couples sérodifférents.
B.- LES CONDITIONS SOCIALES FONT L'OBJET DE DEMANDES D'ÉLARGISSEMENT QUI SEMBLENT POUR CERTAINES SOUHAITABLES
Les conditions sociales de recours à l'AMP font en revanche l'objet de demandes de révision beaucoup plus nombreuses, et pour certaines complexes.
1.- Les conditions tenant à la situation des futurs parents et des donneurs
de gamètes doivent être adaptées
L'article L. 2141-2 du code de la santé publique exige de l'homme et de la femme formant le couple qui recourt à l'AMP qu'ils soient « mariés ou en mesure d'apporter la preuve d'une vie commune d'au moins deux ans ». Cette différence de traitement entre les personnes mariées et celles vivant en concubinage est difficile à justifier. Si le mariage semblait, par le passé, apporter un gage de stabilité plus grande des couples, force est de constater que l'évolution des m_urs et la fragilité croissante des unions ne permettent plus de le privilégier au dépens des autres formes de vie commune. De plus, ainsi que le notait le Conseil d'État dans son rapport de novembre 1999, Les lois de bioéthique : cinq ans après, la règle actuelle pose problème pour le corps médical « qui éprouve parfois un embarras à devoir recueillir des informations de nature administrative telles que la preuve de la communauté de vie d'un couple ».
Il paraît donc souhaitable et plus juste d'unifier le régime, soit en exigeant des couples mariés la même exigence de vie commune, soit en supprimant cette dernière pour les concubins. Les circonstances de plus en plus tardives dans lesquelles la stérilité d'un couple est diagnostiquée - les femmes ayant souvent plus de 38 ans - la longueur des processus d'AMP et les nouvelles données sociologiques relatives aux unions semblent militer en faveur de la suppression de toute exigence de vie commune.
S'agissant des donneurs de gamètes, pour lesquels l'article L. 1244-2 du code de la santé publique impose qu'ils fassent « partie d'un couple ayant procréé », il semble opportun de supprimer la condition d'appartenance du donneur à un couple, ce qui permettrait de ne plus exclure les personnes divorcées, veuves ou ayant vécu maritalement. Ce changement offrirait l'avantage de remédier partiellement à la pénurie de dons de gamètes tout en s'accordant davantage à la réalité sociale. En revanche, la condition liée à la procréation antérieure des donneurs, formule maladroite comme le soulignait devant la Mission le Professeur Pierre Jouannet qui lui préférait celle de parents, doit être maintenue pour préserver la finalité même du don. Le Président de la fédération des CECOS déclarait ainsi : « Ce qui nous semble important, c'est la qualité de parent, c'est d'être soi-même père ou mère. Donc, de bien connaître la paternité ou la maternité. Cela ne conduira pas cet homme ou cette femme à fantasmer sur les enfants qu'ils ont aidé d'autres couples à procréer ».
2.- Le transfert post mortem d'embryon doit être autorisé sous réserve
du consentement du père de son vivant
L'arrêt de la Cour d'appel de Toulouse en date du 18 avril 1994 a fait grand bruit, tant parmi les juristes qu'au sein de l'opinion publique. Il faut avouer que le cas de la plaignante était particulièrement douloureux : après six tentatives échouées de FIV, une septième implantation est entreprise le 12 octobre 1990. À cette occasion, les époux signent un document dans lequel ils déclaraient, conformément à une pratique largement suivie dans les hôpitaux que « le transfert (d'embryons) ne pourra être réalisé qu'en présence de chacun (des parents) » et qu'en cas de dissolution du couple, les embryons seront détruits. Hélas, cette septième tentative conduit à une fausse couche le 10 décembre 1990 ; le mari qui se rend au chevet de son épouse est alors victime d'un accident de la route mortel. Au cours de l'année 1991, la veuve demande à ce que lui soient transférés les deux embryons restants de leur projet parental. Devant le refus que lui oppose l'hôpital, elle intente, au cours de l'année 1992, une procédure judiciaire qui confirme ce refus, le tribunal de première instance considérant, dans son jugement du 11 mai 1993, que le transfert post mortem « constituerait une entreprise d'immortalité contraire à l'ordre public et aux règles fondamentales de la transmission de la vie ». Il s'appuyait par ailleurs sur la volonté du mari, exprimée par écrit, de procréer de son vivant. La cours d'appel de Toulouse a confirmé, dans son arrêt du 18 avril 1994, cette décision et a, au surplus, ordonné la destruction des embryons.
Il convient de rappeler la situation qui prévalait avant les lois de juillet 1994. Les tribunaux se trouvaient alors divisés tant sur la question de l'utilisation du sperme d'un mari décédé que sur celle du transfert d'embryon. Le législateur ayant fait le choix de refuser ces deux pratiques, selon le principe défendu par le Conseil d'État dans son rapport de 1988 (42) : « deux parents, pas un de plus, pas un de moins », l'article L. 2141-2 du code de la santé publique impose donc que « l'homme et la femme formant le couple doivent être vivants ». Selon cette approche, l'AMP ne doit pas conduire à la constitution d'une famille monoparentale au motif qu'il n'est pas souhaitable de faire délibérément venir au monde un enfant sans père, enfant qui serait, en quelque sorte, le fruit du deuil de la mère.
Au Royaume-Uni, ainsi que l'indiquait le 5 juillet 2000, devant la Mission, Mme Françoise Shenfield, membre de l'Agence britannique de la fécondation humaine et de l'embryologie (Human Fertilization and Embryology Authority, HFEA), il est possible d'exprimer par écrit son consentement pour que ses gamètes ou ses embryons soient utilisés, après le décès de l'un des membres du couple, par son conjoint ou compagnon en vue de la poursuite du processus d'AMP, ou soient donnés à un autre couple, ou soient utilisés à des fins de recherche. « Le couple doit alors, avec l'aide d'un conseil psychologique - c'est un point important de la loi anglaise - remplir le formulaire avant que la clinique ait la permission de congeler le sperme ou les embryons ». En revanche, la paternité de l'enfant né du sperme de l'homme décédé ou de l'implantation de l'embryon qui existait avant son décès, n'est pas reconnue par la loi britannique.
Différents arguments militent aujourd'hui en faveur de la révision du choix de 1994. On peut d'abord considérer comme essentielle et prééminente l'existence du projet parental qui a conduit à la conception in vitro d'embryons, « personnes humaines potentielles », dès lors que le père a consenti de son vivant à la poursuite de ce projet dans l'hypothèse de son décès. C'est aussi reconnaître à l'épouse sa qualité de mère et le droit à élever son enfant, à l'instar d'une femme dont le mari ou le compagnon viendrait à décéder au cours de sa grossesse. C'est enfin sortir de la situation quasi absurde où la veuve doit aujourd'hui choisir entre la destruction ou l'accueil par un autre couple de ses embryons surnuméraires, ce qui fait de ces derniers des orphelins de père et de mère biologiques, alors que cette dernière les réclame.
Devant la Mission le 21 juin 2000, Mme Chantal Ramogida, fondatrice de l'association « Pauline et Adrien » qui apporte soutien et information aux couples qui s'engagent dans l'AMP, déclarait ainsi que « s'agissant de l'implantation post mortem, ce n'est ni du sperme ni un ovocyte, mais un embryon. Il y a eu un projet parental qui malheureusement n'a pu être mené à terme en raison du décès du mari. Certains soulèvent le fait que cet enfant n'aura pas de père. Il connaîtra son père car il pourra le voir en photo. Si le mari décède quand sa femme est enceinte, où est la différence ? Quid des femmes mariées et divorcées ? Même si le père est physiquement absent, son image est là ».
On peut aussi considérer avec Mme Yvette Roudy (43), membre de la Mission, que l'embryon étant une partie du corps de la femme, c'est à elle de décider en dernier ressort, dès lors que le projet parental est avancé et que le père était d'accord pour sa poursuite.
Le Professeur Claude Sureau, président de l'Académie nationale de médecine, indiquait devant la Mission le 12 juillet 2000 qu'un grand nombre de praticiens ont été profondément choqués de l'application stricte de la loi qui a interdit à la veuve, dans le cas précité de Toulouse, « l'implantation de ses embryons congelés, en application, et cela est curieux, du rapport du Conseil d'État de 1988 et de l'affirmation péremptoire et difficile à soutenir : deux parents, pas un de plus, pas un de moins, ce qui ne signifie pas grand chose au siècle où nous vivons ».
En revanche, force est de reconnaître que le désaccord est toujours profond à propos du travail de deuil. Mme Martine Aurillac, membre de la Mission, considère (44)que l'autorisation du transfert post mortem permettrait à la veuve de « se remettre autrement bien » de son deuil. M. Jean-Loup Clément, psychologue d'un CECOS, auditionné par la Mission le 13 septembre 2000, est d'un avis tout à fait contraire. Il considère en effet qu'au bout de six mois, délai envisagé par l'avant-projet de loi de révision des lois bioéthiques au cours duquel serait autorisé un transfert posthume, la veuve n'a fait « aucun travail psychologique (...) au sens psychanalytique, réaliser un travail de deuil, c'est manifester une capacité d'élaboration sur un événement traumatique que l'on a vécu afin de faire en sorte qu'il soit le moins douloureux possible. Si une femme parvient à accepter la mort de son mari, elle ne demande plus à vivre dans le souvenir. Il serait sans doute très douloureux pour un enfant de naître plusieurs années après la mort d'un homme qui aurait pu être son père ».
Pour lui, le décès du père « suspend tout, arrête le projet d'enfant ». Il souligne par ailleurs que, du point de vue psychologique, « faire peser sur un enfant le souvenir d'un homme (...) ne peut que nuire à son développement ».
Pour sa part, M. Jean-François Mattei, membre de la Mission, se faisant à son tour l'écho de psychologues et de psychiatres sur les motivations qui animent les veuves dans une telle situation, considère que « dès lors que l'on entre dans une démarche post mortem, on n'est plus du tout dans la correction de processus naturels pathologiques, on est dans la manipulation », mais reconnaît aussi qu'il n'y a pas de solution parfaite à ce problème.
On doit constater que la loi actuelle, dans sa rigueur, est vivement contestée et qu'il y a lieu de s'interroger sur sa révision. Votre Rapporteur est donc favorable, avec la majorité des membres de la Mission, à ce que soit autorisé le transfert post mortem dès lors que le père y a consenti de son vivant. Il convient toutefois d'encadrer cette autorisation pour ne pas créer de situations juridiques, voire humaines, inextricables. D'un côté, le règlement des successions exige que cette possibilité de transfert posthume soit limitée dans le temps. De l'autre, il ne faudrait pas que l'autorisation de transfert post mortem soit irréalisable dans la pratique et ne crée que de faux espoirs. De l'avis de votre Rapporteur, il serait donc opportun, en premier lieu, de préserver un délai de quelques mois après le décès du père afin que la mère ne soit pas contrainte de décider alors qu'elle se trouve dans une situation de fragilité extrême, et, par ailleurs, d'autoriser une interprétation souple du second délai qui serait ouvert pour procéder à l'implantation des embryons, si des raisons médicales le justifient.
Votre Rapporteur est ainsi favorable à l'institution de deux délais : le premier, de six mois au moins après le décès du père, au cours duquel la femme aurait le choix de décider ou non de l'implantation des embryons et le second, de dix-huit mois, afin de permettre plusieurs tentatives de transfert, ce délai pouvant être exceptionnellement prolongé si des raisons médicales le justifient. Avant l'expiration de ce délai, la liquidation de la succession du défunt devrait être suspendue pour préserver les intérêts de l'enfant ou des enfants qui pourraient naître. Il va sans dire que la filiation du père devra être automatiquement reconnue à l'instar de ce que prévoit déjà le code civil dans son article 311-20 dans le cas d'une AMP réalisée avec le consentement du père, « à moins qu'il ne soit soutenu que l'enfant n'est pas issu de la procréation médicalement assistée ou que le consentement a été privé d'effet ».
3.- Les nouvelles demandes exprimées par les personnes homosexuelles
ou transsexuelles semblent difficiles à satisfaire
Ainsi que cela a été rappelé à plusieurs reprises, la loi a exclusivement réservé le recours à l'AMP aux couples hétérosexuels, mariés ou en concubinage, excluant de ce fait les demandes pouvant émaner de célibataires ou de couples homosexuels, suivant l'adage du Conseil d'État « deux parents, pas un de plus, pas un de moins ».
Cependant les débats qui se sont déroulés au Parlement à l'occasion de l'adoption de la loi n° 99-944 du 15 novembre 1999 relative au pacte civil de solidarité (PACS), où la question de l'adoption par les couples homosexuels a été abordée, ont remis en exergue certaines demandes d'élargissement des conditions de recours aux techniques d'AMP. L'association des parents et futurs parents gays et lesbiens (APGL), dont les coprésidents ont été reçus par le Président de la Mission et votre Rapporteur, réclame ainsi la suppression des discriminations qui frappent des « parents potentiels en raison de leur orientation sexuelle. (...) Il y a des techniques de reproduction qui ne sont pas autorisées aux couples de même sexe pour des raisons basées sur des préjugés qui laissent à penser que les gays et les lesbiennes ne seraient pas des parents « convenables » ». L'association revendique ainsi que l'AMP soit ouverte à toute personne en âge de procréer qui justifie d'un projet parental cohérent et s'engage à devenir parent « qu'il s'agisse de personnes seules, de couples de même sexe ou de sexe différent ou encore de paires constituées d'un père gay et d'une mère lesbienne ».
En conséquence, l'APGL demande à ce que soit supprimé l'article 16-7 du code civil qui interdit « toute convention portant sur la procréation ou la gestation pour le compte d'autrui », disposition issue de la loi de 1994. Le législateur avait en effet souhaité donner force de loi à la jurisprudence du Conseil d'État et de la Cour de Cassation à l'encontre de la pratique des « mères porteuses » qui s'était développée au cours des années 1980 et qui avait suscité une vive émotion parmi les praticiens, les juristes et l'opinion publique. À l'appui de cette revendication, l'association souligne que le recours aux mères pour autrui « est autorisé dans plusieurs pays anglo-saxons où l'éthique n'est pas fondée sur des principes préexistants mais sur une morale de la moindre souffrance. Ce qui semble être l'argument le plus souvent invoqué pour justifier l'interdiction de cette pratique est celui de l'instrumentalisation du corps de la femme, avec parfois une forte connotation morale. C'est mettre de côté le fait que certaines femmes peuvent avoir envie de porter un enfant pour autrui, pour des raisons qui leur sont propres, et dans lesquelles les aspects financiers sont secondaires. Leur histoire, leur passé, leur personnalité, d'une part, et leur relation avec des personnes, connues ou inconnues d'elles, désireuses d'être parents, d'autre part, peuvent les pousser à cet acte. Cette pratique, au lieu d'être interdite totalement, ce qui favorise de fait de complexes transactions à l'étranger, devrait être encadrée, selon une procédure se rapprochant de celle de l'adoption. Les demandes devraient ainsi être examinées au cas par cas après accompagnement des demandeurs et de la future mère ; la question d'un délai de rétractation de la mère pourrait être posée ; l'enfant devrait pouvoir accéder à ses origines ».
Votre Rapporteur considère que la légalisation de la pratique des mères de substitution, même encadrée, serait dangereuse ; facilement détournée, elle pourrait aboutir à la violation des principes fondamentaux de non-commercialisation et d'indisponibilité du corps humain. Par ailleurs, il semble que notre société ne soit pas prête à accepter l'abandon de la règle « biologique » choisie par le législateur de 1994 selon laquelle le couple qui s'engage dans une AMP est formé d'un homme et d'une femme.
L'attention de la Mission a également été attirée sur une seconde demande d'élargissement de l'accès à l'AMP. Il s'agit de couples composés sur le plan chromosomique de deux femmes mais, selon l'état civil, d'un homme et d'une femme, l'une d'entre elles ayant changé d'état civil, ainsi que le permet la jurisprudence en vigueur, à la suite de son opération pour changer de sexe.
Les avis sont, à ce sujet, partagés. Mme Brigitte Feuillet-Le Mintier, directeur du Centre de recherches juridiques de l'Ouest (CRJO), auditionnée par la Mission le 5 juillet 2000, considère que l'état de la personne est défini par son état civil. En conséquence, la demande exprimée par ces couples remplit la condition formelle de couple formé d'un homme et d'une femme. Elle déclarait ainsi : « au regard du texte actuel, lorsqu'un transsexuel a fait rectifier son état civil, je ne vois pas comment un médecin pourrait s'y opposer. En pratique, les médecins apprécient au cas par cas. Certes, le nombre de couples concernés est faible, mais les incidences d'un tel choix sur le droit de la famille sont considérables. À mon avis, ce n'est pas au médecin de décider au cas par cas s'il doit ou non apporter son assistance, c'est au législateur de répondre à la question de savoir si un couple de transsexuels peut accéder à l'assistance médicale à la procréation ».
À l'inverse, M. Jean-Loup Clément, au cours de son audition précitée devant la Mission, manifestait son total désaccord en tant que psychologue d'un CECOS. C'est là, selon lui, un sujet « crucial (...) pour les psychologues et les psychiatres qui se heurtent aux médecins, à savoir les demandes d'insémination pour les couples dont l'homme est en fait une femme opérée, c'est-à-dire un transsexuel. Nous avons manifesté notre désaccord. Légalement, c'est un homme et ils sont mariés. Au regard de la loi, ils forment un couple, mais je pense que l'on ne change jamais de sexe. Le sexe anatomique et génétique est immuable ».
III.- L'APPLICATION PRATIQUE DES TECHNIQUES D'AMP
Les naissances permises par l'AMP sont aussi le fruit de recherches et de techniques médicales très perfectionnées, lesquelles exigent des équipes scientifiques et médicales une grande maîtrise et un dévouement exceptionnel ; mais, comme toute technique médicale, elles ne sont pas exemptes de risques : risque en particulier pour les donneuses d'ovocytes, risque de prématurité élevé pour les enfants, comme cela a été indiqué dans le premier chapitre de la présente partie et surtout risque d'échec car l'AMP n'est hélas pas la panacée de tous les cas d'infertilité. S'y ajoutent des risques propres à certaines techniques qui ont pu susciter de sérieuses inquiétudes. Le problème de l'évaluation et de l'encadrement des techniques se pose ainsi avec acuité tandis que restent jusqu'à ce jour incertaines certaines questions éthiques relatives à la conservation des embryons ou au don des gamètes.
A.- L'ÉVALUATION DES NOUVELLES TECHNIQUES D'AMP
FAIT GRAVEMENT DÉFAUT
Le législateur de 1994 pensait-il qu'aucune nouvelle technique d'AMP ne pourrait se développer avant la révision des lois, qui aurait dû intervenir en 1999, ou a-t-il omis de mettre en place un système d'évaluation et d'autorisation préalable des nouvelles techniques ? En tout état de cause, la science a démontré, une fois de plus, qu'elle était aussi rapide qu'inattendue dans ses progrès et ses applications, exigeant de la part du législateur une attention et une capacité de réaction permanentes.
Les circonstances de la découverte de cette nouvelle technique de fécondation, par injection intracytoplasmique d'un seul spermatozoïde en perforant la membrane pellucide de l'ovocyte, sont désormais connues. C'est bien une anomalie de manipulation technique dans le laboratoire d'une université néerlandophone de Bruxelles (45)qui est à l'origine de cette découverte en 1991. Ainsi que l'indiquait M. Yvon Englert, directeur du laboratoire « biologie et psychologie de la fertilité humaine » à l'Université libre de Bruxelles, dans son audition devant la Mission le 20 septembre 2000, « à l'époque tout le monde pensait la chose irréalisable. Des expérimentations avaient été faites sur des modèles animaux, notamment par Lanzendorf en Allemagne à la fin des années quatre-vingt, elles s'étaient toutes soldées par des échecs ».
Parce qu'elle permet de traiter des formes de stérilité masculine sévères, liées au nombre, à la mobilité ou à la morphologie des spermatozoïdes, et qu'elle autorise ainsi des couples à procréer sans recourir à un don de sperme avec des résultats à peu près comparables à la FIV « classique », cette technique s'est rapidement développée en Europe et dans le reste du monde en dépit de l'absence d'expérimentation préalable et d'évaluation. Les premières naissances en France utilisant l'ICSI ont eu lieu à l'Hôpital Américain de Neuilly-sur-Seine en 1995. Le pourcentage de FIV réalisées avec cette technique a ensuite rapidement progressé passant de 35 % en 1996 à 47 % en 1999, avec des taux de succès élevés, comme l'illustre le tableau ci-après.
ÉVOLUTION DES TAUX DE SUCCÈS DE L'ICSI
(en %)
Années | ||||||
1995 |
1996 |
1997 |
1998 |
1999 |
98/99 | |
Ponctions réussies |
99,4 |
99,5 |
99,6 |
99,2 |
99,5 |
NS |
Transferts / Ponction |
88,9 |
91,7 |
92,4 |
91,7 |
91,9 |
NS |
Taux de grossesses |
||||||
par ponction |
21,4 |
24,2 |
24,2 |
24,1 |
24,8 |
NS |
par transfert |
24,0 |
26,4 |
26,5 |
26,2 |
27,0 |
NS |
Naissance d'au moins un enfant normal vivant /Ponction |
17,3 |
19,6 |
19,5 |
19,7 |
|
|
Source : Unité 292 de l'INSERM de M. de Mouzon, sur la base des données fournies par la FIVNAT en 2000. | ||||||
L'émotion au sein de la communauté scientifique de même qu'au sein de l'opinion publique a été forte devant la méconnaissance manifeste des principes posés par le Code de Nuremberg de 1947 sur l'expérimentation biomédicale. Ainsi que le soulignait le Docteur Axel Kahn devant la Mission, au cours de son audition le 7 juin 2001, « personne ne pouvait prédire à quoi cela allait aboutir si l'on forçait le pouvoir fécondant de ces spermatozoïdes, c'est-à-dire, si l'on rendait fécondants ces spermatozoïdes naturellement non fécondants. Quel type de bébé allait-il donner ? Vraiment personne ne pouvait répondre à cette question. Si bien que, d'un point de vue théorique, il y avait au moins toutes les raisons de se poser des questions importantes ».
Le président du Comité consultatif national d'éthique pour les sciences de la vie et de la santé (CCNE.), M. Didier Sicard, notait pour sa part, devant la Mission le 31 mai 2000, le contraste existant « entre l'efficacité de cette technique, qui est mise en pratique, et l'inconnue sur ses conséquences, c'est-à-dire que l'on est face à une situation qui s'est développée de façon complètement laxiste. (...) On est dans une situation typique d'incertitude absolue ».
Mme Nicole Questiaux, rapporteure de la Commission nationale consultative des droits de l'homme (CNCDH) sur les lois bioéthiques considérait, devant la Mission le 24 mai 2000, qu'il y a atteinte au principe d'égalité avec le développement sans autorisation de l'ICSI. Votre Rapporteur considère lui aussi que cette situation place les couples et leurs enfants nés après AMP dans une situation inégale s'agissant, d'une part des risques sanitaires potentiels liés à l'ICSI et, d'autre part, des taux de succès des FIV pratiquées.
Le Professeur Jacques Testart, devant la Mission le 31 mai 2000, soulignait l'absurdité de la situation qui aurait ensuite conduit à inverser la chronologie des expérimentations sur l'homme et sur l'animal : « Je reconnais que c'est désastreux, que nos collègues belges ont d'abord expérimenté (l'ICSI) dans l'espèce humaine (...) j'ai été très véhément sur le sujet. Mais une fois qu'étaient nés quelques centaines d'enfants belges, on nous a demandé de faire l'ICSI chez des souris. C'était aberrant ».
Cette situation place le législateur dans une position délicate qui impliquerait, selon certains juristes, sa responsabilité. Mme Brigitte Feuillet-Le Mintier, dans son audition précitée, s'adressait ainsi aux députés membres de la Mission : « C'est un choix démocratique. Inutile de vous dire que si vous interdisiez l'ICSI, cela provoquerait de nombreuses protestations. Mais imaginons le pire. Puisque des études semblent montrer que l'ICSI favorise la transmission d'anomalies à l'enfant, si l'on s'aperçoit, dans un certain temps, que tous les enfants nés de cette technique sont porteurs d'anomalies, vous aurez une part de responsabilité. On ne pourra pas dire que l'on n'a pas écrit qu'il y avait des risques. Il convient d'agir en ce domaine. Il me paraîtrait extrêmement grave de maintenir la situation actuelle. Dans le cabinet du médecin, les uns et les autres ne peuvent pas être objectifs ».
Sur les risques de l'ICSI, on dispose aujourd'hui de certaines données fiables mais incomplètes. En effet, comme l'indiquait devant la Mission le Professeur Jacques Testart, il faudra sans doute « une ou deux générations, c'est-à-dire encore un siècle (pour) savoir si l'on n'a pas commis une erreur. Si on ne le fait pas, on ne le saura jamais. Personne n'a proposé un protocole qui permettrait de savoir si l'ICSI est dangereuse autrement qu'en faisant l'ICSI ». Ainsi que le précisait le Professeur Claude Sureau devant la Mission, en faisant état d'études belges conduites par le Docteur Van Steirteghem, l'un des « pères » de l'ICSI, on est désormais certain que l'ICSI a pour conséquence de transmettre à l'enfant et à sa descendance l'anomalie génératrice de l'insuffisance spermatique : « Par l'ICSI, la stérilité peut (donc) devenir héréditaire ». L'ICSI conduit également à la « transmission d'anomalies génétiques telles que la mucoviscidose, responsable de la stérilité mais aussi possiblement d'autres pathologies ».
S'y ajoute le constat d'une plus grande fréquence, chez les enfants issus de l'ICSI, d'anomalies chromosomiques en ce qui concerne les chromosomes sexuels X et Y. Le Professeur Claude Sureau notait ainsi : « Alors que le taux général de malformations est de 2 à 2,5 %, on dénombre chez les enfants ainsi conçus 4 à 5 % d'anomalies des gonosomes, c'est-à-dire les chromosomes sexuels. Cela s'explique probablement par des raisons techniques, mais on n'en est pas sûr ».
Enfin, comme l'indiquait M. Didier Sicard dans son audition précitée, il semblerait que l'utilisation de l'ICSI provoque des interruptions de grossesse plus précoces.
Pourtant, les avis selon lesquels les avantages de l'ICSI sont supérieurs aux risques évoqués, sont assez nombreux. Le Professeur Claude Sureau considérait devant la Mission que « le bilan est globalement positif. Auparavant, un couple dont le mari n'avait pratiquement pas de spermatozoïdes devait obligatoirement recourir à l'insémination artificielle avec donneur. On lui offre maintenant la possibilité d'avoir un enfant par ses propres moyens ». C'est aussi la position exprimée par le Docteur Marie-Odile Alnot, médecin en biologie de la reproduction à l'hôpital Necker, le 6 septembre 2000 devant la Mission : « Il s'agit, selon moi, d'une bonne technique car elle évite à de nombreux couples d'avoir recours à un don de gamètes ».
Dans ce contexte, que faire ? Interdire l'ICSI et s'exposer à une vive contestation des patients pour lesquels cette technique est la seule solution leur permettant de procréer à l'intérieur de leur couple, sans devoir recourir au don ? Tolérer cette technique en considérant qu'il est trop tard ? Considérer, avec le Professeur Jacques Testart, que la polémique sur les risques de l'ICSI a été excessive parce qu'elle aurait été organisée « par les généticiens et les banques de sperme qui ont perdu les deux tiers de leur activité grâce ou à cause de l'ICSI, les banques de sperme expliquant aux gens que l'ICSI était très dangereuse et qu'il était préférable de prendre un donneur » ? Se poser avec Mme Brigitte Feuillet-le Mintier la question de l'intérêt de l'enfant, à savoir : « Peut-on accepter qu'un enfant né à la suite d'une assistance médicale à la procréation soit infertile ? ».
Votre Rapporteur considère, avec un grand nombre de personnalités auditionnées par la Mission, qu'une meilleure information doit être apportée aux parents auxquels l'utilisation de l'ICSI est proposée, tandis qu'un suivi médical des enfants nés grâce à cette technique doit être mis en place, en dépit des difficultés qu'il présente. Comme le préconisait le Docteur Marie-Odile Alnot, il serait souhaitable de « laisser aux couples, après une bonne information, le choix de la technique qu'ils souhaitent employer ». L'obligation d'informer, qui existe déjà dans tout processus d'AMP, doit donc être renforcée dans le cas de l'ICSI et strictement appliquée.
Parallèlement, le suivi épidémiologique des enfants nés grâce à l'ICSI doit être organisé, quelles que soient les difficultés juridiques et éthiques qu'il soulèverait et que soulignait devant la Mission le président du CCNE, M. Didier Sicard, tout en reconnaissant par ailleurs sa nécessité : « Cela pose (...) un problème éthique : les enfants nés par ICSI doivent-ils subir des contrôles par des équipes de psychologues ? Sont-ils dépendants ? La femme doit-elle signer un engagement de se conformer à une demande sociale ? Généralement, lorsqu'elle a eu son enfant, la mère considère que cet enfant échappe au contrôle de la société ». Mme Nicole Questiaux indiquait à son tour devant la Mission que « la Commission nationale consultative des droits de l'homme est convaincue de cette nécessité, mais un peu angoissée à l'idée des problèmes d'intimité et de liberté qu'implique le suivi de personnes nées du recours à l'ICSI ou à une autre technique d'AMP ».
Malgré ces réserves, il semble indispensable d'assurer ce suivi, ne serait-ce même que par devoir vis-à-vis des enfants nés après recours à l'ICSI, de leurs parents et des générations futures. Ce suivi ne pose d'ailleurs aucun problème dans d'autres pays, à l'instar du Royaume-Uni où tous les enfants nés grâce à l'AMP font l'objet d'un suivi sans que cela ait jamais été contesté ou critiqué.
Les controverses et inquiétudes entourant l'utilisation de l'ICSI auront au moins eu le mérite de souligner la nécessité d'instituer un dispositif d'évaluation et d'autorisation de toute nouvelle technique d'AMP entendue au sens large. Il convient en effet de se préoccuper également des techniques de prélèvement de gamètes, de précurseurs de gamètes et de tissus germinaux dont certaines sont déjà appliquées en France sans expérimentation préalable.
C'est le danger que soulignait le Docteur Axel Kahn devant la Mission dans les propos suivants : « Ce qui est relativement ennuyeux, c'est que les situations de stérilité masculine auxquelles les biologistes essaient de faire face sont de plus en plus difficiles. On a commencé par prendre un spermatozoïde sur 100 000, alors que normalement un homme en a 200 millions par millilitre. Ensuite, on en a pris un sur mille. Ensuite, on a piqué directement l'épididyme pour pouvoir prendre un spermatozoïde parce qu'il n'y en avait pas ailleurs. Plus tard, on a piqué directement le testicule. En outre, dans le testicule, il y avait des spermatides immatures, on les a fait mûrir. Enfin, récemment, on a même pris des cellules qui n'avaient pas subi la méiose et on leur a fait subir une méiose. Tout cela naturellement, toujours sans expérimentation préalable, alors qu'à chaque fois, les dangers sont importants ».
Des recherches repoussant toujours plus loin les certitudes acquises, sont d'ores et déjà en cours à l'étranger, comme l'indiquait le Professeur Claude Sureau dans son audition devant la Mission : « Des tentatives, dont deux en France, ont consisté en l'utilisation des précurseurs des spermatozoïdes, les spermatides, voire de précurseurs plus en amont, les spermatocytes. À ce niveau, l'incertitude est beaucoup plus grande. Un groupe franco-hispano-turc fait des recherches dans ce domaine, non pas en France où c'est interdit, mais en Espagne ».
Selon les informations recueillies par votre Rapporteur, l'équipe de la biologiste Carmen Mendoza en Espagne a, de fait, réussi à cultiver in vitro du tissu testiculaire pour en obtenir des spermatozoïdes. Quelques enfants seraient déjà nés de cette méthode ; la « pionnière » à Alicante, prénommée Naria, est aujourd'hui âgée de plus d'un an.
S'agissant des ovocytes, les expériences en cours sur l'animal montrent qu'il est possible de prélever par c_lioscopie, plusieurs fois au cours d'un même cycle, des ovocytes dans des follicules et d'obtenir leur maturation in vitro. Il a aussi été possible de récupérer des ovocytes f_taux provenant d'avortement mais on ignore encore si ces ovocytes ont la potentialité d'assurer leur développement interne. Les chercheurs ont aussi réussi à prélever des ovocytes chez des souriceaux impubères de 12 jours, à les faire mûrir et à les féconder in vitro avec cependant un pourcentage de naissance très faible. Le Professeur Jacques Testart, dans son audition devant la Mission, nourrissait à ce sujet certaines craintes : « Je pense que d'ici dix à vingt ans - peu importe le terme, mais c'est évident que l'on va y arriver, disons trente ans pour être sûr - on aura la possibilité de produire pour chaque couple stérile ou non, qui voudra un enfant, 100 ou 200 embryons simultanément, sans que la femme n'ait rien à souffrir d'autre qu'un premier prélèvement de tissu ovarien, qui se ferait autant que possible chez les petites filles, par exemple en même temps qu'une appendicectomie. Un tout petit prélèvement de cortex ovarien lorsqu'il est encore riche de potentiel de procréation, c'est-à-dire d'ovocytes, alors qu'il dégénère très vite puisque ces cellules sont celles qui subissent de plus de dégénérescence. On emploie des mots savants pour le dire comme « apoptose ». Cela signifie qu'il y a au début cinq millions de ces cellules chez un f_tus féminin et, que, dans la vie, cela fera 300 ovules ».
S'agissant des techniques d'AMP, le Professeur Peter Braude, membre de l'HFEA britannique, avouait : « Nous ne connaissons pas les effets de nombreuses techniques nouvelles. Aujourd'hui, il n'y a pas d'alternative. Il y en aura lorsque la science progressera ».
Que penser ainsi de la technique consistant à vider de son noyau l'ovule d'une donneuse pour le remplacer par le noyau de l'ovule d'une femme stérile permettant d'obtenir un ovule « régénéré » qui peut être fécondé in vitro avant d'être implanté dans l'utérus de la mère stérile ? Aux États-Unis, cette technique aurait déjà donné naissance à une trentaine d'enfants sans qu'aucune expérimentation préalable sur l'animal n'ait été réalisée.
En Espagne, l'équipe du Docteur Carmen Mendoza a récemment annoncé avoir réussi l'expérimentation de cette technique, soulevant une vive polémique en l'absence d'autorisation à cet effet. Le ministère espagnol de la santé l'a vivement condamnée en rappelant que la fécondation d'ovules humains à des fins autres que la procréation est formellement interdite par la loi.
Que penser encore de la technique inverse dite du transfert d'ooplasme qui consiste à transférer l'ooplasme (le cytoplasme ovocytaire) d'une donneuse fertile sur l'ovocyte d'une receveuse ? Ces deux techniques permettent certes, soit de résoudre le problème de stérilité de femmes dont l'ooplasme est défectueux (46), soit d'éviter que soient transmises à l'enfant des maladies graves liées à l'ADN mitochondrial, dont certaines sont létales et d'autres entraînent des troubles neuro-musculaires très sévères. Mais on ignore quelles sont les conséquences de la coexistence, au sein d'un même embryon, de l'ADN provenant de deux femmes : l'ADN nucléaire de la mère et l'ADN mitochondrial des deux femmes. D'aucuns vont jusqu'à y voir un risque de dérive vers le clonage. La HFEA britannique a d'ailleurs fait récemment savoir que cette technique ne serait pas autorisée au Royaume-Uni devant les incertitudes qu'elle comporte avec, notamment, le risque d'altération de la lignée germinale.
Ces perspectives de découvertes, et celles qui sont aujourd'hui encore inimaginables, exigent que soit mis en place pour l'avenir un système d'évaluation et d'autorisation des nouvelles techniques d'AMP. Comme le proposait l'Académie nationale de médecine dans sa recommandation du 13 février 1996, il pourrait être opportun de différencier les techniques d'AMP selon qu'elles sont éprouvées, en cours d'évaluation ou encore à l'état de recherches afin d'informer et de protéger au mieux le patient et le futur enfant et d'éviter, comme dans le cas de l'ICSI, un passage trop rapide à la pratique.
Force est de reconnaître cependant qu'aucun système d'évaluation ne pourra être totalement efficace. On ne saurait préconiser, en effet, de tester une technique en autorisant l'implantation d'embryons in vivo et encore moins de détruire l'embryon implanté issu de la technique évaluée si le danger de cette dernière devait être prouvé. Les évaluations ne pourront donc porter que sur les embryons in vitro. Dans ces conditions, on ne peut qu'accepter le caractère partiel de l'évaluation puisqu'il faudrait implanter l'embryon, le laisser se développer voire attendre la naissance de l'enfant sans être certain de l'innocuité totale de la technique évaluée. Il faut donc concilier différents principes. Votre Rapporteur considère en l'espèce que la nécessité d'évaluer toutes les nouvelles techniques d'AMP, poussée à l'extrême, pourrait apparaître éthiquement inacceptable.
Pour compléter le dispositif, il conviendrait de soumettre à autorisation préalable l'application clinique de toute nouvelle technique d'AMP. C'est ce que préconisait d'ailleurs le Conseil d'État dans son rapport de novembre 1999 qui ajoutait que l'organisme chargé du contrôle du secteur de l'AMP devrait aussi avoir la « faculté de s'auto-saisir de la question du développement d'une nouvelle technique de traitement de la stérilité ».
Enfin, pour satisfaire aux exigences de sécurité sanitaire, en particulier en ce qui concerne la possibilité d'identifier la cause d'un problème de santé publique dans la chaîne des événements, il est nécessaire de mettre en place un suivi des enfants nés grâce aux techniques, actuelles et futures, d'AMP. C'est aussi ce que préconisait le Conseil d'État dans le même rapport :
« Longtemps le souci de ne pas mettre en cause la sérénité des enfants a emporté une préférence pour l'absence de « PMA-vigilance » systématique touchant tous les enfants issus de ces techniques de procréation. Ce choix paraît toutefois devoir être réexaminé, d'autant plus que les nouvelles techniques mises en _uvre présenteraient des risques plus sérieux et encore mal évalués. Si l'on a désormais un recul satisfaisant en ce qui concerne la FIV classique, relativement proche du processus naturel, l'ICSI mérite, comme on l'a vu, un suivi attentif. La PMA-vigilance est difficile à deux titres : d'une part, parce que l'on n'a aucune connaissance a priori des risques ou des effets indésirables qu'il s'agit de détecter ; d'autre part, car c'est un suivi à long terme, voire à très long terme, qui est pertinent, afin que puissent être identifiées d'éventuelles maladies à déclenchement tardif. Aussi, quand bien même ces risques, aujourd'hui mal ou non identifiés, peuvent paraître avoir une probabilité d'occurrence très faible, l'organisation d'un système de suivi des techniques utilisées en AMP et des enfants nés grâce à elles est indispensable. Il est d'ailleurs dans l'intérêt même de ce secteur de pouvoir être en mesure de démontrer, si survenait un problème de santé, que ce ne sont pas les techniques et les produits utilisés dans le cadre d'une procréation médicalisée qui sont en cause ».
Le Conseil d'État propose ainsi de procéder, en premier lieu, à un archivage dans chaque centre d'AMP de fiches individuelles comportant l'indication exhaustive et détaillée des techniques, des traitements et des produits utilisés, ce qui permettrait de pouvoir identifier d'éventuels accidents sanitaires, et en second lieu d'organiser des enquêtes épidémiologiques à partir de groupes de parents qui s'engagent dans une procédure d'AMP et qui accepteraient d'y participer avec leur enfant pour suivre médicalement et psychologiquement leur développement.
B.- LES IMPERFECTIONS DE LA TECHNIQUE ONT CRÉÉ DES PROBLÈMES MÉDICAUX OU ÉTHIQUES
1.- Le recours excessif aux traitements de stimulation ovarienne
La définition des activités d'AMP par la loi de juillet 1994 n'est vraisemblablement pas satisfaisante puisque la liste réglementaire de ces activités en a exclu les traitements de stimulation ovarienne (traitements dits « d'induction d'ovulation »). En effet, l'article L. 2141-1 du code de la santé publique définit l'AMP de manière incomplète en la réservant aux « pratiques cliniques et biologiques permettant la conception in vitro, le transfert d'embryons et l'insémination artificielle ainsi que toute technique d'effet équivalent permettant la procréation en dehors du processus naturel ». Cette dernière mention a sans doute empêché que la stimulation ovarienne entre dans le champ de l'AMP bien qu'elle soit pratiquée sur les femmes qui vont recourir à une FIV ou sur celles qui vont faire don de leurs gamètes. Il s'agit en effet d'obtenir la maturation d'un peu moins de dix ovocytes qui seront prélevés avant d'être fécondés in vitro. Le traitement est aussi pratiqué sur un grand nombre de femmes ayant des difficultés à ovuler et pour lesquelles le processus naturel de procréation, sans cette stimulation, est difficile voire impossible.
Ce traitement hormonal n'est pas sans danger. Il comporte en effet des risques immédiats, à moyen et à long terme dont les plus importants sont le syndrome d'hyperstimulation ovarienne et les grossesses multiples. Le premier correspond au développement d'un nombre trop grand de follicules ; sous sa forme bénigne, il donne des douleurs abdominales, des ballonnements et dans 2 % des cas des troubles de la vision. S'y ajoutent, sous sa forme modérée, des nausées, des vomissements et diarrhées avec prise de poids de moins de 6 kg. Enfin, sous sa forme sévère, le syndrome se manifeste par de fortes douleurs abdominopelviennes, l'augmentation de volume des ovaires avec apparition de kystes susceptibles de se rompre ou de se tordre, l'apparition de troubles biologiques « dont les conséquences peuvent être redoutables (47) » et une prise de poids qui atteint ou dépasse les 10 kg. Lorsqu'il y a grossesse, ces symptômes sont aggravés et peuvent nécessiter l'hospitalisation.
S'agissant des grossesses multiples, les risques élevés de fausses-couches et d'accouchements prématurés, avec tous les inconvénients et dangers que présente pour l'enfant la grande prématurité, sont désormais bien connus. Il faut souligner que ces grossesses surviennent aujourd'hui plus fréquemment en dehors des FIV. La pratique a, en effet, considérablement évolué vers le transfert d'un nombre d'embryons moins élevé que par le passé, comme le montrent les statistiques présentées dans le premier paragraphe de la présente partie. La Commission nationale de médecine et de biologie de la reproduction et du diagnostic prénatal (CNMBRDP), dans son rapport d'activités 1996, estime ainsi que la stimulation ovarienne, pratiquée en dehors de la FIV, « a eu comme conséquence, au cours des vingt dernières années, l'augmentation de 25 % des grossesses gémellaires et de 400 % des grossesses triples. Les grossesses triples et de rang supérieur sont principalement dues aux inductions par les gonodostimulines (hMG et FSH) réalisées par des praticiens insuffisamment formés. Cette situation pose le problème de la prévention des grossesses multiples par le contrôle des pratiques d'induction de l'ovulation ».
Il semblerait donc que la possibilité, qui existe actuellement pour tous les médecins, quelles que soient leur formation et leur maîtrise des problèmes de stérilité, de prescrire des inducteurs d'ovulation soit abusivement utilisée. De nombreux traitements sont ainsi prescrits par des médecins généralistes dès qu'apparaît la moindre inquiétude sur la fécondité au sein d'un couple, ainsi que le notait la CNMBRDP dans le rapport précité : « Certains médecins qui se trouvent sollicités par les couples ont des difficultés à refuser des traitements dont ils savent d'ailleurs qu'ils seront d'autant plus efficaces que la patiente n'est pas vraiment stérile. Il y a donc une convergence d'intérêts entre le médecin (qui vient en aide) et la patiente (qui se sent prise en charge) alors même qu'il n'y a ni maladie ni nécessité de traitement. L'accroissement de ces thérapeutiques chez des femmes en fait fécondes explique pour une part l'inflation des grossesses multiples ». De son côté, le Professeur Claude Sureau, président de l'Académie nationale de médecine, indiquait devant la Mission que l'Académie, dans un rapport rendu à l'INSERM sur le sujet, considère que « la stimulation ovarienne est un problème majeur. (...) Il est évident que la plupart des grands prématurés sont liés à des stimulations ovariennes hors PMA (procréation médicalement assistée). Le problème, c'est d'avoir une attitude nuancée sur la limitation apportée à l'exercice médical ».
Il convient donc d'encadrer spécifiquement cette activité, qu'elle intervienne dans le cadre de l'AMP ou en dehors de celle-ci, dès lors qu'elle pose des problèmes de santé publique qui peuvent être graves pour la mère et pour l'enfant. La CNMBRDP propose, outre une information accrue des patientes et des praticiens, la mise en place d'un contrôle de l'usage des indicateurs prescrits par un système de déclaration annuelle de chaque médecin, qui pourrait lui être adressée. Ce système aurait, selon elle, plusieurs mérites :
« - il soulignerait aux yeux des médecins le contrôle de la société sur les effets néfastes de ces traitements lorsqu'ils sont manipulés à la légère ;
- certains renonceraient à ce type de prescriptions en réalisant qu'elles occasionnent une surcharge administrative de plus ;
- la CNMBRDP pourrait intervenir dans une modalité à définir devant les situations anormales et dangereuses qu'elle détecterait ».
Votre Rapporteur craint qu'un tel système soit impossible à gérer compte tenu du nombre de déclarations qu'il faudrait analyser chaque année. Il serait donc préférable de mettre en place des règles de bonne pratiques adéquates, sous l'égide de l'Agence nationale d'accréditation et d'évaluation en santé (ANAES), en distinguant les protocoles hors AMP des autres protocoles. Dans tous les cas, une meilleure information des patientes et une meilleure formation des praticiens doivent être assurées.
2.- Les difficultés liées à la conception d'embryons au-delà des besoins réels
Le législateur de 1994 n'avait sans doute pas prévu que l'on se trouverait confronté à l'existence d'un grand nombre d'embryons surnuméraires, c'est-à-dire ne correspondant plus à un projet parental, soit parce que les parents ont satisfait, à l'occasion d'une FIV réussie ou d'une adoption, leur désir d'enfant, soit parce qu'ils ont abandonné ce projet à la suite d'échecs répétés ou en raison d'un changement de leur situation familiale. Il est nécessaire de rappeler, en effet, que la congélation des embryons, en 1994, avait été envisagée comme une solution transitoire, dans l'attente du procédé de congélation des ovocytes que l'on croyait proche. En effet, à l'inverse du sperme, les ovocytes ne se prêtent pas à la congélation ce qui exige de procéder à leur insémination dans les heures qui suivent leur ponction, de transférer in utero une partie des embryons obtenus in vitro et de congeler les autres pour la poursuite du projet parental. Une expérience a certes été réussie de congélation et de décongélation de deux ovocytes à la fin de l'année 2000, à Singapour, permettant la naissance de jumeaux mais elle n'a pas été, jusqu'à ce jour, validée.
La deuxième raison pour laquelle on se trouve aujourd'hui en présence d'un grand nombre d'embryons surnuméraires tient aux risques que présente la stimulation ovarienne, risques exposés précédemment, qui incitent le praticien à prélever en une seule opération une moyenne de 8,6 ovocytes (48), avant de les inséminer puis de congeler en moyenne 4,5 embryons, plutôt que de procéder à deux opérations de stimulation ovarienne et de ponction des ovocytes.
Il en a résulté la situation suivante : au 31 décembre 1998, date du dernier recensement analysé par la CNMBRDP (49), 73 205 embryons étaient conservés. Pour 45 505 d'entre eux, soit 62 %, existait un projet parental en cours ; 12 782 couples avaient renoncé à leur projet tandis que l'on ignorait la décision sur la poursuite ou l'interruption du projet de 10 991 couples. Au total, on comptait donc, avant l'année 1999, 23 773 embryons surnuméraires.
Le tableau ci-après présente ces données, comparées avec celles de l'année précédente, en distinguant les embryons congelés depuis plus ou moins de cinq ans.
Projet parental | ||||||||
En cours |
Inconnu |
Interrompu | ||||||
1997 |
1998 |
1997 |
1998 |
1997 |
1998 |
1997 |
1998 | |
Embryons conservés depuis moins de 5 ans |
48.146 |
52.507 |
34.634 |
39.377 |
4.167 |
4.359 |
4.754 |
6.309 |
Embryons conservés depuis plus de 5 ans |
12.921 |
17.143 |
3.267 |
2.828 |
4.568 |
6.521 |
4.573 |
6.329 |
Total |
61.067 |
73.205 |
37.901 |
45.505 |
8.735 |
10.991 |
9.327 |
12.782 |
Source : CNMBRDP, rapport d'activité 1998.
Il faut en effet rappeler que l'article 8 de la loi n° 94-654 du 29 juillet 1994 a posé comme règle la destruction des embryons qui existaient avant la date de promulgation de la loi, dont la durée de conservation est au moins égale à cinq ans, dès lors qu'ils ne faisaient plus l'objet d'un projet parental ou d'une demande d'accueil par un couple tiers. Or, selon les informations recueillies par votre Rapporteur, il semble que les centres qui sont agréés pour conserver les embryons humains, actuellement au nombre de 101, n'ont reçu aucune instruction pour procéder à cette destruction. Certains, semble-t-il, en auraient pris l'initiative mais la grande majorité d'entre eux, se trouvant « dans l'embarras » en l'absence de toutes règles établissant les modalités et les conditions sanitaires de cette destruction, ont poursuivi la conservation de ces embryons, ne sachant comment appliquer la loi. On ne peut que regretter en l'espèce que le pouvoir réglementaire n'ait pas pris les dispositions nécessaires au respect de la volonté du législateur, sur un sujet aussi grave et sensible, accentuant ainsi le problème du devenir des embryons surnuméraires.
S'agissant des embryons conçus après la promulgation des lois de 1994 et ne rentrant plus dans le cadre d'un projet parental ni d'un projet d'accueil ou d'étude, le législateur est resté silencieux sur leur devenir, s'en remettant au législateur d'aujourd'hui. L'article L. 2141-3 du code de la santé publique ne se prononce, en effet, que sur la conservation des embryons à laquelle le couple peut consentir « dans l'intention de réaliser (sa) demande parentale dans un délai de cinq ans ». Il précise également que le couple est consulté chaque année pendant ces cinq ans « sur le point de savoir (s'il maintient) sa demande parentale ».
Ainsi le couple est libre d'accepter ou non la conservation de ses embryons pour que soient tentées, après une première tentative, d'autres FIV dans les années suivant leur conception in vitro, mais nul ne peut décider de mettre fin à cette conservation, même s'ils n'entrent plus dans un projet parental. Il en est de même si l'on perd la trace du couple, si ce dernier ne répond pas aux courriers que lui adresse son centre d'AMP ou s'il est dissous à la suite du décès de l'un de ses membres ou d'un divorce. La loi actuelle n'envisage, en effet, que la possibilité, pour le membre survivant, ou pour le couple qui le désire « à titre exceptionnel », de consentir à ce que les embryons conservés soient accueillis par un autre couple.
Le législateur de 1994 a donc renvoyé à plus tard la décision sur le sort des embryons surnuméraires en rendant impossible la destruction de ceux qui ont été conçus après l'entrée en vigueur de la loi et qui viennent ainsi, chaque année, grossir le nombre de ceux qui existaient auparavant. La conservation d'embryons en vue d'un transfert ultérieur est en effet pratiquée pour 90 % des embryons issus d'une FIV.
Cette situation pose plusieurs problèmes : celui de l'opportunité d'un délai fixé pour la conservation des embryons, au-delà duquel il serait mis fin à celle-ci, et celui des modalités de consultation du couple sur la poursuite de son projet parental.
Certains pays ont choisi de limiter dans le temps cette conservation : cinq ans dans le cas de l'Espagne et du Royaume-Uni, trois ans en Norvège et un an en Autriche et en Suède. Ainsi que le notait le Professeur Jacques Montagut (50), une durée trop courte peut favoriser une destruction systématique des embryons, « le couple n'ayant pas toujours la possibilité de tenter une nouvelle grossesse dans le trimestre suivant l'accouchement », après réussite de la première FIV. À l'inverse, une conservation trop longue ne paraît pas souhaitable si la femme approche ou dépasse l'âge de la ménopause, ce qui serait le cas d'un nombre croissant de patientes devant l'âge de plus en plus tardif d'entrée dans un processus d'AMP. C'est l'avis qu'exprimait le Docteur Marie-Odile Alnot devant la Mission, se déclarant favorable à la destruction « après dix ans de garde - ce qui serait, me semble-t-il, le moindre mal - et ce par référence à l'âge de la femme. En effet, les femmes qui ont recours à l'assistance médicale à la procréation ont en moyenne 33-35 ans et ne sont donc plus, au bout de dix ans, en âge de procréer ».
L'argument éthique milite lui-même en faveur de cette limitation. M. Carlos de Sola, responsable de la division bioéthique du Conseil de l'Europe, considérait ainsi devant la Mission le 13 septembre 2000 : « La conservation ad infinitum n'est pas un destin humain. Cela n'a pas de sens humain ». Pour le Docteur Marie-Odile Alnot, la destruction « éviterait de donner l'éternité à ces embryons orphelins, la conservation par congélation pouvant en effet durer des milliers d'années... Or ce n'est pas le propre de notre statut d'humain d'être éternel ! ».
Dans le même temps, l'expérience montre qu'il est extrêmement difficile de procéder à cette destruction, même lorsqu'elle a été légalement et réglementairement décidée. C'est ainsi qu'au Royaume-Uni, devant l'émotion suscitée dans l'opinion publique par l'annonce, le 31 juillet 1996, ainsi que le prévoyait la loi, de la destruction de près de 3 000 embryons qui ne faisaient plus l'objet d'un projet parental, le Gouvernement britannique décida de doubler de cinq à dix ans la durée légale de conservation des embryons.
Se pose enfin la difficulté majeure de la prise de décision par le couple et de son expression. La présidente de la HFEA britannique, Mme Ruth Deech, indiquait qu'« après cinq ou dix ans, les cliniques qui écrivent aux couples n'obtiennent aucune réponse, le plus vraisemblablement par incapacité psychologique à faire face à une telle situation ». Elle considérait pour sa part que devant le silence des couples, passé le délai légal, « il faut permettre d'éliminer les embryons », même s'ils ne donnent pas leur consentement exprès.
Le Docteur Marie-Odile Alnot soulignait également, devant la Mission, cette incapacité de nombreux couples à se prononcer. Interrogés sur leur souhait de poursuivre leur projet parental, de consentir à l'accueil ou de demander la destruction de leurs embryons pour ceux qui existaient à la date de la promulgation de la loi de 1994 et qui sont conservés depuis plus de cinq ans, nombre d'entre eux ne répondent pas. Ainsi « Le CECOS de l'hôpital Necker a en garde plus de 1 500 embryons orphelins, pour lesquels les géniteurs n'ont exprimé aucun choix. Et ce malgré les relances que nous avons mises en place dès 1989. Quatre cent cinquante couples sont concernés : la moitié d'entre eux ne sont plus joignables, car ils n'habitent plus à l'adresse indiquée, et l'autre moitié ne répond pas à nos courriers. Nous pouvons ajouter à ces populations les couples qui se sont séparés, et pour qui l'embryon est devenu l'enjeu de leur désunion, et qui soit ont des avis opposés, soit ne se prononcent pas. (...) Comment interpréter cette situation ? Pour certains couples, c'est seulement l'expression d'un désintérêt. Mais pour d'autres, c'est surtout la manifestation d'une ambivalence devant un choix difficile : c'est la première fois, dans le parcours de l'assistance médicale à la procréation, qu'ils se trouvent confrontés à une réalité, c'est-à-dire à leurs embryons. Et ils doivent, seuls, décider de leur sort. (...) Leur silence est un refuge : ils n'osent pas se prononcer. (...) ils ne nous répondent pas : nous n'obtiendrons donc jamais leur accord ».
Le législateur de 1994 avait bien essayé d'atténuer le problème des embryons surnuméraires en ouvrant la possibilité de leur accueil par un couple tiers, après consentement des géniteurs ou de l'un d'eux en cas de décès de l'autre. Comme l'indiquait M. Claude Huriet, sénateur, devant la Mission le 17 mai 2000, c'est M. Jean Chérioux, le rapporteur devant le Sénat des lois de juillet 1994, qui avait souhaité prévoir cette possibilité, avec d'autres parlementaires, étant « angoissé par le sort des embryons surnuméraires ».
Bien qu'il soit trop tôt pour dresser le bilan de cette procédure d'accueil, en raison de la parution tardive du décret d'application, publié seulement le 2 novembre 1999, il semble que cette solution soit loin d'être la panacée. La procédure, qui nécessite l'intervention d'une décision judiciaire proche de celle applicable en cas d'adoption, est en effet des plus complexes. Elle fait d'ailleurs à ce titre l'objet de vives critiques. La Fédération française des CECOS dénonçait (51), en particulier, l'ambiguïté des rôles respectifs du praticien et du président du tribunal de grande instance (TGI), l'absence de choix du TGI par le couple qui consent à l'accueil
- au risque que la confidentialité et l'anonymat ne soient pas respectés, alors que ce droit existe dans le cas d'un don de gamètes - la multiplication des documents et dossiers à constituer, autre source d'indiscrétions.
C'est également l'une des critiques majeures du Professeur Claude Sureau qui déclarait devant la Mission « être gêné par l'atteinte à la confidentialité inscrite dans cet article de loi. Dans le cas où un couple stérile est décidé, après longue réflexion, à accueillir un embryon abandonné par un couple donneur, le processus judiciaire engagé nous paraît extraordinairement lourd. En particulier, la recommandation faite au président du tribunal de mettre en _uvre une procédure d'investigation psychologique, sociale, éducative, finalement policière, pour savoir si le couple qui l'accepte est à même d'obtenir cet embryon, rend, à l'évidence, la confidentialité nulle. À mon avis, on a commis l'erreur totale d'assimiler l'accueil de l'embryon au processus de l'adoption (...) laquelle ne peut, en aucune manière, être secrète, alors que l'accueil de l'embryon peut, si les parents le décident, être secret. Lorsque l'on demandera au président du tribunal, au greffier, aux secrétaires, voire au commissaire de police, de faire une enquête, tout le monde le saura. Cela va très au-delà de ce que signifie le souci de la responsabilité parentale et cela me choque profondément ».
Enfin, le Docteur Marie-Odile Alnot relevait devant la Mission que le législateur avait omis de préciser la nature des établissements où l'accueil d'embryon peut être pratiqué : « Ni dans la loi de 1994, ni dans les décrets d'application, il n'est écrit que cette activité ne peut être pratiquée que dans les établissements et les organismes de santé publics et privés à but non lucratif. Ce principe a bien été spécifié pour le don de gamètes, mais pas pour l'accueil de l'embryon. On peut donc être surpris, voire choqué, de la non-application de ce principe : ce serait la première fois qu'un élément du corps humain pourrait « entrer » dans le système libéral ».
Outre ces critiques, force est de constater que les couples prêts à consentir à l'accueil de leurs embryons par un couple tiers paraissent peu nombreux. Le Professeur Pierre Jouannet évaluait devant la Mission à un millier le nombre d'embryons pour lesquels les géniteurs consentent aujourd'hui au « don » à un autre couple.
Il est donc impératif d'établir un régime précis et rigoureux pour la congélation et la destruction des embryons qui respecte les principes de transparence et de consentement. Votre Rapporteur considère qu'une durée de conservation limitée à cinq ans semble correspondre le mieux à la réalité des situations rencontrées dans le cadre des AMP. Pour responsabiliser les couples, il conviendrait de renverser la charge de la preuve quant à l'expression de leur volonté de poursuivre leur projet parental. Il reviendrait donc aux parents de se manifester chaque année, interrogés la première fois par courrier par leur centre leur donnant une date limite de décision annuelle, pour manifester leur souhait de poursuivre ce projet ou de l'interrompre. À défaut, au bout de cinq ans, le silence vaudrait décision tacite de destruction de leurs embryons surnuméraires. Un tel dispositif, certes sévère, aurait les mérites de clarifier les responsabilités de chacun, les centres d'AMP n'ayant plus la charge de relancer et de rechercher les parents. Il revêtirait aussi l'avantage de supprimer certaines pratiques abusives de tourisme médical dont faisait état, devant la Mission, Mme Chantal Ramogida de l'association « Pauline et Adrien » qui déclarait : « Le plus important serait de supprimer la congélation d'embryons. Les couples, lorsqu'ils vont voir un médecin, ne lui disent pas qu'ils ont déjà des embryons congelés. De ce fait, on se retrouve avec une quantité d'embryons congelés dont on ne sait que faire. Il faut responsabiliser les patients. Nous disons à nos adhérents qu'avant toute nouvelle tentative, il faut déjà implanter les embryons congelés ». Elle proposait à cet effet la création d'un « carnet de fertilité » dont la présentation conditionnerait la prise en charge par la sécurité sociale des actes d'AMP et sur lequel seraient indiqués, notamment, le nombre de tentatives d'induction ovarienne et celui des embryons congelés.
Reste la question du moment le plus opportun pour interroger le couple sur sa décision quant au sort des embryons surnuméraires : devrait-il se déterminer lorsqu'il entre dans la procédure d'AMP ou seulement lorsqu'il décide d'y mettre un terme ? Il est certain que la psychologie du couple et sa prédisposition à accepter l'accueil, la destruction et peut-être demain la recherche sur ses embryons sont différents à chacun de ces stades. Le Docteur Marie-Odile Alnot notait ainsi, devant la Mission, la difficulté des couples à se projeter dans l'avenir et leur prédisposition, dans de nombreuses décisions, à s'en remettre aux praticiens : « Un couple qui doit avoir recours à une fécondation in vitro, qui désire un enfant et qui est prêt à tout pour l'avoir, est ravi de se soumettre au médecin - lequel dispose d'un pouvoir énorme qui, d'ailleurs, fait peur - et de ne pas avoir à décider. Et si le médecin lui explique qu'en congelant des embryons il aura 10 % de chances supplémentaires - et sans procéder au traitement lourd qu'il redoute et qui n'est pas toujours bien supporté par la femme - il va automatiquement choisir cette solution sans penser à ses conséquences : même si on lui parle, il vit dans le présent et il est incapable de se projeter dans l'avenir. Les entretiens que j'ai avec mes patients sont toujours très longs - ils durent plus d'une heure - et j'ai pu me rendre compte qu'ils avaient un mal fou à se projeter dans l'avenir. J'ai du mal à aborder certains sujets avec eux, tels que le secret ou l'anonymat : ils sont trop pris par leur désir d'enfant ».
Votre Rapporteur considère qu'il serait souhaitable d'interroger le couple sur ses intentions quant au devenir de ses embryons surnuméraires lorsqu'il s'engage dans l'AMP, tout en l'autorisant tout au long de son parcours à revenir sur son consentement initial. De la sorte, son silence éventuel après plusieurs années ne serait plus source de blocage pour les centres. Le Professeur Pierre Jouannet exprimait d'ailleurs devant la Mission le souhait des CECOS d'un dispositif clarifié : « Ce qui importe, c'est que les embryons que nous conservons aient un destin et que cette destinée, ce ne soit pas nous qui en décidions mais les couples qui sont à l'origine des embryons. Il nous importe de pouvoir « gérer » le devenir de ces embryons. Ce devenir est fonction du souhait des couples. C'est cela que nous essayons de faire ».
Quelles que soient les solutions qui seront retenues, il est nécessaire d'ouvrir le débat et de mettre en place des règles claires et transparentes en la matière.
3.- L'aide psychologique apportée aux couples est insuffisante
L'article L. 2141-10 impose aux couples qui souhaitent recourir à l'AMP des entretiens particuliers préalables « avec les membres de l'équipe médicale pluridisciplinaire du centre ». Hélas, le caractère « médical » l'a largement emporté sur le caractère « pluridisciplinaire » dans l'interprétation de cette disposition par l'autorité réglementaire. L'arrêté du 12 janvier 1999 relatif aux règles de bonnes pratiques cliniques et biologiques en AMP dispose ainsi que : « Le couple demandeur doit rencontrer un clinicien et un biologiste de l'équipe pluridisciplinaire. Il doit pouvoir également, sur sa demande ou celle des praticiens, rencontrer tout autre membre de l'équipe, y compris le médecin qualifié en psychiatrie ou le psychologue dont l'établissement s'assure le concours ».
Le recours à un psychologue ou un psychiatre du centre n'est donc pas systématique, ce que regrettent de nombreux observateurs ou acteurs de l'AMP. Le Docteur Marie-Odile Alnot devant la Mission se prononçait ainsi en faveur d'entretiens systématiques et obligatoires : « Les couples ont des deuils à faire et il faut les aider ». Mme Bernadette Isaac-Sibille, membre de la Mission, notait pour sa part que : « Le plus souvent les personnes qui ont subi trois ou quatre tentatives de procréation médicalement assistée sont réticentes à l'adoption car elles n'ont pas fait le deuil d'un enfant « naturel ». Un entretien après trois ou quatre échecs me paraît donc essentiel, voire obligatoire, sinon nous allons avoir affaire à des parents adoptants qui n'auront pas fait le deuil d'une naissance « naturelle » et qui n'adopteront pas dans les meilleures conditions ».
Mme Yvette Roudy, membre de la Mission, regrettait pour sa part que l'on n'explique pas ou très peu aux couples que leur projet parental peut aussi prendre la forme d'une adoption ; elle ajoutait : « Je connais des couples qui sont sortis détruits du parcours de l'assistance médicale à la procréation ; ils ont été pris dans un engrenage pendant dix ans et n'en sont pas sortis. Ils n'osent pas le dire mais on m'a même parlé de harcèlement à certains moments. C'est la raison pour laquelle je pense que des entretiens avec les couples hors du milieu médical concerné seraient une bonne chose ».
Cette situation est regrettable car elle conduit de nombreux couples, en situation de détresse à la suite de plusieurs tentatives d'AMP ayant échoué en France, ou dans l'attente d'un don de gamètes, à pratiquer un « tourisme médical procréatif » dans des conditions difficiles et contestables s'agissant du respect de nos principes éthiques.
La pratique varie, en outre, beaucoup d'un centre à l'autre, comme l'expliquait devant la Mission M. Jean-Loup Clément, psychologue dans un CECOS : « Dans les CECOS comme dans les centres de FIV, il existe deux sortes d'interventions des psychologues et des psychiatres. Certains, comme moi, appartiennent à l'institution, sont statutairement rattachés à l'hôpital et travaillent en étroite collaboration avec les médecins. D'autres CECOS fonctionnent avec des correspondants extérieurs privés. Les psychologues ou les psychiatres ayant un statut dans l'institution agissent en réelle collaboration avec les médecins, ce qui a une incidence sur les couples. Ils peuvent les rencontrer plusieurs fois, les accompagner et les faire cheminer comme ils le souhaitent, alors que dans un cabinet privé, ils n'ont qu'un rôle d'expert beaucoup plus limité. Les gens ont le sentiment que leur interlocuteur n'est pas en lien direct avec les médecins de l'institution. C'est tout à fait regrettable car l'approche psychologique est complémentaire de l'approche médicale, elle ne lui est pas concurrentielle ».
Une enquête réalisée par l'INSERM à l'occasion des journées de la Fédération française d'étude de la reproduction les 23, 24 et 25 septembre 1998 montre que la pluridisciplinarité des équipes des centres est à géométrie variable ; elle n'implique pas systématiquement l'intervention de toutes les spécialités, que l'AMP soit homologue ou hétérologue (52), alors qu'un grand nombre de praticiens souhaitent que la décision d'admettre un couple dans un processus d'AMP soit prise collectivement par l'équipe. L'enquête montre aussi que l'accompagnement psychologique des couples candidats à l'AMP est bien accueilli par les praticiens, surtout dans l'hypothèse d'une AMP avec tiers donneur.
De l'ensemble de ces données, votre Rapporteur conclut à la nécessité de rendre systématique et obligatoire l'accompagnement psychologique des couples dans l'AMP, avant qu'ils s'y engagent et pendant le processus.
C.- LES PROBLÈMES SPÉCIFIQUES LIÉS AU RECOURS À UN TIERS DONNEUR
Le recours à un tiers donneur pose deux problèmes de nature éthique ou médicale : le premier, relatif aux dons de gamètes, pour lequel des solutions semblent possibles, et le second, soulevant la question de l'anonymat pour lequel il ne semble ni opportun ni souhaitable de modifier la règle en vigueur.
1.- La gestion du don de gamètes
Il est difficile de comparer le don de sperme au don d'ovocytes tant ils sont différents. Il n'en demeure pas moins que tous deux sont confrontés au même problème de pénurie qui rend encore plus long et difficile un parcours d'AMP hétérologue.
a) Une situation de pénurie inquiétante
Le délai pour réaliser une insémination artificielle avec sperme de donneur est aujourd'hui supérieur à un an, plus de 700 couples étant chaque année inscrits sur les listes d'attente des 22 CECOS de France. Le président de leur fédération, le Professeur Pierre Jouannet, indiquait aux membres de la Mission que « 372 volontaires se sont présentés dans les centres pour donner leur sperme en 1999. C'est très faible par rapport aux besoins. D'autant plus faible quand on sait que, parmi ces volontaires, seulement 50 à 60 % pourront effectivement être retenus comme donneurs de sperme. Le manque de volontaires est un problème important. En termes d'utilisation, en 1999, il y a eu en tout 9 128 cycles de traitement à partir de sperme de donneurs distribué par les CECOS, dont près de 8 000 par insémination artificielle et un peu plus d'un millier par fécondation in vitro. Ces cycles de traitement ont conduit à l'obtention de 1 279 grossesses. Près d'un millier par insémination artificielle et plus de 300 par fécondation in vitro ».
Pour le don d'ovocytes, la situation est bien plus grave puisque les listes dépassent plus de deux ans d'attente (53), conduisant plusieurs centaines de nos compatriotes, hélas, à pratiquer un « tourisme médical » coûteux, controversé, et moralement destructeur en cas d'échec, dans les pays où il est possible d'acheter des ovocytes, en sélectionnant éventuellement la « donneuse ».
Un véritable marché de gamètes s'est, de fait, développé dans certains pays. Sur Internet, il est désormais possible d'acheter le sperme d'un prix Nobel, en sélectionnant la spécialité du prix décerné (sic) ou encore de choisir sur dossier, photographies à l'appui, un mannequin modèle prête à mettre aux enchères ses ovocytes. L'un de ces sites de vente avance à ce propos l'argument commercial suivant : « Des milliers d'hommes dans le monde rêveraient de combiner leurs gènes à ceux d'une femme belle, en bonne santé et intelligente (...). Un don tel que la beauté, l'intelligence ou les aptitudes sociales aidera votre enfant dans sa quête du bonheur et du succès ». Sans aller jusqu'à ces excès, dans des cliniques canadiennes à la réputation sérieuse, il est possible, ainsi que l'a appris à votre Rapporteur un témoignage émouvant, d'acheter pour plusieurs dizaines de milliers de francs les ovocytes d'une femme, préalablement sélectionnée sur dossier, qui comprend notamment les photographies de la « donneuse » et de ses enfants. Une première tentative est pratiquée de transfert de quelques embryons « frais » après ponction et insémination par le sperme du mari ou du compagnon de la patiente et une seconde est tentée, en cas d'échec de la première, avec les embryons qui auront été congelés.
Cette situation est d'autant plus grave qu'elle conduit à créer un véritable trafic d'ovocytes où les droits et la santé des femmes semblent gravement menacés. Le Professeur Jacques Testart informait ainsi la Mission de l'existence de pratiques très inquiétantes : « À Chypre, un gynécologue américain d'origine russe fait venir des charters de femmes russes dans une clinique pour « pondre » à destination de demandeuses d'ovules européennes. C'est un circuit clandestin. J'ai essayé d'attirer l'attention des journalistes, mais ceux-ci n'aiment pas beaucoup les enquêtes, ils préfèrent les ragots ou le spectacle. (...) En Espagne, à Séville, se trouve une clinique dans laquelle la plupart des praticiens français qui ont besoin de don d'ovules utilisent ce réseau, depuis Paris. Dans un laboratoire autorisé pour l'assistance à la procréation, le biologiste de Séville, qui se trouve être français, remet une note aux personnes demandant de lui payer 19 000 francs, pour le traitement et les femmes donneuses qui seraient rémunérées 4 000 francs. Il est de notoriété publique que certaines donneuses - et en France, ce serait mal vu qu'elles soient payées - sont venues pour une fécondation in vitro et qu'on leur a pris des ovules sans le leur dire ».
En France, si le principe de gratuité est en apparence préservé, une pratique s'est développée dans les centres consistant à accélérer les demandes des couples qui parviennent à recruter un donneur ou une donneuse. Aucun de ces dons n'est « dirigé », les gamètes des amis ou parents sollicités venant alimenter la banque commune mais on peut s'interroger sur le caractère éthique de ces pratiques qui peuvent cacher une rémunération des donneurs ou de fortes pressions, notamment familiales, exercées sur ces derniers. Un praticien rencontré par votre Rapporteur considérait que ces pratiques peuvent cacher des phénomènes de subordination des donneuses d'ovocytes que certains couples avouent rémunérer plus de 10 000 francs. M. Jean-Loup Clément, psychologue du CECOS de Lyon, critiquait pour sa part la fausse motivation de certains donneurs : « Quand je leur demande pourquoi, ils me répondent : « Parce que dans une autre ville un parent ou un ami me l'a demandé pour réduire le délai d'attente ». Si je leur demande s'ils auraient été donneurs spontanément, ils me disent que non. C'est très dommageable ».
Pourtant l'arrêté du 12 janvier 1999 relatif aux bonnes pratiques cliniques et biologiques d'AMP « interdit de subordonner le bénéfice de l'AMP à la désignation d'un donneur par le couple receveur, même en faveur d'un couple tiers anonyme » tout en spécifiant que cette « disposition n'interdit pas d'informer les couples receveurs potentiels sur les difficultés rencontrées pour disposer de donneurs de gamètes et de les encourager à aider l'équipe dans cette recherche ».
Ainsi la situation de pénurie de gamètes conduit-elle à dévoyer le principe du don altruiste, totalement anonyme et gratuit, tandis que certains couples n'hésitent pas à franchir nos frontières pour acheter ce qui ne devrait pourtant ne pas faire l'objet d'un commerce.
Pour M. Jean-Loup Clément, les motivations qui sous-tendent le don peuvent être complexes. Elles expliqueraient en partie le phénomène de pénurie. Il déclarait devant la Mission :
« Le discours manifeste des hommes est de rendre service à un couple stérile, d'aider quelqu'un. Le discours latent est plus compliqué. (...). C'est la raison pour laquelle nous avons une pénurie de donneurs (...) les consultants ne se voient pas avoir des enfants dans la nature. Le même argument démobilise 99,9 % des donneurs potentiels. Les donneurs sont plutôt des gens sains, sympathiques et solides. Il est très difficile de les recruter ».
En revanche, il distinguait nettement la psychologie d'une donneuse : « Pour les femmes, c'est très différent parce que c'est le don d'une femme à une autre femme avec un discours obscur qui consiste à dire que la grossesse va absorber les qualités premières de l'ovocyte. On se met à nier toute notion biologique et génétique de l'ovocyte ».
b) Une information et une sensibilisation insuffisantes en faveur du don
Le phénomène de pénurie des dons de gamètes met en évidence l'insuffisance des actions d'information et de sensibilisation en faveur du don. Ces actions relèvent, d'après la loi de juillet 1994, de la responsabilité du ministère chargé de la santé. Hélas, comme le regrettait le Président Pierre Jouannet devant la Mission, « celui-ci n'a pas les moyens de cette politique, de ce choix ». Il se prononçait alors en faveur d'un « établissement public, une agence ou autre, qui aurait entre autres missions de prendre en charge l'information du public et la sensibilisation des volontaires pour le don de gamètes », sur le modèle de la HFEA britannique.
S'il est impératif de disposer de davantage de moyens pour mieux informer et sensibiliser l'opinion publique sur le don de gamètes, un second obstacle devrait vraisemblablement être levé s'agissant de l'interdiction, posée par l'article L. 1211-2 du code de la santé publique, de toute publicité du « don d'éléments ou de produits du corps humain au profit d'une personne déterminée ou au profit d'un établissement ou organisme déterminé ». L'interprétation stricte de cet article va, en effet, jusqu'à interdire de donner l'adresse d'un CECOS qui prendrait l'initiative d'animer une campagne d'information locale en faveur du don. Il semble pourtant indispensable de relayer sur le plan local les actions d'information qui doivent être menées au niveau national, les premières devant renforcer et nourrir le message des secondes. Dans ce cadre, on ne voit pas pourquoi la simple mention de l'adresse du CECOS le plus proche, qui faciliterait la démarche du donneur potentiel touché par une campagne d'information nationale ou locale, serait une entorse à l'interdiction de publicité dirigée en faveur du don.
L'expérience réussie du CECOS de Lyon qui, par ses actions de proximité, a réussi à réduire à six mois le délai d'attente des demandes de don de sperme, milite en faveur de ce changement que préconisait M. Jean-Louis Clément, en s'adressant ainsi aux membres de la Mission : « Il serait bon que vous leviez dans la loi l'ambiguïté liée à l'information et à la publicité sur le recrutement des donneurs. Les donneurs doivent être très libres dans leur démarche. Personne ne doit faire pression sur eux. C'est pourquoi l'on doit faire une information publique (...). À Lyon, nous faisons de l'information dans les journaux, en ville, des affiches dans les gares (...) je donne toujours l'adresse Internet des services du CECOS, en sorte que les gens peuvent trouver l'adresse du centre le plus proche. Mais arrêtons de faire pression. (...) Les centres hospitaliers de référence doivent pouvoir attribuer un budget à chaque centre procédant aux cessions de gamètes afin qu'une information soit faite localement ».
c) Les problèmes spécifiques liés au don d'ovocytes
L'insuffisance des dons d'ovocytes peut s'expliquer par la pénibilité de l'opération de prélèvement. Cet acte nécessite en effet, après traitement de stimulation ovarienne dont les risques ont été précédemment évoqués, une anesthésie générale et une série d'examens biologiques et échographiques longs et coûteux. S'ajoutent aussi, à la difficulté physique et au risque médical du don, d'éventuels obstacles financiers. L'article L. 1211-4 du code de la santé publique, qui interdit que soit alloué au donneur d'un élément ou d'un produit de son corps, toute forme de paiement, autorise toutefois que lui soient remboursés les frais engagés selon des modalités fixées par décret en Conseil d'État. Ce dernier a été publié le 11 mai 2000 et il est encore trop tôt pour savoir si les dispositions qu'il prévoit sont pleinement appliquées. Il prévoit en effet que l'ensemble des dépenses de soins et de traitement, y compris le forfait hospitalier, ainsi que les frais de déplacement afférents à ces soins, seront désormais pris en charge par l'établissement de santé où le prélèvement est réalisé. Une indemnité pour perte de rémunération pourra en outre être versée « sur présentation des justificatifs nécessaires (...) (qui) ne peut être supérieure au double de l'indemnité journalière maximale de l'assurance maladie du régime général », soit près de 500 francs. Avant la parution de ce décret, la Fédération française des CECOS, dans ses propositions de révision du 24 décembre 1999, notait en effet que les frais engagés pour le don d'ovocytes « sont considérables et ne peuvent être pris en charge par la Sécurité sociale, ni le plus souvent par les hôpitaux (...). Les donneurs, en sus du don de leurs gamètes, couvrent personnellement une grande partie de ces frais ».
Il faudra donc rester très vigilant sur le respect des dispositions établies en espérant que les établissements de santé, en période de maîtrise des dotations régionales d'hospitalisation, n'en feront pas une interprétation trop restrictive. On peut, toutefois, encore s'interroger sur l'opportunité d'indemniser plus généreusement la perte de temps subie par les donneuses, dont le cas devrait être traité spécifiquement.
Les conséquences du difficile recrutement des donneuses se trouvent encore aggravées par les opérations successives que doivent subir les ovocytes une fois ponctionnés, au premier rang desquelles l'obligation de « quarantaine » qui leur est imposée. Le décret du 28 février 1996 oblige, en effet, à congeler les embryons obtenus après insémination des ovocytes ponctionnés pendant six mois afin de renouveler, à l'issue de ce délai, les tests sanitaires sur la donneuse pour vérifier sa séronégativité vis à vis notamment des virus du sida, de l'hépatite B et C.
Il n'est donc plus permis, comme par le passé, de transférer les embryons obtenus sans passer par l'étape de la cryoconservation, laquelle réduit considérablement le taux de grossesse par donneuse ponctionnée. Ainsi, selon le bilan réalisé sur l'année 1999 par le Groupe d'étude sur le don d'ovocytes (GEDO), qui regroupe l'activité de dix-huit centres sur les vingt qui pratiquent en France l'AMP avec don d'ovocytes :
- 194 donneuses se sont présentées mais 150 ont pu être retenues ;
- 150 stimulations ont été pratiquées aboutissant à 141 ponctions ;
- ces 141 ponctions ont permis de prélever 1 401 ovocytes mais seulement 1 308 ovocytes ont été inséminés ;
- sur ces 1 308 ovocytes fécondés, 853 embryons ont été obtenus ;
- sur ces 853 embryons, 79,5 % ont été congelés ;
- la même année 1999, la perte embryonnaire à la décongélation étant égale à 30 %, 360 embryons ont été transférés après décongélation au cours de 171 transferts (environ 3 embryons par transfert) ;
- au final, 67 grossesses ont pu être obtenues (29 grossesses cliniques et 38 débutantes), à comparer avec 40 grossesses réussies en 1998 qui ont permis la naissance de 36 enfants, la différence correspondant aux fausses-couches qui se sont produites.
De nombreux praticiens dénoncent « l'aspect délétère » ou « l'effet désastreux » de cette politique de congélation embryonnaire systématique sur les chances de grossesse avec don d'ovocytes. Certains d'entre eux ont d'ailleurs décidé, le 30 janvier 2001, d'interrompre leurs activités en « jugeant non éthique de proposer un traitement contraignant à des donneuses pour un résultat limité », dénonçant une réglementation « unique en Europe (...) qui pousse le principe de précaution jusqu'à l'excès ».
De fait, la France est le seul pays européen à pratiquer cette quarantaine, en dépit de l'absence de toute preuve scientifique sur l'existence du risque de transmission materno-f_tale des virus précités. L'on sait en effet que ce risque est dépendant de la charge virale mais aucun élément virologique ne permet aujourd'hui de suspecter qu'un ovocyte, qui comprend moins de 5 % de cellules lymphocytaires, serait susceptible d'infecter la receveuse. Par ailleurs, les quatre années d'application de ce décret ont démontré qu'aucun cas de donneuse séropositive n'a été mis en évidence à la suite de la décongélation des embryons.
Le décret de 1996 est donc fortement remis en question. Sa révision paraît nécessaire et souhaitable à la condition que les patients soient pleinement informés de la possibilité d'un risque faible de transmission, les tests étant bien sûr toujours pratiqués sur la donneuse avant et à l'occasion du don de ses ovocytes.
L'ensemble des problèmes spécifiques au don d'ovocytes milite en faveur de la distinction de cette activité par rapport au don de sperme. C'est en particulier l'avis exprimé par le Professeur Pierre Jouannet devant la Mission qui déclarait, en sa qualité de Président de la Fédération française des CECOS : « Notre expérience démontre que toute prise en charge d'assistance médicale à la procréation par don, qu'il s'agisse de don de spermatozoïdes, de don d'ovocytes ou, demain, de don d'embryons, doit répondre à des exigences particulières en termes de sécurité sanitaire, bien entendu, mais aussi en termes d'accueil et d'accompagnement des couples et des donneurs. Elle comporte des aspects techniques, par exemple pour congeler le sperme, pour prélever les ovocytes ou pour conserver les embryons, mais elle a aussi une dimension psychologique et relationnelle majeure. Il me semble que les compétences et les responsabilités nécessaires à l'exercice de cette activité doivent être reconnues de manière spécifique. Ceci favoriserait l'évaluation et serait sûrement un facteur de promotion de la qualité ».
Aujourd'hui, seuls sont agréés le médecin qui pratique la ponction des ovocytes et le biologiste qui en assure la fécondation. Il serait nécessaire, comme le soulignait le Docteur Marie-Odile Alnot devant la Mission, d'agréer des structures avec leurs équipes multidisciplinaires pour le don d'ovocytes, à l'instar des CECOS. Plusieurs d'entre eux (54), qui ont souhaité s'impliquer dans le don d'ovocytes, tel que le CECOS de l'hôpital Necker à Paris, l'ont d'ailleurs fait avec succès, c'est-à-dire avec les avantages que décrivait le Docteur Marie-Odile Alnot :
« Une structure telle que le CECOS n'existe pas pour le don d'ovocytes. Depuis 1986, le CECOS de Necker intervient en matière de don d'ovocytes, exactement comme pour le don de sperme, et il a été l'un des premiers à le faire : nous sommes en dehors des équipes soignantes mais en intime rapport avec elles puisque nous collaborons au quotidien. Nous voyons tous les demandeurs et les donneuses, et nous sommes responsables de l'attribution et de la gestion. Nous avons donc une espèce d'indépendance à l'égard des soignants et cela est important car, dans cette activité que je connais bien, il faut être neutre et ne pas être influencé par des liens affectifs qui s'établissent systématiquement entre patients et soignants ».
Votre Rapporteur partage cet avis en faveur d'un encadrement spécifique du don d'ovocytes qui permettrait d'apporter de meilleures garanties sur la qualité des soins pratiqués, lesquels mettent particulièrement en danger la santé des femmes.
d) L'alternative possible pour répondre au problème de l'insuffisance des dons
de sperme
La situation est beaucoup plus simple pour résoudre le problème de pénurie des donneurs de sperme. Il suffit, en effet, d'étendre l'utilisation des gamètes d'un même donneur pour atténuer le phénomène de pénurie. Actuellement, l'article L. 1244-4 du code de la Santé publique dispose que « le recours aux gamètes d'un même donneur ne peut délibérément conduire à la naissance de plus de cinq enfants ». Cette limitation s'explique par le souci d'éviter le risque de consanguinité entre les enfants nés d'une AMP avec tiers donneurs et leurs « demi-frères » et « demi-s_urs », « enfants » des donneurs. Cependant il apparaît que la limitation posée est excessive au regard de la réalité du risque, les démographes démontrant qu'il est statistiquement accru seulement pour plus de vingt enfants par donneur. La comparaison avec l'étranger montre d'ailleurs que la situation est très variable : l'Espagne, la Suède et la Suisse limitant à six le nombre d'enfants issus des gamètes d'un même donneur, le Royaume-Uni et les Pays-Bas le fixant à dix. La Fédération française des CECOS propose pour sa part de modifier la loi afin qu'elle limite désormais à cinq fratries l'utilisation des gamètes d'un même donneur.
À ce propos, le Professeur Pierre Jouannet déclarait devant la Mission : « Quand on aide des couples à avoir plusieurs enfants, on devrait pouvoir utiliser le sperme du même donneur car ces enfants ne se marieront pas entre eux plus tard. La limitation du nombre d'enfants nous empêche actuellement de répondre à des demandes de familles qui voudraient avoir plus d'enfants. Par exemple, dans mon centre, actuellement, nous mettons en attente des demandes faites par des couples qui ont déjà eu deux enfants par don de sperme et qui en souhaiteraient un troisième. Nous sommes dans une telle situation de pénurie de dons que nous faisons appel à leur compréhension. Nous devons leur expliquer que nous répondons en priorité à la demande des couples qui n'ont pas encore d'enfant. Il n'en serait pas de même si nous pouvions utiliser le sperme d'un même donneur, celui qui a déjà été utilisé pour les premiers enfants de la même famille. Si l'on doit fixer des limites, que ce soit plutôt sur le nombre de fratries ou de familles dans lesquelles ces enfants sont conçus que sur le nombre d'enfants eux-mêmes ».
Le Conseil d'État, dans son rapport de novembre 1999, Les lois de bioéthique : cinq ans après, considère que la notion de fratrie est insuffisamment claire, les demi-frères et demi-s_urs pouvant ou non en faire partie. Il préconise que le législateur augmente le nombre maximum de naissances par donneur.
Devant la Mission, le psychologue Jean-Loup Clément a pour sa part manifesté son désaccord avec tout raisonnement en termes de fratries : « Depuis quinze ans, je répète avec force que, puisqu'il n'y a pas de lien biologique du père à son enfant, pourquoi juxtaposer un lien biologique entre des enfants ? De plus, il y a une pénurie de sperme. On ne va pas demander aux gens s'ils en veulent pour un deuxième, pour un troisième. On veut ramener le biologique là où il n'a pas lieu d'être et on devient incohérent. Si des enfants sont issus d'un même donneur, un même homme est vraiment dans la famille. Il n'y a pas de lien biologique du père à l'enfant. Il n'y a donc pas de lien biologique entre les enfants. En disant cela à la Fédération des CECOS, j'ai dû convaincre la moitié des biologistes, qui me l'ont dit, les autres estimant qu'il est bien de garder un lien biologique entre enfants ».
Le débat est donc ouvert mais ne pourrait-on pas, dans la solution finale qui sera retenue, laisser le choix aux familles qui souhaitent recourir plus d'une fois à l'IAD ?
2.- La question de l'anonymat et du secret
Le principe de l'anonymat du don de gamètes n'est que rarement contesté dans notre pays. Comme le soulignait le Conseil d'État dans son rapport de 1988, Sciences de la vie : de l'éthique au droit, il est « à la fois le gage de l'autonomie de l'épanouissement de la famille qui se fonde et la protection loyale du désintéressement (du donneur) qui y contribue. La convergence de ces deux considérations, dont la première joue également en faveur de l'enfant, explique que dans la hiérarchie des valeurs, elles l'emportent ensemble sur le prétendu droit à la connaissance de son origine ».
Le projet de loi n° 2870 relatif à l'accès aux origines personnelles, qui a été adopté en première lecture à l'Assemblée nationale le 31 mai 2001, a d'ailleurs résolument écarté de son champ d'application les enfants nés grâce au recours à l'une des techniques d'AMP sans que cette exclusion ait été contestée lors des débats. Devant la Mission, plusieurs intervenants se sont prononcés pour le strict maintien de l'anonymat. M. Jean-Loup Clément indiquait ainsi que : « Les psychologues et les psychiatres des CECOS sont unanimes pour estimer que le don doit être réalisé dans l'anonymat. À l'extérieur, les gens proches d'un courant naturaliste sont plutôt hostiles à la méthode palliative. Ceux qui y sont un peu favorables restent partisans de l'anonymat mais souhaitent la levée du secret ».
Le Professeur Pierre Jouannet considérait de son côté que « pour la procréation, un spermatozoïde de donneur est remplaçable par n'importe quel spermatozoïde d'un autre donneur pourvu qu'il corresponde aux critères d'appariement entre donneurs et receveurs. Contrairement au donneur, le père stérile n'est pas remplaçable, c'est en cela qu'il est unique pour l'enfant. C'est cette fonction paternelle de procréation qu'il nous semble important de privilégier, c'est elle qu'il nous semble important de protéger. Lever l'anonymat du don, c'est fragiliser ce lien, c'est privilégier de manière artificielle la dimension génétique de la procréation par rapport à ce que j'appellerai la dimension plus humaine qui est à l'origine de cet enfant. C'est dans ce sens, en tenant compte de la société dans laquelle nous sommes, qu'il nous semble nécessaire de maintenir l'anonymat ».
Par ailleurs, il n'est pas certain que la levée de l'anonymat soit revendiquée par les intéressés eux-mêmes, comme semble le montrer une enquête menée récemment par les CECOS, évoquée par le Professeur Pierre Jouannet devant la Mission : « De temps à autre, on lit dans des organes de presse des articles selon lesquels les enfants conçus par sperme de donneur vont très mal du fait qu'ils ne connaissent pas l'identité du donneur, que certains enfants recherchent « avec frénésie » leur père biologique, le donneur de sperme. Au début de cette année, étonnés par ces déclarations, nous avons mené une enquête dans l'ensemble des CECOS pour savoir si les enfants de plus de quinze ans que nous avions aidés à naître s'étaient présentés dans les centres pour poser des questions sur les donneurs de sperme. D'après cette étude, 2 662 enfants ont été conçus avec l'aide des CECOS avant 1980, ils ont donc plus de vingt ans aujourd'hui, et 8 274 sont nés entre 1980 et 1985, soit un total de près de 11 000 enfants de plus de quinze ans. Aucun d'entre eux, pas un seul, n'a fait une démarche dans un de nos centres pour s'informer sur l'identité du donneur. (...) Un certain nombre d'enfants sont venus dans nos centres pour poser des questions diverses sur l'insémination artificielle, sur le don de sperme, sur les modalités de notre travail mais pas sur l'identité du donneur. Soit ces enfants ne savaient pas comment ils ont été conçus (...) mais un bon nombre d'entre eux le savent, soit ce n'est pas leur préoccupation principale. Lorsque l'on discute avec ces enfants, on se rend compte que c'est leur histoire, leur origine qui leur importent plus que l'identité du donneur. L'origine, ce n'est pas l'identité du donneur, l'origine c'est l'histoire de leurs parents, celle du couple qui, à un moment donné, a fait une démarche pour une procréation par don de sperme ».
Quelques voix se sont cependant élevées pour demander d'« assouplir » la règle afin, notamment, de répondre au problème de pénurie de dons d'ovocytes. D'aucuns défendent ainsi la pratique de dons intra-familiaux en s'appuyant sur les possibilités ouvertes dans le cadre du don d'organes. Dans une étude présentée le 20 février 2001 devant l'Académie nationale de médecine (55), le Professeur Jacques Salat-Baroux et ses confrères s'interrogent ainsi sur l'opportunité « par souci d'efficacité (...) (de) changer les principes « intangibles » que sont la gratuité et l'anonymat (...) dans des conditions exceptionnelles : dans une même famille et pour une raison vitale ».
Un tel assouplissement serait vraisemblablement dangereux et contraire à l'intérêt de l'enfant. M. Jean-Loup Clément, en tant que psychologue, exprimait à cet égard son refus catégorique : « Il convient d'appréhender l'assistance médicale à la procréation en termes de système. Nous appliquons la règle de l'exogamie. Quand un homme et une femme veulent faire des enfants, ils sortent de leur groupe d'origine. On fait donc un enfant avec un étranger. Les travaux de Françoise Héritier sont très intéressants à cet égard. Dans ce qu'elle appelle l'inceste du deuxième type, elle englobe les alliés. Geneviève Delaisi de Parseval a écrit dans « Enfant de personne », qu'elle était très favorable aux IAD intra-familiales puisque le géniteur est connu. Pour ma part, je considère que c'est une aberration. Il faut être cohérent. Cette médecine qui échappe à l'image de la médecine classique est destinée à faire des enfants. Un enfant ne peut pas avoir son oncle comme géniteur et son père pour l'élever. Un beau-frère ne doit pas inséminer sa belle-s_ur par l'entremise d'un médecin ».
Le Professeur Pierre Jouannet, au nom de la Fédération française des CECOS, déclarait pour sa part : « Nous pensons qu'introduire le donneur dans la relation qui s'établit entre les parents et l'enfant risque d'être perturbant pour tout le monde, surtout si le donneur est un proche, un frère, une s_ur, un familier. L'introduction de l'identité du tiers donneur va établir un système d'échanges et de relations extrêmement complexes et peut-être à risque pour tout le monde. J'ai rencontré des couples qui pensaient que ce serait plus simple si l'on utilisait le sperme du frère ou d'un ami. Je leur disais : « Admettons que l'on réponde à votre demande, que l'on vous aide à concevoir un enfant dans ces conditions, que se passera-t-il par la suite ? Quand vous aurez l'enfant, comment allez-vous gérer cette situation ? Comment réagira le donneur ? Comment allez-vous réagir ? Comment l'enfant va-t-il réagir dans ce contexte où vous serez ses parents mais où, par ailleurs, il connaîtra l'oncle ou l'ami qui aura donné le spermatozoïde qui aura permis sa conception ? Après ces entretiens, pratiquement tous les couples que j'ai vus ont préféré l'anonymat parce qu'ils ont eu conscience que cela créerait une situation moins complexe, moins risquée pour l'organisation ultérieure de leur famille. (...). Ma réflexion sur le don d'ovocytes est similaire même si la situation est différente dans la mesure où il est moins facile d'exclure la femme stérile de son rôle de mère puisqu'elle porte l'enfant et accouche alors que l'absence du lien génétique et de tout lien charnel avant la naissance peut conduire certains à penser que l'homme stérile n'est pas le « vrai » père de l'enfant. Je suis frappé que l'on puisse envisager de supprimer l'anonymat uniquement parce que le nombre de donneuses d'ovocytes est insuffisant alors que si peu de choses sont faites pour en trouver. L'AMP par don anonyme dépend de la capacité à sensibiliser suffisamment d'hommes et de femmes à donner leurs gamètes ».
Votre Rapporteur considère lui aussi qu'il serait choquant de renoncer au principe d'anonymat, voire de gratuité, pour résoudre le problème de pénurie de gamètes alors que des solutions simples, qui respectent le fondement éthique de nos choix, existent : renforcement de l'information et des actions de sensibilisation en faveur du don, amélioration de la prise en charge des donneurs, suppression de la quarantaine sanitaire des ovocytes... Il partage cet avis avec de nombreux autres membres de la Mission, tel M. Alain Calmat qui soulignait, le 12 juillet 2000, l'incohérence que revêtirait la levée de l'anonymat des enfants nés après AMP vis à vis des autres : « Je trouve qu'il est complètement aberrant de permettre à tous les enfants qui seraient nés par cette technique de pouvoir accéder, de façon certaine, sous prétexte que vous savez quel est le donneur, à leur paternité, alors que l'enfant qui naît en dehors de cette technique n'a pas les moyens de le savoir à 100 %. J'ai entendu le chiffre, qui me paraît absolument monumental, de 17 % des enfants dont le père ne serait pas le père présumé. Dès lors, pourquoi faire une différence entre ceux qui naissent d'un couple qui n'a pas recouru à cette technique et les autres, parce que cela peut aussi poser des problèmes dans les familles si ce droit à connaître ses origines biologiques était reconnu. Le droit de savoir si l'on est le fils de son propre père est, à mon avis, un faux problème. L'anonymat est absolument nécessaire ».
En revanche, la question du secret du mode de procréation mérite d'être posée dans d'autres termes. Les psychologues et psychiatres s'accordent aujourd'hui pour considérer comme pathogène le maintien du secret pour l'enfant voire pour son entourage. M. Jean-Loup Clément indiquait ainsi qu'il recevait fréquemment des appels téléphoniques désespérés de la part de pères ou de mères d'enfants nés par AMP avec tiers donneur : « Des femmes m'ont dit qu'elles n'en pouvaient plus de vivre dans un mensonge imposé depuis vingt-cinq ans par leur mari (...) ».
De plus, comme le soulignait le Docteur Marie-Odile Alnot devant la Mission, l'enfant pressent souvent la vérité et souffre du fait qu'on la lui cache. Elle déclarait : « Je suis favorable à ce que l'on dise à l'enfant la vérité sur sa conception, non seulement s'il y a eu don mais aussi en cas de fécondation in vitro sans tiers donneur. Or, je pense que ce secret est encore très répandu et que les parents ne disent pas facilement à leur enfant qu'il est né à la suite d'une congélation de X années dans des bonbonnes d'azote liquide et que son aîné est en vérité son jumeau ! Les secrets de famille se distillent, souvent les enfants les perçoivent et, à mon avis, ces secrets de procréation, même quand celle-ci résulte des gamètes du couple, ne sont pas dits ».
Il ne peut cependant qu'appartenir aux parents et à eux seuls de décider de la levée de ce secret. Les CECOS, ainsi que l'indiquait à la Mission le Professeur Pierre Jouannet, s'efforcent de sensibiliser les parents à cette question : « Les praticiens des CECOS, pensent que leur rôle est de faire en sorte que les parents aient le choix de dire ou de ne pas dire à l'enfant comment il a été conçu. La plupart d'entre nous pensons qu'il est sans doute préférable d'informer l'enfant mais nous pensons aussi que ce n'est pas à nous d'en décider, que c'est aux parents de faire ce choix. Nous ne devons pas les obliger à révéler à l'enfant son mode de conception s'ils pensent que ce n'est pas possible pour eux ».
Reste à savoir si la société actuelle est assez libérée des préventions longtemps nourries à l'encontre de l'AMP pour créer un environnement favorable à la levée de ce secret. Pour le Professeur Pierre Jouannet, ces préventions sont encore trop puissantes : « De nombreux couples souhaiteraient pouvoir parler avec l'enfant de ses origines. Beaucoup ne le font pas, tant par crainte des réactions de l'enfant que par crainte de celle de l'entourage. Si l'enfant sait qu'il a été conçu par don de gamètes, il est pratiquement inévitable que toute autre personne puisse le savoir. Quelle serait la réaction de la famille, des amis des voisins, des copains à l'école ? Souvent, les parents craignent des réactions, des attitudes négatives à l'égard de l'enfant et préfèrent garder le secret. Ils se trompent peut-être, mais force est de constater que notre société ne fait pas preuve de beaucoup de tolérance et d'ouverture à l'égard de ce mode de procréation. Les commentaires négatifs de certains, les nombreux articles de presse parlant toujours du donneur comme du père et ignorant le « vrai » père, l'homme stérile qui est à l'origine de la conception de l'enfant, ne favorisent pas l'expression des personnes concernées. Plus que celle de l'anonymat, la question du secret de l'origine est importante. C'est celle dont nous parlons avec tous ceux qui choisissent de devenir parents par don. C'est celle qui devrait être mieux appréhendée dans notre société pour être mieux comprise ».
Actuellement, les informations transmises obligatoirement aux couples qui entrent dans une procédure d'AMP à l'occasion de l'entretien préalable avec l'équipe multidisciplinaire ou par le biais du dossier-guide qui leur est remis, ne traitent pas expressément de la question du secret qui relève donc de la pratique individuelle des praticiens ou des psychologues des centres. Il serait sans doute souhaitable d'étudier le meilleur moyen pour que cette question soit systématiquement abordée avec le couple.
IV.- LE TROP FAIBLE CONTRÔLE DES CENTRES D'AMP
Nul ne remet en cause, parmi les interlocuteurs de la Mission, l'opportunité d'un encadrement rigoureux du secteur de l'AMP. Au contraire, nombreux sont les partisans de son renforcement, d'une part, en raison des questions même qui sont en jeu, au centre desquelles se trouve la vie, et, d'autre part, dans la perspective possible de l'autorisation d'une recherche sur l'embryon. Mme Nicole Questiaux, Présidente de la Commission nationale de Médecine et de Biologie de la Reproduction et du Diagnostic prénatal (CNMBRDP), notait ainsi lors de son audition devant la Mission le 12 juillet 2000 :
« Pour le moment, dans notre pays, les parents et les praticiens acceptent la réglementation de ces activités. (...) Il est clair qu'il existe des attitudes (...) différentes à l'égard de ces problèmes dans d'autres pays, poussés vers une plus grande autonomie, un plus grand libéralisme. (...) L'équilibre un peu paternaliste qui est propre à l'organisation de ces activités en France semble, pour le moment, accepté par les gens qui se mêlent de ces questions. Du côté de la Commission, nous n'avons vu aucune suggestion du genre : « Éloignez de nos lèvres ce calice et réduisez cet encadrement. » Tout le monde raisonne même comme s'il était question de le renforcer, avec l'extension de la recherche sur l'embryon » et sans doute, ajoutait-elle, avec les conséquences du « développement quelque peu anarchique de l'ICSI ».
D'aucuns s'interrogent sur la possibilité de mettre en place un encadrement évolutif qui permette de s'adapter aux progrès de la science et à l'évolution des pratiques sans avoir besoin de toujours réviser la loi ou la réglementation. C'est notamment le v_u exprimé par Mme Nicole Questiaux qui déclarait souhaiter que soit trouvée une certaine souplesse permettant, par exemple, de prendre en compte le développement des réseaux de soins : « Il faut se méfier de tout renvoyer au décret en Conseil d'État et essayer de trouver des mécanismes, des procédures et des personnes qui conservent tout de même une petite marge de jeu, non pas sur les droits des personnes, non sur le point de savoir qui a ou non le droit d'accéder à l'activité, mais, par exemple, sur ce que sera, à l'avenir, le bon réseau, la bonne liaison. Par exemple, nous devons accorder actuellement les autorisations à un établissement déterminé et toute la médecine est en train d'évoluer pour fonctionner en réseau. D'ici cinq ans, nous serons gênés parce que la vision géographique des services ne sera plus appréhendée de la même manière ».
Pour le Professeur Pierre Jouannet, il faudrait réfléchir à une nouvelle structure qui serait chargée non seulement d'encadrer le secteur mais aussi d'anticiper sur les évolutions susceptibles de se produire et sur les problèmes qui peuvent se poser : « L'expérience nous montre que dans un domaine aussi complexe et où les besoins sont nombreux, la loi ne peut ni tout régler ni tout résoudre. Elle peut définir les principes, le cadre qui doivent orienter les pratiques mais ce n'est pas suffisant. Il serait nécessaire de créer une structure qui permette de gérer ces activités mais aussi de répondre aux évolutions possibles et aux questions qui apparaissent ».
En tout état de cause, tous s'accordent pour réclamer un changement des règles et des structures qui encadrent les activités d'AMP au regard des faiblesses constatées de la CNMBRDP et du modèle que représente pour beaucoup la HFEA britannique.
A.- LE RÔLE CONFIÉ À LA COMMISSION NATIONALE DE MÉDECINE ET DE BIOLOGIE DE LA REPRODUCTION ET DU DIAGNOSTIC PRÉNATAL (CNMBRDP)
Initialement, c'est un décret de 1988, Mme Michèle Barzach était alors ministre de la santé, qui créa la Commission nationale de médecine et de biologie de la reproduction afin d'éclairer le Gouvernement dans ses choix. L'encadrement des activités d'AMP, qui regroupent des disciplines « jeunes », a en effet été mis en place progressivement en suivant l'évolution des techniques et des problèmes soulevés par leur application. Jusqu'à l'intervention des lois de juillet 1994, l'encadrement réglementaire concernait essentiellement l'AMP homologue. De cette époque, ainsi que le note le Professeur Jacques Montagut, date la distinction entre les activités « cliniques » et les activités « biologiques » et le double mécanisme d'agrément ministériel nécessaire pour exercer ces activités : agrément des établissements et laboratoires et agrément des praticiens qui ont la responsabilité et la compétence de gestion de ces activités.
La loi n° 94-654 du 29 juillet 1994 a repris et précisé ces notions et procédures tout en consacrant le rôle de la CNMBRDP, dont le champ de compétence fut alors étendu au diagnostic prénatal. La Commission est cependant restée un organisme consultatif chargé de donner un avis sur les demandes d'agrément des centres et des praticiens, leur renouvellement ou retrait, et de participer au suivi et à l'évaluation des activités concernées. À ce titre, elle remet chaque année au ministre chargé de la santé un rapport sur l'évolution de la médecine et de la biologie de la reproduction, et sur la politique du diagnostic prénatal en se basant, notamment, sur les rapports d'activité annuels que tous les centres et tous les laboratoires agréés sont tenus de transmettre eux-mêmes au ministre. À la seule exception des études sur l'embryon, qui ne peuvent être entreprises selon les termes de l'article L. 2141-8 du code de la santé publique, qu'après avis conforme de la CNMBRDP, la Commission ne rend que des décisions de nature consultative.
Le tableau ci-après présente les décisions rendues par la Commission durant l'année 1999 par type d'activité d'AMP :
Activité |
Nombre de demandes |
Nombre D'avis favorables |
Avis positifs Nombre de demandes |
Nombre d'autorisations |
Activités cliniques |
||||
*Ponctions d'ovocytes et transferts d'embryons |
17 |
4 |
23% |
3 |
Ponctions de spermatozoïdes |
14 |
7 |
50% |
5 |
Activités biologiques |
||||
Recueil traitement de sperme |
67 |
36 |
53% |
32 |
*FIV sans micromanip. |
18 |
8 |
44% |
7 |
FIV avec micromanip. |
15 |
10 |
67% |
9 |
* Activité soumise à indice de carte sanitaire.
Pour l'essentiel, le ministère chargé de la santé a suivi ces avis. Les avis défavorables de la Commission rendus sur des demandes d'autorisation ou des demandes d'agrément ont en effet toujours été suivis d'un refus du ministre. Quelques avis favorables donnés sur des demandes d'autorisation n'ont pas été suivis lorsque le Comité national d'organisation sanitaire et sociale (CNOSS) avait donné un avis défavorable, estimant que la création d'un nouveau centre ne répondait pas aux besoins de la population. Il s'agissait principalement de désaccords sur quelques dossiers concernant des centres de recueil et de traitement de sperme que le CNOSS ne souhaitait pas multiplier (56).
S'agissant de la composition de la Commission, la loi renvoyait à un décret le soin de la déterminer après avoir précisé qu'elle devait comprendre : « des praticiens désignés sur proposition de leurs organisations représentatives, des personnalités choisies en raison de leur compétence dans les domaines de la procréation, de l'obstétrique, du diagnostic prénatal, du conseil génétique et du droit de la filiation et des représentants des administrations intéressées et des ordres professionnels ainsi qu'un représentant des associations familiales » (article L. 2113-3 du code de la santé publique).
Le tableau ci-après présente la composition exacte de la Commission, établie par le décret du 6 mai 1995, en distinguant ses membres de droit et les membres de ses deux sections : celle du diagnostic prénatal et celle de l'AMP :
Composition actuelle de la CNMBRDP
Les membres des deux sections de la commission |
Les membres de la section du diagnostic prénatal |
Les membres de la section de l'assistance médicale à la procréation |
1° Membres de droit : |
Nommés par arrêté du ministre chargé de la santé, soit sur proposition d'organisations représentatives, soit en raison de leur compétence. |
Nommés par arrêté du ministre chargé de la santé, soit sur proposition d'organisations représentatives, soit en raison de leur compétence. |
· Dans le premier cas : |
· Dans le premier cas : | |
- 98 - - le directeur général de la santé ou son représentant ; - le directeur des hôpitaux ou son représentant ; - le directeur de la sécurité sociale ou son représentant ; - le directeur général de la recherche et de la technologie au ministère chargé de la recherche ou son représentant ; - le directeur des affaires civiles et du sceau ou son représentant ; - le président du Conseil national de l'ordre des médecins ou son représentant ; - le président du conseil central de la section G de l'ordre national des pharmaciens ou son représentant ; - le directeur de la Caisse nationale d'assurance maladie des travailleurs salariés ou son représentant ; - le directeur de l'Institut national de la santé et de la recherche médicale (INSERM) ou son représentant ; |
- deux praticiens ayant une expérience de diagnostic prénatal, choisis sur une liste de six personnes établie par l'Association française pour le dépistage et la prévention des handicaps de l'enfant ; - un médecin choisi sur une liste de trois personnes établie par l'Association des cytogénéticiens de langue française ; - un gynécologue-obstétricien expérimenté en matière de prélèvements sur le f_tus, choisi sur une liste de trois personnes établie par le Collège national des gynécologues et obstétriciens français ; - un médecin choisi sur une liste de trois personnes établie par la Société francophone de médecine f_tale. |
- un gynécologue-obstétricien et un biologiste, choisis sur une liste de trois gynécologues-obstétriciens et de trois biologistes établie par le groupe de la fécondation in vitro en France ; - un biologiste de la reproduction, choisi sur une liste de trois biologistes établie par la Fédération des biologistes des laboratoires d'études de la fécondation et de la conservation de l'_uf ; - deux praticiens, l'un clinicien et l'autre biologiste, choisis sur une liste de six personnes établie par la Fédération des centres d'études et de la conservation des _ufs et du sperme humains. |
Les membres nommés par arrêté du ministre chargé de la santé |
||
- 99 - - un représentant du Comité consultatif national d'éthique pour les sciences de la vie et de la santé, sur proposition du président du comité ; - un représentant des associations familiales, choisi sur une liste de trois personnes établie par le président de l'Union nationale des associations familiales ; - un médecin inspecteur d'une direction régionale des affaires sanitaires et sociales ; - une haute personnalité scientifique ; - un spécialiste de droit de la filiation ; - un praticien ayant une formation ou une expérience particulière en génétique humaine, choisi sur une liste de trois personnes établie par la Société française de génétique humaine. |
· Dans le second cas : - un pédiatre exerçant son activité en maternité ; - deux médecins expérimentés en échographie f_tale ; - deux biologistes ayant une expérience particulière dans la réalisation d'examens de cytogénétique, dont l'un exerce dans le secteur public et l'autre dans le secteur privé ; - une personnalité scientifique choisie en raison de sa compétence dans la recherche en matière de diagnostic prénatal. |
· Dans le second cas : - un médecin choisi en fonction de sa compétence dans le domaine de l'assistance médicale à la procréation avec tiers donneur ; - un épidémiologiste ayant une expérience en médecine de la reproduction ; - un gynécologue-obstétricien et un biologiste d'un établissement de santé public ayant une expérience dans le domaine de l'assistance médicale à la procréation ; - un gynécologue-obstétricien d'un établissement de santé privé ayant une expérience dans le domaine de l'assistance médicale à la procréation ; - un directeur de laboratoire d'analyses de biologie médicale ayant une expérience dans le domaine de l'assistance médicale à la procréation ; - un médecin choisi en raison de son expérience en andrologie ; - une personnalité scientifique choisie en raison de sa compétence dans la recherche en matière d'assistance médicale à la procréation. |
B.- LES FAIBLESSES ET LACUNES DE LA CNMBRDP
Les critiques et autocritiques de la CNMBRDP sont principalement de deux ordres : les critiques concernant l'absence de moyens de la Commission et celles liées à sa composition qui peut mettre en cause son indépendance ou sa nature excessivement technocratique.
Selon notre collègue M. Jean-François Mattei, membre de la Mission qui fut membre de la Commission, la CNMBRDP « n'est pas en mesure d'accomplir son travail pour des raisons de moyens (...). Comme pour les équipes de l'INSERM ou du CNRS, il faut que six ou huit membres de la Commission se déplacent in situ et passent une ou deux journées à s'entretenir avec les responsables, les patients, le personnel technique. C'est seulement ainsi qu'il est possible de se faire une véritable idée de la façon dont les choses fonctionnent. (...) Cette commission ne fait pas son travail en aval, parce qu'elle n'en a pas les moyens, non seulement en termes de secrétariat et de personnel administratif, mais aussi d'experts ».
Le jugement du Docteur Marie-Odile Alnot, qui témoignait devant la Mission en qualité de praticienne d'un CECOS, est encore plus sévère, soulignant avec plus de force le dénuement dans lequel travaille la Commission : « À l'heure actuelle, la Commission (...) n'a aucun pouvoir de contrôle. Elle ne fonctionne que sur les chiffres que nous lui envoyons, or nous pouvons lui envoyer n'importe quels chiffres, nous ne serons jamais contrôlés. Par ailleurs, cette commission ne dispose d'aucun moyen : j'en ai fait partie lorsqu'elle a été créée et nous ne disposions alors d'aucun moyen, pas même d'un secrétariat, mais je crois que mes collègues qui y siègent actuellement vous donneraient le même écho. Elle est incapable de donner des statistiques sur le nombre d'embryons conservés ; seuls les CECOS peuvent donner quelques chiffres. Elle ne donne jamais de chiffres de résultats ».
Le Professeur Claude Sureau témoignait lui aussi de sa déception :
« En tant que membre, je ne suis pas satisfait par les moyens mis à notre disposition. C'est la raison pour laquelle mon collègue Georges David, l'initiateur des CECOS, qui a joué un très grand rôle dans l'évolution en matière de procréation humaine, a démissionné, il y a quatre ou cinq ans, pour manifester son désaccord avec la manière dont cette commission peut fonctionner. »
La Présidente de la Commission, loin de nier le problème, le reconnaissait devant la Mission avec une grande humilité en précisant qu'elle ne dispose, pour réaliser l'ensemble de ses missions, que de trois fonctionnaires : « Ce qui manque, c'est le travail technique très approfondi qui nourrirait la Commission et nous permettrait de ne donner qu'un avis. Très souvent, nous sentons qu'en réalité, grâce au savoir-faire des uns et des autres, nous instruisons, nous ajoutons des éléments et nous essayons d'être justes. Mais moi, qui suis fonctionnaire de métier, quand je pense que je vais renouveler ces autorisations et que notre responsabilité est le suivi de ces autorisations, je vous dirai très franchement que la Commission avant que je la préside n'avait pas les moyens d'un suivi : « c'était zéro ». J'ai personnellement dit au ministère que je ne voulais pas faire les renouvellements sans une campagne spéciale des DDASS (57). On me l'a promise. Je pense donc que j'aurai un avis des DDASS au moins sur une partie des dossiers à renouveler. Mais c'est vous dire que cela ne ressemble en rien à ce que l'on peut imaginer ».
La collaboration des services de l'État, centraux et déconcentrés, fait en effet gravement défaut. S'y ajoute de plus un problème de qualité ainsi que le soulignait le rapport d'activité de la CNMBRDP de 1996 qui constatait : « Lors de l'étude des dossiers de demande d'autorisation, la Commission a fréquemment rencontré des difficultés liées à la mauvaise qualité de présentation des dossiers ainsi qu'à l'insuffisance des renseignements complémentaires apportés par les avis des médecins inspecteurs de santé publique ». Selon la Commission, ces insuffisances sont la conséquence de l'ampleur et du nombre de tâches qui incombent aux DDASS, qui se trouvent dans l'incapacité d'y faire face, et aussi de la formation insuffisante des médecins inspecteurs de santé publique « sur des sujets techniques pour lesquels ils n'ont pas reçu de formation spécifique ». Devant la Mission, Mme Nicole Questiaux déclarait ainsi : « Vous ne pouvez pas demander au ministère de la santé, avec les forces dont il dispose, de dégager le demi-emploi qui serait consacré à la politique de formation de la CNMBRDP alors que tout le monde veut en faire dans ce ministère ! ».
Elle considérait que l' « on n'a pas besoin d'une énorme agence mais d'une structure spécialisée qui ait son propre corps d'inspection. Je comprend très bien que les DDASS qui doivent déjà aller voir les plages, ne puissent faire plus que ce qu'elles font. Où voulez-vous qu'elles trouvent le temps ? Finalement, nous avons envie de vérifier si les rapports avec les parents sont satisfaisants. Pour cela, il faut bien passer un jour sur deux sur place. Il faut regarder comment cela marche. Tout cela, je ne vois pas comment le faire sans être disponible. Très honnêtement, je considère que l'opération de renouvellement (58) que je dois mener à bien - et malheureusement pour moi, la révision des lois ne va pas arriver en temps utile pour ce renouvellement - n'est pas bonne parce que je ne peux pas me fonder sur une connaissance suffisante et, par conséquent, donner un avis ».
Dans ces conditions, on ne peut guère être étonné de certains jugements extrêmement sévères à l'égard de la CNMBRDP, tel celui de Mme Chantal Ramogida devant la Mission qui considérait comme « très mal fait » le travail d'évaluation, d'agrément et de suivi des centres d'AMP : « Les agréments sont accordés sur des dossiers qui n'ont jamais été vérifiés. (...) On accorde des agréments, mais ces médecins ont-ils été contrôlés une seule fois ? Je ne suis pas favorable à des contrôles systématiques, mais la DDASS ne s'est jamais déplacée une seule fois dans un centre pour vérifier sa conformité avec les dispositions de la loi votée il y a six ans. Nous avons signalé à la commission nationale de médecine et de biologie de la reproduction que certains centres mériteraient d'être visités, notamment deux, car nous considérions comme déplacé que ces centres procèdent toujours à des c_lioscopies pour ponctions ovocytaires à une patiente sur deux ».
L'insuffisance manifeste des moyens à la disposition de la CNMBRDP l'empêche également de mener à bien dans des conditions satisfaisantes la mission d'étude et de proposition qui lui a été confiée par la loi sur l'évolution de la médecine et de la biologie de la reproduction et du diagnostic prénatal. C'est le regret qu'exprimait devant la Mission sa présidente actuelle : « Nous tentons donc, « mais c'est du bricolage », de dépasser le jour le jour parce que nous voyons bien qu'il y a là une connaissance du terrain qu'il ne faut pas gaspiller. Mais nous ne sommes pas en mesure de le faire correctement ».
Enfin, la composition de la Commission est elle-même remise en question devant son ouverture insuffisante à la société civile. La loi de juillet 1994 prévoyait bien la présence, en son sein, d'un représentant des associations familiales (59) mais force est de constater que cet unique représentant, « noyé » au milieu des trente-neuf autres membres de la Commission, ne peut à lui seul se faire l'écho de l'ensemble des opinions exprimées au sein de la société et exprimer aussi les préoccupations des patients. C'est d'ailleurs le constat que faisait Mme Nicole Questiaux devant la Mission : « Ce qui manque peut-être, c'est la composante société civile. Nous n'avons qu'un seul représentant des associations familiales. On pourrait vivre avec quelques laïcs, quelques civils plus nombreux, moins de personnalités et plus de personnes du tout venant... ». D'aucuns considèrent que l'appartenance de certains praticiens de centres d'AMP à la Commission porte atteinte à l'impartialité de cette dernière. C'est la critique qu'exprimait devant la Mission Mme Chantal Ramogida. Toutefois, ainsi que le notait notre collègue M. Jean-François Mattei, la présence de ces praticiens apporte une expertise indispensable à la Commission dans des disciplines récentes où les spécialistes ne sont pas légion : « Pour apprécier les compétences techniques, nous tombons dans la difficulté insurmontable des juges et parties car ce sont les praticiens qui les connaissent. Les exclure ne réglerait en rien le problème et me paraît impensable. Peut-être faut-il élargir sa composition comme cela a été fait en intégrant une autre personne en plus de la représentante de l'UNAF. Toutefois, pour l'avoir vécu, j'admets que cela ressemble parfois à une distribution interne à la commission, même si celui dont il est question quitte la salle avant que la commission ne se prononce. Sans porter aucune accusation, quelle qu'elle soit, il est difficile d'être à la fois juge et partie. Il reste néanmoins à trouver des compétences qui ne soient pas directement impliquées dans la pratique, car sinon les personnes n'ont pas le recul nécessaire ».
Selon les informations recueillies par votre Rapporteur, il semble que la pratique des votes au sein de la Commission, dont sont écartés systématiquement les membres qui y auraient un intérêt direct ou indirect, écarte les soupçons de partialité. Il convient cependant de réfléchir à une composition plus équilibrée de la Commission ou de l'organe qui pourrait lui succéder, ainsi que le défendait M. Philippe Pedrot dans son audition devant la Mission le 5 juillet 2000 : « Ces problèmes ne concernent pas seulement la communauté scientifique. Ce n'est pas aux scientifiques seuls de décider mais à la société civile et à ses représentants. La pluridisciplinarité de l'approche de ces problèmes - anthropologique, philosophique, éthique, juridique - me paraît très importante. En définitive, c'est aux politiques, au sens noble du terme, de trancher ».
C.- LA DEMANDE QUASI-UNANIME DE CRÉATION D'UNE AGENCE DOTÉE
DE RÉELS MOYENS POUR SUPPLÉER LA CNMBRDP
Des faiblesses de la CNMBRDP, constatées par toutes les personnalités auditionnées par la Mission, et de la perspective possible d'autorisation de la recherche sur l'embryon est née l'idée de substituer à la Commission une agence indépendante, dotée de moyens et de pouvoirs conséquents et à la composition élargie, sur le modèle de la HFEA britannique, qui fait de nombreux émules. C'est d'ailleurs en substance la proposition présentée par le Conseil d'État dans son rapport, Les lois bioéthiques : cinq ans après, de novembre 1999, en dépit des réserves connues que nourrit l'institution à l'égard de la multiplication des agences. M. Frédéric Salat-Baroux, maître des requêtes, déclarait ainsi devant la Mission le 24 mai 2000 : « Il faut un corps d'inspection, des contrôles et des moyens pour réaliser ces contrôles. Il faut aussi que les responsabilités soient externalisées et clarifiées pour savoir qui est responsable, de la commission qui donne un avis ou du ministre. En France, on s'est toujours interrogé sur les agences. Le raisonnement logique consiste à dire : « Pourquoi faire faire par un démembrement ce que l'État est parfaitement légitimé à faire ? ». L'expérience montre qu'à partir du moment où l'on crée des établissements publics appelés agences, cette logique d'apport de moyens, d'individualisation des responsabilités s'accélère et répond mieux aux besoins. Le Conseil d'État n'a, certes, jamais été favorable à la logique des agences. Mais il nous est apparu, par pragmatisme, qu'une bonne idée pouvait consister, en nous inspirant de la référence anglaise, à créer, dans un premier temps, une agence ayant une compétence en matière d'assistance médicale à la procréation, voire de recherche sur l'embryon, sans s'interdire qu'un jour puisse intervenir un regroupement de diverses agences existantes ».
Mme Nicole Questiaux, elle-même ancien membre du Conseil d'État, exprimait à titre personnel le même avis : « Je suis très fermement favorable à la création de l'agence. Je voudrais bien que cela ne soit pas interprété par le Parlement comme une autocritique de la Commission (60)mais plutôt comme l'expression de notre insatisfaction à sentir que nous avons des atouts, notamment dans cette participation des professionnels, dans le rapport possible avec les parents, et que, par manque de moyens techniques, nous ne pouvons pas créer autour de cette idée de la procréation assistée, de l'embryon, un petit noyau de compétence, d'intérêt, d'information « qui marche ». Alors, je lorgne évidemment du côté des Britanniques. Pourquoi en Angleterre où ils ne sont pas plus intéressés que nous par ces questions ni plus en avance - c'était plutôt nous qui étions en avance sur eux - peuvent-ils se doter d'une telle structure et nous pas ? ».
Pour notre collègue, M. Jean-François Mattei, ancien membre de la CNMBRDP, la création de cette agence serait « la seule façon de dégager un véritable budget, avec du personnel et des moyens budgétaires, comme peut le faire l'agence du médicament. L'ancienne agence française du sang, aujourd'hui des produits de santé, a été très utile en son temps, mais après dix ans d'exercice, elle avait fait la preuve de ses limites et il fallait changer de structure. Peut-être le moment est-il venu de changer de structure ».
Tous les interlocuteurs de la Mission s'accordent sur la nécessité de prévoir une composition large de cette future agence au sein de laquelle la société civile serait suffisamment représentée. Le Professeur Pierre Jouannet, évoquant la prise de position de la Fédération des CECOS en faveur de la révision des lois bioéhiques, soulignait ainsi devant la Mission leur souhait « en faveur d'un établissement ou d'une agence qui (...) serait sous contrôle de l'État mais qui serait, en même temps, indépendante du ministère de la santé. Elle pourrait être constituée d'un tiers de professionnels, d'un tiers de représentants de l'État ou des autorités sanitaires et des institutions, et d'un tiers de patients et de personnes représentatives de la société ».
Le Docteur Marie-Odile Alnot se prononçait également en faveur d'une « agence indépendante, disposant de moyens importants et, surtout, composée de nombreuses personnes émanant de la société civile, car c'est à la société civile de se prononcer sur ce qui est faisable ou interdit ».
On peut se féliciter, à cet égard, que le projet de loi de révision des lois bioéthiques envisage de créer une « Agence de la procréation, de l'embryologie et de la génétique humaines » dotée d'un Haut conseil ou seraient représentés le Parlement ainsi que des associations de malades et d'usagers du système de santé. Il conviendra de veiller à ce que la société civile soit représentée dans ce cadre dans une proportion satisfaisante.
Pour Mme Brigitte Feuillet-Le Mintier, ce sont les capacités propres de contrôle de l'agence qui devraient primer. Elle déclarait devant la Mission : « L'absence de contrôle sur le terrain me semble anormale. Même si l'on crée une agence dotée de plus de pouvoirs, sans contrôle sur le terrain, on en restera aux bavardages et rien ne se fera. La question principale est de savoir comment, concrètement, assurer des contrôles. Mais elle affirmait parallèlement « qu'en aucun cas, cette agence ne devrait être composée en majorité de scientifiques et de médecins ».
À côté de sa composition élargie, de nombreux observateurs préconisent une ouverture de la future agence au public à la fois pour l'informer davantage des activités d'AMP et de diagnostic prénatal, voire demain de recherche sur l'embryon, et dans le même temps pour consulter l'opinion et animer en permanence le débat public, à l'instar de la HFEA britannique. Mme Nicole Questiaux, devant la Mission, considérait cette évolution inévitable et déclarait à ce sujet : « Évidemment, je pourrais me mettre devant mon ordinateur et faire de la formation toute seule. Mais que devient le rapport avec les parents, le rapport avec les praticiens concernés, la manière de les conduire à comprendre qu'il faut plus de formation, la manière de faire participer à notre activité des gens qui seraient en dehors de la commission ? Pour certaines affaires litigieuses, difficiles ? Pour les problèmes de consentement, par exemple ? On imagine quantité de solutions astucieuses qui pourraient être organisées si nous avions les moyens mais vous savez très bien que la simple édition de la plus petite plaquette est toute une affaire ».
Le Professeur Pierre Jouannet, au nom de la Fédération française des CECOS, se prononçait en faveur d'une agence qui ne se contente pas de compter chaque année le nombre d'inséminations ou de FIV mais qui évalue aussi leurs conséquences sur le plan social, culturel ou individuel. Cette agence pourrait, selon lui, « stimuler et ouvrir le débat dans la société quand de nouvelles questions apparaissent (...), s'occuper de l'information et de la sensibilisation du public, (...) proposer des travaux de recherche dans des domaines qui sont particulièrement sensibles ou qui émergent à un moment donné (...) et enfin être l'interlocuteur des médias pour une information de qualité », à une époque où priorité est donnée aux informations « anecdotiques et parcellaires ».
Seule la question des pouvoirs qui seraient confiés à cette nouvelle agence divise les interlocuteurs de la Mission, une grande majorité d'entre eux se prononçant toutefois pour que lui soit transféré le pouvoir de décision en matière d'agrément des centres. Mme Nicole Questiaux évoquait à ce propos la difficulté pour la CNMBRDP de décider dans les faits, puisque aucun des avis de la Commission, en matière d'agrément, n'est dans la pratique remis en cause par le ministre, sans en avoir la responsabilité pleine et entière. Elle déclarait ainsi : « Cette agence serait-elle consultative ou délivrerait-elle les agréments ? Je sais bien que lorsque vous irez devant le Conseil d'État, pour en revenir à lui, il sera plus facile de dire qu'elle ne donne que des avis. Pour ma part, cela ne m'arrange pas tellement de donner des avis suivis à 100 % par le ministre parce que j'aimerais bien savoir, le jour où nous aurons donné les uns et les autres un mauvais avis, à qui sera attribuée la responsabilité de la faute. C'est assez désagréable. En réalité, je sais bien que si je souhaite « barrer » ou « prendre » une personne, en tant que président de la Commission, si je le veux, je le peux. Je n'aime pas le faire, je ne le fais pas, mais je sais aussi que compte tenu des enjeux professionnels, des gens qui ont investi leur vie dans ce secteur... Dans d'autres secteurs de la médecine, vous pouvez vous livrer à de nombreuses activités beaucoup plus dangereuses que celle-là sans passer par ma commission. C'est tout de même quelque chose de très sérieux « d'embêter » ainsi et les familles qui veulent avoir des enfants et les praticiens qui veulent exercer ce métier. Nous sommes peut-être très satisfaits de ce système à la française. Moi, je trouve qu'il n'est pas moderne ».
M. Claude Evin, membre de la Mission, considérait pour sa part, le 12 juillet 2000, que la technicité croissante des matières concernées milite pour une « agence de moyens », véritable « autorité indépendante » dotée d'un réel pouvoir de décision dans un nouveau partage des rôles avec le ministère, qui conserve l'autorité réglementaire : « (...) quant au rapport entre l'agence et le ministère. Pour avoir été à l'origine de la création de cette commission et en regardant les dix dernières années, je peux dire que l'on a beaucoup évolué sur ces sujets (...). Pas seulement dans le domaine de la représentation des usagers (...), mais aussi en ce qui concerne le rapport avec une administration dont la responsabilité doit être une responsabilité de réglementation. Mais devant l'évolution des techniques qui sont de plus en plus complexes, il est évident que les réponses ne pourront pas seulement être des réponses réglementaires ou législatives. Nous avons donc besoin d'une autorité indépendante car il faut qu'elle puisse prendre une décision qui puisse être opposable si nécessaire, et qu'à ce moment-là, elle le fasse en toute autonomie ».
À l'appui du modèle britannique, est souvent présentée par les partisans d'une telle agence l'expérience des deux agences de sécurité sanitaire créées par la loi n° 98-535 du 1er juillet 1998, en particulier l'Agence française de sécurité sanitaire des produits de santé (AFSSAPS) qui a succédé, avec un champ de compétences élargi, à l'Agence du médicament. Cet établissement public placé sous tutelle du ministre chargé de la santé a notamment les pouvoirs de procéder ou de faire procéder « à toute expertise et à tout contrôle technique relatifs aux produits » de santé, de « prendre ou de demander aux autorités compétentes de prendre les mesures de police sanitaire nécessaires lorsque la santé de la population est menacée ». Elle peut aussi fixer elle-même des conditions particulières d'utilisation des produits de santé « afin de garantir leur sécurité sanitaire ». Dans l'exercice de cette mission, la loi donne en outre à l'AFSSAPS le pouvoir de suspendre « les essais, la fabrication, la préparation, l'importation, l'exploitation, l'exportation, la distribution (...), la conservation, la mise sur le marché (...), la publicité (...), la prescription, la délivrance ou l'administration d'un produit (...), lorsque ce produit soit présente ou est soupçonné de présenter, dans les conditions normales d'emploi ou dans des conditions raisonnablement prévisibles, un danger pour la santé humaine (...) ». Elle dispose, à cet effet, de son propre corps d'inspecteurs habilités et assermentés à rechercher et constater les infractions aux lois et règlements applicables.
Il convient enfin de noter que l'ensemble des décisions qui relèvent de la compétence de l'Agence sont prises par son directeur général « au nom de l'État ». Ces décisions, comme le prévoit l'article L. 5322-2 du code de la santé publique, « ne sont susceptibles d'aucun recours hiérarchique. Toutefois, en cas de menace grave pour la santé publique, le ministre chargé de la santé peut s'opposer, par arrêté motivé, à la décision du directeur général et lui demander de procéder, dans le délai de trente jours, à un nouvel examen du dossier ayant servi de fondement à ladite décision. Cette opposition est suspensive de l'application de cette décision ». Il existe donc un « verrou » à la fois juridictionnel, puisque toute décision de l'Agence peut faire l'objet d'un recours contentieux, et politique.
Votre Rapporteur est convaincu que l'AFSSAPS, dont l'expertise et l'impartialité sont reconnues par l'ensemble de ses interlocuteurs, peut servir de référence dans la perspective de création d'une agence de la reproduction humaine. L'expérience a en effet montré que ses pouvoirs sont exercés avec pertinence et modération sans soulever jusqu'à ce jour aucune polémique ni inquiétude, convainquant jusqu'à ses détracteurs comme le remarquait M. Bernard Charles, Président de la Mission, lors de l'audition de la Présidente de la CNMBRDP :
« Même sur la création d'une agence, l'appréciation de l'administration a changé par rapport à ce qu'elle était lors de la création des premières d'entre elles. On avait alors l'impression que, pour l'administration, il s'agissait seulement de structures qu'on lui enlevait. Je me rappelle d'un ministre des affaires sociales, qui n'était pas de la majorité actuelle, qui y était très opposé et qui, deux ou trois ans après, voyant le professionnalisme avec lequel elles fonctionnent, reconnaissait que cela constituait un progrès et marquait une heureuse évolution, que ce soit en termes de technicité, d'indépendance, etc ».
Deux avis divergents se sont cependant exprimés devant la Mission. Le Professeur Pierre Jouannet, au nom de la Fédération française des CECOS, a exprimé la préférence de la Fédération pour la création d'un organisme indépendant « sans pour autant lui confier le pouvoir de donner les agréments aux centres comme c'est le cas en Grande-Bretagne, c'est-à-dire d'avoir une responsabilité de type administratif ».
Le président du Comité consultatif national d'éthique (CCNE), M. Didier Sicard, déclarait pour sa part devant la Mission, le 31 mai 2000, craindre le « pouvoir sans partage » ; selon lui, « il est préférable d'être en situation dynamique, c'est-à-dire que la commission elle-même aurait besoin d'être renforcée, de monter en charge plutôt que de confier un pouvoir de décision à une nouvelle instance. Il reconnaissait cependant que « certains membres du CCNE sont tout à fait favorables à celle-ci. Il peut y avoir un débat tout à fait ouvert sur la possibilité d'une instance apte à juger, rapidement, de l'opportunité de telle ou telle technique ».
D.- L'EXPÉRIENCE CONCLUANTE DE LA HUMAN FERTILIZATION AND
EMBRYOLOGY AUTHORITY (HFEA) BRITANNIQUE
L'étude des compétences et de l'expérience de la HFEA a renforcé l'opinion de la majorité des membres de la Mission, au premier rang desquels votre Rapporteur et son Président, en faveur de la création en France d'une véritable « agence de moyens », autonome, dotée de véritables pouvoirs et à la composition élargie, qui serait chargée de tout le secteur de l'AMP, du diagnostic prénatal et éventuellement de la recherche sur l'embryon, si celle-ci devait demain être autorisée.
La Mission a donc souhaité recevoir, le 5 juillet 2000, des représentants de la HFEA afin de mieux connaître ses modalités de fonctionnement sans pour autant discuter des choix éthiques qui sont à la base de la législation britannique, tels que la possibilité pour des femmes célibataires, homosexuelles ou hétérosexuelles, y compris à un âge avancé (61), de recourir à une FIV, la reconnaissance, sous réserve d'une décision judiciaire, des maternités de substitution ou encore la création d'embryons à des fins de recherche. Il s'agit donc seulement d'étudier plus particulièrement certaines règles dans la composition et relatives au fonctionnement de la HFEA qui pourraient inspirer notre législation.
« L'Autorité » britannique séduit d'abord par sa composition variée et par l'originalité de son mode de recrutement. La volonté du législateur d'outre-Manche de prendre en compte l'opinion publique, dans ses attentes comme dans ses craintes vis-à-vis des progrès de la science, en matière d'AMP, l'a en effet conduit à privilégier une « commission citoyenne ». La HFEA, dont les vingt et un membres sont nommés par le ministre chargé de la santé, doit ainsi représenter l'ensemble de la société civile, respecter la parité entre les hommes et les femmes et ne compter qu'une minorité de personnalités du monde médical ou scientifique. Actuellement, ainsi que l'indiquait devant la Mission la Présidente de la HFEA, Mme Ruth Deech, l'Autorité est notamment composée d'un cadre de la BBC, d'un rabbin, d'un avocat, de trois juristes, d'un philosophe, d'un psychologue, d'une journaliste, d'une actrice, d'un pédagogue, d'un fonctionnaire et d'un théologien. Elle précisait également leur mode de sélection : « Au départ, il y a une dizaine d'années, des chasseurs de têtes s'en occupaient, mais aujourd'hui, le Gouvernement a changé sa politique de recrutement. On fait paraître des annonces dans la presse nationale pour recruter des membres. La dernière fois qu'une annonce a été publiée, nous avons été très surpris de constater que près de 340 personnes avaient posé leur candidature pour quatre postes à pourvoir ».
Parmi les critères de sélection, outre le sexe, l'appartenance géographique et la profession qui doivent dessiner un certain équilibre, sont exigés la compréhension du fonctionnement de l'agence et le respect des principes prévus par la loi, conditions vérifiées à l'occasion d'un entretien avec le candidat organisé par l'administration centrale. Ainsi, comme l'indiquait la Présidente, Mme Ruth Deech : « Une personne opposée à la recherche sur l'embryon et la fécondation in vitro ne pourrait pas être membre de l'agence. (...) Lorsque l'on devient membre de la HFEA, il faut que l'on accepte les dispositions prévues par la loi. Au préalable, nous devons réunir certaines informations sur la personne qui souhaite rejoindre notre agence : si elle travaille dans un organisme gouvernemental, si elle détient des actions dans des entreprises pharmaceutiques, par exemple. En ce qui concerne les opposants à la recherche sur l'embryon, il faut garder à l'esprit que nous travaillons dans le cadre de la loi. Il y a une loi qui autorise la recherche sur l'embryon et la fécondation in vitro. Si un membre de notre agence était opposé à cela, comment pourrait-il travailler avec nous ? Il ne pourrait s'opposer à rien d'illégal ».
Cette ouverture à la société civile est également présente dans les missions confiées à la HFEA d'information, de sensibilisation et de consultation du public. Outre la publication de guides gratuits à destination des patients et des citoyens en général et l'existence d'un site Internet, l'agence s'emploie en effet à nourrir un débat public large et régulier. Ainsi que l'indiquait à nouveau sa Présidente : « Des réunions sont organisées dans les régions. Nous rencontrons les représentants des professions, des groupes d'intérêt et des visiteurs étrangers. Nous consultons le public sur certaines questions, comme le clonage, le diagnostic préimplantatoire ou la rémunération des donneurs. En général, nous suivons ses observations ».
Mme Suzanne Mc Carthy, directrice générale de la HFEA, ajoutait : « Tous les ans, nous organisons une conférence annuelle, des forums, des discussions avec les membres de l'agence, des inspecteurs, des membres de cliniques et d'autres délégués. Des conférences récentes ont ainsi traité du bien-être de l'enfant, du nombre d'embryons devant être transférés par cycle de traitement ou de la prise en compte du point de vue du patient. Depuis 1997, la HFEA a également engagé un programme de réunions régionales qui se poursuit. Le résumé de toutes ces discussions est consultable sur notre site Web. En outre, la HFEA organise des réunions, deux fois par ans, avec les représentants des organisations professionnelles comme la British Fertility Society ou le Royal College of Obstetricians and Gynaecologists. La HFEA a également des contacts avec d'autres organisations, telles que les associations pro vie ».
La consultation du public permet donc d'alimenter la réflexion de l'agence qui en présente les conclusions au ministre, à charge pour ce dernier d'en tirer les conséquences politiques en proposant éventuellement une modification de la loi ou de la réglementation en vigueur. Des consultations ont été ainsi récemment menées sur le clonage, la sélection des sexes, les mères porteuses ou le consentement. Elles permettent de consulter directement le public, « au-delà des médias » comme le soulignait la Présidente Ruth Deech qui précisait le mode d'intervention de l'agence : « Dans certains domaines, nous lançons dans la presse une conférence avec les journalistes et nous distribuons directement aux organisations ou aux individus des milliers de copies de questionnaires que tout le monde peut se procurer, notamment sur notre site Web. Nous organisons des réunions dans les régions et à Londres. Néanmoins, la barrière de la sous-information existe et il faut être attentif aux réponses organisées car la pression des lobbies reste importante ».
Les pouvoirs de la HFEA sont par ailleurs très étendus en matière d'agrément des centres pratiquant les techniques d'AMP, d'autorisation de la recherche sur l'embryon et de contrôle de l'ensemble de ces activités. Sans aller jusqu'à préciser le détail de ses attributions, il est intéressant de noter que les agréments, donnés pour trois ans renouvelables, peuvent à tout moment être suspendus, retirés ou soumis à des restrictions ou conditions. Le Docteur Françoise Shenfield, membre de la HFEA, précisait ainsi devant la Mission : « S'il s'agit d'une nouvelle clinique, la licence est accordée pour un an. (...) En cas de difficulté, la durée de renouvellement de la licence peut être inférieure à trois ans, voire inférieure à un an. Nous avons le pouvoir de donner une licence, de la révoquer complètement ou de changer les conditions sous laquelle elle est donnée. Nous avons également le pouvoir de demander que la personne responsable soit limogée ».
Le taux de renouvellement est cependant très élevé puisqu'il est proche de 98 %. Il arrive toutefois que des autorisations soient brutalement retirées comme l'indiquait la Présidente Ruth Deech qui mentionnait le cas d'un retrait pour traitements inhumains et non-respect des prescriptions relatives au consentement.
Les pouvoirs et les moyens de contrôle sur pièce et sur place de la HFEA sont également considérables. L'agence dispose en effet, ainsi que le précisait le Docteur Françoise Shenfield, de soixante et un inspecteurs dont vingt-quatre sont des cliniciens (médecins pour la plupart spécialisés dans les techniques de FIV), vingt-trois sont des scientifiques (biologistes, spécialistes de la recherche sur l'embryon...) et quatorze sont infirmiers, psychologues, spécialistes des problèmes éthiques ou membres de la HFEA. L'organisation des contrôles surprend par sa rigueur mais aussi par la préférence donnée aux actions préventives sur les actions répressives. Chaque centre d'AMP est de fait suivi par un coordinateur d'inspection - la HFEA en compte huit pour un total de 117 centres - chargé des relations entre ce centre et la HFEA. Ainsi que l'indiquait le Docteur Françoise Shenfield :
« Si le centre rencontre un problème, il peut se mettre en contact avec le coordinateur en dehors de toute inspection. Il est du devoir des coordinateurs, et de très bonne pratique, d'avoir des liens réguliers avec le centre, notamment en lui téléphonant de temps en temps pour savoir si tout va bien. La philosophie du système consiste à identifier et à prévenir les problèmes plutôt que de les gérer dans l'urgence. (...) les inspections se font presque tous les ans. L'inspection principale, quant à elle, se déroule tous les trois ans. À cette occasion, tous les coins et recoins du centre sont visités, depuis l'accueil jusqu'aux laboratoires où les embryons sont congelés. Cette inspection complète doit être menée par un membre de la HFEA, un clinicien, un scientifique, un psychologue et le coordinateur responsable du suivi de ce centre. Les inspections intermédiaires se déroulent en général tous les ans. (...) Au terme de l'inspection, nous devons rédiger un rapport qui sera présenté, avec le rapport de l'année précédente, au comité de licence. (...) Dans la pratique, nous rencontrons toute l'équipe du centre et le responsable de son fonctionnement. (...) L'inspection des dossiers médicaux constitue une phase essentielle : nous avons le droit d'examiner tous les dossiers médicaux, même s'ils sont protégés par une loi de confidentialité. (...) Leur lecture permet de se rendre compte des manquements à la loi ou à l'esprit du code de pratique. Nous disposons d'une protocole d'inspection qui nous sert de guide pour savoir comment les choses se passent dans les laboratoires, comment est organisé le conseil psychologique (...) aux couples. Nous avons aussi à nous assurer que l'intérêt de l'enfant est bel et bien pris en compte ».
Dans ce cadre, comme le soulignaient les représentants de la HFEA, les contrôles inopinés sont rares, l'agence préférant, dans un esprit constructif, « travailler avec le centre, avec les cliniciens et les biologistes plutôt que contre eux ».
S'agissant du régime des décisions prises par « l'Autorité » et de sa responsabilité, il faut noter la pleine responsabilité qu'en assume l'agence devant le Gouvernement et le Parlement. Toutes ses décisions peuvent en outre faire l'objet d'un appel. Sa présidente indiquait à cet égard qu'une grande partie du budget de la HFEA, qui s'élève à près de 1,6 million de livres sterling (62), est consacrée au conseil juridique et au traitement de litiges.
Enfin, il convient de noter une disposition originale qui permet à l'agence de proposer une révision quinquennale de son fonctionnement, ainsi que l'indiquait Mme Suzanne Mc Carthy :
« À cette occasion, nous devons nous assurer que l'agence demeure la solution la plus efficace pour rendre le service qui lui a été confié. Diverses options peuvent être envisagées : la suppression de l'agence ou le maintien du statu quo, la fusion avec d'autres agences, la privatisation ou la sous-traitance. Même si le choix demeure celui du maintien de l'agence, la révision quinquennale offre l'opportunité d'apprécier sa performance par rapport à ses objectifs essentiels et à ses normes de qualité. L'évaluation porte également sur les relations avec les ministères et sur la réponse de la HFEA aux attentes des différents acteurs ».
La deuxième révision quinquennale, actuellement en cours, a ainsi été confiée à trois consultants indépendants qui ont entrepris de réaliser un audit financier et organisationnel de l'agence.
Sur l'ensemble des points évoqués, il y a donc matière à s'inspirer de la HFEA. Il est frappant de noter à cet égard le contraste existant entre les modalités de suivi et de contrôle des activité d'AMP retenues dans la législation française et dans la loi britannique, comme le notait votre Rapporteur lors de l'audition des représentants de la HFEA. En effet, d'un côté, la loi britannique du 1er novembre 1990 a autorisé la recherche sur l'embryon de moins de quatorze jours, mais elle a mis en place un encadrement extrêmement strict des activités d'AMP et de la recherche, sous l'autorité de la HFEA, créée à cet effet. De l'autre, avec les lois bioéthiques de juillet 1994, la France a refusé que cette recherche soit autorisée et s'est contentée d'un dispositif d'autorisation et de contrôle du secteur de l'AMP, dont on peut douter aujourd'hui qu'il soit suffisant.
Notre pays a sans doute beaucoup à apprendre de l'expérience de l'agence britannique qui a fait la preuve de son indépendance et de sa grande compétence.
E.- LES AMÉNAGEMENTS MINEURS NÉCESSAIRES AU DISPOSITIF D'ENCADREMENT DES CENTRES D'AMP
À côté de la réforme d'envergure que constituerait la création, sur le modèle d'outre-Manche, d'une agence chargée de la reproduction humaine, plusieurs révisions des règles d'encadrement des centres d'AMP sont souhaitables.
La première concerne la clarification des critères en fonction desquels les agréments sont attribués aux centres d'AMP. Une certaine confusion semble de fait exister aujourd'hui entre les critères qualitatifs et les critères géographiques, ainsi que l'avouait Mme Nicole Questiaux en sa qualité de Présidente de la CNMBRDP : « Le double emploi entre nous et la carte sanitaire n'est pas raisonnable parce que dans certains cas, on ne peut s'empêcher d'être influencé par l'idée que ce praticien n'est peut-être pas très bon, mais que dans le fin fond de cette province, il faudrait que des gens fassent des kilomètres pour faire une AMP. On empiète alors sur l'appréciation en termes de carte sanitaire. On essaie naturellement de ne pas le faire trop souvent. (...) Quels sont nos cas de conscience ? Placer de petites équipes dans des zones mal desservies où il y aura peu d'activité alors que tout le monde nous dit que pour être au bon niveau dans ces domaines, il faut avoir beaucoup d'activités. Que décider ? Il faudrait que quelqu'un nous aide sur ce point ».
Pour notre collègue Jean-François Mattei, aucun des critères, appliqué strictement, n'est satisfaisant : « Si on se base sur des critères strictement qualitatifs, on risque d'avoir, sur une estimation de soixante centres (un pour un million d'habitants), vingt centres pour la seule ville de Paris et sa couronne, et aucun dans certaines régions comme la Bretagne. Si on se base sur des critères strictement géographiques, on risque de donner l'agrément à des praticiens qui ont des qualités moindres que celles d'équipes auxquelles l'agrément est refusé. Par ailleurs, lorsque le nombre maximal de soixante agréments à donner a été atteint, cela signifie que le système est fermé et que les nouvelles équipes de jeunes, qui ont acquis une expérience, à l'étranger notamment, ne peuvent mettre en place leurs pratiques ».
Il conviendrait donc de réfléchir à une combinaison des critères qualitatif et géographique, le premier devant vraisemblablement l'emporter sur le second pour assurer un respect égal, sur tout le territoire, du principe de précaution. Cette réflexion devrait également conduire à déterminer l'instance ou l'autorité compétente pour décider en dernier ressort de la primauté du critère retenu entre l'actuelle CNMBRDP et le Comité National d'organisation sanitaire et sociale (CNOSS), qui fixe la carte sanitaire. Dans la perspective d'une nouvelle agence de moyens qui se substituerait à la CNMBRDP, votre Rapporteur est favorable à ce que le pouvoir final lui soit confié pour qu'elle assure la cohérence de l'ensemble des agréments tout en en assumant l'entière responsabilité.
Une deuxième révision des règles d'encadrement devrait conduire à autoriser la Commission, ou l'agence qui pourrait lui succéder, à révoquer un agrément qui aurait été donné à un centre avant la fin des cinq ans, durée pour laquelle l'agrément est donné selon la loi de juillet 1994, si l'activité ou les résultats de ce centre sont insuffisants, à l'instar des maternités. Cette disposition permettrait, ainsi que le notait le Conseil d'État dans son rapport, Les lois de bioéthique : cinq ans après, de renouveler le parc actuel des centres et laboratoires, aujourd'hui figé, tout en donnant un poids accru aux contrôles de ces établissements. C'est aussi l'enseignement que l'on peut retirer de l'exemple britannique évoqué précédemment.
Une troisième modification pourrait concerner le système actuel d'agrément de praticiens « responsables » pour chaque centre agréé. La CNMBRDP, dans son rapport d'activité pour les années 1997 et 1998, soulignait les inconvénients des situations fréquentes où plusieurs praticiens sont agréés pour le même centre sans que le nombre de ces agréments soit proportionnel au volume d'activité de celui-ci. De même, il arrive également que deux praticiens de même compétence ne puissent être agréés en même temps afin de limiter le nombre de responsables. Pour résoudre ces difficultés, la Commission s'est prononcée en faveur d' « un agrément individuel délivré non plus pour être responsable de l'activité mais pour effectuer tout acte d'AMP soumis à autorisation. L'agrément serait donc lié à la qualification du praticien, à son expérience et au nombre de praticiens agréés en rapport avec le volume d'activité du centre ».
Enfin, il y a lieu certainement de s'interroger sur la pertinence de la distinction, dans la réglementation actuelle, entre activités cliniques et activités biologiques d'AMP qui rend difficile la constitution d'équipes pluridisciplinaires imposée par la loi, ou du moins qui ne facilite pas un véritable travail d'équipe pourtant indispensable afin de remettre le patient ou la patiente au centre du système de soins.
TROISIÈME PARTIE : LA RECHERCHE SUR L'EMBRYON
La question de la recherche sur l'embryon est sans doute « la question » fondamentale qui est aujourd'hui posée au législateur à l'occasion de la révision des « lois bioéthiques ». Elle mérite donc que lui soit consacrée une partie exclusive, sachant qu'existent d'autres pistes de recherches concurrentes n'impliquant pas l'embryon ou ses cellules qui devront être examinées parallèlement sans a priori. Il appartient donc à la Représentation nationale de retenir certaines pistes de recherche et, éventuellement, d'en refuser d'autres, ainsi que de fixer, à l'égard des premières, leur périmètre et l'encadrement auquel elles seront soumises.
Le débat sur l'autorisation ou l'interdiction de la recherche sur l'embryon in vitro n'est pas nouveau puisqu'il était déjà ouvert lors de la préparation des lois de bioéthiques adoptées en juillet 1994. Il a cependant totalement changé de nature. À l'époque, plusieurs scientifiques et praticiens, tel le Professeur Charles Thibault (63), défendaient déjà l'opportunité de cette recherche autour de cinq projets :
- mieux connaître les causes de la mortalité embryonnaire humaine, particulièrement élevée, à l'inverse de ce que l'on constate dans la procréation animale, puisqu'elle est proche de 60 % ;
- comprendre pourquoi l'embryon humain peut s'implanter dans la trompe et provoquer ainsi des grossesses tubaires qui sont à l'origine de graves accidents, ce qui constitue une autre spécificité de l'espèce humaine ;
- rechercher de nouvelles voies contraceptives par inhibition de la fécondation en agissant sur les gamètes ;
- étudier l'origine des anomalies phénotypiques du spermatozoïde humain ;
- améliorer les techniques d'intervention à but médical telles que les conditions de culture ou de congélation des embryons in vitro, les nouvelles méthodes de fécondation ou de diagnostic.
Depuis, les progrès de la thérapie génique et cellulaire, les découvertes sur le pouvoir des cellules souches embryonnaires et adultes et la naissance des premiers mammifères clonés ont bouleversé l'état des connaissances scientifiques et ouvert des perspectives jusqu'alors insoupçonnées. Il y a eu, de l'avis même des scientifiques, un véritable « effet Dolly » qui a stimulé la recherche fondamentale en lui ouvrant des perspectives de découvertes tout à fait nouvelles, hier encore inimaginables. Il est ainsi aujourd'hui envisagé de guérir des maladies graves ou fortement invalidantes, dont certaines sont à ce jour incurables, qui frappent tous les stades de la vie, de l'enfant atteint d'une maladie génétique grave à la personne âgée touchée par une maladie neurodégénérative. La recherche sur l'embryon, qui était jusqu'alors cantonnée au secteur de l'AMP, est subitement devenue un enjeu scientifique majeur intéressant l'ensemble de l'humanité et porteuse de tous les espoirs, y compris les plus fous, avec des incidences économiques considérables. Comme le notait le Professeur Claude Sureau, Président de l'Académie nationale de médecine, devant la Mission, lors de son audition du 12 juillet 2000, on assiste à un véritable « changement de décor »: « La société civile française se découvre brutalement un intérêt considérable pour les cellules embryonnaires. (...) D'un problème de procréation humaine qui n'intéressait pas grand monde, on passe à un problème de thérapeutique (...) qui intéresse tout le monde ». Il reste que la recherche sur l'embryon, qui est « la grande question » posée aujourd'hui au législateur, pourrait, si elle était autorisée, être appliquée aussi dans le but d'améliorer les techniques et les résultats de l'AMP et par là même permettre de faire progresser la médecine embryonnaire.
Dès lors, devant l'importance des enjeux et des perspectives qui sont ouvertes par les avancées de la science et les questions qu'elle pose qui restent encore en suspens, il appartient au législateur d'examiner les possibilités qui s'offrent aujourd'hui en s'attachant à préserver le fragile équilibre qu'il a toujours essayé de ménager entre les valeurs éthiques auxquelles nous sommes attachés, l'évolution des données sociales et les possibilités nouvelles offertes ou promises par la science, susceptibles de sauver des vies, de guérir ou d'améliorer les conditions d'existence d'un grand nombre de malades. S'agissant de la recherche sur l'embryon in vitro, force est de constater que le choix retenu en 1994 n'est plus adapté.
I.- L'INADÉQUATION DE LA LÉGISLATION DE 1994
Le compromis atteint par le législateur en 1994 traduit, par son ambiguïté, les controverses suscitées à l'époque autour de la question du statut et de la protection adéquate de l'embryon.
Ainsi que le constatait devant la Mission le Docteur Axel Kahn, le 7 juin 2000, « le législateur (...) voulait ne pas fermer complètement la porte à des études ne portant pas atteinte au développement de l'embryon et bien marquer son souci du respect de la vie humaine dans l'embryon ».
Cette attitude l'a conduit à adopter une position ambivalente que d'aucuns, tel M. Philippe Pedrot, juriste, auditionné par la Mission le 5 juillet 2000, ont pu qualifier d' « hypocrisie positive ». D'un côté, le législateur a, en effet, clairement prohibé toute « expérimentation » sur l'embryon à l'article L. 2141-8 du code de la santé publique qui interdit également la conception in vitro d'embryons « à des fins d'étude, de recherche ou d'expérimentation ». De l'autre, il a permis que soient menées « à titre exceptionnel (...) des études » sur les embryons à la condition que celles-ci aient une « finalité médicale » et qu'elles ne portent pas « atteinte à l'embryon », sous réserve du consentement écrit des géniteurs et de l'avis conforme de la CNMBRDP.
Parallèlement, l'article L. 2131-4 du même code permet la pratique du diagnostic préimplantatoire (DPI) sur les embryons des couples ayant une « forte probabilité de donner naissance à un enfant atteint d'une maladie génétique d'une particulière gravité reconnue comme incurable au moment du diagnostic ».
Ainsi, la recherche est interdite mais l'étude est autorisée sans que l'on sache précisément ce qui distingue ces deux notions, comme le notait le Docteur Axel Kahn qui soulignait devant la Mission l'absence de « différence sémantique entre études et recherches ». Pour M. Jean-Paul Renard, directeur de recherches à l'Institut national de recherches agronomiques (INRA), auditionné par la Mission le 12 juillet 2000, cette distinction n'est pas fondée : « Il faut préciser un point de vocabulaire qui fait problème dans la définition de la loi de 1994, à savoir la distinction entre « études » et « recherches » sur l'embryon. L'emploi du mot « études » renverrait au fait qu'on n'intervient pas sur l'embryon, alors que celui de « recherches » signifierait qu'on le manipule physiquement. Mais on peut, simplement en modifiant le milieu de culture affecter le développement et beaucoup plus tard ! Cette distinction est à mon avis mal fondée ».
Par ailleurs, force est de constater que la condition selon laquelle les études qui seraient autorisées ne peuvent porter atteinte à l'embryon est contradictoire avec l'autorisation du DPI qui a pour conséquence d'éliminer ceux des embryons dont le diagnostic aura été positif, afin de sélectionner le ou les embryons non porteurs de la maladie congénitale recherchée, ce qui n'est pas le moindre des paradoxes.
La CNMBRDP elle-même, dans son rapport d'activités pour les années 1997 et 1998, considère qu'« il existe une contradiction entre le fait que la loi autorise le diagnostic préimplantatoire et interdit la recherche sur l'embryon. En effet, la technique du DPI avant d'être appliquée doit être expérimentée afin de ne pas nuire aux embryons soumis au diagnostic ; elle nécessite donc au préalable une recherche sur l'embryon ».
Dans la pratique, la condition de non-atteinte à l'embryon vide de tout effet la possibilité d'études sur l'embryon autorisées. Elle semble en effet vouloir signifier que l'embryon doit toujours pouvoir être implanté après l'étude menée et poursuivre son développement normal. C'est l'interprétation que donne en effet le décret n° 97-613 du 27 mai 1997 relatif aux études menées sur des embryons humains in vitro et modifiant le code de la santé publique qui précise qu'« aucune étude ne peut être entreprise si elle a pour objet ou risque d'avoir pour effet de modifier le patrimoine génétique de l'embryon, ou est susceptible d'altérer ses capacités de développement ».
Pour le Docteur Axel Kahn, cette condition démontre le caractère inopérationnel du dispositif : « Pour savoir s'il n'est pas porté atteinte au développement de l'embryon, encore faut-il le remettre dans le ventre d'une femme et regarder dans quel état sera, après ces études, le bébé qui naîtra de cet embryon. J'interprète cette loi comme disant : « avec l'avis des géniteurs, des études peuvent être permises, mais le développement doit se poursuivre et, ma foi, on appréciera le succès ou l'insuccès de l'étude à l'état du bébé qui naîtra ». Ce qui naturellement est quelque chose qui ne peut, sous cette forme, persister ».
Cette rédaction interdit même que soient tentées des études portant, par exemple, sur les milieux de culture de l'embryon alors que l'amélioration de ces milieux pourrait améliorer sa viabilité. C'est ce que regrettait le Professeur Claude Sureau lors de son audition précitée devant la Mission, en déclarant : « Si dans « études ne portant pas atteinte à l'embryon », vous incluez des études portant sur les milieux de culture, c'est également contradictoire. Ces études apparemment anodines peuvent parfaitement porter atteinte à l'embryon, mais on ne peut le savoir qu'a posteriori. C'est un des arguments qui nous fait militer en faveur de la recherche sur l'embryon ».
C'est aussi la critique qu'exprimait M. Jean-Paul Renard, dans son audition précitée devant la Mission, qui démontrait l'impossibilité de conduire des recherches sur les milieux de culture des embryons, en dépit de leur intérêt certain : « Les travaux avec l'embryon de souris ont montré qu'occasionnellement, en fonction du milieu de culture, mais pour des facteurs que l'on ne connaît pas, des effets pouvaient se manifester très tardivement dans le développement. On n'y avait pas prêté énormément attention jusqu'au clonage. Avec le clonage, on intervient sur l'embryon et on exacerbe ces effets. On sait donc maintenant que le milieu de culture a une énorme importance et que l'épigénèse existe dès le début du développement.
La loi met d'emblée l'accent sur des différences qui existeraient entre études et recherches. Mais il y a une réelle difficulté pour nous, scientifiques, à faire la distinction entre les deux. Car si des « études », par exemple l'exposition à un nouveau milieu de culture, peuvent être réalisées sur l'embryon humain, on sait aujourd'hui, avec les données chez la souris, qu'elles peuvent avoir des conséquences très importantes à long terme, non seulement sur les organismes issus de ces embryons, mais aussi peut-être sur leurs descendants ! D'où la nécessité de commencer par mieux comprendre la nature de ces phénomènes chez l'animal. Il existe aujourd'hui des pistes, mais elles restent assez floues ».
La mise en culture des cellules souches embryonnaires (cellules « ES » en référence au terme anglais « embryonic stem cells ») est également impossible comme le regrettait devant la Mission, le 28 juin 2000, M. Gilles de Poncins, directeur général d'une entreprise de biotechnologies : « Mettre un embryon en culture est impossible. En revanche, prélever deux cellules pour pratiquer un diagnostic préimplantatoire est possible. C'est permis par la loi. Mais mettre un embryon en culture pour obtenir des cellules ES est impossible. Ou même le mettre en culture, sans milieu de culture, pour qu'il meure est impossible ».
En réalité, c'est au cours des différentes navettes du projet de loi que cette disposition a perdu son sens, la rédaction initiale interdisant de « porter atteinte à l'intégrité génétique » de l'embryon, ce qui avait un tout autre sens puisqu'il s'agissait alors d'interdire la manipulation du patrimoine génétique de l'embryon.
S'agissant de la seconde condition posée à l'autorisation des études sur l'embryon, et relative à leur finalité, le décret précité définit strictement deux finalités possibles : soit présenter un avantage direct pour l'embryon concerné, notamment en vue d'accroître les chances de réussite de son implantation, soit contribuer à l'amélioration des techniques d'assistance médicale à la procréation, notamment par le développement des connaissances sur la physiologie et la pathologie de la reproduction humaine.
Le dispositif manque cependant de clarté s'agissant du moment à partir duquel l'étude est autorisée. On peut, en effet, considérer qu'il y a embryon dès qu'un spermatozoïde pénètre tout ou partie des enveloppes ovocytaires ou alors, comme la majorité des scientifiques, dès que le mélange des chromosomes paternels et maternels aboutissent à la constitution irréversible du génome propre à l'embryon. Si l'on admet cette dernière hypothèse, des recherches sur la fécondation sont possibles dès lors qu'elles s'arrêtent au stade du zygote où les noyaux paternels et maternels dans l'ovule n'ont pas encore fusionné. Dans le cas contraire, il faut admettre que très peu d'études seraient possibles. Une étude sur l'ICSI avant la fusion des noyaux serait de fait interdite puisqu'elle conduirait à mettre en présence les deux noyaux avant leur fusion.
Enfin, le caractère « exceptionnel » des études pouvant être autorisées n'est pas explicite. Aucune indication n'est donnée, y compris dans le décret d'application précité, sur les critères qui justifient l'exception, ni sur la ou les personnes qui en décident, privant ainsi de toute portée cette disposition.
Force est donc de reconnaître l'ambiguïté et l'inadéquation du dispositif actuel qui semble ne satisfaire personne tant d'un point de vue juridique que scientifique.
Ainsi que l'indiquait Mme Nicole Questiaux, Présidente de la CNMBRDP, devant la Mission le 12 juillet 2000, seulement onze demandes d'études sur l'embryon ont été déposées devant la commission, ce qui illustre le champ très restrictif du cadre actuel et son intérêt limité pour les chercheurs. Sur ces onze demandes, cinq projets ont été retenus et sont en cours avec les thèmes d'étude suivants :
- mise au point des techniques nécessaires à la pratique du diagnostic préimplantatoire (Hôpitaux universitaires de Strasbourg) ;
- étude sur les facteurs de blocage cytogénétique et moléculaire du développement embryonnaire préimplantatoire (Hôpital Antoine Béclère en région parisienne /Laboratoire MÉRIEUX) ;
- mise au point des techniques nécessaires à la réalisation du diagnostic préimplantatoire (Centre hospitalier universitaire de Montpellier) ;
- apoptose au cours du développement embryonnaire préimplantatoire : détection, régulation (Centre hospitalier régional E. Herriot à Lyon ) ;
- étude de la méthylation et de l'expression du gène « BRCA1 » au cours de l'évolution du zygote à la morula (Centre hospitalier régional E. Herriot à Lyon ).
Le Professeur Pierre Jouannet, Président de la Fédération française des CECOS précisait devant la Mission, le 12 juillet 2000, que pour permettre les études sur l'embryon, près de 500 d'entre eux ont été donnés par des couples qui ne souhaitaient plus poursuivre leur projet parental.
II.- DES PERSPECTIVES DE RECHERCHE IMMENSES
De récentes découvertes en matière de génie cellulaire et certaines expérimentations sur des mammifères, au premier rang desquelles le clonage de la brebis « Dolly » par l'équipe de Ian Wilmut de l'Institut Roslin d'Edimbourg en février 1997, sont venues bousculer les certitudes acquises et ont ouvert un nouveau champ de recherche insoupçonné il y a encore quelques années par la communauté scientifique elle-même. Se pose ainsi avec acuité le problème de la levée de l'interdiction, dans la législation française, des recherches sur l'embryon et des limites de ces recherches. Comment les définir ? Sur quels choix doit-on se fonder ? Quels principes sont intangibles ? Comment les concilier ? Quel cadre poser à cette recherche ? Quel contrôle mettre en place ?
Les difficultés sont d'autant plus grandes que l'on se trouve dans le domaine du possible, les pistes de recherche s'ouvrant devant nous n'offrant à ce jour que des promesses qui doivent encore être scientifiquement validées pour l'homme.
On est donc confronté au dilemme suivant : soit l'on ne ferme aucune piste de recherche, à l'exclusion du clonage reproductif, unanimement rejeté et condamné, afin de valider l'une ou l'autre et d'abandonner celles qui s'avéreraient décevantes,
- c'est ce que suggérait le Premier ministre Lionel Jospin lors de son allocution devant le CCNE le 28 novembre 2000 -, soit l'on ferme dès aujourd'hui certaines de ces pistes tout en sachant que ces recherches sur l'embryon sont d'ores et déjà en cours à l'étranger.
Le tableau ci-après montre en effet que certains pays autorisent d'ores et déjà la recherche sur l'embryon à des degrés divers et dans la poursuite de différentes finalités.
Quelle attitude adopter alors si les recherches sur l'embryon conduites à l'étranger devaient effectivement aboutir à la mise au point de traitements thérapeutiques innovants ? C'est la question que posait notamment M. Gilles de Poncins, au cours de son audition précitée devant la Mission : « Si l'on refuse d'utiliser les cellules ES humaines, il faut aller jusqu'au terme du raisonnement : quelle réponse apporte-t-on aux médicaments développés ailleurs avec ces technologies ? Quelle réponse apporte-t-on aux thérapies basées sur ce type de cellules ? Si l'on interdit d'utiliser en France ces nouveaux outils, que fera-t-on, dans douze ans, quand Geron (64) offrira des progéniteurs pour soigner les gens victimes d'une leucémie ? La cohérence d'ensemble des choix à faire se trouve là. Par sa portée, ces choix visent plus que d'autoriser ou non les cellules ES ou les cellules souches ».
La grande majorité des personnalités entendues par la Mission sont favorables à une certaine ouverture de la recherche sur l'embryon à la condition que celle-ci soit strictement encadrée et contrôlée. Plusieurs institutions, telles l'Académie nationale de médecine et le CCNE, se sont prononcées fortement en faveur de cette recherche en s'appuyant sur les perspectives thérapeutiques immenses qu'elle ouvrirait. Deux interlocuteurs ont cependant exprimé leur opposition à cette évolution. Pour le Professeur Jacques Testart, Directeur de recherches à l'INSERM (65), auditionné par la Mission le 31 mai 2000, le projet est prématuré et il conviendrait de poursuivre les recherches sur l'animal : « Même si des perspectives nouvelles de traitement apparaissent et qu'elles s'avèrent autrement motivantes que les vagues projets « de recherche » énoncés il y a quelques années, l'implication des embryons humains dans ces programmes semble prématurée. D'une part, il serait éthiquement inacceptable de ne pas procéder préalablement à des essais à partir d'embryons animaux, et en particulier de primates, unique façon de montrer la faisabilité de l'entreprise et de dégrossir les procédures ».
Le Professeur Arnold Munnich, également Directeur de recherches à l'INSERM (66), auditionné par la Mission le 6 septembre 2000, estimait lui aussi qu'il est trop tôt pour autoriser la recherche sur l'embryon devant l'incertitude des résultats espérés, et considérait que le débat doit être poursuivi au niveau national avant de prendre une décision en ce sens : « Il faut (...) aller au bout de la logique et en réalité, à mon sens, on ne peut pas s'engager dans l'autorisation de la recherche sur l'embryon sans ipso facto avaliser demain les bénéfices thérapeutiques de ces recherches : ce n'est pas possible, vous n'y arriverez pas ! La pression de l'opinion, des médias, des compagnies d'assurances et du grand capital vous en empêcheront ! En conséquence, il faut, selon moi, garder à l'esprit que les situations sont changeantes à tout instant et la communauté nationale s'honorerait peut-être à différer cette décision au motif qu'on ne peut pas statuer et qu'il faut encore débattre. Il y a des forums citoyens, des rendez-vous : il faut donc parler et discuter tous ensemble sur ce qu'est un embryon...
(...) Il ne faut pas s'arc-bouter sur l'énoncé du problème d'aujourd'hui car on peut se retrouver pris à contre-pied par un énoncé totalement différent demain. Je préférerais que l'on encourage la recherche sur les cellules souches totipotentes, parce que là nous nous trouvons en réalité, vous comme moi, devant une situation inédite. En matière de dons d'organe, on n'a jamais vu qu'un don remette en cause l'existence du donneur : quand une mère donne un rein à son fils qui souffre d'une insuffisance rénale, cette mère continue de vivre ; quand un frère donne sa moelle à son petit frère qui souffre d'une leucémie, cela ne remet pas en cause l'existence du donneur ! Pour ce qui est des embryons, nous nous trouvons dans une situation complètement inédite puisque, en réalité, elle implique la suppression de l'existence ou de l'existence potentielle de la vie ».
Parmi les personnalités nombreuses qui se sont exprimées devant la Mission, favorables à l'ouverture de la recherche sur l'embryon, des voix se sont, en revanche, élevées à l'encontre de la méthode dite du « clonage thérapeutique » qui soulève certaines inquiétudes et interrogations. Il nous appartient donc d'évaluer chacune des pistes de recherche qui s'ouvrent aujourd'hui, d'en mesurer les risques mais aussi les promesses afin de choisir celles qui paraissent, en l'état des connaissances scientifiques actuelles et dans le respect des valeurs qui nous sont communes, les plus porteuses d'espoirs pour les générations futures.
A.- DES PISTES DE RECHERCHES NOMBREUSES
On ne rappellera que brièvement l'existence d'une recherche sur le clonage reproductif, tant celui-ci fait l'objet d'une condamnation unanime et sans appel en France et au sein des grandes organisations européennes ou mondiales.
On ne peut cependant ignorer que cette méthode, appliquée à ce jour avec succès sur des brebis et des porcs, est considérée malheureusement par quelques « scientifiques marginaux » comme une piste de recherche sérieuse qui devrait être appliquée à l'homme. Il s'agirait soit de permettre la naissance d'être humains clonés, soit de constituer des réserves de cellules, de tissus, voire d'organes parfaitement immuno-compatibles à destination d'un individu qui bénéficierait ainsi d'un véritable « kit de réparation génétique (67) ». Comme le soulignait déjà le CCNE dans son avis n° 54 du 22 avril 1997 sur le clonage reproductif, « on ne peut probablement pas exclure qu'existe un courant social tendant à légitimer le recours à ces techniques, au moins dans la perspective de couples dont l'un des conjoints ne posséderait pas de gamète fécondant. D'ailleurs, le clonage pourrait aussi répondre au désir d'une femme sans gamète de se perpétuer biologiquement par autoclonage, en utilisant des ovocytes énucléés d'une donneuse. Plus récemment, certains commentateurs ont évoqué, dans la revue Nature, la reproduction par clonage d'un enfant mort ou sur le point de mourir. Des déclarations faites dans des cadres variés ont également envisagé la reproduction par ces méthodes de personnes « exceptionnelles »..., d'être chers... ».
Dès lors, la question qui se pose au législateur est de décider ou non de l'interdiction explicite du clonage reproductif humain à l'occasion de la révision des lois bioéthiques. Juridiquement, cette interdiction existe implicitement avec l'article 16-4 du code civil qui prohibe toute « atteinte à l'intégrité de l'espèce humaine ». Toutefois, votre Rapporteur considère, à l'instar du Conseil d'État dans son rapport, Les lois de bioéthique : cinq ans après, qu'il serait éminemment souhaitable de condamner expressément le clonage reproductif dans la loi, comme l'envisage d'ailleurs le Gouvernement britannique.
1.- Le pouvoir des cellules souches
Avant d'étudier en détail chacune des pistes de recherches qui s'offrent à nous aujourd'hui, il convient de rappeler brièvement le pouvoir des cellules souches en général, pouvoir qui laisse augurer des découvertes et des applications extrêmement prometteuses.
Une cellule souche se caractérise par sa capacité de prolifération. Mise dans un environnement tissulaire approprié, elle est en effet capable de se multiplier en cellules spécialisées d'après la morphologie et la fonction spécifique du tissu, selon un processus irréversible dit de « différenciation ». Une cellule souche peut donc être à l'origine d'une multitude de cellules qui peuvent être de nature très différente : cellules nerveuses, sanguines, osseuses, musculaires, pancréatiques, épidermiques... Ce pouvoir de différenciation varie cependant selon la qualité ou le stade de développement de la cellule souche.
Une cellule souche « totipotente » a ainsi la capacité de conduire à la formation de tous les tissus du corps humain, y compris ceux de la lignée germinale. C'est le cas des cellules souches embryonnaires jusqu'à 2 à 3 jours après la fusion des gamètes, soit jusqu'au stade de la troisième division de l'_uf fécondé qui donne huit cellules. Une seule cellule de l'embryon a alors la possibilité de donner à elle seule un embryon viable, normal, fertile et apte à se reproduire.
Au stade suivant, une cellule souche « pluripotente » a la possibilité de se différencier dans tous les tissus à l'exclusion des cellules de la lignée germinale. C'est le cas des cellules embryonnaires présentes dans le blastocyste, soit six jours après la fusion ; on compte alors dans le « bouton embryonnaire », qui donnera le futur embryon, quarante cellules pluripotentes qui vont poursuivre encore leurs divisions.
La cellule souche « multipotente », présente dans l'organisme adulte, n'a pour sa part qu'une capacité limitée de se différencier en plusieurs types de cellules déterminées ; elle est en quelque sorte préprogrammée pour la production de certaines cellules à l'instar des cellules souches mesenchymateuses, présentes dans la moelle osseuse, aptes à produire des cellules osseuses, cartilagineuses et peut-être musculaires et des cellules souches hématopoïétiques, également présentes dans la moelle osseuse, qui produisent toutes les cellules sanguines.
Enfin, la cellule souche « unipotente » ne peut produire qu'un seul type de cellules, comme la cellule de l'épiderme qui ne produit que des kératinocytes. Le schéma ci-après tente d'illustrer cette brève présentation
 |
_ · |
_ _ _ _ _ _ (a) |
· (b) |
1 cellule indifférenciée _ |
pouvant produire une quantité illimitée de cellules, chacune pouvant redevenir un embryon, pouvant donner toutes les cellules possibles, y compris germinales | ||
 |
_ · |
· _ (...) | |
1 cellule indifférenciée _ |
pouvant donner toutes les cellules à l'exclusion de celles de la lignée germinale sans reversibilité | ||
 |
_ · |
· _ | |
1 cellule indifférenciée ne pouvant donner que quelques types de cellules prédéterminées | |||
UNIPOTENCE |
_ · |
· · · · ... | |
1 cellule ne produisant qu'un seul type de cellules différenciées | |||
(a) Autres types de cellules indifférenciées. (b) Et autres types de cellules différenciées. | |||
2.- Les cellules souches embryonnaires
Au quatrième jour suivant la fécondation, l'ovule fécondé, appelé alors blastocyste, est composé de seize cellules pluripotentes. Son feuillet externe donnera naissance au placenta tandis que l'ensemble des cellules de sa masse interne, rassemblées dans le « bouton embryonnaire », donneront les feuillets embryonnaires
- mésoderme, endoderme et ectoderme (68) - à l'origine de l'embryon. In vivo, le blastocyste commence son processus de nidation dans l'utérus de la mère qui a lieu, dans l'espèce humaine autour du septième jour. Lorsqu'il se pose alors sur la membrane utérine, il se libère de sa membrane extérieure pour s'implanter dans la muqueuse tandis que les cellules du bouton embryonnaire, qui ont poursuivi leurs divisions, entament leur processus de différenciation. C'est alors que se développent les premiers échanges hormonaux entre la mère et le blastocyste, début de la grossesse.
À ce stade, les cellules souches embryonnaires (ES) ont donc perdu leur totipotence, ainsi que le soulignait le Docteur Françoise Shenfield, membre de la HFEA britannique, devant la Mission le 20 septembre 2000 : ces cellules « ne pourront donner un enfant puisque si l'on sépare les cellules de « la coquille externe » qui permet à l'embryon de s'accrocher à l'utérus, la masse interne perd tout potentiel de devenir un enfant ». En revanche, une fois le blastocyste dissocié, les cellules ES qui en sont extraites peuvent être cultivées en laboratoire à l'infini tout en conservant leur caractère pluripotent et en conservant un génome intact. C'est ce qu'indiquait devant la Mission le Docteur Axel Kahn dans son audition du 7 juin 2000 : « Lorsque l'on met ces cellules [les cellules de bouton embryonnaire] en culture, suivant les conditions de culture, on peut soit les amener à se diviser sans se différencier, en restant telles qu'elles sont, telles qu'on les a prélevées, ou alors les amener à se différencier pour donner des cellules du cerveau, du foie, de la moelle, de la peau, du pancréas, etc ».
En novembre 1998, deux équipes universitaires américaines ont pour la première fois isolé et mis en culture des cellules souches de l'embryon humain à partir de blastocystes, la recherche privée sur l'embryon n'étant pas interdite aux États-Unis.
Immédiatement, des perspectives thérapeutiques susceptibles de bouleverser les pratiques médicales se sont ouvertes, d'aucuns considérant que la création de lignées cellulaires à partir des cellules ES permettra de soigner par exemple le diabète, en « produisant » des cellules pancréatiques, l'artériosclérose, avec des cellules encothéliales, ou encore le cancer et les maladies neurodégénératives telle que celle de Parkinson.
Ainsi, à partir de quatorze blastocystes humains, des chercheurs américains ont pu récemment obtenir cinq lignées de cellules souches embryonnaires, lesquelles transplantées sous la peau d'une souris, ont été capables de se transformer en divers tissus, représentants les trois feuillets nécessaires au développement : ectoderme, mésoderme et endoderme. Ces travaux confirment donc l'universalité d'observations anciennes effectuées sur la souris, montrant la pluripotence des cellules du bouton embryonnaire. Chez le rat, la culture de cellules ES a permis leur différenciation en cellules nerveuses (oligodendrocytes), lesquelles ont été capables de compenser partiellement un défaut de myélinisation dans le cerveau de jeunes rats.
Il est nécessaire, à ce stade, de donner la définition d'une « lignée » de cellules souches embryonnaires. Selon M. Jean-Paul Renard, on peut la définir comme un ensemble de cellules qui peuvent :
- être maintenues en culture à l'état indifférencié pendant plusieurs semaines, voir plusieurs mois ;
- être congelées puis décongelées sans perdre leur potentiel de multiplication ;
- être différenciées in vitro ou in vivo dans toutes les différentes lignées cellulaires de l'organisme, d'abord celles qui correspondent aux trois premiers types cellulaires de l'embryon, l'ectoderme, le mésoderme et l'endoderme, puis tous les autres, et aussi la lignée germinale ;
- conserver ces propriétés après avoir été isolées et replacées en culture.
Reste cependant à résoudre le problème de la compatibilité entre ces cellules ES, créées à des fins thérapeutiques et le patient qu'elles seraient susceptibles de soigner, ainsi que le notait le Docteur Axel Kahn dans son audition précitée devant la Mission : « Imaginons que l'on commande, que l'on sache commander à ces cellules souches de donner des cellules dopaminergiques du cerveau pour soigner la maladie de Parkinson et que l'on certifie que ces cellules ne déclencheront pas une tumeur cérébrale, ce serait naturellement fantastique. Cela dit, ces cellules souches embryonnaires, isolées par des Australiens et par des Américains, n'auront pas un génome identique au génome du receveur qui va donc développer une réaction de rejet de greffe contre elles, car elles seront considérées comme étrangères ».
Un autre problème doit encore être résolu : celui de la maîtrise du processus de différenciation, afin de garantir l'innocuité des cellules. Les expériences sur la souris ont en effet démontré l'existence d'un risque sanitaire lors du processus de différenciation « commandé » ; il semble en effet que si l'on laisse en culture des cellules non différenciées, ces dernières sont susceptibles de devenir in vivo des cellules tumorales. M. Gilles de Poncins, dans son audition précitée devant la Mission, reconnaissait ce risque dans les termes suivants : « On prend des cellules embryonnaires de souche humaine (...) qui ont néanmoins été cultivées. (...). Vous amplifiez ces cellules, vous les différenciez en neurones. Toutes ces manipulations prennent des semaines pendant lesquelles les cellules sont en culture dans un milieu simplement nutritif ou inducteur de différenciation. Or, vous n'avez aucun système de contrôle de l'introduction de mutations, comme il en existe dans un organisme, et grâce auquel une cellule qui commence à produire une protéine un peu bizarre est automatiquement détectée et éliminée. Ce contrôle n'existe pas in vitro. La question qu'il va falloir régler, une fois que l'on aura réglé toutes les questions scientifiques, sera de savoir, en termes de santé publique, quels tests il faudra faire pour garantir la qualité de la préparation cellulaire réimplantée chez le patient. Le risque est-il suffisamment faible pour que je décide de réimplanter ? (...) Vous prenez des précurseurs, vous les amplifiez in vitro, vous tuez toutes les cellules et, en particulier, toutes les cellules tumorales. Puis, vous réinjectez. L'enjeu majeur, avant de généraliser cette technologie, est de savoir comment faire pour être sûr que, lorsque l'on réinjecte, il ne reste pas de cellules tumorales ».
3.- Les cellules souches adultes
Il existe deux sortes de cellules souches dans l'organisme adulte : celles qui sont présentes dans le sang, l'épiderme et l'intestin qui participent au renouvellement permanent de ces tissus et celles que l'on trouve dans d'autres organes tels que le foie, le cerveau, le pancréas ou les tissus comme le muscle ou l'intestin qui interviennent dans la réparation tissulaire « en coopération » avec les cellules souches de réserve.
Dès 1991, une équipe française (69) mettait en évidence la présence dans le sang de l'homme adulte d'une cellule souche « mésenchymateuse » de nature multipotente. D'autres travaux postérieurs démontrèrent que cette cellule correspond vraisemblablement avec les cellules souches hépatiques, musculaires et endothéliales (70) à une seule cellule souche circulante. Le contrôle et l'activation de cette cellule souche multipotente seraient effectués par des lymphocites qui déclencheraient ainsi le processus de réparation tissulaire. Plus récemment a été mis en évidence l'existence, dans le cerveau de l'homme, de cellules souches multipotentes pouvant donner naissance aux neurones, aux astrocytes et aux oligodendrocytes (71), mettant fin à la croyance d'après laquelle le capital cellulaire du cerveau ne pouvait que décroître avec l'âge. Le Docteur Axel Kahn exposait devant la Mission, lors de son audition précitée, les perspectives ouvertes par ces dernières découvertes :
« On s'est rendu compte récemment que des cellules souches neurales, des cellules souches du système nerveux central, pouvaient, dans certaines conditions, participer à toute l'embryogenèse. On prend un cerveau adulte de souris, on cultive des cellules prélevées dans ce cerveau et on met ensuite ces cellules dans un embryon. On voit que des cellules dérivées de ces cellules neurales participent à la fabrication de la moelle, de la peau, du foie, du muscle. Alors que l'on ne l'imaginait pas du tout. On pensait que ces cellules souches du cerveau ne pouvaient « faire que du cerveau ». En réalité, elles ont une plasticité résiduelle, si bien qu'il se pourrait, dans l'avenir - mais ce n'est pas sûr -, que l'on puisse prendre des cellules de ce type et modifier leur destin de manière à réparer, suivant ce dont nous souffrons, notre vieille arthrite ou bien notre maladie d'Alzheimer ».
Mais ce sont surtout les récents travaux sur les cellules souches présentes dans la moelle osseuse de souris adultes qui laissent entrevoir des possibilités jusqu'alors insoupçonnées.
Des chercheurs américains ont su, en effet, démontrer la nature multipotente de ces cellules capables de donner naissance non seulement à toutes les lignées cellulaires sanguines mais aussi à des cellules épithéliales de foie, de poumon, de la peau et des voies gastro-intestinales alors que l'on pensait, jusqu'à présent, que seules les cellules souches embryonnaires avaient cette multipotence. Si cette découverte est extrapolable à l'espèce humaine, il serait envisageable, demain, de réparer n'importe quel organe en utilisant des cellules multipliées à partir de cette cellule « mère » dont on commanderait le processus de différenciation. Aujourd'hui, il convient en effet de rappeler que seules les cellules de réserve du sang et de l'épiderme peuvent être utilisées en thérapie sans que l'on puisse modifier leur nature et leur destination.
Le rapport établi en novembre 2000 à la demande de M. Roger-Gérard Schwartzenberg, ministre de la recherche, par un groupe de travail présidé par M. François Gros comparant les cellules souches adultes avec les cellules ES, soulignait l'intérêt des premières découvertes tout en notant la nécessité de confirmer ces résultats chez l'homme :
« Les observations récentes indiquent que des tissus d'accès facile pourraient être en fait des réservoirs de cellules souches réparatrices de plusieurs tissus d'utilisation physiologique aisée. Le résultat le plus spectaculaire concerne la moelle osseuse : elle contient non seulement des cellules souches du tissu sanguin (hématopoïétiques), et du tissu osseux et cartilagineux mais aussi des cellules souches du foie et peut-être des cellules souches capables de produire certaines catégories de cellules nerveuses. La moelle osseuse étant facilement accessible chez tout individu, et à tout âge, cette observation peut suggérer que ce tissu serait une source de cellules réparatrices pour nombre de tissus. Deux autres tissus adultes, le muscle et le cerveau, contiennent également plusieurs types de cellules souches : le muscle contient des cellules souches hématopoïétiques et certaines régions du cerveau des cellules souches nerveuses aussi capables de différenciation en cellules musculaires ou en cellules sanguines. Ces derniers résultats ne reposent cependant que sur très peu de données et méritent d'être confirmés (...). Il n'est donc pas exclu que l'on puisse, à partir d'un prélèvement de moelle osseuse adulte, démontrer qu'il est possible d'améliorer certaines pathologies touchant des organes tels que le foie, le muscle ou certaines maladies neurodégénératives, pour lesquelles les thérapeutes sont actuellement assez démunis ».
L'intérêt de ces découvertes, si elles devaient être confirmées et validées chez l'homme, est immense puisqu'elles laissent espérer la mise au point de thérapies parfaitement immuno-compatibles avec l'individu que l'on souhaite soigner. C'est l'intérêt que soulignait devant la Mission M. Gilles de Poncins, dans son audition précitée : « Le principe est de prélever les propres cellules du patient pour les lui réimplanter. Les enjeux sont simplement techniques : comment faire pour prélever ces précurseurs, pour les amplifier en suffisamment grande quantité, avant de les réinjecter ? Mais les avantages sont très importants. Le jour où on saura le faire, toutes les autres techniques deviendront assez peu intéressantes, à la fois du point de vue éthique et au regard des problèmes d'immunité. C'est vraiment une des grandes alternatives. L'enjeu scientifique est ici énorme ».
Toutefois, certaines questions restent encore sans réponse ; on ignore ainsi si le fait de manipuler des cellules souches somatiques (adultes) d'un patient malade pour les lui réinjecter peut avoir pour conséquence de reproduire à nouveau la maladie. C'est la question que posait devant la Mission M. Gilles de Poncins dans son audition précitée :
« Une question est encore source de débat dans la communauté scientifique. Prendre des cellules sur un patient qui est a priori malade, c'est-à-dire aller dans le tissu malade pour prendre ces cellules, les réamplifier, puis les réinjecter, est-ce une stratégie payante à long terme ? La question est celle de savoir quelle est la fréquence d'apparition des maladies et quel est le rôle de l'environnement par rapport à la génétique ? Le facteur génétique est essentiel. Vous allez prendre à quelqu'un ses propres cellules et vous allez les lui réinjecter, spontanément il aura tendance à reproduire cette maladie. Vous pouvez imaginer qu'à moyen terme, cette technique n'est qu'un palliatif. Il s'agit d'une question scientifique complètement ouverte. Il n'y a pas du tout de réponse. Il est clair toutefois que si on arrive à maîtriser cette technique, cela permettra toujours, en termes de santé publique, de soigner les gens, même si ce n'est que pour quelques années ».
D'autres perspectives seraient encore ouvertes si la technique de « transdifférenciation » pouvait être mise au point. Il s'agirait de commander aux cellules souches adultes pluripotentes de se différencier en des cellules autres que celles pour lesquelles elles sont programmées. C'est la piste de recherche qu'exposait devant la Mission M. Jean-Claude Renard, dans son audition précitée :
« Aujourd'hui, les chercheurs proposent d'aller plus loin. S'il existe dans notre organisme des cellules qui ne sont pas complètement différenciées, mais qui ne sont plus des cellules embryonnaires ou germinales, pourrait-on changer leur destin en les plaçant à volonté dans un autre milieu environnant ? Pourrait-on parvenir à contrôler cette transdifférenciation » ? En mettant en culture, dans des conditions appropriées, des précurseurs des cellules sanguines - un prélèvement sanguin suffit pour en disposer en nombre suffisant - ne pourrait-on pas obtenir des cellules musculaires nerveuses, hépatiques par exemple, que l'on pourrait utiliser pour des autogreffes ? ».
M. Gilles de Poncins, lors de son audition devant la Mission, a considéré cependant que cette piste de recherche, si elle est confirmée, ne pourrait aboutir qu'à un horizon lointain :
« La dernière piste est celle de la transdifférenciation. Quelques publications montrent qu'à partir de progéniteurs du sang, on pourrait obtenir des neurones et réciproquement. Je n'entre pas dans la discussion scientifique qui en est encore au stade de savoir jusqu'à quel point cela est reproductible. Du point de vue technique, ce n'est pas une alternative pour développer de nouveaux médicaments dans les cinq à dix ans à venir, c'est un sujet de recherche et cela le demeurera peut-être encore pendant dix ou quinze ans. Il y aurait, là encore, un avantage majeur d'un point de vue éthique ».
4.- Les cellules souches f_tales
Entre cinq à neuf semaines, il est également possible d'isoler, à partir d'embryons ou de f_tus issus d'avortements, deux sortes de cellules souches : des cellules somatiques f_tales présentes dans les mêmes tissus que chez l'adulte et des cellules germinales, issues de l'ébauche du tissu germinal du f_tus de nature pluripotente. Le rapport Gros précité considère que parmi les premières, les cellules souches des zones germinatives du système nerveux central et les hépatocytes f_taux (72) offrent des perspectives thérapeutiques encourageantes, en particulier dans le traitement de certaines pathologies, neurodégénératives, tandis que les cellules germinales semblent pour l'instant inutilisables à des fins thérapeutiques en raison de l'instabilité de leur génome, mais offrent « d'importantes perspectives en recherche fondamentale ».
Dans un récent article de la presse spécialisée (73), le Professeur Jacques Testart faisait part de récents travaux où des cellules embryonnaires germinales ont pu être isolées chez le préf_tus (embryon de six à huit semaines) « au cours de leur migration vers les crêtes germinales et semblent se comporter comme des cellules souches embryonnaires ».
Pour M. Gilles de Poncins, ces cellules présentent l'inconvénient de leur faible disponibilité et quantité. Il déclarait ainsi devant la Mission au cours de son audition précitée : « On a évoqué les cellules souches f_tales, en disant : « Pourquoi prendre les cellules à partir d'embryons avec les problèmes éthiques que cela pose, alors que l'on peut les prendre, à partir de f_tus essentiellement d'avortement ? » C'est possible techniquement. Deux inconvénients majeurs apparaissent, qui sont la disponibilité et la quantité.
Sur les cellules souches embryonnaires, vous avez une lignée (...) à amplifier, il n'y a pas de problème. Dans l'autre cas, il faut (...) un f_tus, il faut (...) quelques cellules souches, essayer de les amplifier et travailler dessus. Vous avec donc un problème de disponibilité et de quantité. C'est très important. En outre, vous avez un saut technologique à réaliser. On ne sait pas actuellement, à partir de quelques f_tus, récupérer des cellules souches des différents tissus, les amplifier et les conserver. Vous seriez perpétuellement obligés de retourner sur des embryons que vous aurez, à chaque fois, en faible quantité, etc. Avantages scientifiques et techniques : aucun. Avantage éthique : oui, mais avantages scientifiques et techniques : non ».
5.- Le « clonage thérapeutique »
Pour pallier le problème d'immuno-compatibilité que poserait l'injection de cellules ES dans un organisme adulte, des chercheurs envisagent de recourir à la méthode du transfert de noyau somatique (TNS), couramment appelée « clonage thérapeutique », selon des termes impropres, comme nous l'avons souligné précédemment.
Pour résumer simplement cette technique, il s'agirait donc d'introduire le noyau d'une cellule adulte - celle du malade dont on recherche la guérison - dans un ovocyte préalablement énucléé, c'est-à-dire privé de son propre noyau.
M. Gilles de Poncins, dans son audition précitée devant la Mission, considère qu'elle constitue la technique d'avenir : « Vous prenez des cellules du patient. C'est très simple, un peu de derme de la bouche ou de sang, vous récupérez le noyau et vous substituez ce noyau à celui des cellules ES humaines provenant d'une lignée que vous avez « en stock ». En fait, vous combinerez le potentiel de différenciation de ces cellules avec le noyau du patient. En simplifiant à l'extrême, vous avez artificiellement créé des cellules souches embryonnaires du patient, même si le cytoplasme et les mitochondries ne viennent pas du patient. Vous avez établi des cellules totipotentes ou multipotentes du patient. Vous les différenciez, vous les greffez. Ce sont des cellules du soi, elles ne sont pas rejetées en tant que telles. A priori, c'est l'avenir de la thérapie cellulaire ».
Par cette technique du « clonage thérapeutique », on obtiendrait génétiquement un embryon sans pour autant passer par une procréation sexuée, ce qui n'est pas sans susciter certaines controverses et interrogations, ainsi que l'indiquait devant la Mission le 20 septembre 2000, M. Carlos de Sola, responsable de la section bioéthique du Conseil de l'Europe :
« La loi allemande, quant à elle, définit l'embryon comme la résultante de l'union des gamètes, mais elle s'est trouvée en porte à faux lorsqu'il s'est agi de savoir comment qualifier un embryon résultant de la technique de transfert nucléaire. C'est pourquoi, dans ces domaines, il faut parfois ne pas trop donner de précisions qui, du reste, dépendent des progrès de la science. Toutefois, je vous signale qu'un parlementaire espagnol a récemment défendu l'idée qu'un embryon ou une entité créée à partir de la technique de transfert nucléaire, « nucléovule », selon lui, n'est pas un vrai embryon. Certains pays avaient déjà employé l'expression de préembryon pour désigner un tel être. Pour ma part, je crois que ce parlementaire a raison jusqu'à un certain point, mais qu'à partir du moment où cette entité commencera à se diviser, elle se comportera comme un embryon véritable. Ne nous voilons donc pas la face. Si un embryon, quelle que soit la technique qui le produit, peut donner un enfant, c'est qu'il s'agit d'un vrai embryon. Il ne faut pas escamoter le problème éthique en imaginant des terminologies nouvelles ».
Cette possibilité de conception asexuée de l'espèce humaine, même si elle ne poursuit pas un but de procréation, pose dans des termes nouveaux la question, qui demeure une perpétuelle énigme, du début de la vie. Notre collègue M. Alain Calmat, lors de la réunion de la Mission le 12 juillet 2000, en concluait que l'on peut désormais être sûr que la vie ne commence pas à la fécondation, « puisque le clonage peut donner une vie à un mammifère par transfert de noyau, c'est-à-dire sans fécondation ».
Le Professeur Claude Sureau déclarait pour sa part devant la Mission le même jour : « La fécondation n'est certainement pas le début de la vie puisque la vie est éternelle et que les spermatozoïdes et les ovocytes sont vivants avant la fécondation. On peut parler du début de la personne mais sûrement pas de la vie » . Mais il reconnaissait cependant que le « clonage thérapeutique » est « un peu perturbant » dans la mesure ou « toutes les cellules d'un organisme sont des personnes humaines potentielles, puisqu'en introduisant leur noyau dans un ovocyte, vous obtenez un individu. À cet égard, les progrès de la biologie devraient conduire à une réflexion plus approfondie sur nos motivations, sur ce que nous pensons de l'individu et de la personne humaine ».
Pour notre collègue Jean-François Mattei, c'est l'implantation qui doit définir l'embryon, ce dernier ne pouvant pas, selon lui, « au sens culturel », résulter d'un processus asexué. Il déclarait ainsi devant la Mission le 6 septembre 2000 (74) : « Je crois que nous devons en effet de plus en plus nous appuyer sur la notion d'implantation. Si l'on acceptait l'idée qu'un transfert nucléaire - comme les Britanniques viennent de l'autoriser - est un clonage embryonnaire, de ce fait l'espèce humaine deviendrait une espèce dont la reproduction n'est plus nécessairement sexuée. Il s'agirait donc d'un saut anthropologique et je ne suis pas sûr que l'on ait le droit d'utiliser le qualificatif d'embryon, au sens historique, traditionnel et culturel de ce terme, l'aspect religieux étant mis de côté. En effet, dans l'approche anthropologique, un embryon est toujours le résultat de la fécondation d'un ovule par un spermatozoïde et c'est d'ailleurs la raison pour laquelle on dit que la vie existe dès la fécondation. Or lorsqu'on procède à un transfert nucléaire, il n'y a plus de fécondation ! ».
Il s'agit là d'une véritable question qui mérite un débat. Et nous devons aller au fond de ce débat. Être pour ou contre le clonage des cellules du bouton embryonnaire, cela veut dire qu'on respecte ou non la structure embryonnaire fécondée qui peut donner un être humain. Avec le transfert nucléaire, c'est totalement différent car, selon que l'on va implanter ou non la cellule, elle va prendre la valeur d'un embryon (...) Je pense qu'au moment où nous discutons de ces questions, nous devons essayer de marier nos nouvelles connaissances scientifiques avec le poids culturel que nous portons. Et je crois que dire demain à la population qu'un embryon n'est pas forcément le résultat d'une fécondation est un acte lourd, beaucoup plus que d'expliquer les effets d'un stérilet ou de la pilule du lendemain pour lesquels personne ne manifeste d'opposition car, en définitive, dans l'esprit des gens la première semaine ne compte pas ».
Une fois obtenu l'embryon issu d'un transfert de noyau somatique (embryon ITNS), ses cellules souches seraient ensuite prélevées, mises en culture et différenciées selon la destination recherchée pour une thérapie précise : cellules neuronales, musculaires, sanguines, hépatiques... lesquelles seraient ensuite introduites dans l'organisme adulte, « père » du noyau originel. Il s'agit donc d'utiliser la méthode du clonage mais en arrêtant le processus de développement de l'embryon au stade du blastocyste.
Le schéma ci-après illustre ce processus.
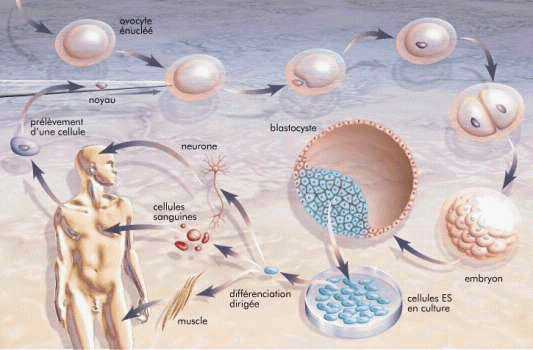
Comme l'indiquait devant la Mission, le 20 septembre 2000, le Docteur Françoise Shenfield, membre de la HFEA, c'est cette technique que vient d'autoriser le Parlement britannique :
« « Le transfert nucléaire », expression que nous préférons scientifiquement au terme de clonage, n'est qu'une partie des différentes méthodes qui permettront, peut-être, d'utiliser ces cellules souches qui ont ce potentiel de se transformer en cellules matures, lesquelles pourront, dans de nombreuses infections, remplacer les cellules défectueuses chez l'adulte malade (...) On pourrait ainsi, à partir des cellules souches de l'embryon, obtenir des cellules pluripotentes, et non pas totipotentes comme le prétendent à tort certaines publications puisque l'adjectif « totipotent » signifie qu'une cellule peut devenir un embryon entier qui, lui-même pourrait, après réimplantation, donner un f_tus qui, comme nous le savons tous, sans entrer dans le débat de savoir s'il serait un être humain depuis sa conception, serait une personne légale à la naissance. Comme ces cellules ne peuvent pas donner une personne légale, ce sont bien des cellules pluripotentes qui, en revanche, peuvent être transformées - certaines données scientifiques nous permettent de le penser - en d'autres cellules matures et différenciées jusqu'à devenir ou des neurones, qui seraient fort utiles dans le traitement de la sclérose en plaques, de la maladie d'Alzheimer, de la maladie de Parkinson, ou encore des cellules cutanées, fort utiles, elles, dans le traitement des grands brûlés, ou des cellules musculaires cardiaques.
Or la différence entre ces cellules issues d'embryons surnuméraires et celles obtenues par transfert nucléaire en appliquant « la méthode Dolly » à l'embryon humain réside dans le fait que ces dernières préviendraient - j'emploie le conditionnel car tout cela n'est encore que de la théorie - le phénomène de rejet. En effet, il ne faut pas oublier que les cellules issues d'un embryon surnuméraire n'auront pas les mêmes gènes de compatibilité que celles du receveur ce qui, comme dans les greffes de rein, de foie, ou de poumon, entraînerait probablement un phénomène de rejet. Cette spécificité permettrait peut-être de traiter certaines affections sans besoin d'avoir recours aux molécules extrêmement puissantes qui peuvent aussi avoir des effets négatifs sur la santé du patient ».
B.- EN L'ÉTAT DES CONNAISSANCES SCIENTIFIQUES ACTUELLES,
IL REVIENT AU PARLEMENT DE DÉTERMINER LE CHAMP
DE LA RECHERCHE AUTORISÉE
Présentant l'avant-projet de loi au Comité consultatif national d'éthique, le 28 novembre 2000, le Premier ministre, M. Lionel Jospin, a clairement mis en évidence que « la découverte de cellules souches, totipotentes ou multipotentes, identifiables avant le stade de la différenciation tissulaire ouvre de réelles perspectives à la recherche thérapeutique ». Évoquant ce que certains ont pu qualifier de « cellules de l'espérance », le Premier ministre s'est dit « convaincu que la société française aspire à ce que la prise en compte de valeurs fondamentales encadre, sans les rendre impossibles, l'avancée des connaissances scientifiques et leurs applications potentielles dans le domaine de la santé humaine ». Souhaitant ainsi ne pas renoncer à certaines pistes de recherche, M. Lionel Jospin a avancé l'idée qu'il ne fallait pas exclure la constitution de cellules souches à partir des embryons surnuméraires et, « si cela s'avérait un jour nécessaire », à partir du transfert de cellules somatiques, sachant que, parallèlement, seraient soutenues les recherches sur les cellules souches provenant du sang du cordon ombilical.
Cette attitude ouverte à la recherche mais, selon « des protocoles strictement définis et encadrés », tient compte du fait qu'aucun scientifique n'est aujourd'hui en mesure de déterminer avec certitude quelle piste de recherche, parmi celles qui ont été précédemment présentées, aboutira effectivement à la mise au point de nouvelles thérapies.
C'est l'avis qu'exprimait devant la Mission, le 6 septembre 2000, Mme Marina Cavazzana-Calvo, chercheur à l'INSERM (75) dans l'équipe qui a réussi, l'an passé, à guérir par thérapie cellulaire des « bébés bulles » (76) : « Sur les cellules souches adultes et les cellules souches embryonnaires, je dirai que les deux voies de recherche sont aujourd'hui ouvertes. À l'heure actuelle, il ne serait pas raisonnable pour un chercheur d'adopter une position tranchée en affirmant qu'il est possible de tout faire avec une cellule souche adulte ou, à l'inverse, avec une cellule souche embryonnaire : il n'y a pas aujourd'hui dans la littérature de résultats suffisamment clairs qui nous autoriseraient à abandonner l'une de ces deux voies de recherche ».
C'est aussi l'avis du Professeur Claude Sureau qui déclarait devant la Mission dans son audition précitée : « Tant que l'on n'aura pas suivi les trois voies et que l'on n'en aura pas comparé les résultats - cellules embryonnaires, cellules clonées, cellules transdifférenciées - on ne pourra pas savoir laquelle est la meilleure ».
En revanche, il est certain que les arguments éthiques peuvent militer pour l'élimination d'une piste de recherche au profit d'une autre. D'aucuns considèrent ainsi que les perspectives offertes par les cellules souches somatiques (cellules souches adultes) pourraient éviter de recourir aux cellules ES ou au « clonage thérapeutique ». Le Sénateur Claude Huriet exprimait cette position devant la Mission en déclarant, le 17 mai 2000 : « On peut trouver, à différents niveaux, des cellules souches chez l'individu adulte. Cette découverte porte en elle-même des espoirs (...) Il va de soi que si ces perspectives, sur lesquelles il est trop tôt pour porter un jugement définitif, se développent, on sort du dilemme qui personnellement me hante : ou bien on interdit l'utilisation des cellules souches de l'embryon humain, et dans ce cas on laisse à leur désespoir des malades atteints de maladies aussi graves que la maladie de Parkinson ou des maladies neurodégénératives, ou bien on privilégie les perspectives thérapeutiques, et, dans ce cas, on est amené à considérer l'embryon humain comme une matière première ».
En revanche, les cellules souches adultes ne sont apparemment disponibles qu'en faible quantité ; enfouies dans les tissus, elles sont en outre plus difficiles d'accès. C'est le constat que faisait, devant la Mission, M. Gilles de Poncins, lors de son audition précitée : « Avec les cellules souches somatiques, on dit « faisons encore plus simple en prélevant des cellules sur des adultes consentants ». Là, nous avons le même problème de disponibilité et de quantité, aggravé même parce qu'il y a très peu de cellules souches chez l'adulte. En outre, c'est plus difficile en termes de conditions (...). C'est techniquement possible, mais il y a un saut de savoir à réaliser (...). Il faut donc entreprendre des programmes de recherche majeurs pour savoir ce que sont les cellules souches chez l'adulte, ce qui est encore un sujet de biologie fondamentale. On va avancer, certes, mais ce n'est pas encore une connaissance disponible. En revanche, cette piste offre, à nouveau, un avantage éthique manifeste ».
Le « clonage thérapeutique » suscite, quant à lui, différentes craintes. Pour certains, telle le Docteur Marie-Odile Alnot, son autorisation entraînerait, tôt ou tard, le franchissement de la frontière avec le clonage reproductif. Elle déclarait ainsi être « très inquiète en ce qui concerne le clonage. En effet, si pour l'instant on envisage le « clonage thérapeutique », vous savez comme moi que, lorsque la technique sera au point, on envisagera le clonage reproductif, d'autant que les barrières éthiques s'effondrent dès qu'apparaissent des intérêts financiers énormes. J'ai donc très peur du « clonage thérapeutique » sur l'embryon, alors qu'il existe des cellules utilisables chez l'adulte et sur lesquelles les chercheurs pourraient travailler ».
Devant la Mission le 31 mai 2000, le Professeur Didier Sicard, Président du CCNE, se déclarait à son tour « préoccupé » en considérant que l'espace entre le « clonage thérapeutique » et le clonage reproductif est « plus étroit qu'on ne le pense ». Selon lui, la technique du transfert de noyau somatique risquerait de conduire à l'instrumentalisation de l'embryon tout en posant un grave problème de protection des femmes, dont les ovocytes sont nécessaires à la réalisation de cette technique :
« Les promesses des cellules souches embryonnaires, si elles se confirment, feront que l'instrumentalisation de l'embryon risquera d'être rapide. Le besoin, en termes cellulaires, de ces embryons va devenir en effet considérable. Si l'on ouvre la porte à cette capacité des embryons de devenir du matériau thérapeutique, cela posera deux problèmes essentiels.
Le premier est bien connu, c'est celui de la place de l'embryon dans notre culture humaine collective. Quelques cellules embryonnaires ne sont pas des cellules animales - il y a tout de même ici une promesse d'homme - et en faire une sorte de matériau thérapeutique apportant la guérison ou la réparation me paraît, dans notre société, quelque chose d'extrêmement grave. Je ne me place pas du point de vue du statut ontologique, sur lequel je me garderai bien d'avoir un avis personnel, parce que je n'en sais pas plus que quiconque. Je ne sais pas ce qu'est un embryon hormis que c'est une possibilité de personne. Et personne n'arrivera à statuer sur ce point car immédiatement quelqu'un sera d'un avis opposé. Je ne pense pas que le problème soit celui du statut juridique de l'embryon, mais du respect a priori qu'on doit lui témoigner.
Le deuxième problème concerne le statut des femmes. Je ne suis pas féministe, mais je suis frappé du silence des femmes dans ce domaine. Pour faire des cellules embryonnaires, il faut des ovocytes, des ovules. Les femmes ont quelques centaines d'ovocytes dans leur vie. Prendre les ovocytes est difficile, il faut faire une c_lioscopie (...). On peut mettre éventuellement, à cette occasion, la vie de la femme en danger. On peut imaginer que si les cellules souches embryonnaires deviennent un matériau thérapeutique, les ovocytes et les ovaires de femmes deviendront presqu'un matériau, une marchandise. Les femmes, dans les pays sans contraintes ou loi répressive, pourront ainsi donner ou vendre des ovocytes ».
Le CCNE dans son avis précité du 18 janvier 2001 sur l'avant-projet de révision des lois de bioéthique, ne cache pas les divergences d'opinion qui se sont manifestées en son sein autour de la question suivante : « Les bénéfices thérapeutiques espérés de l'utilisation des cellules souches obtenues à partir d'embryons ITNS justifient-ils de contrevenir au principe sur lequel repose jusqu'à présent notre législation, selon lequel la création d'embryons humains à toute autre fin que leur propre développement est interdite, fût-ce pour la recherche ? »
Comme il a été indiqué dans la première partie du présent rapport, après avoir exposé les arguments en faveur du « clonage thérapeutique » et ceux qui lui sont opposés, le CCNE indique qu'une majorité s'est dégagée, en son sein, pour l'autorisation « encadrée » de cette technique en souhaitant que des garanties soient apportées aux femmes qui feraient don de leurs ovocytes ou de leur tissu ovarien afin de les protéger des risques qui pèsent sur leur santé.
De son côté, le Groupe européen d'éthique des sciences et des nouvelles technologies auprès de la Commission européenne a adopté, dans son avis du 14 novembre 2000 sur les aspects éthiques de la recherche sur les cellules souches humaines et leur utilisation, une attitude très réservée préférant à la technique du transfert de noyaux somatiques dont les perspectives thérapeutiques lui paraissent éloignées, la recherche sur les cellules souches adultes, les cellules ES à partir d'embryons surnuméraires et les cellules issues du tissu f_tal. Il déclarait ainsi : « Le Groupe tient compte de l'intérêt du transfert de noyaux de cellules somatiques en vue d'étudier les conditions nécessaires à la « reprogrammation » des cellules humaines adultes. Il est également conscient qu'en vue de la thérapie cellulaire future, la création d'embryons par cette technique est peut-être la manière la plus efficace d'obtenir des cellules souches pluripotentes génétiquement identiques à celles d'un patient et, dès lors, des tissus parfaitement histocompatibles, le but étant d'éviter le rejet de tissus après leur transplantation. Toutefois, ces perspectives thérapeutiques éloignées doivent être mises en balance avec d'autres considérations liées au risque que l'utilisation des embryons soit banalisée, que des pressions soient exercées sur les femmes en tant que sources d'ovocytes et que les possibilités d'instrumentalisation de la femme s'accroissent. Étant donné les très mauvais résultats actuellement enregistrés dans le transfert de noyaux de cellules somatiques, la fourniture de lignées de cellules exigerait de très nombreux ovocytes.
Le Groupe est d'avis que sur ce sujet extrêmement sensible, on doit appliquer le principe de proportionnalité et adopter une approche de précaution : il ne suffit donc pas de considérer la légitimité du but poursuivi, à savoir soulager les souffrances humaines, mais il est également essentiel de tenir compte des moyens employés. En particulier, les espoirs attachés à la médecine régénératrice sont encore très hypothétiques et controversés dans les milieux scientifiques. Le Groupe invite à la prudence et estime que, pour l'heure, la création d'embryons par transfert de noyaux de cellules somatiques pour les besoins de la recherche sur la thérapie par les cellules souches serait prématurée, étant donné qu'il existe un vaste champ de recherches à explorer à l'aide d'autres sources de cellules souches humaines (à partir d'embryons surnuméraires, de tissu f_tal et de cellules souches d'adulte) ».
Le Président de la République Jacques Chirac, lors de son allocution à Lyon le 8 février 2001 à l'occasion du forum mondial de biotechnologies, s'était pour sa part prononcé en faveur du renforcement de la recherche sur les cellules souches adultes, de l'ouverture temporaire de la recherche sur les cellules ES issues d'embryons surnuméraires avec l'accord des géniteurs mais contre la technique du transfert de noyau somatique au motif que celle-ci conduit à la création d'embryons à des fins de recherche : « Il faudra mettre à profit ce délai en ouvrant au plus tôt des alternatives. Au plan national comme au plan européen, il est indispensable de lancer et de financer des programmes de recherche portant sur les cellules souches adultes. Bien qu'à leurs débuts, ces recherches permettent d'espérer qu'il sera demain possible d'éviter le recours à des cellules embryonnaires. Par ailleurs, je ne suis pas favorable à l'autorisation du « clonage thérapeutique ». Il conduit à créer des embryons à des fins de recherche et de production de cellules, et, malgré l'interdit, rend matériellement possible le clonage reproductif et risque de conduire à des trafics d'ovocytes ».
De son côté, la Commission nationale consultative des droits de l'homme (CNCDH), dans son avis sur l'avant-projet de loi tendant à la révision des lois relatives à l'éthique biomédicale, adopté, en son sein, le 25 janvier 2001, a fait état des divisions auxquelles a donné lieu la question de la recherche sur l'embryon. Une faible majorité s'est finalement prononcée contre le « clonage thérapeutique » aux motifs qu'il conduirait à la création d'embryons pour la recherche, que les perspectives thérapeutiques ne sont pas « suffisamment mûres pour peser à ce point dans la balance » et enfin que la protection des femmes qui donneraient leurs ovocytes, « contre des pressions faites au nom de la recherche (...) n'apparaît pas encore clairement ».
La Commission nationale consultative des droits de l'homme (CNCDH) s'est donc prononcée, à l'instar du Groupe européen d'éthique, pour l'ouverture de la recherche sur les cellules ES à partir d'embryons surnuméraires et pour la poursuite de la recherche sur les cellules souches adultes, ces deux pistes devant selon elle, permettre « d'avancer dans la voie des connaissances ».
Plus récemment, l'assemblée générale du Conseil d'État du 14 juin 2000 qui se réunissait sur l'avant-projet de révision des lois bioéthiques, s'est prononcée, à une voix de majorité, contre l'autorisation du « clonage thérapeutique » (77).
La technique du transfert de noyau somatique fait donc l'objet à ce jour d'un vif débat. Dans un souci de consensus, le Premier ministre a donc décidé de retirer du projet de loi de révision des lois bioéthiques le dispositif de l'avant-projet qui autorisait cette technique, en confiant au Parlement le soin de se prononcer définitivement. Il est en effet souhaitable que la Représentation nationale examine à son tour cette question, à l'instar du CCNE, de la Commission nationale consultative des droits de l'homme et, plus récemment du Conseil d'État. Dans cette optique, la commission spéciale chargée de l'examen du projet de loi de révision des lois bioéthiques, qui succédera à la présente Mission, devra approfondir cette question au moyen, par exemple, de l'organisation d'une audition publique où les défenseurs et les opposants au « clonage thérapeutique » pourront s'exprimer et éclairer ainsi le choix final du législateur.
La création de lignées de cellules ES, issues d'embryons surnuméraires, à des fins de recherche, semble recueillir quant à elle un large accord. L'intensification de la recherche sur les cellules souches adultes est également souhaitée par de nombreux observateurs et interlocuteurs de la Mission mais il semble qu'une majorité de scientifiques aient la conviction que des résultats seront plus faciles à atteindre avec la recherche sur les cellules ES. Le Docteur Marina Cavazzana-Calvo, dans son audition précitée devant la Mission, résumait ainsi cette position largement répandue : « Je pense que la versatilité de différenciation de la cellule souche embryonnaire est telle qu'elle nous laisse espérer des avantages thérapeutiques majeurs par rapport à la cellule souche adulte, dont on sait aujourd'hui que les capacités de prolifération sont extrêmement limitées. On ignore, par exemple, s'il sera possible d'enrayer certaines maladies atteignant notamment les muscles, le système nerveux central et le foie par une cellule souche de type adulte ».
Votre Rapporteur considère pour sa part que toutes les pistes de recherche doivent être examinées sans a priori particulier par le Parlement auquel doit revenir la responsabilité du choix final des recherches qui seront autorisées demain. S'agissant de sujets de société qui mettent en jeu des questions éthiques fondamentales, il doit en effet revenir à la Représentation nationale de se prononcer après avoir entendu tous les avis et positions qui s'expriment sur ces sujets. À cet égard, il convient de rappeler que le Conseil constitutionnel, dans sa décision du 27 juillet 1994, a jugé que le choix du législateur de ne pas appliquer aux embryons in vitro le principe du respect de tout être humain dès le commencement de sa vie relevait, en l'état des connaissances scientifiques, de son pouvoir d'appréciation et qu'il n'appartenait pas au Conseil Constitutionnel de le remettre en cause. Le débat est donc ouvert et il devra se poursuivre lorsque le projet de loi de révision des lois bioéthiques sera examiné devant notre assemblée. C'est d'ailleurs ce que préconisait la CNCDH à l'issue de ses recommandations dans son avis précité : « Il appartiendra au Parlement, lorsque le dilemme scientifique se sera éclairci, de se prononcer sur une ouverture (de la recherche sur les cellules ES) qui est aujourd'hui, selon le CNCDH, prématurée ».
C.- DES ESPOIRS THÉRAPEUTIQUES CONSIDÉRABLES
Les pistes de recherche présentées précédemment ouvrent des perspectives hier encore inimaginables. Elles permettent d'envisager une véritable révolution dans le traitement de maladies dont certaines sont à ce jour incurables. Ainsi que l'indiquait M. Gilles de Poncins, Directeur général d'une entreprise de biotechnologie dans son audition précitée, « schématiquement, la pharmacie va passer, en quelques années, de l'ère de la découverte de molécules à l'ère de l'exploitation des données de la génétique. Désormais, nous ne voyons une maladie qu'à travers un gène. Nous voulons trouver le gène qui est derrière et, à partir de celui-ci, développer des médicaments. Des gènes sont liés au diabète. Vous cherchez à comprendre quelle est leur fonction. Une fois que vous avez compris comment ils fonctionnent, vous en déduisez, ou vous en comprenez, les dysfonctionnements. Ensuite, vous construisez une stratégie sous forme de médicaments, de thérapie génique, etc. Désormais, le développement de médicaments part des données génétiques. Y compris pour les maladies complexes, multigéniques. (...)
Actuellement, les molécules-médicaments peuvent, à la limite, corriger, mais elles ne peuvent pas réparer. Avec la thérapie cellulaire, vous pourriez a priori réparer. Des neurones meurent - une neuro-dégénérescence - vous pouvez imaginer réimplanter des neurones. Vous restaurez une fonction, ce qui n'est pas possible avec les technologies actuelles.
En terme de potentiel de guérison, et même en termes de qualité de vie des patients, il s'agit bien d'une innovation fantastique. Ceci est le deuxième enjeu des cellules souches embryonnaires ».
Ainsi se nourrissent les espoirs de soigner des maladies neuro-dégératrices, des accidents médullaires (78), des maladies hépatiques, des maladies du sang, ou de la peau... Le Docteur Marina Cavazzana-Calvo, dans son audition précitée devant la Mission, se disait quant à elle convaincue des nombreuses perspectives ouvertes pour les recherches en cours ; elle déclarait à ce propos : « Je ne pense pas que la thérapie génique guérira toutes les maladies mais, néanmoins, nous sommes en droit d'attendre des progrès raisonnables, surtout pour certaines maladies très particulières de type héréditaire qui atteignent le système hématopoïétique. Plus nous serons en mesure de remporter des succès sur des maladies simples de type monogénique, où un seul gène est responsable de la maladie, plus nous aurons l'espoir d'élargir cette approche thérapeutique à des maladies polygéniques ».
Par respect des malades, il faut cependant rappeler et souligner que ces perspectives thérapeutiques appartiennent encore au domaine de l'espoir et des promesses puisque l'on se trouve toujours aujourd'hui au stade de la recherche fondamentale, à de rares exceptions. La presse et les media en général doivent, à cet égard, jouer un rôle pédagogique fondamental pour expliquer à l'opinion publique l'état des recherches et les perspectives qu'elles permettent d'envisager à moyen terme, sans créer de vains espoirs dans le court terme.
1.- Des expérimentations réussies sur l'animal
De récents travaux, en particulier sur les souris et les rongeurs illustrent les espoirs de la thérapie génique et cellulaire de demain. On a ainsi démontré que des cellules souches embryonnaires de souris pouvaient être transformées en cellules sécrétrices d'insuline qui, greffées à une souris diabétique, guérissaient momentanément son diabète. On sait également, depuis plusieurs années, faire régresser les tumeurs chez les rongeurs.
Au début de l'année 2001, une équipe de l'université de médecine de Washington à Saint-Louis aux États-Unis, annonçait son succès dans la réparation de la moelle épinière de rats grâce à l'injection de cellules souches embryonnaires de souris. Ces cellules, préalablement traitées in vitro pour orienter leur différenciation en des cellules du système nerveux, ont été injectées, au niveau de la lésion des moelles épinières d'une cohorte de vingt-deux rats adultes paralysés par cette lésion. Dès le dixième jour suivant la greffe, les animaux traités ont retrouvé une certaine mobilité et coordination de leurs membres antérieurs et postérieurs. On ignore encore cependant comment ont agi les cellules embryonnaires pour reconstituer partiellement le fonctionnement de la moelle épinière de ces rats : se substituent-elles aux neurones détruits ou déclenchent-elles un processus chimique de reconstruction de la moelle ?
Au mois de février 2001, une équipe de chercheurs américains de l'université de Californie a annoncé son intention de passer à la première phase d'essai d'une thérapie génique sur deux sujets atteints de la maladie d'Alzheimer à la suite de la publication de ses travaux menés avec succès sur le singe. Ces derniers ont montré dans un premier temps que le vieillissement des singes choisis pour l'expérience s'accompagnait d'une réduction de 25 % de l'innervation corticale (79) par le système cholinergique (80) de l'animal. Dans une deuxième temps, ce déclin a pu être substantiellement amélioré par l'apport de cellules (81) prélevées initialement sur l'animal et modifiées génétiquement afin de produire un facteur de croissance nerveux.
Par ailleurs, un récent article paru dans la revue Science faisait état d'une régression de certaines affections d'hématologie neurologique chez la souris par implantation de cellules souches embryonnaires murines (de souris), préalablement différenciées en cellules neuronales.
2.- Des essais d'application chez l'homme.
À ce jour, la thérapie génique ou cellulaire à partir de cellules ES n'offre que des espérances dans la mise au point de traitements curatifs efficaces sur l'homme.
Il existe cependant un cas de réussite exemplaire de thérapie génique réussie sur quatre jeunes enfants atteints du déficit immunitaire combiné sévère lié au chromosome X, plus couramment appelés « bébés bulles ».
a) Le succès de la thérapie sur les « bébés bulles »
Le Docteur Marina Cavazzana-Calvo, membre de l'équipe du Professeur Fischer qui a réussi cette expérimentation à l'hôpital Necker de Paris, a décrit devant la Mission le 6 septembre 2000, la technique et le protocole suivis :
« Pour traiter ces enfants qui n'ont pas de lymphocytes, on est parti d'une cellule souche de la moelle osseuse, présente dans les os plats, que l'on sait isoler, manipuler in vitro et faire proliférer, mais qui peut se différencier en tout excepté en cellules protégeant contre les infections (...) ;
- Nous avons donc eu recours à un rétrovirus défectif, c'est-à-dire ne contenant comme seule information génétique que l'information du gène défectueux chez ces malades. Aujourd'hui, nous sommes capables d'introduire ce type de virus défectifs, donc non réplicatifs, à l'intérieur d'une cellule souche qui prolifère. Une fois entrés, lesdits virus substituent à l'information défectueuse, dans le patrimoine génétique de cette cellule somatique, une information génétique nouvelle. Grâce à cette opération, il devient possible de restaurer une fonction déficitaire chez les malades (...).
- Il avait déjà été prouvé en laboratoire, par des tests in vitro, donc précliniques, que si on introduisait dans une cellule souche un virus avec le gène approprié, il était possible de restaurer la population lymphocytaire manquante (...) C'est à la suite de ces travaux précliniques que nous avons obtenu l'autorisation demandée (...) d'une part, du ministère de la santé et, d'autre part, de l'Agence française de sécurité sanitaire des produits de santé. Outre ces deux instances, qui émettent un avis sur les éventuels effets toxiques sur l'organisme des manipulations génétiques de cellules, se prononcent également le Comité consultatif national d'éthique et un assureur prenant en charge les effets négatifs possibles (...) ».
Le schéma ci-après illustre les étapes de l'expérimentation qui vient d'être décrite :
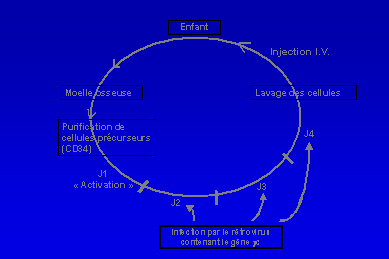
Source : Laboratoire de thérapie cellulaire et génique de l'hôpital Necker Enfants Malades ».
Sur les cinq enfants traités entre janvier 1999 et avril 2000, quatre vivent aujourd'hui normalement au sein de leur famille (82), sans autre traitement que celui qui leur a été administré à l'hôpital Necker. Leur système immunitaire a été complètement restauré ; ils ont ainsi aujourd'hui des lymphocytes capables de les protéger des infections. Pour trois d'entre eux, l'équipe du Professeur Fischer considère que le recul est assez important pour que l'on puisse parler de guérison.
b) Une récente réussite dans l'utilisation de cellules f_tales
À la fin du mois de novembre 2000, l'équipe du Docteur Marc Peschanski à l'hôpital Mondor de Créteil, a annoncé la réussite de la première greffe de neurones f_taux sur trois malades atteints d'une grave affection neuro-dégénérative d'origine génétique, la chorée de Huntington, qui provoque la démence du patient puis sa mort en dix à vingt ans. En France, 6 000 personnes en sont atteintes et 12 000 autres sont porteuses du gène déficient.
c) Le pari des scientifiques pour l'avenir
Cependant force est de constater que ce succès de thérapie génique par manipulation de cellules souches, en l'espèce adultes puisque les cellules souches de la moelle osseuse utilisées ont été prélevées sur les enfants traités, est encore isolé. Le Professeur Jacques Testart soulignait devant la Mission, lors de son audition précitée, les faibles résultats atteints jusqu'à ce jour et exprimait un fort scepticisme à l'égard de ces thérapies : « Il faudrait montrer la faisabilité de ces thérapies à partir de cellules embryonnaires. Or, à ma connaissance, il y a eu manipulations chez le rat et chez la souris, mais sans rien obtenir de concluant pour le moment, c'est-à-dire que l'on a greffé des cellules issues d'embryons, on les a modifiées, elles se sont implantées dans le tissu et multipliées un peu, et après, apparemment, elles dégénèrent.
En tout cas, cela n'a pas permis de soigner vraiment des individus, des animaux malades. C'est une voie sûrement intéressante. J'observe que ceux qui la défendent le plus sont ceux qui depuis dix ans nous promettent une thérapie génique qui a toujours été un échec. On pourrait m'opposer le cas des « bébés bulles », qui est un cas particulier (...) De toute façon, c'est une forme de thérapie cellulaire in vitro, puisque finalement on leur a greffé leurs propres cellules, ce qui n'a rien à voir avec la guérison de la mucoviscidose ou de la myopathie qui me paraît impossible avec cette technologie.
Il est assez ridicule d'entendre, comme je l'ai entendu dire il y a quinze jours, que puisque l'on a fait la carte du chromosome 21 cela va permettre de guérir les « mongoliens ». Pour le moment, la faisabilité des thérapies géniques ou cellulaires (à partir des cellules embryonnaires) n'a pas été démontrée. (...)
Il faut d'ailleurs noter que mes collègues français qui disent que pour le bien de nos patients il faut absolument faire ces recherches, taisent que, depuis des années, des gens qui ne sont pas plus bêtes qu'eux, aux États-Unis, en Écosse ou ailleurs, essaient de le faire et qu'ils n'ont pas encore réussi. Ce n'est pas parce que quelques Français vont s'y mettre que cela va changer la face du monde (...) Autrement dit, cela me paraît prématuré ».
Sans partager ce pessimisme, il faut toutefois reconnaître que les applications thérapeutiques des thérapies génique et cellulaire utilisant des cellules souches ne pourront être mises au point, si les espoirs des scientifiques se réalisent, qu'à moyen ou long terme. On se trouve en effet, à l'exception notable de la thérapie suivie par les « bébés bulles », dans le cadre de la recherche fondamentale, les résultats obtenus sur les animaux devant être poursuivis et validés pour être ensuite confirmés dans l'espèce humaine. Dans le cas du succès obtenu par l'équipe du Professeur Fischer, il faut en effet rappeler que de nombreuses années de recherche ont précédé le stade du protocole clinique précédemment décrit, comme le précisait le Docteur Marina Cavazzana-Calvo dans son audition précitée devant la Mission : « Je tiens cependant à souligner que ces travaux ont duré sept ans, ce qui laisse à penser que ce n'est pas demain que l'on procédera aux clonages thérapeutiques, à l'utilisation des cellules souches embryonnaires et au remplacement sur demande de tous les organes déficients chez un individu : il faudra bien compter une dizaine d'années de recherches avant d'obtenir des résultats cliniques exploitables ! C'est un point d'autant plus important qu'à mon avis ce sont précisément ces dix années de recherche que l'on vous demande d'encadrer de façon très précise ».
S'agissant de la technique du « clonage thérapeutique », M. Gilles de Poncins, dans son audition précitée devant la Mission, considérait que cette technique constitue « a priori l'avenir de la thérapie cellulaire ». Cependant il estimait que les difficultés restant à résoudre laissent entrevoir des résultats seulement à long terme : « Ces avancées interviendront à des horizons très lointains. Scientifiquement, il s'agit de manipulations très risquées, mettant en jeu des questions fondamentales, comme les conséquences d'un transfert de noyau sur la reprogrammation génétique. Les données scientifiques montrent qu'on arrive à atteindre des degrés de pureté qui sont de l'ordre de moins d'une cellule tumorale par million de cellules réinjectées. Sur des malades en phase terminale, on n'hésite évidemment pas. Mais il s'agit d'un risque majeur pour des phénomènes que l'on ne contrôle pas intéressant les cellules normales. Chez la grenouille, le transfert nucléaire se fait depuis le début du siècle. Chez les mammifères depuis beaucoup moins longtemps. On ne dispose pas encore d'un fort recul. Par exemple, à l'INRA, les animaux obtenus par transfert nucléaire ne sont pas en très grande forme. Scientifiquement, il s'agit donc d'un vrai risque ».
De nombreuses questions scientifiques sont en outre encore sans réponse. M. Jean-Paul Renard, Directeur de recherches à l'INRA, dans son audition précitée devant la Mission, faisait état des dernières découvertes qui soulèvent de nombreuses interrogations quant à la manipulation des cellules souches embryonnaires.
Le milieu dans lequel les cellules sont cultivées semble ainsi avoir un effet important, voire déterminant sur leur développement. Selon M. Jean-Paul Renard : « On sait maintenant - surtout grâce aux données obtenues chez l'animal - qu'il existe dès le début du développement une régulation du milieu environnant non directement génétique (on dit alors épigénétique). Cela signifie que le milieu environnant est capable d'inactiver des gènes qui ne s'exprimeront que dans la vie adulte de l'animal. Par exemple, il a été montré chez la souris que la culture d'embryons pendant la journée qui suit la fécondation (stade une cellule) suffit pour entraîner chez les femelles adultes l'inactivation de gènes qui produisent des molécules impliquées dans l'olfaction et dans le comportement sexuel. Et ces modifications peuvent se transmettre à la descendance. Une « épimutation » peut donc être induite par le milieu de culture dès le tout début de l'embryogénèse. Nous sommes loin de n'être que le produit de nos gènes ! »
De récents travaux ont par ailleurs démontré le rôle décisif, mais encore indéterminé, du cytoplasme de l'_uf fécondé, ce qui pourrait avoir des incidences sur les cellules obtenues par « clonage thérapeutique ». Le cytoplasme, c'est-à-dire la cellule sans le noyau, contient ainsi que l'indiquait à nouveau M. Jean-Paul Renard, « des protéines, des facteurs maternels, qui ont été accumulés pendant des mois (et des années chez l'humain) depuis la vie f_tale (...). On s'aperçoit que la façon dont il est organisé autour du noyau est essentielle pour le développement. Par exemple, on s'est aperçu que pour qu'il y ait une vie, les premières divisions doivent être légèrement asymétriques. Si elles sont complètement symétriques, l'embryon n'arrive pas à s'organiser. Qu'est-ce qui détermine cette asymétrie ? Est-ce le hasard ou bien existe-t-il une polarisation de la cellule dès le début du développement ? ».
Ainsi, si l'on procède au clonage de deux ovocytes avec des noyaux identiques, on « obtiendrait », dans l'absolu, deux individus différents, les gènes des mitochondries du cytoplasme de chacun des ovocytes, qui règlent l'énergie cellulaire, étant différents.
Ces dernières découvertes démontrent ainsi le rôle, jusqu'alors sous-estimé, de l'environnement dans le développement d'un organisme vivant, ce qui remet en question, selon M. Jean-Paul Renard, la notion d'inné dicté par les gènes : « L'activité des gènes reste bien entendu déterminante dans l'unicité de chaque individu. Et on sait depuis longtemps que le produit d'expression des gènes, « l'inné » intervient dans les connexions entre les cellules nerveuses. Mais l'évolution des recherches sur l'embryon met en évidence que la notion d'acquis est déjà à l'_uvre au tout début du développement. On pourrait dire en forçant le trait que, s'affranchissant de la logique dictée par les gènes, on redécouvre l'importance de l'épigénèse qui d'Aristote à E. Wolff reste une énigme pour la biologie du développement ! L'environnement du noyau et celui des cellules jouent un rôle déterminant pour mettre en route, puis moduler l'activité des gènes. Il peut induire des changements irréversibles de l'activité - et non de la structure - des gènes en modifiant l'organisation des protéines (la chromatine) du noyau. Cette irréversibilité crée les « épimutants ». Les cellules souches somatiques viennent renforcer l'impression que la vie est un tout sur lequel on peut intervenir non seulement par le programme génétique, mais aussi par l'environnement ».
La fusion d'un ovocyte énucléé avec un noyau « étranger » intervenant dans la technique du « clonage thérapeutique » pourrait, selon M. Gilles de Poncins, créer un problème d'immunologie : « On n'est pas tout à fait sûr - déclarait-il devant la Mission - que ces cellules ne soient pas rejetées à long terme, parce qu'il y a quand même de l'ADN mitochondrial venant d'un autre individu, et qui produit lui-même des protéines qui peuvent être reconnues ».
Selon lui également, les manipulations en jeu dans les techniques pourraient poser des problèmes de santé publique, comme cela était précédemment évoqué, en raison des nombreuses manipulations au cours desquelles les cellules sont en culture : « Or, vous n'avez aucun système de contrôle de l'introduction de mutations, comme il en existe dans un organisme, et grâce auquel une cellule qui commence à produire une protéine un peu bizarre est automatiquement détectée et éliminée. Ce contrôle n'existe pas in vitro ». Il concluait à la nécessité de mettre au point des tests qui permettraient de vérifier l'innocuité de la préparation cellulaire réimplantée chez le patient.
Enfin, et surtout, restent encore à mettre au point les techniques de prolifération et de différenciation des cellules souches. Certaines des variables qui influencent ce processus commencent à être connues grâce aux travaux conduits en particulier sur les cellules ES de souris (addition d'hormones, modification du mode de culture, du milieu de culture ou de sa température), mais beaucoup reste encore à faire pour maîtriser ces processus, a fortiori s'agissant de la transdifférenciation.
De l'ensemble de ces éléments, il faut donc retenir les perspectives immenses et prometteuses de la thérapie cellulaire ou génétique basée sur les cellules souches, qui pourraient faire franchir à la médecine un saut qualitatif majeur. Toutefois, ces perspectives thérapeutiques sont susceptibles de ne déboucher que dans le moyen ou long terme. Il s'agit donc aujourd'hui de franchir la première étape en autorisant et en encadrant la recherche sur les cellules souches puisque l'on se trouve pour l'heure au stade de la recherche fondamentale.
III.- LES CONDITIONS INDISPENSABLES D'ENCADREMENT DE LA RECHERCHE SUR LES CELLULES SOUCHES
Quel que soit le choix des pistes de recherche qui seront retenues par la Représentation nationale, il est impératif de mettre en place un cadre strict d'encadrement des recherches qui seront autorisées. En effet, c'est sans doute dans le domaine de l'infiniment petit que les manipulations sont les plus aisées, voire les plus dangereuses comme le craint le Professeur Jacques Testart qui considérait (83) que «... c'est seulement à ce stade, le plus précoce de l'être humain, que sont possibles les manipulations les plus audacieuses ou les plus folles (clonage, transgenèse, parthénogenèse, etc.). aussi et contrairement aux affirmations courantes, l'encadrement de ces travaux doit-il être beaucoup plus strict que celui qu'on admet pour la recherche sur les personnes ».
Comme l'écrivait le Conseil d'État dans son rapport « Les lois de bioéthique : cinq ans après », le législateur est plus particulièrement confronté à la « question délicate de savoir, s'il doit, pour ne pas entraver des recherches à perspectives thérapeutiques très prometteuses, mais néanmoins encore incertaines, revenir sur l'interdiction des recherches sur l'embryon qu'il avait édictée cinq ans auparavant ». Pour M. Carlos de Sola, dans son audition précitée devant la Mission, les États européens doivent aujourd'hui répondre à la « question principale de savoir si l'on autorise ou interdit, d'une part, la constitution d'embryons à des fins de recherche ou à d'autres finalités qui ne soient pas la procréation et, d'autre part, l'utilisation d'embryons qui ont été constitués à des fins de procréation mais pour lesquels il n'existe pas de projet parental ». Il s'agit, pour le législateur français, de trouver un nouvel équilibre entre le principe de dignité due à l'embryon en sa qualité de personne humaine potentielle et le principe de solidarité vis-à-vis des personnes atteintes de maladies graves, qui nourrissent légitimement des espoirs à l'égard des recherches sur les cellules souches. Il s'agit également de trouver un compromis entre les demandes d'ouverture de la recherche émanant des scientifiques, au nom de la liberté de la recherche, et certains principes éthiques fondamentaux auxquels nous sommes attachés.
Il est impératif de définir le cadre de la recherche qui pourrait être demain autorisée à partir de l'embryon et de ses cellules autour de quelques principes intangibles. M. Philippe Pedrot, juriste auditionné par la Mission le 5 juillet 2000, considérait ainsi indispensable de « poser un certain nombre d'interdits fondateurs (...). Il faudra nécessairement se poser la question des limites et des transgressions possibles. Il ne s'agit pas d'arrêter les recherches, mais il y a des pratiques inadmissibles : le clonage reproductif humain, certaines manipulations entre l'animal et l'homme interdites par des textes dans différents pays ».
1.- L'interdiction d'implanter un embryon ayant fait l'objet de recherches
Il existe une unanimité, parmi les interlocuteurs de la Mission favorables à la recherche sur l'embryon, pour que soit interdite l'implantation d'un embryon qui aurait fait l'objet d'une telle recherche, ou dont les cellules auraient été utilisées dans le cadre d'une recherche, le DPI demeurant bien sûr une exception à cette règle dans les limites strictes posées par les dispositions légales relatives à la médecine prédictive. Le rapport précité du Conseil d'État en fait d'ailleurs l'une des conditions mises à l'ouverture de la recherche sur l'embryon.
M. Carlos de Sola, lors de son audition devant la Mission, indiquait que le protocole additionnel actuellement en préparation au Conseil de l'Europe, devrait également adopter ce principe de non-implantation avec toutefois une forte nuance, puisque les recherches pourraient être néanmoins autorisées sur des embryons destinés à être implantés en l'absence d'une méthode de recherche alternative, ce qui réduit considérablement la portée du principe : « Un des principes (du futur protocole) consistera sans doute à préciser qu'on ne peut effectuer des recherches sur un embryon qui doit être implanté, dès lors qu'il est possible d'obtenir des résultats comparables à partir d'autres recherches, y compris sur des embryons surnuméraires. Deux valeurs sont en jeu : l'une concerne la protection de l'embryon en tant que tel ; l'autre concerne non seulement l'embryon, mais aussi l'enfant à naître, donc l'intégrité d'une personne qui va naître ».
2.- L'interdiction de toute thérapie germinale
Il est indispensable, également, d'interdire toute manipulation des cellules germinales d'un embryon, ce qui aurait pour incidence de modifier son patrimoine génétique et celui de sa descendance.
Le Docteur Marina Cavazzano-Calvo, dans son audition précitée devant la Mission, en sa qualité de chercheur, exprimait ainsi le souhait d'interdire formellement « tout essai de thérapie génique sur des cellules germinales ». Elle ajoutait : « Il s'agit également de bien encadrer les essais de thérapie génique pour éviter la dispersion des organismes génétiquement modifiés parmi les cellules de la reproduction. Cette interdiction doit être bien encadrée et maintenue ».
M. Carlos de Sola estimait nécessaire, lui aussi, d'interdire les expérimentations de thérapie germinale au stade actuel des connaissances scientifiques, pour se protéger des risques de dérive de la recherche : « Tout récemment (...) une association américaine s'est prononcée pour l'interdiction de la thérapie germinale. Une telle attitude est indispensable, notamment lorsqu'on prend en compte l'enfant qui va naître, mais aussi pour protéger la recherche d'éventuels dérapages. Cela n'empêche cependant pas que l'on puisse revenir sur les interdictions avec les développements de la recherche. Aujourd'hui, il est préférable de poser une interdiction pour, ensuite, l'aménager, plutôt que de laisser faire, au risque de s'exposer à des dérapages qu'on ne pourrait pas contrôler ».
Il indiquait par ailleurs que le protocole additionnel à la convention d'Oviedo précédemment évoqué devrait poser deux principes forts : « Le premier (...) concerne les tests et le génome. Il précise que l'on peut réaliser des tests uniquement pour des raisons médicales et que les modifications apportées au génome ne peuvent avoir qu'une finalité de santé. Le second (...) a été probablement adopté pour des raisons de sécurité. Il interdit la thérapie germinale, en particulier la thérapie sur les premiers stades de l'embryon. On ne peut donc modifier le génome de la descendance. Ce principe n'interdit cependant pas les recherches sur les embryons surnuméraires, en particulier celles visant à modifier le génome d'un embryon surnuméraire qui ne serait pas destiné à être implanté, dans la mesure où le terme descendance est compris comme l'enfant à naître ».
Une telle attitude serait en effet des plus sages devant le désir de certains chercheurs de pousser sans cesse plus loin les limites de leurs études. Cependant, cette position n'est pas unanimement partagée. Un biologiste français écrivait ainsi dans une revue spécialisée (84) : « Soigner c'est bien, guérir complètement, y compris la lignée germinale, c'est l'idéal. Seul permettra d'y parvenir le clonage intracouple par transfert dans un ovocyte de la femme de cellules ES issues d'un blastocyste du couple, corrigées génétiquement ou de cellules EG provenant d'un avortement thérapeutique et également corrigées ».
Notre collègue Jean-François Mattei, lors d'une audition de la Mission le 6 septembre 2000, considérait pour sa part qu'il faut dès à présent envisager la thérapie germinale et refuser « l'anathème » dont elle fait l'objet : « (qui) conduit à une sorte d'incohérence médicale consistant à faire un diagnostic en s'interdisant éventuellement de soigner à terme (...) En clair, lorsque l'on a un _uf sur lequel on voit un gène de la mucoviscidose ou un gène de la myopathie, est-ce que notre but ultime - lorsque les techniques seront plus au point - n'est pas de corriger l'anomalie plutôt que d'éliminer l'_uf ? (...) On ne peut pas considérer comme acquise la discussion sur la thérapie germinale, parce que la logique médicale est bien de soigner plutôt que d'éliminer ».
Votre Rapporteur considère qu'en l'état actuel des connaissances, il est encore trop tôt pour permettre cette recherche au regard des risques d'eugénisme qu'elle présenterait.
B.- DES LIMITES ET DES CONDITIONS CLAIRES À POSER
1.- Le consentement des parents au don de leurs embryons
surnuméraires à la recherche
Un large accord s'est dessiné parmi les interlocuteurs de la Mission en faveur de la recherche sur les embryons surnuméraires dès lors que les parents, qui renonceraient à la poursuite de leur projet parental, y consentiraient expressément. C'est la position qu'exprimaient notamment le CCNE dans son avis portant sur l'avant-projet de loi tendant à la révision des lois relatives à l'éthique biomédicale, ainsi que Conseil d'État dans le rapport précité, l'Académie nationale de médecine, la CNCDH dans son avis du 29 juin 2000 et la CNMBRDP. Pour le Docteur Axel Kahn, cette possibilité serait le témoignage d'une solidarité transgénérationnelle ; il posait ainsi devant la Mission, lors de son audition précitée , la question suivante :
« Est-il moins respectueux de la dignité que l'on doit à l'embryon de le détruire sans autre forme de procès ou bien, avec avis de ses géniteurs, de l'utiliser dans une recherche dont on attend la manifestation d'une solidarité envers le traitement de vies futures qui pourront advenir et en vue desquelles on veut acquérir de nouvelles informations ? (...)
Quant à moi, qui suis attaché au principe du respect de l'embryon, personne n'a pu encore me convaincre du fait que décongeler un embryon et le jeter dans l'évier serait lui manifester de la dignité. Alors que l'intégrer dans un protocole de recherche - du type de celui que j'ai évoqué - me semble quelque chose de tout à fait acceptable. Cela traduit une forme réelle de solidarité, que je rapprocherai de celle s'exprimant dans le don d'un organe d'un homme mort à un homme vivant, c'est-à-dire manifestant une solidarité au-delà de la mort, dans le cas de l'embryon, au-delà du non-développement ».
Le Sénateur Claude Huriet, lors de son audition devant la Mission, regrettait une attitude quelque peu utilitariste de la part de certains chercheurs : « On part des embryons surnuméraires ne faisant plus l'objet d'un projet parental et on dit : « il vaudrait mieux qu'ils servent à quelque chose ». Cette expression est tout à fait révélatrice : si on dit d'un embryon humain qu'il doit servir à quelque chose, on n'a pas besoin de définir ce qu'est l'instrumentalisation de l'embryon ».
Votre Rapporteur considère pour sa part que la possibilité d'ouvrir la recherche sur les embryons surnuméraires pourrait être reconnue dès lors que le consentement des parents, préalablement informés des objectifs de cette recherche, serait clairement et préalablement exprimé, que les limites à ces recherches seraient strictement établies, notamment par la loi, et qu'un suivi et un contrôle seraient assurés par une autorité indépendante.
Selon Mme Chantal Ramogida, fondatrice de l'association « Pauline et Adrien », auditionnée par la Mission le 21 juin 2000, de nombreux couples engagés dans une procédure d'AMP seraient disposés à faire don de leurs embryons surnuméraires à la recherche : « Nous avons fait une enquête (au sein de l'association) : 68 % des patientes ont répondu favorablement à la recherche sur les embryons qu'elles ne désiraient plus ».
Certains craignent toutefois qu'à terme, si les espoirs de la recherche devaient se confirmer, le nombre des embryons surnuméraires disponibles pour les applications thérapeutiques ne soit insuffisant. C'est la question que posait devant la Mission M. Frédéric Salat-Baroux, l'un des auteurs du rapport du Conseil d'État précité lors de son audition le 24 mai 2000 : « Est-ce que ce sera suffisant ? Si cette technique fonctionne, n'aura-t-on pas besoin de plus de matériau, ce qui conduira à produire des embryons pour créer la base de ces thérapies cellulaires ? ». Le CCNE lui-même, dans son avis précité, exprimait la même crainte en rappelant que : « Le nombre des embryons surnuméraires potentiellement disponibles pour la recherche est normalement appelé à décroître dans l'avenir, compte tenu de la meilleure maîtrise des techniques et de la diminution des besoins liée à la réduction du nombre moyen des embryons transférés à chaque intervention. Il ne faudrait pas, dans ces conditions, que l'assistance médicale à la procréation soit mise à profit pour constituer volontairement des embryons surnuméraires en vue de les utiliser ultérieurement pour la recherche ».
On peut regretter qu'à ce sujet, la convention d'Oviedo soit muette et s'en remette aux choix de chaque État membre du Conseil de l'Europe, ainsi que l'indiquait M. Carlos de Sola, lors de son audition précitée devant la Mission : « S'agissant des embryons surnuméraires, la convention reste muette et les points de vue sont divergents. Autrement dit, un État qui autorise la recherche, y compris la recherche destructive sur des embryons surnuméraires, peut ratifier la convention sans être en difficulté, au même titre qu'un État qui interdit toute recherche destructive.
Un protocole additionnel est actuellement en préparation sur la protection de l'embryon et du f_tus humains. Sa position sera probablement la même, le choix du principe de l'utilisation des embryons surnuméraires pour la recherche sera laissé à chaque État. Ensuite, pour les États qui auraient choisi d'autoriser une telle recherche, des normes et des principes fondamentaux devront être respectés ».
2.- Le stade de développement de l'embryon sur lequel la recherche serait autorisée doit être précis et limité
Si le législateur décide lors de la révision des lois bioéthiques de permettre la recherche sur les embryons et leurs cellules, il lui faudra déterminer avec précision le stade de développement de l'embryon jusque auquel la recherche serait autorisée. Les Britanniques ont choisi, quelque peu arbitrairement, le stade du quatorzième jour après la fécondation en s'appuyant sur une notion de « préembryon » qu'ils ont depuis plusieurs années abandonnée (85) en reconnaissant son absence de signification. Il correspond cependant au moment où apparaît la gouttière primitive, ébauche du système nerveux.
Le Docteur Peter Braude, membre de la HFEA indiquait que les recherches prévues par la grande majorité des protocoles autorisés au Royaume Uni ont lieu beaucoup plus tôt :
« Il faut bien savoir que la limite de quatorze jours est théorique. La plupart des projets de recherche ne vont pas au-delà de cinq jours, du stade du blastocyste. Nous ne cultivons pas des embryons au-delà de ce stade ; il existe certains projets où l'on essaie de voir, de manière artificielle, comment a lieu le processus d'implantation du blastocyste dans l'utérus. Il serait important de mener ces recherches pour avoir plus de connaissances sur les fausses couches, mais cela n'est pas encore fait de manière active ».
Il rappelait par ailleurs qu'au-delà de dix jours, un embryon in vitro ne peut poursuivre son développement s'il n'a pas été implanté.
L'embryogenèse, c'est-à-dire l'organisation de la grossesse in vitro étant loin d'être à ce jour envisageable, ce dont on peut se réjouir, la véritable question se situe donc à propos du stade de développement de l'embryon qu'il serait opportun de définir avant ces dix jours.
L'avant-projet de loi de révision des lois bioéthiques proposait de choisir le stade précédant la différenciation tissulaire, laquelle commence environ entre le sixième et le huitième jour de développement. De l'avis de plusieurs scientifiques et du CCNE, ce processus est continu et son commencement ne peut être clairement défini ; le CCNE considère que cette référence est « abstraite et ambiguë ». Il écrivait ainsi, dans son avis précité : « Selon qu'on considère le moment où les tissus qui donneront le placenta se différencient de ceux qui donneront le bouton embryonnaire, ou celui auquel se différencie tel ou tel tissu de l'embryon, on se situe à des stades de développement très éloignés dans le temps. L'implantation de l'embryon dans l'utérus constitue en revanche un événement ponctuel considérable. Le Comité recommande donc de substituer à la référence proposée une référence à la fin du stade préimplantatoire, c'est-à-dire au moment où l'embryon acquiert la capacité à s'implanter dans l'utérus ».
Le Docteur Axel Kahn considérait lui aussi, lors de son audition précitée devant la Mission, que le stade de l'implantation, à partir duquel la recherche ne serait plus possible, serait un bon choix : « Pour toute une série de raisons, il serait raisonnable, de toute façon, de limiter ces recherches jusqu'au stade normal de l'implantation qui se situe aux alentours du blastocyste, c'est-à-dire de sept jours, en tout cas avant l'éclosion. Vous savez probablement qu'il n'y a pas que les _ufs de poule qui éclosent, les _ufs humains également, tous les _ufs de mammifères. Il y a un moment où l'embryon sort de la membrane pellucide vers le huitième jour. Il faudrait impérativement qu'on limite la recherche à la période antérieure. On peut en discuter les raisons, mais cela me semble vraiment raisonnable. De toute façon, même si l'embryon ne change pas de nature, à partir du moment où il y a implantation, il est bien vrai que les relations entre la mère et son embryon sont tout autres ».
3.- La nécessité absolue de protéger les femmes
qui donneraient leurs ovocytes à la recherche
Si le « clonage thérapeutique » devait être autorisé, il nécessitera sans doute un grand nombre d'ovocytes. La deuxième partie du présent rapport a exposé les risques que fait peser, sur la santé des femmes, la stimulation ovarienne et le prélèvement des ovocytes, qui nécessite une intervention chirurgicale loin d'être bénigne.
Le CCNE, dans son avis précité, s'est vivement inquiété des risques auxquelles seraient exposées les femmes, dénonçant un « vide juridique » autour de cette question. On peut par ailleurs craindre que des pressions excessives soient exercées à l'encontre des femmes, pressions des médias, voire pressions familiales, pour les inciter à faire don de leurs ovocytes.
Le Professeur Ian Wilmut, « père » de la brebis Dolly, se disait ainsi persuadé, lors des auditions publiques de l'Office parlementaire d'évaluation des choix scientifiques et technologiques du 25 novembre 1999, que les femmes seraient nombreuses à donner leurs ovocytes dans la perspective de sauver des vies : « Comme des personnes donnent leurs cellules de moelle osseuse, par une intervention chirurgicale mineure mais douloureuse, dans bien des cas, les femmes seraient prêtes à donner leurs ovules si cela devait permettre de soigner des maladies graves. S'il était possible de prendre l'ovule d'une mère dont l'enfant est atteint de leucémie, pour produire un embryon génétiquement identique à l'enfant malade, il serait possible d'obtenir les cellules nécessaires pour remplacer les leucocytes endommagés. Beaucoup de femmes seraient certainement très heureuses de pouvoir sauver ainsi leur enfant. Elles seraient sans doute également prêtes à aider de la même manière un de leurs parents, un ami, un proche. Vous connaissez tous des personnes atteintes des maladies que nous avons évoquées. Envisageriez-vous de donner un ovule pour aider un ami atteint ? »
Votre Rapporteur considère que cette question est l'une des plus importantes que pose le « clonage thérapeutique ». Il considère que l'on ne peut assimiler le don d'ovocytes à un quelconque don de cellules dans la mesure où les gamètes contiennent le patrimoine génétique de la personne et où le prélèvement des ovocytes met en péril la santé des femmes. Il faut donc apporter des garanties maximales au don d'ovocytes, si celui-ci devenait nécessaire dans le cadre d'une recherche autorisée, en s'interrogeant sur le risque de créer deux filières, l'une pour la recherche et l'autre pour l'AMP, la seconde étant en situation permanente de pénurie à l'inverse vraisemblablement de la première qui est susceptible de faire l'objet de vastes campagnes d'information au regard des enjeux de la recherche.
Pourquoi, dans ce cas, ne pas imaginer un recrutement commun des donneuses qui pourraient exercer librement leur choix en donnant leurs ovocytes pour la recherche ou pour l'AMP ou pour les deux finalités concurremment ?
4.- La transparence des recherches doit être assurée
La transparence des recherches est un devoir vis à vis de l'opinion publique et une nécessité pour s'assurer du respect du cadre législatif qui serait défini. Mme Chantal Ramogida, dans son audition précitée, exigeait pour les parents qui feraient don de leurs embryons à la recherche, un droit à l'information étendu : « Toute recherche sur l'embryon ne peut se faire qu'avec nos gamètes, nous demandons donc au corps médical un droit d'information. Ces recherches doivent être faites selon un protocole précis et contrôlé, et donner lieu à une publication. Nous demandons une information régulière, pour éviter toute dérive. (...) En tant qu'association de patients, nous devons être tenus informés de ces recherches et de leur date d'aboutissement ».
La législation britannique est particulièrement précise et intéressante à ce sujet. Le Professeur Peter Braude, membre de la HFEA, indiquait ainsi devant la Mission le 5 juillet 2000 que le consentement des couples doit être très spécifique : « Lorsqu'ils remplissent les formulaires, les couples doivent exprimer leur accord pour que les embryons soient utilisés pour la recherche, donnés à d'autres personnes ou détruits. Ce sont les embryons surnuméraires. Les couples doivent savoir quel projet de recherche est autorisé. Les informations doivent toujours leur être données par écrit ».
M. Philippe Pedrot, dans son audition précitée devant la Mission, prônait quant à lui la transparence des recherches sur l'embryon en considérant qu'elles ne doivent pas relever uniquement de la responsabilité scientifique.
5.- La finalité précise de la recherche et l'absence d'alternative à l'utilisation de l'embryon ou de ses cellules doivent être prouvées
L'avant-projet de loi manque de clarté en utilisant différents termes pour qualifier la finalité des recherches qui peuvent être autorisées. En conséquence, la distinction entre les notions de « finalité médicale », « finalité scientifique » et « finalité thérapeutique » appelle un effort de clarification et une utilisation rigoureuse. Mme Noëlle Lenoir, Présidente du Groupe européen d'éthique des sciences et des nouvelles technologies auprès de la Commission européenne notait ainsi, dans son audition devant la Mission le 31 janvier 2001, que « l'on parle aujourd'hui volontiers de « clonage thérapeutique », alors qu'il n'a encore rien de thérapeutique, et de thérapie génique alors que, pour l'instant, ses vertus thérapeutiques sont assez limitées sinon presque inexistantes en dehors de la guérison des bébés bulles (...) ».
S'agissant de la recherche sur les cellules ES, qui se trouve aujourd'hui et encore pour quelques années, au stade de la recherche fondamentale afin de comprendre puis de maîtriser les processus d'induction de leur prolifération et de différenciation, votre Rapporteur propose de parler de « recherches scientifiques à visée thérapeutique ». Cette formule permettait de montrer que l'on se trouve toujours dans une phase de recherche fondamentale, visant à accroître le champ des connaissances, tout en limitant celle-ci à un but thérapeutique afin que n'importe quelle manipulation sur l'embryon ou ses cellules ne soit pas permise.
Les protocoles de recherche impliquant l'utilisation d'embryons ou de cellules ES devraient de plus n'être autorisés qu'à la condition de prouver qu'il n'existe aucune méthode alternative permettant de poursuivre la même finalité de recherche sans devoir recourir à l'utilisation d'un embryon ou de cellules embryonnaires, en utilisant par exemple des cellules souches adultes.
C.- GARANTIR LE RESPECT DE CES INTERDITS ET DE CES LIMITES
PAR UN ENCADREMENT RIGOUREUX
C'est sans doute l'élément essentiel qui peut fonder l'autorisation de la recherche sur l'embryon par le législateur et au-delà par la société qui doit bénéficier de solides garanties sur l'encadrement de la recherche sur l'embryon.
Pour de nombreux interlocuteurs de la Mission, le contrôle, le suivi et l'évaluation des recherches doivent être assurés par une autorité indépendante, experte sur ces questions, dotée de réels moyens et de pouvoir. C'est ainsi que pour, le Docteur Marie-Odile Alnot, la création de cette instance est une condition sine qua non pour l'autorisation de cette recherche.
La HFEA, dont le rôle et la composition ont été précédemment analysés, constitue à cet égard un modèle intéressant puisque sa compétence est étendue à l'autorisation et au suivi des recherches sur l'embryon. Le Professeur Peter Braude, membre de l'Autorité britannique, expliquait aux membres de la Mission, lors de son audition le 12 juillet 2000, la procédure extrêmement précise suivie pour examiner les demandes d'autorisation de recherche sur l'embryon :
« Le formulaire de demande porte sur les objectifs du projet, la méthodologie que l'on a l'intention de suivre. S'agissant des embryons, il faut donner une estimation du nombre d'ovocytes que l'on a l'intention d'utiliser, l'origine de ces embryons, c'est-à-dire s'ils sont donnés par les patients ou créés en vue de la recherche. Le dossier est soumis à l'appréciation d'experts qui statuent sur le domaine, l'importance de la recherche et son originalité, sur le point de savoir si l'utilisation d'embryons humains est justifiée, si la méthodologie est raisonnable. Il faut également s'assurer de la réputation de la personne qui demande l'autorisation et savoir si elle aura la capacité de mener cette recherche à bien. Voilà ce que l'on demande aux experts. Le comité peut alors octroyer une autorisation, la refuser dans sa forme actuelle ou encore demander de nouvelles informations à la personne qui dépose une candidature. Un centre qui n'a pas la licence pour un traitement (86) sera obligatoirement inspecté en tant que tel, sinon le suivi de la recherche se fera à l'occasion de l'inspection annuelle. Chaque année, un rapport est fait sur la progression du projet de recherche. Après trois ans, un rapport final doit être présenté ».
La question se pose enfin de savoir quel type de pouvoir devrait être confié à l'agence qu'il est envisagé de créer pour assurer le contrôle des activités d'AMP, agence à laquelle seraient également confiés l'autorisation, le contrôle et le suivi des recherches sur l'embryon. L'avant-projet de loi n'envisage qu'un pouvoir consultatif. Certes, il est prévu que les avis de cette agence seront publics et que les ministres chargés de la santé et de la recherche, à qui reviendrait le pouvoir d'autorisation de la recherche, devraient en tenir compte.
Votre Rapporteur est convaincu, avec plusieurs des interlocuteurs de la Mission et certains de ses collègues, qu'il est nécessaire d'aller plus loin et de confier un véritable pouvoir de décision à la future agence, à l'instar de la HFEA britannique. Il ne s'agit pas, bien sûr, de confisquer en l'espèce le pouvoir au bénéfice d'experts, d'autant que cette agence devrait, comme on l'a précédemment souligné, refléter dans sa composition l'ensemble de la société civile. Il ne s'agit pas, non plus, de déposséder le politique de ses responsabilités. En revanche, il paraît souhaitable que le législateur détermine précisément la finalité des recherches sur l'embryon et les conditions de son autorisation. À l'agence reviendrait ensuite le pouvoir de se déterminer, à partir de ce cadre législatif, en assurant la transparence de ses décisions, lesquelles pourraient toujours faire l'objet d'un recours devant le juge. Les ministres de tutelle de l'agence pourraient cependant disposer d'un pouvoir de suspension temporaire des décisions de l'agence par arrêté motivé, en lui demandant de statuer une seconde fois sur une demande d'autorisation, sur le modèle de ce que prévoit l'article L. 5322-2 du code de la santé publique pour les décisions de l'AFSSAPS (87), créée par la loi du 1er juillet 1998. Cet article dispose en effet que « le directeur général de l'agence, prend, au nom de l'État, les décisions qui relèvent, en ce qui concerne les produits (...) de la compétence de celle-ci (...) ainsi que des mesures réglementaires prises pour l'application de ces dispositions. Les décisions prises par le directeur général en application du présent article ne sont susceptibles d'aucun recours hiérarchique. Toutefois, en cas de menace grave pour la santé publique, le ministre chargé de la santé peut s'opposer, par arrêté motivé, à la décision du directeur général et lui demander de procéder, dans le délai de trente jours, à un nouvel examen du dossier ayant servi de fondement à ladite décision. Cette opposition est suspensive de l'application de cette décision ».
Ainsi le législateur serait assuré que le cadre qu'il aura fixé pour l'autorisation de la recherche sur l'embryon sera respecté sans que les autorisations données puissent faire l'objet de décisions qui pourraient être subjectives si elles ne dépendaient que de la volonté des ministres concernés. On ne peut en effet écarter l'hypothèse où un ministre ayant une position a priori sur cette recherche viderait de son contenu la volonté du législateur.
QUATRIÈME PARTIE :
DON ET UTILISATION DES ÉLÉMENTS DU CORPS HUMAIN
Le 22 juin 2000, Mme Martine Aubry, ministre de l'emploi et de la solidarité, annonçait, dans son « Plan greffes », vingt-cinq mesures destinées à remédier à la pénurie de greffons. Elle lançait la journée nationale de la greffe, qui se tiendra désormais tous les 22 juin.
Ce Plan, dont le but est de « construire une véritable politique publique du prélèvement et de la greffe », s'est donné quatre priorités :
- favoriser l'accès à la greffe, c'est-à-dire avant tout au greffon ;
- réduire les inégalités régionales d'accès à la greffe ;
- renforcer la solidarité et soutenir la générosité de nos concitoyens ;
- enfin, accompagner les efforts de recherche pour améliorer les résultats.
Ce plan a été présenté dans un climat de relative sérénité ; après la crise du début des années quatre-vingt-dix, la confiance est, semble-t-il, revenue.
Comme l'a rappelé le Professeur Didier Houssin, Directeur général de l'Établissement français des Greffes, lors de son audition, par la Mission, le 10 janvier 2001, l'activité de la greffe se trouvait, à l'époque, dans un climat de crise : l'inquiétude était vive « en matière de sécurité sanitaire sur les tissus, de suspicion de trafic de priorité, d'un pourcentage très élevé de patients non résidents sur certaines listes d'attente, d'un dysfonctionnement notable en matière de consentement à l'occasion de l'affaire dite d'Amiens ». Il est donc utile de rappeler dans quelles circonstances a dû travailler le législateur de 1994.
I.- LES LOIS DE 1994 ONT ÉTÉ ADOPTÉES DANS UN CONTEXTE DÉFAVORABLE À L'ACTIVITÉ DE TRANSPLANTATION
Lors de leur audition par la Mission le 10 janvier 2001, les Professeurs Didier Houssin et Dominique Durand l'ont rappelé : « le climat était un climat de crise. » (Professeur Didier Houssin) ; « ...l'Établissement français des greffes a été installé dans une période difficile pour la transplantation, difficile parce qu'il y avait incontestablement, à cette époque, une rupture de confiance entre la société et le monde médical de la transplantation » (Professeur Dominique Durand).
Malgré de réels progrès dans l'efficacité thérapeutique de la greffe grâce à l'amélioration des méthodes chirurgicales, à une meilleure connaissance des systèmes d'incompatibilité des tissus et à l'apparition de nouveaux médicaments, les années 1991 à 1993 sont marquées par une chute considérable des prélèvements (30 % pour la cornée ; 20 % pour les organes).
En outre, les chiffres dont nous disposons actuellement montrent que le déclin allait se poursuivre : 1 095 personnes prélevées en 1991 contre 889 en 1995 ; 202 personnes se sont opposées au prélèvement d'organe en 1991, contre 484 en 1995. Ce sont ainsi plus de trois cents malades qui sont décédés en attente d'organes vitaux pendant cette période (88).
L'histoire de la greffe montre que les premières greffes réussies ont été effectuées dans les années cinquante pour les transplantations rénales, la première greffe du c_ur remontant à 1968. Mais ces résultats, portant sur de très petits nombres de greffes, n'étaient pas statistiquement probants. Les besoins en organes se sont développés dans le courant des années 1970.
LES GRANDES DATES DE LA TRANSPLANTATION | |
1959 |
première transplantation réussie du rein |
1967 |
première transplantation réussie du c_ur |
1972 |
première transplantation du foie |
1976 |
première transplantation du pancréas |
1981 |
première transplantation du bloc c_ur-poumons |
1987 |
première transplantation du poumon |
Les premières greffes de moelle osseuse au sein de familles (entre frères et s_urs au système Human Leucocyte Antigen identique) ont été réalisées au cours des années 1970. Initialement tentée autour du concept de l'histocompatibilité, la greffe fera un progrès déterminant grâce à l'utilisation de drogues immunosuppressives. Ces progrès font que désormais la greffe d'organes s'est banalisée, et que son indication se multiplie, entraînant de facto un besoin accru en organes.
Initialement, la transplantation d'organe reposait sur la seule générosité du don. La loi du 7 juillet 1949 relative à la greffe de la cornée, faisait dépendre le don des yeux d'une démarche volontaire. C'est le principe du consentement explicite. Lors de son audition par la Mission le 4 octobre 2000, M. Henri Caillavet a expliqué comment la confidence du Professeur Hamburger sur le manque de greffons serait à l'origine de la loi n° 76-1181 du 22 décembre 1976 sur les prélèvements d'organes. Son article premier autorise le prélèvement en vue d'une greffe ayant un but thérapeutique « sur une personne vivante majeure et jouissant de intégrité mentale, y ayant librement et expressément consenti », et son article 2 dispose que « des prélèvements peuvent être effectués à des fins thérapeutiques ou scientifiques sur le cadavre d'une personne n'ayant pas fait connaître de son vivant son refus d'un tel prélèvement ». C'est le principe du consentement présumé.
À l'usage, la loi s'est révélée difficilement applicable, la règle du consentement présumé étant juridiquement contestable, et les hésitations des pouvoirs publics n'ont pas contribué à clarifier la situation, d'autant que l'on a pu constater des pratiques contestables. La multiplication des indications, de même que l'extension des prélèvements à la quasi-totalité du corps humain, y compris les gamètes, ont rendu nécessaire une refonte de la législation.
Plusieurs événements peuvent expliquer la crise qui sévissait avant l'intervention des lois de 1994.
1.- Le cas du coma dépassé d'Amiens (89)
Au cours d'un procès, un anesthésiologiste fait état d'une expérimentation simulant les conditions qui ont précédé la mort d'une patiente. Faite quelques jours auparavant sur un malade en état de « coma dépassé », cette expérimentation a fait l'objet d'un « testament scientifique » de la part de son auteur, dans le Quotidien du médecin du 29 février 1988 dans lequel il précise que « La France s'honorerait à être le premier pays dans lequel l'expérimentation humaine sur des volontaires soit possible en cas de coma dépassé aussi bien que dans celui d'état végétatif chronique, étant admis que la différence fondamentale entre ces deux états pathologiques est qu'un coma dépassé est une mort cérébrale alors qu'un état végétatif est une personne humaine très diminuée, sans aucune communication avec l'extérieur et dont le degré de conscience reste jusqu'ici problématique ».
Une plainte avec constitution de partie civile pour coups et blessures volontaires sur une personne hors d'état de se protéger elle-même a été déposée. Dans un communiqué, Mme Michèle Barzach, ministre de la santé, précisait : « Cette affaire ne doit pas conduire à jeter le discrédit dans l'esprit du public sur la médecine hospitalière et sur les dons d'organes ».
2.- Le cas des multi-prélèvements (90)
Les parents d'un jeune homme de 19 ans, décédé en juillet 1991 des suites d'un accident de bicyclette, reçoivent du CHU un avis des sommes à payer adressé à leur fils, lequel document mentionne en outre qu'une série d'actes de chirurgie a été réalisée sur le corps. La mère du jeune homme apprendra que la facturation du forfait journalier résulte d'une « erreur de manipulation informatique par un personnel de remplacement pendant une période de vacances » et le détail des prélèvements effectués : le c_ur, le foie, les deux reins et les deux cornées. Les quatre bénéficiaires de ces prélèvements, deux hommes et deux femmes âgés de vingt-cinq à cinquante ans, vont bien.
Toutefois, comme les circonstances du décès de leur fils sont mal connues, les parents ont porté plainte pour homicide involontaire contre le responsable de l'accident. Ayant à ce titre accès au dossier d'instruction, ils ont entre les mains le compte rendu opératoire résumant les gestes pratiqués par les chirurgiens sur le cadavre de leur fils. Ce document révèle que les prélèvements comprenaient, outre le c_ur, le fois et les reins, l'aorte descendante avec les gros troncs supérieurs de la crosse, l'artère iliaque et fémorale droite, les deux veines saphènes internes et la veine fémorale droite. Les parents apprennent également que des globes oculaires ont été placés sur le cadavre après prélèvement des cornées, (qui relevait de la loi du 7 juillet 1949 prévoyant le don des yeux sous forme testamentaire) et que toutes les incisions ont été refermées en deux plans, « avec fil métallique au niveau du thorax ».
Bien que militant pour le don d'organes, les parents de ce jeune homme ont porté plainte pour viol et violation de sépulture.
3.- La rumeur des voleurs d'organes
Les affaires évoquées ci-dessus touchaient à plusieurs principes éthiques aux fondements anthropologiques : respect du mort et de sa dépouille, importance accordée, par la famille qui vient de perdre un être cher, à l'observation de cette prescription.
Une autre affaire a contribué à atteindre l'image de la transplantation. Il s'agit d'une rumeur selon laquelle il existerait une « mafia » des voleurs d'organes. Dans un document intitulé « La greffe, la rumeur et les médias : les récits de vols d'organes » (91), rédigé à la demande de l'Établissement français des Greffes, Véronique Campion-Vincent, ingénieur de recherche au CNRS, a fait une enquête sur les rumeurs persistantes selon lesquelles une mafia internationale du trafic des organes et tissus humains organiserait, pour le compte de chirurgiens indélicats et cupides, un système de prélèvement sous contrainte de reins, de c_urs et de globes oculaires sur des enfants ou des adultes du tiers-monde, en destination de riches patients des pays industrialisés. Le point d'orgue de cette rumeur a été l'adoption, par le Parlement européen le 15 septembre 1988, d'une motion condamnant « les trafics de bébés du tiers-monde ».
Les psychosociologues définissent la rumeur (92) comme étant le phénomène de propagation d'une nouvelle qui n'a pas de rapport avec la question de la vérité et de l'erreur - elle est, indifféremment, messagère de l'une ou de l'autre - et qui répond à autre chose que le besoin de connaissance, et qui serait à chercher du côté de l'inconscient collectif. Elle s'impose par le fait de sa diffusion et subit, dans sa diffusion, un type de déformation qui accroît son impact, car elle se nourrit d'elle-même.
Préférant parler de récit et de légende, Véronique Campion-Vincent met en évidence que, bien que cumulant force détails, les récits relatifs aux prélèvements contraints d'organes ne résistent pas à une vérification plus approfondie. Cependant, il existe incontestablement des faits qui rendent ces récits plausibles, comme la vente de reins dans des pays du tiers-monde ou l'exploitation illicite des tissus cadavériques. De nombreux pays maltraitent leurs enfants de bien des façons, et, en effet, « ceux qui tuent sans autre motif que de faire régner l'ordre, ceux qui réduisent les enfants en servitude et les utilisent pour la prostitution, hésiteraient-ils à tuer leurs victimes pour en tirer un profit ? certes pas... ». L'inconscient collectif le pense également. Elle voit dans cette rumeur une « métaphore aggravée de l'exploitation du tiers-monde et des ses matières premières par les pays riches ».
Mais d'autres données contredisent ces rumeurs, qu'il s'agisse des contraintes techniques concernant la circulation des organes, de l'importance de l'équipement chirurgical nécessaire, du suivi des greffes et du nombre nécessaire de personnes impliquées dans ces activités.
Par ailleurs, qu'aurait à gagner l'élite médicale à se compromettre dans de tels trafics ?
Comment de tels récits deviennent-ils convaincants dans nos pays ? Rappelant qu'en chaque être le mode de pensée rationnelle et réflexive alterne avec le mode de la pensée emblématique, Véronique Campion-Vincent explique que ces récits « réactivent la fable du massacre des innocents ». En outre, « ils permettent d'exprimer, par le détour d'une histoire, des réticences ressenties, mais non explicitées face aux avancées de la médecine et à la greffe ». Ces réticences trouveraient leurs racines dans le rejet croissant de l'utilisation thérapeutique des éléments, causé par quelques scandales retentissants comme l'expérience sur des comas dépassés, bien relayés par les médias, et par la difficulté à accepter la notion de mort cérébrale, qui donne une nouvelle apparence à la mort alors que le corps respire et que le c_ur continue de battre.
Même si la finalité des prélèvements d'organes bénéficie d'une appréciation positive, car il s'agit de sauver des vies, réfléchir sereinement au don n'est pas chose aisée, car cela oblige chacun à envisager sa propre mort.
L'étude commandée par l'Établissement français des greffes a le mérite de souligner les réticences qui s'expriment par l'intermédiaire des rumeurs et montre qu'il serait vain de stigmatiser ces réticences pour promouvoir le don d'organes.
II. - LA RÉPONSE DU LÉGISLATEUR :
DONNER UN STATUT AU CORPS HUMAIN
Il est acquis désormais que les progrès de la science et de la médecine ont profondément modifié la fonction thérapeutique (93) : initialement centrée sur la personne soignée, la médecine est amenée à multiplier les interventions « à caractère manipulatoire » (94) sur le corps humain qui posent la question de leur légitimité. En outre, le développement de ces nouvelles technologies modifie les frontières traditionnelles de la vie et de la mort.
Après la publication d'un nombre important d'études relatives à la bioéthique, le Gouvernement dépose, en 1992, trois projets de loi, en vue de « reconnaître un statut au corps humain tout en autorisant certaines interventions aussi bien pour autrui que dans l'intérêt de la recherche » (95). Le premier, relatif au corps humain, modifie le code civil et pénal. Le second complète le code de la santé publique, et le troisième la loi « informatique et libertés ».
Après deux lectures dans chaque Assemblée et réunion d'une commission mixte paritaire - la première lecture à l'Assemblée nationale ayant eu lieu au cours de la neuvième législature - les lois n° 94-653 du 29 juillet 1994 relative au respect du corps humain, et n° 94-654 du 29 juillet 1994 relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal ont posé des principes généraux afin de donner un statut juridique au corps humain. Elles ont affirmé les principes régissant le don et l'utilisation de ses éléments et produits et les modalités particulières de ce don et de cette utilisation.
Par ailleurs, les lois précitées de 1949 (dite Loi Lafay), relative à la greffe de cornée à l'aide de donneurs d'yeux volontaires, et de 1976 (dite Loi Caillavet), relative aux prélèvements d'organes, ont été abrogées.
A.- L'INSCRIPTION DU RESPECT DU CORPS HUMAIN DANS LE CODE CIVIL :
DES PRINCIPES GÉNÉRAUX
Les articles 16 à 16-9 sont des dispositions d'ordre public qui définissent les principes généraux garantissant le respect du corps humain. En outre, le Conseil constitutionnel a reconnu une valeur constitutionnelle au principe de la sauvegarde de la dignité de la personne humaine.
Le législateur a inscrit dans le code civil la primauté de la personne, l'interdiction de toute atteinte à sa dignité et le respect de l'être humain dès le commencement de la vie (article 16).
L'inviolabilité et l'indisponibilité du corps humain figurent à l'article 16-1 du code précité ; l'inviolabilité n'interdit pas l'atteinte à l'intégrité physique de la personne en cas de nécessité médicale (96) pour elle-même ou pour autrui, sous réserve de son consentement (article 16-3).
L'article 16-4 consacre l'intégrité de l'espèce humaine et interdit toute pratique eugénique tendant à la sélection des personnes, les articles 16-5, 16-6, 16-7 et 16-8 affirment respectivement la non-patrimonialité du corps humain, le principe de gratuité en interdisant toute rémunération du donneur, la nullité de toute convention sur la procréation ou la gestation pour le compte d'autrui, et le principe d'anonymat.
B.- L'APPLICATION DE CES PRINCIPES AU DON ET À L'UTILISATION DES ÉLÉMENTS ET PRODUITS DU CORPS HUMAIN.
Ces principes sont repris dans le code de la santé publique et figurent, depuis la nouvelle codification, au titre premier du livre II relatif au don et à l'utilisation des éléments et produits du corps humain (article L. 1211-1 à L. 1211-9). Après référence au code civil (article L. 1211-1), sont donc successivement réaffirmés le principe du consentement au prélèvement y compris après la mort, et sa révocabilité (article L. 1211-2), l'interdiction de la publicité du don (article L. 1211-3), la gratuité (article L. 1211-4), l'anonymat (article L. 1211-5), et les règles de sécurité sanitaire et de vigilance (articles L. 1211-6 et L. 1211-7). L'article L. 1211-8 exclut de ces dispositions certains éléments : les cheveux, poils, ongles, dents, urines et autres produits d'excrétion, ainsi que le lait maternel.
Le code de la santé publique complète le code civil en matière de transparence et de sécurité sanitaire et organise, par ailleurs, le prélèvement d'organes sur les personnes décédées.
A.- UNE AMÉLIORATION GLOBALE DE LA SITUATION
1.- L'activité de prélèvement et de greffe a retrouvé son niveau de départ
Lors de son audition par la Mission le 10 janvier 2001, le Professeur Didier Houssin a indiqué que la situation était redevenue « normale » : « l'activité de prélèvements et de greffes de cornées a retrouvé son niveau de départ, et l'a même largement dépassé aujourd'hui. L'activité de prélèvement d'organes a remonté une partie de la chute : nous sommes à 1 016 prélèvements d'organes effectués sur des personnes décédées pour l'année 2000 ; au pic, en 1991, il y en avait eu 1 090 [...] ».
Si la situation s'est améliorée sur le plan quantitatif, les personnalités auditionnées par la Mission ont également souligné que la société avait, à l'égard des greffes d'organes, une attitude plus pacifiée. Pour le Professeur Didier Houssin, « Sur le plan médiatique, la situation est plus sereine. L'analyse très régulière et le suivi très précis que nous faisons des productions médiatiques, écrites ou audiovisuelles, montrent que le climat est transformé. Les présentations sont beaucoup plus positives. Dans certains cas, nous voyons même se développer une forme de participation des médias au soutien de cette activité. » Lors de son audition par la Mission le 7 juin 2000, Mme Martine Allain-Régnault a confirmé cette évolution et témoigné de l'état d'esprit de l'opinion : « Concernant les greffes, je pense qu'il y a deux problèmes et que ce n'est pas la réforme des lois de bioéthique qui changera quelque chose. Le premier point, c'est que les indications se sont élargies et que, en tout citoyen, il y a un receveur qui sommeille. En revanche, même parmi nous, peu de personnes sont prêtes à donner spontanément les organes de leur enfant dans les heures qui suivent sa mort. Elles sont prêtes maintenant, mais au moment des faits, tout parent - puisqu'on cherche particulièrement les organes d'enfants - est encore dans une situation latine d'émotion et de stress. Cela viendra, mais il faudra des années. Il est vrai aussi que les scoops racontant des drames, qui sont le fait de médecins, consistant à prélever des yeux clandestinement - surtout lorsque la famille avait donné le reste des organes - ont beaucoup joué. [...] Je pense que cela va bouger. Ce sera très lent parce que les indications de greffes augmentent et les freins ne sont pas là où on le croit. Il faudra un jour avoir un débat sur ces freins au don d'organes et on constatera alors que c'est toute la chaîne des professionnels qui ne fonctionne pas : le jour où elle fonctionnera, je vous assure que la population suivra ».
Le tableau suivant résume les principaux résultats des activités de prélèvement et de greffe d'organes dans notre pays en 2000.
RÉSULTATS PRÉLIMINAIRES NON CONSOLIDÉS DES ACTIVITÉS DE PRÉLÈVEMENT ET DE GREFFE D'ORGANES EN FRANCE EN 2000
(ÉTABLISSEMENT FRANÇAIS DES GREFFES)
L'activité de prélèvement
Le recensement des sujets en état de mort encéphalique est en augmentation constante depuis 1996. L'année 2000 se traduit en effet par une augmentation de 5 % du recensement mais aussi du prélèvement des sujets en état de mort encéphalique. Malgré les efforts qui ont été engagés, la baisse des prélèvements d'organes constatée à partir de l'année 1992 n'est cependant pas encore totalement corrigée. Ce déséquilibre tient au niveau encore bas du prélèvement dans certaines régions.
L'opposition reste une cause importante de non-prélèvement. Ce facteur semble cependant parvenu à un plateau après l'augmentation des oppositions constatée au cours des années 1993 et 1994.
L'augmentation des prélèvements observée en 2000 doit être confrontée à la baisse relative qui se poursuit des morts encéphaliques de cause traumatique, mais aussi à la baisse de 5 % des accidents mortels de la voie publique en 2000. L'accident vasculaire cérébral est ainsi devenu la cause principale des morts encéphaliques, et ceci peut être relié à l'augmentation de l'âge moyen des donneurs. Ce dernier reste encore très inférieur à celui observé en Espagne, le pays ayant l'activité de prélèvement la plus élevée au monde. La possibilité existe d'augmenter en France les prélèvements, à condition de prêter une attention plus soutenue à l'éventualité de prélever les personnes de plus de 60 ans.
L'activité de greffe
L'analyse de la liste d'attente confirme l'accroissement lent mais progressif des besoins, en particulier pour le foie, vraisemblablement en raison d'un élargissement des indications. Elle révèle une stabilité du nombre de décès avant la greffe.
On peut constater une discordance dans l'évolution du nombre de greffes selon le type d'organe. La stabilité des greffes d'organes thoraciques, malgré l'augmentation du nombre de prélèvements, est sans doute liée en partie au vieillissement des donneurs et à la difficulté d'obtenir des greffons de qualité fonctionnelle suffisante. L'augmentation des greffes de rein est directement liée à l'augmentation des prélèvements sur personne décédée, car on constate une faible augmentation des prélèvements chez les donneurs vivants apparentés. Dans l'augmentation importante du nombre de greffes de foie interviennent trois facteurs : l'augmentation des prélèvements sur donneur décédé, l'augmentation du partage des greffons hépatiques et l'augmentation du prélèvement chez les donneurs vivants apparentés.
En conclusion, l'évolution observée en 2000 est encourageante. Elle permet d'espérer, avec les moyens mis en _uvre dans le cadre du plan « greffe » à partir de 2001, que les années à venir voient se prolonger cette tendance.
Source : Information presse du 30 janvier 2001.
ÉVOLUTION DE L'ACTIVITÉ DE GREFFE D'ORGANES ENTRE 1996 ET 2000
SELON LE TYPE D'ORGANE
1996 |
1997 |
1998 |
1999 |
2000 | |
Coeur |
397 |
366 |
370 |
321 |
328 |
C_ur-Poumons |
27 |
25 |
26 |
28 |
25 |
Poumons |
69 |
65 |
88 |
71 (1) |
70 (2) |
Foie |
626 (11) |
621 519° |
693 (28) |
699 (33) |
806 (56) |
Reins |
1638 (57) |
1688 (70) |
1882 (73) |
1842 (77) |
1924 (84) |
Pancréas total |
48 |
63 |
47 |
50 |
54 |
Intestin |
2 |
10 |
9 |
7 |
4 |
Total |
2807 (68) |
2839 (89) |
3116 (101) |
3023 (111) |
3211 (149) |
() dont donneur vivant.
Source : Établissement français des Greffes, Information presse du 30 janvier 2001.
B.- LA PERSISTANCE D'UNE PÉNURIE D'ORGANES
1.- La pénurie : une inadéquation entre des besoins et des ressources
Comme se le demande M. Jean-Michel Dubernard, membre de la Mission, « cette révision des lois bioéthiques pourrait-elle être utilisée pour mettre un terme à la pénurie d'organes, qui va en s'aggravant, et qui fait que, chaque année - nous avons eu des chiffres récemment - des gens meurent, de plus en plus nombreux, parce qu'ils n'ont pas reçu d'organes ? » (97).
Bien que l'indication de greffe se soit élargie, il est une réalité bien connue des transplanteurs qu'en ce domaine, la loi de l'offre et de la demande est paradoxale, car l'augmentation de la demande ne permet pas une augmentation de l'offre (98).
Une évaluation des besoins passe par l'étude des indicateurs utilisés pour mesurer la pénurie qui sont la taille des listes de patients en attente d'une greffe, le nombre de décès avant la greffe et le temps passé par les patients en attente d'une greffe.
LISTE D'ATTENTE
Évolution du nombre de patients restant inscrits en attente de greffe d'organes au 31 décembre de chaque année entre 1996 et 2000 | |||||
1996 |
1997 |
1998 |
1999 |
2000 | |
C_ur |
259 |
247 |
270 |
336 |
341 |
C_ur-Poumons |
78 |
69 |
68 |
67 |
58 |
Poumons |
101 |
113 |
109 |
116 |
122 |
Foie |
238 |
239 |
261 |
344 |
403 |
Reins |
4.113 |
4.424 |
4.494 |
4.827 |
4.893 |
Pancréas |
10 |
121 |
133 |
179 |
202 |
Intestin |
11 |
12 |
10 |
10 |
17 |
Total |
4.903 |
5.225 |
5.345 |
5.879 |
6.036 |
Évolution du nombre de patients nouvellement inscrits en attente de greffe d'organes chaque année entre 1996 et 2000 | |||||
1996 |
1997 |
1998 |
1999 |
2000 | |
C_ur |
550 |
517 |
521 |
507 |
477 |
C_ur-Poumons |
68 |
52 |
52 |
56 |
33 |
Poumons |
129 |
121 |
123 |
131 |
121 |
Foie |
2.284 |
2.293 |
2.255 |
2.485 |
2.303 |
Évolution du nombre de décès avant greffe enregistrés sur la liste d'attente entre 1996 et 2000 selon le type d'organe | |||||
1996 |
1997 |
1998 |
1999 |
2000 | |
C_ur |
113 |
108 |
82 |
87 |
93 |
C_ur-Poumons |
27 |
23 |
20 |
20 |
13 |
Poumons |
37 |
36 |
25 |
45 |
34 |
Foie |
96 |
81 |
84 |
86 |
94 |
Total |
273 |
248 |
211 |
238 |
234 |
Source : Établissement français des Greffes, Information presse du 30 janvier 2001.
Les chiffres communiqués par l'Établissement français des Greffes sont éloquents. On est passé de 4 903 à 6 036 patients restant inscrits en attente de greffe d'organes entre le 31 décembre 1996 et le 31 décembre 2000. Ce sont, depuis 1996, à peu près 4 000 nouveaux patients qui s'inscrivent chaque année. Et l'évolution du nombre de décès enregistrés sur la liste d'attente entre 1996 et 2000 donne un chiffre désespérément élevé, supérieur à deux cents personnes.
Lors de son audition par la Mission le 10 janvier 2001, le Professeur Dominique Durand a rappelé que « l'augmentation du nombre d'organes disponibles se fait par deux voies ». La première concerne le don d'organes, et « passe par l'information de la société. C'est une voie difficile où l'on n'avance qu'à petits pas et qui est, finalement, assez peu productive. Il est probable que les résultats seront enregistrés bien plus tard. Il va falloir laisser le temps à la société, devant un progrès technique très rapide, de mûrir sa réflexion. ». Mais, ajoutait-il, « il est très important de rendre toutes les procédures de prélèvement efficientes ».
Pour sa part, le Professeur Didier Houssin a estimé que le manque de greffons est bien la question la plus difficile. « En matière de tissus, c'est une question d'organisation. On peut facilement parvenir, dans les deux ans qui viennent, à résoudre totalement le manque de greffons cornéens. C'est en revanche beaucoup plus difficile pour les organes, pour une raison très simple et méconnue : la rareté des greffons tient avant tout à la rareté de l'état de mort encéphalique ».
Mort encéphalique : cessation totale et définitive de toutes les fonctions du cerveau (destruction des hémisphères cérébraux et du tronc cérébral). Ce terme est plus précis, médicalement parlant, que ses synonymes - « mort cérébrale » (terme officiellement recommandé en 1988) et « coma dépassé » (d'après P. Mollaret et M. Goulon, 1959) - qu'il serait souhaitable d'abandonner afin d'éviter des confusions dans le public. La survie apparente (respiration et corps chaud) n'est assurée que par l'emploi permanent d'un respirateur artificiel et de perfusions de vasopresseurs. On parle ainsi de « décès à c_ur battant ». Cette mort apparaît, dans des cas assez rares, à la suite de syncope cardio-respiratoire prolongée, ou après des lésions destructrices des centres nerveux (accidents de la route, suicides ou ruptures d'anévrisme). La constatation de la mort encéphalique est un préalable nécessaire aux prélèvements d'organes destinés à la transplantation. Elle s'opère soit par l'aspect plat de deux électroencéphalogrammes d'une durée de trente minutes espacés de quatre heures, soit par une angiographie objectivant l'arrêt de la circulation encéphalique (décret du 2 décembre 1996).
Aux termes du premier alinéa de l'article L. 1232-1 du code de la santé publique, le prélèvement d'organes sur une personne décédée ne peut être effectué qu'à des fins thérapeutiques ou scientifiques et après que le constat de la mort a été établi.
Le constat de la mort est défini par le décret n° 96-1041 du 2 décembre 1996 relatif au constat de la mort préalable au prélèvement d'organes, de tissus et de cellules à des fins thérapeutiques ou scientifiques et modifiant le code de la santé publique. Le constat de la mort ne peut être établi que si les trois critères cliniques énoncés, à savoir l'absence totale de conscience et d'activité motrice spontanée, l'abolition de tous les réflexes du tronc cérébral et l'absence totale de ventilation spontanée, sont simultanément présents.
La mort encéphalique est une notion « un peu méconnue », dont le Professeur Didier Houssin observe qu'il constitue « un état tout à fait exceptionnel qui n'est observé que dans 2 000 à 3 000 décès hospitaliers [...] Pourquoi ? Parce que les circonstances qui conduisent à l'état de mort encéphalique sont exceptionnelles. En fait, la grande raison du manque de greffons, et ceci est vrai dans tous les pays, tient au fait que cette rareté est presque structurelle [...], inhérente à la pathologie qui conduit à cette atteinte cérébrale » (99).
La rareté des greffons est donc liée au faible nombre de morts encéphaliques constatées (2 004 en 2000 pour 3 959 patients nouvellement inscrits sur la liste des malades en attente de greffe). On a dit que ces morts proviennent d'accidents de la circulation, de suicides et de ruptures d'anévrisme.
« Fort heureusement, les accidents de la voie publique ont diminué [...] et les greffons seront de plus en plus souvent prélevés sur des sujets en état de mort encéphalique liée à des accidents vasculaires cérébraux et non plus à des accidents de la voie publique et à des traumas crâniens ».
ÉVOLUTION DE L'ÂGE DES SUJETS EN ÉTAT DE MORT ENCÉPHALIQUE PRÉLEVÉS ENTRE 1997 ET 2000
1997 |
1998 |
1999 |
2000 | |
0-1 an |
0,3 |
0,9 |
0,5 |
0,2 |
2-10 ans |
2,9 |
2,5 |
2,6 |
1,5 |
11-17 ans |
7,8 |
6,5 |
5,5 |
5,2 |
18-29 ans |
22,1 |
22,8 |
19,5 |
18,7 |
30-45 ans |
48,4 |
34,0 |
28,8 |
30,0 |
46-55 ans |
23,0 |
20,1 |
27,9 |
26,3 |
56-65 ans |
13,4 |
11,6 |
12,0 |
12,8 |
Plus de 65 ans |
2,1 |
1,6 |
3,2 |
5,3 |
Total |
100 % |
100 % |
100 % |
100 % |
Âge moyen (ans) |
38,4 |
37,8 |
40,2 |
41,5 |
Source : Établissement français des Greffes, Information presse du 30 janvier 2001.
Lors de son audition, le Professeur Didier Houssin n'a pas mentionné une autre éventualité qu'il évoquait cependant dans son ouvrage (100) : « Tout va très vite. Aussitôt née, la « nouvelle mort » pourrait bien disparaître. Si un traitement judicieusement appliqué peu après la lésion cérébrale pouvait éviter qu'elle y conduise, qui ne s'en réjouirait ? Il est certainement de la responsabilité des médecins et des chercheurs d'envisager cette éventualité et de chercher sans relâche d'autres solutions, car le greffeur et ses malades sont, quant à eux, maintenant condamnés à rester toujours avec leurs rêves de greffons ».
À l'heure où les cellules souches laissent espérer de réelles avancées thérapeutiques, cette perspective mérite en effet d'être envisagée.
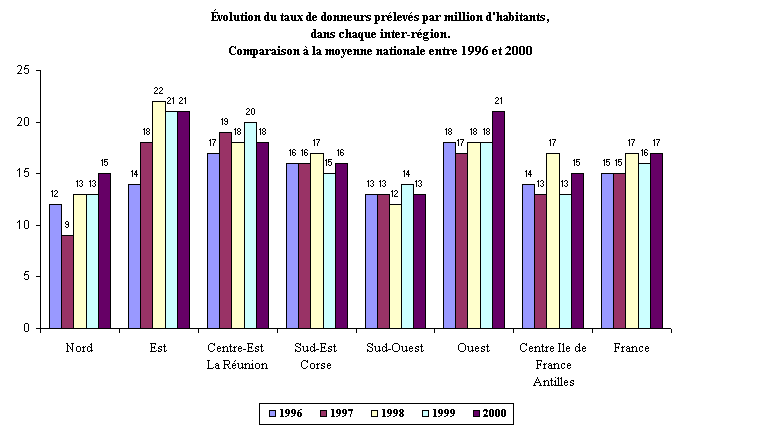
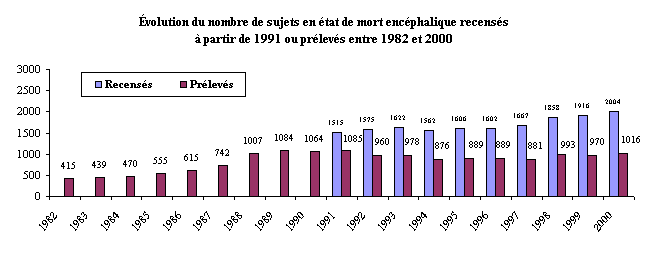
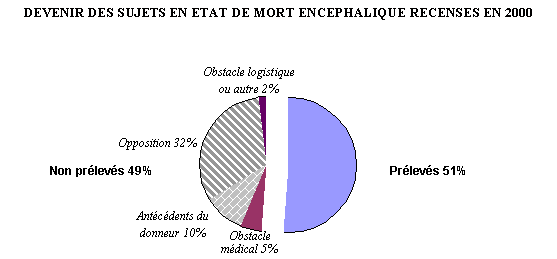
Source :Établissement français des Greffes, Information presse du 30 janvier 2001.
ÉVOLUTION DES CAUSES DE DÉCÈS DES SUJETS EN ÉTAT DE MORT ENCÉPHALIQUE
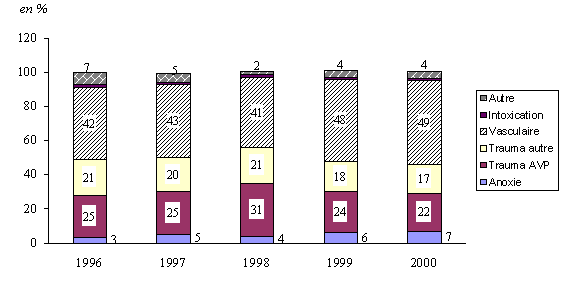
Source : Établissement français des Greffes, Information presse du 30 janvier 2001.
La deuxième cause nettement identifiée, et perçue par le plus grand nombre comme étant la principale cause de la pénurie, est le nombre des refus. Dans ce domaine, le Professeur Dominique Durand précise, lors de son audition par la Mission le 10 janvier 2001, que la France ne se distingue pas des autres pays européens : « le pourcentage de refus n'a pas diminué depuis de longues années, non seulement en France mais aussi dans des pays qui ont fait des efforts plus importants que le nôtre, comme l'Espagne, par exemple. En Espagne, qui est un modèle pour nous puisque c'est un pays dans lequel on prélève énormément d'organes, le pourcentage de refus est presque aussi important qu'en France, malgré tous les efforts consentis. Il y a peut-être là une image d'un refus très profond de la société vis-à-vis d'une activité difficile ».
Pour rappel, l'Espagne est le premier pays au monde en matière de transplantation d'organes, avec un taux de prélèvements par million d'habitants de 33,6.
ÉVOLUTION DE LA PART DE L'OPPOSITION AU PRÉLÈVEMENT CHEZ LES SUJETS EN ÉTAT DE MORT ENCÉPHALIQUE ENTRE 1996 ET 2000
Deux autres facteurs interviennent pour expliquer la rareté des greffons : « l'incapacité de certains hôpitaux à dépister ou identifier l'état de mort cérébrale [...] et la mobilisation du tissu hospitalier français ». Car il est un constat rapporté par de nombreuses personnalités auditionnées par la Mission, le Professeur Didier Houssin, le Professeur Dominique Durand, Mme Martine Allain-Régnault : à la difficulté d'identifier la mort encéphalique s'ajoute une certaine réticence à l'égard du prélèvement d'organe dans le corps médical.
4.- Le recours aux donneurs vivants
Plus restrictif depuis la loi de 1994, le recours aux donneurs vivants n'est possible à l'heure actuelle que dans le cadre familial (père, mère, fils ou fille, frère ou s_ur du donneur). En cas d'urgence, le donneur peut être le conjoint. Il est fait exception à ces restrictions pour la moelle osseuse.
Il nécessite le consentement explicite du donneur.
La technique de transplantation avec donneur vivant s'est développée très tôt dans le domaine rénal, avec succès. Le recul permet d'en évaluer les avantages et les inconvénients. Du côté des inconvénients, le risque vital péri-opératoire (évalué à 2,6/10 000), les complications postopératoires précoces ainsi que le caractère douloureux et pénible de la période postopératoire immédiate, des complications chirurgicales tardives possibles, et, dans une minorité de cas, des conséquences psychologiques négatives (dépressions, deux cas de suicide après échec de la greffe).
Les avantages reconnus sont la possibilité de programmer l'intervention, des résultats bien meilleurs, des conséquences psychologiques très positives pour le donneur en termes d'estime de soi, et un avantage économique puisque une greffe de rein réussie est beaucoup moins coûteuse pour l'assurance maladie que la dialyse.
Les situations particulières qui nécessitent le recours à un donneur vivant concernent la pédiatrie, le cas des non-résidents et de certains malades hyperimmunisés.
Il faut rappeler que la pratique de transplantation avec donneur vivant va à l'encontre de plusieurs principes éthiques : le primum non nocere, l'anonymat et l'inviolabilité du corps humain. C'est dire l'importance de l'information qui doit être donnée au donneur potentiel avant de recueillir son consentement car son autonomie et sa liberté de décision peuvent être soumises à des pressions extérieures de l'équipe médicale, dans sa façon de donner l'information, et des pressions « intérieures » qu'engage le processus psychologique lié à la question de secourir un proche si c'est possible.
Cette pratique est diversement appréciée par les professionnels dont certains y sont par principe réticents. Selon le Professeur Dominique Durand, « Il faut savoir qu'en France, à l'heure actuelle, les possibilités de la loi, qui est finalement assez restrictive, ne sont pas pleinement utilisées, parce que les médecins et les chirurgiens montrent toujours une profonde réticence à prendre un organe chez quelqu'un en bonne santé, avec les risques que cela comporte. Nous avons connu des accidents. Et encore récemment. Et en augmentant le nombre de prélèvements, nous aurons des accidents. » (101). Ce que confirme notre collègue Jean-Michel Dubernard : « Quel est le risque médical pour le donneur ? Nous venons de vivre à Lyon un drame : la perte d'un donneur vivant de foie. Nous avions vécu, il y a trente ans, un drame similaire pour un donneur de rein », avant d'exprimer une autre inquiétude « Nos décisions doivent prendre en compte le risque de développement d'un commerce d'organes. Quand on parle de commerce d'organes, cela ne veut pas forcément dire paiement, ce peuvent être aussi des pressions psychologiques directes ou indirectes. On se retrouve alors dans des situations d'échange qui ne sont pas dramatiques en soi, mais qui renvoient aux sources de la bioéthique, [...] parce qu'elles mettent en cause la dignité de l'homme, celle du corps qui peut être considéré comme le véhicule de la dignité humaine ».
En outre, le Professeur Didier Houssin a constaté que la demande n'est pas très forte, ni de la part des malades et de leurs familles, ni de la part des professionnels de santé.
D'autres arguments ont été avancés qui insistent sur le risque de ne plus recourir d'abord aux organes prélevés sur donneurs décédés. Par exemple, le Professeur Dominique Durand a insisté sur ce point : « Je crois profondément que le manque d'organes ne doit pas être un moteur du don familial. Le moteur doit être une véritable volonté de don qu'il faut identifier. En revanche, le manque d'organes doit nous rendre extrêmement dynamique pour prélever des patients en état de mort cérébrale ».
C.- L'ORGANISATION DE L'ACTIVITÉ DE PRÉLÈVEMENT PEUT ÊTRE AMÉLIORÉE
La chaîne mise en place depuis le prélèvement de l'organe jusqu'à l'activité de greffe sur un receveur représente une véritable course contre la montre. Pour le Professeur Dominique Durand (102), « Cette chaîne fragile, qui part du moment où une équipe de réanimation prend en charge un grand blessé sur une route, qu'elle amène dans un centre de réanimation, et qui va jusqu'au prélèvement doit être renforcée ».
Lors de son audition par la Mission, Mme Martine Allain-Régnault a témoigné de ce que : « l'analyse dans le détail montre que les freins à la transplantation relèvent plus des courroies intermédiaires que de l'opinion. Il faut que le généraliste, qui est à côté d'une famille endeuillée, soit favorable au don d'organes... Ensuite, vous avez des hôpitaux qui sont donneurs mais pas préleveurs, ce qui est une source de complication... Un greffeur est plus intéressant qu'un préleveur et le préleveur ne voit pas pourquoi il se démènerait... Je crois que le jour où les procédures, et notamment les rémunérations seront clarifiées, le don d'organes ne sera plus une complication pour les gens ».
Cette chaîne fait intervenir de nombreux acteurs aux compétences très spécifiques (103) :
- Le service de réanimation. L'activité de réanimation d'un patient en état de mort cérébrale et de prélèvement est extrêmement lourde. Une fois effectué le diagnostic de mort encéphalique, les médecins doivent entretenir la fonction des organes, rechercher la volonté du défunt, les contre-indications médicales à l'utilisation des organes et proposer le prélèvement à l'Établissement français des Greffes ;
- Le prélèvement d'organes : « Le premier obstacle est qu'il n'est pas certain que tous les médecins réanimateurs et neurochirurgiens en France considèrent que le prélèvement d'organes est un acte thérapeutique aussi simple, au moins dans sa conception, qu'une intervention ». D'après le Professeur Dominique Durand, le second obstacle se situe à ce niveau : « Le deuxième obstacle, ce sont les établissements publics, les hôpitaux, en particulier les hôpitaux généraux [...]. Il faut donc que l'institution, que ces hôpitaux généraux adhèrent au projet [...] C'est une activité extrêmement contraignante [...] S'il y a de nouveaux moyens à donner, ce sont certainement des moyens de motivation pour les équipes des hôpitaux généraux ».
Il est donc suggéré de revaloriser l'activité de prélèvement. Ce type d'intervention dure huit heures environ. Selon le Professeur Didier Houssin, la considérer comme une activité médicale à part entière, « [...] restaurerait l'image que l'on en a dans l'hôpital », elle serait ainsi considérée comme « une activité noble qui aboutit au traitement de malades » (104) ;
- La transplantation sur un receveur désigné par l'Établissement français des Greffes.
Pour le Professeur Dominique Durand, la segmentation a été sans doute un peu trop poussée. Selon lui, « elles se sont segmentées, mais elles se sont segmentées d'une façon qui a dépassé l'objectif poursuivi. La loi indique, et c'est une évidence, que les médecins et chirurgiens responsables des équipes de transplantation ou de prélèvement ne doivent pas participer au diagnostic de mort cérébrale. Ce doivent être des médecins indépendants. Mais dès que la mort cérébrale est déclarée, dès cet instant, il me paraîtrait extrêmement important qu'une coordination des équipes de réanimation et des médecins chirurgiens transplanteurs, lesquels peuvent venir soit comme experts soit pour renforcer les équipes au moment de la réanimation, participent à cette activité.[...] Il faut qu'en périphérie se mette en place une organisation interne à l'hôpital qui intègre le dynamisme des transplanteurs. Cela, aucun texte ne l'interdit. C'est une piste complémentaire parce que c'est en ajoutant des éléments les uns aux autres que nous gagnerons du terrain ».
C'était notamment l'une des principales mesures du Plan Greffes du 22 juin 2000.
LES 25 MESURES DU « PLAN GREFFES »
FAVORISER L'ACCÈS À LA GREFFE
Renforcer les coordinations hospitalières
· Créer 120 postes de coordonnateurs sur trois ans
· Améliorer l'organisation hospitalière du prélèvement
· Faire entrer la qualité de l'accueil des familles dans les procédures d'accréditation
Valoriser le prélèvement
· Donner toute sa place au prélèvement dans l'activité hospitalière
· Favoriser le développement de réseaux de prélèvement
· Valoriser le prélèvement dans le PMSI
· Rembourser les frais de prélèvements
Former les professionnels
· Augmenter la taille du fichier français
· Rembourser toutes les demandes de typage
· Améliorer la qualité du fichier en typage
· Développer une communication spécifique dans les établissements de transfusion sanguine
· Poursuivre le développement des banques de sang placentaire
· Intégrer France Greffe de Moelle à l'Établissement français du sang
RÉDUIRE LES INÉGALITÉS D'ACCÈS
· Adapter les règles de répartition
· Mieux organiser l'orientation des patients entre les régions
· Réviser la carte sanitaire
· Enrichir le fichier français de donneurs de moelle osseuse en groupes rares
ACCOMPAGNER LA RECHERCHE
· Donner une priorité à la recherche en thérapie cellulaire dans le programme hospitalier de recherche clinique (PHRC)
· Créer 40 postes d'assistants de recherche clinique
SOUTENIR LA GÉNÉROSITÉ ET LA SOLIDARITÉ
· Développer l'information sur le libre choix
· Lancer une journée annuelle de réflexion sur le don d'organe et une campagne médiatique sur le don
· Inscrire dans la loi une mention valorisant le don
· Développer la solidarité nationale
D.- LA TRANSPARENCE EST ASSURÉE PAR L'ÉTABLISSEMENT FRANÇAIS DES GREFFES
On l'a compris, une telle tension entre l'offre et la demande nécessite que les règles d'attribution soient précises et transparentes. Cet objectif était précisément identifié, comme le rappelle le Professeur Dominique Durand : « Le deuxième objectif a été d'encadrer parfaitement les listes d'attente. Aujourd'hui, pour qu'un malade soit inscrit, il faut que cette inscription soit validée par l'Établissement français des Greffes » (105).
1.- La liste des patients en attente d'une transplantation
ou d'une greffe
L'article L. 1251-1 du code de la santé publique précise que « peuvent seules bénéficier d'une greffe d'organes, de moelle, de cornée ou d'autres tissus [...] les personnes, quel que soit leur lieu de résidence, qui sont inscrites sur une liste nationale », cette inscription étant assurée par les établissements de santé autorisés à pratiquer des transplantations.
2.- La répartition et l'attribution des greffons
C'est le titre V du code de la santé publique qui traite de l'Établissement français des Greffes, établissement public de santé créé par l'article 56 de la loi n° 94-43 du 18 janvier 1994 relative à la santé publique et à la protection sociale. Sa mission principale est l'enregistrement de l'inscription sur la liste, la gestion de celle-ci et de l'attribution des greffons. La nécessité d'un tel établissement avait été soulignée par un rapport de l'IGAS, en raison de l'importance de certaines missions confiées à des associations telles que France-Transplant, France-Tissus ou bien France greffe de moelle.
Selon le Professeur Dominique Durand, la question de la distribution des organes est essentielle : « Dans un contexte de pénurie, il est clair que l'attribution des organes est extrêmement difficile. [...] Les textes ont été notamment élaborés après des réunions des praticiens de la cellule de transplantation qui avaient auparavant fait des propositions. Ils sont donc le fruit d'une collaboration et d'un compromis entre ce qui peut apparaître comme la nécessité d'une égalité parfaite vis-à-vis de la transplantation et des nécessités médicales de gestion des organes. Le choix, qui n'est pas celui fait dans tous les pays, a été de laisser un certain degré de liberté aux équipes à l'échelon local. »
L'Établissement français des Greffes est chargé de cette répartition, de l'attribution des greffons prélevés sur une personne décédée en vue de la transplantation d'organe, et de l'attribution des greffons tissulaires prélevés sur une personne décédée ou recueillis au cours d'une intervention médicale en vue de greffe, suivant des règles homologuées par arrêté du ministre de la santé.
Une Commission nationale de consultation publique créée à cet effet a insisté, dans un rapport remis au secrétaire d'État à la santé en juillet 1996, sur la nécessité que les règles de répartition et d'attribution des greffons s'inspirent des principes d'égalité dans le traitement des malades en attente de greffe et de liberté thérapeutique, nécessaire à la qualité des soins.
Quatre échelons de répartition sont prévus : local, interrégional, national et international, une priorité nationale étant accordée à l'enfant.
L'Établissement français des Greffes est en outre chargé de coordonner les activités de prélèvement et de greffe, de recueillir les informations permettant une évaluation des activités de prélèvement et de greffe, et de gérer un fichier national de donneurs volontaires non apparentés de moelle osseuse.
3.- Un accompagnement de sécurité sanitaire
« Dans le contexte de l'époque, cet aspect était essentiel car la sécurité sanitaire en matière de transplantation d'organe a été organisée à un très haut niveau, selon des critères extrêmement contraignants », a rappelé le Professeur Dominique Durand lors de son audition par la Mission le 10 janvier 2001.
L'Établissement français des Greffes est chargé d'élaborer les règles de bonnes pratiques applicables à l'utilisation des organes du corps humain, après avis de l'AFSSAPS. Depuis sa création par la loi du 1er juillet 1998 relative au renforcement de la veille sanitaire et au contrôle de la sécurité sanitaire des produits destinés à l'homme, il revient à l'AFSSAPS d'édicter les règles de bonnes pratiques qui s'appliquent au prélèvement, à la conservation, au transport et à la transformation des tissus et cellules autres que ceux destinés aux thérapies génique ou cellulaire et des produits du corps humain utilisés à des fins thérapeutiques, après avis de l'Établissement français des Greffes.
Dans les deux cas, ces règles sont homologuées par arrêté du ministre chargé de la Santé.
Lors de son audition du 10 janvier 2001, le Professeur Dominique Durand a émis quelques réserves sur la lourdeur des contraintes en matière de sécurité sanitaire : « C'est extrêmement onéreux pour, dans de nombreux cas, un résultat minime. Maintenant que le système est en place, que l'atmosphère autour de la transplantation est apaisée, il serait sans doute bon de revoir si la sécurité sanitaire après transplantation doit être aussi pesante. Le suivi de ces patients ne nécessite peut-être pas autant de surveillance. »
4.- Les autres missions de l'EFG
Selon le Professeur Didier Houssin, l'Établissement français des Greffes est « un organisme qui doit tenir la crête entre des intérêts contradictoires : ceux ces malades, et il a un rôle important de promotion à jouer pour obtenir des greffons [...], et ceux de la société, qui a besoin d'être informée et rassurée sur le fait qu'elle est invitée à participer jusque dans sa chair, dirai-je, à cette activité. Tenir la crête, cela signifie qu'il faut répondre à ces deux exigences : agir en priorité pour les malades mais tout en tenant compte de l'intérêt et de la position de la société sur cette question » (106).
Qualifié par lui d'« agence qui renforce l'État dans un domaine où, pour des raisons sanitaires mais surtout de justice, de solidarité et d'information du public, l'État a besoin d'être renforcé », l'Établissement français des Greffes est investi d'une mission de surveillance des activités relevant de sa compétence, d'une mission d'information du public et de promotion du don d'organes, il est chargé de promouvoir et de favoriser l'innovation scientifique et de participer à l'enseignement et à la recherche dans le domaine des greffes.
Le directeur général, nommé par décret sur proposition du ministre chargé de la santé, remplit un rôle essentiel d'harmonisation de la relation avec les tutelles, d'harmonisation avec les interlocuteurs, au premier rang desquels les hôpitaux, les agences régionales d'hospitalisation et, évidemment, les équipes médicales. Il saisit également le conseil médical et scientifique.
Ce conseil scientifique, composé pour l'essentiel de praticiens représentant les différentes activités de greffe, est l'instance d'expertise, de conseil et de proposition de l'Établissement français des Greffes. Il procède à des expertises et émet des avis, portant notamment sur l'évaluation des résultats des greffes. L'importance de cette évaluation a été soulignée par le Professeur Didier Houssin : « En ce qui concerne l'évaluation des résultats des greffes, l'Établissement français des Greffes a conduit une action originale - en tout cas, une première en France - d'évaluation d'une thérapeutique équipe par équipe, avec publication des résultats. [...] Celle-ci a permis de faire ressortir des éléments rassurants. Premièrement, les résultats des équipes de greffes d'organes en France sont finalement assez voisins d'un centre à l'autre et ne justifient pas, en tout cas, que les malades se précipitent à l'autre bout de la France au prétexte d'un meilleur résultat. Ce résultat rassurant peut s'expliquer par le fait qu'il s'agit d'une thérapeutique assez spécifique, dans un monde hospitalier assez bien ciblé. En tout cas, cela a permis de montrer que l'on peut, en France, mener des comparaisons par équipes et en rendre publics les résultats, voire les commenter dans la presse ».
De telles pratiques de transparence et d'évaluation ne peuvent que contribuer à restaurer la confiance de l'opinion publique dans l'activité de prélèvement et de transplantation d'organes. A cet égard, lors de son audition par la Mission le 7 juin 2000, Mme Martine Allain-Régnault soulignait l'importance de mener une action continue et pédagogique : « il faut aller doucement et favoriser les campagnes d'information ».
IV.- LA QUESTION DU CONSENTEMENT, CONDITION DE
L'AMÉLIORATION DU DISPOSITIF ?
A.- LA PLACE CROISSANTE DU CONSENTEMENT EN MATIÈRE MÉDICALE
Les lois de 1994 se sont inscrites dans une logique de promotion du don.
Concilier le droit du patient à accéder au traitement qui le sauve avec le droit de la personne sur laquelle on procède au prélèvement était bien le but du législateur de 1994. Mais le recours de plus en plus fréquent aux éléments du corps humain pour soigner risque cependant de faire considérer le corps humain comme une source de matériaux biologiques, c'est-à-dire de conduire à une représentation segmentée du corps réduit à un ensemble de « pièces détachées ». Comme le relevait le philosophe Jean-François Mattéi, le corps est désormais abordé de manière analytique par le scientifique et le médecin, alors qu'il est vécu quotidiennement comme une unité (107). La loi de 1994 le reconnaît implicitement, qui inscrit l'inviolabilité du corps humain dans le code civil.
Selon le sociologue Patrick Pharo (108), « La structure de la demande d'organes consiste en effet à mettre sur un tiers, qui est le donneur ou son témoin, la responsabilité d'un dilemme qui ne le concernait pas initialement, mais qui devient le sien dès lors qu'on s'adresse à lui. Toutefois, rien ne prouve qu'un tel transfert soit toujours légitime. [...] Lorsqu'on cherche à transférer immédiatement sur l'obligation de don les exigences qui portent sur la demande, on risque de nier à la fois l'idée de la demande et celle du don. Car une demande dont la réponse est obligatoire devient un commandement, et un don rendu obligatoire devient un commandement, et un don rendu obligatoire jusqu'à la nature des objets à donner risque de s'apparenter davantage à un impôt ou à une dette qu'à un don ».
Le choix a été fait, en France, de considérer le don d'organe comme un acte de solidarité entre les individus, que ce soit dans le cas du don d'organes en vue de soigner une autre personne ou dans celui à visée de connaissance scientifique, dont on considère qu'elle sera bénéfique aux malades à venir.
Cette logique de solidarité, mise en avant dans notre pays, contraste avec la logique de liberté que certains souhaitent voir mise en _uvre par l'instauration d'une manifestation explicite en faveur du don. Selon eux, cette manifestation pourrait prendre la forme d'un registre des personnes favorables au don, et du port d'insignes particuliers dont le plus fréquemment suggéré est l'insertion d'une pastille sur des documents officiels (carte d'identité, livret de santé, etc.).
Même si la terminologie utilisée dans la loi relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale et à la procréation et au diagnostic prénatal mentionne « le prélèvement » et la « collecte » d'éléments ou de produits du corps humain, le législateur s'est inscrit dans une logique de don, que traduisent clairement les principes de gratuité et d'anonymat. Mais la manifestation principale de cette volonté réside évidemment dans la place donnée au consentement : tout prélèvement est soumis au consentement présumé, la liberté de l'individu de refuser ce don est garantie par la création du registre des refus. C'est donc la reconnaissance que cet acte de prélèvement ne se fonde pas sur une norme imposée, mais sur l'exercice de la libre volonté de chacun.
Enfin, afin que cette liberté puisse s'exercer en toute connaissance de cause, la bonne information de chacun a une valeur pédagogique évidente : l'ignorance est sans doute l'un des principaux freins à l'expression du consentement en faveur du prélèvement. L'ignorance relative à la notion de mort cérébrale, l'ignorance sur la destination des organes prélevés constituent autant de causes de la réticence marquée à l'encontre de l'activité de prélèvement. C'est pourquoi il a été prévu que l'information des personnes est assurée par l'État. Nous y reviendrons.
Soucieux de donner à cette nouvelle forme de solidarité les moyens de se développer en permettant à chacun de s'exprimer personnellement et explicitement, votre Rapporteur a souhaité mener une réflexion sur le consentement, véritable clé de voûte des lois de bioéthique. En effet, en matière de don d'organe ou de tissus, il est acquis que le donneur, ou sa famille, consent au prélèvement, le receveur consent à subir une greffe, le tout après bonne information sur les risques qu'il encourt.
Les progrès scientifiques et médicaux peuvent donner l'impression que le corps humain est devenu un réservoir à matériaux, à organes ou un objet d'expérimentation lorsque la personne se prête à des recherches biomédicales. L'évolution de certaines thérapies, notamment les thérapies cellulaires, peut aussi renforcer cette impression. Comment s'assurer de la réalité d'un consentement libre et éclairé des personnes concernées ? C'est en effet le consentement qui permet de lever l'interdit d'atteinte à l'intégrité physique de la personne. Il est évoqué à l'article 16-3 du code civil qui dispose qu'il ne peut être porté atteinte à l'intégrité du corps humain qu'en cas de nécessité médicale pour la personne, et que le consentement de l'intéressé doit être recueilli préalablement hors le cas où son état rend nécessaire une intervention thérapeutique à laquelle il n'est pas à même de consentir. Il est l'expression du droit de chacun à refuser, lié à l'autonomie de la personne.
L'article 36 du code de déontologie médicale fait obligation au médecin de rechercher le consentement de la personne examinée ou soignée dans tous les cas. Cette obligation ne se comprend qu'au regard de l'obligation d'informer énoncée à l'article 35 selon lequel « Le médecin doit à la personne qu'il examine, qu'il soigne ou qu'il conseille une information loyale, claire, appropriée à son état, sur les investigations et les soins qu'il propose. Tout au long de la maladie, il tient compte de la personnalité du patient dans ses explications et veille à leur compréhension ».
Ces deux articles sont à la base de la notion de consentement éclairé qui permet au patient d'approuver les prescriptions proposées par le médecin après qu'il en a expliqué, de façon simple, compréhensible et loyale, les motifs, les risques éventuels et les modalités de son intervention.
La Cour de Cassation, dans un arrêt en date du 25 février 1997, a imposé au médecin la charge de la preuve quant au respect de son obligation d'information. Elle a en outre précisé, dans un arrêt du 14 octobre 1997, que le médecin pourra faire la preuve d'avoir donné cette information par tout moyen (témoignage, mention au dossier, compte rendu au médecin traitant, etc).
1.- Qui s'exprime ? (109)
Lors de son audition par la Mission le 4 octobre 2000, M. Francis Kernaleguen a relevé que « La plupart des textes visent « la personne concernée », selon l'expression utilisée. On comprend que c'est la personne qui est directement intéressée. Elle n'est pas la seule personne concernée puisqu'elle est en général en situation familiale, donc, des proches sont aussi concernés de manière moins directe, disons indirecte. Dans la loi française, c'est, en général, la personne concernée, et elle seule, qui consent ».
Si la personne est hors d'état d'exprimer sa volonté, l'article 9 du code de déontologie prévoit que le médecin est tenu de lui porter assistance ou de s'assurer qu'il reçoit les soins nécessaires. En outre, l'article 36 indique au médecin qu'il « ne peut intervenir sans que (les) proches aient été prévenus et informés, sauf urgence ou impossibilité », cette précision n'exigeant pas le recueil du consentement.
En revanche, lorsqu'il s'agit de personnes protégées - mineurs ou majeurs en tutelle - « le consentement est exprimé par la personne habilitée à consentir à leur place, en termes d'autorité parentale ou en termes de régime de protection ». Pour les mineurs, il s'agit des parents ; selon l'article 372-2 du code civil, chacun des parents est réputé agir avec l'accord de l'autre à l'égard des tiers de bonne foi. Dans le cas des majeurs en tutelle, c'est le représentant légal qui est appelé à consentir.
« Mais le code de déontologie médicale prévoit expressément qu'une information adaptée doit être donnée à la personne concernée - donc au mineur et au majeur protégé - et que, dans la mesure du possible, il doit être tenu compte de son avis, non pas le suivre mais en tenir compte », a précisé M. Francis Kernaleguen lors de son audition précitée.
La question se pose toutefois, concernant l'incapacité : « Faut-il prévoir des zones franches dans lesquelles l'incapacité ne produirait pas d'effet ? ». Il s'agit notamment du cas des mineures en matière d'IVG, pour lequel « la question a été posée de savoir quoi faire dans l'hypothèse où l'opposition entre les représentants de l'incapable et l'incapable lui-même se fait dans l'autre sens [...], quand le mineur souhaite une intervention que le représentant refuse. La loi n'était pas très explicite. Dans la pratique judiciaire, il s'est trouvé des juges, notamment des juges des enfants, qui, par le biais de l'assistance éducative, ont trouvé un moyen de permettre de passer outre à l'opposition des parents ».
Il faut rappeler que depuis l'intervention de cette audition, le projet de loi relatif à l'interruption volontaire de grossesse et à la contraception, adopté définitivement le 30 mai 2001, a prévu la possibilité pour une femme mineure non émancipée d'avoir accès à l'interruption volontaire de grossesse dans les cas où elle ne veut pas que ses parents ou son représentant légal soient consultés pour consentement, ou si le consentement n'est pas obtenu. Elle se fait alors accompagner dans sa démarche par la personne majeure de son choix (110).
Par ailleurs, M. Francis Kernaleguen ajoutait que « la difficulté réside, dans l'hypothèse, qui n'est pas rare, des personnes majeures qui ne sont pas soumises à un régime de protection et qui, cependant, sont atteintes de troubles mentaux faisant craindre pour elles ou douter de leur lucidité [...] On peut craindre que cette personne, ne bénéficiant pas d'une protection légale et étant renvoyée à son seul consentement, puisse être exposée davantage que les autres. On peut supposer qu'en raison de son trouble, ce seul consentement n'est pas une protection suffisante ». Il est vrai que le malade mental n'est exclu que s'il est protégé ; dans le cas contraire, il appartient au médecin d'apprécier la portée du consentement. Que vaut, auprès de ces personnes, l'information qui leur aura été délivrée ?
Ainsi qu'on a pu le dire, « en cas de maladie mentale, voici que la notion de consentement se dérobe » (111). Et pourtant, cette situation ne doit pas être totalement exceptionnelle. Car, même si l'on ne peut affirmer que les conditions sociales sont la cause première d'un certain nombre de maladies mentales, il est reconnu qu'on trouve plus de malades mentaux dans les milieux urbains que ruraux, dans les sociétés en rapide développement que dans celles ayant une relative stabilité. Le niveau d'exigence de la société engendrerait des conditions révélatrices des incapacités adaptatives qui seraient, sans cela, restées pratiquement inexistantes. Par ailleurs, les personnes socialement très défavorisées ne subissent-elles pas également de nombreuses pressions ?
Pour autant, dans son audition précitée, M. Francis Kernaleguen se demandait s'il fallait « imaginer un régime intermédiaire instituant pour les personnes en cause soit une procédure légale, par exemple, devant un comité d'experts, soit des mesures d'accompagnement particulières ? ». Selon lui, « on risquerait (...) de créer une discrimination difficile à justifier. Comment dire que telle personne doit suivre une procédure que le droit commun n'impose pas de suivre aux majeurs non incapables ? Je vois là une difficulté d'articulation et, finalement, d'acceptation ».
2.- Pour quelle expression ? (112)
L'acceptation est la décision souhaitée par le médecin, mais le refus, par une personne capable, de se soumettre à un acte médical doit être respecté, à condition pour le médecin d'avoir expliqué au patient les conséquences de sa décision.
L'acceptation vaut pour la durée du traitement entrepris : l'accord donné au départ sur un objet précis n'a pas à être renouvelé, mais la question se pose en cas d'évolution de la situation requérant une réaction adéquate, comme cela peut se produire au cours d'une intervention chirurgicale. S'il y a risque à surseoir, la nécessité justifie l'extension de l'intervention, mais en l'absence d'urgence, il faut solliciter un nouveau consentement.
Enfin, le consentement est révocable, ce qui expose le médecin aux mêmes exigences d'information qu'en cas de refus initial.
3.- Les autres cas d'exigence du consentement
L'exigence du consentement se trouve énoncée dans de nombreux articles. Elle est requise notamment pour les études génétiques (article 16-10 du code civil), l'identification par les empreintes génétiques (article 16-11 du code civil), le prélèvement des éléments et produits du corps humain (article L. 1211-2 du code de la santé publique), le prélèvement d'organes sur les personnes vivantes (article L. 1231-1 du code de la santé publique) et les personnes décédées (article L. 1232-1) ; elle figure également aux articles L. 2142-2 relatif à l'assistance médicale à la procréation et L. 2141-4 et L. 2141-5 relatifs à l'accueil d'embryon. Il est requis également pour la recherche biomédicale (articles L. 1211-1 et L. 1211-2).
Le consentement de la personne est requis pour toute atteinte à son intégrité physique. Toutefois, il se manifestera différemment selon que la personne est vivante, ou décédée.
4.- Le consentement au prélèvement d'organe : cas du donneur vivant
Les articles L. 1231-1 à L. 1231-5 du code de la santé publique énoncent les conditions du prélèvement sur une personne vivante : il ne peut s'agir que d'une finalité thérapeutique pour un receveur, qui doit avoir la qualité de père ou de mère, de fils ou de fille, de frère ou de s_ur du donneur, sauf en cas de prélèvement de moelle osseuse en vue d'une greffe. En cas d'urgence, le donneur peut être le conjoint.
Le consentement du donneur est exprimé devant le président du tribunal de grande instance ; en cas d'urgence, il est recueilli par tout moyen par le procureur de la République. Il est révocable sans forme et à tout moment.
Aucun prélèvement ne peut être effectué sur une personne vivante majeure faisant l'objet d'une mesure de protection légale. En revanche, seul un prélèvement de moelle osseuse peut être opéré sur un mineur au bénéfice de son frère ou de sa s_ur, sous réserve du consentement de chacun des titulaires de l'autorité parentale ou de son représentant légal, exprimé devant le président du tribunal de grande instance. En cas d'urgence, le consentement est recueilli par tout moyen par le procureur de la République. Un comité d'experts, composé de deux médecins dont un pédiatre, et d'une personnalité n'appartenant pas aux professions médicales, accorde l'autorisation d'effectuer le prélèvement après avoir vérifié que le mineur a été informé du prélèvement envisagé en vue d'exprimer sa volonté, s'il y est apte.
Outre les risques médicaux, faibles (en statistique, mais pouvant être graves du point de vue individuel) mais réels, existant pour le donneur vivant, le don d'organe entre personnes vivantes pose la question de l'autonomie du donneur potentiel, qui s'exprime par son consentement. Devant la Mission, le Professeur Dominique Durand a souligné l'importance de cet enjeu (113) : « D'après notre expérience, la difficulté est de savoir si la volonté de donner est profondément libre. Lorsqu'un frère ou une s_ur est donneur, nous faisons tous les examens. Une fois que nous savons que la transplantation est possible, nous convoquons le donneur potentiel dans l'intimité d'un bureau en lui disant « Si vraiment vous ne voulez pas, nous dirons que c'est impossible pour des raisons médicales ». Un nombre important de personnes prennent cette brèche ».
a) L'élargissement de la catégorie des donneurs vivants et la notion de proche
Comment s'assurer de l'absence de pressions extérieures, comment s'assurer qu'il n'y a pas commerce ? Notre collègue, Jean-Michel Dubernard a exprimé cette inquiétude à plusieurs reprises, notamment lors de l'audition de M. Henri Caillavet, le 4 octobre 2000 : « Ce risque de mort existe, mais il n'y a pas que lui ; il peut aussi y avoir des complications psychologiques. Ce dernier risque est aussi très sérieux pour le donneur et encore très mal documenté sur le plan scientifique [...] Le risque de dérive commerciale est extraordinaire. Vous avez parlé de dignité. Mais le corps est le véhicule de la dignité humaine. Mon propos est un peu solennel mais à partir du moment où vous vendez une partie de votre corps, la dignité humaine est détruite. [...] Je comprends bien, mais alors pourquoi pas l'ami, la cousine de l'ami et ainsi de suite ? ». Lors de l'audition de M. Francis Kernaleguen, il a ajouté que « la question qui se pose est de savoir si ce consentement est libre dans la mesure où, toujours dans le cas du don d'organe, des pressions, qui sont des pressions affectives très précises, ne manquent pas de s'exercer qui ne sont pas sans conséquences parfois à terme. Tous les transplanteurs connaissent des histoires de frère donneur qui fait chanter ultérieurement le frère receveur, lui demandant des avantages en échange de son don ou ne cessant de lui rappeler qu'il lui a fait un don magnifique ».
Indépendamment des réticences exprimées par les médecins et les chirurgiens, eu égard aux risques encours par le donneur, il ne semble pas qu'une demande pressante existe vraiment. Lors de son audition du 10 janvier 2001, le Professeur Didier Houssin a précisé à la Mission que « L'Établissement français des greffes a été peu sollicité sur cette question, peu interrogé. Nous avons effectivement reçu deux ou trois courriers de personnes disant qu'étant mariées depuis trente ans et aimant leur conjoint, elles ne comprenaient pas pour quelles raisons on leur interdisait de leur donner un rein, pourquoi on rendait le don si difficile. Mais cela reste un nombre de cas très limité. La réflexion et le débat qui auront lieu sur cette question seront certainement intéressants. Nous pouvons imaginer un élargissement mais à la condition d'instaurer un dispositif de contrôle car il serait dangereux d'ouvrir de manière incontrôlée la possibilité de greffes avec donneur vivant à des personnes non apparentées ». Il rappelait par ailleurs « qu'en Inde, où on pratique beaucoup de greffes avec donneur vivant, les donneurs sont souvent des femmes. C'est la déviation que l'on peut toujours craindre ».
Lors de l'audition du Professeur Dominique Durand, notre collègue Mme Yvette Roudy, s'interrogeant à propos de la notion de proche, relevait : « Faut-il définir ce terme à tout prix ? Faut-il d'ailleurs que ce soit un proche ? Je me dis qu'après tout, il peut se trouver une personne qui, dans un geste de grande générosité, ait envie de faire un don sans préciser pour autant à qui. Même si c'est rare, cela peut se produire. Pourquoi écarter cette possibilité ? ».
Le recours aux donneurs vivants est très peu pratiqué en France, alors que les pays nordiques, les États-Unis, la Belgique, le Japon y recourent plus fréquemment, notamment pour les transplantations rénales (114). Il est souligné qu'en Norvège par exemple, les problèmes posés par le prélèvement ne sont pas exprimés en terme d'opposition entre donneurs vivants et donneurs cadavériques.
Témoin de l'incertitude du milieu médical, le Professeur Dominique Durand a exprimé ses doutes devant la Mission : « Faut-il élargir ? Chacun le ressent de façon personnelle. Mon avis personnel est que je ne vois pas quelle règle éthique pourrait empêcher quelqu'un de donner à un ami.[...] Les pays anglo-saxons, notamment l'Angleterre, ont résolu ce problème en nommant une commission d'experts qui comprend des médecins non experts, qui essaient de valider la réalité d'une volonté de don ».
L'élargissement de la catégorie des donneurs vivants pourrait être envisagée avec un encadrement permettant de s'assurer de la qualité du consentement, à l'instar de ce qui existe déjà pour les mineurs donneurs de moelle osseuse.
Créés par la loi n° 94-654 du 29 juillet 1994 relative au don et à l'utilisation des éléments et produits du corps humain, à l'assistance médicale à la procréation et au diagnostic prénatal, les comités d'experts chargés d'autoriser les prélèvements de moelle osseuse sur une personne vivante mineure se sont mis en place conformément au décret n° 96-375 du 29 avril 1996.
Il existe actuellement sept comités d'experts pour sept interrégions déterminées en fonction des centres de transplantation. Quel que soit le lieu du prélèvement, le comité d'experts compétent pour autoriser le prélèvement est celui dans le ressort duquel demeure le mineur donneur, en France métropolitaine, ce qui peut poser des problèmes aux familles dont l'enfant receveur se trouve dans un hôpital situé hors de l'interrégion du lieu de résidence. Il serait d'ailleurs opportun de réfléchir à une modification de cette règle afin de laisser le choix du comité d'experts aux familles.
Le comité d'experts est composé de trois membres titulaires et trois membres suppléants, bénévoles, désignés pour trois ans par arrêté du ministre chargé de la santé :
- un médecin non pédiatre désigné sur proposition du directeur général de l'Établissement français des Greffes et choisi au sein du personnel de cet établissement ;
- un pédiatre, qui donne des informations sur la maladie et l'indication de la greffe ;
- et une personnalité n'appartenant pas aux professions médicales, en raison de sa compétence et de son expérience dans le domaine de la psychologie ou de la défense des droits de l'enfant. Un comité ne peut délibérer valablement que si ses trois membres, titulaires ou suppléants, sont présents.
Au cours de la période allant du 1er juillet 1996 au 31 décembre 1998, ces comités ont procédé à 265 entretiens de donneurs mineurs, pour lesquels aucune décision de refus n'a été prise. Selon des informations recueillies par votre Rapporteur, les membres des comités d'experts reconnaissent que « la conséquence d'un veto serait d'une gravité telle pour le receveur que l'analyse du rapport bénéfice/risque est d'emblée très orientée ». Toutefois, évoquant les termes de la loi selon lesquels « le refus du mineur fait obstacle au prélèvement », ces mêmes membres sont d'avis que si une décision de refus d'autorisation devait être prise par le comité en réponse à ce principe, il est important que cette décision ne soit pas motivée, comme le prévoit la loi, afin de ne pas mettre l'enfant qui était potentiellement donneur en difficulté et rappellent que c'est le comité qui prend la décision, et non l'enfant.
La réunion d'un comité en urgence s'est imposée à vingt reprises en raison de l'urgence de l'indication de la greffe.
S'il a pu être fait état de certaines difficultés avec quelques équipes de greffes, le comité ayant le sentiment d'être mis devant « le fait accompli », il faut souligner qu'elles sont apparues au tout début de l'activité de ces comités. Désormais, l'expérience et la légitimité acquises auprès des équipes de transplantation ne font plus de doute.
Le rôle des comités est de mettre en balance le bénéfice/risque par rapport au donneur, et de s'assurer de l'information donnée à celui-ci.
Selon son âge, le mineur est vu avec ou sans ses parents ; dans ce dernier cas, il est vu dans un deuxième temps avec ses parents dont la présence apparaît très souhaitable, bien que cela ne soit pas imposé par la loi.
Le risque à éviter était que le prélèvement de moelle sur l'enfant donneur soit considéré comme un pur acte médical par l'équipe médicale, laquelle peut ne pas toujours penser aux conséquences psychologiques pour le mineur. Il appartient au comité d'experts de répondre aux questions que celui-ci peut se poser sur la maladie de son frère ou de sa s_ur, sur la douleur pouvant accompagner les gestes médicaux, sur l'échec éventuel de la greffe et le sentiment de culpabilité qui pourrait survenir. L'opportunité d'établir une évaluation des prélèvements en effectuant un suivi des donneurs, sur le plan psychologique, a été évoquée.
Le tableau suivant liste les questions abordées par les membres du comité.
Les points abordés ou analysés par les membres du comité
· La loi bioéthique et le rôle du comité,
· L'information reçue par le donneur et ses parents,
· La compréhension de cette information,
· L'attitude du donneur vis à vis de la procédure médicale,
· La possibilité d'un choix dans la fratrie,
· Le risque d'échec de la greffe et la non-responsabilité de l'enfant donneur en cas d'échec,
· La relation donneur/parents,
· La relation donneur/receveur,
· L'éventualité d'un suivi psychologique pour l'enfant donneur avant et/ou après le don,
· Les questions et les inquiétudes éventuelles de l'enfant ou des parents.
Les principaux thèmes abordés par les enfants et les familles
· Perte d'identité ou transfert d'identité de l'enfant donneur,
· Confusion entre identité HLA et risque identique vis-à-vis de la maladie du frère ou de la s_ur,
· Perte d'un élément du corps pas toujours perçu comme régénérable,
· Modification du lien fraternel avec la création d'un couple donneur-sauveur/receveur-débiteur,
· Difficulté pour le donneur à assumer sa nouvelle position dans la famille,
· Place de la responsabilité du donneur dans l'efficacité du traitement,
· Peur de la douleur,
· Peur de l'anesthésie,
· Appréhension vis-à-vis de la période suivant le don et la greffe pendant laquelle le receveur sera séparé de sa famille et en particulier du donneur,
· Valorisation de l'acte du donneur par sa famille.
Source : Établissement français des Greffes.
Selon des informations recueillies par votre Rapporteur, les membres des comités d'experts ont proposé des aménagements pour améliorer leur efficacité, résumés dans le tableau ci-dessous :
Questions soulevées par les membres de comités d'experts et propositions
· Apport de plus de souplesse à l'organisation territoriale,
· Renfort des effectifs des comités d'experts,
· Encadrement des indications urgentes,
· Redéfinition du rôle des comités dans l'appréciation de la « justification médicale de l'opération »,
· Redéfinition des moyens donnés aux comités,
· Le choix des membres des comités parmi des médecins non impliqués dans la prise en charge du donneur ou du receveur,
· Mise en place d'une évaluation,
· Création d'un registre des donneurs mineurs de moelle osseuse.
Sources : Établissement français des greffes.
Les membres des comités d'experts estiment que leur intervention contribue à garantir le respect des principes éthiques, qu'ils apportent une protection accrue du donneur mineur, grâce à leur nature de médiateur ne faisant pas partie des équipes de prélèvement ou de greffe, intervenant dans un lieu neutre, donnant la possibilité aux enfants et aux parents de s'exprimer dans un contexte différent. Ils garantissent ainsi qu'un nouveau regard soit porté sur le donneur par les familles et les équipes de prélèvement et de greffe.
NOMBRE D'ENFANTS VUS EN COMITÉ D'EXPERTS
(CHIFFRES COMMUNIQUÉS PAR L'EFG)
1996 |
1997 |
1998 |
1999 |
2000 | |
Inter Région I (Nord) |
5 |
16 |
8 |
13 |
19 |
Inter Région II (Est) |
2 |
13 |
9 |
12 |
8 |
Inter Région III |
12 |
20 |
21 |
10 |
24 |
Inter Région IV |
5 |
13 |
14 |
12 |
8 |
Inter Région V |
4 |
11 |
7 |
9 |
5 |
Inter Région VI |
5 |
17 |
14 |
18 |
20 |
Inter Région VII |
15 |
19 |
35 |
23 |
26 |
Total |
48 |
109 |
108 |
97 |
110 |
L'expérience qu'ils ont acquise justifie d'envisager une extension du champ de leur intervention pour tout prélèvement sur donneur vivant mineur ou majeur, apparenté ou non apparenté.
Le projet de loi relatif à l'interruption volontaire de grossesse et à la contraception récemment adopté (115) a d'ores et déjà créé un nouveau comité d'experts chargé d'apprécier la justification médicale de la stérilisation à visée contraceptive sur une personne majeure dont l'altération des facultés mentales constitue un handicap (article 27). Ce comité, composé de personnes qualifiées sur le plan médical et de représentants d'associations de personnes handicapées, devra apprécier, outre la justification médicale de l'acte envisagé, ses conséquences normalement prévisibles sur les plans physique et psychologique.
Conscient de la nécessité de légiférer sur cette question pour éviter que des dérives n'apparaissent, votre Rapporteur s'inquiète cependant de la multiplication des actes dérogatoires d'atteinte à l'intégrité physique accompagnés, certes, de la création de comités ad hoc chargés de veiller à la protection des mineurs et des majeurs protégés. La garantie des droits apportée par la loi bioéthique a d'autant plus de sens lorsqu'elle concerne des personnes qui, par elles-mêmes, n'ont pas ou ont difficilement la capacité de faire respecter leurs droits. Qu'en sera-t-il des personnes particulièrement fragiles, socialement très défavorisées, face à une pression sociale de plus en plus forte de rejet de la différence et de demande d'un certain confort ? Que vaudrait le refus des faibles, face à cette pression ?
Il convient donc de s'assurer que l'intervention de tels comités garantisse une protection réelle, c'est-à-dire que leur institution ne soit pas seulement une forme de « réflexe » en vue de tranquilliser la conscience du corps social, mais expriment le souci d'une démarche attentive à l'effectivité de la protection accordée aux plus fragiles.
À l'annonce du décès d'une adolescente âgée de 14 ans atteinte de mucoviscidose, la veille de la greffe de ses poumons, votre Rapporteur s'est étonné d'apprendre par la presse (116), alors que non seulement aucune disposition légale ne l'autorisait ni même qu'aucun projet de loi n'avait encore été déposé sur le bureau de l'Assemblée nationale, qu'une dérogation avait été donnée aux médecins pour réaliser un prélèvement de poumon sur une tante de la jeune fille.
Cette affaire, marquée de nombreuses entorses à la loi et à la déontologie médicale, montre à quelle confusion la notion d'urgence peut parfois conduire.
En outre, le Parlement est d'autant plus surpris que le principe de l'élargissement des prélèvements sur donneur vivant puisse être considéré comme acquis, alors même que la réflexion n'a pas été menée à son terme et que les auditions de la Mission ont montré les doutes et les réserves que cet élargissement suscite.
5.- Le consentement au prélèvement d'organe : cas du donneur décédé
Le prélèvement d'organes sur personne décédée est régi par l'article L. 1232-1 du code de la santé publique aux termes duquel le prélèvement peut avoir lieu dès lors que la personne concernée n'a pas fait connaître, de son vivant, son refus d'un tel prélèvement. Le refus peut être exprimé par l'indication de sa volonté sur un registre national automatisé, appelé Registre des refus. Toutefois, le dernier alinéa de l'article précité oblige le médecin à s'efforcer de recueillir le témoignage de la famille du défunt.
Lors de son audition par la Mission, le 10 janvier 2001, le Professeur Didier Houssin a dressé un constat positif : « Quant au consentement, la procédure mise en place de recueil du consentement sous la forme d'un régime de consentement présumé avec la mise en place d'un registre national des refus et du recueil du témoignage de la famille sur la position du défunt est un dispositif nuancé, subtil, de mon point de vue, de très bonne qualité, mais difficile à expliquer et sans doute assez mal compris par la population. Il n'en reste pas moins qu'aucun dysfonctionnement n'a été noté dans cette procédure de consentement ».
Cette procédure a néanmoins suscité de nombreuses interrogations au cours des auditions de la Mission.
a) Consentement présumé ou autorisation de la famille ?
De nombreux intervenants se sont plaints, en effet, de ce que la règle du consentement présumé était détournée. À plusieurs reprises, notre collègue Jean-Michel Dubernard a fait le constat qu'en réalité « la notion de consentement présumé n'est pas appliquée ». Ainsi relevait-il le 4 octobre 2000 : « Ce système fait dévier le consentement présumé [...] En réalité, on demande l'autorisation aux familles. Comment faire, sans être trop schématique et dictatorial, pour que le consentement présumé, qui a beaucoup de sens, soit respecté ? » Ce que confirmait notre collègue Mme Catherine Génisson, le 10 janvier 2001 : « en fait, non seulement nous informons les familles, mais nous leur demandons même leur autorisation [...] Cela se passe comme cela partout, parce qu'actuellement, le registre des refus n'est pas « grand public » ».
Lors de son audition, le Professeur Dominique Durand observait que « la famille, confrontée à la pire situation qui soit, ne sait pas faire la nuance entre témoigner d'une volonté et avoir une attitude patrimoniale. Elle considère qu'elle est l'héritière de la volonté du défunt ».
Comment une famille, sous le choc du décès d'un être cher, peut-elle prendre la distance nécessaire ? « Il est vraiment douloureux de parler aux familles de ces questions pour la première fois alors qu'elles vivent un drame terrible [...] Poser cette question, pour la première fois, à une personne qui n'y a peut-être jamais pensé, dans ces circonstances, c'est vraiment se mettre dans les pires conditions pour obtenir un accord », observait notre collègue Mme Jacqueline Fraysse, le 7 juin 2001.
Pour sa part, notre collègue Mme Yvette Roudy, présidente de la commission spéciale chargée d'examiner les projets de loi sur la bioéthique en 1992, a rappelé aux membres de la Mission, le 4 octobre 2000, les circonstances de l'institution de ce registre : « Nous parlions du consentement libre et éclairé de la personne avant sa mort et de l'impression que pouvait avoir la famille [...] Cela nous semblait une question difficile à poser à la famille endeuillée et, en même temps, on nous avait dit qu'il fallait faire très vite pour les transplantations. Nous avions donc pensé à ce registre, mais il est vrai que celui-ci ne donne pas satisfaction ».
De fait, le registre des refus a été l'objet de nombreuses critiques, surtout de la part des associations militant en faveur du don d'organes.
b) Le Registre des refus : un bilan prématuré
« Pour ma part, le registre du refus me semble dépassé », estimait notre collègue Mme Yvette Benayoun-Nakache, comme notre collègue Mme Martine Aurillac : « Le registre ne marche pas », lors de l'audition de M. Henri Caillavet, le 4 octobre 2000.
Mais interrogé sur le prétendu trop faible nombre des inscriptions sur le registre des refus pour qu'il puisse vraiment jouer son rôle, le Professeur Didier Houssin a insisté sur la nécessité de tempérer ces critiques. Pour lui, si le nombre de refus est faible en pourcentage, ce que ne manquent pas de souligner toutes les associations militant pour le don d'organes, il n'est pas nul, en valeur absolue : « 50 000 personnes, cela représente tout de même une bonne ville de France ».
Ce registre, objet de nombreuses critiques, a d'abord souffert d'une mise en place très tardive, puisqu'il a été institué par le décret n° 97-704 du 30 mai 1997, sa mise en service ayant eu lieu le 15 septembre 1998, soit quatre ans après promulgation de la loi.
Son fonctionnement et sa gestion sont confiés à l'Établissement français des Greffes, qui doit prendre toutes mesures destinées à garantir la sécurité et la confidentialité de l'ensemble des informations contenues. Toute personne désireuse de s'inscrire doit adresser sa demande datée, signée et accompagnée de la photocopie de tout document susceptible de justifier de l'identité de son auteur.
Le décret précise que toute personne majeure ou mineure âgée de 13 ans au moins peut s'inscrire sur le registre afin d'exprimer son refus d'un prélèvement sur son corps après son décès, soit à des fins thérapeutiques, soit pour rechercher les causes du décès, soit à d'autres fins scientifiques.
Ce refus peut être révoqué à tout moment par l'intéressé, selon les mêmes modalités.
Mais l'inscription sur le registre des refus n'est pas le seul moyen de faire connaître son opposition au prélèvement, puisque les médecins doivent également s'efforcer de recueillir le témoignage de la famille du défunt. Là réside toute la difficulté.
Lors de son audition par la Mission le 4 octobre 2000, M. Henri Caillavet a suggéré de revoir les dispositions légales en supprimant l'alinéa 2 qui fonde l'autorisation familiale: « Il existe un registre des refus sur lequel vous pouvez vous inscrire [...]. L'inscription faite dans ce registre est révocable. Ce registre apporte la preuve matérielle - il n'est plus nécessaire d'écouter autrui - que vous ne voulez pas donner vos organes. A contrario, si vous n'êtes pas sur le registre, personne ne peut contester votre engagement ». C'est aussi ce que souhaite l'Académie de médecine qui a proposé de faire de ce registre le seul moyen d'expression des refus de prélèvement.
Une telle proposition ne risquerait-elle pas d'aller à l'encontre du but poursuivi ? Votre Rapporteur s'interroge en effet sur les conséquences, à plus long terme d'une telle proposition si elle était mise en _uvre. Ne risquerait-on pas de faire prévaloir une approche en termes trop exclusivement institutionnels, au nom de l'efficacité immédiate, qui pourrait avoir pour conséquence une renaissance d'inquiétude de l'opinion ? Une totale prise en compte de la dimension humaine de cette question est nécessaire. Il est certain que la souffrance et l'inquiétude des malades en attente d'un greffon et de leur famille ne peut laisser personne indifférent. Pourtant, il faut également éviter qu'elles ne conduisent à méconnaître l'attention nécessaire dans la relation avec les personnes en état de choc causé par le traumatisme de la perte brutale d'un être cher. Et d'ailleurs, le Professeur Dominique Durand a douté, devant la Mission, que la société soit prête à affronter une telle démarche (117).
Le Professeur Didier Houssin a présenté le bilan de ce registre au cours de son audition du 10 janvier 2001 : « À l'analyse, le registre national des refus joue son rôle. Il répond à une attente. Il serait dommage de le remettre en cause. Il rassemble aujourd'hui près de 50 000 personnes. Celles-ci étaient suffisamment opposées pour aller se faire inscrire sur un registre informatique, ce qui est une initiative assez lourde. [...] Il est aujourd'hui consulté sans difficulté à partir des hôpitaux, avec un délai de réponse de l'ordre de quelques minutes. En l'an 2000, il a été interrogé 7 426 fois pour un prélèvement thérapeutique, 1 032 fois pour un prélèvement à visée d'autopsie et 366 fois pour un prélèvement dans le cadre d'une recherche à caractère scientifique. Le nombre d'inscrits était hier de 46 852. C'est un registre qui existe dans d'autres pays, comme le Portugal, la Pologne et l'Autriche qui ont aussi adopté le principe de consentement présumé. Il rend service et me paraît tout à fait congruent avec l'adoption d'un principe de consentement présumé ».
« La loi est parfaitement cohérente : nous sommes généreux, et nous sommes donneurs ; nous sommes libres, donc, l'expression de cette liberté est de pouvoir dire que l'on refuse d'être donneur », observait pour sa part le Professeur Dominique Durand lors de son audition par la Mission le 10 janvier 2001. Voilà pourquoi les campagnes d'information insistent particulièrement sur la nécessité pour chacun de prendre position, afin d'éviter que la famille ou les proches n'aient à affronter un tel dilemme.
En outre, compte tenu du retard même apparu dans sa mise en place, il faut certainement attendre encore quelque peu avant de porter une appréciation sur ce registre qui n'a pas trois ans d'existence.
6.- Les tissus, cellules et produits d'origine humaine
En l'état actuel de la législation, aucune disposition spécifique n'entoure le consentement requis pour le prélèvement de tissus, cellules ou produits du corps humain en ce qui concerne le donneur vivant, alors que l'article 5 de la convention d'Oviedo énonce : « Une intervention dans le domaine de la santé ne peut être effectuée qu'après que la personne y a donné son consentement libre et éclairé. Cette personne reçoit préalablement une information adéquate quant au but et à la nature de l'intervention ainsi que quant à ses conséquences et ses risques. La personne concernée peut, à tout moment, librement retirer son consentement ».
a) Sécurité sanitaire et vigilance
Pratique en pleine expansion, les greffes de cellules et tissus aux conséquences thérapeutiques prometteuses bénéficient des progrès accomplis dans différentes disciplines médicales, et de l'apport des techniques biologiques de culture in vitro des cellules humaines.
Les utilisations de tissus peuvent être à des fins diagnostiques, thérapeutiques (en vue de greffe le plus souvent) et à des fins de recherche fondamentale, épidémiologique ou clinique. Les allogreffes de cellules et de tissus ne sont habituellement pas à l'origine de réactions de rejet.
Pour la plupart, les prélèvements sont soumis aux principes généraux relatifs au respect du corps humain (consentement, gratuité, anonymat, interdiction de la publicité et sécurité sanitaire) bien que leur application soit écartée en tout ou partie dans certains cas, en considération de la nature ou de l'utilisation envisagée de ces éléments et produits.
Compte tenu de l'accroissement des risques de transmission de maladie lié à l'expansion de ces greffes, la sécurité sanitaire et la vigilance sont désormais des principes énoncés aux articles L. 1211-6 et L. 1211-7 du code de la santé publique. Ce sont les banques de tissus qui assurent la sécurité, la traçabilité et la distribution des tissus. Un important corpus réglementaire énonce les principes généraux de sécurité sanitaire applicables au don et à l'utilisation des éléments et produits du corps humain, tant en ce qui concerne les prélèvements que l'utilisation à des fins thérapeutiques de tout élément ou produit prélevé ou collecté sur le corps humain (art. R. 665-80-1 à R. 665-80-11 du code de la santé publique).
Enfin, la création de l'Agence française de sécurité sanitaire des produits de santé par la loi n° 98-535 du 1er juillet 1998 relative au renforcement de la veille sanitaire et au contrôle de la sécurité sanitaire des produits destinés à l'homme a doté cette nouvelle agence d'une compétence en matière de contrôle de la sécurité sanitaire pour tous les produits de santé. Cette compétence s'étend aussi aux éléments et produits du corps humain, ainsi qu'aux produits thérapeutiques annexes (art. L. 1263-1 à 4 du code précité) destinés à entrer en contact avec des organes, tissus, cellules ou produits issus du corps humain lors de leur conservation, de leur préparation, de leur transformation, de leur conditionnement ou de leur transport avant leur utilisation thérapeutique chez l'homme. Elle joue un rôle prépondérant dans l'élaboration des règles de bonnes pratiques en la matière et dispose de pouvoirs de police sanitaire.
Le deuxième alinéa de l'article R. 665-80-8-I du code précité précise qu'en cas d'urgence vitale pour le receveur, appréciée en tenant compte de l'absence d'alternatives thérapeutiques et si le risque prévisible encouru par le receveur en l'état des connaissances scientifiques n'est pas hors de proportion avec le bénéfice escompté, il peut être dérogé à l'interdiction de toute transplantation d'organe ou de tissu dont l'analyse révèle un risque de transmission d'infection, après information du receveur par le médecin. Communément désignée sous le nom de balance bénéfice/risque, cette notion impose, in fine, au receveur de prendre une décision à un moment où il peut être particulièrement fragile. Aussi nécessite-t-elle d'être accompagnée d'une information sérieuse et appropriée.
« La moelle osseuse est un tissu qui a pour fonction d'assurer l'hématopoïèse, c'est-à-dire la fabrication des cellules du sang. Les cellules intéressantes pour la greffe sont des cellules très minoritaires, capables de multiplication et d'auto-renouvellement : les cellules souches hématopoïétiques. Ces cellules se retrouvent non seulement dans la moelle, mais aussi dans le sang placentaire, et dans le sang de tous les individus. Le recours à la greffe de cellules souches hématopoïétiques est justifié par l'existence de maladies hématologiques [...] pour lesquelles cette greffe est parfois l'unique traitement efficace » (118).
L'ensemble des professionnels s'accordent sur le fait qu'il ne s'agit pas d'un organe et que les modalités de prélèvement ne peuvent être comparées à celles d'un prélèvement d'organe. Ils demandent que la moelle osseuse soit considérée comme un tissu.
Par ailleurs, les sources des cellules hématopoïétiques se sont diversifiées (119) : il est désormais possible de prélever les cellules souches hématopoïétiques chez l'adulte en procédant par cytaphérèse (technique consistant à prélever le sang d'un donneur pour en extraire un type de cellules et à restituer le reste), ce qui évite au donneur le risque d'une anesthésie générale. Cette technique n'est pas prévue par la loi de 1994.
Enfin, le sang de cordon ombilical constitue la troisième source : spontanément présentes à la naissance dans le sang, dans le placenta et le cordon ombilical, les cellules hématopoïétiques ont alors une capacité de prolifération et d'expansion plus importante que chez les adultes. « Le prélèvement est simple, sans danger pour le donneur ; il peut être congelé dans l'azote liquide à - 180 °C pour une période excédant dix ans. Les autres avantages sont la rapidité de la recherche, les cellules préalablement testées sont immédiatement utilisables, les cellules de nouveau-né n'ont jamais été en contact avec des agents infectieux, dont la probabilité de transmettre un agent infectieux est très faible » (120). L'accord de la mère est indispensable.
De nombreuses banques ont été créées en Europe ; il en existe trois en France. Si cette méthode ne pose pas de problème technique particulier, il faut savoir qu'il existe des problèmes financiers, le coût de chaque prélèvement s'élevant à 10 000 francs environ, et surtout éthiques, concernant notamment la gratuité et l'anonymat du don. La communauté des hématologistes se préoccupe de plusieurs questions : la propriété du sang placentaire, la protection du donneur, la nécessité d'un consentement informé, d'autant qu'il s'agit d'un déchet opératoire. Elle constate aussi la rapidité avec laquelle la naissance de nouvelles pratiques se heurte à un cadre juridique contraignant et très vite caduque, nécessitant que la loi évolue rapidement pour s'adapter à ces nouvelles techniques (121).
c) Le statut des déchets opératoires
L'utilisation des résidus opératoires est indispensable dans certaines indications très précises. Le Docteur Perrin (122) en donne les particularités :
- ces résidus sont recueillis à l'occasion d'un acte thérapeutique qui n'a aucun rapport avec la greffe pour laquelle ils doivent être utilisés,
- ils sont par ailleurs reconnus comme étant désormais sans utilité pour le donneur,
- ils sont enfin destinés à des greffes de tissus, certainement utiles, mais dont le caractère indispensable n'est pas toujours établi.
Le recueil et l'utilisation des résidus opératoires est une activité fréquente des chirurgiens qui ne pratiquent alors plus un acte thérapeutique résultant d'un contrat tacite avec le patient. Le chirurgien agit alors « dans l'intérêt de la banque de tissus, dans le cadre d'un contrat avec cette banque, et engage à ce titre sa responsabilité personnelle. Celle-ci pourrait en particulier être recherchée en cas de transmission de maladie infectieuse au receveur [...] La responsabilité de la banque de tissus n'est engagée que pour les opérations qu'elle effectue elle-même (stockage, sécurisation, distribution, et traçabilité des produits) ». Le Docteur Perrin considère que la licéité de l'utilisation des résidus opératoires nécessite que la greffe soit absolument justifiée, que cette pratique soit aussi sûre que possible pour le receveur, compte tenu de ce que l'existence toujours possible de facteurs contaminants.
Comme le rappelle le CCNE (123) dans son avis sur la constitution de collections de cellules embryonnaires humaines et leur utilisation à des fins thérapeutiques ou scientifiques, « depuis 1960 ont été développées au Wistar Institute de Philadelphie (lignée W138) puis au Medical Research de Londres (lignée MRC5) des lignées fibroblastiques embryonnaires développées chacune à partir d'un embryon d'IVG ». Ces lignées sont utilisées comme systèmes cellulaires pour des diagnostics, la préparation de réactifs et comme substrat pour la production industrielle de vaccins. Il existe aussi des collections de cellules humaines provenant de sujets atteints de maladies ou des embryons après interruption de la grossesse à la suite du diagnostic prénatal de ces maladies.
Ces tissus peuvent être greffés en vue du traitement de la maladie de Parkinson et offrent de réelles perspectives thérapeutiques.
Dans son avis précité, le CCNE considère que les collections de tissus ou organes d'embryons humains pathologiques ne présentent pas de problème éthique particulier. Pour les recherches à visée cognitive, elles doivent faire l'objet d'un protocole. Le CCNE rappelle en outre que des tissus et organes normaux ne peuvent provenir que d'embryons après interruptions volontaires de la grossesse dans le cadre de la loi.
De fait, les tissus, cellules ou produits d'embryons ou de f_tus humains morts rentrent actuellement dans la catégorie des déchets opératoires. En 1984, dans son avis n° 1 sur les prélèvements de tissus d'embryons et de f_tus humains morts, à des fins thérapeutiques, diagnostiques et scientifiques, le CCNE, se fondant sur la reconnaissance non équivoque du caractère humain de l'embryon, reconnaissait qu'« Il n'est ni évident ni opportun de considérer l'embryon mort avant la 22ème semaine gestationnelle comme le cadavre d'un enfant mineur. D'une part, il n'a jamais vécu et n'a jamais pu vivre d'une façon autonome par rapport à la mère, d'autre part, il convient d'éviter que le consentement de la mère, s'il était nécessairement requis, ne soit interprété, en pratique, comme un don de l'embryon légitimant l'avortement ou comme le prix à payer pour obtenir l'interruption de la grossesse ». Il préconisait déjà l'information de la « mère » et la possibilité d'exercer un droit de veto, réfutant l'argument de ceux qui estiment qu'en décidant de la mort de l'embryon, la mère se prive de tout droit à son égard. Cette position lui paraissait excessive, la faculté de refus devait, selon lui, être préservée s'agissant d'utiliser des tissus humains. Selon lui, la mère pourrait être informée de sa faculté de refus lors de la première visite en vue de l'IVG.
La loi de 1994 ne l'a pas prévu, ce qui a amené votre Rapporteur et le Sénateur Claude Huriet à soulever à nouveau la question dans le rapport de l'Office parlementaire d'évaluation des choix scientifiques et technologiques. En effet, la non-application du régime du consentement parental aux prélèvements sur embryon et f_tus en vue d'une greffe conduit à traiter le f_tus expulsé comme s'il appartenait de plein droit à la communauté médicale (124).
« On parle du produit de l'avortement ? Faut-il s'adresser à la mère qui, seule, a le pouvoir de demander et d'obtenir l'IVG ? Faut-il mieux associer dans ce consentement le mari ou le compagnon, disons, le père potentiel ? » s'est interrogé M. Francis Kernaleguen lors de son audition par la Mission le 4 octobre 2000. « Faut-il demander ? Il y a une certaine ambiguïté - je ne sais si je serai suivi par tout le monde - à recourir à l'interruption de grossesse, ce qui veut dire que concrètement, on a décidé que l'on n'est pas en présence d'un projet parental ou d'un enfant, et, une fois l'interruption de grossesse opérée, traiter le produit non comme un déchet hospitalier mais comme si cela avait été un enfant qui ne serait plus [...]. S'il s'agit de personnes qui ont quitté la position parentale, qui ne l'ont pas acceptée, la question ne se pose pas dans les mêmes termes et il n'y a rien à demander. Les choses sont claires. [...] Pourquoi interroger des personnes pour qui ce n'est pas un enfant puisqu'ils n'avaient pas adopté la position de parents ? ».
Notre collègue, Mme Yvette Roudy, s'interrogeait dans le même sens : « En fait, faut-il demander ce que l'on en fait, dans la mesure où il y a rejet ? L'acte lui-même prouve que l'on n'en veut pas. Venir en plus demander à la femme ce qu'elle veut que l'on en fasse, je trouve que cela ajoute au traumatisme. On ne ménage rien », en ajoutant « faut-il répondre à des questions que l'on ne nous pose pas ? Faut-il tout encadrer ? ».
À cet égard, on peut relever que la recommandation 1100 du 2 février 1989 du Conseil de l'Europe stipule que l'utilisation de matériels biologiques provenant d'embryons ou de f_tus morts, à des fins scientifiques, préventives, diagnostiques, thérapeutiques, pharmaceutiques, cliniques ou chirurgicales, doit être autorisée dans le cadre des règles qu'elle prévoit concernant la recherche, l'expérimentation, le diagnostic et le traitement. Pour votre Rapporteur, il appartient au législateur de se saisir de cette question, à la fois par cohérence avec les décisions prises sur le plan international et, pour qu'elle soit envisagée au-delà des seuls cercles du débat éthique institutionnalisé.
V.- FAVORISER LE DON D'ORGANES
L'un des principaux obstacles en ce qui concerne les dons d'organes tient à la résistance persistante à l'application de la règle du consentement présumé. L'équipe médicale doit ainsi rencontrer la famille et, dans des circonstances extrêmement traumatisantes et douloureuses, lui demander son témoignage dans la perspective d'un prélèvement d'organe. De fait, comme les auditions de la Mission l'ont montré, ce n'est pas un témoignage qu'elles sont amenées à donner, mais bien une autorisation - ou un refus, dans environ 30 % des cas (ce qui signifie donc que 70 % donnent leur accord). Les médecins suivent cet avis, contra legem, dans l'impossibilité qu'ils sont de vérifier qu'il s'agit bien de la volonté de la personne décédée.
Mais la consultation des familles ne participe-t-elle pas aussi, à sa façon, à l'enracinement de la confiance du public à l'égard de l'activité de transplantation ?
A.- COMMUNIQUER EN FAVEUR DU DON D'ORGANES : TENIR COMPTE
DES RÉALITÉS HUMAINES
Selon une enquête nationale réalisée à la demande de l'Établissement français des Greffes, alors que tous les Français (125) s'accordent sur le fait que la greffe d'organes sauve des vies humaines, que 94 % d'entre eux reconnaissent que le prélèvement est un geste de solidarité, et 95 % que la greffe d'organes est un progrès médical indiscutable, pourquoi le taux de prélèvements par million d'habitants plafonne-t-il à seize en France, pour vingt-cinq aux États-Unis et trente-trois en Espagne ?
Plusieurs raisons ont été évoquées. Le refus vient d'abord à l'esprit. Comme l'a expliqué le Professeur Didier Houssin lors de son audition par la Mission, le 10 janvier 2001 : « Évidemment, le refus est l'un des obstacles au prélèvement. Ce refus est, bien sûr, la résultante de l'opposition du défunt, qui peut d'ailleurs s'être inscrit sur le registre national des refus, mais il peut aussi être la conséquence des circonstances dans lesquelles la famille est invitée à dialoguer, circonstances suffisamment dramatiques pour qu'elles opposent parfois un refus catégorique lié au contexte dans lequel elles vivent ce drame ».
Tout au long des auditions, les membres de la Mission se sont heurtés à cette question : pourquoi une telle persistance des refus de la part des familles, alors que, comme le souligne notre collègue Jean-Michel Dubernard (126)« des gens meurent, de plus en plus nombreux, parce qu'ils n'ont pas reçu d'organes ? ».
« Je pense, pour ma part, que l'opinion publique ne sait pas suffisamment ce que cela peut représenter. Elle ne sait pas que l'on a besoin de dons d'organes », a souligné notre collègue Mme Yvette Roudy, ajoutant « On pourrait développer l'instruction civique en faisant davantage appel à la solidarité : une fois que l'on n'est plus là, on peut encore être utile et même en n'étant plus là, on peut aider la vie à continuer ». Pour sa part, notre collègue Mme Yvette Benayoun-Nakache s'est demandé « comment parvenir à toucher réellement toute la population de façon informative et citoyenne (127) ? ».
1.- Le prélèvement et la greffe : une dimension symbolique très forte
« Que les Français l'ignorent, ou qu'ils le refusent, le droit de la bioéthique n'est pas passé dans la conscience collective », constate Marie-Angèle Hermitte (128). Beaucoup s'accordent en effet à reconnaître l'existence d'un contexte général d'ignorance à l'égard de ces problèmes.
Évoqué comme manifestation de cohésion sociale, le don d'organes fait référence à la nécessité d'un gisement d'organes où puiser pour sauver des malades. Cette référence heurte la représentation que l'homme garde de lui-même, de son corps comme siège de l'expérience primordiale de la vie, comme totalité rebelle à une approche analytique. Or il est désormais confronté à une médecine qui recourt de plus en plus à la science laquelle, par définition, analyse, segmente et parcellise son champ d'investigation.
La prescription d'une greffe pour un malade revient, en dernier ressort, à attendre la mort d'une personne inconnue. Mort violente, puisque le prélèvement ne peut avoir lieu que sur une personne en état de mort encéphalique, laquelle n'est pas encore très connue du public et dont les fondements scientifiques, définis par quelques savants, ne sont pas partagés par l'ensemble des sociétés.
D'ailleurs, ce prélèvement résulte-t-il d'une nécessité thérapeutique, ou d'une « impasse thérapeutique » (129) qui oblige à trouver une solution dans le recours à des « pièces détachées » ? Les anthropologues (130) constatent qu'au regard des différences de culture, le projet de transplantation peut apparaître comme celui de la science occidentale par excellence, le triomphe de l'homme machine par remplacement illimité des organes.
Enfin, dans la réalité des faits, la pratique des multi-prélèvements est devenue la norme, réalité très souvent occultée par tous les intervenants de la médiation en faveur du don.
2.- Les circonstances du refus : un moment traumatisant
Les auditions de la Mission ont mis en évidence que la règle du consentement présumé, élaborée en 1976 et confirmée dans les lois de 1994, n'a pu s'émanciper du recueil par les médecins du témoignage de la famille au moment de l'annonce du décès, survenu dans des circonstances violentes. La plupart du temps, ce témoignage se transforme en refus de prélèvement, et donne à la famille un rôle que n'avait pas prévu le législateur. Est-ce parce que, comme le souligne le civiliste Jean Carbonnier, le droit admet un certain prolongement de la personnalité après la mort, une protection posthume de la personne ? C'est ce que semble confirmer le Professeur Dominique Durand lors de son audition par la Mission, le 10 janvier 2001 : « la famille, confrontée à la pire situation qui soit, ne sait pas faire la nuance entre témoigner d'une volonté et avoir une attitude patrimoniale. Elle considère qu'elle est l'héritière de la volonté du défunt. [...] Il y a peut-être là une image d'un refus très profond de la société vis-à-vis d'une activité difficile ».
Activité difficile certes, qui peut expliquer la persistance d'une forte réticence du corps social qu'exprime le taux de refus au prélèvement de 30 % environ. Bien que souvent passée sous silence, cette réticence persiste aussi au sein des milieux hospitaliers, en raison de la complexité de la médiation médicale entre donneurs et receveurs, sachant que cette médiation intervient pour un receveur inconnu et non encore choisi. Lors de son audition par la Mission le 7 juin 2001, Mme Martine Allain-Régnault observait que « l'analyse dans le détail montre que les freins à la transplantation relèvent plus des courroies intermédiaires que de l'opinion. Il faut que le généraliste, qui est à côté d'une famille endeuillée, soit favorable au don d'organes. Or, il ne l'est pas. [...] Le don d'organes passe par un abandon d'organes et, comme pour l'adoption, c'est la partie désagréable des choses ».
Réticence des familles, aussi. Mme Martine Allain-Régnault rappelait : « J'ai ainsi rencontré une famille qui attendait un don d'organe pour sa fille, qui finalement est morte cérébralement à la suite d'une hépatite fulminante. On a alors dit aux parents : Vous attendiez de la terre entière un don d'organe, êtes-vous prêts à donner le c_ur et les poumons de votre enfant ? Et, dans ces circonstances-là, vous voyez très bien toute la difficulté qu'il y a à accepter de donner, bien plus qu'à recevoir ». Elle reconnaissait toutefois que « bien que la loi autorise à prélever sans l'autorisation de la famille, dans l'immense majorité des cas on lui demande son autorisation. Si l'on veut avancer dans ce domaine, qui me tient très à c_ur, il faut aller doucement et favoriser les campagnes d'information, notamment à l'école ».
Lors de son audition par la Mission, le Professeur Didier Houssin a insisté sur ce point : « C'est ici que l'information du public hors tout contexte émotionnel prend toute son importance », le Professeur Dominique Durand ayant confirmé que « l'information de la société (...) est une voie difficile où l'on n'avance qu'à petit pas et qui est, finalement, assez peu productive. Il est probable que les résultats seront enregistrés bien plus tard. Il va falloir laisser le temps à la société, devant un progrès technique très rapide, de mûrir sa réflexion ».
Mûrir sa réflexion nécessite un certain recul car il s'agit d'envisager sa propre mort et de se déterminer en conscience. M. Francis Kernaleguen, s'exprimant à ce sujet devant la Mission le 4 octobre 2000, reconnaissait : « J'ai quatre enfants, deux d'entre eux sont d'accord pour un prélèvement, deux autres non. Il est vrai que je me disais, en attendant mon audition, que je respecterais leur choix, cela va de soi. Mais que se passerait-il si, leur choix étant un refus, celui-ci avait pour conséquence de mettre en danger la vie de quelqu'un qui est dans la chambre d'à côté ? Un problème de conscience se poserait à moi. En même temps, je n'imagine pas refuser de respecter la parole de mes enfants. [...] Le registre de refus est une facilité de ce point de vue. Quand quelqu'un s'y est inscrit, la famille n'a plus rien à dire, et je n'ai plus mon problème moral à régler. [...] Je pense que le choix de donner doit être respecté absolument ».
C'est le thème sur lequel l'Établissement français des Greffes a judicieusement choisi de mener son information : insister sur la nécessité de faire connaître sa position.
B.- L'INFORMATION DU PUBLIC : ASSOCIER LE PLUS GRAND NOMBRE D'ACTEURS
1.- Une responsabilité des pouvoirs publics
L'article L. 1211-3 du code de la santé publique interdit la publicité en faveur d'un don d'éléments ou de produits du corps humain au profit d'une personne déterminée ou d'un établissement ou organisme déterminé, cette interdiction ne faisant pas obstacle à l'information du public en faveur du don d'éléments et produits du corps humain. Cette information est réalisée sous la responsabilité du ministre chargé de la santé par l'Établissement français des Greffes (article L. 1251-1 du code précité). C'est ainsi qu'à l'occasion de la mise en place du registre automatisé des refus de prélèvement, une brochure, intitulée « Pour ou contre, prenez position », a été diffusée à dix millions d'exemplaires distribués dans les officines pharmaceutiques et les cabinets médicaux. Cette brochure était destinée à donner une information générale sur l'activité de greffe et sur son encadrement juridique.
L'encadré ci-joint reproduit le message publié sur le site Internet de l'Établissement français des Greffes à l'occasion de la première Journée nationale de réflexion sur le Don et la Greffe, instituée lors de la présentation du Plan Greffe en juin 2000 :
En de nombreuses occasions, lors de démarches en mairie par exemple, une information en faveur du don d'organes pourrait être donnée. Notre collègue Mme Jacqueline Fraysse (131) a suggéré cette possibilité : « Je me demande si nous ne devrions pas envisager de poser la question tranquillement aux jeunes eux-mêmes, par exemple à l'occasion de la délivrance d'une carte d'identité... ».
Votre Rapporteur suggère pour sa part que l'on puisse réfléchir à transmettre ce message de civisme et de solidarité à l'occasion des journées d'appel de préparation à la défense, organisées par le ministère de la défense. Désormais, l'obligation du recensement concerne en effet tous les jeunes, filles et garçons de nationalité française âgés de seize ans, qui sont tenus de se faire recenser à la mairie de leur domicile ou au consulat s'ils résident à l'étranger. Pourquoi ne pas insérer l'information sur le don d'organe et le registre des refus dans la brochure qui leur est remise à la fin de la journée ? De plus, le bilan d'activité (132) des premières journées a mis en évidence chez les participants que les valeurs à défendre sont l'égalité (67 %), la liberté (65 %), la solidarité (56 %), la fraternité (55 %) et la tolérance (50 %), toutes valeurs requises quant à la réceptivité d'un message comme celui-ci.
Une note d'information sur le don d'organes et les modalités de consentement et de refus pourrait être ainsi insérée dans le dossier qui leur est remis à l'occasion de cette journée. L'encadré ci-après récapitule les enjeux pratiques de la participation à la journée d'appel à la préparation à la défense. Il en ressort que l'information sur la solidarité à travers le don d'organes pourrait accompagner des rubriques de nature à retenir l'attention des jeunes participants, auxquelles ils sont sans doute très réceptifs.
LE RECENSEMENT A 16 ANS, UN DEVOIR QUI DONNE DES DROITS
L'attestation du recensement remise au jeune lui permet
- de poursuivre sa scolarité,
- d'être convoqué à la journée d'appel de préparation à la défense (JAPD),
- d'effectuer des tests de lecture.
Au terme de la JAPD, le jeune reçoit un certificat de participation qui lui permet :
- de se présenter à un examen scolaire,
- de se présenter à l'examen du permis moto ou auto,
- d'obtenir un document officiel à la mairie,
- de se présenter aux concours et emplois de la fonction publique.
Source : Ministère de la défense, Défense et Citoyenneté, Journée d'appel de Préparation à la défense, 1ère participation des filles, 8 avril 2000.
M. Dominique Becquart, membre du Lions Club de Mouvaux le Bosquiel qui a été à l'origine de la campagne d'information réalisée par cette association, a fait une autre suggestion lors de son audition par la Mission le 10 janvier 2001 : « Une action qui me paraît intéressante serait l'édition d'un timbre. En 1996, j'avais écrit à l'organisme qui s'en occupe et j'avais reçu une réponse, très correcte, m'indiquant que d'autres actions passeraient avant notre suggestion. On voit La Poste éditer aujourd'hui des timbres Bon anniversaire ! ou C'est un garçon ou C'est une fille. Ce ne serait pas mal si on pouvait diffuser un message équivalent pour le don ».
2.- Associer l'Éducation nationale
Lors de son audition du 10 janvier 2001, le Professeur Didier Houssin, directeur général de l'Établissement français des Greffes, a considéré que « l'idée de confier au ministère de l'Éducation nationale une tâche dans ce domaine serait [...] extrêmement utile. [...] L'année dernière, le rectorat de Lyon a organisé un concours de philosophie sur ce thème, qui a montré l'appétit des jeunes et des enseignants pour ce type d'enseignement. [...] il pourrait être effectivement très utile qu'un texte de haut niveau accorde une certaine responsabilité à l'Éducation nationale dans ce domaine ».
Le rôle de l'Éducation nationale a en effet été évoqué à plusieurs reprises devant la Mission, que ce soit, par exemple, par notre collègue, Mme Jacqueline Fraysse : « Aussi, je me demande si nous ne devrions pas envisager de poser la question tranquillement aux jeunes eux-mêmes, par exemple à l'occasion de la délivrance d'une carte d'identité ou à l'école de manière systématique » ou par Mme Martine Allain-Régnault lors de son audition du 7 juin 2000 : « il faut aller doucement et favoriser les campagnes d'information, notamment à l'école ».
Lors de son audition par la Mission d'information, le 10 janvier 2001, M. Dominique Becquart a également soutenu ce type de proposition : « [...] Nous pensons qu'il serait intéressant qu'il y ait davantage d'informations dans les lycées, en direction des seize-dix-huit ans, des classes de terminale. Pas dans les collèges. Cette information pourrait se faire dans le cadre d'une instruction civique, en s'appuyant sur des thèmes de solidarité et de fraternité, ou dans celui des sciences de la vie, en insistant très fortement sur la notion de mort encéphalique [...] ».
3.- Les associations participent à l'information
Le centième anniversaire de la loi sur les associations a été l'occasion de leur rendre un hommage justifié.
De nombreuses associations militent depuis longtemps en faveur du don d'organes, bien avant la création de l'Établissement français des Greffes. La plus connue est la Fédération des associations pour le don d'organes et de tissus humains, France Adot, créée en août 1969 par le Professeur Jean Dausset, après la création de France Transplant, structure médicale de transplantation au niveau des centres hospitalo-universitaires répartis dans toute la France. Ces associations ont une vocation nationale ou régionale ; certaines se sont spécialisées en fonction des organes prélevés.
On peut relever en outre la création de l'Organisation Mondiale pour le Don d'Organes et de Tissus humains (OMIDOT), afin de promouvoir « l'Union des États pour sauver leurs malades » car « un échange loyal, concerté, chacun faisant passer la guérison avant toute chose, permettra aux médecins de faire face à leur mission de soigner ».
Toutes ces associations affichent le même but : une mission de promotion en faveur du don d'organe et d'actions en faveur de la recherche. Leurs activités varient autour de l'organisation de manifestations nationales, la coordination et le soutien d'actions locales, l'édition de revues et la production et diffusion de documents, en lien avec les médias. Les associations spécialisées donnent une information plus spécifique et proposent un accompagnement moral et social aux personnes en attente de greffes et aux personnes greffées.
En règle générale, l'information délivrée par ces associations porte sur le contenu de la loi, les règles applicables en matière de greffes d'organes, et les modalités pour s'engager en faveur du don ou pour manifester son opposition. Le message insiste sur la nécessité de s'informer, de prendre position et d'en témoigner auprès de la famille.
Lors de leur audition par la Mission le 10 janvier 2001, MM. Dominique Becquart et Pierre Duchatelle du Lions Club de Mouvaux le Bosquiel, ont présenté la campagne d'information en faveur du don d'organes, qu'ils ont menée depuis 1995, sur le thème « J'aime la vie, je dis oui au don d'organes, s'informer est essentiel », à l'issue d'une réflexion à laquelle ils avaient associé l'Établissement français des Greffes.
Cette campagne a été diffusée à l'ensemble du territoire national. Elle comportait l'envoi en nombre d'un argumentaire à tous les présidents de Lions clubs qui devaient le distribuer à l'occasion d'une réunion statutaire consacrée à ce sujet. Cet envoi comportait des outils de communication destinés à informer le grand public, et particulièrement les jeunes : une bande dessinée, des tracts, des dépliants co-rédigés avec l'Établissement français des Greffes, des banderoles, une cassette vidéo, des affiches, des auto-collants. En clôture de cette action nationale, les membres du Lions Club ont planté un « arbre de la reconnaissance » (un gingko biloba) dans l'enceinte du centre hospitalo-universitaire de Lille.
M. Dominique Becquart a insisté, devant la Mission, sur l'importance d'être très attentif au message devant être délivré : un message de tolérance, insistant sur la nécessité de s'informer « afin de pouvoir prendre une décision en toute connaissance de cause et adhérer ou non au projet [...]. Nous avons démarré, en ayant quelques idées bien claires à l'esprit. [...] Un message qui ne devait pas être diffusé par des médecins. [...] En présentant nous-mêmes ce message, et sans relation avec le corps médical, nous pouvions avoir plus d'impact. [...] Nous avons toujours pensé qu'il nous appartenait à nous, « civils », de le délivrer à des jeunes, [...] et, si possible, en cercle restreint ».
Toutefois, cette campagne, menée avant l'instauration du registre des refus, n'a pas pu mentionner la possibilité de s'y inscrire.
C.- LA FINALITÉ DE L'INFORMATION : DIMINUER LE NOMBRE
DE REFUS
Concernant l'activité d'information à destination du grand public, le but poursuivi est de contribuer à diminuer le taux de refus des familles au prélèvement d'organes sur un donneur décédé, afin de donner toute sa consistance à la règle du consentement présumé. La pertinence du message sur le contenu de la loi, relatif à la règle du consentement présumé, revêt donc une grande importance.
Il s'agit de convaincre la population française que la transplantation n'est pas une mutilation, mais un geste de solidarité destiné à sauver des vies. En aucun cas, cette solidarité ne doit donner l'impression de s'exercer sous une forme de pression ; elle doit être réfléchie même si pour cela, elle demande à chacun d'affronter l'idée de sa propre mort.
1.- À qui s'adresse le message ?
Il peut paraître surprenant de poser la question, tant la réponse semble évidente : au grand public. Mais, à travers lui, il s'adresse de fait aux donneurs potentiels.
Les auditions réalisées par la Mission ont souvent témoigné de la volonté d'atteindre les jeunes, les enfants. Une telle démarche vaut plus dans un processus éducationnel qu'informationnel et demande donc du temps.
En réalité, « les donneurs potentiels sont dans le circuit, irréel, idéel, institutionnel de la communication, soit en tant que cibles des campagnes de promotion, soit en tant que lecteurs ou auditeurs des médias, ou encore en tant que citoyens auxquels s'adressent lois et règlements [...] De la prise de position idéale - en faveur ou contre le don, voire contre le principe même des thérapies substitutives - à un comportement effectif de don ou de refus de don de matériaux, il existe de nombreuses médiations psychologiques (133) ».
Ce contenu est double :
- d'une part, convaincre du bien-fondé du don d'organes afin de donner toute sa portée au consentement présumé, tel que l'avait envisagé son concepteur, M. Henri Caillavet. Pour lui, lors de son audition par la Mission le 4 octobre 2000, « tout individu qui, conscient, n'avait pas dit non, était donneur. Le don était la règle » ;
- d'autre part, communiquer sa décision d'accord ou de refus de prélèvement à son entourage.
D'autres points, relatifs au contenu de la loi, sont abordés :
- Il s'agit d'abord du refus et du registre des refus :
Il faut bien reconnaître la difficulté soulevée par ce registre dans le cadre d'une telle communication. Le fait d'institutionnaliser la possibilité de refus ne favorise-t-il pas cette possibilité ? Ainsi que le constate notre collègue, Mme Catherine Génisson (134), « nous sommes dans une logique de consentement présumé et cette notion est difficile à expliquer au grand public. Elle passe par une connaissance, par tous, de la mise en place du registre du refus mais c'est une campagne qui est plus difficile à réaliser que ce ne serait le cas pour la logique de consentement explicite ». Comment faire campagne en faveur du don et signaler l'existence de ce fichier ? M. Dominique Becquart, qui a conduit la campagne menée par les Lions Clubs, a expliqué : « Nous concluons en leur conseillant, puisque ce sont des jeunes, d'en parler d'abord en famille et, ensuite, s'ils désirent donner leurs organes si, malheureusement, il leur arrivait quelque chose, de l'indiquer à leurs parents et, s'ils le souhaitent, de porter une carte attestant de cette volonté. Dans le cas contraire, nous leur conseillons pareillement de l'indiquer, de porter un papier sur eux l'indiquant et, s'ils le souhaitent, de s'inscrire au registre des refus à Marseille. C'est très clair ».
- Une autre question abordée est celle des différentes manifestations de la volonté de donner :
Le choix du législateur de se fonder sur le constat d'une absence de refus pour pouvoir prélever a en fait pour conséquence l'absence de procédure officielle de recueil des intentions de don. La volonté de donner peut se concrétiser par le port d'une carte de donneur proposée par l'Établissement français des Greffes ou les associations, et par le port d'un insigne. Mais instituer par la loi un tel insigne est loin de faire l'unanimité. Par exemple, M. Henri Caillavet, lors de son audition du 4 octobre 2000, s'interrogeait : « Peut-être pourrait-on imaginer une marque... Cela me rappelle pourtant de mauvais souvenirs mais, en effet, pourquoi ne pas envisager une marque placée [...], sur la carte d'identité ou le passeport lorsqu'on vous les délivre ». Notre collègue, M. Jean-Michel Dubernard, a clairement indiqué aux membres de la Mission que, « les cartes de donneurs [...] posent un énorme problème parce que les gens croient que si on possède la carte, on peut être prélevé et que si on ne l'a pas, on ne peut pas être prélevé. Les familles pensent que nous sommes sous le régime du consentement explicite. J'ai eu à ce sujet de nombreuses discussions avec les membres de l'ADOT. Les cartes de donneur sèment une confusion ».
Il faut rappeler que le port d'une carte de donneur témoigne de la volonté de la personne mais n'exempte pas l'équipe médicale de la consultation du fichier des refus. Cette carte, pour l'Établissement français des Greffes, permet un meilleur dialogue entre la famille et le médecin.
- La question des témoignages en faveur du don est également au centre des réflexions pour déterminer les meilleures façons de communiquer :
Les avis sont nuancés sur le fait de savoir si des patients ayant reçu une greffe d'organe ou des familles qui ont accepté un prélèvement doivent participer à cette information. « Faut-il favoriser cette situation ou même la laisser se produire spontanément dans la mesure où, me semble-t-il, c'est un acte très délicat ? [...] Il ne faut pas entacher ce message de voyeurisme ni laisser s'exprimer des témoignages qui sont trop douloureux à obtenir. Il faut rester pudique en la matière », insistait notre collègue Mme Catherine Génisson, devant la Mission le 10 janvier 2001.
Le recours au levier émotionnel est un moyen très fort et bien connu des publicitaires anglo-saxons mais l'information, par définition, impose une certaine mise à distance. Il est délicat de parier sur l'émotion dans des messages dont le contenu concerne des moments extrêmement douloureux, où il est demandé à chacun, justement, de prendre du recul. De fait, les témoignages exprimés dans la brochure d'information de l'Établissement français des Greffes, portent essentiellement sur le fait que les familles avaient déjà eu des échanges sur l'attitude à l'égard du don d'organes.
- Faire le choix d'informer pour éduquer suppose de mener une entreprise de longue durée :
Le processus d'éducation prend du temps, qui nécessite l'enseignement, l'exemple, l'apprentissage d'autrui. Il est probable que comme les individus, les sociétés doivent être éduquées. En cela, l'extrême rapidité avec laquelle les techniques apparaissent et envahissent notre vie quotidienne demandent du temps pour être assimilées par les individus. « Nous manquons de pédagogie. Lorsque l'on sait que le don est un geste de générosité et de fraternité [...], il faudrait mettre en place une pédagogie plus systématique et permanente. Nous devrions faire en sorte que les enfants, qui sont réceptifs, sachent qu'ils sont donneurs d'organes pour sauver quelqu'un. Donner, c'est sauver une vie », soulignait ainsi M. Henri Caillavet lors de son audition par la Mission le 4 octobre 2000.
Il est en effet probable que la réticence à l'égard du don d'organes ira en s'amenuisant à proportion de la confiance retrouvée et de la qualité de l'information qui sera donnée.
D.- LA RECONNAISSANCE DE LA SOCIÉTÉ : LE DONNÉ ET LE RENDU
L'éthique du don
Différents acteurs interviennent dans le don : le donneur, le receveur, mais aussi un certain nombre d'intermédiaires (familles, médecins). Pour chacun d'eux, l'éthique du don se pose en des termes différents. Pour le receveur, la situation peut sembler relativement simple puisque, en général, son choix est de continuer à vivre ou pas ou du moins d'accéder à des conditions de vie décentes. Cependant, on s'aperçoit que, après avoir reçu cet élément étranger qu'est l'organe transplanté, certains traumatismes psychologiques peuvent se faire jour liés à la difficulté à accepter un fonctionnement du corps totalement dépendant d'un organe étranger vital. Le cas du don de donneur vivant apparenté peut également engendrer de véritables bouleversements psychologiques et relationnels. Il peut en effet créer une situation de forte dépendance de la part du receveur à l'égard du donneur et, en cas de rejet du greffon, entraîner une forte culpabilité de la part de celui-ci. Pour les familles des défunts donneurs potentiels, la difficulté est de faire face, dans le même temps et dans des circonstances de profond choc émotionnel, au décès d'un proche et à la décision d'autoriser le prélèvement. Cette décision est d'autant plus difficile à prendre que le maintien artificiel en fonction des organes ne favorise pas l'acceptation de la mort. Des enquêtes montrent que le comportement des familles, à cet égard, est très lié à leur degré d'information sur la transplantation [...].
D'un point de vue religieux enfin, l'ensemble des confessions est aujourd'hui d'accord pour ne pas s'opposer au don et au prélèvement d'organes et de tissus, considérant que la greffe n'est pas une simple technique médicale mais également une pratique sociale qui fait appel à la solidarité et au combat pour la vie.
Source : Fondation pour la recherche médicale.
Lors de son audition par la Mission le 10 janvier 2001, le Professeur Didier Houssin a rappelé que : « Le don est le geste qui consiste à donner, à recevoir... et à rendre. Vous connaissez la réflexion conduite par Marcel Mauss sur la question. Je pense qu'il est souhaitable de mener cette logique à son terme et de faire apparaître dans la loi que le « rendre » existe aussi. Cette reconnaissance exprimée par la Nation et la société à ceux qui ont donné, même si elle s'exprime de manière symbolique, serait un complément très utile et ouvrirait la porte à des manifestations de caractère symbolique ».
Marcel Mauss (135), s'étant appuyé sur les travaux de Malinowski et Boas, a montré que le don présente un caractère doublement ambivalent : libéral et gracieux, il est régulièrement suivi d'un contre-don tout aussi unilatéral et arbitraire, mais tacitement perçu comme la réponse adéquate à la première prestation. Il serait, sous le point de vue de la pratique, voire de la contrainte sociale, l'amorce d'une relation réciproque, un échange différé. Il recouvrirait un second paradoxe : l'action de donner aurait en fait une dimension agressive, car le cadeau crée une dette. En « obligeant » son partenaire, le donateur acquiert sur lui de l'ascendant, sinon du pouvoir, car donné, un objet précieux garde toujours en soi la présence du propriétaire d'origine, la possession et l'usage étant transférés au donataire. « Il est probable que l'on pourrait concevoir que l'organe donné par un donneur vivant à un malade qui le reçoit, et que son corps ne rejette pas, est en quelque sorte un organe qui reste la propriété du donneur tout en devenant la possession du receveur. De sorte qu'entre ces deux personnes une relation d'endettement direct et ineffaçable s'instituerait. [...] N'oublions pas non plus que dans beaucoup de cultures, découper un corps, prélever un organe, le greffer sur un autre corps, cela se fait tous les jours dans l'imaginaire. C'est ainsi que l'on se représente les pratiques de sorcellerie. [...] Toute campagne pour prélever des organes doit donc se faire en ayant une certaine connaissance des contextes culturels. [...] Si les médecins désirent avoir plus de succès dans la pratique des dons de mort à vivant... il faudrait que cette pratique prenne une dimension civique et symbolique nationale ; que les familles qui y consentent soient célébrées pour leur solidarité « humaine », écrit l'anthropologue Maurice Godelier (136).
La sociologue Simone Novaes (137), que votre Rapporteur a auditionnée, souligne en outre que « le traitement que le don rend possible a non seulement un coût, mais un prix que le patient, et à travers lui le système de santé, aura à payer. Or ce coût de la démarche du donneur, mais aussi la réelle dimension économique de ces pratiques médicales, sont mis hors champ dans tout discours sur la gratuité du don. [...] Cet appel au don anonyme et gratuit traite le geste altruiste du donneur comme acte « pur », isolé du contexte qui le sollicite et des effets qu'il produit. Il met ainsi entre parenthèses toute considération sur la nature des situations censées engager les donneurs et sur le type de liens que leur geste crée (ou ne crée pas) ».
Aucune campagne d'information ne pourra supprimer cette réalité, comme d'autres relatives à l'accueil des familles, à la réticence du corps médical dont les fondements inconscients sont peut-être du même ordre que ceux des familles. Le Professeur Didier Houssin indiquait, d'ailleurs, aux membres de la Mission, que « l'accompagnement des familles qu'assurent les coordinations hospitalières est aussi une manière de rendre et d'être à l'écoute. Si cela figurait dans la loi, sans être pompeux naturellement, ce serait précieux ». C'est aussi le constat que fait Mme Martine Allain-Régnault : « J'ai interviewé des généralistes, qui ont accompagné des parents, afin de démontrer l'importance de leur rôle en la matière ».
Sa réflexion sur le don avait incité le Lions Club à clore sa campagne par la plantation d'un arbre de la reconnaissance, témoin de « l'infinie reconnaissance de ceux qui ont reçu envers ceux qui ont donné (138) ». Cette réflexion a conduit M. Dominique Becquart à manifester le souhait, devant la Mission, le 10 janvier 2001 « que la loi puisse, si elle le peut, davantage reconnaître la famille de ceux qui donnent. Par l'intermédiaire d'un témoignage de reconnaissance très difficile à concrétiser. Nous avons nous-mêmes longtemps cherché et nous avons pensé à la plantation d'un arbre de la reconnaissance. Nous en avons planté un dans l'enceinte du CHU de Lille. Cette initiative a été reprise ailleurs puisque les Lions clubs en ont planté sept dans sept autres villes de France, en accord, bien entendu, avec l'Établissement français des Greffes. C'est une manifestation hautement symbolique, merveilleuse, qu'il faudrait développer. Planter un gingko biloba, cet arbre qui a résisté à la bombe atomique, est un symbole fort ».
Pour votre Rapporteur, il convient d'ores et déjà de préciser que cette reconnaissance, si le principe en était retenu, devrait évidemment s'exprimer en faveur des donneurs seuls, car exprimer de la reconnaissance aux familles « de ceux qui donnent » mettrait à bas tout l'édifice du consentement présumé reposant sur l'intégrité du donneur et le respect de son libre arbitre. Même si la réalité des faits tend à le laisser croire, ce ne sont pas les familles qui donnent, elles ne sont que porteuses, selon la loi, de la parole du défunt.
Le CCNE n'est pas favorable à l'expression d'une reconnaissance, quelle qu'elle soit ; selon son avis sur l'avant-projet de révision des lois de bioéthique, une telle « disposition relève d'une rhétorique dépassée et mal accordée avec l'éthique individuelle du don. Le CCNE rappelle que la valorisation excessive du don a, dans un passé récent, été désignée comme l'une des causes du dysfonctionnement du dispositif français de don du sang. Au surplus, si cette disposition devait se traduire par des manifestations concrètes de reconnaissance (diplôme, décoration, médaille), elle serait contraire au principe fondamental qui inspire la loi, et qui est celui de l'anonymat du don ».
Votre Rapporteur considère que, loin de transgresser le principe d'anonymat, une manifestation de reconnaissance, de manière symbolique et collective, serait à même de recréer du lien social. Par ailleurs, l'éthique du don, comme l'éthique en général, ne peut faire abstraction de certaines réalités humaines et sociales énoncées plus haut. Ce n'est pas faire du sentimentalisme que reconnaître la part d'humanité, avec sa part de souffrance et d'espoir, qui est au c_ur de la démarche éthique. Pour autant, dans une société dite évoluée, une telle éthique relève d'abord de l'éducation dont il a été reconnu qu'elle demande du temps. Enfin, la prise en charge collective d'une manifestation de reconnaissance ne soulagerait-elle pas également les receveurs, pour lesquels pourrait s'être créé inconsciemment un sentiment de dette ?
Les enquêtes révèlent que l'intention de don est étroitement corrélée au niveau d'éducation, ainsi qu'à l'estime de soi (139). Elles mettent en évidence que « communiquer des matériaux, c'est accepter de faire partie du même corps social. Or, le sentiment d'appartenance et d'unité sociale est aujourd'hui en crise [...] Ce projet thérapeutique ne trouve pas les conditions de son exercice dans la seule efficacité des gestes techniques (la première greffe du c_ur eut lieu en Afrique du Sud, au pays de l'apartheid), mais aussi dans l'affirmation, plus ou moins militante, d'un projet social de solidarité et de communication humaines universelles » (140). Aussi votre Rapporteur pense-t-il que tout ce qui peut créer et préserver ce lien doit être encouragé.
VI.- FINALITÉ SCIENTIFIQUE, MÉDICALE OU THÉRAPEUTIQUE
Le 3 juin 1999, le Sénat adoptait un amendement modifiant l'article 16-3 du code civil afin, selon les termes de son auteur, M. François Autain, de rectifier une « coquille » et de préciser qu'il ne peut être porté atteinte à l'intégrité du corps humain qu'en cas de nécessité médicale, au lieu de thérapeutique. M. François Autain défendait ainsi son amendement : « Le code civil prévoit en effet, depuis le vote de cette loi (141), qu'il ne peut être porté atteinte à l'intégrité du corps humain qu'en cas de nécessité thérapeutique. Dans la mesure où le terme thérapeutique exclut la prévention, cette disposition conduit à rendre illégale toute intervention portant atteinte au corps humain dès lors qu'il s'agit, non pas de soigner, mais de prévenir une maladie. Ces interventions peuvent être en effet considérées comme des blessures volontaires imputables au praticien et sanctionnées en application des articles 222-9 et 222-10 du code pénal » (142).
S'ensuivait un débat pour déterminer si la finalité médicale incluait ou non la recherche et la prévention, d'autant que l'article 16-10 du code civil, par exemple, précise que l'étude génétique des caractéristiques d'une personne ne peut être entreprise qu'à des fins médicales ou de recherche scientifique. M. François Autain indiquait que « (cette) modification [...] est attendue avec impatience par un certain nombre de spécialistes, et pas seulement les chirurgiens esthétiques, qui ont été évoqués, mais également par les gynéco-obstréticiens ». L'amendement a été adopté, contre l'avis du secrétaire d'État.
A.- UNE NÉCESSAIRE CLARIFICATION
Quelles différences distinguent la finalité thérapeutique, la finalité médicale et la finalité scientifique ? Que recouvrent exactement les trois termes ? La prévention, la recherche, certaines interventions chirurgicales ?
1.- Finalité thérapeutique, finalité médicale
Le droit et la jurisprudence admettent que le consentement d'une personne à une intervention sur son corps n'est pas un fait justificatif suffisant. Il faut à l'acte médical une cause (ou finalité) thérapeutique, le principe thérapeutique, lequel n'est pas sans limite mais tempéré par la règle de proportionnalité de l'atteinte à l'intérêt de santé à satisfaire. La rédaction de l'article 16-3 du code civil résultant de la loi de 1994 revenait à consacrer dans la loi civile ce principe comme l'un des principes généraux garantissant le respect du corps humain. Beaucoup plus « élastique », la notion de nécessité médicale ne permettrait pas d'en cerner les critères et les contours. Car au-delà de l'intérêt médical du patient, le terme médical recouvre à lui seul « tous les objets de la médecine », d'après le Littré.
Ce même dictionnaire définit la thérapeutique comme « l'ensemble des actions et pratiques destinées à guérir, à traiter les maladies ».
La loi de 1994, en n'autorisant l'atteinte à l'intégrité du corps qu'en cas de nécessité thérapeutique, incluait de fait la notion de nécessité thérapeutique pour autrui, et non pas seulement pour la personne subissant l'atteinte. On peut se demander s'il en va de même pour la nécessité médicale. Certains juristes (143) en effet considèrent que la qualification des situations sera opérée par référence aux critères médicaux, dont le contenu est déterminé par les médecins eux-mêmes.
Que faut-il alors déduire de cette modification instamment désirée par le corps médical ? Par exemple, l'autorisation de stérilisation figure désormais dans la loi relative à l'interruption volontaire de grossesse et la contraception ; elle concerne également la stérilisation des personnes handicapées mentales, alors qu'il existe de nouvelles méthodes contraceptives à effet prolongé. Comment apprécier la nécessité médicale ?
Les textes se réfèrent à « l'intérêt thérapeutique » (article L. 1231-1 du code de la santé publique), aux « fins thérapeutiques ou scientifiques » (article L. 1232-1 du code de la santé publique), au « but thérapeutique ou scientifique » (article L. 1241-1 du code de la santé publique), aux « fins thérapeutiques ou scientifiques » (article L. 1241-3 du code de la santé publique), aux « fins médicales ou de recherche scientifique » (article L. 1131-1 du code de la santé publique), à la « nécessité thérapeutique » (article L. 1211-5 du code de la santé publique), « fins thérapeutiques » (article L. 1211-6 du code de la santé publique), « utilisation thérapeutique » (article L. 1221-2 du code de la santé publique), aux « recherches biomédicales » (article L. 1121-1 du code de la santé publique) (144)sans préciser qui en sont les bénéficiaires.
Dans son avis n° 60 du 25 juin 1998 sur le réexamen des lois de bioéthique en ce qui concerne le don d'organe, le CCNE demandait de bien distinguer, dans le texte de la loi, les trois finalités des prélèvements :
« i) finalité thérapeutique, pour un don d'organe susceptible de sauver une vie menacée, véritable exigence prioritaire en matière de santé publique ;
« ii) finalité médicale, pour l'autopsie, dernier acte médical susceptible de rechercher les causes de la mort ;
« iii) finalité scientifique, concernant non seulement les prélèvements d'organes mais aussi de tissus, de cellules et de produits du corps humain ».
Toutefois, il semble ne pas tirer les conséquences de ces distinctions lorsqu'il préconise, dans la phrase suivante : « Le consentement présumé doit être maintenu pour les prélèvements à finalité thérapeutique et médicale, et étendu aux prélèvements à visée de recherche médicale ».
Que recouvre la notion de finalité scientifique ? La recherche, selon des pratiques expérimentales précises et pertinentes, semble-t-il. Encore faudrait-il savoir s'il s'agit de recherche fondamentale, encadrée par un protocole précis, ou de recherche appliquée. Inscrite à plusieurs reprises dans le code de la santé publique, cette notion viserait la recherche médicale, ou la recherche scientifique à visée médicale.
À l'heure où l'on réfléchit à l'opportunité de la recherche sur l'embryon, ces notions que les spécialistes des disciplines concernées maîtrisent sans doute parfaitement devraient être clairement définies pour éviter que ne se développe une nouvelle « crispation », à l'instar de ce qui s'est produit en matière de pratique de l'autopsie. Les scientifiques et les médecins feraient _uvre utile en éclairant la Représentation nationale.
B.- L'AUTOPSIE, UN RÉVÉLATEUR DE CETTE CONFUSION
Base d'une méthode de raisonnement anatomo-clinique, l'autopsie était ainsi définie en 1902 par M. Letulle, Professeur d'anatomie pathologique à l'hôpital Boucicaut : « L'autopsie est l'étude détaillée d'un être mort dans le but d'y rechercher, et si possible, d'y reconnaître les causes du décès et leurs conséquences. Elle est le complément indispensable des investigations poursuivies sur le vivant par le clinicien désireux d'établir un diagnostic impeccable, sans lequel toute la science pronostique demeurerait un aléa et la thérapeutique, une force aveugle ».
Les partisans de cette pratique indiquent qu'elle donne des informations essentielles aux familles des personnes décédées et à la population en général, en fournissant une explication rationnelle au décès, en contribuant à l'assurance de qualité des soins et en offrant l'opportunité de participer au progrès des connaissances médicales, et notamment de permettre la découverte de maladies nouvelles. Elle fournit aux médecins cliniciens des données objectives sur la maladie et la cause du décès, permet la découverte de maladies nouvelles, constitue un mode d'évaluation de diverses pratiques médicales et constitue un outil de santé publique apportant des renseignements sur la morbidité et la mortalité. Elle représente le meilleur moyen d'apprentissage pour l'enseignement de la médecine et, au niveau de la recherche, elle contribue à une meilleure connaissance de maladies dont la physiopathologie est encore mal élucidée (145).
Toutefois, le milieu médical ne semble pas unanime à son égard. Certains considèrent que les nouvelles méthodes d'imagerie moderne et la biologie moléculaire la rendent « archaïque, inutile..., coûteuse, dangereuse (146) », alors que les anatomo-pathologistes signalent que des dossiers sanitaires brûlants comme l'existence d'une nouvelle forme de la maladie de Creutzfeldt-Jakob, révélée grâce à cet examen, apportent la preuve de son utilité.
2.- L'application de la loi de 1994 serait la cause de son déclin
Dans les faits, on assiste au déclin de cette pratique, ce qu'a souligné Mme Monique Hérold lors de son audition devant la Mission le 8 novembre 2000 : « ... Je voudrais dire un mot sur les autopsies, très précisément sur ce que l'on a coutume d'appeler l'expertise médico-scientifique . Il serait bien que la loi française permette que ces expertises médico-scientifiques reprennent un cours normal alors qu'elles ont chuté de façon considérable sous le fallacieux prétexte de l'avis de la famille. C'est une vraie question, qui se pose aujourd'hui et elle se posera de plus en plus, ne serait-ce qu'en raison de la variante de la maladie de Creutzfeldt-Jakob. Il devient urgent de prendre position sur ce point (147) ».
Quant au Professeur Dominique Durand (148), il considère que c'est un problème majeur : « Le fait que les textes réglementaires imposent strictement le consentement direct pour procéder à des prélèvements scientifiques a conduit à ce que l'on ne fasse plus de tels prélèvements. C'est une catastrophe. Il faut absolument modifier ces textes. Il est terrifiant de voir tout simplement que, lorsqu'un transplanté du rein a une artère rénale malade, si l'on veut prélever une autre artère pour faire une recherche, nous n'en avons pas le droit. [...] En ce qui concerne l'autopsie, elle reste possible si l'on veut rechercher la cause de la mort mais, à titre scientifique, en pratique, c'est impossible ».
L'article L. 1232-3 du code de la santé publique utilise pour la qualifier les termes de « prélèvement à des fins scientifiques [...] ayant pour but de rechercher les causes du décès ». Cet article, en requérant un consentement exprès, contredit ouvertement la règle du consentement présumé affichée à l'article L. 1232-1 du même code : « Le prélèvement d'organes sur une personne décédée ne peut être effectué qu'à des fins thérapeutiques ou scientifiques et après que le constat de la mort a été établi. Ce prélèvement peut être effectué dès lors que la personne concernée n'a pas fait connaître de son vivant, son refus d'un tel prélèvement ».
La contradiction a été soulignée par le Professeur Didier Houssin lors de son audition par la Mission le 10 janvier 2001 : « Je dirai que la perspective d'une harmonisation du recueil du consentement, c'est-à-dire d'un alignement du consentement pour un prélèvement à visée d'autopsie, destiné à connaître les raisons de la mort, sur le prélèvement à visée thérapeutique, nous serait précieuse ». Cette contradiction serait à l'origine de la chute de la pratique de l'autopsie, notamment de l'adulte, constatée à partir des années 1970 et brutalement aggravée depuis 1994 (149).
Dans son avis sur l'avant-projet de révision des lois de bioéthique (150), le CCNE propose d'aborder la question de l'autopsie dans ses contradictions mêmes, en inscrivant explicitement le terme d'« autopsie » dans la loi et en distinguant de façon claire les différentes finalités des prélèvements :
« - l'autopsie médico-légale et l'autopsie justifiée par un danger pour la santé publique ou la nécessité du suivi épidémiologique des maladies dont la liste est précisée par circulaire ministérielle ne nécessitent aucun consentement du défunt ou des proches ni aucune information de l'Établissement français des Greffes ;
« - l'autopsie médicale, dont il faut rappeler qu'elle s'accompagne généralement de prélèvements, ne doit pas être opposée à l'autopsie à des fins scientifiques qui a toujours une visée, à la fois rétrospective et prospective. Elle devrait être réalisable sur la base du consentement présumé, en l'absence d'un refus exprimé sur le registre, et sans qu'il soit nécessaire d'aviser l'Établissement français des Greffes de la nature des prélèvements, ce qui ne doit évidemment pas conduire à s'interdire d'examiner scientifiquement ces prélèvements s'ils révèlent un intérêt pour la recherche médicale. Le médecin doit, dans les limites du secret médical, répondre à toute demande de la famille ou des proches ;
« - lorsque l'autopsie poursuit d'emblée une finalité scientifique qui s'inscrit dans le cadre d'un protocole de recherche préalablement transmis à l'Établissement français des Greffes, elle devrait en revanche être soumise au consentement explicite du défunt ou à défaut à celui de la famille ou des proches, dûment informés des objectifs spécifiques dudit protocole et de son intérêt pour la collectivité ;
« - les prélèvements d'organes à des fins thérapeutiques sont réalisés sur la base du consentement présumé. La famille doit dans la mesure du possible en être préalablement informée et bénéficier d'un accompagnement spécialisé dans ces circonstances éprouvantes. L'Établissement français des Greffes en est préalablement avisé ».
La discussion du projet de loi relatif à la révision de la loi bioéthique donnera l'opportunité d'examiner cette distinction. Votre Rapporteur estime en effet que toute disposition visant à clarifier la finalité des prélèvements et l'expression d'un consentement éclairé doit être encouragée.
3.- La finalité thérapeutique ne doit pas devenir un alibi éthique
La proclamation d'une nécessité thérapeutique repose sur l'appréciation du degré de gravité de la maladie, du stade de l'affection en cours et porte sur l'urgence, ce qui relève du strict domaine médical. Dans ce domaine, exercé par des hommes soumis aux passions comme les autres hommes, la crainte a pu être exprimée devant la Mission, d'une tentation de la performance pour la performance, certes non condamnable en soi mais à proscrire lorsque les enjeux concernent des individus affaiblis par la maladie.
Lors de son audition par la Mission le 22 novembre 2000, M. le Pasteur Olivier Abel constatait : « Nous voyons [...] combien les prouesses techniques augmentent l'angoisse a posteriori. Il y a quand même une sorte d'illusion : croire qu'il y aurait une réponse technique à tous les problèmes. Souvent, les réponses aux problèmes soulèvent elles-mêmes de nouveaux problèmes. Le sachant à l'avance, il faut prévoir des procédures de pondération non seulement dans les m_urs, mais aussi dans la morale, tout au moins la morale spontanée qui permet aux gens de « se débrouiller » dans les problèmes quotidiens et dans les dilemmes de leur vie, pour « faire le moins mal possible ». Cette exigence demande à être soutenue car tous ces pouvoirs sont très forts et très vertigineux ».
Mme Francine Gomez (151), estimant que la lutte contre la souffrance est au c_ur du débat de la bioéthique, expliquait que « La souffrance, c'est non seulement la douleur mais plus largement le mal de vivre, la difficulté existentielle. Tout cela apparaît aujourd'hui inacceptable, voire scandaleux [...] Cette lutte contre la souffrance justifie, finalement, toutes les audaces thérapeutiques [...] Un professeur de philosophie écrivait récemment : « La réduction des souffrances, le recul de la mort et l'accroissement de l'autonomie universelle des hommes sont les seuls critères rationnels qui doivent être invoqués pour réguler les applications de la génétique à l'homme ». Cela me paraît assez représentatif de tout un courant. Ces tendances fortes - refus de la souffrance, montée de l'utilitarisme et volonté de maîtrise sur les événements - se conjuguent pour produire un mouvement d'opinion qui finalement est globalement prêt à accepter l'idée que la recherche s'affranchisse de certaines limites pour soigner... ».
Pourtant, le regard des anthropologues vis-à-vis de la question des transplantations d'organes prend une certaine distance à l'égard de notre conception de la souffrance et de la mort. « Il semble parfois évident que la technologie médicale doive poursuivre sans trêve la marche à l'immortalité : réduire la souffrance, retarder la mort... Cependant les choix qui président à l'affirmation de cette nécessité thérapeutique, autrement dit l'indication de la thérapeutique greffe, sont largement influencés par notre métaphysique, et pas seulement par nos conceptions physiologiques de la souffrance et de la mort. La technique de la transplantation ne s'impose pas comme une évidence, elle est solidaire de l'affirmation de valeurs contingentes [...] La mort n'étant pas un événement purement biologique, la nécessité thérapeutique peut être relativisée de plusieurs points de vue : les points de vue économique et anthropologique notamment. D'un pays à l'autre, la hiérarchie peut varier, qui donne le pas sur l'enfant ou l'adulte, le cancer du foie ou la cirrhose éthylique. L'accent peut être mis sur la transplantation ou sur d'autres stratégies, en particulier préventives de l'insuffisance rénale ou hépatique, modifiant en amont les indications de la greffe. Au regard des différences de culture, le projet de transplantation peut apparaître ainsi comme celui de la science occidentale par excellence (152) ».
En outre, comme le constate la sociologue Simone Bateman-Novaes, l'approche médicale ne constitue qu'une perspective socialement déterminée parmi d'autres sur les usages possibles du corps humain (153) : « Le cadre érigé par le législateur admet comme démarche conforme, mais aussi comme exception légitime à une règle donnée, toute conduite ayant une justification thérapeutique. Mais comment le but thérapeutique d'une opération transformerait-il le recours à un procédé technique controversé en acte moralement acceptable ? Même érigée en norme juridique, la justification thérapeutique repose, pour son application, sur une logique normative autre que légale : en l'espèce, celle mise en _uvre dans l'activité médicale. Une législation ainsi conçue ne fait alors que transférer à la profession médicale la responsabilité de l'orientation normative des pratiques. [...] ».
Par ailleurs, l'étude des comportements (154) face à la santé et à la maladie a mis en évidence que c'est dans les classes sociales où le risque sanitaire est le plus élevé que la consommation médicale est le plus faible. Comme si les membres des milieux les moins favorisés étaient dans notre société moins attentifs que les autres aux sensations morbides, à la douleur, qui doivent être explicitement constituées comme « symptômes » pour entraîner le recours au médecin. Deux raisons seraient à l'origine de ce constat : ils ne disposeraient pas des catégories savantes et du vocabulaire du corps et de la maladie qui sont nécessaires pour déchiffrer les messages du corps, c'est-à-dire pour sélectionner, dans le flux des sensations corporelles, les signes morbides légitimes que les médecins seront disposés à reconnaître et à traiter. D'autre part, contraints de faire un usage professionnel de leur corps et soumis à des conditions matérielles d'existence plus dures que dans les autres groupes, ils adhéreraient à un système de valeurs qui privilégie la « dureté au mal », la résistance et la force physiques et s'interdiraient de prêter trop fortement attention au corps.
Ainsi, soumis aux pressions diffuses de la demande, de l'offre, des circonstances, le critère de la nécessité thérapeutique perdrait sa dimension objective. Il convient de garder cela présent à l'esprit chaque fois que l'on autorise une nouvelle intervention sur le corps humain.
4.- La relation malade-médecin
La personne qui s'adresse à un médecin se trouve toujours en position de demande. Quelle qu'en soit l'origine, malaise, douleur, sa démarche est ce qui fonde sa qualité de patient, voire de malade. Le signe de la maladie réside dans cette démarche qui de facto instaure une relation asymétrique et met le médecin en position d'autorité.
La technicité de la médecine ne devrait pas réduire la rencontre malade-médecin à un inventaire des performances objectives des fonctions biologiques essentielles. Le malade attend de son médecin qu'il ne soit pas indifférent aux souffrances de son corps, à la menace qu'une maladie fait planer sur son avenir et celui de son entourage. Et il demande au médecin de lui apprendre à vivre avec et après la maladie. Le médecin, doté d'une formation scientifique solide que requièrent des méthodes de plus en plus complexes, n'est sans doute pas suffisamment préparé à entendre cette demande, ni prêt à y répondre.
Par ailleurs, le médecin doit lutter contre tout risque d'identification au malade qui l'amènerait à se reconnaître dans une personne souffrante, infirme, menacée. Pour repousser ce risque d'identification, il peut alors considérer alors le patient comme un objet d'étude, un objet de science derrière les écrans de la technique et l'écran du savoir, et non plus un sujet semblable à soi.
Dans ces conditions, n'y a-t-il pas une exigence excessive à demander au patient qu'il prenne toujours des décisions importantes, comme un consentement en toute connaissance de cause ? Le patient ne risque-t-il pas de craindre qu'un refus de consentement à participer à une recherche lui ôte l'accès au soin ? Ne cherchera-t-il pas, en toute hypothèse, à plaire à son médecin ?
S'interrogeant sur la notion de responsabilité individuelle, M. le Pasteur Olivier Abel témoignait de ses doutes, lors de son audition par la Mission le 22 novembre 2000 :
« Il ne faut peut-être pas trop valoriser à l'excès et donner trop de poids à la responsabilité des individus : ils peuvent être parfois dépassés par les décisions qu'ils ont à prendre, trop angoissantes quelquefois. Parfois même, le consentement est une farce : il n'y a pas de consentement. Les gens sont faibles, fragiles, confrontés à des dispositifs d'une immense force sur le plan de l'efficacité et sur celui de la modification des représentations. « Cela est possible ? Pourquoi pas moi, si les autres le font ? ». Il y a une fragilité de l'individu responsable. Le législateur doit donc trouver le moyen terme entre cette place laissée à la responsabilité et cette sollicitude pour la fragilité des individus. Par fragilité, j'entends aussi la corruptibilité, les ambitions, en quelque sorte toutes ces passions du savoir, du pouvoir, qui demandent à être arrêtées ou limitées. [...] Nous ne sommes pas tout le temps des adultes majeurs, vaccinés, consentants, capables de comprendre tout ce qui leur arrive et tout ce qu'ils font ».
CINQUIÈME PARTIE : LA MÉDECINE PRÉDICTIVE
Dans le débat portant sur les avancées de la médecine, la faculté supposée de prévoir les maladies qu'une personne pourrait développer à terme ou les malformations ou handicaps dont un nouveau-né pourrait souffrir, concentre de formidables attentes mais aussi de terribles angoisses.
Les nombreuses découvertes que la presse relate, sur la possibilité de détecter les risques de cancers ou de maladies dégénératives chez certaines personnes, sont, en apparence, porteuses d'espoirs. Mais au-delà de cette première vision des progrès de la science, apparaissent des tensions, qui placent les individus et la société devant de nouvelles « situations-limites », pour reprendre l'expression du philosophe Karl Jaspers, c'est-à-dire des cas où l'homme se retrouve face à sa destinée et à sa condition, sans échappatoire, devant assumer un choix qui est tout sauf simple.
Dans ce cadre, le médecin ou le généticien peut faire figure d'oracle, délivrant la bonne ou la mauvaise nouvelle. Peut-il assumer seul ce rôle qui apparaît terrifiant ? De l'autre côté, la personne à qui l'on assène la prédiction qu'il développera à terme une terrible maladie, dont ses enfants risquent également de souffrir, peut-elle supporter sans le soutien d'un tiers ce type de nouvelle ?
Dans son avis n° 46 du 30 octobre 1995 « Génétique et médecine : de la prédiction à la prévention », le Comité consultatif national d'éthique a mis en évidence ce nouveau questionnement. Il a montré que la notion de prévention, connotée positivement, peut être contestée dans certaines hypothèses. Mais la prédiction peut ne pas avoir seulement pour objet de mettre en _uvre des traitements préventifs. Les examens génétiques peuvent aussi trouver une « utilité » pour les employeurs ou les compagnies d'assurance, avec le risque que des discriminations s'opèrent sur la base du patrimoine génétique. De même, la possibilité d'identifier les caractéristiques des individus avant même leur naissance, peut alimenter la tentation d'établir une sélection des embryons afin de donner vie à un enfant qui serait dénué de tout ce que la société - de moins en moins tolérante sur ce sujet - peut qualifier de tare.
Plus largement, la capacité de prévoir le développement d'une maladie ou l'apparition d'une malformation met l'homme face à une situation inédite, qui remet en cause l'essentiel de nos conceptions philosophiques et sociales. Car il faut être conscient que la réflexion sur la condition humaine s'est toujours référée à la question du temps, à l'incapacité à prévoir son destin et à appréhender la mort. La médecine prédictive risque de bouleverser notre manière d'aborder ces questionnements essentiels. Il ne nous appartient pas de traiter ce problème d'un point de vue philosophique, mais de mettre en lumière les enjeux qui affleurent, contre lesquels on peut se sentir démunis, mais pas totalement cependant (155).
I.- LA MÉDECINE PRÉDICTIVE AU CARREFOUR DES CRAINTES ET DES ESPOIRS
A.- DE LA SÉLECTION À LA DISCRIMINATION
1.- Vers un nouvel eugénisme ?
La notion de « médecine prédictive » renvoie à des enjeux divers qu'il convient de distinguer. Elle concerne, tout d'abord, les examens pratiqués sur les embryons et les f_tus, aux fins de déterminer leurs caractéristiques, avant qu'ils ne viennent au monde. Ces examens pratiqués avant la naissance peuvent avoir pour objet de détecter d'éventuelles malformations ou maladies. Ils peuvent aussi - et certains le craignent - répondre à une fin « plus futile » - la détermination du sexe, des caractères physiques - qui pourrait conduire à l'élimination du f_tus, pour des raisons, à l'évidence, choquantes.
Les possibilités nouvelles de pratiquer ces examens génétiques avant l'implantation de l'embryon dans le corps de la mère multiplient les préventions à l'égard de ces pratiques. En effet, le diagnostic préimplantatoire pourrait laisser planer le spectre d'un nouvel eugénisme - on reviendra sur l'emploi controversé de ce terme - mené dans des conditions beaucoup moins traumatisantes, donc plus simples à accomplir, pour les parents. La tentation de « l'enfant parfait », la détermination de nouveaux critères de viabilité ouvrent la voie à des formes de discriminations intervenant avant même la naissance. Ce qui, jusqu'à maintenant, semblait relever de la seule science fiction paraît à portée de main.
Dans ce cadre, il importe de distinguer le diagnostic préimplantatoire (DPI) du diagnostic prénatal (DPN). Le DPI est la technique qui permet d'identifier une anomalie sur l'embryon in vitro - c'est-à-dire avant l'implantation - alors que le DPN est l'ensemble des techniques permettant de mettre en évidence ces anomalies, chez l'embryon ou le f_tus ; son champ est donc plus large.
Ces techniques nouvelles ont pu susciter des critiques argumentées de personnalités reconnues. C'est le cas, en particulier, du Professeur Jacques Testart, directeur de recherches à l'INSERM, connu pour son activité pionnière dans le domaine de la procréation assistée et auteur de nombreux essais sur ce sujet tels L'_uf transparent et Le désir du gène. Avec force, le Professeur Jacques Testart a présenté devant la Mission le 31 mai 2000 une perspective que l'on a pu qualifier de pessimiste. Il semble craindre que le DPI n'entraîne la résurgence d'un courant eugéniste, compatible, cette fois, avec les systèmes politiques démocratiques. Mais avant de revenir sur l'argumentation développée par le Professeur Jacques Testart, il importe de rappeler rapidement ce que fut le courant eugéniste, à la fin du XIXe siècle et lors de la première moitié du siècle précédent.
L'eugénisme a pour objet d'améliorer la race humaine. Comme le souligne le Professeur Testart, ce terme est fréquemment accolé à celui de « nazisme », mais, s'il est vrai que les nazis ont poussé l'eugénisme à son comble, ils n'ont cependant inventé ni le concept ni la pratique. Jacques Testart observe d'ailleurs que l'un et l'autre ont survécu au nazisme (156). C'est le Docteur Galton, cousin de Darwin, qui a défini le premier, en 1883, le terme d'eugénisme comme « la science de l'amélioration de la race qui ne se borne nullement aux questions d'unions judicieuses, mais qui, particulièrement dans le cas de l'homme, s'occupe de toutes les influences susceptibles de donner aux races les mieux douées un plus grand nombre de chances de prévaloir sur les races les moins bonnes ». Le dictionnaire nous en livre une définition plus claire : « L'eugénisme (ou eugénique) est la science qui étudie et met en _uvre les moyens d'améliorer l'espèce humaine, en cherchant soit à favoriser l'apparition de certains caractères (eugénisme positif), soit à éliminer les maladies héréditaires (eugénisme négatif), fondée sur les progrès de la génétique (157) ».
Sans revenir sur les conditions historiques de mise en _uvre de la doctrine eugénique, on remarquera que l'eugénisme est une réalité ancienne. Pratiqué par exemple à Sparte, il conduisait à l'élimination des nouveaux nés qui, selon les sages et les canons spartiates, ne correspondaient pas à l'idéal du citoyen. Avec le développement de la science au XIXe siècle et l'engouement pour la philosophie du progrès, la question de la sélection des individus aux fins d'améliorer l'humanité retrouva une certaine audience. Au-delà d'un discours pseudo-scientifique, la mise en _uvre de programmes eugéniques dans certains États comme l'Allemagne nazie mais aussi aux États-Unis et dans les pays scandinaves, dans une moindre proportion, conduisit à des pratiques de stérilisations, de castrations, voire d'élimination physique dans le cas allemand, des personnes considérées comme anormales.
A l'issue de la Seconde guerre mondiale, la doctrine a semblé totalement discréditée, même si des pratiques inavouées se perpétuèrent dans les pays nordiques et outre-Atlantique. Des chiffres contradictoires sont avancés sur le nombre de personnes stérilisées en Suède, en Norvège ou aux États-Unis, mais la réalité de telles pratiques semble incontestable (158).
La question semble cependant aujourd'hui resurgir de manière insidieuse, avec le développement de la génétique. La menace d'une sélection négative non plus des individus venus au monde mais des embryons apparaît de plus en plus présente dans les esprits.
Pour autant, si l'on consent à porter un regard moins superficiel sur les évolutions récentes de la science, les termes du débat sont différents de ceux qui existent autour de la question eugénique. Si l'eugénisme est la volonté « d'améliorer » la race humaine, la tentation actuelle de procéder à un tri génétique des embryons ne relève pas tout à fait de la même optique.
Cette perspective ne suscite d'ailleurs pas nécessairement une réaction unanime de rejet, comme on pourrait s'y attendre. Certains scientifiques ont exprimé leur intérêt pour le tri génétique et la possibilité, par exemple, de choisir le sexe de son enfant. C'est le cas du Professeur Bernard Debré qui déclarait récemment que, si le tri des embryons n'était pas encore réellement pratiqué, il espérait qu'on y recourrait afin d'éviter le traumatisme que peut éprouver une femme stérile ayant subi un véritable parcours du combattant pour procréer (159).
Si l'apparition de ce nouveau mode de sélection fait naître des interrogations, des craintes, il ne semble pas susciter une réprobation sociale aussi vive qu'on pourrait l'attendre. On peut expliquer ce phénomène par deux facteurs principaux.
Tout d'abord, cette tentation sélective, qui est condamnable, n'est pas le fait de l'État ou de la société mais des individus eux-mêmes. Refusant d'assumer ce que l'on pourrait qualifier d'accidents de la vie, nos contemporains entendent s'abstraire des épreuves que la fatalité peut leur infliger. De là naît la volonté de ne pas donner la vie à un enfant pouvant souffrir d'une anomalie physique ou d'un handicap.
Un tel choix désormais possible incite à une forme de compréhension. Il est certain que donner naissance à un enfant handicapé est une épreuve à laquelle chacun souhaiterait échapper. En outre, certaines maladies ou malformations peuvent entraîner des souffrances pour l'enfant qu'on peut juger intolérables. Partant de là, peut-on rejeter, sans réflexion préalable, toute tentation de recourir à un examen génétique avant de donner naissance à un enfant ? Une certaine tolérance sociale à l'égard de l'idée d'une sélection génétique peut apparaître. Elle n'est pas acceptable et doit être condamnée fermement. Il nous appartient d'en souligner le caractère pernicieux et dangereux.
Le second facteur qui peut expliquer que la société ne rejette pas brutalement la pratique du tri, tient au mode opératoire de cette sélection. Elle intervient avant la naissance, selon des modalités moins traumatisantes que celles qui consisteraient, dans l'extrême, à éliminer des individus venus au monde.
Mais le problème du tri génétique ne peut se résoudre de manière simple et convenue. La question relève moins, selon votre Rapporteur, de l'ordre du principe que de sa mise en _uvre. Peut-on, en effet, véritablement contester que l'on puisse éliminer les embryons porteurs d'une grave anomalie génétique lorsque celle-ci a été détectée ? Peut-on pour autant accepter que l'on procède de la même manière lorsqu'on déterminera que l'embryon en question risque, sans aucune certitude, de devenir un individu qui pourrait ou non développer à plus ou moins long terme une maladie grave ? De même ce tri serait-il tolérable pour des raisons pouvant apparaître futiles comme le sexe ou la couleur des yeux de l'enfant ? On le voit, la même technique employée, selon des objectifs différents, peut conduire à adopter des positions radicalement contraires. La technique n'est pas en soi mauvaise. Elle peut simplement être dévoyée. Il appartient alors au législateur de fixer le cadre permettant de parer ces dérives.
La perspective du tri génétique et le mythe de « l'enfant parfait » se heurtent à l'idée que la richesse de l'humanité, en tant qu'espèce, réside dans sa diversité. Comme le souligne, le Professeur François Jacob, la diversité est l'une des grandes règles du jeu biologique. Cette combinatoire infinie qui rend unique chacun de nous, on ne peut la surestimer. C'est elle qui fait la richesse de l'espèce et lui donne ses potentialités (160).
Le risque d'un glissement entre la recherche d'un trait héréditaire à incidence morbide et d'un trait héréditaire non conforme à une norme d'ordre social n'est pas de nature fantasmatique. Dans une société où, paradoxalement, la diversité est portée aux nues dans les discours, mais où finalement l'uniformisation gagne du terrain, la perspective d'un passage de la prévention médicale à la sélection sociale apparaît.
La question de la tolérance au handicap et, plus généralement, à la différence est posée. L'arrêt Perruche de la Cour de cassation, en date du 17 novembre 2000, a mis en évidence cette forme de rejet social, dont la reconnaissance juridique par cette cour n'est qu'un épiphénomène. En condamnant le médecin à verser une indemnité à un enfant handicapé au titre du préjudice subi par celui-ci, alors même que la seule alternative à son handicap était l'avortement de sa mère, la Cour de cassation a suscité de nombreuses réactions qui montrent qu'un sursaut est possible et qu'il appartient sans doute au législateur de trouver une solution à ce cas de figure extrême.
Le 29 mai dernier, le Comité consultatif national d'éthique a ainsi eu l'occasion de réaffirmer sa position en ce domaine. Dans son avis n° 68, cette instance a observé que « la personne souffrant d'un handicap profond, plus qu'une autre, ne peut donc trouver sa place dans une société soucieuse de son image au point de ne pas supporter la différence et, trop souvent, de manifester une attitude « handiphobe ». Dans ce contexte de soumission à la norme, le couple qui choisit de ne pas interrompre la grossesse à l'origine d'un enfant « différent » est de plus en plus fréquemment mal compris, ce qui altère l'aide et l'accompagnement qu'il est en droit d'attendre de la société. Ce couple encourt même le risque d'être critiqué, considéré comme « irresponsable », et de ce fait il peut être l'objet de discriminations ».
Face à l'arrêt Perruche, le Comité consultatif national d'éthique appelle à une mobilisation de moyens pour l'accueil des personnes handicapées. Il insiste également sur la nécessité de laisser aux parents la liberté de choix en améliorant les techniques de dépistage des malformations prénatales mais aussi en rendant réellement envisageable la décision parentale de poursuite de la grossesse, malgré le risque de handicap pour l'enfant à naître. L'arrêt Perruche ouvre la voie à un éventuel « réflexe normatif d'autoprotection des professionnels et des familles ». De la sorte, on pourrait voir apparaître une « tendance à une définition sociale des critères, médico-scientifiques ou autres, de la « bonne naissance » [qui] peut être étymologiquement et historiquement qualifiée d'eugénique ».
Devant ce risque, que la jurisprudence contestée de la Cour de cassation laisse apparaître, le Parlement se doit d'envisager les solutions législatives pour que ne soit pas inscrit dans notre système juridique un droit à ne pas naître handicapé, droit qui serait contraire aux principes éthiques - tels que le Comité consultatif les a exposés - et socialement dangereux, en ce qu'il donnerait un fondement juridique et, de ce fait, une reconnaissance sociale, au rejet des personnes handicapées.
2.- La crainte et la condamnation de la discrimination génétique
Le refus de toute discrimination repose sur l'idée fondamentale que les hommes sont tous égaux en dignité et en droits. On a pu mesurer dans le passé les conséquences désastreuses d'une théorie raciste de l'humanité, fondée sur des présupposés faussement scientifiques. La question d'une discrimination selon le patrimoine génétique ne serait pas radicalement différente de celle basée sur la race. Elle apparaîtrait cependant encore plus grosse de catastrophes, puisque s'appuyant sur un fondement scientifique, apparemment plus convaincant. De cette pseudo-objectivité pourrait émerger une théorie sociale et politique fondée sur la discrimination et la classification sociale des individus en fonction de leur patrimoine génétique et de leurs supposées qualités intrinsèques.
C'est pour parer ce type de dérives, que les textes internationaux ont pris soin de condamner toute forme de discrimination fondée sur le patrimoine génétique.
L'article 11 de la Convention sur les droits de l'homme et la biomédecine, signée à Oviedo, dans le cadre du Conseil de l'Europe, le 4 avril 1997, prohibe toute forme de discrimination à l'encontre d'une personne en raison de son patrimoine génétique.
Dans ce cadre, l'article 12 de la convention d'Oviedo stipule qu'il ne pourra être procédé à des tests prédictifs de maladies génétiques ou permettant soit d'identifier le sujet comme porteur d'un gène responsable d'une maladie soit de détecter une prédisposition ou une susceptibilité génétique à une maladie qu'à des fins médicales ou de recherche médicale, et sous réserve d'un conseil génétique approprié.
L'article 22 de la loi n° 94-654 du 29 juillet 1994 a introduit dans le code de la santé publique des dispositions limitant également la possibilité de recourir à l'examen des caractéristiques génétiques d'une personne.
Ainsi, l'article L. 1131-1 de ce code restreint la possibilité de recourir à ces examens aux seules fins médicales ou de recherche scientifique, en dehors du cas particulier des besoins d'une procédure judiciaire. Le consentement de la personne est requis hormis, à titre exceptionnel, le cas où il ne peut être recueilli, dans son intérêt et dans le respect de sa confiance. L'article L. 1131-3 prévoit que sont seules habilitées à procéder à des identifications pour empreintes génétiques à des fins médicales ou de recherche scientifique les personnes ayant fait l'objet d'un agrément.
Par ailleurs, le fait de détourner de son objet les finalités de tels examens est réprimé, aux termes de l'article 226-26 du code pénal. Examinant cette question, le Conseil d'État a estimé que ces dispositions ne présentaient sans doute pas un caractère suffisant. Dans son rapport, il écrit ainsi : « Cette prohibition est donc claire mais, en pratique, son efficacité pourrait être limitée. En effet, des individus connaissant le résultat favorable des tests génétiques pratiqués sur eux ne seraient-ils pas enclin à en faire usage face à un employeur potentiel ou pour se ménager un accès à une assurance dans des conditions contractuelles plus favorables ? N'y a-t-il pas lieu de craindre, dans la vie sociale et économique, une utilisation réductionniste des résultats de ces tests, qui conduirait par exemple à un refus d'assurance ou à un refus d'embauche ? Après la lutte contre les inégalités sociales, comment prévenir une prise en compte dangereuse des inégalités biologiques ? » (161). Ces questions n'appellent pas une réponse simple et le législateur devra, sans doute, y revenir.
B.- LES ESPOIRS LIÉS À LA DÉTECTION DES MALADIES D'ORIGINE GÉNÉTIQUE
Si des craintes peuvent exister concernant la pratique des examens génétiques, elles ne doivent pas occulter les avancées extraordinaires de la science dans la prévention de certaines maladies d'origine génétique. La capacité de détecter les risques de développement de ces pathologies avant qu'elles n'apparaissent constitue un espoir pour des milliers de familles.
Les exemples les plus saisissants en ce domaine sont évidemment ceux qui concernent des maladies pour lesquels un traitement existe. C'est le cas du diagnostic de la phénylcétonurie. Comme le montre l'avis du Comité consultatif national d'éthique du 30 octobre 1995, à partir d'une goutte de sang prélevée dans les jours qui suivent la naissance de l'enfant, on a pu déceler, grâce à un programme national jugé excellent, l'ensemble des cas d'enfants atteints de cette maladie, soit 1 sur 15 000. Or la prévention de cette maladie est possible grâce à un régime alimentaire strict, dès les premières semaines de vie et poursuivi pendant plusieurs années. Ainsi, une cinquantaine de retards mentaux auraient été évités grâce à ce programme.
Les examens génétiques peuvent permettre une surveillance accrue de personnes risquant de développer une maladie d'origine génétique. Ainsi, la Caisse nationale d'assurance-maladie a annoncé en décembre 2000 un programme national de dépistage néonatal systématique de la mucoviscidose, maladie génétique la plus fréquente. Ce dépistage est pratiqué grâce au prélèvement de quelques gouttes de sang sur le talon du nourrisson environ trois jours après sa naissance. Ce diagnostic permet une prise en charge adaptée, avec de la kinésithérapie respiratoire, un régime alimentaire, etc., qui pourrait permettre de prévenir ou de retarder les symptômes les plus handicapants, afin d'augmenter l'espérance de vie des personnes atteintes de cette maladie, qui serait actuellement de trente-cinq ans.
L'examen génétique peut également intervenir avant la naissance dans le cadre d'un DPN ou DPI. On y reviendra plus précisément, par la suite. On notera simplement que, là aussi, des avancées considérables ont pu être observées, comme, par exemple, pour détecter le gène de la mucoviscidose par voie de DPI. Deux enfants sont ainsi nés indemnes de cette maladie en janvier 2001, à la suite d'un tel diagnostic (162). La première naissance après un DPI, en France, était intervenue quelques semaines auparavant, en novembre 2000. Le petit Valentin est né indemne d'une maladie métabolique grave qui avait déjà entraîné le décès de trois enfants dans sa famille (163).
2.- Relativiser la portée prédictive des examens génétiques
La portée prédictive des examens doit cependant être relativisée à un double titre.
Elle doit l'être, tout d'abord, parce qu'elle ne permet pas toujours d'être certain que la personne ayant subi le test développera la maladie en cause. Celle-ci peut relever de plusieurs facteurs qui ne sont pas tous d'origine génétique. L'environnement peut également se révéler déterminant dans le développement de la maladie. La connaissance de la présence du gène favorisant l'affection peut parfois conduire à prendre les dispositions nécessaires pour faire en sorte que la personne porteuse de ce gène n'évolue pas ensuite dans un environnement susceptible de permettre à la maladie de se développer. Dans d'autres cas, cette connaissance n'apporte rien, parce que l'on ignore les autres facteurs déclenchant.
Le cas de la polykystose rénale illustre la difficulté mais également l'intérêt de recourir à ce type de tests génétiques. Cette affection est une maladie dominante qui est responsable d'environ 10 % des déficits rénaux nécessitant une dialyse. En 1985, on a découvert le gène mutant responsable de cette maladie sur le chromosome 16. Or il est apparu ensuite que, dans certaines familles, la maladie n'était pas liée aux marqueurs situés sur le chromosome 16. Il semble qu'existe au moins un autre gène localisé sur le chromosome 4. L'examen génétique ne donne pas la certitude que la maladie se développera. En revanche, il permet de rassurer des sujets indemnes de la maladie dans des familles où le gène responsable a pu être identifié.
La portée de ces examens demeure également relative lorsqu'ils ne peuvent conduire à des traitements adaptés. C'est le cas de la maladie de Huntington, dont le gène a été localisé en 1983. Cette affection se développe à l'âge adulte, autour de quarante ans, se manifestant par une affection dégénérative du système nerveux central évoluant de manière progressive et irréversible, sans qu'aucune thérapie n'existe.
On ne doit donc pas tout attendre des examens génétiques. Les diagnostics ne sauraient d'ailleurs être livrés bruts aux patients, dans la mesure où leur interprétation est délicate et met à jour de grandes difficultés humaines. Le caractère finalement moins prédictif que présomptif de ces tests doit être pris en compte, une réflexion devant être menée sur l'accompagnement des diagnostics réalisés.
3.- Une réflexion nécessaire sur l'accompagnement des diagnostics
Dans son avis n° 46 du 30 octobre 1995 « Génétique et médecine : de la prédiction à la prévention », le Comité consultatif national d'éthique a proposé des orientations en matière de tests génétiques. Il s'est inspiré à cet effet des travaux du Hasting center présentés en 1972 ainsi que de ceux contenus dans le rapport établi en 1983, par la President' Commission for the study of ethical problems in medecine and biomedical and behavioral research qui a énoncé des directives pour le dépistage et le conseil en matière de maladies génétiques.
Les orientations proposées par le Comité consultatif national d'éthique tiennent compte de l'impact de tels examens sur la vie de la personne s'y prêtant mais aussi sur les membres de sa famille. Comme le note le Comité dans son rapport : « Tout autant, et même plus qu'un autre examen médical, un test génétique comporte une entrée dans l'intimité d'une personne, à savoir son intimité corporelle et les significations qu'elle y attache en rapport avec son identité psychique. De plus, peuvent être relevées des fragilités quasiment « constitutives », innées et non accidentelles, dont l'interprétation pour la représentation de soi-même ainsi que les conséquences pour sa vie présente et future ont une importance majeure. Le respect de ces réalités invite à prendre des précautions toutes particulières dans la proposition d'un test, dans l'annonce du résultat et dans la confidentialité de ce résultat. D'autant plus qu'à cet aspect d'intimité s'ajoute l'aspect relationnel et familial des réalités génétiques : reçues des parents, elles sont transmissibles à des enfants, et partagées de diverses manières avec des frères et s_urs et avec des collatéraux ; enfin, elles pourront concerner également le conjoint avec lequel la procréation est envisagée. Le respect de ces relations familiales conduit à renforcer encore les précautions concernant l'usage des tests génétiques ».
Il est vrai que le résultat d'examens génétiques peut conduire à un bouleversement de la vie de la personne concernée mais aussi de son entourage. Les individus sont-ils en mesure de supporter cette connaissance du plus intime de soi, connaissance qui peut prendre la forme d'une sorte de malédiction génétique ? Or, comme le note le Comité consultatif national d'éthique dans son avis, le développement des tests génétiques précédera celui des thérapies permettant de soigner les maladies ainsi identifiées. On imagine alors la situation des personnes apprenant qu'elles présentent des risques importants de développer une maladie à laquelle on ne peut opposer aucun traitement.
Dans cette hypothèse, peut-on laisser les personnes assumer cette révélation et le choix de recourir à un tel test ? Les orientations proposées par le Comité consultatif national d'éthique entendent permettre de mieux cerner cette problématique en fixant un cadre concret, une procédure, qui offre au médecin les moyens d'accompagner le patient dans cette démarche.
Le Comité consultatif national d'éthique suggère, tout d'abord, les conditions dans lesquelles la proposition d'examen doit s'opérer. Elle doit, en premier lieu, respecter l'autonomie de l'individu. Celui-ci doit exercer sa faculté de choix sans pression et coercition. Il doit pouvoir comprendre l'ensemble des implications d'une telle décision, ce qui suppose qu'il connaisse la nature de l'examen prescrit, la signification des résultats, la prévention et la thérapie existante. En outre, le patient doit disposer de la capacité juridique de donner un consentement libre et éclairé.
Plus difficile est sans doute la recherche d'une forme d'accord de la part de la famille de la personne concernée. Le Comité consultatif national d'éthique invite à une telle démarche sans méconnaître les obstacles qui peuvent s'y opposer. Le rapporteur du Comité s'interroge : « Lorsque l'examen exige un prélèvement biologique à l'ensemble de la famille, comment sera faite l'information à chacun des sujets ? Comment persuader ceux qui sont réticents ? (...) Le sujet demandeur doit prendre contact lui-même avec les membres de sa famille, cette demande pouvant être délicate et exiger de longs délais. De plus, elle ne sera pas obligatoirement couronnée de succès en raison du respect dû à la liberté de choix de ceux qui sont sollicités ».
À côté du principe de l'autonomie de la personne, doit apparaître le principe de son consentement. Cette directive est conforme à l'article L. 1131-1 du code de la santé publique qui impose le consentement de la personne concernée pour tout examen des caractéristiques génétiques. Toutefois, on rappellera que le dernier alinéa de cet article permet, à titre exceptionnel, de ne pas recueillir le consentement de la personne concernée dans son intérêt et dans le respect de sa confiance. Ce dispositif permet d'éviter le dévoilement éventuel de la nature extraconjugale d'une filiation méconnue ou cachée, mais dont la connaissance sur le plan biologique est indispensable pour l'interprétation des résultats, et, éventuellement, pour leur utilisation dans un but diagnostic.
Selon le Comité consultatif national d'éthique, l'information doit être directe, orale et accompagnée d'un document d'explication. Cela implique l'intervention d'un professionnel bien formé. Cette information doit également être délivrée après l'examen génétique afin d'expliquer clairement la signification et les implications des résultats, pour pallier les répercussions psychologiques d'une telle connaissance.
Si des précautions doivent être prises lors de la prescription de l'examen, il est également important d'accompagner la personne lors de l'annonce des résultats, dont la révélation peut être traumatisante. Le Comité consultatif national d'éthique considère, à juste titre, que la révélation d'un test génétique peut « limiter l'autonomie morale d'un sujet ». Ce peut être particulièrement le cas dans les maladies monogéniques, liées au sexe. On imagine la situation difficile d'une femme apprenant qu'elle risque de transmettre une maladie comme une myopathie ou l'hémophilie. Le sujet peut légitimement refuser de connaître ces résultats, après avoir accepté de pratiquer l'examen.
Comme le note le rapport du Comité consultatif national d'éthique, plus complexe est la connaissance d'une simple probabilité. C'est le cas des gènes dits de susceptibilité, par exemple, à un cancer ou à une maladie neuro-psychiatrique. Cette difficulté devient encore plus aiguë lorsque n'existe aucun traitement contre la maladie identifiée.
L'accent est mis, dans l'avis du Comité, sur la responsabilité du laboratoire dans la communication du résultat. Il doit être délivré par un médecin, avec le souci de ne délivrer aucune information relative à la vie privée du sujet testé et de sa famille, comme, par exemple, une fausse paternité.
Au regard de ces directives, la commercialisation de tests génétiques par correspondance risque de conduire à de graves dysfonctionnement, selon le Comité consultatif national d'éthique, qu'on ne peut que suivre dans ses préventions.
Les études génétiques peuvent concerner une personne mais aussi s'appliquer à une famille. C'est le cas lorsqu'a été détecté le risque d'une maladie génétique qui pourrait affecter les membres de la famille. Pour le Comité consultatif national d'éthique, le recours à un examen génétique ne doit pas être envisagé comme une forme de routine. La question revêt d'ailleurs un aspect plus délicat lorsqu'il s'agit de réaliser ce type de tests sur des enfants mineurs qui ne peuvent donner leur consentement. Les parents peuvent-ils prendre la décision de recourir à un tel test, auquel leur enfant, une fois devenu adulte, aurait peut-être refusé de se soumettre ? Le Comité consultatif national d'éthique a déjà eu l'occasion d'aborder cette question dans son avis n° 25 du 24 juin 1991, insistant sur le fait que les parents ne devaient pouvoir demander de telles analyses que dans le cas où la maladie liée au génotype de leur enfant pourrait se déclarer avant 18 ans ou pourrait bénéficier de mesures préventives avant cet âge. Une telle prescription interdirait, par exemple, de pratiquer ces tests pour des maladies comme celle de Huntington.
Dans ce domaine, les familles ont à assumer une lourde responsabilité. Il appartient aux parents d'informer leur enfant du résultat des tests dès qu'il est en mesure de le comprendre et quand il est en état de procréer lorsque l'examen met en évidence des caractéristiques génétiques qui conduirait à certains risques pour la propre descendance de cet enfant. De fait, l'information des parents est indispensable avant toute décision de pratiquer un tel examen chez l'enfant. Comme le souligne le Comité consultatif national d'éthique, « cette décision ne peut pas uniquement reposer sur le désir de savoir des parents ou du médecin traitant ».
Si le recours aux examens génétiques soulève des difficultés dans le domaine médical lorsqu'ils sont pratiqués sur des personnes nées, l'enjeu est sensiblement différent en matière de diagnostic prénatal ou préimplantatoire.
II.- LA PRATIQUE RASSURANTE DU DIAGNOSTIC
PRÉIMPLANTATOIRE ET PRÉNATAL
A.- UN DÉBAT RÉCURRENT AUTOUR DE LA QUESTION DES FINALITÉS ET DES CONSÉQUENCES DU DIAGNOSTIC PRÉIMPLANTATOIRE
Le DPN et le DPI interviennent, comme cela a été précédemment rappelé, à différents stades du développement de l'embryon ou du f_tus. L'origine de ces deux techniques est tout aussi différente puisque la pratique du DPN existe depuis plus de vingt cinq ans et que certaines de ses techniques, telle l'échographie, sont d'ores et déjà intégrées dans le suivi médical obligatoire et régulier des grossesses. Le DPI est, en revanche, une pratique récente, puisque le premier eut lieu en 1990 en Angleterre, et très sophistiquée dans son procédé. Comme le soulignait devant la Mission le 6 septembre 2000, le Professeur Arnold Munnich, chercheur à l'INSERM et responsable de l'un des trois centres de DPI existant en France, il s'agit en effet d'une véritable « prouesse » technologique : « Ces tests nécessitent une mise au point prolongée. Alors qu'un diagnostic prénatal standard, qui se fait sur des millions et des millions de cellules, est techniquement simple, un diagnostic préimplantatoire se fait sur une seule cellule et constitue donc une prouesse technologique nécessitant, à ce titre, une mise au point qui est également très lourde. En conséquence, il ne faut pas s'étonner si, pendant qu'une technicienne fait une quinzaine de diagnostics prénatals, un ingénieur ne fait qu'un seul diagnostic préimplantatoire car ce dernier nécessite des investissements considérables en personnel et en temps ».
La question du DPN n'a été abordée par aucun des interlocuteurs de la Mission. Elle ne semble en effet poser aucun problème particulier à l'exclusion de l'insuffisance des crédits hospitaliers et de la configuration de la carte sanitaire (164) qui créent certaines insuffisances ou insatisfactions. En revanche, le DPI, qui conduit à opérer un tri génétique entre les embryons in vitro porteurs d'un gène malade et ceux qui sont sains, soulève plus de controverses autour de la crainte d'un eugénisme individuel. La Professeur Jacques Testart, au cours de son audition du 31 mai 2000 devant la Mission, faisait état de son grand pessimisme à ce propos en estimant que le DPI débouchera, tôt ou tard, sur des pratiques d'eugénisme menaçant l'humanité elle-même :
« Il y a des pulsions chez tous les gens, par exemple, en ce qui concerne le DPI, celle d'avoir des enfants "normaux". Contre cela, il faut arriver à mettre des garde-fous. J'attends que l'on me montre où sont des garde-fous. Depuis seize ans, personne ne m'a montré de garde-fous, sauf à dire que les médecins sont des gens sérieux et que l'on trouvera bien des parades. Cela ne paraît par suffisant. Je suis donc pessimiste, car les gens sont ce qu'ils sont, ils sont fragiles (...).
Très vite, je voudrais dire en quoi cela va changer. Je pense que d'ici dix ou vingt ans - peu importe le terme, mais c'est évident que l'on va y arriver, disons trente ans pour être sûr - on aura la possibilité de produire pour chaque couple stérile ou non, qui voudra un enfant, 100 ou 200 embryons simultanément, sans que la femme n'ait rien à souffrir d'autre qu'un premier prélèvement de tissu ovarien (...). Ensuite, on appliquera des tests génétiques. Avec les biopuces qui se développent, on peut avoir des milliers de caractérisations. On peut faire tout le génome. On peut multiplier les cellules, faire des clones cellulaires à partir d'une cellule embryonnaire, ce qui permet d'appliquer cette batterie de tests sur le même embryon (...). Ensuite, on se retrouve avec un embryon qui, suivant le profil génétique qu'appliquera l'ordinateur, sera défini comme étant " le meilleur ". Je voudrais dire tout de suite que, d'un couple à l'autre, il n'y aura pas beaucoup de variations du choix, car personne ne souhaitera un enfant ayant un risque de diabète ou de cancer du sein, ou autres choses désagréables. Il n'y aura jamais de bébés parfaits, mais on essaiera de tendre vers un enfant de meilleure qualité biologique, c'est-à-dire génétique. Je vois mal qu'on y résiste, que les couples y résistent. Tout le monde a envie d'avoir un enfant en bonne santé, qui puisse subir la compétition de la société libérale, et s'en sortir assez bien. C'est en même temps intéressant pour les professionnels : généticiens, gynécologues, biologistes de la procréation assistée, industrie des biotechnologies. Et je crois qu'un jour cela intéressera beaucoup la Sécurité sociale, car comme vous le savez, les enfants handicapées coûtent cher ».
Le Docteur Axel Kahn, lors de son audition devant la Mission le 7 juin 2000, considérait lui aussi que le risque existe d'élargir le recours au DPI à des critères qualitatifs et non plus médicaux devant le caractère plus « acceptable » de ce diagnostic, à l'inverse du DPN : « Pour autant, le diagnostic préimplantoire pose un problème difficile : il est relativement plus indolore, plus acceptable, de se mettre à faire des tris d'embryons sur des caractéristiques peu pathologiques. On ne va pas provoquer un avortement facilement sur des arguments peu pathologiques alors qu'un tri d'embryons sur des arguments qui, à terme, pourraient être qualitatifs, est quelque chose qui peut se concevoir. Si bien que le diagnostic préimplantatoire doit, selon moi, être extrêmement encadré et ne doit être offert que comme une alternative à des situations où, aujourd'hui, on admet un diagnostic prénatal suivi, en cas de détection de la maladie, d'une interruption dite médicale de grossesse. Il faut donc limiter le diagnostic préimplantatoire à la détection de la maladie que la famille est susceptible de transmettre, afin d'éviter cette dérive d'une utilisation du diagnostic préimplantatoire à des fins de sélection de l'embryon, c'est-à-dire d'eugénisme de convenance ».
Cette menace de recherche de « l'enfant parfait » existe sans doute dans le futur et il convient d'en être averti afin de résister aux pressions qui pourraient s'exercer sur le législateur dans le sens d'une trop grande pratique du DPI qui l'assimilerait à une pratique « de confort ». Toutefois, comme le reconnaissait lui-même le Professeur Jacques Testart, le cadre législatif actuel pose des limites claires dont la pratique semble tout à fait rassurante : « Si personne n'envisage de le modifier, restons en là, sauf que l'on vit en France, c'est-à-dire parmi d'autres en Europe et dans le monde, et que cette loi qui, je le répète, est d'une rédaction parfaite, va s'user assez vite. Pour le moment, ce diagnostic en est aux balbutiements ».
B.- L'ACTIVITÉ DES CENTRES DE DIAGNOSTIC PRÉIMPLANTATOIRE
Mme Nicole Questiaux, Présidente de la CNMBRDP, précisait, lors de son audition du 12 juillet 2000, que sur onze demandes d'autorisation pour pratiquer le DPI, trois ont été à ce jour accordées à des unités de Paris, Montpellier et Strasbourg. Comme l'indiquait le Professeur Arnold Munnich, il s'agit d'activités très pointues qui exigent des compétences précises et une expérience reconnue en génétique médicale qui sont peu répandues : « Je ne crois pas qu'il faille envisager une augmentation du nombre des centres habilités à pratiquer le diagnostic préimplantatoire. Je pense que pour pratiquer ce diagnostic, il faut d'abord avoir bien entendu la demande des couples et bien compris la situation des familles, ce qui suppose de s'adosser à une structure de génétique médicale, avec un service de génétique habituer à écouter et à analyser les situations (...) Il n'est donc pas possible d'envisager le diagnostic préimplantatoire en dehors d'une structure de génétique très forte, capable d'examiner avec attention les réalités, d'identifier la maladie et son mode de transmission ainsi que la demande exacte du couple, le risque réel de récidive... Cela représente un gros travail préalable de génétique médicale qui, dans bien des cas, avait été purement et simplement scotomisé, des gynécologues ou des obstétriciens adressant directement le couple au centre de DPI. Je considère donc qu'une solide armature de génétique est indispensable pour faire du bon diagnostic préimplantoire... ».
Ce nombre limité de centres en France semble tout à fait suffisant au regard de la demande qui reste peu élevée ; il n'existe d'ailleurs dans le monde qu'une vingtaine de centres spécialisés dans cette pratique. Deux raisons expliquent le besoin à ce jour peu élevé de DPI : en premier lieu, la loi de 1994 en a limité la pratique aux seuls cas où une maladie génétique d'une particulière gravité, reconnue incurable au moment du diagnostic, a été préalablement diagnostiquée chez l'un des parents. Cette limitation sur des critères médicaux existe dans d'autres législations qui autorisent le DPI : la Norvège l'autorise ainsi dans les cas exceptionnels d'une maladie héréditaire incurable ; l'Allemagne autorise seulement le diagnostic sur le sperme en faveur de la détection de maladies liées aux chromosomes sexuels afin d'éviter la transmission d'une maladie héréditaire liée au sexe. En second lieu, on ne dispose pas aujourd'hui de tests pour toutes les maladies génétiques graves. Comme l'indiquait la CNMBRDP dans son rapport d'activité pour les années 1997 et 1998, seize pour cent des anomalies récessives sont liées au sexe, et pour la grande majorité d'entre elles, il n'existe pas de diagnostic génétique possible. La détermination du sexe des embryons dans le but de ne replacer in utero que les embryons de sexe féminin est donc réalisée chez des couples où existe un risque de transmettre par exemple la myopathie de Duchenne, la paraplégie spastique liée à l'X, l'agammaglobulinémie, l'hémophilie A, la maladie de Tay-Sachs, le syndrome de Lesch-Nyhan, l'adrénoleucodystrophie...
Le Professeur Arnold Munnich devant la Mission témoignait ainsi de son impuissance en l'absence de test génétique pour certaines maladies graves, qui entrent pourtant dans le champ du DPI, en particulier pour les maladies orphelines :
« Dans certains cas, nous sommes amenés à répondre par la négative, alors que l'affection est effectivement d'une particulière gravité, car techniquement, ou faute de moyens, nous ne sommes pas en mesure de satisfaire la demande. C'est là une situation dramatique : c'est un coup terrible du destin que d'avoir une maladie génétique - cela concerne 30.000 naissances par an en France et touche 25 millions d'Européens - mais c'est un second coup d'avoir une maladie génétique rare car elle est un peu laissée pour compte, y compris de notre part. Quand nous avons trente femmes myopathes qui attendent et une femme qui présente un risque de maladie de Menkes, nous nous occupons d'abord des premières : c'est dramatique mais c'est comme ça ! ».
Pour la mucoviscidose, la technique permet aujourd'hui d'identifier trois groupes d'embryons in vitro : les non-porteurs de la mutation, les porteurs hérézygotes et les homozygotes atteints. Seuls les embryons des deux premiers groupes sont transférés in utero ; le risque de transmettre la maladie n'est donc pas totalement écarté mais est considérablement réduit. Il faut à cet égard souligner l'impossibilité d'assurer la fiabilité totale des tests de DPI. Les embryons sont, en effet, constitués de plusieurs lignées cellulaires génétiquement différents, appelées « mosaïques ». Le diagnostic n'étant pratiqué que sur une seule cellule ou quelques-unes, il existe donc un risque irréductible d'erreur puisque l'on n'est jamais assuré d'avoir testé toutes les mosaïques de l'embryon. Cet aléa explique que l'on propose aux couples de réaliser un DPN en cours de grossesse pour confirmer ou informer le DPI. En cas d'erreur, il lui est alors proposé de pratiquer une interruption médicale de grossesse, ce qui explique les faibles résultats atteints puisqu'à ce jour par les centres de DPI en terme de naissances. À ce sujet, le Professeur Arnold Munnich fournissait à la Mission les indications suivantes :
« Aujourd'hui, le bilan officieux est le suivant : à l'hôpital Necker, nous avons actuellement cinq femmes enceintes et j'espère avoir, sinon dans quelques semaines, du moins dans un petit nombre de mois, la joie de vous annoncer les premières naissances (165). Mais au prix de quelles difficultés ! Cinq femmes enceintes, cela signifie pratiquement cinquante grossesses débutantes car, malheureusement, toutes les grossesses débutantes ne se traduisent pas par l'arrivée d'un bébé à la maison : il y a un très grand déficit entre le nombre de grossesses débutantes et le nombre d'enfants qui rentrent à la maison dans les bras de leur mère. Les médecins de la reproduction sont optimistes quand ils parlent de 30 % de succès. Si j'en crois mon expérience de praticien généticien, nous n'avons pas eu plus de 10 % de succès et la femme aujourd'hui la plus avancée dans sa grossesse, qui en est au cinquième ou sixième mois, a fait l'objet de plusieurs tentatives et a dû attendre plusieurs cycles avant d'être enfin enceinte ».
Selon lui également, le nombre de demandes de DPI devrait augmenter à l'avenir dans des proportions qui resteront limitées car peu de couples remplissent les critères restrictifs retenus par la loi. Une sélection des dossiers est ainsi assurée par les centres de DPN à qui le décret d'application n° 98-216 du 24 mars 1998 a confié un rôle de filtre. À l'issue de la procédure, peu de couples recourent effectivement au DPI. Le Professeur Arnold Munnich distingue ainsi : « un premier groupe, composé d'environ 25 % des couples, ne s'est pas présenté le jour de la consultation, leur demande étant l'expression d'une révolte contre le destin et l'injustice dont ils étaient l'objet ; un deuxième groupe correspondait aux couples victimes d'une erreur d'orientation faute d'avoir bénéficié d'une consultation de génétique préalable ; le troisième groupe était constitué de couples présentant de très bonnes indications et « référés » par des généticiens ».(...) En réalité, on peut chiffrer la demande à une trentaine ou une quarantaine de couples par an dans notre centre et autant à Strasbourg, ce qui n'est pas gigantesque. Je ne pense pas que ces demandes progresseront beaucoup : ce sont des demandes fondées, qui portent sur des maladies d'une particulière gravité, des maladies mortelles, sans aucune tentation eugénique. »
C.- L'EFFICACITÉ CONSTATÉE DE LA LÉGISLATION EN VIGUEUR CONTRE TOUTE DÉRIVE EUGÉNIQUE
Le cadre strict délimité par la loi de 1994, qui ne s'adresse qu'à un nombre restreint de couples, et la pratique, respectueuse de ces limites auxquelles s'ajoutent celles de la science, témoignent de l'absence de risque eugénique. La réalité montre, en effet, des cas qui restent rares de couples amenés à recourir à des fécondations in vitro et au DPI après des parcours familiaux toujours douloureux ainsi que l'attestait le Docteur Axel Kahn dans son audition précitée :
« Comme généticien, j'ai été confronté deux fois au moins dans ma vie à des situations particulières. Une fois, il s'agissait d'une famille où l'homme de ce couple était l'animateur d'une association de malades. Ils avaient eu un enfant qui était mort d'une maladie qui tue les enfants à deux ans, tragique, que l'on appelle une leuco-dystrophie métachromatique. Un autre enfant était atteint. Leurs deux enfants sont morts. Deux fois, la grossesse avec un diagnostic prénatal avait abouti à un diagnostic de la maladie et à l'avortement. Bien évidemment, cette femme ne pouvait pas envisager de recommencer une grossesse. Si un couple vient me voir en disant : « Docteur, maintenant vous pouvez faire un diagnostic avant de transplanter cet _uf, faites-le parce que nous ne pouvons plus recommencer une grossesse avec la perspective d'avorter ». Que vais-je lui répondre ? Vais-je dire : « Madame, non. C'est mal de faire cela. Avortez donc » ? Moralement, je ne me sens pas la capacité de ne pas lui offrir, si je sais le faire, la possibilité de redémarrer une grossesse, qu'en étant à peu près assuré que cette grossesse ne sera pas à nouveau vouée à l'avortement après un diagnostic prénatal ».
Pour le Professeur Arnold Munnich, la détresse des couples qui s'adressent au centre de DPI qu'il dirige est telle qu'on ne peut les soupçonner d'aucune tentation eugénique : « Lorsque l'on interroge ces couples et qu'on les écoute comme je le fais dans les consultations de génétique, on constate qu'ils ne veulent à aucun pris tenter l'épreuve de la grossesse sans diagnostic préimplantatoire car ils refusent catégoriquement d'envisager une nouvelle interruption médicale de grossesse. Par conséquent, leur demande me paraît véritablement profonde, sincère, dénuée de toute tentation eugénique et j'ai l'impression de faire une bonne action médicale en pratiquant le diagnostic préimplantatoire (...) On ne peut pas se présenter dans n'importe quel centre de DPI sans une histoire lourde justifiant cette démarche.
En écoutant les couples, je dois vous dire que je n'ai pas une seconde perçu une tentation eugénique de leur part. J'entends des couples qui ont vécu des histoires dramatiques : des mères qui ont perdu un enfant, puis un autre et qui, à la suite de ces décès à répétition, ont fait l'expérience d'une, deux, trois, voire quatre interruptions médicales de grossesse. En réalité, ces couples sont dans une grande souffrance et n'ont nullement l'intention de faire une demande autre que celle portant sur la maladie qui fait l'objet de la consultation. Jamais je n'ai entendu un couple dire qu'il préférerait un garçon ou une fille, un grand ou un petit, un blond ou un brun... Ce n'est vraiment pas le problème : le problème, hélas, c'est bien la récidive de maladies lourdement invalidantes ».
Il ne paraît donc pas opportun de modifier la loi s'agissant des conditions de recours au DPI. Tout au plus pourrait-on lever la contradiction qui existe dans l'article L. 2131-4 du code de la santé publique entre le risque de « donner naissance à un enfant atteint d'une maladie génétique d'une particulière gravité reconnue comme incurable au moment du diagnostic » et la finalité fixée au DPI qui « ne peut avoir pour objet que de rechercher cette affection ainsi que les moyens de la prévenir et de la traiter ». Il conviendrait donc de supprimer cette dernière finalité relative au traitement de la maladie ou tout au moins de distinguer la recherche de la pathologie des moyens de la prévenir ou de la guérir en décidant que ces différentes finalités soient alternatives et non pas cumulées. Cette seconde solution serait sans doute préférable dans la perspective d'une évolution de la science qui pourrait permettre, demain, non plus d'éliminer les embryons atteints de la maladie recherchée mais de les guérir.
En revanche, certaines voix se sont élevées pour élargir le recours au DPI aux femmes de plus de 38 ans dont la grossesse tardive expose leurs enfants à un risque de maladie génétique telles que les trisomies. C'est le regret qu'exprimait le CNMBRDP dans son rapport d'activité précité pour les années 1997 et 1998 : « La loi en France ne permet pas la réalisation d'un DPI pour âge maternel élevé et n'autorise à rechercher sur l'embryon que la pathologie liée à l'anomalie génétique parentale, ce qui est regrettable pour le cas des femmes de plus de 38 ans chez lesquelles une fois la grossesse initiée, un diagnostic prénatal sera proposé ».
C'est aussi la position que défendait devant la Mission le Professeur Claude Sureau au nom de l'Académie nationale de médecine le 12 juillet 2000 : « Nous sommes très nettement en faveur d'une extension du diagnostic préimplantatoire. Si l'on fait un diagnostic préimplantatoire, en raison du risque de transmission d'une anomalie génétique, mucoviscidose ou myopathie, il faudra éventuellement, notamment en fonction de l'âge ou du résultat des marqueurs biologiques, pratiquer secondairement une amniocentèse, parce que l'on n'aura pas pu analyser la situation chromosomique au moment de cet examen, ce qui est tout à fait illogique ».
Il reconnaissait cependant que cette extension du recours au DPI aurait un « aspect eugénique » en déclarant : « Cela étant, il faut reconnaître que l'extension de ce dépistage conduit à une attitude qui, dans une certaine mesure, présente un aspect eugénique directement lié au fait que les enfants trisomiques ne sont pas encore suffisamment pris en charge par la collectivité. Il existe un problème de fond qui est plus philosophique et politique que strictement biologique ».
Votre Rapporteur considère qu'il faut examiner avec prudence cette demande d'extension car il s'agirait de conduire des milliers de femmes qui n'ont pas de problèmes de stérilité à s'engager dans un processus d'AMP, puisque le DPI ne peut être pratiqué que sur des embryons in vitro, avec tous les risques et toutes les difficultés que présente l'AMP, qui ont été exposés dans la deuxième partie du présent rapport. Il lui semble donc plus raisonnable de ne pas étendre les possibilités de recours au DPI en dehors des risques de transmission à l'enfant d'une maladie génétique familiale.
III.- LES EXAMENS GÉNÉTIQUES ET LES ASSURANCES
En dehors de l'application des examens génétiques en matière médicale, existent aujourd'hui des interrogations sur le recours à ces tests dans deux domaines : celui des assurances et celui du marché de l'emploi. Le Conseil d'État s'est saisi de ces questions dans son rapport de novembre 1999, rendant, sur ce sujet, des conclusions décevantes.
On peut, sans grande imagination, percevoir tout l'intérêt que les assureurs auraient à connaître le patrimoine génétique des candidats à la souscription d'une assurance. Face à des personnes risquant, à terme, de développer ou non une maladie d'origine génétique, ils pourraient moduler la police d'assurance, voire refuser à certains l'autorisation de souscrire.
Le caractère discriminatoire que revêtirait une telle mesure est choquant. Face à la crainte de voir se développer une méfiance et une critique sociales fortes à leurs égards, les professionnels de l'assurance ont souhaité prendre des mesures conservatoires, en fonction de considérations qui ne nous ont pas toujours semblé totalement convaincantes.
A.- DES INTÉRÊTS CONTRADICTOIRES
1.- Les contraintes propres au marché de l'assurance
Sur ce sujet, la mission a reçu le 13 septembre 2000, M. Claude Fath, président de la commission plénière des assurances de personnes et M. François Ewald, directeur de la recherche et de la stratégie, de la Fédération française des sociétés d'assurance (FFSA). Ces intervenants ont mis en avant les contraintes propres au marché de l'assurance.
Selon les termes de M. Claude Fath, l'assurance-vie recouvre trois types d'activités. Le premier est la collecte d'épargne en vue de la retraite, qui représente un gros volume de chiffre d'affaires et des couvertures de risques décès relativement limitées. Exceptées quelques garanties planchers, on note ainsi que peu de garanties complémentaires fonctionnent en cas de décès de l'assuré. Le deuxième secteur important est la garantie de capitaux ou de revenus si l'assuré vient à décéder en cours de contrat. Ce sont des opérations de prévoyance qui permettent à une personne de garantir des emprunts, sa famille ou son entreprise. Cette activité obéit également à des règles normales de souscription et de sélection. Le troisième secteur est celui des assurances destinées à se protéger contre les conséquences d'un arrêt d'activité par suite d'incapacité de travail ou d'invalidité. Toutes ces opérations sont facultatives. Elles relèvent d'un souci de protection et de prévoyance. C'est dans le cadre de ces assurances que se pose la question des tests génétiques.
D'après M. Claude Fath, l'assureur doit, dans l'intérêt de la mutualité des assurés qu'il garantit, être extrêmement attentif aux phénomènes d'anti-sélection, c'est-à-dire aux comportements des assurés qui ont tendance à vouloir se garantir lorsqu'ils connaissent la réalité de leur situation pathologique et non pas lorsqu'ils sont encore en pleine forme. Les assureurs ont donc besoin de demander à l'assuré des informations de caractère médical, d'autant plus approfondies que les capitaux à garantir sont élevés et/ou que l'âge de l'assuré est avancé. Selon les termes des représentants des professionnels des assurances, les différents examens médicaux suffisent aujourd'hui pour garantir et fixe le tarif des risques proposés.
Les représentants de la FFSA ont insisté sur le fait que l'assurance ne peut fonctionner équitablement et durablement que dans l'égalité d'information entre le souscripteur et l'assureur. Aux termes de l'article 113-8 du code des assurances, l'opération d'assurance doit être de bonne foi. En cela, elle repose sur la déclaration exacte et complète des éléments connus de l'assuré, de nature à faire apprécier par l'assureur le risque à garantir. Selon M. Claude Fath, si l'assuré a connaissance d'éléments aggravants concernant sa santé, il faut que l'assureur dispose de la même information pour pouvoir établir équitablement le tarif du risque à garantir. Les assureurs insistent sur le fait que l'égalité d'information entre l'assureur et l'assuré est la condition du maintien d'un tarif équitable pour l'ensemble des assurés.
On comprend cette logique qui, sur le plan théorique, n'appelle que peu de remarques. Elle méconnaît cependant la réalité du face-à-face entre assureurs et souscripteurs, qui est foncièrement inégal.
2.- Le face-à-face inégal entre assureurs et souscripteurs
Comme ont pu le souligner certains membres de la Mission lors de l'audition des représentants de la FFSA, les assureurs n'ont pas aujourd'hui recours aux tests génétiques parce que ceux-ci ne leur permettent pas, pour l'heure, de disposer d'une information exploitable. Mais on peut supposer qu'à l'avenir les professionnels de l'assurance envisageront sérieusement l'emploi de ces tests. Face à cela, les souscripteurs n'auront que peu de choix. Même s'il s'agit de contrats d'assurances volontaires et non obligatoires - comme le soulignent à dessein les assureurs - ils sont importants dans la vie de chacun. Or quels moyens seraient offerts aux candidats à la souscription pour résister à l'exigence, formalisée ou non dans un contrat, de fournir les résultats d'un test génétique ? Peut-on réellement penser que les individus pourront résister aux compagnies d'assurance sur ce sujet ?
Le législateur ne peut accepter que les acteurs soient ainsi livrés à eux-mêmes dans une confrontation dont le résultat est acquis d'avance, au profit des compagnies.
La pratique actuelle des assureurs répond temporairement aux craintes exprimées sur cette question. Mais il appartient au Parlement de fixer les règles en la matière.
B.- LA PRATIQUE ACTUELLE DES ASSUREURS
Conscients des réactions qui se faisaient jour et du risque réel de voir monter la méfiance à l'égard de leur profession, les assureurs ont décidé à deux reprises, en 1994 et en 1999, d'adopter un moratoire sur l'usage des tests génétiques.
Il s'agit là d'un triple engagement quinquennal de ne pas faire de la soumission à un test génétique une condition d'assurance, de ne pas demander les résultats de tests éventuellement subis par le souscripteur et de ne pas en tenir compte si ces résultats sont spontanément fournis.
L'adoption de ce moratoire va de pair avec une revendication des assureurs. Tant devant le Conseil d'État que la mission parlementaire, elle a été exprimée de la manière suivante : il est trop tôt pour légiférer étant donné qu'on ne connaît pas encore de manière exacte les conséquences de ces tests sur le marché de l'assurance.
Devant le Conseil, les assureurs ont exprimé cette interrogation : « L'angoisse qui fait craindre des dérives est-elle un motif suffisant de l'intervention du législateur ? » A cette question, il nous sera permis de répondre par l'affirmative.
2.- La mise en avant par les assureurs du modèle britannique
Lors de leur audition par la mission, les représentants de la FFSA ont appelé à ce que soit examiné avec attention le modèle britannique.
Le gouvernement du Royaume-Uni a demandé, il y a quelques temps, aux assureurs de lui faire des propositions. Ceux-ci ont répondu par un code de conduite extrêmement contraignant pour les assureurs. Ce code interdit ainsi la sous-tarification suite à la présentation d'un test génétique. Il interdit aussi de demander des tests génétiques comme condition d'assurance. Ce corpus de règles qui a été repris par le gouvernement anglais, prévoit également qu'un assureur ne pourra avoir accès à un test génétique dont un assuré connaîtrait le résultat que dans la mesure où le test est « relevant », c'est-à-dire pertinent, du point de vue scientifique - ce qui veut dire qu'en la matière la connaissance est acquise et consolidée - et d'autre part au regard du risque couvert. Car on pourrait disposer d'un test qui aurait une valeur prédictive pour une maladie à développement très long. Or pour une garantie relativement brève, il est clair que l'utilisation d'une telle donnée n'est pas pertinente puisqu'elle n'a pas d'incidence sur le risque.
Selon les représentants de la FFSA, ce type de régulation montre que l'on peut très bien avoir des règles déontologiques encadrées et contrôlées par des institutions publiques. En Angleterre, il n'appartient pas aux assureurs de décider qu'un test est « relevant ». Ils peuvent seulement, au vu des connaissances scientifiques, exprimer leur souhait d'avoir la possibilité de demander à un assuré s'il a subi tel test. Une commission indépendante décide si ce test est « relevant » ou non en la matière. Ce point est très important dans la pratique de l'assurance parce qu'il confère à l'examen de santé génétique un caractère objectif et public.
Pour séduisante qu'elle soit, cette solution ne nous paraît pas correspondre à l'objectif qui est, pour nous, essentiel, celui de non-discrimination des personnes en fonction de leur patrimoine génétique.
1.- Une question âprement débattue
Lors de son déplacement aux États-Unis, votre Rapporteur a pu noter que, contrairement aux idées reçues, la question de l'usage des tests génétiques en matière d'assurance mais aussi d'emploi était débattue, tant dans les milieux politiques que de l'entreprise. Il est difficile de distinguer aux États-Unis les problèmes de l'assurance et de l'emploi puisque, étant donné le mode de couverture sociale, les deux champs apparaissent très imbriqués.
Les membres du National Human Genome Research Institute, ont clairement souligné que la discrimination génétique était un véritable sujet de préoccupation. C'est pourquoi au sein de cet organe public de recherche a été créé un pôle consacré aux problèmes éthiques, le ELSI (Ethical, legal, social implications of human genetic research), doté d'un budget important.
On ignore actuellement le nombre de tests génétiques réalisés sur le lieu de travail aux États-Unis. Une affaire récente a néanmoins montré que ces tests pouvaient être un réel enjeu économique. Ainsi une compagnie de chemins de fer a tenté d'obtenir une réduction de ses versements au titre de l'assurance couvrant les maladies professionnelles, en montrant que certains de ses employés étaient prédisposés génétiquement à ces pathologies. Il apparaît que l'armée a également procédé à des tests pour limiter le paiement de pensions. Un membre du corps des Marines atteint d'un cancer s'était vu opposer l'argument selon lequel il était prédisposé à une certaine forme de cancer. En conséquence de quoi, l'armée avait refusé de lui verser une pension. Ayant saisi les tribunaux, la victime a obtenu gain de cause.
On note que les syndicats sont très préoccupés par ces questions. Dans l'affaire de la compagnie des chemins de fer, ils ont immédiatement porté plainte, comme la commission fédérale chargée de l'égalité et de l'emploi. La demande de législation - qui n'est pas le réflexe premier aux États-Unis - est patente. Elle est aujourd'hui examinée avec attention.
2.- Vers une législation fédérale ?
Actuellement, sur cinquante États, une trentaine ont légiféré pour limiter l'emploi des tests génétiques par les assureurs et les employeurs. Le Président Clinton avait envisagé le dépôt d'un projet de loi en 2000, mais faute de majorité possible, il a préféré limiter son intervention aux emplois fédéraux, sur lesquels le Président des États-Unis a une autorité réglementaire directe. L'idée était que cette décision, interdisant toute discrimination génétique dans ces emplois fédéraux, servirait de modèle. Le résultat est, à ce jour, mitigé. On ne décèle pas un vaste mouvement vers une généralisation des tests génétiques. Mais les quelques exemples cités plus haut montrent qu'aujourd'hui la tentation existe. Or, dans un pays où la couverture sociale dépend, pour l'essentiel, de l'assurance privée, un tel système est socialement difficilement acceptable.
Des initiatives récentes ont été prises dans la lutte contre la discrimination génétique. Des sénateurs démocrates ont, en effet, déposé au début du mois de juin 2001 une proposition de loi contre cette forme de discrimination au travail ou par des compagnies d'assurance médicale. Cette proposition interdirait aux employeurs d'utiliser des informations génétiques pour prendre des décisions en matière d'embauche, et aux compagnies d'assurance médicale de recourir à ces informations pour écarter un client ou établir sa police d'assurance. Cette nouvelle législation prohiberait également la communication à des tiers d'informations génétiques. Selon le chef de la majorité démocrate au Sénat, M. Tom Daschle, les sénateurs démocrates soutenant cette proposition sont nombreux et une initiative équivalente à la Chambre des représentants aurait recueilli l'appui de 250 députés, démocrates et républicains.
Face à ce mouvement outre-Atlantique, on comprendrait mal que le Parlement français reste en retrait.
D.- L'INTERVENTION DU LÉGISLATEUR
1.- Les préconisations insatisfaisantes du Conseil d'État
Abordant cette question, le Conseil d'État s'est livré à une analyse très sophistiquée mais peu convaincante. S'appuyant sur la théorie économique des jeux, dans une démonstration qui relève plus de l'exercice de style que d'un traitement réel de la question, le Conseil d'État propose d'obliger les assurés à révéler à la compagnie d'assurance à laquelle ils s'adressent, les résultats des tests génétiques pratiqués par ailleurs ou révélés à une autre compagnie.
Reprenons les termes mêmes du rapport du Conseil d'État pour mieux juger du caractère abstrait de cette analyse. « Si un assureur B, étranger ou national, a le droit d'exploiter les résultats de tests prédictifs, il appliquera une tarification individuelle proportionnelle au niveau du risque estimé. Si sa population d'assurés est la même que celle de l'assureur A, sa tarification moyenne sera égale au coût marginal de son activité, c'est-à-dire légèrement supérieure à la tarification de l'assureur A du fait du coût des tests. Mais il y a de fortes chances pour que la population d'assurés de A et de B ne soit pas identique. En effet, les assurés devraient être incités à subir des tests afin de pouvoir éventuellement bénéficier d'un tarif plus bas. Si les tests se révélaient peu favorables pour un individu, il se retournerait évidemment vers l'assureur « vertueux », A, qui lui propose un tarif moyen ne tenant pas compte de ces tests. Par un mécanisme de contre-sélection, les mauvais risques afflueraient vers A, dont la situation économique serait compromise. D'où la revendication des assureurs et leur raisonnement : « Si l'autre fait des tests, je fais des tests ; si l'autre ne fait pas de tests, je fais des tests » » (166).
La démonstration du Conseil d'État suit sa logique pour montrer, in fine, qu'il est impossible d'atteindre un équilibre de Pareto, c'est-à-dire la seule situation dans laquelle on ne peut améliorer la situation d'un acteur sans détériorer la situation d'un autre acteur. En l'espèce, cet équilibre ne pourrait être atteint que si toutes les compagnies ne pratiquaient pas de test. Dans cette hypothèse, elles ne subiraient pas le risque de voir affluer vers elles les personnes dotées d'un patrimoine génétique jugé « défavorable » et les assurés ne souffriraient pas d'une discrimination fondée sur les gènes. Cet équilibre de Pareto ne peut être atteint faute d'un accord international empêchant l'ensemble des compagnies d'assurances de tous les pays d'exiger de tels tests. Le meilleur moyen de s'approcher de cet « optimum social », consisterait alors à exiger des personnes ayant subi un test génétique pour l'accès à une assurance d'en dévoiler le contenu à la compagnie à laquelle elles s'adressent ensuite, après avoir été une première fois refoulées.
Selon le Conseil d'État, « une telle mesure devrait dissuader les futurs assurés qui ne savent rien de leur génome de courir le risque de subir des tests pour obtenir auprès d'un assureur étranger de meilleures conditions contractuelles ». Le meilleur moyen d'éviter l'utilisation des tests serait donc de les soumettre totalement au principe de loyauté contractuelle.
Cette démonstration se fonde notamment sur l'idée que les individus pourraient en quelque sorte vendre leurs tests favorables aux assureurs. Pourrait alors exister une rupture de concurrence entre les compagnies, étrangères, acceptant ces tests ou les sollicitant, et les autres. Les premières pourraient ainsi sélectionner leurs souscripteurs, assumant de la sorte un risque financier moindre, alors que les secondes accueilleraient, sans autre choix, les « recalés » de ce système de sélection et seraient ainsi contraintes de supporter des risques plus importants, mettant en péril leur équilibre économique.
Selon votre Rapporteur, ce raisonnement ne prend que peu en compte l'idée selon laquelle procéder à un test génétique ne serait pas un geste banalisé. Accepter de connaître son « destin génétique » - avec tout ce que cette expression a d'approximatif - n'est pas une démarche anodine. Seront-ils alors nombreux ceux qui entreprendront de procéder à de tels tests pour simplement obtenir la baisse d'une prime d'assurance, au risque d'apprendre qu'ils sont porteurs d'un gène inquiétant ?
Parallèlement, il convient de distinguer celui qui pourrait décider de subir un tel test dans une logique rationnelle économique et celui qui, au contraire, doit subir cet examen pour des raisons proprement médicales. Doit-on lui imposer de délivrer une information qui touche à son intimité la plus secrète, parce que certains utilisent de telles informations pour bénéficier d'une diminution de leur prime d'assurance ? Une telle démarche ne nous semble pas conforme à l'éthique.
Dans son rapport de novembre 1999, le Conseil d'État présente trois solutions possibles.
La première consisterait à interdire totalement l'exploitation des données génétiques conformément à l'avis du Comité consultatif national d'éthique du 30 octobre 1995. Pour le Conseil d'État, cette interdiction ferait purement et simplement abstraction du principe de loyauté contractuelle. Il serait alors illégitime d'empêcher les assureurs de demander aux souscripteurs les résultats des tests qu'ils auraient subis par ailleurs. Aux yeux du Conseil d'État, apparemment convaincu par les professionnels des assurances, cette solution serait extrême.
La deuxième voie consisterait à n'interdire que les discriminations reposant sur l'utilisation de tests génétiques visant des maladies qui ne sont pas déclarées ou des prédispositions génétiques à des maladies. Les résultats d'un test diagnostique servant à préciser la pathologie dont souffre un malade resteraient quant à eux communicables. Selon le Conseil d'État, cette option ménagerait un équilibre entre le principe de non-discrimination et celui de loyauté contractuelle. Cette solution paraît effectivement pouvoir être étudiée avec attention.
Le Conseil d'État juge cependant plus cohérente une dernière possibilité : celle qui consiste à contraindre le souscripteur à dévoiler le résultat d'un test de prédisposition qu'il aurait subi volontairement avant la conclusion de son contrat. Cette troisième solution serait, selon les termes du Conseil d'État, plus cohérente avec la législation actuelle et plus conforme aux principes de l'équité actuarielle. Elle permettrait, en outre, de dissuader les souscripteurs potentiels de « s'engager dans des stratégies opportunistes ».
On a indiqué, plus haut, en quoi ce raisonnement abstrait ne pouvait nous convaincre. Il est de notre devoir de législateur d'intervenir sur cette question. Les deux premières solutions - celle de l'interdiction totale ou partielle des résultats de tests génétiques - peuvent être, selon votre Rapporteur, discutées parallèlement. Mais, en tout état de cause, ces problèmes ne peuvent être traités par les seuls professionnels, dans le cadre d'un face-à-face inégal avec les souscripteurs.
IV.- LES EXAMENS GÉNÉTIQUES ET LES EMPLOYEURS
Le Conseil d'État a proposé une analyse claire de la problématique relative aux examens génétiques et au marché de l'emploi. Dans son rapport, il montre que les questions qui apparaissent ici sont assez proches de celles touchant le marché des assurances : les entreprises peuvent-elles utiliser des tests génétiques pour engager leurs employés ? Une telle sélection lors de l'embauche est-elle possible, utile, souhaitable ? Parallèlement, une personne à la recherche d'un emploi peut-elle trouver profit à présenter à une entreprise les résultats d'examens génétiques pour prouver son aptitude à exercer certains postes ?
En dépit d'une problématique proche de celle du marché de l'assurance, la question de la discrimination à l'embauche sur le fondement de critères génétiques semble moins plausible et moins immédiatement préoccupante selon le Conseil d'État.
A.- L'ÉVALUATION DU RISQUE DE DISCRIMINATION
1.- Les tests génétiques au service des employeurs ?
Dans son avis n° 46 du 30 octobre 1995 « Génétique et médecine : de la prédiction à la prévention », le Comité consultatif national d'éthique a montré quels étaient les risques induits par l'utilisation par des employeurs des résultats de tests génétiques. Deux types d'utilisation sont envisageables : lors de la sélection des candidats à l'embauche ; dans la gestion du déroulement de carrière.
Les employeurs pourraient trouver un intérêt objectif à connaître les résultats des tests génétiques de leur personnel. Ils pourraient ainsi sélectionner les personnes présentant le moins de risque de développer des maladies de sorte à limiter les coûts liés à l'absentéisme et à une baisse de productivité. Ils pourraient également mieux cibler les actions de formation ou les promotions en fonction de ces résultats.
Comme le souligne le Comité consultatif national d'éthique dans son avis du 30 octobre 1995, le fait que ces tests ne soient pas totalement fiables ne serait pas forcément de nature à conduire les employeurs à les négliger. Il est vrai que les entreprises ont souvent recours à des méthodes plus incertaines encore.
On mesure en quoi tous ces éléments d'appréciation pourraient éventuellement améliorer la gestion de l'entreprise. Mais on distingue aussi tout ce qu'aurait de choquant la mise à disposition des employeurs de ce type d'informations ainsi que leur utilisation dans une visée exclusivement économique. L'atteinte à la dignité humaine et à la vie privée serait manifeste.
2.- La position du Conseil d'État : relativiser le risque de discrimination
Le Conseil d'État relativise ce risque en se fondant sur la législation actuelle. Ainsi, l'article L. 122-45 du code du travail interdit les pratiques discriminatoires lors des procédures d'embauche, de sanction ou de licenciement :
« Aucune personne ne peut être écartée d'une procédure de recrutement, aucun salarié ne peut être sanctionné ou licencié en raison de son origine, de son sexe, de ses m_urs, de sa situation de famille, de son appartenance à une ethnie, une nation ou une race, de ses opinions politiques, de ses activités syndicales ou mutualistes, de ses convictions religieuses ou, sauf inaptitude constatée par le médecin du travail dans le cadre du titre IV du livre II du présent code, en raison de son état de santé ou de son handicap.
Aucun salarié ne peut être sanctionné ou licencié en raison de l'exercice normal du droit de grève.
Toute disposition ou tout acte contraire à l'égard d'un salarié est nul de plein droit ».
Par ailleurs, dans ses articles 225-1 et suivants, le code pénal définit et sanctionne les discriminations illicites :
« Art. 225-1. - Constitue une discrimination toute distinction opérée entre les personnes physiques à raison de leur origine, de leur sexe, de leur situation de famille, de leur état de santé, de leur handicap, de leurs m_urs, de leurs opinions politiques, de leurs activités syndicales, de leur appartenance ou de leur non-appartenance, vraie ou supposée, à une ethnie, une nation, une race ou une religion déterminée.
Constitue également une discrimination toute distinction opérée entre les personnes morales à raison de l'origine, du sexe, de la situation de famille, de l'état de santé, du handicap, des m_urs, des opinions politiques, des activités syndicales, de l'appartenance ou de la non-appartenance, vraie ou supposée, à une ethnie, une nation, une race ou une religion déterminée des membres ou de certains membres de ces personnes morales ».
L'article 225-2 du code pénal sanctionne de deux ans d'emprisonnement et de 200 000 francs d'amende ces discriminations, notamment quand elles consistent à refuser d'embaucher, à sanctionner ou à licencier une personne. L'article 225-3 du même code prévoit cependant quelques cas de dérogations à ce principe, principalement lorsque ces dérogations visent à protéger les personnes subissant ce traitement discriminant.
« Art. 225-3. - Les dispositions de l'article précédent ne sont pas applicables :
1° Aux discriminations fondées sur l'état de santé, lorsqu'elles consistent en des opérations ayant pour objet la prévention et la couverture du risque décès, des risques portant atteinte à l'intégrité physique de la personne ou des risques d'incapacité de travail ou d'invalidité ;
2° Aux discriminations fondées sur l'état de santé ou le handicap, lorsqu'elles consistent en un refus d'embauche ou un licenciement fondé sur l'inaptitude médicalement constatée soit dans le cadre du titre IV du livre II du code du travail, soit dans le cadre des lois portant dispositions statutaires relatives à la fonction publique (...) ».
L'exception instituée au 2° de cet article permet de ne pas sanctionner un refus d'embauche lorsque l'inaptitude de la personne est médicalement constatée. La question suivante a pu se poser : cette exception vaut-elle pour les inaptitudes futures détectées par voie d'examen génétique ?
Les juridictions n'ont jamais reconnu cette possibilité. Pour justifier un refus d'embauche, l'inaptitude doit être présente et non incertaine et future. Elle doit conduire à une incapacité immédiate d'exécuter la prestation de travail. Cette position juridique a été renforcée par l'avis du Comité consultatif national d'éthique du 30 octobre 1995 précité. Se fondant sur les dispositions légales prohibant toute discrimination à l'égard des personnes séropositives qui n'ont pas développé la maladie du SIDA, le Comité considère qu'une discrimination fondée sur les résultats d'un test génétique présymptomatique ou probabiliste serait illicite.
Le Comité consultatif national d'éthique ajoute que les cas où l'examen des caractéristiques génétiques pourrait être utile pour prévenir une maladie professionnelle sont rares, en l'état actuel des connaissances. Pourtant, dans cette hypothèse, le médecin du travail pourrait prescrire des tests permettant de détecter une prédisposition à une maladie, dont le développement serait favorisé dans l'environnement professionnel en cause. De tels examens ne pourraient cependant être réalisés sans le consentement de l'employé, les résultats demeurant confidentiels et ne pouvant en aucun cas être communiqués à l'employeur. Le Comité consultatif national d'éthique a cependant souligné, dans son avis de 1995, le rôle ambigu des médecins du travail, salariés de l'entreprise.
Le Conseil d'État estime dans son rapport que la remise en cause de la jurisprudence, interdisant la pratique de discrimination fondée sur le potentiel développement d'une maladie, ne paraît concevable que dans des hypothèses circonscrites, où « il serait démontré qu'une prédisposition génétique contribue au déclenchement d'une maladie professionnelle, ou pour laquelle il s'avérerait que la réalisation inopinée d'un risque génétiquement déterminé met la vie d'autrui en danger. Aussi le maintien en l'état des dispositions précitées paraît-il suffisamment protecteur ».
B.- LA NÉCESSITÉ D'INTERDIRE DANS LA LOI TOUTE DISCRIMINATION À L'EMBAUCHE FONDÉE SUR LE PATRIMOINE GÉNÉTIQUE
1.- Une position du Conseil d'État peu convaincante
L'attitude prudente du Conseil d'État ne semble pas avoir été partagée par l'ensemble des acteurs en cause. Ainsi, selon le rapport de la Haute juridiction, le ministère de la justice a estimé qu'il était nécessaire d'inscrire dans la loi un principe de non-discrimination, à raison du patrimoine génétique des personnes dans le domaine de l'emploi. Le Conseil d'État examinant cette proposition s'interroge sur le caractère « excessif » d'une telle mention dans le code pénal ou le code du travail au même titre que les discriminations en raison de la race ou du sexe (167).
Le Comité consultatif national d'éthique a souligné, dans son avis de 1995, les pressions que les employeurs pourraient exercer sur les candidats à l'embauche, pour obtenir d'eux le passage d'examens génétiques. Il note également qu'on ne pourrait exclure la présentation spontanée de telles informations par les candidats, dans le cas où celles-ci leur seraient favorables. Pour le Comité, il serait difficile de s'opposer à de telles démarches. La seule solution consisterait à encadrer strictement les modalités de prescriptions des examens par les médecins.
2.- Affirmer le principe de non-discrimination
Face à cela, la prévention du Conseil d'État à l'égard d'une inscription dans la loi d'un principe de non-discrimination à raison du patrimoine génétique, paraît peu fondée. Il semble, au contraire, utile à votre Rapporteur de consacrer dans la loi ce principe de non-discrimination, qui apparaît, d'ailleurs, dans les textes internationaux. Même si ce principe ne sera invoqué que dans quelques cas rares, l'introduire dans notre droit interne permet de marquer nettement la limite de ce qui est ou non acceptable. La discrimination à raison du patrimoine génétique ne paraît pas moins tolérable que celle fondée sur le sexe ou la race, contrairement à ce que le Conseil d'État laisse entendre dans une formulation sans doute maladroite.
La réflexion menée aux États-Unis sur ce point montre qu'une telle position de principe ne saurait se voir opposer des arguments économiques, auxquels les dirigeants américains sont pourtant souvent sensibles.
SIXIÈME PARTIE :
LA BREVETABILITÉ DU GÉNOME HUMAIN :
DES ORIENTATIONS POUR TRAITER D'UN SUJET GRAVE
Parallèlement à la révision des lois bioéthiques, la question de la brevetabilité du génome humain s'est imposée à la Mission, pour deux raisons. Les avancées rapides du décryptage du génome et les revendications des sociétés américaines sur les gènes récemment découverts ont mis en lumière des enjeux scientifiques et économiques qu'il est difficile de passer sous silence. Parallèlement, la question de transposition de la directive 98/44/CE du 6 juillet 1998 relative à la protection juridique des inventions biotechnologiques a été l'occasion de l'ouverture d'un débat animé qui agite l'Europe et intéresse au plus haut point les observateurs américains.
La question de la brevetabilité du génome humain a été sujette à une certaine confusion et à des raccourcis. On a pu parler d'appropriation de l'homme par les grandes sociétés en violation du principe de non-commercialisation de l'humain. En effet, les grandes sociétés participant au décryptage du génome humain tentent d'obtenir des brevets non seulement sur la fonction attachée à un gène qu'elles ont découvert mais également sur le gène ou la séquence génétique eux-mêmes. Le traitement d'un problème aussi grave ne saurait souffrir de sensationnalisme et de simplification. La mise en avant médiatique de principes auxquels nous adhérons tous ne peut suffire pour résoudre une question et clore une discussion dont les enjeux sont marqués par un enchevêtrement de principes et d'intérêts.
La difficulté de transposition de la directive 98/44/CE du 6 juillet 1998 relative à la protection juridique des inventions biotechnologiques montre combien il est tout sauf simple de transcrire en droit une situation complexe où s'opposent plusieurs principes. C'est ici que se rencontrent, en effet, une technique juridique
- le droit des brevets -, des impératifs économiques et scientifiques - rémunérer l'invention ce qui stimule la recherche - et des principes éthiques - la non-commercialisation du corps humain, la liberté de recherche, l'accès à la connaissance.
Autoriser la brevetabilité des gènes ou des séquences de gènes : y a-t-il là un saut dans l'inconnu et la transgression d'une barrière éthique essentielle ? Quelle est la part du risque avéré dans les craintes suscitées par la directive de 1998 ? C'est autour de ces questions que se noue actuellement le débat sur la brevetabilité du génome humain.
Mais il importe, avant tout, d'aborder ce débat sereinement, tout en ayant aussi clairement conscience des limites d'une approche exclusivement nationale de ce sujet. La brevetabilité du génome humain peut être entendue comme une question corollaire à celle, plus large, de la brevetabilité du vivant. Néanmoins, elle appelle évidemment une attention et un traitement particuliers. Le préalable à toute réflexion sur ce sujet grave est d'en éclairer les enjeux sérieusement pour les aborder avec responsabilité.
I.- LE DROIT DES BREVETS FACE AU VIVANT
A.- LE DROIT DES BREVETS :
UN PROCÉDÉ CLASSIQUE POUR PROTÉGER LES INVENTEURS
1.- L'objet paradoxal du droit des brevets :
protéger les inventeurs en rendant publiques les inventions
a) La fonction du droit des brevets
Le brevet est le titre de propriété industrielle qui confère à son titulaire, inventeur ou entreprise, un droit exclusif sur une invention pour une période de vingt ans. La justification de l'octroi de ce monopole réside dans l'enrichissement de la technique et de la connaissance auquel l'inventeur contribue.
Il existe plusieurs types de brevets. Certains portent sur des procédés permettant d'obtenir un produit : il s'agit des brevets de procédés. Dans ce cas, les inventeurs réussissant à mettre à jour un autre moyen inédit d'obtenir le même produit pourront détenir un brevet sur ce nouveau procédé. D'autres concernent directement un produit. Lorsque ces brevets de produit sont déposés, un inventeur découvrant une nouvelle application de ce produit devra obtenir l'accord du premier inventeur. Son brevet est alors qualifié de dépendant.
Le brevet ne confère pas un droit de propriété matériel sur l'invention, ni un droit inconditionnel d'exploiter. Ainsi, un médicament breveté ne peut être exploité qu'après une autorisation de mise sur le marché. Le brevet reconnaît principalement un droit d'interdire à des tiers, pendant vingt ans à compter du dépôt de la demande, d'utiliser l'invention à des fins commerciales sans l'accord de l'inventeur. Il est important de noter que cette interdiction ne s'étend pas aux activités de recherche ou d'expérimentation. Dans certains cas, la justice peut ordonner la délivrance d'une licence obligatoire, soit pour satisfaire la demande du titulaire d'un brevet dépendant, soit dans certains cas plus rares, lorsqu'à la demande de l'État, des raisons de santé publique sont excipées.
Pour obtenir un brevet, il convient de déposer une demande auprès d'un organe en charge de l'instruire ; en France, l'Institut national de la propriété industrielle (INPI) est compétent pour ce faire. De ce fait, l'inventeur rend l'invention publique en même temps qu'il la protège. Le brevet permet à son titulaire d'interdire à tout autre d'exploiter l'invention sans son autorisation et, le cas échéant, de poursuivre les contrefacteurs. Le brevet lui garantit donc la jouissance du fruit de sa recherche et de ses investissements.
b) La construction de ce droit
Les origines du droit des brevets sont anciennes, mais son développement véritable date de la fin du XVIIIe siècle et des révolutions française et américaine qui mirent en avant le droit de propriété et le principe de liberté du commerce et de l'industrie. La Constitution américaine de 1787 donne ainsi pour mission au Congrès des États-Unis de « favoriser le développement de la science et des arts utiles, en garantissant pour une période de temps déterminée aux auteurs et inventeurs le droit exclusif à leurs livres et à leurs inventions ». Peu après, en 1790 est adoptée, dans ce pays, une loi organisant un système de délivrance des brevets avec un examen d'utilité, d'importance et de nouveauté de l'invention.
C'est par une loi du 7 janvier 1791 qu'en France, la Constituante a adopté un régime juridique de protection des brevets, le système choisi étant celui de l'octroi du brevet sans examen préalable. Les brevets étaient alors délivrés sans garantie du gouvernement, les juridictions étant compétentes pour trancher les litiges éventuels a posteriori. Le brevet ainsi délivré par l'État conférait à son titulaire un droit exclusif mais temporaire d'exploitation de l'invention, la loi de 1791 consacrant des principes encore en vigueur aujourd'hui : la brevetabilité des perfectionnements apportés à une invention, ou l'exigence de nouveauté de l'invention.
C'est par une loi du 5 juillet 1844 que furent révisées les règles adoptées en 1791. Elle maintint le système de non-examen préalable de l'invention, alors que d'autres pays en Europe, telle l'Allemagne, optaient pour un mécanisme inverse. C'est alors sous le double effet de la révolution industrielle et de l'harmonisation du droit des brevets au plan international, que le droit français a évolué.
L'internationalisation du commerce a conduit à repenser le droit des brevets, dans la mesure où la protection d'une invention, limitée à un seul pays, n'avait alors guère de sens. A la suite de plusieurs conférences internationales, la convention de Paris du 20 mars 1883 a institué une union, dont l'objet était d'atténuer les inconvénients du principe de territorialité des brevets. Lors de la construction européenne au XXe siècle, cette question a été également abordée. Après la création de l'Institut international des brevets en 1947 entre la France et le Bénélux, une organisation de plus grande dimension a vu le jour en Europe, dans les années soixante dix : l'Office européen des brevets.
2.- La protection européenne des brevets
a) L'Office européen des brevets
L'Office européen des brevets (OEB) est une administration internationale, née de la Convention sur le brevet européen, signée le 5 octobre 1973 à Munich et entrée en vigueur le 7 octobre 1977 (168). Cette instance, qui n'est pas une institution de l'Union européenne, jouit d'une large indépendance sur le plan administratif. Elle s'autofinance entièrement par les taxes de procédure perçues par ses soins, ainsi que par un pourcentage des taxes annuelles acquittées au titre des brevets européens délivrés.
La création de l'OEB est le fruit de la coopération entre les États européens dans le domaine de la protection de la propriété industrielle. L'Organisation européenne des brevets, dont l'OEB est l'organe exécutif, compte actuellement vingt États membres, à savoir tous les États de l'Union européenne ainsi que le Liechtenstein, la Principauté de Monaco, la Suisse, Chypre et la Turquie. Il est, par ailleurs, actuellement possible d'étendre la protection conférée par les demandes et les brevets européens à plusieurs États d'Europe centrale et orientale.
L'OEB a pour mission de délivrer des brevets européens selon une procédure uniforme et centralisée. Pour obtenir une protection par brevet dans tous les États membres de l'Organisation européenne des brevets, il suffit aujourd'hui de déposer une demande dans l'une des trois langues officielles de l'Office, à savoir l'allemand, l'anglais et le français. En 1998, environ 113 400 demandes de brevet ont été déposées auprès de l'OEB.
Après leur délivrance, les brevets européens sont administrés par les États que le titulaire du brevet a désignés dans sa demande. La durée de validité des brevets européens est de vingt ans. Il est possible de proroger la protection conférée par les brevets portant sur des médicaments et des produits phytosanitaires.
L'OEB applique des critères stricts pour délivrer un brevet : l'invention doit être nouvelle, impliquer une réelle activité inventive et être susceptible d'application industrielle.
Selon les termes de l'OEB, le système du brevet européen offre une grande sécurité juridique. Les brevets européens ne sont, en principe, délivrés qu'après un examen approfondi et une recherche de nouveauté exhaustive dans une documentation comportant plus de 31 millions de documents. En effet, en contrepartie du droit d'exploitation exclusif de son invention, le demandeur en divulgue le contenu. Les demandes de brevet représentent donc les documents les plus actuels sur les innovations techniques. Selon l'Office, les technologies de pointe donnant lieu à une vive concurrence, la fonction informative des brevets revêt une importance sans cesse croissante. Grâce à des publications par voie informatique, opérant en étroite collaboration avec les offices nationaux, l'OEB permet aux utilisateurs d'accéder aux connaissances techniques contenues dans les brevets. Avec 31 millions de documents pouvant être consultés dans les langues officielles de tous les États membres de l'Organisation européenne des brevets, le site dédié à cette fonction est le service gratuit d'information sur les brevets le plus complet dans le monde.
La question de la délivrance des brevets renvoie à celle du territoire sur lequel la protection du brevet s'exerce. Lorsque l'Office européen des brevets accorde un brevet, la protection corollaire ne s'applique que dans les pays membres de la convention de Munich. Si l'inventeur souhaite obtenir le respect de ses droits dans d'autres États, il lui appartient d'engager des démarches similaires auprès des organes de chacun des pays, au premier rang desquels figurent, à l'évidence, les États-Unis.
En d'autres termes, un droit de propriété industrielle français ou européen ne saurait conférer une quelconque protection aux États-Unis. Un brevet n'est valable que sur le territoire pour lequel il a été attribué, que ce soit la France, l'Europe, les États-Unis ou le Japon, et chaque territoire requiert un dépôt spécifique. Par exemple, le brevet américain n'est valable que sur le territoire des États-Unis et la protection ne peut pas être étendue à l'ensemble de l'ALENA (Accord de Libre Échange Nord-Américain). Le brevet devra ainsi être déposé dans chacun des membres de l'ALENA - Canada, États-Unis, Mexique - séparément. Compte tenu du poids des États-Unis en ce domaine, il paraît utile de s'arrêter un instant sur la place des brevets dans ce pays.
3.- Les brevets aux États-Unis
a) L'United States Patent and Trademark Office :
une agence fédérale puissante et respectée
Les brevets sont au c_ur de la vision américaine de l'économie, puisque, comme on l'a souligné, la Constitution de 1787 le mentionne explicitement dans l'article 1er de sa section 8. La propriété intellectuelle est ainsi manifestement un élément de stratégie commerciale aux États-Unis, notamment dans un secteur à fort potentiel de développement comme l'industrie des biotechnologies. L'organe qui délivre les brevets, l'United States Patent and Trademark Office (USPTO), est fort logiquement considéré comme l'une des agences fédérales les plus importantes aux États-Unis.
Cet organe, qui est l'une des quatorze agences du Département du commerce, emploie 5 000 personnes en son sein. Son budget avoisine le milliard de dollars par an, les ressources provenant des droits perçus lors de la procédure d'examen des demandes de brevets. C'est une agence respectée pour son professionnalisme et sa rigueur déontologique. Elle ne compte pas moins de 300 personnes chargées plus spécialement de traiter les questions de biotechnologie, soit 10 % de l'effectif global des employés procédant à l'instruction des demandes. Parmi ces personnes, figurent 150 docteurs ou post doctorants en sciences ou en médecine dont la compétence est reconnue.
Le nombre de demandes de brevets enregistrées dans le domaine des biotechnologies augmente régulièrement de plus de 10 % par an depuis 1990, et l'USPTO a accordé 6 503 brevets de biotechnologie en 1997, dont 224 d'origine française. Pour l'année 1997, l'USPTO a examiné 17 003 nouvelles demandes, et les demandes en cours d'examen (pending applications) ont atteint le chiffre record de 20 249.
b) La règle du premier inventeur, une spécificité du droit des brevets américain
Le droit américain des brevets connaît une particularité qui le distingue nettement des droits applicables dans les autres pays. Le brevet y est délivré au premier inventeur (first to invent) et non au premier déposant (first to file). La priorité est donc donnée à l'acte d'invention et non à la démarche juridique qui consiste à faire reconnaître cette invention par un office des brevets. Cette approche spécifiquement américaine est protectrice des inventeurs mais elle leur impose le respect de procédures lourdes dans leur activité quotidienne. Ils doivent ainsi tenir des carnets de recherche au jour le jour pour être en mesure ultérieurement de prouver qu'ils ont inventé un procédé ou un produit à une date certaine. Cette spécificité s'impose à tous les scientifiques, américains ou non, qui souhaitent voir reconnaître leur invention aux États-Unis. Des négociations sont d'ailleurs envisagées dans le cadre de l'OMC pour convaincre les Américains de modifier leur approche originale. Les chances de voir aboutir une telle démarche ne semblent pas, cependant, évaluées de manière unanime par les interlocuteurs que votre Rapporteur a pu rencontrer lors de son déplacement à Washington.
Les États-Unis sont souvent apparus en pointe en matière de délivrance des brevets. Cette remarque prend tout son sens dans le domaine du vivant.
B.- LA PROBLÉMATIQUE ANCIENNE DE LA BREVETABILITÉ DU VIVANT
1.- Des précédents historiques et des initiatives américaines
dans les années 80
La brevetabilité du vivant suscite, au premier abord, interrogation, incrédulité, voire répulsion. Sans que cela soit forcément de nature à la justifier, cette pratique n'est pourtant pas inédite puisque Louis Pasteur obtint, dès 1873, le premier brevet sur une matière vivante, la levure de bière. Pourtant, pendant longtemps, la position des autorités chargées de délivrer les brevets fut de considérer que les produits de la nature ne pouvaient pas faire l'objet d'un brevet. Le raisonnement est simple et fondé. Si la matière est naturelle, la mettre en évidence constitue une découverte et non une invention. L'évolution des sciences et le développement des biotechnologies a bouleversé l'application de ce principe, la distinction traditionnelle en droit des brevets, entre découverte - non brevetable - et invention - brevetable - devenant particulièrement ténue.
En 1981, un tournant est pris puisque la Cour Suprême des États-Unis a considéré alors que, si les phénomènes naturels dans leur état naturel n'étaient pas brevetables, les organismes génétiquement modifiés, eux, l'étaient bien. Ainsi une souche bactérienne génétiquement modifiée capable de dégrader un hydrocarbure n'était pas considérée comme étant le fruit de la nature mais bien celui de l'ingéniosité et de la recherche humaines (affaire Diamond vs Chakrabarty).
Basé sur des considérations économiques, l'objectif de cette décision était de stimuler le processus d'innovation dans le secteur biologique, sans pénaliser les entreprises américaines. La prise en compte de valeurs, autres qu'économiques, dans un jugement portant sur un brevet ne relevait pas de la Cour Suprême, comme elle l'a clairement indiqué, mais de celui du Congrès. Ce brevet était le premier d'une longue série à l'origine de nombreuses avancées dans le domaine médical, de l'environnement et de l'agriculture.
En 1987, l'USPTO rendait brevetables « les organismes vivants multicellulaires non naturels et non humains, incluant les animaux » et, l'année suivante, était brevetée la première « oncosouris », c'est-à-dire une souris génétiquement modifiée pouvant développer des pathologies cancéreuses, et qui allait servir de modèle à l'étude de cette maladie chez l'homme.
2.- Les critères de brevetabilité du vivant
Les critères de brevetabilité du vivant sont ceux appliqués aux autres types d'inventions. Il s'agit du critère de nouveauté (novelty), celui d'utilité (utility), et enfin celui de non-évidence (non-obviousness). Ces critères utilisés aux États-Unis le sont également - à quelques nuances près - en Europe.
La loi américaine impose que l'invention soit nouvelle afin d'être brevetée. L'invention ne doit pas être déjà connue, soit qu'elle ait déjà fait l'objet d'un brevet, soit qu'elle ait été connue ou utilisée par d'autres personnes aux États-Unis, soit qu'elle ait été décrite dans une publication aux États-Unis ou à l'étranger. La loi subordonne aussi la brevetabilité à la non-évidence de l'invention, c'est-à-dire que celle-ci ne doit pas paraître évidente à l'homme de métier, c'est-à-dire, en biotechnologie, au chercheur.
Dans ce domaine, la condition de nouveauté n'appelle aucune remarque particulière par rapport aux autres inventions, le test utilisé par l'USPTO au moment de l'examen est celui de la « main de l'homme » (the man's hand test) : à partir du moment où l'invention suppose une intervention de l'homme et qu'elle n'est pas déjà connue, elle remplira la condition de nouveauté. C'est ici que se distinguent les découvertes des inventions proprement dites, les découvertes n'étant pas le résultat d'une intervention humaine.
La question de la non-évidence est, en revanche, plus délicate. Si l'invention faisait partie de l'art antérieur et si l'homme du métier de compétence normale avait pu la découvrir, selon le droit commun des brevets, l'invention ne satisfait pas la condition de non-évidence. L'USPTO a relevé que l'art antérieur dans le domaine de la biotechnologie évoluait si rapidement qu'il était difficile d'établir des critères objectifs pour juger de l'évidence d'une invention. En matière de génétique, il a abandonné, le 28 mars 1995, la règle, qu'il avait instaurée en 1994, selon laquelle un nouveau gène ne satisfait pas la condition de non-évidence lorsqu'une petite partie de sa séquence appartenait à l'art antérieur. Désormais, les examinateurs de l'USPTO déterminent le caractère évident d'une invention au cas par cas en se référant aux circonstances spécifiques de la demande.
Afin d'éviter la prolifération de brevets qui ne porteraient que sur de simples curiosités, ou qui auraient un objet illégal ou immoral, la loi américaine impose que l'invention présente un caractère utile.
La Cour suprême estime qu'une invention remplit cette condition d'utilité lorsque l'inventeur démontre que cette dernière possède une utilité substantielle ou pratique (substantial or practical utility) et qu'elle est opérationnelle (operative). L'imprécision de cette expression la rend difficilement applicable aux inventions issues des biotechnologies.
L'USPTO a été amené à exprimer, dans des directives (guidelines), sa volonté d'assurer que la condition d'utilité repose sur des critères qui ne freinent pas la recherche, le développement et la commercialisation des avancées technologiques dans le domaine de la biotechnologie. En revanche, la nature de l'utilité n'a pas été définie. On ignore toujours si celle-ci doit être thérapeutique, ou simplement commerciale. Pour l'USPTO, la demande de l'inventeur sera rejetée pour défaut d'utilité, si ce dernier ne fournit pas un argument crédible de l'utilité de son invention. La commission d'examen de l'USPTO considérera que la condition d'utilité est remplie à moins qu'un « homme du métier de compétence ordinaire » dans sa spécialité ne puisse douter de la crédibilité de cet argumentaire.
L'utilité est également appréciée par rapport au bénéfice immédiat que l'invention peut procurer au public. En présence d'inventions pharmaceutiques, on peut très bien imaginer qu'un gène ne possède aucune utilité au moment de sa découverte, et que dix ans plus tard, il se révèle d'une utilité révolutionnaire. L'inventeur n'ayant pas pu breveter son gène pour défaut de bénéfice immédiat ne bénéficiera d'aucune retombée que lui aurait conféré un brevet d'invention. L'USPTO a résolu cette contradiction en créant une présomption d'utilité. La personne qui affirme que son invention possède une utilité scientifiquement crédible, pourra bénéficier d'une telle présomption, même en l'absence de bénéfice immédiat stricto sensu.
Le droit français des brevets s'appuie sur des critères qui, sans être strictement équivalents, ressortissent à la même logique.
L'article L. 611-10 du code de la propriété intellectuelle dispose que les découvertes ne sont pas brevetables et que pour l'être une invention doit être nouvelle, impliquer une activité inventive et être susceptible d'application industrielle. L'article L. 611-11 prévoit, pour sa part, qu'une invention est considérée comme nouvelle si elle n'est pas comprise dans l'état de la technique, celui-ci étant constitué par tout ce qui est rendu accessible au public avant la date de dépôt de la demande de brevet par une description orale ou écrite, un usage ou tout autre moyen. Par ailleurs, l'invention doit présenter un caractère inventif. L'article L. 611-14 du code de la propriété intellectuelle exige qu'elle ne découle pas de manière évidente, pour un homme du métier, de l'état de la technique. Le critère de non-évidence est donc présent ici comme aux États-Unis. Enfin, une invention doit être susceptible d'application industrielle. L'article L. 611-15 du code indique qu'il est alors nécessaire que cette invention puisse être utilisée dans tout genre d'industrie, y compris l'agriculture.
L'ensemble de ces critères sont donc appliqués aux inventions relatives au vivant. Leur mise en _uvre pour les gènes humains soulève des difficultés à la fois juridiques, pratiques et surtout éthiques.
II.- LE DROIT DES BREVETS FACE À L'HUMAIN
A.- LA PROBLÉMATIQUE NOUVELLE DU GÉNOME HUMAIN : DE LA DÉMARCHE SCIENTIFIQUE À LA LOGIQUE ÉCONOMIQUE
1.- La logique de valorisation des découvertes par les entreprises américaines
Pour les dirigeants des principales entreprises américaines de ce secteur mais aussi pour bon nombre de responsables publics rencontrés par votre Rapporteur lors de son déplacement outre-Atlantique, la question des brevets est essentielle pour l'avenir de l'industrie des biotechnologies. Selon les termes du Docteur William Haseltine, président de Human Genome Science, sans brevets il n'y aurait pas de médicaments, les entreprises n'acceptant de prendre des risques financiers que dans la perspective de les voir aboutir à des brevets.
Du fait des coûts importants de la recherche dans le secteur des biosciences et de la quasi-absence de produit sur le marché pour le moment, une part importante de la valorisation des sociétés du secteur repose sur la propriété intellectuelle. En effet, pour mener à bien leurs recherches, les entreprises ont besoin de moyens considérables, notamment en matériels informatiques, la visite du siège de Celera Genomics permettant d'en juger. Pour obtenir les financements nécessaires, les sociétés de biotechnologies sont contraintes de valoriser leurs recherches. Le seul moyen à leur disposition est le dépôt de brevets qui rend tangible leur activité de recherche et laisse espérer aux investisseurs un retour financier à plus ou moins long terme. Ce n'est pas un hasard si, comme votre Rapporteur a pu l'observer, la salle de conférence de Human Genome Science, l'une des principales entreprises de biotechnologies, est emplie de cadres où apparaissent des fac-similés des brevets délivrés à la société. La possibilité pour les sociétés de biotechnologies de breveter leurs inventions est une condition indispensable de leur développement et d'une visibilité économique qui attire les investisseurs.
2.- Le dépôt massif de demandes de brevets
En 1991 et 1992, alors que le Programme Génome Humain (PGH) - le pôle public chargé du décryptage du génome - avait débuté en 1988, les NIH (National Institutes of health) déposaient successivement des dossiers de demande de brevet, d'abord sur 350 fragments de gènes, puis sur 2 375 fragments, et enfin dans une troisième demande sur 4 000 fragments. Ces demandes n'ont pas été retenues par l'USPTO qui les a jugées non conformes au critère de nouveauté et à celui d'utilité. Les NIH, qui n'ont pas fait appel de cette décision, ont alors déclaré que les séquences de gènes humains obtenues dans le cadre du PGH ne feraient désormais plus l'objet de demandes des brevets et resteraient à la disposition de la communauté scientifique.
La controverse sur la brevetabilité de fragments de gènes a resurgi en 1997 lorsque l'USPTO a accepté de breveter des EST (169) déposés par la société Incyte. L'USPTO reconnaissait ainsi qu'un EST pouvait être utile à condition que les applications potentielles qui en découlent soient suffisamment décrites. Cette nouvelle décision a fait craindre, d'une part un affaiblissement des critères d'éligibilité pour les nouveaux brevets sur des fragments de gènes, d'autre part la perspective de nombreux problèmes juridiques résultant de la portée des brevets sur le gène complet séquencé, grâce à l'outil que constitue l'EST.
Lors de son déplacement aux États-Unis, votre Rapporteur a rencontré les dirigeants des principales sociétés de biotechnologies, en particulier celles engagées dans le décryptage du génome humain. Les représentants de Celera Genomics lui ont fait part des objectifs qu'ils s'étaient fixés en termes de brevets. La procédure de demande de brevets coûte cher à une société. C'est pourquoi Celera Genomics semble s'orienter finalement vers l'exploitation d'un nombre plus limité de brevets en ciblant ses requêtes selon des critères scientifiques et économiques. Environ 300 gènes pourraient être concernés par la stratégie de Celera Genomics.
Ces informations apparaissent contradictoires avec celles délivrées par d'autres sources. Depuis 1990, le nombre de demandes de brevets liées au génome ne cesserait d'augmenter régulièrement. Fin 1999, l'USPTO avait accordé 2 330 brevets relatifs à des séquences de gène. Selon l'Human Genome Project Information, plus de 3 millions de demandes relatives au génome auraient été déposées, que ce soit sur les gènes, leurs fragments, des tests génétiques ou des protéines. Parmi ces demandes figurent des « patents applications » - c'est-à-dire des demandes classiques d'octroi d'un brevet - mais surtout des « provisional patents applications » qui sont des demandes de brevet provisoire au formalisme simplifié, sans revendication et au coût modique. Ce type de demande simplifiée vaut priorité sur une demande complète pourvu qu'une telle demande soit déposée dans un délai de douze mois.
Face à cette poussée du droit des brevets dans le champ de l'humain, l'Europe a tenté de fixer un cadre dont l'élaboration et, aujourd'hui, la mise en place sont sujettes à débat.
B.- LE DÉBAT EUROPÉEN AUTOUR DE LA DIRECTIVE 98/44/CE
1.- La genèse mouvementée de la directive
Le 16 mars 1989, le Parlement européen a adopté une résolution sur les problèmes éthiques et juridiques liés aux manipulations génétiques, dans laquelle il était fait référence au principe de dignité humaine, visant notamment le clonage et la création de chimères. C'est ce principe qui va fonder toute la démarche communautaire en la matière.
En 1993, le Groupe de conseillers pour l'éthique de la biotechnologie auprès de la Commission - précurseur du Groupe européen d'éthique - a rendu un avis sur cette question. Admettant la validité du dépôt de brevets sur des inventions portant sur la matière vivante, comme les médicaments dérivés du sang ou du plasma humains, le Groupe a alors mis l'accent sur le fait que soit reconnu, au plan communautaire, le principe selon lequel les parties et produits du corps humain ne devaient pas faire l'objet d'exploitation commerciale, notant que devaient être exclus de la brevetabilité les gènes et leurs séquences. Le Groupe a appelé alors également de ses v_ux la conclusion d'un accord international sur les conditions de brevetabilité des inventions résultant de programmes de recherche sur le génome humain.
Le 1er mars 1995, le Parlement européen a usé, pour la première fois, de son pouvoir de veto dans le cadre de la procédure de codécision, pour rejeter le projet de directive, initié par la Commission sur ce sujet. Le motif principal de ce rejet reposait sur l'ambiguïté d'une disposition de la directive sur les conditions de brevetabilité des gènes humains. On doit noter que ce texte reprenait mot pour mot la rédaction de l'article L. 611-17 du code de la propriété intellectuelle français, introduit par la loi du 29 juillet 1994 : « Le corps humain, ses éléments et ses produits ainsi que la structure totale ou partielle d'un gène humain ne peuvent, en tant que tels, faire l'objet de brevets ». Les termes « en tant que tels » ont été jugés peu précis et donc peu protecteurs par les parlementaires européens.
Le Groupe de conseillers pour l'éthique de la biotechnologie a alors été saisi par la Commission. Dans son avis du 25 septembre 1996, sur le projet de directive relative à la protection juridique des inventions biotechnologiques, ce Groupe a fixé les principes auxquels on ne peut qu'adhérer : la non-commercialisation du corps humain, le libre consentement des personnes sur lesquelles les prélèvements sont faits, la subordination des impératifs économiques aux exigences éthiques.
Il y est également affirmé la nécessité de distinguer la découverte - non brevetable - de l'invention, sachant, que pour qu'un brevet soit délivré pour une invention réalisée à partir de la connaissance d'un gène ou d'une séquence génétique, il est indispensable d'en identifier la fonction et d'en décrire l'application avec précision.
A cette occasion, l'un des membres du Groupe - le Professeur Dietmar Mieth - a adopté l'avis en souhaitant y apporter une nuance : il a estimé que le brevet pouvait couvrir l'application technique issue de la connaissance du gène mais, en aucun cas, le gène lui-même. Cette opinion, qu'on ne peut pas vraiment qualifier de dissidente, met en lumière la principale problématique de ce débat.
La complexité du droit des brevets appliqué aux biotechnologies suppose une grande précision des termes. La directive 98/44/CE, fruit d'un compromis entre les États membres et les institutions communautaires, n'a sans doute pas su répondre totalement à cette exigence, ce qui explique le débat actuel sur sa transposition.
2.- Les ambiguïtés de la rédaction
L'analyse de la directive 98/44/CE nécessite de mettre en parallèle les articles de ce texte mais aussi les considérants qui ouvrent cette directive. C'est l'article 5 de la directive qui fait l'objet d'une contestation. Il convient d'en rappeler les termes.
L'article 5 de la directive est le suivant :
« Article 5. - 1. Le corps humain, aux différents stades de sa constitution et de son développement, ainsi que la simple découverte d'un de ses éléments, y compris la séquence ou la séquence partielle d'un gène, ne peuvent constituer des inventions brevetables.
« 2. Un élément isolé du corps humain ou autrement produit par un procédé technique, y compris la séquence ou la séquence partielle d'un gène, peut constituer une invention brevetable, même si la structure de cet élément est identique à celle d'un élément naturel.
« 3. L'application industrielle d'une séquence ou d'une séquence partielle d'un gène doit être concrètement exposée dans la demande de brevet. »
Cet article dispose donc, dans son premier alinéa, que la simple découverte de la séquence ou de la séquence partielle d'un gène ne peut constituer une invention brevetable. Mais dans son deuxième alinéa, l'article 5 prévoit que de telles séquences isolées ou autrement produites par un procédé technique peuvent être brevetées, même si leur structure est identique à celle d'un élément nature. On ne peut pas dire que cette rédaction souffre d'une grande clarté.
Il est vrai que ses alinéas 1 et 2 semblent contradictoires. Le dernier alinéa de l'article 5 peut permettre de résoudre cette difficulté. Car, en fait, il convient de lire les alinéas 2 et 3 de l'article 5 ensemble, comme imposant une condition cumulative aux demandeurs de brevets : le gène ou la séquence de gène ne peut être brevetée (art. 5.2) que si est décrite son application industrielle concrètement exposée dans la demande de brevet (art. 5.3). Les considérants n° 22 et 23 de la directive confirment cette analyse en exigeant que la fonction et l'application industrielle soient décrites, conformément au droit classique des brevets.
La directive 98/44/CE s'articule donc autour de trois axes principaux : le principe de non-brevetabilité du gène, la question de la fonction de ce gène et le respect de l'ordre public et des bonnes m_urs.
Concernant la non-brevetabilité du gène, le considérant n° 16 de la directive prévoit que : « Le droit des brevets doit s'exercer dans le respect des principes fondamentaux garantissant la dignité et l'intégrité de l'Homme ; qu'il importe de réaffirmer le principe selon lequel le corps humain, dans toutes les phases de sa constitution et de son développement, cellules germinales comprises, ainsi que la simple découverte d'un de ses éléments ou d'un de ses produits, y compris la séquence ou séquence partielle d'un gène humain, ne sont pas brevetables ; que ces principes sont conformes aux critères de brevetabilité prévus par le droit des brevets, critères selon lesquels une simple découverte ne peut faire l'objet d'un brevet ». Ce considérant doit se lire en complément du premier alinéa de l'article 5. Le principe de non-brevetabilité du gène semble donc solidement établi par la directive.
Ce texte aborde aussi la question de la fonction et celle de l'application industrielle du gène. Dans son considérant n° 23, il indique que : « une simple séquence d'ADN sans indication d'une fonction ne contient aucun enseignement technique ; qu'elle ne saurait, par conséquent, constituer une invention brevetable ». Ce texte doit être mis en rapport avec le considérant précédent : « Considérant que le débat sur la brevetabilité de séquences ou de séquences partielles de gènes donne lieu à des controverses, que, aux termes de la présente directive, l'octroi d'un brevet à des inventions portant sur de telles séquences ou séquences partielles doit être soumis aux mêmes critères de brevetabilité que pour tous les autres domaines technologiques, nouveauté, activité inventive et application industrielle ; que l'application industrielle d'une séquence ou d'une séquence partielle doit être exposée de façon concrète dans la demande de brevet telle que déposée ». Le considérant n° 24 précise ces termes : « Considérant que, pour que le critère d'application industrielle soit respecté, il est nécessaire, dans le cas où une séquence ou une séquence partielle d'un gène est utilisée pour la production d'une protéine ou d'une protéine partielle, de préciser quelle protéine ou protéine partielle est produite ou quelle fonction elle assure. » Ces considérants annoncent le dernier alinéa de l'article 5.
Parallèlement, l'article 6 de la directive interdit aussi certains brevets portant atteinte à l'ordre public et aux bonnes m_urs.
« Article 6. - 1. Les inventions dont l'exploitation commerciale serait contraire à l'ordre public ou aux bonnes m_urs sont exclues de la brevetabilité, l'exploitation ne pouvant être considérée comme telle du seul fait qu'elle est interdite par une disposition légale ou réglementaire. »
« 2. Au titre du paragraphe 1 ne sont notamment pas brevetables :
« a) les procédés de clonage des êtres humains ;
« b) les procédés de modification de l'identité génétique germinale de l'être humain ;
« c) les utilisations d'embryons humains à des fins industrielles ou commerciales ;
« d) les procédés de modification de l'identité génétique des animaux de nature à provoquer chez eux des souffrances sans utilité médicale substantielle pour l'homme ou l'animal, ainsi que les animaux issus de tels procédés ».
Les termes de la directive étant rappelés, on observe qu'elle comporte des ambiguïtés de rédaction relevées par plusieurs observateurs. Les notions de découverte et d'invention, la distinction entre les gènes, leurs fonctions, leurs applications industrielles, le procédé ayant permis d'isoler le gène n'apparaissent pas clairement dans la directive de 1998, entraînant ainsi une réelle confusion et des inquiétudes légitimes.
À quoi sert-il d'affirmer qu'on ne peut breveter le gène alors qu'au total, l'application et la fonction étant décrites, le gène ou sa séquence seront bel et bien inclus dans le brevet ? Par ailleurs, une fois ce type de brevet acquis, l'inventeur initial a-t-il le droit de percevoir des redevances sur toute invention née de la mise en évidence d'une nouvelle fonction et application du gène concerné ? La directive ne le dit pas.
Sans doute la pratique de l'Office européen des brevets qui applique la directive permettra-t-elle d'acquérir une meilleure lisibilité de ce dispositif. Mais peut-on attendre cette mise en _uvre pour juger de la pertinence de la directive ? Cette question semble d'autant moins appeler une réponse affirmative que la pratique récente de l'OEB est apparue contestable.
3.- La pratique contestable de l'Office européen des brevets
Le 8 décembre 1994, l'OEB a autorisé un brevet sur le gène humain codant la relaxine, hormone produite par l'utérus et le placenta, pendant la grossesse, et qui permet d'atténuer les contractions. La protéine et la séquence de gène correspondante ont donc fait l'objet d'un brevet, bien qu'existant dans la nature, parce que ces substances ne sont pas accessibles au public à l'état pur et isolé. Il s'agissait là d'une première application anticipée des règles qui, ensuite, ont été consignées dans la directive 98/44/CE.
Plus récemment, en décembre 1999, l'OEB a été sévèrement mis en cause après avoir délivré un brevet à une université écossaise, affiliée à un groupe industriel australien, sur un procédé s'avérant couvrir le prélèvement de cellules embryonnaires humaines, la manipulation génétique de ces cellules et la fabrication d'embryons génétiquement modifiés, en contravention avec la plupart des lois des États européens. Les réactions, en particulier allemandes, ont conduit l'OEB à s'expliquer et reconnaître qu'il s'agissait là d'une faute grave. Seule l'opposition d'un tiers permet d'obtenir l'annulation d'un brevet délivré. Il semble que Greenpeace, qui a révélé l'affaire, se soit engagé dans cette procédure.
Ce dérapage conduit à s'interroger sur la qualité du fonctionnement de cette institution. Elle apparaît aborder le problème de la brevetabilité du vivant et de l'humain sous un angle purement technique sans autre considération. Il s'agit là d'un état d'esprit qui ne peut nous satisfaire. On peut le regretter d'autant plus qu'il semble d'ailleurs qu'un bon nombre des questions posées sur ces sujets auraient déjà pu trouver une réponse dans une pratique plus stricte de la part de l'OEB.
En 2000, l'OEB a inséré dans son règlement les dispositions de la directive européenne 98/44/CE sur la protection des inventions biotechnologiques. D'ores et déjà, les critères contenus dans ce texte s'appliquent donc aux brevets européens ce qui amène également à s'interroger sur les effets d'une transposition ou d'une absence de transposition au plan national. C'est autour de cette question que le débat s'est engagé en France.
a) L'appel contre la brevetabilité des gènes humains
En avril 2000, nos collègues, MM. Jean-François Mattei et Wolfgang Wodarg, député au Bundestag, ont lancé sur Internet un « Appel contre la brevetabilité des gènes humains ». Ce texte, qui semble avoir récolté quelques milliers de signatures, est le suivant :
« Je souhaite par ce message attirer l'attention de la communauté internationale et plus particulièrement de l'Union Européenne sur la question de la brevetabilité des gènes humains.
« Considérant que le génome humain est un patrimoine commun de l'Humanité, je refuse l'appropriation des séquences géniques qu'induit la logique des brevets.
« Je m'oppose donc à la transposition en l'état de la directive européenne 98/44/CE du 6 juillet 1998 et demande un moratoire immédiat permettant sa renégociation ainsi que la suspension de toute attribution de brevets sur le génome.
« Le corps humain, y compris ses gènes, n'est pas une marchandise. La gravité de cette question nécessite un débat public et transparent impliquant les citoyens. Il est urgent que chaque État l'organise avant d'arrêter une décision lourde de conséquences. Il en va de l'avenir de l'Homme. »
Les principes énoncés par ce texte ne peuvent que recueillir une adhésion de la plupart, mais cette pétition ne rend pas tout à fait compte de la complexité du débat et de la directive. Cette approche cursive, sans doute liée à la forme d'une telle initiative, doit être complétée par un débat parlementaire, le plus à même de traiter publiquement cette question de manière nuancée et précise. L'avis rendu par le Comité consultatif national d'éthique ne fait que confirmer cette nécessité.
b) La position du Comité consultatif national d'éthique
En rendant son avis le 8 juin dernier sur la directive relative à la protection juridique des inventions biotechnologiques, le Comité consultatif national d'éthique (CCNE), saisi par le secrétaire d'État à l'industrie, a, en quelque sorte, institutionnalisé les critiques émises par MM. Jean-François Mattei et Axel Kahn vis-à-vis de cette directive. En appelant à une renégociation de celle-ci, l'avis du Comité consultatif national d'éthique a connu un certain écho dans la presse. Il importe de s'interroger sur la portée de cet avis et les présupposés sur lesquels il se fonde.
Le Comité consultatif national d'éthique a rappelé les principes auxquels il convient de se conformer d'un point de vue éthique.
Il a, tout d'abord, énoncé le principe de non-commercialisation du corps humain. Ce principe est affirmé dans les articles 16-1 et 16-5 du code civil et reconnu par le Conseil constitutionnel. Le Comité consultatif national d'éthique indique qu'il est le premier principe qui doit prévaloir lorsque l'on s'interroge sur la brevetabilité du génome humain. On peut néanmoins se demander si ce principe peut s'appliquer directement au génome. Comme le reconnaît le Comité consultatif national d'éthique, « avec le gène, nous sommes au niveau moléculaire, où qualifier d'humaine la réalité n'a guère de sens ».
Le Comité consultatif national d'éthique n'en conclut pourtant pas moins que, par extension, ce principe doit s'appliquer à la brevetabilité du génome dans une démonstration qui peut ne pas emporter l'adhésion. Une séquence du génome humain - c'est-à-dire une simple séquence moléculaire - doit-elle être assimilée à l'homme ?
Plus convaincant est l'argument développé par le Comité d'éthique, selon lequel, si l'on cédait sur ce sujet, un seuil serait alors franchi et le maintien du corps humain hors commerce paraîtrait beaucoup plus difficile à justifier. Ce risque n'est pas négligeable ; il convient d'en être conscient et de s'en prémunir.
Le Comité consultatif national d'éthique fait également référence au principe du libre accès à la connaissance du génome. Il s'appuie notamment sur la notion de patrimoine commun de l'humanité mise en _uvre par l'UNESCO en 1997. Sur ce point, le Comité d'éthique fait écho à la déclaration commune de Bill Clinton et Tony Blair en mars 2000.
Mais le droit des brevets s'oppose-t-il vraiment à ce libre accès à la connaissance ? N'est-il pas plutôt de nature à limiter la rétention de l'information dans la mesure où, octroyant une protection juridique à l'invention, il en permet l'exploitation au grand jour ? L'absence de brevet ne conduirait-elle pas, à l'inverse, les entreprises à maintenir le secret sur leurs découvertes ?
Un risque plus indirect semble apparaître. Si, comme l'a revendiqué Craig Venter, de la société Celera Genomics, on acceptait de breveter en masse des séquences du génome humain associées à de vastes domaines d'application mal définis, il est clair qu'un certain nombre d'autres chercheurs hésiteraient à poursuivre leurs études dans ces différents domaines puisqu'ils n'auraient pas la possibilité d'exploiter leurs découvertes, qui entreraient dans le champ très large de brevets déposés auparavant.
Le Comité consultatif national d'éthique s'inquiète aussi des conséquences économiques de la brevetabilité du génome humain qui pourraient conduire des sociétés, en très grande majorité américaines, à disposer d'un monopole sur l'exploitation de l'essentiel du génome humain.
L'avis du Comité consultatif national d'éthique présente un intérêt essentiel. Il met en évidence les limites du droit des brevets pour appréhender les découvertes en matière de biotechnologie. Ces limites touchent évidemment la difficile distinction entre l'invention et la découverte. Toutefois, le Comité consultatif national d'éthique souhaite que l'on ne fasse pas le procès du droit des brevets. Celui-ci est en mesure d'adapter ses procédures et de délivrer des brevets avec les contrôles nécessaires. Mais pour que ces vérifications s'opèrent dans des conditions acceptables, il est essentiel que l'on introduise des principes éthiques en la matière. Les offices qui délivrent les brevets doivent pouvoir juger du caractère moral ou non de l'invention présentée. Ce n'est pas le moindre mérite de l'avis du Comité consultatif national d'éthique que d'inciter à introduire réellement dans ce droit de tels principes éthiques.
En conclusion, le Comité consultatif national d'éthique a souhaité que la notion de découverte soit entendue le plus largement possible en matière de gène, afin de maintenir l'accès à la connaissance. Or l'article 5 de la directive manque de précision et son ambiguïté peut engendrer, selon le Comité consultatif national d'éthique, toutes les dérives. Il a donc préconisé l'absence de transposition de la directive de 1998 et une nouvelle discussion de ce texte au plan européen.
c) La position du ministre de la recherche
M. Roger-Gérard Schwartzenberg, ministre de la recherche, a exprimé la position française sur la question de la brevetabilité du génome, lors de la réunion du G8 « Recherche » à Bordeaux, en 2000.
Le ministre de la recherche a, tout d'abord, insisté sur l'attachement du Gouvernement à trois principes :
- le principe de la non-commercialisation du corps humain ;
- le principe du libre accès à la connaissance du gène ;
- le principe du partage de cette connaissance.
De ces principes on peut, en premier lieu, déduire la nécessité de maintenir l'accès de tous aux données brutes du séquençage. Le génome humain est un patrimoine commun de l'humanité et les prétentions de certains de breveter les données brutes issues du séquençage sont irrecevables du point de vue éthique. Il importe qu'elles le soient aussi sur le plan juridique.
Le ministre a également déclaré très clairement que la simple découverte ne saurait être brevetable. C'est d'ailleurs ce que rappelle la directive européenne de 1998 : une simple découverte ne peut faire l'objet d'un brevet (considérant n° 16). S'opère ici la distinction classique, en droit des brevets, entre la découverte et l'invention véritable.
Mais au-delà des données brutes issues du séquençage, se pose la question de l'identification de la fonction d'un gène et de son caractère ou non brevetable. L'identification, la caractérisation de la fonction d'un gène doivent-elles être brevetables ? Comme l'a noté le ministre de la recherche, beaucoup de chercheurs répondent oui, à condition que cette fonction ait été déterminée, par une méthode expérimentale. D'autres chercheurs estiment cependant que l'identification de la fonction d'un gène n'est pas à elle seule brevetable et qu'elle doit s'accompagner, en outre, de la détermination des applications potentielles à des fins diagnostiques ou thérapeutiques. C'est la question de l'applicabilité. Il faudrait donc mettre au point des tests diagnostiques ou des outils thérapeutiques - par exemple un gène utilisé comme médicament - définir et mettre au point les effets diagnostiques ou les effets thérapeutiques de ce gène pour pouvoir déposer un brevet.
Cela dit, certains chercheurs font observer qu'entre la phase 2
- identification de la fonction d'un gène - et la phase 3 - mise au point d'un médicament par exemple - la distance intellectuelle est réduite.
Il y a donc débat entre les scientifiques, sur le point de savoir si la caractérisation de la fonction d'un gène, démontrée expérimentalement, doit permettre le dépôt d'un brevet ou si elle doit s'accompagner, en outre, de la mise au point de ses applications diagnostiques ou thérapeutiques.
On l'a vu, la directive de 1998 n'apporte pas, sur ce point, une réponse totalement satisfaisante.
Pour le Gouvernement, qui s'est exprimé par la voie de son ministre de la recherche, le dépôt de brevets sur de véritables inventions biotechnologiques à partir de gènes, sur l'identification de la fonction d'un gène débouchant sur la mise au point de nouveaux outils diagnostiques ou thérapeutiques, ne semble pas contredire les principes éthiques de non-commercialisation du corps humain, de libre accès à la connaissance et de partage de cette connaissance.
Pour autant la rédaction de la directive n'est pas satisfaisante. Observant que le débat sur ces questions était opaque et que la frontière entre séquençage génétique et thérapie n'était pas évidente, le Conseil d'État a relevé la nécessité de lever ces ambiguïtés dans son rapport d'évaluation des lois de 1994. Il a proposé que la transposition de la directive subordonne explicitement la brevetabilité d'une séquence d'un gène à la précision de sa fonction au sens de l'application trouvée, par exemple la production d'une protéine ayant une portée thérapeutique. Il a suggéré qu'en substance les articles du code de la propriété intellectuelle concernés soient modifiés en ce sens.
Si cet éclaircissement s'impose, il importe qu'il se produise de manière coordonnée entre les États. Quel sens auraient, en effet, des droits nationaux qui interpréteraient dans des sens divergents la directive ?
La position allemande sur cette directive revêt pour la France une importance considérable, quand on connaît les exigences éthiques de notre partenaire sur ces sujets. De plus, une position commune franco-allemande ne manquerait pas de poids en Europe, alors que la question demeure en débat.
Une délégation de la Mission s'est rendue à Berlin le 25 janvier 2001, pour tenir une réunion avec les députés allemands, membres de la commission d'étude du Bundestag « droit et éthique de la médecine moderne ». Cette rencontre a montré à quel point le débat était loin d'être clos dans ce pays. Si on laisse de côté les effets de perspective liés au fait que les députés allemands rencontrés sont les plus actifs sur ce dossier et ne représentent pas forcément la moyenne des opinions de leurs collègues, il apparaît cependant que la question des brevets et des biotechnologies fait l'objet d'une attention particulière en Allemagne, dans l'opinion comme dans les milieux institutionnels.
a) Le dépôt d'un projet de loi par le Gouvernement fédéral
Après un long processus d'examen, le gouvernement allemand a présenté le 18 octobre 2000 un projet de loi sur la brevetabilité des inventions biotechnologiques visant à transposer la directive européenne 98/44/CE. Ce projet est le fruit d'un compromis entre les ministères fédéraux de la justice, de la santé et de la recherche.
Selon le ministre fédéral de la justice, le projet de loi vise à créer des conditions économiques favorables aux innovations dans le domaine des biotechnologies et à protéger la propriété intellectuelle des inventions en relation avec les gènes et les autres matières biologiques, tout en posant les frontières éthiques nécessaires.
Ainsi, aux termes de ce texte, la découverte d'un gène ou le fait d'isoler une séquence génétique ne seront pas brevetables en tant que tels. Une séquence génétique humaine ne pourra être brevetée - parce qu'elle sera alors considérée comme une invention - que dans l'hypothèse où ses propriétés et une fonction particulière, comme une application médicale, seront clairement définies. D'autre part, se référant à la loi allemande sur la protection de l'embryon et reprenant les dispositions de l'article 6 de la directive, le projet interdit de breveter certaines applications : les procédés de clonage de l'être humain, les procédés de modification de l'identité génétique des lignées germinales humaines ou l'utilisation d'embryons humains à des fins commerciales ou industrielles.
Par ailleurs - ce point est important - le projet prévoit que, si une nouvelle fonction d'une séquence génétique déjà brevetée est découverte, aucune retombée financière ne sera exigible par le premier détenteur du brevet.
Lors d'un entretien au magazine Die Woche le 21 décembre 2000, le chancelier Schroeder a insisté sur le fait que les coûts de développement et de recherche pour les entreprises de biotechnologie étaient importants, la rentabilité de ces sociétés dépendant de la protection de leurs inventions. Mais il a également mis en balance avec cet impératif la nécessité de lier l'octroi des brevets au respect de règles éthiques claires. Pour lui, la simple découverte d'un gène ne doit ainsi pas être brevetable tandis que la description précise de la fonction d'un gène qui mène à un nouveau produit ou un nouveau procédé pourra l'être.
Conformément à la procédure législative allemande, le Bundesrat a été saisi du projet de loi, émettant à cette occasion de très nettes réserves. Après avoir réaffirmé la nécessité de protéger les inventions dans un cadre européen, la chambre haute du Parlement a constaté, en janvier 2001, que de nombreuses questions demeuraient en suspens, notamment au regard des principes éthiques. Le Bundesrat a demandé en conséquence au gouvernement fédéral d'initier un processus de modification de la directive 98/44/CE en vue de l'améliorer et de la préciser.
La chambre haute a aussi insisté sur l'importance de préserver les droits des inventeurs lorsque ceux-ci mettent en lumière, à partir d'une même matière naturelle, une fonction nouvelle de celle qui a déjà fait l'objet d'un brevet. Elle a également mis en avant la nécessité de ne breveter que le trio « substance, fonction, application ». L'accent est mis aussi sur le consentement des personnes concernées par le brevet, faute de quoi celui-ci ne pourrait être délivré. Enfin, un suivi de l'application de la directive est instamment demandé au Gouvernement fédéral.
La chambre haute du Parlement a appelé le gouvernement fédéral à faire pression au plan européen pour que soit engagée une nouvelle négociation de la directive, estimant que celle-ci soulevait des problèmes éthiques non résolus.
c) La position de la commission « droit et éthique de la médecine moderne »
au Bundestag
La commission d'enquête parlementaire « droit et éthique de la médecine moderne », créée au mois de juin 2000, s'est également prononcée contre le projet de loi gouvernemental au cours du mois de décembre dernier. Elle a rejeté la possibilité d'obtenir des brevets sur les gènes ou les séquences génétiques, exprimant la crainte de voir ces brevets ouvrir la voie à une utilisation commerciale du patrimoine génétique humain.
Cette commission estime qu'un cadre juridique destiné à l'usage du brevet biologique se doit de remplir les conditions minimales suivantes :
« 1. Les séquences d'ADN ne peuvent être brevetables. Elles ne constituent pas une invention mais une découverte, et ne répondent pas aux critères attachés au brevet de produits.
« 2. Les cellules, tissus et organes ne sont pas brevetables, même s'ils sont porteurs de modifications biotechniques. Seuls sont brevetables les procédés de fabrication de ces cellules, tissus et organes.
« 3. Le travail d'invention doit faire l'objet d'une rémunération appropriée. Il convient donc de limiter celle-ci aux applications pratiques. Le dépôt stratégique de brevets doit être exclu de manière efficace.
« 4. Pour ce qui concerne le risque de voir les brevets limiter les activités de recherche, la commission se félicite de la place accordée par la législation allemande, après jugement rendu par la Cour constitutionnelle, à la liberté de la recherche. Il convient de le souligner dans le cadre de la transposition de la directive en droit national. Le cas échéant, l'octroi de licences obligatoires doit être facilité.
« 5. La brevetabilité d'inventions technologiques doit trouver ses limites dans l'ordre public. Ces limites doivent être concrétisées grâce notamment au maintien de la législation en vigueur. »
Lors de la rencontre avec les membres de cette commission le 25 janvier dernier, plusieurs hypothèses ont alors été évoquées, sans que l'une se dégage nettement :
- La première était celle d'une transposition complète et sans condition de la directive.
- La deuxième était celle d'une transposition moyennant un engagement du Gouvernement fédéral de mener de nouvelles négociations européennes sur cette question. C'est la position exprimée majoritairement par les Länder au Bundesrat.
- La troisième était celle d'une transposition modifiant en partie le contenu de la directive, ou tout du moins, lui conférant une interprétation plus nuancée et plus respectueuse des principes éthiques, avec le risque qu'une telle transposition ne soit pas acceptée in fine par la Commission européenne et que l'Allemagne soit déférée devant la Cour de justice des communautés européennes.
- La dernière hypothèse, peu probable, est celle d'un refus de transposer pur et simple et d'une demande de renégociation.
Malgré les critiques nombreuses, émanant pour l'essentiel de membres de la coalition au pouvoir, le gouvernement fédéral devrait inscrire ce projet de loi à l'ordre du jour du Bundestag pour une première lecture avant l'interruption estivale de la session parlementaire. Dans cette perspective, un groupe de travail commun aux sociaux-démocrates et aux élus verts a été mis en place en vue de rechercher un compromis sur la transposition de la directive européenne 98/44/CE.
Parallèlement, le gouvernement fédéral a annoncé qu'il saisirait la Commission européenne des problèmes éthiques posés par cette directive. Pour autant, la position officielle de l'Allemagne demeure la suivante : une transposition préalable de la directive puis une renégociation au plan européen.
Le débat demeure donc ouvert en Allemagne. Les différentes forces politiques, notamment au sein de la majorité gouvernementale, présentent des analyses qui ne sont pas toutes convergentes. Ce débat existe également aux États-Unis où il revêt plus une dimension économique et juridique qu'éthique, même si cet aspect n'est pas évacué. Les nombreuses demandes de brevets sur le génome humain ont imposé aux autorités américaines des prises de position dont l'importance n'a pas besoin d'être relevée, les États-Unis ayant une avance considérable en ce domaine, et la question des brevets ne pouvant se résoudre dans le seul cadre national ou même européen.
1.- La nature spécifique de ce débat
On aurait tort de penser que la question de la brevetabilité du génome humain ne fait l'objet d'aucun débat aux États-Unis. Votre Rapporteur a pu observer qu'existaient des voix divergentes dans un concert, il est vrai, plutôt favorable à la délivrance de brevets dans ce domaine.
Se faisant le porte-parole d'un mouvement au sein duquel apparaissent, à titre principal, des organisations non gouvernementales, l'essayiste Jeremy Rifkin, qui préside The Foundation on Economic Trends, est l'un des plus virulents opposants à l'octroi de brevets sur les séquences génétiques. Lors d'une rencontre avec cet acteur reconnu du débat public aux États-Unis, votre Rapporteur a pu mesurer sa vision très critique de l'action de l'USPTO. M. Jeremy Rifkin considère que cet organisme a failli à sa mission, en ouvrant notamment la voie à la constitution de monopoles. Il estime également que la directive 98/44/CE est un recul par rapport à la position traditionnelle de l'Europe sur ces questions, souvent plus sensible au respect de certains principes contre une logique totalement économique.
La conseillère du Président Clinton sur ces questions, Mme Rachel Levinson, que votre Rapporteur a rencontrée à Washington, a estimé, à l'inverse de M. Jeremy Rifkin, que la question des brevets sur le génome humain était plutôt bien accueillie aux États-Unis. Elle a considéré que ce sujet ne devait pas soulever de polémiques, les difficultés qui apparaîtraient pouvant être résolues par les juridictions, le cas échéant, et surtout par la négociation de licences entre les entreprises. Elle a simplement jugé que le principal, en ce domaine, était d'éviter la constitution de monopoles ou de positions dominantes.
Cette vision clémente à l'égard de l'USPTO est partagée par Me Jeffrey Kushan ainsi que par le Docteur Ari Patrinos, l'un des promoteurs du programme public de décryptage du génome humain. Ces deux spécialistes de ces questions ont fait part à votre Rapporteur de leur jugement favorable sur les critères exigés par l'office des brevets américains, considérant qu'en l'espèce, on ne brevetait pas vraiment l'humain mais uniquement une information explicitant une fonction. Cette approche a été présentée comme raisonnable et fonctionnelle par le Docteur Ari Patrinos.
Il apparaît qu'aujourd'hui le débat sur la brevetabilité du génome existe aux États-Unis. Mais il se tient selon des modalités propres à ce pays. Il a principalement lieu en dehors des sphères institutionnelles classiques comme le Parlement, les Républicains et les Démocrates ne présentant pas de divergences de vues réelles sur ce sujet. La place reconnue des brevets dans la société et même la culture américaine rendent difficile une critique institutionnelle des mécanismes juridiques de reconnaissance de la propriété intellectuelle. La discussion ne peut se dérouler qu'en dehors de ces instances traditionnelles. C'est au sein de la société civile que la critique se déploie. Quand on connaît le poids de cette société civile en Amérique, on mesure que ce débat n'est nullement marginal. L'enjeu est de savoir s'il prendra une proportion telle que les politiques aient à s'en emparer.
2.- La pratique juridique plus stricte de l'USPTO
Face à ce débat, l'USPTO a cherché à apaiser les critiques en modifiant ses critères d'octroi d'un brevet. Le nouveau dispositif d'ordre général a une portée particulière en matière de gènes humains.
Pour l'USPTO, il est évident qu'une séquence génétique brute ne peut répondre aux critères d'obtention d'un brevet, alors qu'une séquence ou partie de séquence intégrant des éléments de régulation, d'expression, de variabilité en liaison avec une pathologie, sont autant d'éléments plus à même de justifier la défense des critères de brevetabilité.
L'aboutissement ou l'échec d'un grand nombre de demandes de brevets portant sur 20 000 gènes ou portions de gènes est attendu dans les prochains mois. Nombre de ces dossiers ne sont constitués que de séquences brutes décrivant des similitudes entre des familles de gènes d'une même espèce ou d'espèces différentes, ainsi que de spéculations sur des fonctions possibles. La position de l'USPTO sur ces demandes est donc attendue.
Récemment, les conséquences de l'octroi d'un brevet pour un gène, dont la fonction était insuffisamment définie, ont été illustrées par la polémique qui a surgi après que la recherche eut montré le rôle essentiel d'une protéine dans l'infection par le VIH. Or, le gène codant pour cette protéine avait déjà fait l'objet d'une demande de brevet en 1995, accordée en février 2000 à la société Human Genome Science, sans comporter de définition précise des fonctions possibles des protéines codées par ce gène.
Récemment l'USPTO a donc entrepris de préciser sa politique en matière de brevets par la publication en décembre 1999 d'une série de cas pratiques illustrant un projet de nouvelles recommandations ciblées sur le critère d'utilité, celle-ci devant être crédible, spécifique, substantielle, et bien établie.
Afin de remédier à ce type de situation, l'USPTO a donc clarifié - à la demande de la Maison Blanche et du Congrès - sa politique en matière de brevet. Au cours de l'élaboration de ces directives, qui ont fait l'objet d'auditions et de commentaires publics, trois types principaux de remarques ont été formulés :
- Les gènes ne peuvent pas être brevetables au motif qu'il s'agirait de simples découvertes et non d'inventions. L'USPTO n'octroiera pas de brevet pour un gène en tant que tel, tel qu'il apparaît dans son état naturel, mais uniquement pour la séquence isolée et purifiée d'un gène présentant une utilité spécifique, substantielle et crédible. De même, le fait que la structure de cet élément isolé puisse être identique à celle d'un élément naturel importe peu dès lors qu'un tel élément isolé est le résultat de procédés techniques l'ayant identifié, purifié, multiplié et caractérisé en dehors du corps humain, techniques que seul l'être humain est capable de mettre en _uvre et que la nature est incapable d'accomplir par elle-même.
- Les gènes font partie du patrimoine commun de l'humanité et en conséquence, ils ne pourront faire l'objet d'un droit de propriété privatif tel que celui conféré par un brevet. Selon l'USPTO, le brevet ne conférait pas un monopole sur les informations génétiques contenues dans un gène. En effet, le brevet implique que l'inventeur révèle au public le contenu de son invention en échange du droit temporaire d'empêcher les autres de fabriquer, d'utiliser, d'offrir à la vente, de vendre et d'importer l'objet de l'invention. Par conséquent, l'accès à l'information génétique ne serait pas bloqué et le seul élément protégé est un élément isolé et purifié du corps humain présentant une utilité spécifique, substantielle et crédible.
- Les gènes ne sont pas brevetables lorsque l'information génétique qu'ils contiennent ne serait pas en soi utile, l'utilité étant, avec la nouveauté et la non-évidence, une des trois conditions nécessaires à la brevetabilité d'une invention. Le texte publié par l'USPTO dans le Federal Register du 5 janvier 2001 rehausse les exigences liées à la condition d'utilité et précise la procédure correspondante qui doit être suivie lors de l'examen d'une demande de brevet.
En dépit de cet effort de clarification, des questions demeurent. L'évolution des critères de brevetabilité des gènes de la part de l'USPTO, basée en partie sur une jurisprudence qui ne cesse de s'étoffer, parviendra-t-elle à limiter le nombre des contentieux juridiques qui pourraient constituer un frein à la découverte et au développement pour l'ensemble des acteurs de la recherche aussi bien publics que privés ?
La situation concernant les inventions en matière biotechnologique est jugée encore trop mouvante par les personnes concernées par ces questions outre-Atlantique. Me Jeffrey Kushan, avocat spécialiste des brevets, a ainsi fait part à votre Rapporteur du caractère encore trop incertain des règles sur ce sujet, jugeant que cette situation était contraire au principe selon lequel les normes de propriété intellectuelle devaient être lisibles et stables pour permettre de fonder un réel progrès économique en rassurant ainsi les investisseurs. Ce point a été confirmé par le Docteur Ari Patrinos, directeur au département fédéral de l'énergie et responsable du Programme Génome Humain, qui a estimé que les professionnels manquaient d'informations sur ces sujets, en dépit des efforts de l'USPTO.
Une forme de confusion persiste donc. Pour votre Rapporteur, il n'est pas souhaitable que seuls les organes délivrant les brevets fixent les règles en la matière. La question est suffisamment grave pour que le législateur avance des pistes amenant à la résolution de ces problèmes.
III.- QUATRE ORIENTATIONS POUR LE RÉGLEMENT DE CETTE QUESTION
A.- INTRODUIRE DES LIMITES ÉTHIQUES DANS LE DROIT DES BREVETS
1.- Réaffirmer les règles actuelles en matière d'ordre public
et de bonnes m_urs
La technique du droit des brevets prévoit déjà des motifs d'exclusion du champ de la brevetabilité pour certaines inventions (170). Aux termes de l'article L. 611-17 du code de la propriété intellectuelle, en sont ainsi exclues « les inventions dont la publication ou la mise en _uvre serait contraire à l'ordre public ou aux bonnes m_urs, la mise en _uvre d'une invention ne pouvant être considérée comme telle du seul fait qu'elle est interdite par une disposition légale ou réglementaire ». Deux cas de figure sont donc envisagés : celui où la publication à elle seule est contraire aux bonnes m_urs et à l'ordre public ; celui où l'utilisation de l'invention produit cet effet.
La notion d'ordre public et de bonnes m_urs est classique en droit. Elle est intimement liée à l'évolution de la société et à sa tolérance à certains comportements. Elle a pour objet de préserver les intérêts essentiels de cette société autour de valeurs qui fédèrent les individus en son sein.
L'article 27 de l'Accord sur les aspects des droits de propriété intellectuelle qui touchent au commerce (Accord ADPIC de l'Organisation mondiale du commerce) de 1994 renvoie à ces notions. Il prévoit ainsi que les membres parties à l'accord pourront exclure de la brevetabilité les inventions dont il est nécessaire d'empêcher l'exploitation commerciale sur leur territoire pour protéger l'ordre public ou la moralité, y compris pour protéger la santé et la vie des personnes et des animaux ou préserver les végétaux, ou pour éviter de graves atteintes à l'environnement, à condition que cette exclusion ne tienne pas uniquement au fait que l'exploitation est interdite par leur législation
La directive 98/44/CE a également repris, dans son article 6, cette restriction, comme on l'a souligné précédemment. Le deuxième paragraphe de cet article présente quatre exemples d'inventions qui seraient considérées comme contraires à l'ordre public et aux bonnes m_urs : « Les procédés de clonage des êtres humains ; les procédés de modification de l'identité génétique germinale de l'être humain ; les utilisations d'embryons humains à des fins industrielles ou commerciales ; les procédés de modification de l'identité génétique des animaux de nature à provoquer chez eux des souffrances sans utilité médicale substantielle pour l'homme ou l'animal, ainsi que les animaux issus de tels procédés. » Cette liste n'est pas exhaustive comme le souligne le considérant 38 de la directive. Ce considérant indique d'ailleurs que les notions d'ordre public et de bonnes m_urs correspondent notamment à des principes éthiques ou moraux reconnus dans un État membre, dont le respect s'impose tout particulièrement en matière de biotechnologie en raison de la portée potentielle des inventions en ce domaine.
Ces dispositions sont de nature à assurer un certain nombre de protections contre les demandes de brevets les plus choquantes. Pour autant, il conviendrait sans doute de les renforcer, par l'introduction des préoccupations éthiques dans le cadre de la procédure d'attribution des brevets.
2.- Introduire des préoccupations éthiques dans le cadre de la procédure d'attribution des brevets
Lors du déplacement de votre Rapporteur aux États-Unis, les responsables de l'USPTO ont, à plusieurs reprises, insisté sur le fait que leur mission ne consistait pas à examiner les inventions d'un point de vue éthique mais de mettre en _uvre des critères de brevetabilité de nature purement technique. Cette approche est d'ailleurs contestée par une association comme Genetic Alliance, luttant contre les maladies rares d'origine génétique.
Sans doute faudrait-il sortir de cette logique - au plan européen et national - en introduisant un dispositif permettant de faire valoir les arguments éthiques lors de l'examen des demandes de brevets. On pourrait ainsi prévoir, aux côtés de l'Office européen des brevets mais aussi des organismes équivalents nationaux, des structures chargées d'une mission de veille éthique et d'alerter l'office compétent des risques éthiques d'une invention. Il appartiendrait ensuite à cet office d'arguer des dispositions existantes en matière de respect d'ordre et de bonnes m_urs, pour, le cas échéant, refuser la délivrance du brevet.
L'article 7 de la directive 98/44/CE confère au Groupe européen d'éthique des sciences et des nouvelles technologies de la Commission le soin d'évaluer tous les aspects éthiques liés à la biotechnologie. Le considérant n° 44 de ce texte prévoit, par ailleurs, que la consultation de ce groupe peut intervenir en matière de droit des brevets, celui-ci statuant au regard des principes éthiques fondamentaux. On peut voir ici les prémisses d'une introduction des préoccupations éthiques dans la procédure d'examen des brevets, objectif vers lequel on doit tendre.
B.- PRÉSERVER LE LIBRE ACCÈS À LA CONNAISSANCE
1.- La déclaration Clinton-Blair
L'obtention de brevets ne doit pas conduire à interdire l'accès à la connaissance et empêcher la recherche d'avancer. Ce principe est d'autant plus important que l'on touche à l'humain. C'est le sens de la déclaration commune du Président Bill Clinton et du Premier ministre Tony Blair du 14 mars 2000.
Lors d'une conférence commune, MM. Clinton et Blair ont demandé aux scientifiques du monde entier de placer dans le domaine public toutes les informations concernant le décodage du génome humain. Cet appel visait en particulier les sociétés privées impliquées dans le décryptage du génome, qui ne donnent accès aux informations que moyennant le paiement d'un abonnement annuel à une base de données. La portée de la déclaration Clinton-Blair n'est évidemment pas juridique. Mais pour symbolique qu'elle soit, elle a eu un impact réel dans le monde des biotechnologies.
Le droit des brevets ne s'oppose pas nécessairement au principe d'accès aux connaissances. En effet, la recherche fondamentale est toujours possible sur une invention brevetée pour quiconque le souhaite. La seule limite est factuelle et financière. Est-il rationnel de se lancer dans des recherches où l'essentiel est déjà breveté, ce qui signifie qu'on ne pourra pas tirer un réel bénéfice de son travail ? Ce raisonnement pourrait dissuader certains laboratoires privés ou publics, se finançant par le dépôt de brevets, de s'engager dans certaines voies de recherche.
On rappellera aussi que la demande de brevet fait l'objet d'une publicité
- généralement dans un délai de dix-huit mois - qui permet d'informer la communauté scientifique de cette invention dans ses principes essentiels. Il s'agit là d'une contrepartie essentielle au droit d'exclusion dont bénéficie l'inventeur titulaire d'un brevet. C'est en cela que le droit de la propriété intellectuelle a pu contribuer à l'amélioration des techniques.
Devant votre Rapporteur, l'accent a été mis par certaines personnalités très engagées sur ces questions, comme M. William Haseltine, président de Human Genome Science, sur le fait que l'obtention d'un brevet est gage de transparence de l'information. Cet argument a également été développé par l'American Association for the Advancement of Science, la plus grande association scientifique américaine éditrice de la célèbre revue Science.
Pourtant, même si dans son principe cette affirmation est avérée, elle connaît dans la pratique une traduction plus nuancée.
2.- Vers un domaine public en génomique ?
La possibilité de faire progresser la recherche passe par l'accès aux données issues du séquençage du génome humain. C'est pourquoi le décryptage qui a abouti au début de 2001 a relancé la discussion autour d'une mise en commun de ces données.
La notion de libre accessibilité aux données est partagée par le secteur public et universitaire. Elle suit ainsi la position défendue par le Programme Génome Humain, dont l'objectif est d'encourager la recherche et l'innovation de la part de l'ensemble des acteurs publics et privés. Cette position peut paraître en contradiction avec le Bayh-Dole Act qui encourage la commercialisation des recherches financées par des fonds publics, et avec le fait que les secteurs public et universitaire américains sont encore actuellement les premiers détenteurs de brevets en matière de séquences génétiques. Toutefois, c'est autour de la position du Programme Génome Humain que s'est développée la notion de « domaine public en génomique », fruit des réflexions éthiques menées par différents groupes. Celles-ci ont été reprises lors de la déclaration commune de Bill Clinton et de Tony Blair, à la suite de l'échec des tentatives de rapprochement entre les laboratoires du Programme Génome Humain et Celera Genomics afin d'offrir à la communauté scientifique l'ensemble des données du génome humain.
En effet, les sociétés de génomique constituées sur un modèle de développement différent de celui de l'industrie pharmaceutique entrent en compétition avec le secteur public en termes de rapidité de constitution de leur base de données, qui leur permettrait de revendre l'information génétique et de mettre ainsi à disposition de leurs clients un accès et un service exclusifs pour l'interprétation des bases de données privées intégrant les données publiques.
La course de vitesse actuelle entre le Programme Génome Humain et Celera Genomics aurait probablement à gagner sur le plan scientifique d'une collaboration entre les deux groupes, comme cela fut d'ailleurs le cas entre eux pour le génome de la drosophyle. Mais pour sa survie financière, Celera se doit de conserver son avantage concurrentiel vis-à-vis du secteur public le temps d'élargir la gamme de ses prestations comme cela est annoncé en protéomique.
Face à une certaine pression politique, les sociétés participant au décryptage du génome sont, semble-t-il, en quête d'un compromis entre le droit à l'accès à l'information et la possibilité pour elles de percevoir des gains issus de leur activité de recherche.
Lors du déplacement de votre Rapporteur aux États-Unis, le Docteur Paul Gilman, directeur dans la société Celera Genomics, a clairement mis en avant la volonté de sa compagnie de devenir une société d'informations, à l'égal de Bloomberg, dans le secteur financier. Pour diffuser les données qu'elle produit, Celera Genomics utilise les voies électroniques, comme Internet. Le génome de la mouche est ainsi accessible sur CD-ROM. La mise à disposition de ces données peut être gratuite ou non, selon les cas et la stratégie de l'entreprise. Celera Genomics propose aussi des abonnements à ces bases de données, les abonnés étant libres de réaliser toutes les recherches qu'ils souhaitent à partir de ces données. Ils ne peuvent, en revanche, diffuser eux-mêmes les données qui proviennent de Celera Genomics, un tel acte serait alors considéré comme un piratage, susceptible de poursuites judiciaires.
La diffusion des données issues du décryptage du génome est aujourd'hui au c_ur du débat sur la brevetabilité. Car plus que de produits, il est question ici de procédés et d'informations, notions que le droit de brevet peut permettre d'appréhender. L'accès libre aux données est l'une des conditions essentielle pour limiter la constitution des situations monopolistiques, et empêcher l'appropriation privée des informations relatives au génome humain.
C.- S'OPPOSER À LA CONSTITUTION DE MONOPOLES
1.- La pratique des licences et des brevets dépendants
La juste rémunération apportée par le brevet ne doit pas conduire à une forme de monopole au bénéfice du premier inventeur. Cette règle générale s'impose plus encore pour les biotechnologies. Il ne faudrait pas, en effet, que l'inventeur d'une fonction, disposant ainsi d'un brevet sur l'application, la fonction et - dans la logique actuelle de la directive -, au final, sur le gène lui-même, puisse verrouiller le dispositif en percevant des droits sur toutes les applications et fonctions nouvelles inventées à partir d'un même gène.
Une demande de brevet comporte deux parties : une description de l'invention et une liste de revendications. La description permet d'apprécier si les conditions de brevetabilité sont remplies. Les revendications sont ensuite examinées par l'office des brevets qui les agrée en tout ou partie. L'accord donné par l'office sur-le-champ, plus ou moins large, des revendications est, à cet égard, déterminant dans la lutte contre l'établissement de monopoles en la matière.
Deux cas de figure se présentent. Lorsqu'un inventeur améliore un produit ou un procédé, il peut obtenir un brevet sur ce qui est considéré comme une nouvelle invention. Si la seconde invention ne constitue pas une amélioration de la première mais simplement un corollaire - c'est-à-dire que la seconde ne pourrait exister sans la première - alors le deuxième inventeur se trouve dans la dépendance de l'inventeur initial. Le titulaire du brevet corollaire doit alors obtenir l'accord du titulaire du brevet de base pour exploiter cette nouvelle application.
L'enjeu consiste donc à mettre en place les moyens juridiques de contraindre les titulaires des premiers brevets à accepter l'octroi de brevets dépendants dans des conditions économiques non prohibitives.
Un exemple est souvent cité à ce sujet. La société Human Genome Science s'est vu attribuer un brevet sur le gène CCR5 et la protéine qui y est liée. Ce gène code un récepteur membranaire. Le champ de ce brevet est large et couvre tout usage de ce récepteur. Or lorsque la société en question a présenté sa demande de brevet, elle ignorait que ce récepteur jouait un rôle important dans la pénétration du virus du SIDA dans les cellules. En droit, toute utilisation de ce gène et de sa protéine dans une action de prévention du SIDA serait dépendante du brevet accordé à la société initiale. Votre Rapporteur estime qu'une telle situation n'est pas acceptable.
2.- Les moyens de s'opposer à la constitution des monopoles
Les interlocuteurs rencontrés aux États-Unis par votre Rapporteur se sont révélés plus sensibles à la question de la constitution d'éventuels monopoles qu'aux questions proprement éthiques. Il semble que ce point préoccupe les responsables américains, qui finalement n'envisagent l'adoption d'une loi sur les brevets en matière de biotechnologies que dans le cas où apparaîtraient de tels monopoles. On ne peut cependant attendre passivement que ces monopoles se constituent. L'affaire Microsoft montre combien la résolution a posteriori de ces problèmes est longue et chaotique.
Une solution pourrait consister à limiter les brevets dépendants par l'octroi de licences obligatoires, permettant au titulaire d'un brevet dépendant d'obliger le titulaire du brevet antérieur à lui accorder une licence. Ces licences obligatoires sont décidées par le juge à la demande d'une partie privée. Il existe aussi des licences d'office décidée par les gouvernements en cas de nécessité publique. Il faudrait encourager ces pratiques en étudiant la possibilité de limiter le coût de ces licences, de sorte que les entreprises titulaires de brevets dépendants puissent exploiter leur invention et faire progresser les techniques.
Cette question revêt évidemment une importance particulière dans le domaine des inventions relatives à la santé. Mais, au-delà, ne faut-il pas engager une réflexion plus large sur la brevetabilité du vivant qui semble à l'origine des difficultés que nous connaissons aujourd'hui ?
D.- REMETTRE EN CAUSE LES BREVETS DE PRODUITS SUR LE VIVANT PAR UNE DÉMARCHE EUROPÉENNE ET INTERNATIONALE
1.- Remettre en cause les brevets de produits sur le vivant
Votre Rapporteur estime qu'il n'est pas acceptable que des entreprises puissent finalement s'approprier par des brevets de produits des éléments de la nature. Revenir sur cette possibilité aujourd'hui ouverte par le droit des brevets ne remet pas en cause la propriété intellectuelle car il existe des mécanismes à même de préserver les intérêts des sociétés qui ont engagé des investissements lourds, tout en assurant le respect du principe selon lequel la nature ne peut être un objet de propriété.
Les revendications de ces entreprises doivent être limitées aux procédés qu'elles ont réellement inventés pour permettre d'exploiter les éléments naturels ainsi mis en évidence. Cette limite apportée aux revendications ne pourra cependant être opérante que dans le cadre d'un accord européen et international qui unifierait les principes et les pratiques.
Pour certains, un obstacle d'ordre juridique pourrait s'opposer à cette solution. La Commission européenne soutient, en effet, que l'article 27 des accords ADPIC, auquel le douzième considérant de la directive de 1998 se réfère, prévoit que la protection conférée par un brevet doit être la même dans tous les domaines technologiques. Or, par exemple, en matière pharmaceutique, la première description d'une molécule qui préexiste dans la nature ou issue d'un procédé de synthèse suffit à conférer des droits sur le produit. Une telle solution s'imposerait donc pour les séquences génétiques selon ce raisonnement.
Elle n'emporte pas la conviction de votre Rapporteur. Plus largement, elle montre même la nécessité d'aller plus loin dans ce débat. Le droit des brevets est utile mais il ne doit pas se réduire à un corpus normatif totalement intangible.
2.- Engager une nouvelle politique à l'échelon européen et international
La transposition de la directive 98/44/CE a suscité des réactions dans plusieurs pays européens, au premier rang desquels l'Allemagne et la France. Il est clair que ce texte soulève des difficultés qui rendent difficile sa transposition en l'état.
C'est pourquoi notre pays, par la voix du Président de la République, en accord avec le Premier ministre, a pris l'initiative, l'an passé puis à nouveau en février dernier, d'interroger la Commission européenne sur l'interprétation à donner de la directive de 1998, mettant en avant les problèmes que soulève notamment son article 5.
La réponse de la Commission n'a pas été rendue publique mais elle ne semble pas avoir donné satisfaction aux responsables français. En conséquence, il paraît utile à votre Rapporteur de soutenir toute initiative politique, française ou non, tendant à éclaircir le texte de la directive dans le sens des orientations qui viennent d'être présentées.
Mais au-delà de la seule question de la transposition de la directive de 1998 s'impose la nécessité de remettre en débat le droit de délivrer des brevets de produits sur les éléments naturels.
Cette question au sein de laquelle la brevetabilité du génome humain tient une place particulière ne peut se régler dans le seul cadre national alors que l'exploitation de ces recherches est évidemment mondiale. Il importe donc d'observer des règles communes, ce à quoi les Européens consentent, mais pas nécessairement les Américains. En dehors du seul cadre européen, c'est donc au niveau de l'OMC que ces sujets devraient être abordés. L'accord ADPIC conclu à Marrakech en 1994 devrait être révisé sur ce point, ce qui, votre Rapporteur ne l'ignore pas, n'est pas chose aisée.
Au-delà des positions singulières de chaque pays, se profile sans doute une bataille commerciale sans précédent. Les décisions prises aujourd'hui engagent l'avenir pour des décennies et la prise de position isolée d'un pays - en dehors des États-Unis - ne saurait infléchir un mouvement, largement imprimé outre-Atlantique, par des acteurs non étatiques. La coopération européenne est, dès lors, une nécessité impérieuse. Elle doit conduire à une position équilibrée qui préserve ces principes essentiels que sont la dignité humaine, la libre recherche et l'accès à la connaissance. Sans cette coopération, l'alignement se fera, au plan mondial, sur un seuil minimum où les principes ainsi énoncés auront été, depuis longtemps, évacués des pratiques, des textes et - qui sait ? - des esprits.
La Mission a examiné le présent rapport d'information lors de sa séance du 27 juin 2001.
Le Président Bernard Charles a considéré que le dépôt, par le Gouvernement, du projet de loi relatif à la bioéthique ouvrait une nouvelle étape du processus de révision des lois de 1994 imposant la clôture des travaux de la Mission et la création d'une commission spéciale chargée d'examiner ce projet, les différents groupes politiques ayant accepté ce principe d'organisation. Il a indiqué avoir lui-même demandé la constitution de cette commission le 25 juin dernier.
Après avoir fait état de ses démarches insistantes auprès du Gouvernement pour voir le texte soumis à l'examen de l'Assemblée nationale avant les prochaines élections législatives, il a rappelé que la Mission avait un rôle exclusivement d'information des membres de la future commission spéciale sur les principaux points en discussion lors de la révision des lois bioéthiques. Il a fait savoir que la Mission avait travaillé à un rythme soutenu, ayant tenu 25 réunions, pour une durée totale de 53 heures. Il a constaté que la Mission avait respecté son plan de travail, rappelant que, par souci d'efficacité le périmètre de réflexion délimité par l'étude du Conseil d'État avait été retenu. Il a indiqué que la Mission avait tenu des auditions à caractère institutionnel - comme par exemple, le Comité consultatif nationale d'éthique et la Commission nationale consultative des droits de l'homme - plusieurs « grands témoins », des journalistes, des associations fortement impliquées dans la réflexion en matière de bioéthique ou des représentants des différentes familles spirituelles.
Il a conclu en précisant que la présente réunion avait pour objet d'autoriser la publication du rapport présenté par M. Alain Claeys, en application de l'article 145 du Règlement.
Après que M. Jean-Luc Préel se fut interrogé sur le sort réservé à un projet de loi déposé en fin de législature, prenant l'exemple des lois de 1994 dont l'adoption avait été repoussée du fait d'un dépôt tardif, le Président Bernard Charles a observé qu'il existait de nombreux précédents en ce domaine, se déclarant assuré que le travail accompli au Parlement avant les élections de 2002 ne serait pas inutile.
Après avoir jugé que le temps écoulé depuis le dépôt du rapport du Conseil d'État avait permis d'examiner ces questions dans un climat apaisé, propice à la révision des lois bioéthiques, M. Alain Claeys, Rapporteur, a tout d'abord observé que des événements récents avaient montré combien la science, dont les progrès ouvrent des perspectives thérapeutiques sans précédent, pouvait aussi conduire à des situations étonnantes ou inquiétantes. Il a cité, à ce propos, le cas de cette femme de 62 ans qui s'est rendue à l'étranger dans des conditions obscures pour avoir la possibilité de donner naissance à un enfant, ainsi que l'audition par le Congrès des États-Unis de la secte raëlienne qui vient de former une société pour pratiquer à terme le clonage reproductif humain. Il a considéré que, face à cela, il convenait d'adopter une démarche de responsabilité en s'appuyant sur un principe connu mais revisité : le principe de précaution. Il a insisté sur le fait que cette démarche ne devait pas être assimilée à une méfiance ou à une frilosité vis-à-vis de la science, mais qu'au contraire, le principe de précaution, dont le corollaire est une méthode - celle de l'évaluation - devait conduire à un soutien en faveur de la recherche. Il a ajouté que plus nos connaissances en matière fondamentale seraient avancées, plus le législateur pourrait décider et agir avec justesse, car ce qui nuit à la décision et à l'action, c'est l'absence de connaissances et non l'inverse.
Le Rapporteur a jugé qu'il appartenait au Parlement de fixer un cadre nouveau pour une recherche et une pratique médicale maîtrisées, tout en estimant essentiel de prendre la mesure de l'aspect humain des questions bioéthiques. Il a considéré que derrière les règles, les normes, les procédures, apparaissaient des femmes et des hommes, des médecins, des scientifiques, des patients qui ont à appliquer et à vivre ces questions quotidiennement. Il a estimé qu'un effort devait donc être engagé vers eux pour leur permettre de mieux faire face à ces enjeux.
Le Rapporteur a également considéré que la mise en _uvre du principe de précaution devait conduire à mesurer la demande sociale, tout en s'interrogeant sur sa nature. Il a ajouté que ce principe devait aussi amener le législateur à préserver parallèlement les principes éthiques devant être respectés dans notre société. Il a insisté sur le fait qu'il appartiendrait au législateur de concilier des principes et des aspirations parfois divergentes, en ayant à l'esprit que soigner les malades, contribuer à atténuer leurs souffrances est aussi un principe éthique de premier rang.
Il a observé que des enjeux nouveaux étaient apparus depuis 1994, évoquant d'abord les innovations scientifiques et prenant l'exemple du clonage et de la révolution génétique, avec les perspectives thérapeutiques qu'elle ouvre. Il a indiqué que ces enjeux étaient aussi économiques, notre monde étant celui de la concurrence et les biotechnologies tenant désormais dans nos économies une place importante alors que les États-Unis confortent leur rôle dominant dans ce secteur. Il a précisé que cette concurrence était aussi celle des chercheurs, des médecins, avec des effets induits nouveaux comme ce que l'on qualifie désormais couramment de tourisme médical. Il a ajouté, enfin, que ces enjeux étaient aussi ceux du droit, les normes internationales et européennes occupant une place de plus en plus importante dans ce domaine. Il a rappelé, à cet égard, que plusieurs textes avaient été adoptés dernièrement comme la Déclaration de l'Unesco sur le génome humain et les droits de l'homme et la Convention d'Oviedo du Conseil de l'Europe de 1997. Il a souhaité que le législateur mesure l'impact du droit international dans notre droit interne et qu'il profite de la révision des lois bioéthiques pour ratifier la Convention d'Oviedo.
Le Rapporteur a observé que la Mission s'était inscrite dans le cadre d'un débat résolument citoyen, cet esprit ayant présidé à ses travaux mais aussi à la démarche transparente entreprise par le Gouvernement qui a consulté le Conseil d'État, le Comité consultatif national d'éthique et la Commission nationale consultative des droits de l'homme. Il a estimé qu'à l'issue de cette année d'auditions, il apparaissait possible de livrer l'état du débat, sachant qu'il appartiendrait ensuite à la commission spéciale et à l'Assemblée dans son entier de définir un nouveau cadre légal.
Abordant la question de l'assistance médicale à la procréation, il a jugé que les conditions médicales de recours à l'AMP étaient globalement satisfaisantes, l'assouplissement de quelques règles étant néanmoins souhaitable pour permettre expressément l'autoconservation des gamètes ou des tissus germinaux de personnes qui subissent des traitements ou des opérations chirurgicales stérilisants ainsi que pour autoriser l'AMP avec tiers donneur en première indication, si l'on sait que l'AMP intraconjugale n'a que peu de chance d'aboutir.
Il a ajouté que les conditions sociales faisaient l'objet de demandes d'élargissement qui semblaient pour certaines souhaitables, comme l'harmonisation de la règle applicable aux personnes mariées et à celles vivant en concubinage. Il a avancé l'idée qu'il fallait soit imposer la même exigence de vie commune préalable, soit supprimer cette dernière pour les concubins. Il a ajouté que, s'agissant des donneurs de gamètes, qui doivent aujourd'hui faire « partie d'un couple ayant procréé », il semblait opportun de supprimer la condition d'appartenance du donneur à un couple, ce qui permettrait de ne plus exclure les personnes divorcées, veuves ou ayant vécu maritalement. Enfin, il a indiqué que le transfert post mortem d'embryon ferait l'objet d'un débat.
Le Rapporteur a ensuite constaté que l'application pratique des techniques d'AMP posait différents problèmes qui exigeaient une réponse du législateur. Il a noté que l'évaluation des nouvelles techniques d'AMP faisait gravement défaut et devait être mise en place, en dépit du caractère forcément incomplet d'un tel dispositif. Il a évoqué, à ce sujet, les problèmes posés par l'ICSI.
Il a ajouté que, pour compléter le dispositif, il conviendrait de soumettre à autorisation préalable l'application clinique de toute nouvelle technique d'AMP et que, pour satisfaire aux exigences de sécurité sanitaire, il était nécessaire de mettre en place un suivi des enfants nés grâce aux techniques, actuelles et futures, d'AMP.
Il a enfin jugé que le recours excessif aux traitements de stimulation ovarienne et les dangers qu'il représente pour la santé des femmes et des enfants exigeait d'encadrer spécifiquement cette activité qu'elle intervienne dans le cadre de l'AMP ou en dehors de celle-ci.
Evoquant la nécessité de définir un cadre précis pour la destruction des embryons surnuméraires et pour le mode d'expression du consentement des parents, le Rapporteur a fait savoir que la situation actuelle était la suivante : au 31 décembre 1998, 73 205 embryons étaient conservés ; pour 45 505 d'entre-eux, soit 62 %, existait un projet parental en cours ; 12 782 couples avaient renoncé à leur projet tandis que l'on ignorait la décision sur la poursuite ou l'interruption du projet de 10 991 couples ; au total, on comptait donc, avant l'année 1999, 23 773 embryons surnuméraires.
Il a considéré qu'il fallait un régime précis et rigoureux pour la congélation et la destruction des embryons qui respecte les principes de transparence et de consentement, une durée de conservation limitée à cinq ans semblant correspondre le mieux à la réalité des situations rencontrées dans le cadre des AMP. Pour responsabiliser les couples, il a estimé qu'il conviendrait de renverser la charge de la preuve quant à l'expression de leur volonté de poursuivre leur projet parental. Il a précisé qu'il reviendrait donc aux parents de se manifester chaque année, interrogés la première fois par courrier par leur centre, celui-ci leur donnant une date limite de décision annuelle, pour manifester leur souhait de poursuivre ce projet ou de l'interrompre. Il a ajouté qu'à défaut, au bout de cinq ans, le silence vaudrait décision tacite de destruction de leurs embryons surnuméraires, un tel dispositif, certes sévère, ayant le mérite de clarifier les responsabilités de chacun.
Le Rapporteur a également indiqué que l'aide psychologique apportée aux couples était insuffisante et devrait systématiquement leur être apportée. Il a noté, par ailleurs, que les problèmes spécifiques liés au don d'ovocytes exigeaient de revoir les règles applicables et d'envisager en particulier l'abrogation du décret qui exige leur mise en quarantaine.
Abordant la question du trop faible contrôle des centres d'AMP, il a constaté que l'accord était général pour réclamer un changement des règles et des structures qui encadrent les activités d'AMP au regard des faiblesses constatées de la Commission nationale de médecine et de biologie de la reproduction et du diagnostic prénatal et du modèle que représente pour beaucoup la Human Fertilization Embryology Authority (HFEA) britannique. Il a précisé que les critiques et autocritiques de la Commission nationale de médecine et de biologie de la reproduction et du diagnostic prénatal étaient principalement de deux ordres : celle concernant l'absence de moyens de la commission et celle liée à sa composition qui pourrait mettre en cause son indépendance ou sa nature excessivement technocratique. Il a jugé que la collaboration des services de l'État, centraux et déconcentrés, faisait en outre gravement défaut et que la composition de la commission était elle-même remise en question à cause de son ouverture insuffisante à la société civile.
Il a observé que tous les interlocuteurs de la Mission s'accordaient sur la nécessité de prévoir une composition large de cette future agence au sein de laquelle la société civile serait suffisamment représentée, d'autres insistant cependant plus sur les capacités propres de contrôle de l'agence.
Le Rapporteur a conclu que seule la question des pouvoirs qui seraient confiés à cette nouvelle agence divisait les interlocuteurs de la Mission, une grande majorité d'entre eux se prononçant toutefois pour que lui soit transféré le pouvoir de décision en matière d'agrément des centres, à l'instar de la HFEA dont l'expérience, développée dans le rapport, apparaît concluante.
Observant que jusqu'alors le débat sur la recherche sur l'embryon était cantonné au champ de l'AMP, il a constaté qu'elle était subitement devenue un enjeu scientifique majeur intéressant l'ensemble de l'humanité et porteuse de tous les espoirs. Le compromis atteint par le législateur en 1994 pouvant sembler manquer de cohérence a posteriori en n'autorisant que des « études », il a estimé qu'il n'était manifestement plus adapté.
Le Rapporteur a donc jugé que se posait aujourd'hui avec acuité le problème de la levée de l'interdiction de recherches sur l'embryon et des limites de ces recherches : Comment les définir ? Sur quels choix doit-on se fonder ? Quels principes sont intangibles ? Comment les concilier ? Quel cadre poser à cette recherche ? Quel contrôle mettre en place ?
Il a également considéré que nous étions confrontés au dilemme suivant : soit l'on ne ferme aucune piste de recherche, à l'exclusion du clonage reproductif, unanimement rejeté et condamné, afin de valider l'une ou l'autre et d'abandonner celles qui s'avéreraient décevantes, soit l'on renonce dès aujourd'hui à certaines de ces pistes tout en sachant que ces recherches sur l'embryon sont d'ores et déjà en cours à l'étranger. Il s'est alors interrogé sur les attitudes à adopter si les recherches sur l'embryon conduites à l'étranger devaient effectivement aboutir à la mise au point de traitements thérapeutiques innovants. Il a rappelé que la grande majorité des personnalités entendues par la Mission étaient favorables à une certaine ouverture de la recherche sur l'embryon à la condition que celle-ci soit strictement encadrée et contrôlée. Il a fait savoir que le rapport présentait les différentes pistes de recherches aujourd'hui ouvertes et les avantages et inconvénients de chacune d'entre elles dans l'état des connaissances scientifiques actuellement disponibles.
Constatant qu'aucun scientifique n'était aujourd'hui en mesure de déterminer avec certitude quelle piste de recherche, parmi celles qui ont été précédemment présentées, aboutirait effectivement à la mise au point de nouvelles thérapies, il a noté que certains considéraient que les perspectives offertes par les cellules souches adultes pourraient éviter de recourir aux cellules souches embryonnaires ou au clonage thérapeutique, les cellules souches adultes cependant n'étant apparemment disponibles qu'en faible quantité et difficiles d'accès. Il a ajouté que le clonage thérapeutique faisait, quant à lui, l'objet de différentes interrogations. Il a jugé que quelle que soit la ou les pistes de recherche que l'on déciderait de retenir, il faudrait impérativement mettre en place un dispositif strict d'encadrement, de suivi et de contrôle des recherches qui seraient autorisées autour de plusieurs principes pouvant être :
- l'interdiction d'implanter un embryon ayant fait l'objet d'une recherche, ou dont les cellules auraient été utilisées dans le cadre d'une recherche ;
- l'interdiction de toute manipulation des cellules germinales d'un embryon ;
- l'obligation de recueillir le consentement écrit et éclairé des parents qui accepteraient de donner leurs embryons surnuméraires à la recherche ;
- le choix d'un stade de développement de l'embryon, sur lesquels la recherche serait autorisée, précis et limité, qui pourrait être la fin du stade préimplantatoire ;
- la nécessité absolue de protéger les femmes qui donneraient leurs ovocytes à la recherche dans un cadre approprié ;
- l'obligation d'assurer la transparence des recherches entreprises ;
- l'autorisation des recherches à la double condition d'une absence d'alternative à l'utilisation d'un embryon et d'une finalité « scientifique à visée thérapeutique » ;
- et enfin la mise en place d'une autorité indépendante chargée d'appliquer le cadre strict des recherches fixé par le législateur, qui pourrait être la même agence que celle qui contrôlerait le secteur de l'AMP, à l'instar de la HFEA britannique. Cette agence pourrait décider d'autoriser les protocoles de recherches sur l'embryon ; ses décisions pourraient toujours faire l'objet d'un recours contentieux ou d'une demande de seconde délibération des ministres qui en contesteraient l'opportunité. Il a indiqué qu'en qualité de Rapporteur, il prenait position pour l'indépendance de cette agence. Il a conclu qu'il s'agissait sans doute là d'un élément essentiel pouvant permettre l'autorisation de la recherche sur l'embryon par le législateur et au-delà par le sentiment public, celui-ci devant être assuré de l'existence de solides garanties sur l'encadrement de cette recherche. Il a ajouté qu'il appartenait au Parlement de fixer le champ des recherches autorisées.
Abordant la question du don et de l'utilisation des éléments du corps humain, il a constaté que les nombreux progrès réalisés faisaient que l'indication de la greffe s'élargissait régulièrement, grâce notamment aux prélèvements d'organes sur personnes vivantes. Il a observé que subsistait toutefois une pénurie permanente de greffons, dont les causes sont bien identifiées, le Plan greffe, présenté en juin 2000, ayant d'ores et déjà apporté des aménagements afin de remédier à cette situation. Il a indiqué être convaincu que l'activité de transplantation ne pourrait se développer que dans un climat de confiance réciproque entre le corps médical et la société, cette confiance, toujours fragile, reposant sur trois principes fondamentaux qui sont le corollaire de l'indisponibilité et de l'inviolabilité du corps humain : gratuité, anonymat et consentement.
Observant que la publication tardive du décret de mise en place du registre des refus ne permettait pas aujourd'hui de tirer de conclusion sur son fonctionnement, il a indiqué que la Mission avait pris acte de ce que l'indication croissante de la greffe conjuguée avec les mutations de la société permettaient d'élargir toujours plus le cercle des donneurs vivants au sein des familles mais aussi à ceux qui deviennent proches par les circonstances de la vie. Il a insisté sur la nécessité de s'assurer de la réalité du consentement éclairé de ces donneurs qui risquent d'être soumis à des pressions financières et/ou psychologiques non négligeables. Il a rappelé que l'Etablissement français des greffes avait souhaité travailler sous le signe de la transparence : attribution des greffons, évaluation des résultats, information auprès du public font partie de ses missions. Il a estimé que son action, qui pourrait être relayée par d'autres acteurs comme l'éducation nationale notamment, contribuait à améliorer cette grande cause qui permet de sauver des vies.
Le Rapporteur a, ensuite, évoqué la question de la médecine prédictive, qui, renvoyant à des enjeux multiples, posait d'abord le problème d'une éventuelle tentation de sélection des embryons lors du DPI et du DPN. Il a constaté qu'en effet, s'agissant d'une technique indolore, le risque existait que l'on passe de la recherche d'une maladie d'une particulière gravité à celle de caractéristiques peu pathologiques, et de transformer cette technique en pratique de confort, voire en tentative de recherche de « l'enfant parfait », le cadre posé par la loi de 1994 ayant permis toutefois d'éviter cette dérive. Il a insisté sur le fait que la crainte, ainsi apparue de voir émerger une forme d'intolérance vis-à-vis des personnes nées avec un handicap ou une maladie, dans la mesure où l'on accepterait de moins en moins ce qui ne correspondrait plus à une forme de « normalité », méritait la vigilance du Parlement.
Il a ajouté que la question de l'utilisation des examens génétiques à des fins autres que médicales posait aussi des difficultés, notamment en matière d'assurance et d'emploi. Il lui a semblé inacceptable, contrairement aux préconisations timides du Conseil d'État dans son rapport de novembre 1999, de laisser aux professionnels le soin de déterminer s'ils utiliseraient ou non ces tests génétiques, pour fixer le montant d'une prime d'assurance ou accepter d'embaucher une personne, la loi devant affirmer le principe de non-discrimination sur la base du patrimoine génétique.
Enfin, il a informé les membres de la Mission que la question de la brevetabilité du corps humain et de ses éléments, connexe à la révision des lois de 1994, serait abordée dans le cadre du rapport, un état des lieux de ce débat, complexe pour le moins, en France et en Europe étant présenté.
En conclusion, le Rapporteur a jugé que les travaux de la Mission constituaient un fondement utile à ceux de la commission spéciale, dans la mesure où ils montraient le besoin d'encadrement d'une pratique médicale et d'une recherche dont le champ s'élargit sans cesse. Il a observé que, de vastes débats étant ouverts, on devait reconnaître au Gouvernement une volonté très nette de laisser libre cours à la discussion au sein du Parlement. Il a conclu qu'il appartiendrait au législateur de se livrer à cet exercice ardu dans quelques mois.
Mme Yvette Roudy s'est félicitée que le rapport présenté couvre tout le champ des questions abordées par la Mission. Elle a vivement regretté la longueur des délais avant l'inscription à l'ordre du jour de l'Assemblée nationale du projet de loi relatif à la bioéthique. Elle a jugé que l'opinion risquait de ne pas comprendre ce retard tandis que la législation adoptée pourrait rapidement devenir obsolète. Elle a estimé qu'il revenait au Parlement de proposer un cadre assez large pour la recherche afin de ne fermer aucune piste mais qu'il fallait admettre que le législateur ne pouvait ni tout encadrer ni tout prévoir. Elle a ajouté qu'on pouvait ainsi s'interroger sur ce qu'il convenait de faire s'agissant de l'ICSI.
Elle s'est ensuite déclarée « plutôt séduite » par le clonage thérapeutique bien que ce terme soit mal choisi par l'analogie qu'il crée avec le clonage reproductif. Elle a considéré qu'il pourrait, en effet, ouvrir d'immenses bénéfices thérapeutiques et que l'on comprenait mal les prétendus dangers qu'il présenterait et qui pourraient justifier son abandon.
Elle a ajouté qu'il fallait débattre, par ailleurs, de la possibilité d'autoriser le transfert post mortem, qui a été retiré du projet de loi alors que les cas seraient dans la réalité extrêmement rares. Elle a estimé qu'il faudrait veiller à ouvrir davantage la future agence de la procréation et de l'embryologie à la société civile en s'assurant que les membres choisis soient régulièrement renouvelés. Enfin, elle a souhaité que l'on s'interroge sur les meilleurs moyens de populariser le débat bioéthique et sur la crainte qu'une législation trop restrictive ne conduise à la fuite des chercheurs de notre pays.
Après avoir félicité le rapporteur pour son travail considérable sur des sujets éminemment difficiles, M. Jean-François Mattei a rappelé que la présente Mission n'avait pas pour rôle de prendre position sur le fond, ce qui reviendra à la commission spéciale et à l'Assemblée tout entière. Il a ensuite présente les remarques suivantes :
- Le plan du rapport qualifie de concluante l'expérience de l'HFEA alors que ce jugement apparaît discutable.
- L'intitulé de la partie du rapport relative à la recherche sur l'embryon ne paraît pas adapté. Les Britanniques parlent d'ailleurs de la recherche sur le « préembryon ». On ne peut éluder le statut de l'embryon qui est l'un des principaux éléments du débat. Ainsi peut-on s'interroger sur le nom qu'il convient de donner à l'ovocyte énucléé dans lequel on a transféré un noyau somatique si l'on considère qu'il ne peut s'agir d'un embryon en l'absence de reproduction sexuée. Il est regrettable également de ne pas faire mention dans ce titre des développements que permettrait la recherche sur l'embryon en faveur de la médecine embryonnaire.
- Une contradiction semble apparaître entre l'utilisation du terme de « clonage thérapeutique » dans le rapport et la mise en en garde qu'il semble contenir contre l'alibi de la thérapeutique.
- S'agissant des conditions de recherche sur les cellules ES, la recherche ne se réduit pas à ces cellules, la compréhension des milieux de culture et des autres cellules souches étant essentielle.
- L'interdiction, prônée par le Rapporteur, de toute thérapie germinale, apparaît contestable, compte tenu de l'intérêt qu'il y aurait à modifier le patrimoine génétique d'un embryon atteint d'une grave maladie. La science doit tendre non plus à éliminer l'embryon malade mais plutôt à le guérir.
- L'intitulé de la quatrième partie du rapport ne parle pas explicitement des organes.
- Le choix du titre relatif à la médecine prédictive peut également être contesté dans la mesure où il n'inclut pas les tests génétiques utilisés dans les procédures judiciaires dans le cadre de la recherche d'un lien de filiation.
- En revanche, on peut se féliciter du titre choisi pour le chapitre relatif à la brevetabilité, le sujet, apparaissant en effet d'une particulière gravité.
En résumé, M. Jean-François Mattei a noté qu'il existait beaucoup de points de rencontre mais également des divergences. Il a conclu qu'il fallait se féliciter du travail accompli, le plus important restant à faire.
M. Jean-Michel Dubernard a également considéré que le travail de préparation de la révision des lois bioéthiques avait bien avancé, mais a regretté que l'on ait perdu du temps avant le dépôt du projet de loi et son inscription encore repoussée à l'ordre du jour de l'Assemblée nationale. Il a souhaité que le rapport ne soit pas trop synthétique sur des questions fondamentales qui méritent d'être approfondies.
Souhaitant qu'il ne soit pas donnée une trop grande importance au principe de précaution, il a considéré qu'il fallait, par ailleurs, se préserver des pressions institutionnelles et des lobbies qui sont particulièrement puissants, citant l'exemple du « clonage thérapeutique », auquel M. Jean-Michel Dubernard s'est déclaré favorable en raison des perspectives de recherche qu'il permettrait d'ouvrir, mais qui a été pourtant retiré du projet de loi.
Il a souhaité qu'on ne distingue pas excessivement les organes et les tissus, les tissus composites, utilisés de manière croissante dans les greffes, ne répondant à la définition d'aucune de ces deux catégories. Là encore, il a estimé qu'il fallait savoir résister à la puissance du lobby favorable à l'élargissement du cercle des donneurs vivants, malgré un risque médical élevé pour les donneurs et une possible dérive vers des pratique commerciales.
Mme Yvette Benayoun-Nakache et M. Alain Calmat ont souligné qu'il appartenait au Parlement, et à lui seul, de décider s'il convenait ou non de retenir la piste de recherche offerte par la méthode du clonage thérapeutique.
Le Président Bernard Charles a observé que, lors de l'adoption en Conseil des ministres, le 20 juin 2001, du projet de loi relatif à la bioéthique, le Premier ministre avait expressément confié au Parlement le soin de se prononcer sur cette question.
M. Alain Claeys, Rapporteur, a insisté sur la conception nouvelle qu'il entendait retenir du principe de précaution. Il a considéré que dans un monde ou l'inconnu dominait, seule l'amélioration des connaissances pouvait lever les incertitudes et permettre d'agir en connaissance de cause. Il a insisté sur le fait que le principe de précaution était un principe d'action qui permet, en disposant de davantage de données scientifiques, de faire des choix éclairés et, dans un deuxième temps, de les évaluer. Il a ajouté que, sur le clonage thérapeutique, le débat n'était certainement pas clos et devrait se poursuivre au sein de l'Assemblée nationale.
La Commission a ensuite autorisé la publication du rapport d'information et des comptes rendus des auditions.
ANNEXE 1
CONVENTION POUR LA PROTECTION DES DROITS DE L'HOMME ET DE LA DIGNITÉ DE L'ÊTRE HUMAIN À L'ÉGARD DES APPLICATIONS DE LA BIOLOGIE ET DE LA MÉDECINE (CONVENTION SUR LES DROITS DE L'HOMME ET LA BIOMÉDECINE SIGNÉE À OVIEDO LE 4 AVRIL 1997)
Préambule
Les États membres du Conseil de l'Europe, les autres États et la Communauté européenne signataires de la présente Convention,
Considérant la Déclaration universelle des Droits de l'Homme, proclamée par l'Assemblée générale des Nations Unies le 10 décembre 1948 ;
Considérant la Convention de sauvegarde des Droits de l'Homme et des Libertés fondamentales du 4 novembre 1950 ;
Considérant la Charte sociale européenne du 18 octobre 1961 ;
Considérant le Pacte international sur les droits civils et politiques et le Pacte international relatif aux droits économiques, sociaux et culturels du 16 décembre 1966 ;
Considérant la Convention pour la protection de l'individu à l'égard du traitement automatisé des données à caractère personnel du 28 janvier 1981 ;
Considérant également la Convention relative aux droits de l'enfant du 20 novembre 1989 ;
Considérant que le but du Conseil de l'Europe est de réaliser une union plus étroite entre ses membres, et que l'un des moyens d'atteindre ce but est la sauvegarde et le développement des droits de l'homme et des libertés fondamentales ;
Conscients des rapides développements de la biologie et de la médecine ;
Convaincus de la nécessité de respecter l'être humain à la fois comme individu et dans son appartenance à l'espèce humaine et reconnaissant l'importance d'assurer sa dignité ;
Conscients des actes qui pourraient mettre en danger la dignité humaine par un usage impropre de la biologie et de la médecine ;
Affirmant que les progrès de la biologie et de la médecine doivent être utilisés pour le bénéfice des générations présentes et futures ;
Soulignant la nécessité d'une coopération internationale pour que l'Humanité tout entière bénéficie de l'apport de la biologie et de la médecine;
Reconnaissant l'importance de promouvoir un débat public sur les questions posées par l'application de la biologie et de la médecine, et sur les réponses à y apporter ;
Désireux de rappeler à chaque membre du corps social ses droits et ses responsabilités ;
Prenant en considération les travaux de l'Assemblée parlementaire dans ce domaine, y compris la Recommandation 1160 (1991) sur l'élaboration d'une convention de bioéthique ;
Résolus à prendre, dans le domaine des applications de la biologie et de la médecine, les mesures propres à garantir la dignité de l'être humain et les droits et libertés fondamentaux de la personne,
Sont convenus de ce qui suit :
Chapitre I - Dispositions générales
Article 1 - Objet et finalité
Les Parties à la présente Convention protègent l'être humain dans sa dignité et son identité et garantissent à toute personne, sans discrimination, le respect de son intégrité et de ses autres droits et libertés fondamentales à l'égard des applications de la biologie et de la médecine.
Chaque Partie prend dans son droit interne les mesures nécessaires pour donner effet aux dispositions de la présente Convention.
Article 2 - Primauté de l'être humain
L'intérêt et le bien de l'être humain doivent prévaloir sur le seul intérêt de la société ou de la science.
Article 3 - Accès équitable aux soins de santé
Les Parties prennent, compte tenu des besoins de santé et des ressources disponibles, les mesures appropriées en vue d'assurer, dans leur sphère de juridiction, un accès équitable à des soins de santé de qualité appropriée.
Article 4 - Obligations professionnelles et règles de conduite
Toute intervention dans le domaine de la santé, y compris la recherche, doit être effectuée dans le respect des normes et obligations professionnelles, ainsi que des règles de conduite applicables en l'espèce.
Chapitre II - Consentement
Article 5 - Règle générale
Une intervention dans le domaine de la santé ne peut être effectuée qu'après que la personne concernée y a donné son consentement libre et éclairé.
Cette personne reçoit préalablement une information adéquate quant au but et à la nature de l'intervention ainsi que quant à ses conséquences et ses risques.
La personne concernée peut, à tout moment, librement retirer son consentement.
Article 6 - Protection des personnes n'ayant pas la capacité de consentir
1. Sous réserve des articles 17 et 20, une intervention ne peut être effectuée sur une personne n'ayant pas la capacité de consentir, que pour son bénéfice direct.
2. Lorsque, selon la loi, un mineur n'a pas la capacité de consentir à une intervention, celle-ci ne peut être effectuée sans l'autorisation de son représentant, d'une autorité ou d'une personne ou instance désignée par la loi.
L'avis du mineur est pris en considération comme un facteur de plus en plus déterminant, en fonction de son âge et de son degré de maturité.
3. Lorsque, selon la loi, un majeur n'a pas, en raison d'un handicap mental, d'une maladie ou pour un motif similaire, la capacité de consentir à une intervention, celle-ci ne peut être effectuée sans l'autorisation de son représentant, d'une autorité ou d'une personne ou instance désignée par la loi.
La personne concernée doit dans la mesure du possible être associée à la procédure d'autorisation.
4. Le représentant, l'autorité, la personne ou l'instance mentionnés aux paragraphes 2 et 3 reçoivent, dans les mêmes conditions, l'information visée à l'article 5.
5. L'autorisation visée aux paragraphes 2 et 3 peut, à tout moment, être retirée dans l'intérêt de la personne concernée.
Article 7 - Protection des personnes souffrant d'un trouble mental
La personne qui souffre d'un trouble mental grave ne peut être soumise, sans son consentement, à une intervention ayant pour objet de traiter ce trouble que lorsque l'absence d'un tel traitement risque d'être gravement préjudiciable à sa santé et sous réserve des conditions de protection prévues par la loi comprenant des procédures de surveillance et de contrôle ainsi que des voies de recours.
Article 8 - Situations d'urgence
Lorsqu'en raison d'une situation d'urgence le consentement approprié ne peut être obtenu, il pourra être procédé immédiatement à toute intervention médicalement indispensable pour le bénéfice de la santé de la personne concernée.
Article 9 - Souhaits précédemment exprimés
Les souhaits précédemment exprimés au sujet d'une intervention médicale par un patient qui, au moment de l'intervention, n'est pas en état d'exprimer sa volonté seront pris en compte.
Chapitre III - Vie privée et droit à l'information
Article 10 - Vie privée et droit à l'information
1. Toute personne a droit au respect de sa vie privée s'agissant des informations relatives à sa santé.
2. Toute personne a le droit de connaître toute information recueillie sur sa santé. Cependant, la volonté d'une personne de ne pas être informée doit être respectée.
3. À titre exceptionnel, la loi peut prévoir, dans l'intérêt du patient, des restrictions à l'exercice des droits mentionnés au paragraphe 2.
Chapitre IV - Génome humain
Article 11 - Non-discrimination
Toute forme de discrimination à l'encontre d'une personne en raison de son patrimoine génétique est interdite.
Article 12 - Tests génétiques prédictifs
Il ne pourra être procédé à des tests prédictifs de maladies génétiques ou permettant soit d'identifier le sujet comme porteur d'un gène responsable d'une maladie soit de détecter une prédisposition ou une susceptibilité génétique à une maladie qu'à des fins médicales ou de recherche médicale, et sous réserve d'un conseil génétique approprié.
Article 13 - Interventions sur le génome humain
Une intervention ayant pour objet de modifier le génome humain ne peut être entreprise que pour des raisons préventives, diagnostiques ou thérapeutiques et seulement si elle n'a pas pour but d'introduire une modification dans le génome de la descendance.
Article 14 - Non-sélection du sexe
L'utilisation des techniques d'assistance médicale à la procréation n'est pas admise pour choisir le sexe de l'enfant à naître, sauf en vue d'éviter une maladie héréditaire grave liée au sexe.
Chapitre V - Recherche scientifique
Article 15 - Règle générale
La recherche scientifique dans le domaine de la biologie et de la médecine s'exerce librement sous réserve des dispositions de la présente Convention et des autres dispositions juridiques qui assurent la protection de l'être humain.
Article 16 - Protection des personnes se prêtant à une recherche
Aucune recherche ne peut être entreprise sur une personne à moins que les conditions suivantes ne soient réunies :
i. il n'existe pas de méthode alternative à la recherche sur des êtres humains, d'efficacité comparable ;
ii. les risques qui peuvent être encourus par la personne ne sont pas disproportionnés par rapport aux bénéfices potentiels de la recherche ;
iii. le projet de recherche a été approuvé par l'instance compétente, après avoir fait l'objet d'un examen indépendant sur le plan de sa pertinence scientifique, y compris une évaluation de l'importance de l'objectif de la recherche, ainsi que d'un examen pluridisciplinaire de son acceptabilité sur le plan éthique ;
iv. la personne se prêtant à une recherche est informée de ses droits et des garanties prévues par la loi pour sa protection ;
v. le consentement visé à l'article 5 a été donné expressément, spécifiquement et est consigné par écrit. Ce consentement peut, à tout moment, être librement retiré.
Article 17 - Protection des personnes qui n'ont pas la capacité de consentir à une recherche
1. Une recherche ne peut être entreprise sur une personne n'ayant pas, conformément à l'article 5, la capacité d'y consentir que si les conditions suivantes sont réunies :
i. les conditions énoncées à l'article 16, alinéas i à iv, sont remplies ;
ii. les résultats attendus de la recherche comportent un bénéfice réel et direct pour sa santé ;
iii. la recherche ne peut s'effectuer avec une efficacité comparable sur des sujets capables d'y consentir ;
iv. l'autorisation prévue à l'article 6 a été donnée spécifiquement et par écrit ; et
v. la personne n'y oppose pas de refus.
2. A titre exceptionnel et dans les conditions de protection prévues par la loi, une recherche dont les résultats attendus ne comportent pas de bénéfice direct pour la santé de la personne peut être autorisée si les conditions énoncées aux alinéas i, iii, iv et v du paragraphe 1 ci-dessus ainsi que les conditions supplémentaires suivantes sont réunies :
i. la recherche a pour objet de contribuer, par une amélioration significative de la connaissance scientifique de l'état de la personne, de sa maladie ou de son trouble, à l'obtention, à terme, de résultats permettant un bénéfice pour la personne concernée ou pour d'autres personnes dans la même catégorie d'âge ou souffrant de la même maladie ou trouble ou présentant les mêmes caractéristiques ;
ii. la recherche ne présente pour la personne qu'un risque minimal et une contrainte minimale.
Article 18 - Recherche sur les embryons in vitro
1. Lorsque la recherche sur les embryons in vitro est admise par la loi, celle-ci assure une protection adéquate de l'embryon.
2. La constitution d'embryons humains aux fins de recherche est interdite.
Chapitre VI - Prélèvement d'organes et de tissus sur des donneurs vivants à des fins de transplantation
Article 19 - Règle générale
1. Le prélèvement d'organes ou de tissus aux fins de transplantation ne peut être effectué sur un donneur vivant que dans l'intérêt thérapeutique du receveur et lorsque l'on ne dispose pas d'organe ou de tissu appropriés d'une personne décédée ni de méthode thérapeutique alternative d'efficacité comparable.
2. Le consentement visé à l'article 5 doit avoir été donné expressément et spécifiquement, soit par écrit soit devant une instance officielle.
Article 20 - Protection des personnes qui n'ont pas la capacité de consentir au prélèvement d'organe
1. Aucun prélèvement d'organe ou de tissu ne peut être effectué sur une personne n'ayant pas la capacité de consentir conformément à l'article 5.
2. A titre exceptionnel et dans les conditions de protection prévues par la loi, le prélèvement de tissus régénérables sur une personne qui n'a pas la capacité de consentir peut être autorisé si les conditions suivantes sont réunies :
i. on ne dispose pas d'un donneur compatible jouissant de la capacité de consentir ;
ii. le receveur est un frère ou une s_ur du donneur ;
iii. le don doit être de nature à préserver la vie du receveur ;
iv. l'autorisation prévue aux paragraphes 2 et 3 de l'article 6 a été donnée spécifiquement et par écrit, selon la loi et en accord avec l'instance compétente,
v. le donneur potentiel n'y oppose pas de refus.
Chapitre VII - Interdiction du profit et utilisation d'une partie du corps humain
Article 21 - Interdiction du profit
Le corps humain et ses parties ne doivent pas être, en tant que tels, source de profit.
Article 22 - Utilisation d'une partie du corps humain prélevée
Lorsqu'une partie du corps humain a été prélevée au cours d'une intervention, elle ne peut être conservée et utilisée dans un but autre que celui pour lequel elle a été prélevée que conformément aux procédures d'information et de consentement appropriées.
Chapitre VIII - Atteinte aux dispositions de la Convention
Article 23 - Atteinte aux droits ou principes
Les Parties assurent une protection juridictionnelle appropriée afin d'empêcher ou faire cesser à bref délai une atteinte illicite aux droits et principes reconnus dans la présente Convention.
Article 24 - Réparation d'un dommage injustifié
La personne ayant subi un dommage injustifié résultant d'une intervention a droit à une réparation équitable dans les conditions et selon les modalités prévues par la loi.
Article 25 - Sanctions
Les Parties prévoient des sanctions appropriées dans les cas de manquement aux dispositions de la présente Convention.
Chapitre IX - Relation de la présente Convention avec d'autres dispositions
Article 26 - Restrictions à l'exercice des droits
1. L'exercice des droits et les dispositions de protection contenus dans la présente Convention ne peuvent faire l'objet d'autres restrictions que celles qui, prévues par la loi, constituent des mesures nécessaires, dans une société démocratique, à la sûreté publique, à la prévention des infractions pénales, à la protection de la santé publique ou à la protection des droits et libertés d'autrui.
2. Les restrictions visées à l'alinéa précédent ne peuvent être appliquées aux articles 11, 13, 14, 16, 17, 19, 20 et 21.
Article 27 - Protection plus étendue
Aucune des dispositions de la présente Convention ne sera interprétée comme limitant ou portant atteinte à la faculté pour chaque Partie d'accorder une protection plus étendue à l'égard des applications de la biologie et de la médecine que celle prévue par la présente Convention.
Chapitre X - Débat public
Article 28 - Débat public
Les Parties à la présente Convention veillent à ce que les questions fondamentales posées par les développements de la biologie et de la médecine fassent l'objet d'un débat public approprié à la lumière, en particulier, des implications médicales, sociales, économiques, éthiques et juridiques pertinentes, et que leurs possibles applications fassent l'objet de consultations appropriées.
Chapitre XI - Interprétation et suivi de la Convention
Article 29 - Interprétation de la Convention
La Cour européenne des Droits de l'Homme peut donner, en dehors de tout litige concret se déroulant devant une juridiction, des avis consultatifs sur des questions juridiques concernant l'interprétation de la présente Convention à la demande :
- du Gouvernement d'une Partie, après en avoir informé les autres Parties ;
- du Comité institué par l'article 32, dans sa composition restreinte aux Représentants des Parties à la présente Convention, par décision prise à la majorité des deux tiers des voix exprimées.
Article 30 - Rapports sur l'application de la Convention
Toute Partie fournira, sur demande du Secrétaire Général du Conseil de l'Europe, les explications requises sur la manière dont son droit interne assure l'application effective de toutes les dispositions de cette Convention.
Chapitre XII - Protocoles
Article 31 - Protocoles
Des protocoles peuvent être élaborés conformément aux dispositions de l'article 32, en vue de développer, dans des domaines spécifiques, les principes contenus dans la présente Convention.
Les protocoles sont ouverts à la signature des signataires de la Convention. Ils seront soumis à ratification, acceptation ou approbation. Un signataire ne peut ratifier, accepter ou approuver les protocoles sans avoir antérieurement ou simultanément ratifié, accepté ou approuvé la Convention.
Chapitre XIII - Amendements à la Convention
Article 32 - Amendements à la Convention
1. Les tâches confiées au «comité» dans le présent article et dans l'article 29 sont effectuées par le Comité directeur pour la bioéthique (CDBI), ou par tout autre comité désigné à cette fin par le Comité des Ministres.
2. Sans préjudice des dispositions spécifiques de l'article 29, tout État membre du Conseil de l'Europe ainsi que toute Partie à la présente Convention qui n'est pas membre du Conseil de l'Europe peut se faire représenter au sein du comité, lorsque celui-ci accomplit les tâches confiées par la présente Convention, et y dispose d'une voix.
3. Tout État visé à l'article 33 ou invité à adhérer à la Convention conformément aux dispositions de l'article 34, qui n'est pas Partie à la présente Convention, peut désigner un observateur auprès du comité. Si la Communauté européenne n'est pas Partie, elle peut désigner un observateur auprès du comité.
4. Afin de tenir compte des évolutions scientifiques, la présente Convention fera l'objet d'un examen au sein du comité dans un délai maximum de cinq ans après son entrée en vigueur, et par la suite à des intervalles que le comité pourra déterminer.
5. Toute proposition d'amendement à la présente Convention ainsi que toute proposition de protocole ou d'amendement à un protocole, présentée par une Partie, par le comité ou le Comité des Ministres, est communiquée au Secrétaire Général du Conseil de l'Europe et transmise par ses soins aux États membres du Conseil de l'Europe, à la Communauté européenne, à tout signataire, à toute Partie, à tout État invité à signer la présente Convention conformément aux dispositions de l'article 33, et à tout État invité à y adhérer conformément aux dispositions de l'article 34.
6. Le comité examine la proposition au plus tôt deux mois après qu'elle a été transmise par le Secrétaire Général conformément au paragraphe 5. Le Comité soumet le texte adopté à la majorité des deux tiers des voix exprimées à l'approbation du Comité des Ministres. Après son approbation, ce texte est communiqué aux Parties en vue de sa ratification, son acceptation ou son approbation.
7. Tout amendement entrera en vigueur, à l'égard des Parties qui l'ont accepté, le premier jour du mois qui suit l'expiration d'une période d'un mois après la date à laquelle cinq Parties, y compris au moins quatre États membres du Conseil de l'Europe, auront informé le Secrétaire Général qu'elles l'ont accepté.
Pour toute Partie qui l'aura accepté ultérieurement, l'amendement entrera en vigueur le premier jour du mois qui suit l'expiration d'une période d'un mois après la date à laquelle ladite Partie aura informé le Secrétaire Général de son acceptation.
Chapitre XIV - Clauses finales
Article 33 - Signature, ratification et entrée en vigueur
1. La présente Convention est ouverte à la signature des États membres du Conseil de l'Europe, des États non membres qui ont participé à son élaboration et de la Communauté européenne.
2. La présente Convention sera soumise à ratification, acceptation ou approbation. Les instruments de ratification, d'acceptation ou d'approbation seront déposés près le Secrétaire Général du Conseil de l'Europe.
3. La présente Convention entrera en vigueur le premier jour du mois qui suit l'expiration d'une période de trois mois après la date à laquelle cinq États, incluant au moins quatre États membres du Conseil de l'Europe, auront exprimé leur consentement à être liés par la Convention, conformément aux dispositions du paragraphe précédent.
4. Pour tout Signataire qui exprimera ultérieurement son consentement à être lié par la Convention, celle-ci entrera en vigueur le premier jour du mois qui suit l'expiration d'une période de trois mois après la date du dépôt de son instrument de ratification, d'acceptation ou d'approbation.
Article 34 - États non membres
1. Après l'entrée en vigueur de la présente Convention, le Comité des Ministres du Conseil de l'Europe pourra, après consultation des Parties, inviter tout État non membre du Conseil de l'Europe à adhérer à la présente Convention par une décision prise à la majorité prévue à l'article 20, alinéa d, du Statut du Conseil de l'Europe et à l'unanimité des voix des représentants des États contractants ayant le droit de siéger au Comité des Ministres.
2. Pour tout État adhérent, la Convention entrera en vigueur le premier jour du mois qui suit l'expiration d'une période de trois mois après la date du dépôt de l'instrument d'adhésion près le Secrétaire Général du Conseil de l'Europe.
Article 35 - Application territoriale
1. Tout Signataire peut, au moment de la signature ou au moment du dépôt de son instrument de ratification, d'acceptation ou d'approbation, désigner le territoire ou les territoires auxquels s'appliquera la présente Convention. Tout autre État peut formuler la même déclaration au moment du dépôt de son instrument d'adhésion.
2. Toute Partie peut, à tout moment par la suite, par une déclaration adressée au Secrétaire Général du Conseil de l'Europe, étendre l'application de la présente Convention à tout autre territoire désigné dans la déclaration et dont elle assure les relations internationales ou pour lequel elle est habilitée à stipuler. La Convention entrera en vigueur à l'égard de ce territoire le premier jour du mois qui suit l'expiration d'une période de trois mois après la date de réception de la déclaration par le Secrétaire Général.
3. Toute déclaration faite en vertu des deux paragraphes précédents pourra être retirée, en ce qui concerne tout territoire désigné dans cette déclaration, par notification adressée au Secrétaire Général. Le retrait prendra effet le premier jour du mois qui suit l'expiration d'une période de trois mois après la date de réception de la notification par le Secrétaire Général.
Article 36 - Réserves
1. Tout État et la Communauté européenne peuvent, au moment de la signature de la présente Convention ou du dépôt de l'instrument de ratification, d'acceptation, d'approbation ou d'adhésion, formuler une réserve au sujet d'une disposition particulière de la Convention, dans la mesure où une loi alors en vigueur sur son territoire n'est pas conforme à cette disposition. Les réserves de caractère général ne sont pas autorisées aux termes du présent article.
2. Toute réserve émise conformément au présent article comporte un bref exposé de la loi pertinente.
3. Toute Partie qui étend l'application de la présente Convention à un territoire désigné par une déclaration prévue en application du paragraphe 2 de l'article 35 peut, pour le territoire concerné, formuler une réserve, conformément aux dispositions des paragraphes précédents.
4. Toute Partie qui a formulé la réserve visée dans le présent article peut la retirer au moyen d'une déclaration adressée au Secrétaire Général du Conseil de l'Europe. Le retrait prendra effet le premier jour du mois qui suit l'expiration d'une période d'un mois après la date de réception par le Secrétaire Général.
Article 37 - Dénonciation
1. Toute Partie peut, à tout moment, dénoncer la présente Convention en adressant une notification au Secrétaire Général du Conseil de l'Europe.
2. La dénonciation prendra effet le premier jour du mois qui suit l'expiration d'une période de trois mois après la date de réception de la notification par le Secrétaire Général.
Article 38 - Notifications
Le Secrétaire Général du Conseil de l'Europe notifiera aux États membres du Conseil, à la Communauté européenne, à tout Signataire, à toute Partie et à tout autre État qui a été invité à adhérer à la présente Convention :
a. toute signature ;
b. le dépôt de tout instrument de ratification, d'acceptation, d'approbation ou d'adhésion ;
c. toute date d'entrée en vigueur de la présente Convention, conformément à ses articles 33 ou 34 ;
d. tout amendement ou protocole adopté conformément à l'article 32, et la date à laquelle cet amendement ou protocole entre en vigueur ;
e. toute déclaration formulée en vertu des dispositions de l'article 35 ;
f. toute réserve et tout retrait de réserve formulés conformément aux dispositions de l'article 36 ;
g. tout autre acte, notification ou communication ayant trait à la présente Convention.
En foi de quoi, les soussignés, dûment autorisés à cet effet, ont signé la présente Convention.
ANNEXE 2
PROTOCOLE ADDITIONNEL DU 12 JANVIER 1998 À LA CONVENTION POUR LA PROTECTION DES DROITS DE L'HOMME ET DE LA DIGNITÉ DE L'ÊTRE HUMAIN À L'ÉGARD DES APPLICATIONS DE LA BIOLOGIE ET DE LA MÉDECINE, PORTANT INTERDICTION DU CLONAGE D'ÊTRES HUMAINS
Les États membres du Conseil de l'Europe, les autres États et la Communauté européenne, signataires du présent Protocole additionnel à la Convention pour la protection des Droits de l'Homme et de la dignité de l'être humain à l'égard des applications de la biologie et de la médecine,
Prenant acte des développements scientifiques intervenus en matière de clonage de mammifères, en particulier par la division embryonnaire et par le transfert de noyau ;
Conscients des progrès que certaines techniques de clonage peuvent, en elles-mêmes, apporter à la connaissance scientifique ainsi qu'à ses applications médicales ;
Considérant que le clonage d'êtres humains pourrait devenir une possibilité technique ;
Ayant noté que la division embryonnaire peut se produire naturellement et donner lieu parfois à la naissance de jumeaux génétiquement identiques ;
Considérant cependant que l'instrumentalisation de l'être humain par la création délibérée d'êtres humains génétiquement identiques est contraire à la dignité de l'homme et constitue un usage impropre de la biologie et de la médecine ;
Considérant également les grandes difficultés d'ordre médical, psychologique et social qu'une telle pratique biomédicale, employée délibérément, pourrait impliquer pour toutes les personnes concernées ;
Considérant l'objet de la Convention sur les Droits de l'Homme et la biomédecine, en particulier le principe énoncé à l'article 1 visant à protéger l'être humain dans sa dignité et son identité,
Sont convenus de ce qui suit :
Article 1
1. Est interdite toute intervention ayant pour but de créer un être humain génétiquement identique à un autre être humain vivant ou mort.
2. Au sens du présent article, l'expression être humain «génétiquement identique» à un autre être humain signifie un être humain ayant en commun avec un autre l'ensemble des gènes nucléaires.
Article 2
Aucune dérogation n'est autorisée aux dispositions du présent Protocole au titre de l'article 26, paragraphe 1, de la Convention.
Article 3
Les Parties considèrent les articles 1 et 2 du présent Protocole comme des articles additionnels à la Convention et toutes les dispositions de la Convention s'appliquent en conséquence.
Article 4
Le présent Protocole est ouvert à la signature des Signataires de la Convention. Il sera soumis à ratification, acceptation ou approbation. Un Signataire ne peut ratifier, accepter ou approuver le présent Protocole sans avoir antérieurement ou simultanément ratifié, accepté ou approuvé la Convention. Les instruments de ratification, d'acceptation ou d'approbation seront déposés près le Secrétaire Général du Conseil de l'Europe.
Article 5
1. Le présent Protocole entrera en vigueur le premier jour du mois qui suit l'expiration d'une période de trois mois après la date à laquelle cinq États, incluant au moins quatre États membres du Conseil de l'Europe, auront exprimé leur consentement à être liés par le Protocole conformément aux dispositions de l'article 4.
2. Pour tout Signataire qui exprimera ultérieurement son consentement à être lié par le Protocole, celui-ci entrera en vigueur le premier jour du mois qui suit l'expiration d'une période de trois mois après la date du dépôt de l'instrument de ratification, d'acceptation ou d'approbation.
Article 6
1. Après l'entrée en vigueur du présent Protocole, tout État qui a adhéré à la Convention pourra adhérer également au présent Protocole.
2. L'adhésion s'effectuera par le dépôt, près le Secrétaire Général du Conseil de l'Europe, d'un instrument d'adhésion qui prendra effet le premier jour du mois qui suit l'expiration d'une période de trois mois après la date de son dépôt.
Article 7
1. Toute Partie peut, à tout moment, dénoncer le présent Protocole en adressant une notification au Secrétaire Général du Conseil de l'Europe.
2. La dénonciation prendra effet le premier jour du mois qui suit l'expiration d'une période de trois mois après la date de réception de la notification par le Secrétaire Général.
Article 8
Le Secrétaire Général du Conseil de l'Europe notifiera aux États membres du Conseil de l'Europe, à la Communauté européenne, à tout Signataire, à toute Partie et à tout autre État qui a été invité à adhérer à la Convention :
a. toute signature ;
b. le dépôt de tout instrument de ratification, d'acceptation, d'approbation ou d'adhésion ;
c. toute date d'entrée en vigueur du présent Protocole conformément à ses articles 5 et 6 ;
d. tout autre acte, notification ou communication ayant trait au présent Protocole.
En foi de quoi, les soussignés, dûment autorisés à cet effet, ont signé le présent Protocole.
ANNEXE 3
DÉCLARATION UNIVERSELLE SUR LE GÉNOME HUMAIN
ET LES DROITS DE L'HOMME DU 11 NOVEMBRE 1997
La Conférence générale,
Rappelant que le Préambule de l'Acte constitutif de l'UNESCO invoque "l'idéal démocratique de dignité, d'égalité et de respect de la personne humaine" et rejette tout "dogme de l'inégalité des races et des hommes", qu'il précise "que, la dignité de l'homme exigeant la diffusion de la culture et l'éducation de tous en vue de la justice, de la liberté et de la paix, il y a là, pour toutes les nations, des devoirs sacrés à remplir dans un esprit de mutuelle assistance", qu'il proclame que "cette paix doit être établie sur le fondement de la solidarité intellectuelle et morale de l'humanité", et qu'il indique que l'Organisation cherche à atteindre "par la coopération des nations du monde dans les domaines de l'éducation, de la science, et de la culture, les buts de paix internationale et de prospérité commune de l'humanité en vue desquels l'Organisation des Nations Unies a été constituée, et que sa Charte proclame",
Rappelant solennellement son attachement aux principes universels des droits de l'homme affirmés, en particulier, par la Déclaration universelle des droits de l'homme du 10 décembre 1948 et les deux Pactes internationaux des Nations Unies relatifs aux droits économiques, sociaux et culturels, et aux droits civils et politiques du 16 décembre 1966, la Convention des Nations Unies pour la prévention et la répression du crime de génocide du 9 décembre 1948, la Convention internationale des Nations Unies sur l'élimination de toutes les formes de discrimination raciale du 21 décembre 1965, la Déclaration des Nations Unies sur les droits du déficient mental du 20 décembre 1971, la Déclaration des Nations Unies sur les droits des personnes handicapées du 9 décembre 1975, la Convention des Nations Unies sur l'élimination de toutes les formes de discrimination à l'égard des femmes du 18 décembre 1979, la Déclaration des Nations Unies sur les principes fondamentaux de justice relatifs aux victimes de la criminalité et aux victimes d'abus de pouvoir du 29 novembre 1985, la Convention des Nations Unies relative aux droits de l'enfant du 20 novembre 1989, les Règles des Nations Unies pour l'égalisation des chances des handicapés du 20 décembre 1993, la Convention sur l'interdiction de la mise au point, de la fabrication et du stockage des armes bactériologiques (biologiques) ou à toxines et sur leur destruction du 16 décembre 1971, la Convention de l'UNESCO concernant la lutte contre la discrimination dans le domaine de l'enseignement du 14 décembre 1960, la Déclaration de l'UNESCO des principes de la coopération culturelle internationale du 4 novembre 1966, la Recommandation de l'UNESCO concernant la condition des chercheurs scientifiques du 20 novembre 1974, la Déclaration de l'UNESCO sur la race et les préjugés raciaux du 27 novembre 1978, la Convention de l'OIT (Nº 111) concernant la discrimination en matière d'emploi et de profession du 25 juin 1958 et la Convention de l'OIT (Nº 169) concernant les peuples indigènes et tribaux dans les pays indépendants du 27 juin 1989,
Ayant à l'esprit, et sans préjudice de leurs dispositions, les instruments internationaux susceptibles d'intéresser les applications de la génétique dans le domaine de la propriété intellectuelle, notamment, la Convention de Berne pour la protection des oeuvres littéraires et artistiques du 9 septembre 1886 et la Convention universelle de l'UNESCO sur le droit d'auteur du 6 septembre 1952, révisées en dernier lieu à Paris le 24 juillet 1971, la Convention de Paris pour la protection de la propriété industrielle du 20 mars 1883, révisée en dernier lieu à Stockholm le 14 juillet 1967, le Traité de Budapest de l'OMPI sur la reconnaissance internationale du dépôt des micro-organismes aux fins de procédure en matière de brevets du 28 avril 1977, et l'Accord relatif aux aspects des droits de propriété intellectuelle qui touchent au commerce (ADPIC) annexé à l'accord établissant l'Organisation mondiale du commerce entré en vigueur le 1er janvier 1995,
Ayant également à l'esprit la Convention des Nations Unies sur la diversité biologique du 5 juin 1992 et soulignant à cet égard que la reconnaissance de la diversité génétique de l'humanité, ne doit donner lieu à aucune interprétation d'ordre social ou politique de nature à remettre en cause "la dignité inhérente à tous les membres de la famille humaine et de leurs droits égaux et inaliénables", conformément au Préambule de la Déclaration universelle des droits de l'homme,
Rappelant ses résolutions 22 C/13.1, 23 C/13.1, 24 C/13.1, 25 C/5.2, 25 C/7.3, 27 C/5.15, 28 C/0.12, 28 C/2.1 et 28 C/2.2 engageant l'UNESCO à promouvoir et développer la réflexion éthique et les actions qui s'y rattachent, en ce qui concerne les conséquences des progrès scientifiques et techniques dans les domaines de la biologie et de la génétique, dans le cadre du respect des droits de l'homme et des libertés fondamentales,
Reconnaissant que les recherches sur le génome humain et leurs applications ouvrent d'immenses perspectives d'amélioration de la santé des individus et de l'humanité tout entière, mais soulignant qu'elles doivent en même temps respecter pleinement la dignité, la liberté et les droits de l'homme, ainsi que l'interdiction de toute forme de discrimination fondée sur les caractéristiques génétiques,
Proclame les principes qui suivent et adopte la présente Déclaration.
A. LA DIGNITÉ HUMAINE ET LE GÉNOME HUMAIN
Article premier
Le génome humain sous-tend l'unité fondamentale de tous les membres de la famille humaine, ainsi que la reconnaissance de leur dignité intrinsèque et de leur diversité. Dans un sens symbolique, il est le patrimoine de l'humanité.
Article 2
a) Chaque individu a droit au respect de sa dignité et de ses droits, quelles que soient ses caractéristiques génétiques.
b) Cette dignité impose de ne pas réduire les individus à leurs caractéristiques génétiques et de respecter le caractère unique de chacun et leur diversité.
Article 3
Le génome humain, par nature évolutif, est sujet à des mutations. Il renferme des potentialités qui s'expriment différemment selon l'environnement naturel et social de chaque individu, en ce qui concerne notamment l'état de santé, les conditions de vie, la nutrition et l'éducation.
Article 4
Le génome humain en son état naturel ne peut donner lieu à des gains pécuniaires.
B. DROITS DES PERSONNES CONCERNÉES
Article 5
a) Une recherche, un traitement ou un diagnostic, portant sur le génome d'un individu, ne peut être effectué qu'après une évaluation rigoureuse et préalable des risques et avantages potentiels qui leur sont liés et en conformité avec toutes autres prescriptions prévues par la législation nationale.
b) Dans tous les cas, le consentement préalable, libre et éclairé de l'intéressé(e) sera recueilli. Si ce(tte) dernier(e) n'est pas en mesure de l'exprimer, le consentement ou l'autorisation seront obtenus conformément à la loi et seront guidés par son intérêt supérieur.
c) Le droit de chacun de décider d'être informé ou non des résultats d'un examen génétique et de ses conséquences devrait être respecté.
d) Dans le cas de la recherche, les protocoles de recherche doivent être soumis, de plus, à une évaluation préalable, conformément aux normes ou lignes directrices nationales et internationales applicables en la matière.
e) Si conformément à la loi une personne n'est pas en mesure d'exprimer son consentement, une recherche portant sur son génome ne peut être effectuée qu'au bénéfice direct de sa santé, sous réserve des autorisations et des mesures de protection prescrites par la loi. Une recherche ne permettant pas d'escompter un bénéfice direct pour la santé ne peut être effectuée qu'à titre exceptionnel, avec la plus grande retenue, en veillant à n'exposer l'intéressé(e) qu'à un risque et une contrainte minimums, et si cette recherche est effectuée dans l'intérêt de la santé d'autres personnes appartenant au même groupe d'âge ou se trouvant dans les mêmes conditions génétiques, et sous réserve qu'une telle recherche se fasse dans les conditions prévues par la loi et soit compatible avec la protection des droits individuels de la personne concernée.
Article 6
Nul ne doit faire l'objet de discriminations fondées sur ses caractéristiques génétiques, qui auraient pour objet ou pour effet de porter atteinte à ses droits individuels et à ses libertés fondamentales et à la reconnaissance de sa dignité.
Article 7
La confidentialité des données génétiques associées à une personne identifiable, conservées ou traitées à des fins de recherche ou dans tout autre but, doit être protégée dans les conditions prévues par la loi.
Article 8
Tout individu a droit, conformément au droit international et au droit interne, à une réparation équitable du dommage qu'il aurait subi et dont la cause directe et déterminante serait une intervention portant sur son génome.
Article 9
Pour protéger les droits de l'homme et les libertés fondamentales, des limitations aux principes du consentement et de la confidentialité ne peuvent être apportées que par la loi, pour des raisons impérieuses et dans les limites du droit international public et du droit international des droits de l'homme.
C. RECHERCHES SUR LE GÉNOME HUMAIN
Article 10
Aucune recherche concernant le génome humain, ni aucune de ses applications, en particulier dans les domaines de la biologie, de la génétique et de la médecine, ne devrait prévaloir sur le respect des droits de l'homme, des libertés fondamentales et de la dignité humaine des individus ou, le cas échéant, de groupes d'individus.
Article 11
Des pratiques qui sont contraires à la dignité humaine, telles que le clonage à des fins de reproduction d'êtres humains, ne doivent pas être permises. Les États et les organisations internationales compétentes sont invités à coopérer afin d'identifier de telles pratiques et de prendre, au niveau national ou international, les mesures qui s'imposent, conformément aux principes énoncés dans la présente Déclaration.
Article 12
a) Chacun doit avoir accès aux progrès de la biologie, de la génétique et de la médecine, concernant le génome humain, dans le respect de sa dignité et de ses droits.
b) La liberté de la recherche, qui est nécessaire au progrès de la connaissance, procède de la liberté de pensée. Les applications de la recherche, notamment celles en biologie, en génétique et en médecine, concernant le génome humain, doivent tendre à l'allégement de la souffrance et à l'amélioration de la santé de l'individu et de l'humanité tout entière.
D. CONDITIONS D'EXERCICE DE L'ACTIVITÉ SCIENTIFIQUE
Article 13
Les responsabilités inhérentes aux activités des chercheurs, notamment la rigueur, la prudence, l'honnêteté intellectuelle et l'intégrité, dans la conduite de leurs recherches ainsi que dans la présentation et l'utilisation de leurs résultats, devraient faire l'objet d'une attention particulière dans le cadre des recherches sur le génome humain, compte tenu de leurs implications éthiques et sociales. Les décideurs publics et privés en matière de politiques scientifiques ont aussi des responsabilités particulières à cet égard.
Article 14
Les États devraient prendre les mesures appropriées pour favoriser les conditions intellectuelles et matérielles propices au libre exercice des activités de recherche sur le génome humain et pour prendre en considération les implications éthiques, juridiques, sociales et économiques de ces recherches, dans le cadre des principes prévus par la présente Déclaration.
Article 15
Les États devraient prendre les mesures appropriées pour fixer le cadre du libre exercice des activités de recherche sur le génome humain dans le respect des principes prévus par la présente Déclaration, afin de garantir le respect des droits de l'homme, des libertés fondamentales et de la dignité humaine et la protection de la santé publique. Ils devraient chercher à s'assurer que les résultats de ces recherches ne servent pas à des fins non pacifiques.
Article 16
Les États devraient reconnaître l'intérêt de promouvoir, aux différents niveaux appropriés, la création de comités d'éthique indépendants, pluridisciplinaires et pluralistes, chargés d'apprécier les questions éthiques, juridiques et sociales soulevées par les recherches sur le génome humain et leurs applications.
E. SOLIDARITÉ ET COOPÉRATION INTERNATIONALE
Article 17
Les États devraient respecter et promouvoir une solidarité active vis-à-vis des individus, des familles ou des populations particulièrement vulnérables aux maladies ou handicaps de nature génétique, ou atteints de ceux-ci. Ils devraient notamment encourager les recherches destinées à identifier, à prévenir et à traiter les maladies d'ordre génétique ou les maladies influencées par la génétique, en particulier les maladies rares ainsi que les maladies endémiques qui affectent une part importante de la population mondiale.
Article 18
Les États devraient s'efforcer, dans le respect des principes prévus par la présente Déclaration, de continuer à favoriser la diffusion internationale de la connaissance scientifique sur le génome humain, sur la diversité humaine et sur les recherches en génétique et, à cet égard, à favoriser la coopération scientifique et culturelle, notamment entre pays industrialisés et pays en développement.
Article 19
a) Dans le cadre de la coopération internationale avec les pays en développement, les États devraient s'efforcer d'encourager des mesures visant à :
1. évaluer les risques et les avantages liés aux recherches sur le génome humain et prévenir les abus ;
2. étendre et renforcer la capacité des pays en développement de mener des recherches en biologie et en génétique humaines, compte tenu de leurs problèmes spécifiques ;
3. permettre aux pays en développement de bénéficier des avancées de la recherche scientifique et technologique, de façon à favoriser le progrès économique et social au profit de tous ;
4. favoriser le libre échange de la connaissance et de l'information scientifiques, dans les domaines de la biologie, de la génétique et de la médecine, soit encouragé.
5. b) Les organisations internationales compétentes devraient soutenir et promouvoir les initiatives prises par les États aux fins énumérées ci-dessus.
6. F. PROMOTION DES PRINCIPES DE LA DÉCLARATION
7. Article 20
8. Les États devraient prendre les mesures appropriées pour promouvoir les principes énoncés dans la Déclaration, par l'éducation et les moyens pertinents, notamment par la conduite de recherches et de formations dans des domaines interdisciplinaires et par la promotion de l'éducation à la bioéthique à tous les niveaux, en particulier à l'intention des différents responsables de politiques scientifiques.
9. Article 21
10. Les États devraient prendre les mesures appropriées pour encourager toutes autres actions de recherche, de formation et de diffusion de l'information de nature à renforcer la prise de conscience des responsabilités de la société et de chacun de ses membres face aux problèmes fondamentaux au regard de la défense de la dignité humaine que peuvent soulever la recherche dans les domaines de la biologie, de la génétique et de la médecine, et les applications qui en découlent. Ils devraient favoriser sur ce sujet un débat largement ouvert sur le plan international, assurant la libre expression des différents courants de pensée socioculturels, religieux et philosophiques.
11. G. MISE EN OEUVRE DE LA DÉCLARATION
12. Article 22
13. Les États devraient s'efforcer de promouvoir les principes énoncés dans la présente Déclaration et, par toutes mesures appropriées, favoriser leur mise en _uvre.
14. Article 23
15. Les États devraient prendre les mesures appropriées pour promouvoir, par l'éducation, la formation et la diffusion de l'information, le respect des principes ci-dessus énoncés et favoriser leur reconnaissance et leur application effective. Les États devraient également encourager les échanges entre les comités d'éthique indépendants, quand ils existent, et leur mise en réseaux, afin de favoriser la coopération entre eux.
16. Article 24
17. Le Comité international de bioéthique de l'UNESCO devrait contribuer à la diffusion des principes énoncés dans la présente Déclaration et à l'approfondissement des questions que posent leurs applications et l'évolution des techniques en cause. Il devrait organiser toute consultation utile avec les parties concernées telles que les groupes vulnérables. Il devrait formuler, suivant les procédures statutaires de l'UNESCO, des recommandations à l'intention de la Conférence générale et des avis quant au suivi de la Déclaration, en particulier quant à l'identification des pratiques qui pourraient être contraires à la dignité humaine, telles que les interventions sur la lignée germinale.
18. Article 25
19. Aucune disposition de la présente Déclaration ne peut être interprétée comme pouvant être invoquée de quelque façon par un État, un groupement ou un individu pour se livrer à une activité ou accomplir un acte à des fins contraires aux droits de l'homme et aux libertés fondamentales, y compris aux principes énoncés dans la présente Déclaration.
ANNEXE 4
DIRECTIVE 98/44/CE DU PARLEMENT EUROPÉEN ET DU CONSEIL DU 6 JUILLET 1998 RELATIVE À LA PROTECTION JURIDIQUE
DES INVENTIONS BIOTECHNOLOGIQUES
LE PARLEMENT EUROPEEN ET LE CONSEIL DE L'UNION EUROPEENNE,
vu le traité instituant la Communauté européenne, et notamment son article 100 A,
vu la proposition de la Commission,
vu l'avis du Comité économique et social,
statuant conformément à la procédure visée à l'article 189 B du traité
(1) considérant que la biotechnologie et le génie génétique jouent un rôle croissant dans un nombre considérable d'activités industrielles ; que la protection des inventions biotechnologiques revêtira certainement une importance essentielle pour le développement industriel de la Communauté ;
(2) considérant que, notamment, dans le domaine du génie génétique, la recherche et le développement exigent une somme considérable d'investissements à haut risque que seule une protection juridique adéquate peut permettre de rentabiliser ;
(3) considérant qu'une protection efficace et harmonisée dans l'ensemble des États membres est essentielle en vue de préserver et d'encourager les investissements dans le domaine de la biotechnologie ;
(4) considérant que, à la suite du rejet par le Parlement européen du projet commun, approuvé par le comité de conciliation, de directive du Parlement européen et du Conseil relative à la protection juridique des inventions biotechnologiques, le Parlement européen et le Conseil ont constaté que la protection juridique des inventions biotechnologiques avait besoin d'être clarifiée ;
(5) considérant qu'il existe des divergences, dans le domaine de la protection des inventions biotechnologiques, entre les législations et pratiques des différents États membres ; que de telles disparités sont de nature à créer des entraves aux échanges et à faire ainsi obstacle au fonctionnement du marché intérieur ;
(6) considérant que ces divergences risquent de s'accentuer au fur et à mesure que les États membres adopteront de nouvelles lois et pratiques administratives différentes ou que les interprétations jurisprudentielles nationales se développeront diversement ;
(7) considérant qu'une évolution hétérogène des législations nationales relatives à la protection juridique des inventions biotechnologiques dans la Communauté risque de décourager encore plus les échanges commerciaux, au détriment du développement industriel de ces inventions et du bon fonctionnement du marché intérieur ;
(8) considérant que la protection juridique des inventions biotechnologiques ne nécessite pas la création d'un droit particulier se substituant au droit national des brevets ; que le droit national des brevets reste la référence essentielle pour la protection juridique des inventions biotechnologiques, étant entendu qu'il doit être adapté ou complété sur certains points spécifiques pour tenir compte de façon adéquate de l'évolution de la technologie faisant usage de matière biologique, mais répondant néanmoins aux conditions de brevetabilité ;
(9) considérant que, dans certains cas, comme celui de l'exclusion de la brevetabilité des variétés végétales et des races animales ainsi que des procédés essentiellement biologiques d'obtention de végétaux ou d'animaux, certaines notions des législations nationales, fondées sur les conventions internationales relatives aux brevets et aux variétés végétales, ont suscité des incertitudes concernant la protection des inventions biotechnologiques et de certaines inventions microbiologiques ; que, dans ce domaine, l'harmonisation est nécessaire pour dissiper ces incertitudes ;
(10) considérant qu'il convient de prendre en compte le potentiel de développement des biotechnologies pour l'environnement et en particulier l'utilité de ces technologies pour le développement de méthodes culturales moins polluantes et plus économes des sols ; qu'il convient d'encourager, par le système des brevets, la recherche et la mise en oeuvre de tels procédés ;
11) considérant que le développement des biotechnologies est important pour les pays en voie de développement, tant dans le domaine de la santé et de la lutte contre les grandes épidémies et endémies que dans le domaine de la lutte contre la faim dans le monde ; qu'il convient d'encourager de même, par le système des brevets, la recherche dans ces domaines ; qu'il convient par ailleurs de promouvoir des mécanismes internationaux assurant la diffusion de ces technologies dans le tiers monde et au profit des populations concernées ;
(12) considérant que l'accord sur les aspects des droits de propriété intellectuelle qui touchent au commerce (ADPIC), signé par la Communauté européenne et ses États membres est entré en vigueur ; que cet accord prévoit que la protection conférée par un brevet doit être assurée pour les produits et les procédés dans tous les domaines de la technologie ;
(13) considérant que le cadre juridique communautaire pour la protection des inventions biotechnologiques peut se limiter à la définition de certains principes applicables à la brevetabilité de la matière biologique en tant que telle, principes avant notamment pour but de déterminer la différence entre inventions et découvertes à propos de la brevetabilité de certains éléments d'origine humaine, à l'étendue de la protection conférée par un brevet sur une invention biotechnologique, à la possibilité de recourir à un système de dépôt complétant la description écrite et, enfin, à la possibilité d'obtenir des licences obligatoires non exclusives pour dépendance entre des variétés végétales et des inventions, et inversement ;
(14) considérant qu'un brevet d'invention n'autorise pas son titulaire à mettre l'invention en _uvre, mais se borne à lui conférer le droit d'interdire aux tiers de l'exploiter à des fins industrielles et commerciales ; que, dès lors, le droit des brevets n'est pas susceptible de remplacer ni de rendre superflues les législations nationales, européennes ou internationales, fixant d'éventuelles limitations ou interdictions, ou organisant un contrôle de la recherche et de l'utilisation ou de la commercialisation de ses résultats, notamment par rapport aux exigences de santé publique, de sécurité, de protection de l'environnement, de protection des animaux, de préservation de la diversité génétique et par rapport au respect de certaines normes éthiques ;
(15) considérant que ni le droit national ni le droit européen des brevets (convention de Munich) ne comportent, en principe, d'interdiction ou d'exclusion frappant la brevetabilité de la matière biologique ;
(16) considérant que le droit des brevets doit s'exercer dans le respect des principes fondamentaux garantissant la dignité et l'intégrité de l'Homme ; qu'il importe de réaffirmer le principe selon lequel le corps humain, dans toutes les phases de sa constitution et de son développement, cellules germinales comprises, ainsi que la simple découverte d'un de ses éléments ou d'un de ses produits, y compris la séquence ou séquence partielle d'un gène humain, ne sont pas brevetables ; que ces principes sont conformes aux critères de brevetabilité prévus par le droit des brevets, critères selon lesquels une simple découverte ne peut faire l'objet d'un brevet ;
(17) considérant que des progrès décisifs dans le traitement des maladies ont d'ores et déjà pu être réalisés grâce à l'existence de médicaments dérivés d'éléments isolés du corps humain et/ou autrement produits, médicaments résultant de procédés techniques visant à obtenir des éléments d'une structure semblable à celle d'éléments naturels existant dans le corps humain ; que, dès lors, il convient d'encourager, par le système des brevets, la recherche tendant à obtenir et à isoler de tels éléments précieux pour la production de médicaments ;
(18) considérant que, dans la mesure où le système des brevets s'avère insuffisant pour inciter à la recherche et à la production de médicaments issus de biotechnologies et nécessaires pour lutter contre les maladies rares ou dites « orphelines », la Communauté et les États membres ont l'obligation d'apporter une réponse adéquate à ce problème ;
(19) considérant que l'avis n°8 du groupe de conseillers pour l'éthique de la biotechnologie de la Commission européenne a été pris en compte ;
(20) considérant, en conséquence, qu'il est nécessaire d'indiquer qu'une invention qui porte sur un élément isolé du corps humain ou autrement produit par un procédé technique, et qui est susceptible d'application industrielle, n'est pas exclue de la brevetabilité, même si la structure de cet élément est identique à celle d'un élément naturel, étant entendu que les droits conférés par le brevet ne s'étendent pas au corps humain et à ses éléments dans leur environnement naturel ;
(21) considérant qu'un tel élément isolé du corps humain ou autrement produit n'est pas exclu de la brevetabilité puisqu'il est, par exemple, le résultat de procédés techniques l'ayant identifié, purifié, caractérisé et multiplié en dehors du corps humain, techniques que seul l'être humain est capable de mettre en _uvre et que la nature est incapable d'accomplir par elle-même ;
(22) considérant que le débat sur la brevetabilité de séquences ou de séquences partielles de gènes donne lieu à des controverses, que, aux termes de la présente directive, l'octroi d'un brevet à des inventions portant sur de telles séquences ou séquences partielles doit être soumis aux mêmes critères de brevetabilité que pour tous les autres domaines technologiques, nouveauté, activité inventive et application industrielle ; que l'application industrielle d'une séquence ou d'une séquence partielle doit être exposée de façon concrète dans la demande de brevet telle que déposée ;
(23) considérant qu'une simple séquence d'ADN sans indication d'une fonction ne contient aucun enseignement technique ; qu'elle ne saurait, par conséquent, constituer une invention brevetable ;
(24) considérant que, pour que le critère d'application industrielle soit respecté, il est nécessaire, dans le cas où une séquence ou une séquence partielle d'un gène est utilisée pour la production d'une protéine ou d'une protéine partielle, de préciser quelle protéine ou protéine partielle est produite ou quelle fonction elle assure ;
(25) considérant, pour l'interprétation des droits conférés par un brevet, que lorsque des séquences se chevauchent seulement dans les parties qui ne sont pas essentielles à l'invention, le droit des brevets considère chacune d'entre elles comme une séquence autonome ;
(26) considérant que, si une invention porte sur une matière biologique d'origine humaine ou utilise une telle matière, dans le cadre du dépôt d'une demande de brevet, la personne sur laquelle le prélèvement est effectué doit avoir eu l'occasion d'exprimer son consentement éclairé et libre à celui-ci, conformément au droit national ;
(27) considérant que, si une invention porte sur une matière biologique d'origine végétale ou animale ou utilise une telle matière, la demande de brevet devrait, le cas échéant, comporter une information concernant le lieu géographique d'origine de cette matière, si celui-ci est connu ; que ceci est sans préjudice de l'examen des demandes de brevet et de la validité des droits résultant des brevets délivrés ;
(28) considérant que la présente directive n'affecte en rien les fondements du droit des brevets en vigueur selon lequel un brevet peut être accordé pour toute nouvelle application d'un produit déjà breveté ;
(29) considérant que la présente directive ne concerne pas l'exclusion de la brevetabilité des variétés végétales et des races animales. que, en revanche, les inventions portant sur des plantes ou des animaux sont brevetables si leur application n'est pas techniquement limitée à une variété végétale ou à une race animale ;
(30) considérant que la notion de variété végétale est définie par la législation relative à la protection des obtentions végétales ; que, selon ce droit, une obtention est caractérisée par l'intégralité de son génome et qu'elle est par conséquent individualisée et se différencie nettement d'autres obtentions ;
(31) considérant qu'un ensemble végétal caractérisé par un gène déterminé (et non par l'intégralité de son génome) n'est pas soumis à la protection des obtentions ; que, de ce fait, il n'est pas exclu de la brevetabilité, même lorsqu'il englobe des obtentions végétales ;
(32) considérant que, si l'invention se borne à modifier génétiquement une variété végétale déterminée et si une nouvelle variété végétale est obtenue, elle reste exclue de la brevetabilité, même lorsque cette modification génétique n'est pas le résultat d'un procédé essentiellement biologique mais d'un procédé biotechnologique ;
(33) considérant qu'il est nécessaire de définir aux fins de la présente directive quand un procédé d'obtention de végétaux ou d'animaux est essentiellement biologique ;
(34) considérant que la présente directive n'affecte pas les notions d'invention et de découverte telles que déterminées par le droit des brevets, que celui-ci soit national, européen ou international ;
(35) considérant que la présente directive n'affecte pas les dispositions des législations nationales en matière de brevets selon lesquelles les procédés de traitement chirurgical ou thérapeutique du corps humain ou animal et les méthodes de diagnostic pratiquées sur l'organisme humain ou animal sont exclus de la brevetabilité ;
(36) considérant que l'accord ADPIC prévoit, pour les membres de l'Organisation mondiale du commerce, la possibilité d'exclure de la brevetabilité les inventions dont il est nécessaire d'empêcher l'exploitation commerciale sur leur territoire pour protéger l'ordre public ou la moralité, y compris pour protéger la santé et la vie des personnes et des animaux ou préserver les végétaux, ou pour éviter de graves atteintes à l'environnement, à condition que cette exclusion ne tienne pas uniquement au fait que l'exploitation est interdite par leur législation ;
(37) considérant que la présente directive se doit d'insister sur le principe selon lequel des inventions dont l'exploitation commerciale serait contraire à l'ordre public ou aux bonnes moeurs doivent être exclues de la brevetabilité ;
(38) considérant qu'il importe aussi de mentionner dans le dispositif de la présente directive une liste indicative des inventions exclues de la brevetabilité afin de donner aux juges et aux offices de brevets nationaux des orientations générales aux fins de l'interprétation de la référence à l'ordre public ou aux bonnes moeurs ; que cette liste ne saurait bien entendu prétendre à l'exhaustivité ; que les procédés dont l'application porte atteinte à la dignité humaine, comme par exemple les procédés de production d'êtres hybrides, issus de cellules germinales ou de cellules totipotentes humaines et animales, doivent, bien évidemment, être exclus eux aussi de la brevetabilité ;
(39) considérant que l'ordre public et les bonnes moeurs correspondent notamment à des principes éthiques ou moraux reconnus dans un État membre, dont le respect s'impose tout particulièrement en matière de biotechnologie en raison de la portée potentielle des inventions dans ce domaine et de leur lien inhérent avec la matière vivante ; que ces principes éthiques ou moraux, complètent les examens juridiques normaux de la législation sur les brevets, quel que soit le domaine technique de l'invention ;
(40) considérant qu'un consensus existe au sein de la Communauté quant au fait que l'intervention génique germinale sur l'homme et le clonage de l'être humain sont contraires à l'ordre public et aux bonnes moeurs ; qu'il importe par conséquent d'exclure sans équivoque de la brevetabilité les procédés de modification de l'identité génétique germinale de l'être humaine et les procédés de clonage des êtres humains ;
(41) considérant que les procédés de clonage des êtres humains peuvent se définir comme tout procédé, y compris les techniques de scission des embryons, ayant pour but de créer un être humain qui aurait la même information génétique nucléaire qu'un autre être humain vivant ou décédé ;
(42) considérant, en outre, que les utilisations d'embryons humains à des fins industrielles ou commerciales doivent également être exclues de la brevetabilité ; que, en tout état de cause, une telle exclusion ne concerne pas les inventions ayant un objectif thérapeutique ou de diagnostic qui s'appliquent à l'embryon humain et lui sont utiles ;
(43) considérant que l'article F, paragraphe 2, du traité sur l'Union européenne prévoit que l'Union respecte les droits fondamentaux, tels qu'ils sont garantis par la Convention européenne de sauvegarde des droits de l'Homme et des libertés fondamentales, signée à Rome le 4 novembre 1950, et tels qu'ils résultent des traditions constitutionnelles communes aux États membres, en tant que principes généraux du droit communautaire ;
(44) considérant que le Groupe européen d'éthique des sciences et des nouvelles technologies de la Commission évalue tous les aspects éthiques liés à la biotechnologie ; que, à cet égard, il convient de remarquer que la consultation de ce groupe, y compris en ce qui concerne le droit des brevets, ne peut se situer qu'au niveau de l'évaluation de la biotechnologie au regard des principes éthiques fondamentaux ;
(45) considérant que les procédés de modification de l'identité génétique des animaux de nature à provoquer chez eux des souffrances sans utilité médicale substantielle dans le domaine de la recherche, de la prévention, du diagnostic ou de la thérapeutique, pour l'homme ou l'animal, ainsi que les animaux issus de tels procédés, doivent être exclus de la brevetabilité ;
(46) considérant que, le brevet ayant pour fonction de récompenser l'inventeur par l'octroi d'un droit exclusif, mais limité dans le temps, au titre de sa créativité, et d'encourager ainsi l'activité inventive, le titulaire du brevet doit avoir le droit d'interdire l'utilisation d'une matière autoreproductible brevetée dans des circonstances analogues à celles où l'utilisation de produits brevetés non autoreproductibles pourrait être interdite, c'est-à-dire la production du produit breveté lui-même ;
(47) considérant qu'il est nécessaire de prévoir une première dérogation aux droits du titulaire du brevet lorsque du matériel de reproduction incorporant l'invention protégée est vendu à un agriculteur à des fins d'exploitation agricole par le titulaire du brevet ou avec son consentement ; que cette première dérogation doit autoriser l'agriculteur à utiliser le produit de sa récolte pour reproduction ou multiplication ultérieure sur sa propre exploitation et que l'étendue et les modalités de cette dérogation doivent être limitées à l'étendue et aux modalités prévues par le règlement (CE) n°2100/94 du Conseil du 27 juillet 1994 instituant un régime de protection communautaire des obtentions végétales ;
(48) considérant que seule la rémunération envisagée par le droit communautaire des obtentions végétales en tant que modalité d'application de la dérogation à la protection communautaire des obtentions végétales peut être exigée de l'agriculteur ;
(49) considérant, cependant, que le titulaire du brevet peut défendre ses droits contre l'agriculteur abusant de la dérogation ou contre l'obtenteur qui a développé la variété végétale incorporant l'invention protégée si celui-ci ne respecte pas ses engagements ;
(50) considérant qu'une deuxième dérogation aux droits du titulaire du brevet doit autoriser l'agriculteur à utiliser le bétail protégé à un usage agricole ;
(51) considérant que l'étendue et les modalités de cette deuxième dérogation doivent être réglées par les lois, les dispositions réglementaires et les pratiques nationales, en l'absence de législation communautaire concernant l'obtention de races animales ;
(52) considérant que, dans le domaine de l'exploitation des nouvelles caractéristiques végétales issues du génie génétique, un accès garanti moyennant rémunération doit être accordé sous forme de licence obligatoire lorsque, par rapport au genre ou à l'espèce concerné, la variété végétale représente un progrès technique important d'un intérêt économique considérable par rapport à l'invention revendiquée dans le brevet ;
(53) considérant que, dans le domaine de l'utilisation en génie génétique de nouvelles caractéristiques végétales issues de nouvelles variétés végétales, un accès garanti moyennant rémunération doit être accordé sous forme de licence obligatoire lorsque l'invention représente un progrès technique important d'un intérêt économique considérable ;
(54) considérant que l'article 34 de l'accord ADPIC contient une réglementation détaillée de la charge de la preuve qui s'impose à tous les États membres ; que, par conséquent, il n'y a pas lieu de prévoir dans la présente directive une disposition à ce sujet ;
(55) considérant que la Communauté, à la suite de la décision 93/626/CEE, est partie à la convention sur la diversité biologique du 5 juin 1992 ; que, à cet égard, les États membres, dans le cadre de la mise en vigueur des dispositions législatives, réglementaires et administratives nécessaires pour se conformer à la présente directive, tiennent compte notamment de l'article 3, de l'article 8, point j), et de l'article 16, paragraphe 2, deuxième phrase, et paragraphe 5, de ladite convention ;
(56) considérant que la troisième conférence des parties signataires de la convention sur la diversité biologique, qui s'est tenue en novembre 1996, a reconnu, dans la décision III/17, que «des travaux supplémentaires sont nécessaires pour contribuer au développement d'une appréciation commune de la relation entre les droits de propriété intellectuelle et les dispositions afférentes de l'accord sur les aspects commerciaux des droits de propriété intellectuelle et de la convention sur la diversité biologique, notamment sur les questions relatives aux transferts de technologies, la conservation et l'utilisation durable de la biodiversité et le partage équitable des bénéfices de l'utilisation des ressources génétiques, y compris la protection des connaissances, innovations et pratiques des communautés indigènes et locales incarnant des modes de vie traditionnels importants pour la conservation et l'utilisation durable de la biodiversité»,
ONT ARRÊTÉ LA PRÉSENTE DIRECTIVE :
CHAPITRE I - Brevetabilité
Article premier
1. Les États membres protègent les inventions biotechnologiques au moyen de leur droit national des brevets. Ils adaptent leur droit national des brevets, si nécessaire, pour tenir compte des dispositions de la présente directive.
2. La présente directive n'affecte pas les obligations découlant, pour les États membres, des conventions internationales, et notamment de l'accord ADPIC et de la convention sur la diversité biologique.
Article 2
1. Aux fins de la présente directive, on entend par :
a) «matière biologique» : une matière contenant des informations génétiques et qui est autoreproductible ou reproductible dans un système biologique ;
b) «procédé microbiologique» : tout procédé utilisant une matière microbiologique, comportant une intervention sur une matière microbiologique ou produisant une matière microbiologique.
2. Un procédé d'obtention de végétaux ou d'animaux est essentiellement biologique s'il consiste intégralement en des phénomènes naturels tels que le croisement ou la sélection.
3. La notion de variété végétale est définie à l'article 5 du règlement (CE) n°2100/94.
Article 3
1. Aux fins de la présente directive, sont brevetables les inventions nouvelles, impliquant une activité inventive et susceptibles d'application industrielle, même lorsqu'elles portent sur un produit composé de matière biologique ou en contenant, ou sur un procédé permettant de produire, de traiter ou d'utiliser de la matière biologique.
2. Une matière biologique isolée de son environnement naturel ou produite à l'aide d'un procédé technique peut être l'objet d'une invention, même lorsqu'elle préexistait à l'état naturel.
Article 4
1. Ne sont pas brevetables :
a) les variétés végétales et les races animales ;
b) les procédés essentiellement biologiques pour l'obtention de végétaux ou d'animaux.
2. Les inventions portant sur des végétaux ou des animaux sont brevetables si la faisabilité technique de l'invention n'est pas limitée à une variété végétale ou à une race animale déterminée.
3. Le paragraphe 1, point b), n'affecte pas la brevetabilité d'inventions avant pour objet un procédé microbiologique, ou d'autres procédés techniques, ou un produit obtenu par ces procédés.
Article 5
1. Le corps humain, aux différents stades de sa constitution et de son développement, ainsi que la simple découverte d'un de ses éléments, y compris la séquence ou la séquence partielle d'un gène, ne peuvent constituer des inventions brevetables.
2. Un élément isolé du corps humain ou autrement produit par un procédé technique, y compris la séquence ou la séquence partielle d'un gène, peut constituer une invention brevetable, même si la structure de cet élément est identique à celle d'un élément naturel.
3. L'application industrielle d'une séquence ou d'une séquence partielle d'un gène doit être concrètement exposée dans la demande de brevet.
Article 6
1. Les inventions dont l'exploitation commerciale serait contraire à l'ordre public ou aux bonnes moeurs sont exclues de la brevetabilité, l'exploitation ne pouvant être considérée comme telle du seul fait qu'elle est interdite par une disposition légale ou réglementaire.
2. Au titre du paragraphe 1 ne sont notamment pas
brevetables :
a) les procédés de clonage des êtres humains ;
b) les procédés de modification de l'identité génétique germinale de l'être humain ;
c) les utilisations d'embryons humains à des fins industrielles ou commerciales ;
d) les procédés de modification de l'identité génétique des animaux de nature à provoquer chez eux des souffrances sans utilité médicale substantielle pour l'homme ou l'animal, ainsi que les animaux issus de tels procédés.
Article 7
Le Groupe européen d'éthique des sciences et des nouvelles technologies de la Commission évalue tous les aspects éthiques liés à la biotechnologie.
CHAPITRE II - Etendue de la protection
Article 8
1. La protection conférée par un brevet relatif à une matière biologique dotée, du fait de l'invention, de propriétés déterminées s'étend à toute matière biologique obtenue à partir de cette matière biologique par reproduction ou multiplication sous forme identique ou différenciée et dotée de ces mêmes propriétés.
2. La protection conférée par un brevet relatif à un procédé permettant de produire une matière biologique dotée, du fait de l'invention, de propriétés déterminées s'étend à la matière biologique directement obtenue par ce procédé et à toute autre matière biologique obtenue, à partir de la matière biologique directement obtenue, par reproduction ou multiplication sous forme identique ou différenciée et dotée de ces mêmes propriétés.
Article 9
La protection conférée par un brevet à un produit contenant une information génétique ou consistant en une information génétique s'étend à toute matière, sous réserve de l'article 5, paragraphe 1, dans laquelle le produit est incorporé et dans laquelle l'information génétique est contenue et exerce sa fonction.
Article 10
La protection visée aux articles 8 et 9 ne s'étend pas à la matière biologique obtenue par reproduction ou multiplication d'une matière biologique mise sur le marché sur le territoire d'un État membre par le titulaire du brevet ou avec son consentement, lorsque la reproduction ou la multiplication résulte nécessairement de l'utilisation pour laquelle la matière biologique a été mise sur le marché, pourvu que la matière obtenue ne soit pas utilisée ensuite pour d'autres reproductions ou multiplications.
Article 11
1. Par dérogation aux articles 8 et 9, la vente ou une autre forme de commercialisation de matériel de reproduction végétal par le titulaire du brevet ou avec son consentement à un agriculteur à des fins d'exploitation agricole implique pour celui-ci l'autorisation d'utiliser le produit de sa récolte pour reproduction ou multiplication par lui-même sur sa propre exploitation, l'étendue et les modalités de cette dérogation correspondant à celles prévues à l'article 14 du règlement (CE) n°2100/94.
2. Par dérogation aux articles 8 et 9, la vente ou une autre forme de commercialisation d'animaux d'élevage ou autre matériel de reproduction animal par le titulaire du brevet ou avec son consentement à un agriculteur implique pour celui-ci l'autorisation d'utiliser le bétail protégé à un usage agricole. Ceci inclut la mise à disposition de l'animal ou autre matériel de reproduction animal pour la poursuite de son activité agricole, mais non la vente dans le cadre ou le but d'une activité de reproduction commerciale.
3. L'étendue et les modalités de la dérogation prévue au paragraphe 2 sont régies par les lois, les dispositions réglementaires et les pratiques nationales.
CHAPITRE III - Licences obligatoires pour dépendance
Article 12
1. Lorsqu'un obtenteur ne peut obtenir ou exploiter un droit d'obtention végétale sans porter atteinte à un brevet antérieur, il peut demander une licence obligatoire pour l'exploitation non exclusive de l'invention protégée par ce brevet, dans la mesure où cette licence est nécessaire pour l'exploitation de la variété végétale à protéger, moyennant une redevance appropriée. Les États membres prévoient que, lorsqu'une telle licence est accordée, le titulaire' du brevet a droit à une licence réciproque à des conditions raisonnables pour utiliser la variété protégée.
2. Lorsque le titulaire d'un brevet concernant une invention biotechnologique ne peut exploiter celle-ci sans porter atteinte à un droit d'obtention végétale antérieur sur une variété, il peut demander une licence obligatoire pour l'exploitation non exclusive de la variété protégée par ce droit d'obtention, moyennant une redevance appropriée. Les États membres prévoient que, lorsqu'une telle licence est accordée, le titulaire du droit d'obtention a droit à une licence réciproque à des conditions raisonnables pour utiliser l'invention protégée.
3. Les demandeurs des licences visées aux paragraphes 1 et 2 doivent établir :
a) qu'ils se sont vainement adressés au titulaire du brevet ou du droit d'obtention végétale pour obtenir une licence contractuelle ;
b) que la variété ou l'invention représente un progrès technique important d'un intérêt économique considérable par rapport à l'invention revendiquée dans le brevet ou à la variété végétale protégée.
4. Chaque État membre désigne la ou les autorités compétentes pour octroyer la licence. Lorsqu'une licence sur une variété végétale ne peut être octroyée que par l'Office communautaire des variétés végétales, l'article 29 du règlement (CE) n°2100/94 s'applique.
CHAPITRE IV - Dépôt d'une matière biologique, accès à une telle matière et nouveau dépôt
Article 13
1. Lorsqu'une invention porte sur de la matière biologique non accessible au public et ne pouvant être décrite dans la demande de brevet pour permettre à une personne du métier de réaliser l'invention, ou implique l'utilisation d'une telle matière, la description n'est réputée suffisante pour l'application du droit des brevets que si :
a) la matière biologique a été déposée au plus tard le jour du dépôt de la demande de brevet auprès d'une institution de dépôt reconnue. Sont reconnues au moins les institutions de dépôt internationales ayant acquis ce statut conformément à l'article 7 du traité de Budapest du 28 avril 1977 sur la reconnaissance internationale du dépôt de micro-organismes aux fins de la procédure en matière de brevets, ci-après dénommé «traité de Budapest» ;
b) la demande déposée contient les informations pertinentes dont dispose le déposant sur les caractéristiques de la matière biologique déposée ;
c) la demande de brevet mentionne l'institution de dépôt et le numéro de dépôt.
2. L'accès à la matière biologique déposée est assuré par la remise d'un échantillon :
a) jusqu'à la première publication de la demande de brevet, uniquement aux personnes autorisées en vertu du droit national des brevets ;
b) entre la première publication de là demande de brevet et la délivrance du brevet, à toute personne qui en fait la requête ou, si le déposant le demande, uniquement à un expert indépendant ;
c) après la délivrance du brevet et nonobstant une révocation ou annulation du brevet, à toute personne qui en fait la requête.
3. La remise n'a lieu que si le requérant s'engage, pour la durée des effets du brevet :
a) à ne communiquer à des tiers aucun échantillon de la matière biologique déposée ou d'une matière qui en serait dérivée
et
b) à n'utiliser aucun échantillon de la matière biologique déposée ou d'une matière qui en serait dérivée, sauf à des fins expérimentales, à moins que le demandeur ou le titulaire du brevet ne renonce expressément à un tel engagement.
4. En cas de rejet ou de retrait de la demande, l'accès à la matière déposée est limité, à la demande du déposant, à un expert indépendant pendant vingt ans à compter de la date de dépôt de la demande de brevet. Dans ce cas, les dispositions du paragraphe 3 sont applicables.
5. Les demandes du déposant visées au paragraphe 2, point b), et au paragraphe 4 ne peuvent être introduites que jusqu'à la date où les préparatifs techniques de la publication de la demande de brevet sont réputés achevés.
Article 14
1. Lorsque la matière biologique déposée conformément à l'article 13 cesse d'être disponible auprès de l'institution de dépôt reconnue, un nouveau dépôt de là matière est autorisé dans les mêmes conditions que celles prévues par le traité de Budapest.
2. Tout nouveau dépôt doit être accompagné d'une déclaration signée par le déposant certifiant que la matière biologique qui fait l'objet du nouveau dépôt est la même que celle qui faisait l'objet du dépôt initial.
CHAPITRE V - Dispositions finales
Article 15
1. Les États membres mettent en vigueur les dispositions législatives, réglementaires et administratives nécessaires pour se conformer à la présente directive au plus tard le 30 juillet 2000. Ils en informent immédiatement la Commission.
Lorsque les États membres adoptent ces dispositions, celles-ci contiennent une référence à la présente directive ou sont accompagnées d'une telle référence lors de leur publication officielle. Les modalités de cette référence sont arrêtées par les États membres.
2. Les États membres communiquent à la Commission le texte des dispositions de droit interne qu'ils adoptent dans le domaine régi par la présente directive.
Article 16
La Commission transmet au Parlement européen et au Conseil :
a) tous les cinq ans à compter de la date prévue à l'article 15, paragraphe 1, un rapport sur la question de savoir si la présente directive a soulevé des problèmes au regard des accords internationaux sur la protection des droits de l'homme, auxquels les États membres ont adhéré ;
b) dans un délai de deux ans après l'entrée en vigueur de la présente directive, un rapport tendant à évaluer les implications dans le domaine de la recherche fondamentale en génie génétique de la non-publication ou publication tardive de documents dont l'objet pourrait être brevetable ;
c) tous les ans à compter de la date prévue à l'article 15, paragraphe 1, un rapport sur l'évolution et les implications du droit des brevets dans le domaine de la biotechnologie et du génie génétique.
Article 17
La présente directive entre en vigueur le jour de sa publication au Journal officiel des Communautés européennes.
Article 18
Les États membres sont destinataires de la présente directive.
ANNEXE 5
PROGRAMME DE LA MISSION DU RAPPORTEUR
AUX ÉTATS-UNIS DU 9 AU 11 AVRIL 2001
LUNDI 9 AVRIL
7h45 - 8h45 : Réunion à l'Ambassade de France : la propriété intellectuelle aux États-Unis
Jeffrey KUSHAN, Avocat, Cabinet Powell, Goldstein, Frazer & Murphy LLP
Marie-Hélène FORGET, responsable du service juridique (poste d'expansion économique de l'Ambassade)
Olivier MANDEL, chargé des problèmes de propriété intellectuelle (poste d'expansion économique de l'Ambassade)
9h30 - 11h00 : Center for Biologics Evaluation and Research (CBER) - U.S. Food and Drug Administration : la réglementation des nouvelles techniques en biologie
Dr. Kathryn ZOON, Director of CBER
11h30 - 15h00 : National Institutes of Health (NIH) - Office of Technology Transfer (OTT)
11h45 : Ethical, Legal, Social Implications of Human Genetic Research (ELSI), National Human Genome Research Institute (NHGRI) : enjeux scientifiques, juridiques, sociaux et éthiques de la recherche en génomique
Alan GUTTMACHER, Elizabeth THOMSON
12h45 : Office of Biotechnology Activities, Office of Research Policy : les tests génétiques
Susanne HAGA, Amy PATTERSON
13h45 : Office of Technology Transfer (OTT) : la brevetabilité des gènes
Ted ROUMEL, Jack SPIEGEL
16h00 - 17h00 : U.S. Department Of Energy - Human Genome Program - Office of Biological and Environmental Research
Dr. Ari PATRINOS, Associate Director
20h00 : Dîner à la Résidence du Ministre Conseiller
Jean-François BOITTIN, Ministre Conseiller pour les Affaires Economiques et Commerciales
Jacques BESNAINOU, Président d'Emergent BioNet (entreprise de biotechnologie)
MARDI 10 AVRIL
9h00 - 10h30 : Genetic Alliance : les maladies génétiques
Mary DAVIDSON, Executive Director
Sharon TERRY, Vice President for Consumers
11h00 - 12h30 : American Association for the Advancement of Science
Albert H. TEICH, Director, Science & Policy Programs
Mark FRANKEL, Scientific Freedom, Responsibility and Law
Audrey CHAPMAN, Dialogue on Science, Ethics and Religion
13h00 - 14h30 : Déjeuner Ambassade
Jeremy RIFKIN, President of the Foundation on Economic Trends
15h00 - 16h00 : Clairus Technologies (entreprise de biotechnologie)
Martha J. CONNOLLY, Ph.D., Senior VP of Licensing
20h00 : Dîner : Les post-docs en biotechnologies aux États-Unis
Franck LEMIALE
Francois BOUDSOCQ
MERCREDI 11 AVRIL
9h00 - 10h00 : Human Genome Science
Dr. William HASELTINE, chairman
10h30 - 12h00 : Celera Genomics
Dr. Paul GILMAN, Director of Public Policy
13h30 - 15h00 : United States Patent and Trademark Office (USPTO)
John DOLL, Director, Examining Groups 1630 & 1650, Biotechnology, Organic Chemistry & Designs
15h45 - 17h00 : BIO
Michael WERNER, Bioethics Counsel
17h20 - 18h20 : WHITE HOUSE (Administration Clinton)
Rachel LEVINSON, Assistant Director for Life Sciences
_____________
N° 3208.- Rapport d'information de M. Alain Claeys, déposé en application de l'article 145 du Règlement par la mission d'information commune préparatoire au projet de loi de révision des « lois bioéthiques » de juillet 1994 (tome I, rapport).
() Rapport n° 2198 de MM. Alain Claeys, député, et Claude Huriet, sénateur.
() Rapport n° 1407 de MM. Alain Claeys, député, et Claude Huriet, sénateur.
() Conseil d'État, Les lois bioéthiques : cinq ans après, La documentation française, 1999, 337 pages.
() Max Weber, Le savant et le politique, Plon, 1959 (1ère édition 1919).
() Voir Claude Huriet, Rapport d'information n° 267 du 6 avril 2001, sur le fonctionnement des comités consultatifs de protection des personnes dans la recherche biomédicale.
() Axel Kahn, Et l'homme dans tout ça ? Plaidoyer pour un humanisme moderne, Nil éditions, 2000.
() Voir « Clonage humain : la machine à fantasmes », Le Point, 17 janvier 1998, p. 64-71.
() Les scientifiques contestent d'ailleurs le fait qu'un être cloné soit totalement identique à celui dont la cellule provient, l'influence du cytoplasme de l'ovule receveur pouvant être importante bien qu'elle soit encore mal définie.
() Rapport du Conseil d'État, op. cit., p. 14.
() Sur les différentes techniques de clonage, on renverra au rapport n° 2198 de l'Office parlementaire d'évaluation des choix scientifiques et technologiques.
() Libération, 17 février 1998.
() Libération, 12 février 2001.
() Noëlle Lenoir et Bertrand Mathieu, Les normes internationales de la bioéthique, PUF, « Que sais-je ? », 1998. Les développements suivants doivent beaucoup à cet ouvrage auquel on renverra volontiers.
() Expression que l'on peut traduire par « droit souple ou mou ».
() Sur l'influence de la « soft law » dans l'élaboration du droit international : voir René-Jean Dupuy, Droit déclaratoire et droit proclamatoire : de la coutume sauvage à la « soft law », colloque de la société française de droit international, 1975.
() Noëlle Lenoir et Bertrand Mathieu, op. cit., p. 32 et suivantes.
() Voir Noëlle Lenoir et Bertrand Mathieu, op. cit., p. 115 et suivantes.
() On renverra pour plus de détails à l'étude du Conseil d'État, La norme internationale en droit français, La Documentation française, 2000.
() Cass. chambre mixte, 24 mai 1975, Société des cafés Jacques Vabre ; CE, Assemblée, 20 octobre 1989, Nicolo.
() Pour les règlements communautaires : CE, 24 septembre 1990, Boisdet ; pour les directives : CE, Assemblée, 28 février 1992, SA Rothmans International France et SA Philip Morris.
() Source : Le Bilan du monde 2000, Éditions Le Monde, p. 33.
() Source : Rapport annuel du génopole d'Evry, 1999, p. 6.
() Source : Pharmaceutical Research and Manufacturers Association.
() Source : Le Bilan du monde 2000, Éditions Le Monde, p. 24.
() Sur ce sujet, on se reportera au rapport présenté au Conseil économique et social en 1999 par MM. Philippe Rouvillois et Guy Le Fur, La France face au défi des biotechnologies : quels enjeux pour l'avenir ?
() Les Échos, lundi 8 janvier 2001, p. 51.
() Source : Rapport annuel du génopole d'Évry, 1999, p. 4.
() La FIVNAT représente par ses adhérents plus de 90 % de l'ensemble des activités d'AMP.
() Limite qui était jusqu'à présent fixée au 43ème anniversaire de la femme.
() Nomenclature des actes professionnels des médecins, chirurgiens-dentistes, sages-femmes et auxilliaires médicaux et nomenclature des actes de biologie médicale.
() Étude conduite par A. Bachelot et J. de Mouzon, de l'Unité 292.
() Technique de fécondation in vitro par micro-injection intracytoplasmique.
() Rapport précité intitulé : Sciences de la vie : de l'éthique au droit.
() Voir le procès-verbal de l'audition de Mme Chantal Ramogida, p. 12.
() Dans l'équipe des professeurs A. Van Steirteghem et Paul Devroey.
() Ce qui correspondrait à environ 10 % des cas d'infertilité féminine.
() Selon le Docteur Charles Nahmanovici du Centre de FIV Saint Georges à Nice, dans sa communication du 9 décembre 2000 lors de la journée nationale de la fertilité organisée par l'association « Pauline et Adrien ».
() Voir les statistiques présentées dans le premier chapitre de la présente partie.
() Les données du recensement 1999 sont en cours d'analyse et celles de l'an 2000 sont en cours de recensement.
() Dans son ouvrage : Concevoir l'embryon, Éd. Masson, 2000.
() Dans ses commentaires sur le décret le 8 décembre 1999.
() Respectivement à l'intérieur du couple ou avec recours à un tiers donneur.
() En cas d'échec des tentatives d'implantation des embryons, la patiente doit renouveler sa demande d'ovocytes et attendre à nouveau deux ans.
() 82 % des membres du GEDO sont des CECOS.
() « Le don d'ovocytes depuis la loi de bioéthique. Implications médicales, éthiques et juridiques déduites d'une série de 300 cas à l'hôpital de Tenon ».
() Sachant que pour cette activité précise, il n'existe pas d'indice de besoins.
() Directions départementales des affaires sanitaires et sociales.
() Renouvellement des autorisations d'activité des centres d'AMP qui a lieu tous les cinq ans.
() Choisi par le ministre chargé de la santé sur une liste de trois personnes établie par le président de l'Union nationale des associations familiales (UNAF).
() Au Royaume-Uni, la femme la plus âgée à avoir mis au monde en recourant à l'AMP avait cinquante-neuf ans.
() Soit 17,43 millions de francs ou 2,66 millions d'euros.
() Voir son article « Peut-on, doit-on expérimenter sur les gamètes et l'embryon humain ? », Ethique, n° 12, 1994.
() Laboratoire pharmaceutique.
() Directeur de l'Unité de recherche en maturation gamatique et fécondation.
() Directeur de l'Unité de recherche sur les handicaps génétiques de l'enfant.
() Selon les termes mêmes de la secte des Raëliens, évoquée dans la première partie, qui préconise la mise au point du clonage reproductif humain.
() Mésoderme : feuillet moyen du blastoderme qui formera le tissu de soutien, les muscles, les organes géronto-urinaires, le système cardiovasculaire, le sang et l'éphithélium de la cavité coelonique. Endoderme : feuillet interne du blastoderme qui formera la muqueuse intestinale et les glandes annexes. Ectoderme : feuillet externe du blastoderme qui formera le revêtement cutané et les organes des sens d'une part, le système nerveux central et les nerfs périphériques, d'autre part.
() Il s'agit de l'équipe du Professeur Gérard Michaud à l'Université Pierre et Marie Curie.
() Relatives à l'endothélium, tissu recouvrant la paroi interne des vaisseaux et du c_ur.
() Cellules de soutien du système nerveux central.
() Cellules du parenchyme (tissu fonctionnel de l'organe) hépatique qui assure les fonctions exocrine et endocrine du foie.
() Article paru dans Le Quotidien du Médecin du 3 avril 2000.
() Lors de l'audition du Docteur Marie-Odile Alnot.
() Unité 429 de développement normal et pathologique du système immunitaire.
() Enfants atteints du déficit immunitaire combiné sévère lié au chromosome X.
() Relatives à la moelle épinière.
() Relatif au cortex, partie supérieure du cerveau.
() Qui agit par libération d'un médiateur chimique transmettant l'influx nerveux au niveau des synapses.
() Il s'agit en l'espèce de fibroblastes.
() Selon le Docteur Marina Cavazzano-Calvo, l'enfant sur lequel la thérapie a échoué était trop sévèrement malade lorsqu'il a subi le traitement.
() Dans son article paru le 3 avril 2000 dans le Quotidien du médecin.
() Charles Thibault, « Cellules ES, cellules EG, cellules souches. Où en est-on ?, Bull. de la Soc. franç. d'étude de la stérilité, 8, 1, 2000.
() Cette notion étai présente dans les travaux préparatoires à la loi de 1991, en particulier dans le rapport de Lady Warnock.
() C'est-à-dire un agrément pour une activité d'AMP.
() Agence française de sécurité sanitaire des produits de santé.
() Gérard Benoît in Robert Carvais (dir), La greffe humaine, (in)certitudes éthiques : du don de soi à la tolérance de l'autre), PUF, 2000.
() Le Monde (J-Y Nau, 24 février 1988 ; Franck Nouchi, 25 février 1988, 1er mars 1988).
() D'après J-Y Nau, La mort violée (Le Monde du 18 mai 1992).
() Les récits et la légende des vols d'organes, expression des réticences face à la greffe, in Robert Carvais (dir), La greffe humaine, (In)certitudes éthiques : du don de soi à la tolérance de l'autre, PUF, p. 357 et sq.
() Encyclopaedia Universalis, 1999.
() Dominique Thouvenin, Les lois n° 94-548 du 1er juillet 1994, n° 94-653 et n° 94-654 du 29 juillet 1994, ou comment construire un droit de la bioéthique.
() Et non plus thérapeutique depuis l'adoption de la loi n° 99-641 du 27 juillet 1999 portant création d'une couverture maladie universelle.
() Audition de Mmes Allain-Régnault, Hélène Cardin et Marianne Gomez, 7 juin 2000.
() Gérard Benoît, op- cit, p. 276.
() Audition par la Mission le 10 janvier 2001.
() Didier Houssin, L'aventure de la greffe, Denoël, p. 102.
() Audition du 10 janvier 2001.
() Audition du 10 janvier 2001.
() Gérard Benoît, op.cit, p. 220.
() Audition du 10 janvier 2001.
() Audition du 10 janvier 2001.
() Audition du 10 janvier 2001.
() In Robert Carvais (dir.), op. cit., p. 773.
() Francis Kernaleguen, « Le consentement à l'acte médical », Bulletin juridique de l'Ouest, 1999.
() Article 7, texte adopté n° 675, Assemblée nationale.
() Thierry Fossier, « Malades mentaux et éthique médicale », in Jean-François Mattéi (dir.), Philosophie, éthique et droit de la médecine, Thémis Philosophie, PUF, p. 305.
() Francis Kernaleguen, op.cit.
() Audition du 10 janvier 2001.
() Michel Broyer, « Le paradoxe du don dans les transplantations avec donneur vivant », in Robert Carvais (dir.), La greffe humaine, (In)certitudes éthiques : du don de soi à la tolérance de l'autre, PUF, p. 422 et sq.
() Assemblée nationale, texte adopté n° 675.
() Libération du 20 mars 2001 ; Le Figaro du 21 mars 2001.
() Audition du 10 janvier 2001.
() Professeur Vernant, Colloque Greffe et donneur vivant, 13 et 14 mars 1997.
() Eliane Gluckman, in Robert Carvais (dir.), op.cit., p. 243 et sq.
() Greffe des cellules-souches hématopoïétiques : besoins et perspectives, le Greffe humain, op.cit., p. 248.
() Colloque Greffe et donneur vivant, 13 et 14 mars 1997, p. 41.
() Colloque Greffe et donneur vivant, 13 et 14 mars 1997, p. 43.
() L'argumentation a été bien évoquée par des chercheurs : « Si par rapport à l'embryon, « être en devenir », la situation est désormais bien précisée, il est indispensable de déterminer avec rigueur l'attitude de l'expérimentateur par rapport à la femme qui a décidé d'interrompre volontairement sa grossesse.[...] Enfin, une fois l'interruption réalisée avec les répercussions psychologiques et morales qui en résultent toujours, il n'est pas imaginable que le médecin ayant réalisé l'acte sollicite un accord éventuel pour prélever des parties de l'embryon. Le corps de l'embryon n'appartient qu'à lui-même, ce n'est pas à celle, seule ou avec son conjoint, qui a voulu ne pas lui accorder la vie, de décider du devenir des fragments de sa dépouille corporelle, souvent très mutilée d'ailleurs. Ce sont donc aux médecins qui ont réalisé l'IVG et à leurs confrères qui entreprennent l'expérimentation que revient le choix. La responsabilité leur incombe et ils ne peuvent en aucun cas se donner une éventuelle bonne conscience en obligeant celle qui souffre déjà moralement et physiquement à partager les responsabilités qu'ils ne parviendraient pas à assumer seuls » C. et A. Duprez, 9e Congrès mondial de droit médical, Gand, août 1991, Actes, I., p. 281, cité in Dictionnaire permanent de la bioéthique, « Utilisation des cellules, tissus et produits d'embryons ou de f_tus humains ».
() Enquête nationale sur le don et la greffe d'organes, du 18 juin au 12 juillet 1997, in Robert Carvais (dir.), op. cit.
() Réunion du 10 janvier 2001.
() R. Carvais (dir.), op. cit., p. 852.
() D'après Jean-Paul Moatti, Ibid, p. 767.
() Anne-Marie Moulin, Ibid, p. 760.
() Ministère de la défense, direction du service national, Les Journées d'appel de préparation à la défense, Bilan d'activités octobre 1998 à juin 1999.
() Philippe Oliviéro, Intersubjectivité matérielle et disponibilité des matériaux substitutifs in R. Carvais (dir), op. cit., p. 539.
() Audition du 10 janvier 2001.
() Encyclopaedia Universalis, 1999.
() R. Carvais (dir.), op. cit., p. 568.
() « L'appel au don : vers un droit de préemption sur le corps humain ? », Revue Autrement, La négociation.
() Intervention de M. Dominique Becquart au congrès de Sfax, 13 au 17 avril 2000.
() Enquête nationale sur le don et la greffe d'organes, du 18 juin au 12 juillet 1997, La greffe humaine, (In)certitudes éthiques : du don de soi à la tolérance de l'autre, R. Carvais (dir), PUF.
() Philippe Oliviéro, in R. Carvais (dir.), op. cit., p. 558.
() Loi n° 94-653 du 29 juillet 1994 relative au respect du corps humain.
() J.O. Débats Sénat, séance du 3 juin 1999, pp. 3670-3671.
() Dominique Thouvenin, « Les avatars de l'article 16-3, alinéa 1er, du code civil », Recueil Dalloz 2000, Chroniques, p. 485.
() Il convient de noter que les recherches biomédicales sont définies au premier alinéa de l'article L. 1121-1 du code de la santé publique : essais ou expérimentations organisés et pratiqués sur l'être humain en vue du développement des connaissances biologiques ou médicales ; qu'est-ce qui différencie alors la recherche biomédicale sur une personne en état de mort cérébrale d'un prélèvement à des fins scientifiques d'organe (art. L. 1232-1 ) ou de tissus (art. L. 1241-3) ?
() L'autopsie médico-scientifique, Bernard Gosselin, in Les lois « bioéthique » à l'épreuve des faits, Réalités et perspectives (Droit et justice).
() Philippe Coste, « La disparition des autopsies médico-scientifiques, un frein à la recherche », Médecine, 22 mai 2001.
() Audition de M. Michel Tubiana, président, et de Mme Monique Hérold, responsable de la commission santé-bioéthique de la Ligue des droits de l'homme, le 8 novembre 2000.
() Audition du 10 janvier 2001.
() Loi n° 67 du 18 janvier 2001.
() L'autopsie médico-scientifique, Bernard Gosselin, in Les lois « bioéthique » à l'épreuve des faits, réalités et perspectives (Droit et justice) .
() Anne-Marie Moulin in Robert Carvais (dir.) op. cit, p. 761.
() Simone Bateman-Novaes, « De la thérapeutique comme norme », La Pensée, n° 312 octobre-novembre-décembre 1997, pp. 21-32.
() Encyclopédia universalis, 1999.
() A ce stade de l'exposé, on rappellera simplement que les tests génétiques peuvent être utilisés aussi dans le cadre de procédures judiciaires. En matière civile, l'article 16-11 du code civil prévoit que l'identification d'une personne ne peut être recherchée qu'en exécution d'une mesure d'instruction ordonnée par le juge saisi d'une action tendant soit à l'établissement ou à la constatation d'un lien de filiation soit à l'obtention ou à la suppression de subsides. Cet article précise que le consentement de l'intéressé doit être préalablement et expressément recueilli. Dans l'affaire concernant une action en reconnaissance en paternité contre Yves Montand, la Cour d'appel de Paris est cependant passée outre la volonté du défunt et a ordonné une expertise imposant une exhumation de son corps. Il conviendrait sans doute que les dispositions de l'article 16-11 du code civil soient renforcées pour empêcher qu'une telle décision se renouvelle.
() Jacques Testart, Des hommes probables. De la procréation aléatoire à la reproduction normative, Seuil, 1999, p. 47.
() Jacques Testart, op. cit., p. 48-49.
() Le Figaro, supplément magazine, 25 novembre 2000, p. 61.
() Cité dans l'avis n° 46 du Comité national consultatif d'éthique du 30 octobre 1995.
() Rapport du Conseil d'État, op. cit., p. 115-116.
() Le Monde, 1er janvier 2001.
() Voir sur ce point le rapport n° 1407 de l'Office parlementaire d'évaluation des choix scientifiques et technologiques sur l'application de la loi n° 94-654 du 24 juillet 1994 présenté par MM. Alain Claeys, député et Claude Huriet, sénateur.
() Quelques semaines après l'audition du Professeur Munnich devant la Mission, le premier enfant né en France après recours au DPI, prénommé Valentin, a vu le jour le 13 novembre 2000, indemne d'une grave maladie enzymatique dont ses trois frères aînés sont atteints, grâce à l'équipe des Professeurs René Frydman et Arnold Munnich.
() Conseil d'État, Les lois de bioéthique : cinq après, 1999, p. 121.
() Rapport du Conseil d'État, Les lois de bioéthique : 5 ans après, p. 126.
() Pour plus d'informations sur ce sujet, on se reportera au site de l'Office européen des brevets : http://www.european-patent-office.org.
() Expressed Sequence Tags : courtes séquences d'ADN permettant d'étiqueter des gènes et de les séquencer ultérieurement.
() Sur cette question, on se reportera à Frédéric Pollaud-Dulian, Droit de la propriété industrielle, Montchrestien, Paris, 1999, p. 94 et suivantes.