

N° 1281
——
ASSEMBLÉE NATIONALE
CONSTITUTION DU 4 OCTOBRE 1958
QUATORZIÈME LÉGISLATURE
Enregistré à la Présidence de l'Assemblée nationale le 17 juillet 2013.
RAPPORT D’INFORMATION
DÉPOSÉ
en application de l’article 145-7, alinéa 1, du Règlement
PAR LA COMMISSION DES AFFAIRES SOCIALES
sur la mise en œuvre de la loi du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé,
ET PRÉSENTÉ
PAR Mme Catherine LEMORTON et M. Arnaud ROBINET,
Députés.
——
INTRODUCTION 5
I.- LA LUTTE CONTRE LES CONFLITS D’INTÉRÊTS ET LA TRANSPARENCE DES AVANTAGES CONSENTIS PAR LES ENTREPRISES 8
A. L’EXTENSION DES DÉCLARATIONS D’INTÉRÊTS PAR LES PROFESSIONNELS 8
B. LA « CHARTE DE L’EXPERTISE SANITAIRE » 14
C. LA MISE EN œUVRE DU « SUNSHINE ACT » À LA FRANÇAISE : UN RENDEZ-VOUS MANQUÉ ? 18
II.- LA RESPONSABILISATION DES ACTEURS INSTITUTIONNELS DU SYSTÈME DE SÉCURITÉ SANITAIRE : UNE NÉCESSAIRE RÉVOLUTION CULTURELLE 25
A. LES PRÉROGATIVES DE LA NOUVELLE AGENCE NATIONALE DE SÉCURITÉ DU MÉDICAMENT ET DES PRODUITS DE SANTÉ 25
B. L’ARCHITECTURE DE L’AGENCE ET LA TRANSPARENCE DE SES TRAVAUX 29
III.- L’ENCADREMENT DU « HORS-AMM » : ALLIER ACCÈS AUX PROGRÈS THÉRAPEUTIQUES ET SÉCURITÉ DES PATIENTS 34
A. L’ENCADREMENT DES AUTORISATIONS TEMPORAIRES D’UTILISATION (ATU) 34
B. LES RECOMMANDATIONS TEMPORAIRES D’UTILISATION 37
IV.- LA PHARMACOVIGILANCE ET LE SUIVI POST-AMM DES PRODUITS DE SANTÉ 43
A. LA RÉÉVALUATION SYSTÉMATIQUE DES MÉDICAMENTS APRÈS LEUR MISE SUR LE MARCHÉ 43
B. LA CONSOLIDATION DU SYSTÈME DE PHARMACOVIGILANCE 46
C. LE GROUPEMENT D’INTÉRÊT PUBLIC DESTINÉ À PRODUIRE DES ÉTUDES DE VIGILANCE ET D’ÉPIDÉMIOLOGIE 53
D. LA PRESCRIPTION ET LA DÉLIVRANCE DES PRODUITS DE SANTÉ 55
V.- LA FORMATION ET L’INFORMATION DES PROFESSIONNELS ET DU PUBLIC 64
A. LA BASE DE DONNÉES PUBLIQUE SUR LES PRODUITS DE SANTÉ ET LEUR USAGE 64
B. LE CONTRÔLE DE LA PUBLICITÉ POUR LES MÉDICAMENTS ET LES DISPOSITIFS MÉDICAUX 66
C. L’OBLIGATION DE PRESCRIPTION EN DÉNOMINATION COMMUNE INTERNATIONALE 72
D. LES LOGICIELS D’AIDE À LA PRESCRIPTION ET À LA DISPENSATION 72
E. L’ENCADREMENT DE LA VISITE MÉDICALE 73
TRAVAUX DE LA COMMISSION 77
ANNEXE 1 : LISTE DES PERSONNES AUDITIONNÉES 107
ANNEXE 2 : TEXTES RÉGLEMENTAIRES ET AUTRES MESURES D’APPLICATION 110
ANNEXE 3 : RÉSUMÉ DES REMARQUES ET PRÉCONISATIONS DU RAPPORT 117
En application de l’article 145-7 du Règlement de l’Assemblée nationale, le présent rapport fait état des textes réglementaires publiés et des circulaires édictées pour la mise en œuvre de la loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé.
● Les modalités d’application de la loi du 29 décembre 2011 ont été définies principalement au travers de 21 décrets d’application :
– décret n° 2012-745 du 9 mai 2012 relatif à la déclaration publique d’intérêts et à la transparence en matière de santé publique et de sécurité sanitaire ;
– décret en Conseil d’État n° 2013-413 du 21 mai 2013 portant approbation de la charte de l’expertise sanitaire prévue à l’article L. 1452-2 du code de la santé publique ;
– décret en Conseil d’État n° 2013-414 du 21 mai 2013 relatif à la transparence des avantages accordés par les entreprises produisant ou commercialisant des produits à finalité sanitaire et cosmétique destinés à l’homme ;
– décret n° 2012-597 du 27 avril 2012 relatif à l’Agence nationale de sécurité du médicament et des produits de santé ;
– décret n° 2012-1244 du 8 novembre 2012 relatif au renforcement des dispositions en matière de sécurité des médicaments à usage humain soumis à autorisation de mise sur le marché et à la pharmacovigilance ;
– décret n° 2012-1095 du 28 septembre 2012 relatif à diverses pénalités financières encourues par des entreprises exploitant des médicaments et des fabricants ou des distributeurs de dispositifs médicaux ;
– décret en Conseil d’État n° 2012-742 du 9 mai 2012 relatif aux recommandations temporaires d’utilisation des spécialités pharmaceutiques ;
– décret n° 2013-31 du 9 janvier 2013 fixant les conditions de l’expérimentation relative à la consultation du dossier pharmaceutique par les médecins exerçant dans certains établissements de santé ;
– décret n° 2012-743 du 9 mai 2012 relatif à la publicité pour les dispositifs médicaux ;
– décret n° 2012-744 du 9 mai 2012 relatif à la publicité pour les dispositifs médicaux de diagnostic in vitro ;
– décret n° 2012-1135 du 8 octobre 2012 relatif au contrôle des spécifications techniques et à la pénalité financière prévus à l’article L. 165-1-2 du code de la sécurité sociale ;
– décret n° 2012-1033 du 7 septembre 2012 relatif à la procédure de contrôle sur pièces et sur place des agents assermentés des organismes locaux d’assurance maladie ;
– décret n° 2012-1096 du 28 septembre 2012 relatif à l’approvisionnement en médicaments à usage humain ;
– décret n° 2012-1051 du 13 septembre 2012 relatif à l’évaluation et à la prise en charge de certains produits de santé financés dans les tarifs des prestations d’hospitalisation ;
– décret du 1e mai 2012 portant nomination du directeur général de l’Agence nationale de sécurité du médicament et des produits de santé ;
– décret n° 2012-1131 du 5 octobre 2012 relatif à la consultation et à l’alimentation du dossier pharmaceutique par les pharmaciens exerçant dans les pharmacies à usage intérieur ;
– décret n° 2013-66 du 18 janvier 2013 relatif aux autorisations temporaires d’utilisation des médicaments ;
– décret n° 2012-740 du 9 mai 2012 relatif à la prise en charge dérogatoire par l’assurance maladie des spécialités pharmaceutiques bénéficiant d’une recommandation temporaire d’utilisation ou de certains produits et prestations ;
– décret n° 2012-741 du 9 mai 2012 portant dispositions relatives à la publicité pour les médicaments à usage humain ;
– décret n° 2012-910 du 24 juillet 2012 relatif à la délivrance de médicaments indiqués dans la contraception d’urgence dans les services universitaires et interuniversitaires de médecine préventive et de promotion de la santé ;
– décret n° 2012-1562 du 31 décembre 2012 relatif au renforcement de la sécurité de la chaîne d’approvisionnement des médicaments et à l’encadrement de la vente de médicaments sur internet.
● On notera également que la loi du 29 décembre 2011 a prévu le dépôt par le Gouvernement de huit rapports :
– un rapport à remettre avant le 30 juin 2012 sur l’application de la loi six mois après sa publication (article 67 de la loi du 9 décembre 2004) ;
– un rapport sur le financement des associations d’usagers du système de santé et de leurs besoins (article 3);
– un rapport à remettre avant le 1e janvier 2013 dressant le bilan de l’expérimentation sur l’information par démarchage ou la prospection pour les produits de santé réalisé à partir d’une évaluation conduite par la Haute Autorité de Santé (article 30) ;
– un rapport annuel d’activité décrivant le résultat des études menées et formulant des recommandations (article 33) ;
– un rapport à remettre avant le 1er janvier 2013, formulant des propositions en matière de réparation des dommages quand le risque lié à un médicament se réalise (article 41) ;
– dans le cadre de l’examen du projet de loi de financement de la sécurité sociale pour 2014, un rapport dressant le bilan de l’expérimentation de la prise en charge du traitement des patients bénéficiant d’une autorisation temporaire d’utilisation avant obtention de l’autorisation de mise sur le marché) notamment au regard de son impact sur les dépenses et du bon usage des produits concernés (article 24) ;
– un rapport établi par l’ANSM à remettre avant le 30 juin 2012 dressant le bilan des règles applicables à la sécurité des dispositifs médicaux et présentant les mesures susceptibles de l’améliorer (article 41) ;
– un rapport annuel d’activité mentionnant les modalités et principes selon lesquels les commissions spécialisées mettent en œuvre les critères d’évaluation des produits de santé en vue de leur prise en charge par l’assurance maladie (article 15).
I.- LA LUTTE CONTRE LES CONFLITS D’INTÉRÊTS ET LA TRANSPARENCE DES AVANTAGES CONSENTIS PAR LES ENTREPRISES
Le regain de confiance dans notre système de santé et de sécurité sanitaire passe d’abord et avant tout par une amélioration de la transparence et de l’indépendance des décisions prises par les autorités concernées.
Le simple fait de travailler en lien avec les industries de santé n’affecte pas en soi l’impartialité du travail des experts. Mais l’absence de transparence sur ces liens nuit considérablement à la crédibilité de leur analyse, et par suite, de l’expertise publique.
C’est pourquoi les deux premiers articles de la loi du 29 décembre 2011 visaient à généraliser les déclarations d’intérêts et à instaurer toute la transparence nécessaire sur les avantages consentis par les industriels aux professionnels de santé. Force est de constater que sur cet aspect de la réforme, les textes d’application ne sont pas satisfaisants sur certains points.
A. L’EXTENSION DES DÉCLARATIONS D’INTÉRÊTS PAR LES PROFESSIONNELS
L’article 1er de la loi du 29 décembre 2011 prévoit la remise d’une déclaration d’intérêts lors de leur prise de fonctions par un nombre étendu de professionnels travaillant au sein des autorités et des organismes sanitaires. Il s’agit d’uniformiser la pratique des déclarations d’intérêts afin d’en faciliter le contrôle, mais aussi de créer une véritable culture de la transparence en matière d’expertise.
Le décret en Conseil d’État n° 2012-745 du 9 mai 2012 relatif à la déclaration publique d’intérêts et à la transparence en matière de santé publique et de sécurité sanitaire précise le champ et le contenu des déclarations d’intérêts, en définit le modèle et les conditions de publication.
● Les personnes soumises à l’obligation de déclaration d’intérêts
L’article R.1451-1 du code de la santé publique, issu de l’article 1er du décret précité, fixe la liste des personnes soumises à l’obligation de déclaration d’intérêts, en application du I de l’article L. 1451-1 du même code. Les personnes concernées sont :
– les membres des cabinets des ministres chargés de la santé et de la sécurité sociale (I de l’article R.1451-1) ;
– les membres des commissions et conseils siégeant auprès de ces ministres chargés par la loi ou un texte règlementaire de rendre des avis sur des questions de santé publique et de sécurité sanitaire (I de l’article R.1451-1) ;
– le personnel dirigeant des autorités, établissements et groupement public visés au I de l’article L. 1451-1 du code de la santé publique (1) (I de l’article R.1451-1) ;
– les personnes non membres des commissions et conseils siégeant auprès des ministres chargés de la santé et de la sécurité sociale ou des autorités précitées, mais appelées à fournir une expertise (II de l’article R.1451-1) ;
– les agents participant à la préparation des décisions, recommandations et avis des établissements et autorités précitées, ainsi que les agents exerçant des fonctions de contrôle, d’inspection et de surveillance (III de l’article R.1451-1).
Le IV de l’article R. 1451-1 prévoit enfin que pour chaque administration, autorité ou établissement, le ministre, le président de l’autorité, le directeur de l’établissement établit la liste des fonctions et des instances collégiales « remplissant les critères » définis aux I et III de l’article, nécessitant une déclaration d’intérêts.
Ce choix permet certes de responsabiliser les différentes autorités concernées. Cependant, il ne facilite pas la mise en œuvre harmonisée de la déclaration d’intérêts car il peut être fait une interprétation plus ou moins extensive des critères établis par l’article R. 1451-1.
● Le champ et le contenu de la déclaration d’intérêts
L’article 1er de la loi ne définit pas la notion de lien ou de conflit d’intérêts. Il est cependant précisé que la déclaration publique d’intérêts concerne toute personne entretenant des « liens directs ou indirects avec les entreprises, établissements ou organismes dont les activités, les techniques ou les produits entrent dans le champ de compétence de l’instance au sein de laquelle ils siègent, ainsi qu’avec les sociétés ou organismes de conseil intervenant dans ces secteurs ».
Le nouvel article R. 1451-1 du code de la santé publique, fixe le contenu obligatoire des déclarations d’intérêts.
En outre, afin d’en faciliter la lecture et le contrôle, il était essentiel de mettre au point un modèle harmonisé de déclaration d’intérêts. Il existe désormais un formulaire unique, appelé document-type, fixé par l’arrêté du 5 juillet 2012.
La déclaration remise au ministre, au président de l’autorité ou au directeur ou directeur général de l’établissement ou du groupement d’intérêt public doit obligatoirement comporter les informations suivantes :
1° Les nom et prénom du déclarant ;
2° La qualité au titre de laquelle le déclarant est tenu d’établir la déclaration ;
3° L’activité principale actuelle, rémunérée ou non ;
4° Les activités principales et accessoires, rémunérées ou non, exercées au cours des cinq années précédentes dans des sociétés, établissements, organismes et associations dont les activités, les techniques ou les produits entrent dans le champ de compétence en matière de santé publique et de sécurité sanitaire. Sont également déclarés à ce titre et dans les mêmes conditions :
– les activités exercées auprès de sociétés ou organismes de conseil intervenant dans les mêmes secteurs ;
– la participation à une instance décisionnelle d’un organisme public ou privé ;
– l’exercice d’une activité de consultant, de conseil ou d’expertise auprès d’un organisme ;
– les travaux scientifiques et études pour des organismes publics ou privés;
– la rédaction d’article et les interventions, rémunérées ou prises en charge, dans des congrès, des conférences, des colloques, des réunions publiques ou des formations organisées ou soutenues financièrement par des entreprises privées ;
– la détention ou l’invention d’un brevet ou l’invention d’un produit, procédé ou tout autre forme de propriété intellectuelle non brevetée, en relation avec le champ de compétence mentionné ci-dessus.
Le déclarant précise, le cas échéant, les rémunérations perçues soit à titre personnel, soit par un organisme dont il est membre ou salarié ;
5° Les activités que le déclarant dirige ou a dirigées au cours des cinq années précédentes et qui ont bénéficié d’un financement par un organisme à but lucratif dont l’objet social entre dans le champ de compétence mentionné au 4° ainsi que le montant de ce financement ;
6° Les participations financières directes, sous forme d’actions ou d’obligations détenues et gérées directement ou de capitaux propres, dans le capital d’une société dont l’objet social entre dans le champ de compétence mentionné au 4°. Le déclarant en précise le montant en valeur absolue et en pourcentage du capital ;
7° Si elle est connue du déclarant, toute activité mentionnée au 4° et au 5°, exercée ou dirigée actuellement ou au cours des cinq années précédentes par ses parents et enfants, par son conjoint, concubin ou partenaire lié par un pacte de solidarité ou par les parents et enfants de ce dernier ainsi que toute participation financière supérieure à un montant de 5 000 euros ou à 5 % du capital détenue par les mêmes personnes. Le déclarant identifie le tiers concerné par la seule mention de son lien de parenté ;
8° Les autres liens dont le déclarant estime qu’ils sont de nature à faire naître des situations de conflits d’intérêts ainsi que les sommes reçues à ce titre.
La déclaration d’intérêts devra être, à l’initiative des personnes concernées, actualisée dès lors qu’une évolution intervient concernant les liens d’intérêts. L’arrêté du 5 juillet 2012 précise qu’a minima, même sans modification, la déclaration d’intérêts doit être actualisée tous les ans.
Les montants des rémunérations perçues ou des participations financières ne sont pas accessibles au public.
● Les modalités de publication des déclarations d’intérêts
La création d’une instance dédiée au recueil, à la publication et au contrôle des déclarations d’intérêts a été écartée par le Gouvernement à l’époque de l’adoption de la loi.
Cependant, aux termes du nouvel article R. 1451-3 du code de la santé publique, issu du décret du 9 mai 2012, les déclarations d’intérêts sont établies et actualisées par télé déclaration sur un site internet unique ou par la remise d’un formulaire conforme au document type établi par l’arrêté du 5 juillet 2012.
Selon l’article R. 1451-4 du même code, la publicité de toutes les déclarations d’intérêts est assurée, pendant la durée des fonctions ou de la mission au titre desquelles elles ont été établies et les cinq années suivant la fin de ces fonctions. Les déclarations d’intérêts sont conservées pendant une durée de dix ans, à compter de leur dépôt ou de leur actualisation, par l’administration, l’autorité, l’établissement ou le groupement auquel elles sont remises.
L’article R. 1451-3 prévoit qu’un arrêté des ministres chargés de la santé et de la sécurité sociale, pris après avis de la Commission nationale de l’informatique et des libertés, détermine :
– les conditions de fonctionnement du site,
– l’autorité qui en est responsable ;
– les modalités d’établissement, d’authentification et de transmission sécurisée des télédéclarations ;
– les conditions dans lesquelles les administrations, les autorités, les établissements ou le groupement d’intérêt public chargé de réaliser des études de vigilance et d’épidémiologie impliquant des produits de santé, chacun pour ce qui le concerne, à la déclaration d’intérêts ;
– les modalités selon lesquelles les proches des personnes soumises à une déclaration d’intérêts sont informées du recueil et de la publicité des données les concernant.
Il est en outre précisé que l’autorité responsable du site prend les mesures techniques nécessaires pour assurer son intégrité, la sécurité des données, leur protection contre l’indexation par des moteurs de recherche et la confidentialité de celles qui ne sont pas rendues publiques. Elle se conforme aux dispositions de la loi n° 78-17 du 6 janvier 1978 relative à l’informatique, aux fichiers et aux libertés en accomplissant auprès de la Commission nationale de l’informatique et des libertés les formalités nécessaires pour le traitement de données qu’elle met en œuvre pour l’application de la présente section.
Le site internet unique n’a à ce jour pas été créé. Selon le ministère de la santé, il le serait au deuxième semestre 2014. Dans l’attente de sa mise au point, chaque agence ou opérateur est chargé d’assurer la publicité des déclarations sur son propre site.
Selon les informations fournies aux rapporteurs par le ministère de la santé, les douze agences sanitaires ou opérateurs concernés ont désormais mis en place un système de déclaration publique d’intérêts. Neuf agences sur douze disposent d’un comité de déontologie, et toutes sont dotées d’un référent en la matière. La quasi-totalité des déclarations attendues sont publiées sur le site des agences, dont les rapporteurs souhaitent saluer le travail accompli pour appliquer au plus vite les dispositions de la loi.
Vos rapporteurs souhaitent insister sur la nécessaire accessibilité et lisibilité des déclarations sans lesquelles la tentative de prévention des conflits d’intérêts restera lettre morte et ne constituera qu’une contrainte administrative de plus pour les agences et autres institutions sanitaires.
De ce point de vue, il est pour le moins étonnant et peu judicieux d’avoir prévu deux modalités de déclaration, la télé déclaration, solution à privilégier pour faciliter le traitement et la communication des données, et le formulaire papier.
Vos rapporteurs s’étonnent de la non-communication, même encadrée, des rémunérations et participations des personnes soumises à déclaration d’intérêts. Celles-ci devraient pouvoir être accessibles, dans des conditions qui permettent le respect des données personnelles des individus concernés, à tout citoyen qui en fait la demande.
De plus, en l’absence de site unique permettant de regrouper les différentes déclarations, l’accès aux données publiées est particulièrement problématique.
Dans la délibération du 2 mai 2012 n° 2012-125 portant avis sur le projet de décret en Conseil d’État relatif aux règles déontologiques et à la déclaration publique d’intérêts, la Commission nationale de l’informatique et des libertés (CNIL) insiste sur la nécessité d’empêcher l’indexation des informations publiées au nom de la protection des données personnelles.
L’interdiction d’indexation ne devrait concerner que les données directement identifiantes et ne s’appliquer qu’aux moteurs de recherche externes, et non aux moteurs internes propres aux sites des laboratoires, des conseils de l’ordre, ou à celui du site que le ministère mettrait en place et qui permettrait d’avoir l’ensemble des informations compilées pour une personne donnée.
Enfin, le nouvel article R. 1451-5 du code de la santé publique étend l’application de l’article R. 4113-110 aux personnes soumises à déclaration publique d’intérêts mentionnées à l’article L. 1451-1. Cela signifie que l’information du public sur l’existence de liens entre les professionnels de santé et des entreprises ou établissements produisant des produits de santé est faite, à l’occasion de la présentation de ce professionnel, soit de façon écrite lorsqu’il s’agit d’un article destiné à la presse écrite ou diffusé sur internet, soit de façon écrite ou orale au début de son intervention, lorsqu’il s’agit d’une manifestation publique ou d’une communication réalisée pour la presse audiovisuelle.
Vos rapporteurs souhaitent insister sur le fait que cette disposition est trop rarement contrôlée et sanctionnée par les ordres professionnels compétents et qu’aucune sanction n’est prévue pour les personnes n’exerçant pas une profession de santé.
● Les conséquences sur la participation aux décisions en matière de sécurité sanitaire
Il est interdit aux personnes concernées par la déclaration publique d’intérêts de prendre part aux délibérations et aux votes de ces instances tant qu’elles n’auront pas souscrit ou actualisé leur déclaration d’intérêts et, sous peine de cinq ans d’emprisonnement et de 75 000 euros d’amende, de prendre part aux délibérations et aux votes si elles ont un intérêt direct ou indirect à l’affaire examinée.
Cette disposition est particulièrement importante, puisqu’elle garantit l’usage des déclarations d’intérêts comme véritable outil de prévention des conflits d’intérêts, non seulement dans la prise de décision en matière d’expertise sanitaire, mais aussi au stade des délibérations.
B. LA « CHARTE DE L’EXPERTISE SANITAIRE »
L’article 1er de la loi prévoit la création d’une charte de l’expertise sanitaire, approuvée par décret en Conseil d’État. Aux termes de l’article L. 1452-1 du code de la santé publique, cette charte précise les modalités de choix des experts, le processus d’expertise et ses rapports avec le pouvoir de décision, la notion de lien d’intérêts, les cas de conflits d’intérêts, les modalités de gestion d’éventuels conflits et les cas exceptionnels dans lesquels il peut être tenu compte des travaux réalisés par des experts présentant un conflit d’intérêts.
● La définition de l’expertise sanitaire
Le décret en Conseil d’État n° 2013-413 du 21 mai 2013 portant approbation de la charte, se réfère à la norme NF X 50-110 (2) pour définir l’expertise. Il s’agit d’un ensemble d’activités ayant pour objet de fournir à un commanditaire, en réponse à la question posée, une interprétation, un avis ou une recommandation aussi objectivement fondés que possible, élaborés à partir des connaissances disponibles et de démonstrations, accompagnées d’un jugement professionnel. Cette définition s’applique lorsque l’organisme d’expertise et le commanditaire font partie de la même organisation et lorsque l’organisme d’expertise se saisit lui-même d’une question et émet de son propre chef une interprétation, un avis ou une recommandation. La charte s’applique dont aussi bien à l’expertise dite « interne » qu’ « externe ».
Les activités d’expertise sanitaire soumises à la charte sont celles qui ont pour objet d’éclairer le décideur et d’étayer sa prise de décision en fournissant une interprétation, un avis ou une recommandation aussi objectivement fondés que possible, élaborés à partir de l’analyse critique des meilleures connaissances disponibles et de démonstrations argumentées reposant sur des critères explicites, accompagnées d’un jugement professionnel fondé sur l’expérience des experts.
L’expertise sanitaire doit donc être distinguée des activités qui visent à produire des connaissances nouvelles, que ce soit à partir du recueil de données nouvelles ou de l’analyse secondaire de données existantes. L’expertise sanitaire doit également être distinguée des expertises scientifiques réalisées pour contribuer à la sélection de projets d’étude ou de recherche et des expertises médicales portant sur des cas individuels qui ne sont pas destinés à éclairer une décision sanitaire.
La charte s’applique aux expertises sanitaires réalisées à la demande des commissions, conseils, autorités ou organismes mentionnés au I de l’article L. 1451-1 du code de la santé publique ainsi qu’aux expertises sanitaires réalisées à la demande du ministre chargé de la santé. Tous ces organismes veillent, chacun pour ce qui le concerne, à ce que les expertises soient réalisées dans le respect de la charte.
● Les modalités de choix des experts
Chaque organisme chargé de la réalisation d’une expertise rend public son processus de désignation ou de sélection des experts. Il peut procéder à la publication d’appels à candidatures pour leur sélection. L’organisme chargé de la réalisation de l’expertise s’assure que les experts retenus disposent des compétences, de l’expérience ainsi que de l’indépendance nécessaires pour réaliser les travaux d’expertise demandés, en s’appuyant notamment sur l’analyse de leurs curriculum vitae, de leurs compétences professionnelles, de leurs productions scientifiques et de leurs déclarations d’intérêts.
Un expert ne doit pas accepter une mission pour laquelle il n’est pas ou ne s’estime pas être compétent, ou pour laquelle il n’est pas ou n’estime pas être suffisamment indépendant au regard de l’objet de l’expertise. L’organisme chargé de la réalisation de l’expertise s’assure que chaque expert a pris connaissance de la charte. Lorsque, dans l’accomplissement de sa mission, un expert se trouve confronté à une question qui échappe à sa compétence, il doit en informer l’organisme qui l’a désigné pour que celui-ci prenne les mesures appropriées.
● Le processus d’expertise
Dans le cas où l’expertise est réalisée sur demande, l’objet, le calendrier et les conditions de réalisation de l’expertise donnent lieu à une concertation entre l’organisme chargé de la réalisation de l’expertise et le commanditaire de l’expertise, selon des modalités adaptées au contexte et au degré d’urgence de la saisine.
L’accord écrit qui résulte de cette concertation précise notamment, si le commanditaire et l’organisme estiment que l’objet de l’expertise le justifie, les modalités d’association ou de consultation des parties prenantes. Cet accord prévoit également, le cas échéant, les modalités selon lesquelles les conclusions de l’expertise pourront faire l’objet de présentations au commanditaire de l’expertise ou aux parties prenantes, afin d’identifier les éléments qui peuvent nécessiter une clarification au regard des questions posées et des décisions à prendre par le commanditaire.
Quel que soit le type d’expertise, l’organisme chargé de la réalisation de l’expertise est responsable du choix et de la mise en œuvre des méthodes appropriées pour répondre aux questions posées. L’expertise collective est une modalité à privilégier lorsque l’objet de l’expertise est particulièrement complexe ou nécessite une approche pluridisciplinaire. En toute hypothèse, y compris dans le cas où il est recouru à un expert unique, l’expertise doit s’appuyer sur :
– la complétude des données ou de l’état des connaissances existant sur la question posée ;
– la confrontation de différentes opinions, thèses ou écoles de pensées ;
– l’expression d’éventuelles positions divergentes.
Chaque expert est libre d’exprimer son opinion, dans le cadre de l’expertise, sur tout point qu’il juge utile de commenter, même si celui-ci déborde le champ strict de la question initialement posée.
Enfin, l’expertise doit caractériser autant que possible la robustesse qui peut être attribuée à ses conclusions en fonction de la qualité des éléments sur lesquels elles s’appuient et identifie explicitement les points que l’état des connaissances disponibles ne permet pas de trancher avec une certitude suffisante. Il est également fait état des avis divergents ou minoritaires.
● La notion de lien d’intérêts, les cas de conflits d’intérêts et les modalités de gestion des conflits d’intérêts
La notion de lien d’intérêts recouvre les intérêts ou les activités, passés ou présents, d’ordre patrimonial, professionnel ou familial, de l’expert en relation avec l’objet de l’expertise qui lui est confiée. Les liens d’intérêts que l’organisme chargé de la réalisation de l’expertise demande aux experts de déclarer sont détaillés dans le document type de la déclaration publique d’intérêts prévu par l’article R. 1451-2 du code de la santé publique.
Un conflit d’intérêts naît d’une situation dans laquelle les liens d’intérêts d’un expert sont susceptibles, par leur nature ou leur intensité, de mettre en cause son impartialité ou son indépendance dans l’exercice de sa mission d’expertise au regard du dossier à traiter.
Il revient à l’organisme chargé de l’expertise d’analyser les liens d’intérêts déclarés par les experts et d’évaluer les risques de conflits d’intérêts. Au regard d’un dossier précis, l’expert qui suppose en sa personne un risque de conflit d’intérêts, ou estime en conscience devoir s’abstenir, le signale à l’autorité concernée afin qu’elle puisse prendre les mesures appropriées.
L’identification d’un conflit d’intérêts au regard d’une expertise donnée conduit en principe l’organisme à exclure la participation de cet expert. Cependant, la mise en œuvre de ce principe suppose une certaine souplesse. C’est pourquoi, en présence d’un lien d’intérêts qu’il ne juge pas de nature ou d’intensité susceptible de faire mettre en doute l’indépendance ou l’impartialité de l’expert pour l’expertise considérée, l’organisme peut associer cet expert en fonction de son degré d’implication dans l’analyse et du mode d’expertise (individuel ou collégial).
L’étude produite par l’expertise doit en outre préciser si l’analyse des liens d’intérêts déclarés par les experts a identifié ou non des conflits d’intérêts potentiels au regard des points traités, en décrivant, le cas échéant, les mesures mises en œuvre pour les gérer.
Enfin, la charte fait état des cas exceptionnels dans lesquels il peut être tenu compte des travaux réalisés par des experts présentant un conflit d’intérêts :
– si cette expertise présente un intérêt scientifique ou technique indispensable ;
– si l’organisme chargé de la réalisation de l’expertise n’a pas pu trouver d’expert de compétence équivalente dans le domaine concerné et qui n’ait pas de conflit d’intérêts.
Dans ces circonstances exceptionnelles et motivées, cet expert ou ces experts peuvent apporter leur expertise selon des modalités arrêtées par l’organisme chargé de la réalisation de l’expertise et portées à la connaissance du commanditaire.
Cet expert ou ces experts peuvent, par exemple, être auditionnés par l’organisme chargé de la réalisation de l’expertise ou par un groupe de travail qu’il met en place à cette fin, ou apporter une contribution écrite. Ils ne peuvent toutefois en aucun cas participer à la rédaction des conclusions ou des recommandations de l’expertise. Les motivations et les modalités de ces contributions éventuelles sont décrites explicitement en annexe de l’avis, de la recommandation ou du rapport produit par l’expertise.
Force est de constater que la rédaction de cette charte ne constitue pas en soi une avancée majeure. Dans une certaine mesure, elle marque même un recul de la transparence sur les liens d’intérêts, particulièrement sur les deux points suivants :
– la charte définit l’expertise comme un avis « aussi objectivement fondé que possible », alors qu’aux termes de l’article L. 1452-1, l’expertise « répond aux principes d’impartialité, de transparence, de pluralité et de contradictoire ». Vos rapporteurs sont conscients de la difficulté à recruter des experts de qualité et ne prétendent pas écarter ceux qui ont des liens d’intérêts avec les entreprises pharmaceutiques. Mais la définition de l’expertise figurant dans la charte est si imprécise qu’elle pourrait à l’avenir empêcher tout recours contre des décisions motivées par un expert manquant d’impartialité ;
– la charte prévoit explicitement l’audition et l’association étroite d’experts en situation de conflits d’intérêts. L’article L. 1452-2 prévoyait la simple possibilité de « tenir compte des travaux réalisés par des experts présentant un conflits d’intérêts ». La différence est majeure entre la consultation des travaux d’un individu et sa participation active à la prise de décision.
C. LA MISE EN œUVRE DU « SUNSHINE ACT » À LA FRANÇAISE : UN RENDEZ-VOUS MANQUÉ ?
L’article 2 de la loi prévoit la mise en place d’un véritable Sunshine Act à la française. C’est un point fondamental tant on connaît l’influence que peuvent parfois avoir les avantages consentis par les entreprises aux professionnels sur leur prescription. Afin de lever toute suspicion sur ce type de pratique, la transparence doit être totale.
Le nouvel article L. 1453-1 du code de la santé publique prévoit en premier lieu que les entreprises produisant ou commercialisant des produits de santé à usage humain ou assurant des prestations associées à ces produits sont tenues de rendre publique l’existence des conventions qu’elles concluent avec :
– les professionnels de santé (professions médicales, pharmaciens, auxiliaires médicaux) ;
– les associations de professionnels de santé ;
– les étudiants se destinant aux professions de santé et les associations les représentant ;
– les associations de patients ;
– les associations d’usagers du système de santé ;
– les établissements de santé privés et publiques ;
– les fondations, les sociétés savantes et les sociétés ou organismes de conseil intervenant dans le secteur des produits ou prestations mentionnés au premier alinéa ;
– les éditeurs de logiciels d’aide à la prescription et à la délivrance ;
– les personnes morales assurant la formation initiale des professionnels de santé ;
– les organes de presse spécialisée ainsi que les éditeurs de services de radio et télévision.
Un doute a pu exister sur la nature des associations représentant les étudiants se destinant aux professions de santé. Selon la circulaire n° DGS/PP2/2013/224du 29 mai 2013, sont concernées les « associations chargées de défendre les intérêts catégoriels d’une profession ou d’un groupe d’étudiants qui les composent ». Ne sont pas visées les associations reconnues d’utilité publique qui non seulement sont des associations inter-catégorielles, mais qui poursuivent aussi un but d’intérêt général, ni celles réunissant des professionnels de santé et dont l’objet est d’exercer des activités de recherche en santé ou d’y participer ou de formation médicale. Enfin, n’entrent pas non plus dans ce cadre les sociétés savantes qui ont notamment pour objet, dans un champ disciplinaire donné, de rendre compte de l’état de l’art, d’améliorer la connaissance et d’assurer la formation et la recherche du secteur considéré.
● Le seuil de publication des avantages consentis par les entreprises
Par ailleurs, le même article prévoit qu’au-delà d’un seuil fixé par décret en Conseil d’État, les avantages en nature ou en espèces que les mêmes entreprises procurent directement ou indirectement aux personnes précitées doivent être intégralement publiés. Le rapporteur, M. Arnaud Robinet, avait plaidé, de même que l’ensemble des membres de la commission des affaires sociales, pour la mise en place d’un seuil minimal, comme cela a été fait aux États-Unis. Le rapporteur avait également insisté sur la nécessité de centraliser la publication des conventions, afin de faciliter leur consultation par le public.
L’article 1er du décret n° 2013-414 du 21 mai 2013 complète le titre V du livre IV de la première partie du code de la santé publique par un chapitre III consacré aux avantages consentis par les entreprises pharmaceutiques.
Le nouvel article D. 1453-1 du code de la santé publique prévoit la publication des avantages consentis par les entreprises au-delà d’un seuil supérieur ou égal à dix euros.
La section sociale du Conseil d’État, dans une note du mardi 2 avril 2013, a jugé que « le seuil d’un euro pourrait être considéré comme ne constituant pas le seuil prévu par le législateur et qu’un seuil fixé à dix euros prémunissait mieux le projet du risque contentieux à cet égard ». La question avait été largement débattue à l’Assemblée nationale. Le ministre de la santé de l’époque, M. Xavier Bertrand, avait déclaré (3) : « puisque vous me dites qu’il n’y aura pas d’exagération, mon idée demeure de fixer le seuil au premier euro ».
Comme l’a indiqué la ministre de la santé et des affaires sociales lors de son audition, c’est la référence à un seuil dans la loi qui fait obstacle à sa fixation à un euro, vos rapporteurs proposent donc de la modifier afin de supprimer toute référence à un seuil de publication des avantages. Il s’agit ici de ne pas négliger l’impact sur les professionnels de santé des avantages et autres cadeaux, même jugés dérisoires, offerts par les entreprises, afin d’enclencher une réforme des pratiques.
● La publication des conventions et des avantages consentis par les entreprises
Le nouvel article R. 1453-2 du code de la santé publique prévoit que les entreprises produisant ou commercialisant les produits de santé mentionnés au II de l’article L. 5311-1 du même code rendent publique l’existence des conventions qu’elles concluent avec les personnes, associations, établissements, fondations, sociétés, organismes ou organes précédemment cités (article L. 1453-1).
Il est précisé que cette obligation ne s’applique pas aux conventions régies par les dispositions des articles L. 441-3 et L. 441-7 du code de commerce, qui ont pour objet l’achat de biens ou de services entre ces mêmes entreprises et ces personnes, associations, établissements, fondations, sociétés, organismes ou organes. Vos rapporteurs s’étonnent de cette disposition, peu cohérente avec l’objectif de transparence poursuivi par l’article. En effet, et comme cela a été souligné par le Conseil national de l’ordre des médecins en audition « on pourra ainsi savoir le prix d’un billet d’avion offert à un praticien pour se rendre à un congrès mais pas les sommes qui lui sont versées en contrepartie de la présentation qu’il y fera ».
Les entreprises produisant ou commercialisant des lentilles oculaires non correctrice, des produits cosmétiques et des produits de tatouage (14°, 15° et 17° de l’article L. 5311-1) sont soumises à des obligations similaires, prévues par les articles R. 1453-8 et R. 1453-9, pour les conventions relatives à la conduite de travaux d’évaluation de la sécurité, de vigilance ou de recherches biomédicales.
L’article R. 1453-3 précise que, pour ces conventions, chaque entreprise rend publiques les informations suivantes :
– l’identité des parties à chaque convention, soit :
a) Lorsqu’il s’agit d’un professionnel de santé, le nom, le prénom, la qualité, l’adresse professionnelle et, le cas échéant, la qualification, le titre, la spécialité, le numéro d’inscription à l’ordre ou l’identifiant personnel dans le répertoire partagé des professionnels de santé ;
b) Lorsqu’il s’agit d’un étudiant se destinant à l’une des professions relevant de la quatrième partie du code, le nom, le prénom, l’établissement d’enseignement et, le cas échéant, l’identifiant personnel dans le répertoire partagé des professionnels de santé ;
c) Lorsqu’il s’agit d’une personne morale : la dénomination sociale, l’objet social et l’adresse du siège social ;
d) L’identité de l’entreprise concernée ;
– la date de signature de la convention ;
– l’objet de la convention, formulé dans le respect des secrets protégés par la loi, notamment du secret industriel et commercial ;
– lorsque la convention a pour objet une manifestation de promotion ou un colloque professionnel, le programme de cette manifestation.
La circulaire d’interprétation n° DGS/PP2/2013/224 du 29 mai 2013 relative à l’application de l’article 2 de la loi précise que l’objet de la convention s’entend comme l’objet catégoriel (ex : une recherche impliquant la personne humaine, une manifestation à caractère professionnel et scientifique, une collaboration scientifique), afin de respecter le secret industriel et commercial. Cela n’implique donc pas de publier l’objet exact de la convention. Vos rapporteurs estiment qu’il est essentiel de revenir sur cette disposition, afin d’assurer la publicitié du contenu précis des conventions.
Le même article R. 1453-2 organise les modalités de publicité du montant des avantages en nature ou espèces consentis directement ou indirectement par les entreprises.
L’article R. 1453-3 prévoit que chaque entreprise rend publiques les informations suivantes :
– l’identité de la personne bénéficiaire et de l’entreprise ;
– le montant, toutes taxes comprises, arrondi à l’euro le plus proche, la date et la nature de chaque avantage perçu par le bénéficiaire au cours d’un semestre civil ;
– le semestre civil au cours duquel les avantages ont été consentis.
● Les modalités de publication des informations sur le site internet créé à cet effet
Les articles R. 1453-4 à R. 1453-7 du code de la santé publique issus du décret du 21 mai 2013, fixent les conditions de publication des informations relatives aux avantages consentis par les entreprises.
Aux termes de l’article R. 1453-4, les informations sont rendues publiques, en langue française, sur un site internet public unique et sont transmises à l’autorité responsable de ce site.
Un arrêté du ministre chargé de la santé pris après avis de la Commission nationale de l’informatique et des libertés détermine les conditions de fonctionnement du site mentionné au présent article, notamment l’autorité qui en est responsable, les modalités d’établissement, d’authentification et de transmission sécurisée des déclarations électroniques à distance. Cet arrêté n’a pas encore été publié.
Dans un communiqué de presse du 22 mai 2013, la ministre de la santé a annoncé la publication centralisée de ces informations sur un site internet public unique. Le site pourrait être lancé au deuxième semestre 2014.
Cependant, « dans l’attente de la mise en place de ce site, ces informations seront publiées sur le site internet des ordres professionnels concernés et sur le site internet des entreprises. »
L’article 3 du décret précité prévoit en effet que jusqu’à la création du site internet unique, les informations relatives au montant des avantages consentis par les entreprises sont rendues publiques, d’une part, sur le site internet du conseil national de l’ordre de la profession de santé dont relève la personne concernée, et, d’autre part, sur le site internet de l’entreprise ou sur un site commun à deux ou plusieurs entreprises partagé à cet effet. Un syndicat professionnel d’entreprises produisant ou commercialisant des produits de santé dont l’entreprise est adhérente peut rendre publiques les informations mentionnées au précédent alinéa pour le compte de ses adhérents.
Ces informations seront disponibles sur chacun de ces sites, en langue française au sein d’une rubrique dédiée, identifiable et accessible librement et gratuitement. Elles demeurent accessibles au public pendant cinq ans à compter de leur mise en ligne. Si une convention est applicable au-delà d’une durée de cinq ans, elle est à nouveau rendue publique au terme de ce délai. Les données recueillies sont conservées pendant dix ans à compter de la date à laquelle est intervenue leur dernière modification.
L’article R. 1453-5 du code de la santé publique prévoit que l’entreprise transmet les informations à l’autorité responsable du site internet public unique :
– dans un délai de quinze jours après la signature de la convention ;
– au plus tard le 1er août pour les avantages alloués ou versés au cours du premier semestre de l’année en cours et au plus tard le 1er février de l’année suivante pour les avantages alloués ou versés au cours du second semestre de l’année en cours.
L’article R. 1453-6 précise que les informations relatives aux conventions conclues et aux avantages consentis au cours du premier semestre civil doivent être mises en ligne au plus tard le 1er octobre de l’année en cours et au cours du second semestre civil au plus tard le 1er avril de l’année suivante.
Aux termes de l’article 3 du décret précité, dans l’attente de la création du site internet unique prévu à cet effet, le même calendrier s’applique. Ces dispositions sont valables pour les informations relatives aux conventions conclues et aux avantages consentis au cours de l’année 2012 qui doivent être transmises aux conseils nationaux des ordres des professions de santé au plus tard le 1er juin 2013 et publiées par ces conseils et par les entreprises au plus tard le 1er octobre 2013.
Ce calendrier pose un certain nombre de difficultés, dans la mesure où le dépôt des conventions se concentre autour de la date limite prévue par le décret. Les ordres professionnels, qui dans l’attente de la création du site internet sont chargés de la publication des informations, n’ont pas les moyens de traiter ces déclarations disparates dans des conditions satisfaisantes. Il conviendrait à tout le moins d’établir un modèle de déclaration unique et dématérialisé. La création d’un site public unique devrait être l’occasion d’une réflexion plus globale sur la simplification des procédures de publication des liens d’intérêts.
De plus, le délai fixé pour une première publication au 1er octobre prochain, avec une reprise rétroactive des données de 2012, est excessivement court pour permettre aux entreprises et aux ordres de s’organiser, compte tenu du fait que la nature des données à publier n’était pas connue l’année dernière.
Quant aux données rendues publiques, dans son avis du 21 mars 2013 sur le projet de décret, la Commission nationale de l’informatique et des libertés (CNIL) a invité à un juste équilibre entre l’objectif de transparence et la protection des données personnelles.
La CNIL note tout d’abord que toutes les données visées à l’article R. 1453-3 du code de la santé publique n’étaient pas prévues expressément par le législateur. Elle relève dans le même avis que selon la jurisprudence de la Cour de justice européenne « aucune prééminence automatique ne saurait être reconnu à l’objectif de transparence sur le droit à la protection des données à caractère personnel » (4).
Par conséquent, la CNIL a estimé nécessaire de préciser que « les responsables de traitement qui procèdent à la mise en ligne des données à caractère personnels sont tenus de mettre en place des mesures visant à empêcher les moteurs de recherche externes de procéder à une indexation des données directement identifiantes ». Ces mesures peuvent, selon la CNIL, consister en l’utilisation de règles d’indexation à destination des moteurs de recherche correctement définis, ou de mécanismes s’assurant que l’émetteur d’une requête concernant un document est bien un internaute et non un programme informatique.
Conformément à ces préconisations, aux termes de l’article R. 1453-7, l’autorité responsable du site internet public unique devra prendre les mesures techniques nécessaires pour assurer l’intégrité du site et « la protection des seules données directement identifiantes contre l’indexation par des moteurs de recherche ». Précisons cependant que cette interdiction ne s’appliquera pas aux données publiées sur le futur site internet unique.
Il est précisé que l’autorité en charge du site doit se conformer aux dispositions de la loi n° 78-17 du 6 janvier 1978 relative à l’informatique, aux fichiers et aux libertés en accomplissant auprès de la Commission nationale de l’informatique et des libertés les formalités nécessaires pour les traitements de données qu’elle met en œuvre pour l’application de la présente section. Elle indique sur le site internet la possibilité pour la personne d’exercer son droit de rectification des informations les concernant et l’absence d’application du droit d’opposition.
● Les avantages consentis aux étudiants
Enfin, le décret procède à l’actualisation des dispositions réglementaires prises pour l’application de la loi n° 93-121 du 27 janvier 1993 dite « loi anti-cadeaux », la loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé ayant étendu son application aux étudiants ainsi qu’aux associations représentant ces étudiants.
Ainsi les étudiants ne peuvent pas recevoir d’avantages en nature ou en espèces des entreprises sauf, à l’instar des professionnels de santé, dans le cadre de conventions ayant pour objet des activités de recherche ou d’évaluation scientifique, ou à l’occasion de manifestations de promotion ou à caractère exclusivement professionnel et scientifique.
Le décret a soulevé des inquiétudes parmi certaines associations ou sociétés savantes craignant d’être coupées d’une partie de leur financement. La circulaire précitée du 29 mai 2013 précise la nature des associations concernées. Il s’agit des associations chargées de défendre les intérêts catégoriels d’une profession ou d’un groupe d’étudiants qui les composent. Ne sont pas visées les associations reconnues d’utilité publique qui non seulement sont des associations inter-catégorielles, mais qui poursuivent aussi un but d’intérêt général, ni celles réunissant des professionnels de santé et dont l’objet est d’exercer des activités de recherche en santé ou d’y participer ou de formation médicale. Les sociétés savantes qui ont notamment pour objet de rendre compte de l’état de l’art, d’améliorer la connaissance et d’assurer la formation et la recherche du secteur considéré ne seront pas concernées par l’interdiction de financement.
II.- LA RESPONSABILISATION DES ACTEURS INSTITUTIONNELS DU SYSTÈME DE SÉCURITÉ SANITAIRE : UNE NÉCESSAIRE RÉVOLUTION CULTURELLE
La création d’une nouvelle agence chargée de la sécurité des produits de santé est un des enjeux majeurs de la loi du 29 décembre 2011. L’Agence nationale de sécurité du médicament et des produits de santé a été créée le 1er mai 2012, pour succéder à l’Afssaps. Le choix a été fait au moment de l’examen de la loi du 29 décembre 2011 de ne pas remettre radicalement en cause l’architecture de nos agences sanitaires mais d’en rénover la structure et le fonctionnement. L’ANSM doit désormais être clairement identifiée par nos concitoyens comme étant chargée de la « police » du médicament.
Par ailleurs, pour améliorer le pilotage de la politique du médicament et renforcer l’autorité du politique, M. Xavier Bertrand avait annoncé la création d’un comité stratégique de la politique des produits de santé et de la sécurité sanitaire. Celui-ci devait se réunir chaque semaine en comité opérationnel, avec un représentant du ministre, et en comité stratégique, tous les trimestres, sous la présidence du ministre lui-même, en présence de toutes les agences et les directions d’administration centrale concernées. En réalité, les réunions hebdomadaires ne se sont pas tenues. Une réunion a eu lieu pour préparer l’installation du comité en décembre 2011 présidée par les directeurs d’administration centrale. Le comité stratégique sous la présidence du ministre a été installé en mars 2012.
A. LES PRÉROGATIVES DE LA NOUVELLE AGENCE NATIONALE DE SÉCURITÉ DU MÉDICAMENT ET DES PRODUITS DE SANTÉ
● Les moyens de l’ANSM au regard de ses nouvelles missions
De l’Afssaps, l’agence a repris l’intégralité des missions qui s’étaient déjà progressivement élargies au fil du temps.
Néanmoins, à ces missions déjà importantes et diversifiées se sont immédiatement ajoutées de nombreuses responsabilités complémentaires induites par la loi du 29 décembre 2011, dont les mesures visaient à répondre aux enjeux mis en évidence par la crise du Mediator, ainsi que par la transposition de nouvelles directives dans le domaine des produits de santé.
Parmi ces nouveaux champs d’intervention, on peut ainsi citer, de façon non exhaustive :
– encadrer l’utilisation fondée de médicaments hors AMM (autorisation de mise sur le marché) par la possibilité d’instruire et de délivrer des RTU (recommandations temporaires d’utilisation) ;
– assurer un suivi des produits de santé dans la vraie vie par la mise en œuvre d’un pôle d’épidémiologie et contribuer à un futur groupement d’intérêt public (GIP) sur le sujet ;
– gérer un répertoire regroupant les essais cliniques ayant abouti à une autorisation de mise sur le marché (AMM) pour l’ensemble des produits commercialisés en France ;
– mener des études scientifiques indépendantes dans le cadre des missions d’encouragement à la recherche introduites par la loi par le biais d’appel à projets financés sur un budget dédié ;
– encadrer des activités de courtage de médicaments ;
– passer d’un système de déclaration à un système d’autorisation pour les activités de fabrication, d’importation ou de distribution de substances actives entrant dans le champ de compétence de l’Agence ;
– contribuer activement aux travaux du comité européen de pharmacovigilance des médicaments de l’Agence européenne du médicament depuis septembre 2012, comité symétrique de celui d’enregistrement ;
– contrôler a priori la publicité professionnelle pour le médicament ;
– contrôler la publicité pour certains dispositifs médicaux et dispositifs médicaux de diagnostic in vitro ;
– contrôler les spécificités techniques des dispositifs médicaux remboursés ;
– mettre en œuvre une base de données publiques sur les produits de santé sous le pilotage du ministère de la Santé ;
– renforcer les exigences en termes de déontologie vis-à-vis des agents et des experts externes ;
– augmenter les exigences en matière de transparence en passant notamment par le renforcement de l’expertise interne et d’autres mesures telles que l’enregistrement systématique des instances consultatives de l’Agence qui concourent à sa prise de décision, la mise en ligne des comptes rendus des séances, de l’ordre du jour et des vidéos.
Aux missions légales de l’ANSM s’ajoutent les priorités politiques fixées par le Gouvernement en matière de politique sanitaire.
Une lettre de mission adressée le 17 septembre 2012 par la ministre au directeur général de l’agence, qui précise ses orientations pour les années 2012-2015. S’y ajoute le contrat de performance (COP), qui sera élaboré pour la période 2013-2016, et qui permettra de définir et de structurer les priorités de l’Agence pour les années à venir. Ce COP sera établi en tirant les enseignements de celui qu’avait passé l’Afssaps avec l’État pour la période 2007-2010.
Selon son rapport annuel de 2012, sur la base de la lettre de mission, l’agence s’est engagée dans un programme de travail ambitieux s’articulant autour de trois axes :
– favoriser un accès rapide, sécurisé et large à l’innovation et à l’ensemble des produits de santé pour les patients ;
– garantir la sécurité des produits de santé tout au long de leur cycle de vie ;
– informer et communiquer de façon transparente sur les décisions et leur processus d’élaboration.
Chacun de ces axes se décline en actions concrètes, parmi lesquelles :
– le renforcement de la présence européenne de l’Agence sur l’enregistrement des nouvelles molécules innovantes. En effet, comme l’a rappelé M. Dominique Maraninchi en audition, en matière de contribution aux travaux européens, les années 2011 et 2012 ont été marquées par un fort recul du nombre de dossiers attribués à la France en tant que rapporteur ou co-rapporteur. De fait, les règles d’attribution des dossiers de l’Agence européenne du médicament (EMA) se sont renforcées en prenant en compte la capacité des Agences des États-membres à respecter les délais fixés par la règlementation pour la remise des rapports. En 2011, l’EMA a publié pour la première fois les données sur ces retards, qui mettaient la France en dernière position des pays habituellement désignés comme rapporteurs ;
– la conversion progressive des autorisations temporaires d’utilisation (ATU) nominatives en ATU de cohorte pour un accès plus équitable aux traitements concernés par les patients ;
– la mise en œuvre d’un programme de réévaluation des anciennes AMM (d’avant 2005) qui s’avère extrêmement consommateur de ressources, surtout quand il débouche sur des arbitrages européens ;
– un programme de travail sur certaines classes de dispositifs médicaux particulièrement à risque (prothèses mammaires, prothèses de hanche métal/métal, prothèses de genoux, sondes de défibrillation, …) ;
– la mise en œuvre de dispositifs d’information par voie d’abonnement pour les professionnels de santé ;
Les ressources complémentaires nécessaires à la mise en œuvre de ces nouvelles obligations réglementaires avaient été chiffrées à 126 équivalent temps plein (ETP). Les pouvoirs publics avaient ainsi arrêté, en juillet 2011, un plan de renforcement volontariste des moyens de l’agence. Celui-ci était établi sur 2 ans et prévoyait la création de 80 emplois (40 en 2012 et 40 en 2013), l’agence s’engageant à en assumer le tiers (46) par redéploiement. Ces engagements étaient inscrits dans la loi de finances pour 2012. Le solde net réalisé sur 2012 et 2013 n’a finalement été que de 10 postes complémentaires.
Il conviendra de veiller à l’adéquation entre les missions et les moyens de l’ANSM.
● Le développement d’une politique de partenariats conventionnels
Au-delà des relations tutélaires avec le ministère de la santé, il demeure nécessaire de structurer et de renforcer les relations de l’ANSM avec ses partenaires institutionnels, notamment l’assurance maladie, la Haute autorité de la santé (HAS) l’Institut national du cancer (INCa), les autres agences sanitaires mais aussi les structures de coordination de la recherche en santé afin de coordonner les travaux sur des sujets d’intérêt commun et d’échanger des données complémentaires sur la sécurité des produits de santé. L’ANSM doit renforcer ses relations avec les agences régionales de santé, acteur désormais déterminant de l’ensemble de la politique de santé en région. Le partenariat avec la CNAMTS est structurant pour la mise en œuvre de programmes d’études pharmaco-épidémiologiques indépendantes de l’industrie des produits de santé.
La convention avec la HAS sera structurée en priorité autour de la participation de l’ANSM à la Commission de la transparence et à la production (si possible et opportune) de recommandations de pratiques quand elles impliquent des produits de santé. La convention avec l’INCa trace les travaux conjoints menés par les RTU, les ATU de cohorte en oncohématologie et les échanges de données sur les essais cliniques.
Dans le prolongement de la convention-cadre signée par le Conseil national de l’Ordre des pharmaciens (CNOP) le 3 novembre 2011, qui a déjà donné lieu à la mise en place d’un système d’alerte pour les retraits et rappels du marché des médicaments (visant à joindre les officines, les pharmacies des établissements de santé, les distributeurs en gros et les médecins propharmaciens), l’ANSM renforce par d’autres conventions sa collaboration avec le CNOP.
En vue de renforcer l’information des professionnels de santé et des opérateurs, il est prévu la mise en place d’un système d’alerte pour les retraits et rappels du marché des dispositifs médicaux et dispositifs médicaux de diagnostic in vitro distribués dans les officines et les établissements de santé, la diffusion par le CNOP en relais de celle faite par l’ANSM, des informations de sécurité émanant de l’ANSM et des informations sur les ruptures de stock de médicaments. Afin de renforcer la surveillance des produits de santé à l’Agence des données anonymisées seraient recueillies par le CNOP au moyen du dossier pharmaceutique concernant les médicaments.
● Les pouvoirs de sanction de l’agence
La nouvelle agence est dotée de pouvoirs de sanctions administratives et financières étendus, qui lui permettront d’exercer une véritable pression sur les entreprises dans les cas suivants : absence de mise en œuvre du système de pharmacovigilance, absence de déclaration d’un effet indésirable, défaut de transmission dans les délais impartis des résultats des études effectuées après la délivrance de l’autorisation de la mise sur le marché, défaut de communication d’un arrêt de commercialisation, diffusion d’une publicité pour un médicament non autorisé ou pour un dispositif médical pris en charge ou non autorisé.
Cependant, le texte fixant les sanctions applicables en cas de refus des industriels de fournir des réponses à l’ANSM n’a pas encore été publié. En l’état actuel du droit, cette disposition est donc restée lettre morte.
L’article 39 de la loi dispose en effet que le Gouvernement est autorisé à prendre par ordonnance, dans un délai de vingt-quatre mois à compter de la promulgation, les mesures destinées à harmoniser et mettre en cohérence les dispositions relatives aux sanctions pénales et aux sanctions administratives relatives aux produits de santé.
Selon le ministère de la santé, des discussions seraient en cours avec le ministère de la justice pour mettre au point un texte commun.
Vos rapporteurs insistent sur l’urgence de la publication de ces sanctions, qui conditionne la mise en œuvre effective des contrôles effectués par la nouvelle agence.
B. L’ARCHITECTURE DE L’AGENCE ET LA TRANSPARENCE DE SES TRAVAUX
Le décret n° 2012-597 du 27 avril 2012 relatif à l’Agence nationale de sécurité du médicament et des produits de santé, fixe la nouvelle organisation, les missions et le fonctionnement de l’ANSM. Le décret précise les compétences, la composition et les modalités de fonctionnement du conseil d’administration. Il définit les attributions du directeur général de l’agence ainsi que les missions, la composition et les modalités de fonctionnement du conseil scientifique. Le décret fixe en outre le régime financier et comptable de la nouvelle agence ainsi que les règles applicables à son personnel.
● La composition du conseil d’administration
Pour mémoire, l’article 7 de la loi fixe la composition du conseil d’administration de l’agence. Sont représentés, outre le président :
– des représentants de l’État ;
– trois députés et trois sénateurs ;
– des représentants des régimes obligatoires de base d’assurance maladie ;
– des représentants des professionnels de santé autorisés à prescrire et à dispenser des produits mentionnés au même article L. 5311-1 ;
– des représentants d’associations de patients ;
– des personnalités qualifiées ;
– des représentants du personnel de l’agence.
Les droits de vote sont répartis pour moitié entre les représentants de l’État et pour moitié entre les autres membres du conseil d’administration. Le président a voix prépondérante en cas de partage égal des voix.
Le décret précité du 27 avril 2012 prévoit que le conseil d’administration fixe par ses délibérations les orientations de la politique de l’agence, notamment stratégiques pluriannuelles, le programme de travail annuel ainsi que le rapport d’activité, le règlement intérieur, le budget, les subventions ou marchés dont le seuil est supérieur à celui qu’il détermine, les programmes d’investissement, les acquisitions, aliénations et autorisations d’ester en justice, les dons et legs, les participations à un GIP et les conditions relatives au recrutement (article R. 5322-11).
Les fonctions des membres du conseil d’administration sont exercées à titre gratuit (article R. 5322-7). Le conseil d’administration se réunit au moins trois fois par an sur convocation de son Président (article R. 5322-8). Elle est de droit dans les trente jours suivants la demande qui en est faite par le ministre chargé de la santé, le directeur général ou par le tiers au moins des membres du conseil. Sauf urgence, les membres reçoivent, huit jours au moins avant la date de réunion, l’ordre du jour et les documents nécessaires à l’examen des sujets qui y sont inscrits. Le Président arrête l’ordre du jour sur proposition du directeur général (article R. 5322-9).
Le même décret fixe les missions du conseil scientifique de l’agence. Il est composé de 12 membres nommés par le ministre chargé de la santé pour une durée de trois ans renouvelable. Il se réunit au moins trois fois par an sur convocation de son président ou à la demande du directeur général, ou à l’initiative motivée d’au moins un tiers de ses membres. L’ordre du jour est arrêté par son président.
Il doit veiller à la cohérence de la stratégie scientifique de l’agence, donner un avis sur les orientations de recherche ainsi que sur la politique de partenariat, assister le directeur général dans les procédures d’appels à projets, formuler des avis auprès du directeur général ou du conseil d’administration.
Il peut également formuler des recommandations sur toute question technique ou scientifique (article R. 5322-17).
Le directeur général, M. Dominique Maraninchi depuis le 1er mai 2012, est nommé pour une durée de trois ans, renouvelable une fois (article R. 5322-14). Il dirige l’établissement. Il accomplit tous les actes qui ne sont pas réservés au conseil d’administration en vertu de l’article R. 5322-11 du code de la santé publique. Il représente l’ANSM en justice et dans tous les actes de la vie civile. Il passe au nom de l’ANSM les contrats, conventions et marchés ; les actes d’acquisition, de vente et de transaction. Il recrute, nomme et gère les agents contractuels, les personnels scientifiques et techniques de laboratoire.
Il propose au conseil d’administration qui en délibère la création de commissions, comités ou groupes de travail nécessaires à la conduite des missions de l’agence. Il détermine, après avis du conseil d’administration et du conseil scientifique, l’étendue et la durée des missions, la composition et les modalités de consultation de ces commissions et en nomme les membres. Il est enfin l’ordonnateur des recettes et des dépenses du budget de l’agence (article R. 5322-15).
● La nouvelle architecture de l’ANSM
La nouvelle organisation de l’agence est entrée en vigueur le 3 octobre 2012 et son premier Conseil d’administration s’est tenu le 26 octobre 2012. Mise en œuvre le 3 octobre 2012 après un processus de construction et de sélection des nouvelles équipes managériales et de repositionnement d’une majorité des agents, la nouvelle organisation est entrée, au dernier trimestre 2012, en phase de montée en puissance.
Un très important effort de formation et de tutorat interne a été initié au dernier trimestre 2012 afin de permettre aux agents, dont le périmètre fonctionnel a évolué dans le cadre de la mise en place de la nouvelle organisation, de sécuriser leurs actes professionnels dans leur nouveau poste.
Après l’avis du Conseil scientifique du 4 juillet 2012, le Conseil d’administration a délibéré le 26 octobre 2012 sur la création des commissions, comités techniques et groupes de travail nécessaires à la conduite des missions de l’Agence.
Les quatre commissions sont les suivantes :
– commission d’évaluation initiale du rapport bénéfice/risque des produits de santé ;
– commission de suivi du rapport bénéfice/risque des produits de santé ;
– commission des stupéfiants et psychotropes ;
– commission de prévention des risques liés à l’utilisation des catégories de produits.
Elles sont composées de personnalités choisies en raison de leur expertise scientifique et technique dans les domaines de compétences de l’Agence, de spécialistes en médecine générale, de pharmaciens d’officine, de pharmaciens hospitaliers et d’associations d’usagers du système de santé agréées au niveau national.
Concernant la prévention des conflits d’intérêts, on note un changement progressif des pratiques. Ainsi, dans les anciennes commissions de l’agence, les experts avaient été sélectionnés en fonction de leur compétence. En cas de risque de conflit d’intérêts, ils devaient quitter la salle. Depuis la promulgation de la loi, un appel à candidature a été lancé pour renouveler tous les experts, en tenant compte de leur profil déontologique, avec des critères stricts d’affectation en commission. La logique a été inversée pour plus d’efficacité.
Les groupes de travail sont opérationnels depuis le début de l’année 2013. Ils sont au nombre de 36 et l’ordre du jour, la préparation des dossiers, et les comptes rendus afférents sont préparés et rédigés par l’ANSM qui assure l’organisation, l’animation et le secrétariat de ces groupes.
Les experts, au nombre maximal de 20, sont nommés pour une durée de trois ans, renouvelable une fois. Le règlement intérieur établit les modalités de déroulement des séances dont la participation éventuelle de personnes extérieures aux groupes et celles d’instruction des dossiers. Ce règlement rappelle également les règles applicables en matière de déontologie, de confidentialité et de transparence.
Dans le domaine des vigilances, l’activité de mobilisation de l’expertise externe et des réseaux de vigilance repose aussi sur l’organisation de comités techniques. Ils rassemblent les échelons territoriaux des réseaux de vigilance. Deux de ces comités techniques préexistaient à la création de l’ANSM : le comité technique de pharmacovigilance et le comité technique des centres d’information et d’évaluation sur la pharmacodépendance. Au cours du premier semestre 2013, ce mode d’organisation sera étendu à la matériovigilance et à l’hémovigilance.
● La transparence des travaux de l’agence
L’article 7 de la loi crée l’article L. 5324-1 du code de la santé publique, aux termes duquel l’agence rend publics l’ordre du jour et les comptes rendus, assortis des détails et explications des votes, y compris les opinions minoritaires, à l’exclusion de toute information présentant un caractère de confidentialité industrielle ou commerciale ou relevant du secret médical, des réunions des commissions, des comités et des instances collégiales d’expertise, dont les avis fondent une décision administrative.
Les modalités d’application de cette mesure devaient être précisées par décret en Conseil d’État. Les articles R. 1451-6 à R. 1451-9 issus du décret précité n° 2011-745 du 9 mai 2012 précisent que font l’objet d’un enregistrement intégral et de la diffusion de procès-verbaux les débats des commissions, conseils et instances collégiales mentionnés au I de l’article R. 1451-1 du code de la santé publique conduisant à l’adoption d’un avis sur une question de santé publique ou de sécurité sanitaire recueilli, à titre obligatoire ou facultatif, par l’autorité compétente préalablement à une décision administrative.
Les participants sont informés de l’enregistrement au plus tard au début des débats concernés.
Les procès-verbaux sont mis en ligne dans les meilleurs délais, dans le respect des secrets protégés par la loi, sur le site internet de l’administration, de l’autorité, de l’établissement ou du groupement concerné, sous l’autorité, selon le cas, du ministre, du président de l’autorité, du directeur ou du directeur général de l’établissement ou du groupement. Ils demeurent accessibles en ligne pour une durée fixée par ce dernier et qui ne peut être inférieure à un an.
La mise en ligne, dans le respect des secrets protégés par la loi, des enregistrements, peut être décidée, selon le cas, par le ministre, le président de l’autorité, le directeur ou le directeur général de l’établissement ou du groupement.
Ils demeurent alors accessibles en ligne pour une durée fixée par ce dernier et qui ne peut être inférieure à un an. Les participants aux débats concernés en sont informés au plus tard au moment de la mise en ligne.
Les procès-verbaux et les enregistrements mentionnés sont conservés par l’administration, l’autorité, l’établissement ou le groupement concerné pendant une durée de dix ans.
Vos rapporteurs tiennent à saluer le travail effectué dans des délais limités par les dirigeants et le personnel de l’agence. Ils attirent l’attention du Gouvernement sur la nécessité de lui accorder les moyens nécessaires au bon accomplissement de ses nouvelles fonctions. Enfin, l’application des mesures relatives à l’agence prévues par la loi du 29 décembre 2011 supposent des changements profonds dans la pratique, et non seulement dans les textes. Il conviendra d’être particulièrement attentif à la lutte contre les conflits d’intérêts et aux mesures tendant à simplifier et à alléger les travaux de l’agence, afin qu’elle se concentre sur sa vocation première, à savoir la surveillance indépendante des produits de santé.
III.- L’ENCADREMENT DU « HORS-AMM » : ALLIER ACCÈS AUX PROGRÈS THÉRAPEUTIQUES ET SÉCURITÉ DES PATIENTS
L’encadrement des prescriptions « hors AMM » constitue une avancée importante du texte. Celle-ci représenterait entre 15 et 20 % de la totalité des prescriptions, bien d’avantage dans certains domaines comme la pédiatrie, la gérontologie ou la cancérologie.
Le principal objectif de la loi est de bien distinguer l’usage légitime du « hors AMM », faisant l’objet d’une recommandation de la nouvelle agence et d’un suivi des malades, et son usage illégitime, dont l’affaire du Mediator a montré la dangerosité – faut-il rappeler en effet que ce médicament était prescrit à 78 % « hors AMM » en 2008. Cet usage doit être limité et sanctionné si besoin. Le « hors AMM » doit demeurer une exception encadrée.
A. L’ENCADREMENT DES AUTORISATIONS TEMPORAIRES D’UTILISATION (ATU)
L’article 26 de la loi tend à renforcer la procédure de délivrance des autorisations temporaires d’utilisation (ATU) afin de mieux contrôler son usage.
Pour pouvoir être commercialisés, tous les médicaments doivent obtenir une autorisation de mise sur le marché (AMM). L’article L. 1521-12 du code de la santé publique prévoit, à titre dérogatoire, que certains produits peuvent en être dispensés et ce, par le biais d’une ATU, pour traiter des maladies graves ou rares, en l’absence de traitement approprié et lorsque la mise en œuvre du traitement ne peut être différée. Ces médicaments ne peuvent être délivrés qu’à l’hôpital et leur délivrance est régie par des conditions strictes. L’utilisation des ATU a connu des dérives, les laboratoires les ayant parfois utilisées pour commercialiser les médicaments avant l’obtention de l’AMM.
Il existe deux types d’ATU : les ATU de cohorte sont sollicitées directement par le laboratoire titulaire des droits d’exploitation des médicaments ; les ATU nominatives sont attribuées aux médicaments prescrits sous la responsabilité d’un médecin à un patient nommément désigné et ne pouvant participer à une recherche biomédicale dès lors que le médicament est susceptible de présenter un bénéfice pour lui et que son efficacité et sa sécurité sont présumées en l’état des connaissances scientifiques. Le médecin doit justifier que le patient a reçu une information adaptée à sa situation sur l’absence d’alternatives thérapeutiques, les risques courus, les contraintes et le bénéfice susceptible d’être apporté par le médicament.
Désormais, aux termes de l’article L. 1521-12 du code de la santé publique tel qu’issu de la loi du 29 décembre 2011, les ATU sont octroyées par l’ANSM pour une durée limitée, éventuellement renouvelable, à la demande du titulaire des droits d’exploitation pour les ATU de cohorte, ou du médecin prescripteur pour les ATU nominatives ; cette disposition ayant pour but d’éviter que l’autorisation temporaire ne devienne une alternative à la demande d’autorisation de mise sur le marché.
Pour être recevable, la demande d’ATU nominative doit remplir les conditions suivantes :
– le médicament fait l’objet d’une demande d’ATU de cohorte ;
– l’entreprise pharmaceutique s’est engagée à entreprendre la procédure de demande d’AMM ou l’a déjà entamée ;
– des essais cliniques sont conduits en France ou la demande d’essais cliniques a été déposée ;
– le titulaire des droits d’exploitation s’engage à demander une ATU de cohorte ou une AMM dans un délai déterminé par l’agence.
Afin de ne pas limiter l’accès à l’innovation thérapeutique des patients, des exceptions permettent de demander une ATU nominative dérogatoire si :
– en l’état des thérapeutiques disponibles, des conséquences graves pour le patient sont très fortement probables ;
– le médicament a fait l’objet d’un arrêt de commercialisation mais que l’indication thérapeutique sollicitée est différente que la raison pour laquelle l’arrêt de la commercialisation a été décidé et il existe de fortes présomptions d’efficacité et de sécurité du médicament dans l’indication thérapeutique sollicitée ;
– le titulaire des droits d’exploitation s’est vu refuser une ATU de cohorte ou une demande d’autorisation d’essais cliniques, à condition que le patient et le praticien aient reçu une information suffisante sur les motifs du refus de la demande et sous réserve d’un bénéfice individuel pour le patient.
Sauf si elle est accordée dans les conditions dérogatoires précédemment exposées, l’autorisation est subordonnée à la conclusion entre l’agence et le titulaire des droits d’exploitation du médicament d’un protocole d’utilisation thérapeutique et de recueil d’informations concernant l’efficacité, les effets indésirables, les conditions réelles d’utilisation ainsi que les caractéristiques de la population bénéficiant du médicament.
Les médicaments faisant l’objet d’ATU nominative ne sont pas soumis à la conclusion d’un protocole d’utilisation thérapeutique mais le médecin doit transmettre à l’agence des données de suivi des patients à l’expiration ou à la demande de renouvellement de l’autorisation. Toutefois, ces autorisations peuvent faire l’objet d’un protocole et d’un recueil d’information. Le protocole n’étant pas obligatoire pour les ATU nominatives, celles-ci demeurent toujours moins protectrices pour les patients que les autorisations de cohorte, ce qui doit inciter l’ANSM à en limiter le nombre.
Le décret n° 2013-66 du 18 janvier 2013 relatif aux autorisations temporaires d’utilisation des médicaments est venu préciser les conditions fixées par l’article L.5121-12. Codifié aux R. 5121-68 à R. 5121-77-1 du code de la santé publique, il réécrit la section 7 du chapitre 1er du titre II du livre 1er de la cinquième partie du code de la santé publique consacrée aux ATU.
Le décret précise que les ATU de cohorte sont accordées pour une durée d’un an, et les ATU nominatives, pour la durée du traitement mais dans la limite maximale d’un an. Les autorisations sont renouvelables mais le décret ne limite par le nombre des renouvellements, ce qui va à l’encontre de l’esprit initial du texte qui souhaitait mieux encadrer le dispositif d’autorisation afin que celui-ci ne soit détourné de son objet.
L’article 1er du décret qui modifie les articles R. 5121-68 et R. 5121-69 du code de la santé publique précise les éléments que doit contenir le dossier de demande d’ATU de cohorte ou nominative qui doit être adressé à l’ANSM :
– les éléments permettant d’affirmer l’absence de traitement approprié pour traiter la maladie grave ou rare dont les patients sont atteints ; l’impossibilité de différer la mise en œuvre du traitement et prouvant que l’efficacité et la sécurité de ce médicament sont fortement présumées ;
– une copie de la demande d’AMM adressée à l’ANSM ou, le cas échéant, une lettre du demandeur de l’AMM attestant qu’il s’engage à formuler une telle demande dans un délai déterminé ;
L’article R. 5121-70 du même code détaille les éléments qui doivent apparaître dans le protocole d’utilisation thérapeutique. Ce dernier devra obligatoirement définir les modalités de recueil d’informations sur l’efficacité du médicament et ses éventuels effets secondaires.
Le directeur général de l’ANSM peut, à tout moment, demander qu’on lui transmette des données prouvant que le rapport bénéfices/risques reste favorable. Le titulaire des droits d’exploitation a quant à lui une obligation de transmettre sans délai toute donnée susceptible de modifier ce rapport et toute information utile telle que l’interdiction ou la restriction imposée dans un autre pays.
Les protocoles d’utilisation thérapeutiques, les résumés des rapports de synthèse ainsi que les motifs justifiant la suspension ou le retrait d’une ATU de cohorte doivent être publiés sur le site de l’ANSM.
Le titulaire des droits d’exploitation devra régulièrement adresser un rapport de synthèse des informations recueillies à l’ANSM. Il devra faire état des conditions d’utilisation du médicament, de son efficacité, de toute information pouvant entraîner une modification de l’évaluation du rapport bénéfices/risques du médicament.
En cas de suspension de l’autorisation par le directeur général de l’agence, le décret précise que celle-ci ne peut avoir lieu que pour une durée maximale de trois mois.
L’article 41 de la loi précise que les anciennes dispositions continuent de régir les autorisations temporaires d’utilisation, y compris pour leur renouvellement, pendant les trois ans qui suivent la publication de la loi, soit jusqu’à fin 2014. Ces mêmes dispositions s’appliquent aux nouvelles demandes si des autorisations de même nature avaient été accordées pour la même indication pour le médicament concerné.
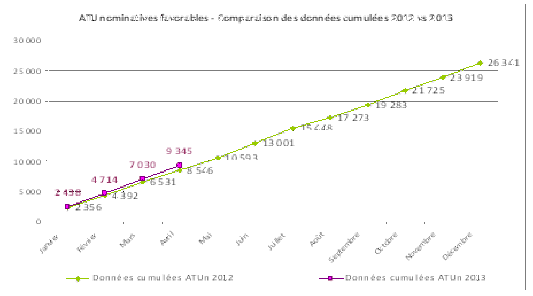
Il est encore trop tôt pour dresser un bilan qualitatif de la mesure. D’ores et déjà, l’ANSM, dans son rapport d’activité montre que, malgré la mise en œuvre d’une démarche de diminution des ATU nominatives au profit de ATU de cohorte auprès des industriels, le nombre d’ATU nominatives délivrées en 2013 a augmenté comparativement à 2012 (+ 9 %), comme le montre le graphique présenté ci-dessus. L’agence avait affiché un objectif de réduction de 20 à 30 % du nombre de demandes d’ATU nominative qui n’est, pour le moment, pas suivi d’effet, ce qui semble logique dans la mesure où les dispositions de la loi concernant les ATU ne commenceront à s’appliquer qu’à la fin de l’année 2014.
B. LES RECOMMANDATIONS TEMPORAIRES D’UTILISATION
L’article 18 de la loi vise à sécuriser les prescriptions médicales faites « hors AMM », c’est-à-dire pour une visée thérapeutique différente de celle prévue dans l’AMM.
Ainsi, l’article L. 5121-12-1 du code de la santé publique, issu de la loi du 29 décembre 2011, prévoit qu’une spécialité pharmaceutique peut faire l’objet d’une prescription non conforme à son autorisation de mise sur le marché en l’absence d’alternative médicamenteuse appropriée et sous réserve :
– que l’indication ou les conditions d’utilisation aient fait l’objet d’une recommandation temporaire d’utilisation (RTU) établie par l’ANSM. Il faut noter ici que les RTU se distinguent des autorisations temporaires d’utilisation (ATU) car les produits concernés bénéficient déjà d’une autorisation de mise sur le marché ;
– ou que le prescripteur juge indispensable, au regard des données acquises de la science, le recours à cette spécialité pour améliorer ou stabiliser l’état clinique du patient.
Les RTU peuvent s’appliquer à tous les médicaments ayant une AMM en France, elles sont temporaires et ne peuvent excéder trois ans. Elles peuvent porter sur une indication ou des conditions différentes de celles de l’AMM.
L’initiative d’engager la procédure d’élaboration d’une RTU relève de la compétence exclusive de l’ANSM qui peut s’autosaisir ou recevoir des signalements. Pour décider d’engager une telle procédure, l’Agence prend en considération plusieurs critères comme le niveau des preuves scientifiques d’efficacité, le caractère innovant et le profil de sécurité du médicament, le pronostic et la fréquence de la maladie ainsi que l’existence d’essais cliniques dans l’indication concernée.
Ces recommandations sont assorties d’un recueil des informations concernant l’efficacité, les effets indésirables et les conditions réelles d’utilisation de la spécialité par le titulaire de l’autorisation de mise sur le marché ou l’entreprise qui l’exploite, dans des conditions précisées par une convention conclue avec l’agence. La convention peut comporter l’engagement, par le titulaire de l’autorisation, de déposer dans un délai déterminé une demande de modification de cette autorisation.
Il est prévu que les RTU soient mises à disposition des prescripteurs. En outre, le médecin a désormais l’obligation de porter sur l’ordonnance la mention « prescription hors autorisation de mise sur le marché ». Notons que cette mention permettra aux pharmaciens d’exercer à leur tour leur responsabilité. De plus, le médecin devra motiver sa prescription dans le dossier médical du patient, ce qui devrait améliorer l’information sur les prescriptions « hors AMM ».
Le décret en Conseil d’État n° 2012-742 du 9 mai 2012 relatif aux recommandations temporaires d’utilisation des spécialités pharmaceutiques précise les conditions de mise en œuvre d’une RTU par l’ANSM.
L’article R. 5121-76-3 du code de la santé publique, issu du décret précité, prévoit que les personnes habilitées à solliciter l’ANSM sont:
– les ministres chargés de la santé et de la sécurité sociale ;
– la Haute Autorité de Santé (HAS) ;
– l’Union nationale des caisses d’assurance maladie (Uncam) ;
– l’Institut National du Cancer (Inca) ;
– les centres de référence/compétence en charge des maladies rares ;
– les associations agréées de patients.
Par ailleurs, une société savante peut alerter l’ANSM. Par contre, un laboratoire pharmaceutique n’a pas vocation à demander à l’ANSM la mise en œuvre d’une RTU. Lorsqu’il identifie une situation de prescription non conforme à l’AMM, il en avise l’ANSM et :
– soit cette situation relève d’un besoin de santé et il appartient au laboratoire d’envisager un dossier d’extension d’indication d’AMM ;
– soit cette situation est injustifiée et il appartient au laboratoire d’informer les prescripteurs du caractère inutile voire dangereux de la prescription.
Lorsqu’elle envisage d’élaborer une RTU, l’ANSM demande au(x) laboratoire(s) concerné(s) de lui transmettre dans un délai de trois mois toute information utile et notamment : toutes données cliniques et non cliniques permettant de contribuer à l’évaluation de l’efficacité et la sécurité du médicament utilisé dans la situation identifiée ; la liste et l’état d’avancement des essais cliniques en cours et programmés en France ou à l’étranger dans l’indication visée et les sites de recherche en France ; une estimation du nombre de patients potentiellement concernés ; un projet de protocole de suivi des patients ; une copie de toute AMM délivrée dans un autre État dans cette indication ; le cas échéant, une copie de tout refus ou retrait d’AMM dans cette indication par un autre État ; le cas échéant, une copie de tout avis scientifique de l’Agence européenne du médicament. Ces avis portent notamment sur :
– l’analyse du besoin d’une RTU au regard des pratiques et des recommandations existantes de prise en charge ;
– les données disponibles en termes d’efficacité et de sécurité dans la situation concernée ;
– le cas échéant, les travaux conduits par le centre de référence de la pathologie concernée.
La RTU mentionne :
– l’indication dérogatoire ;
– la posologie et le mode d’administration ;
– si besoin, des précautions d’emploi, mises en garde ou contres indications spécifiques au cadre de la RTU ;
– les effets indésirables ;
– le cas échéant, le classement de la spécialité si la RTU implique qu’il diffère de celui indiqué dans l’AMM à la rubrique « conditions de prescriptions et délivrance » ;
– sa durée de validité.
L’ANSM évalue le rapport bénéfice/risque présumé de la situation pouvant relever d’une RTU à partir des données dont elle dispose et des données recueillies auprès du laboratoire et, le cas échéant de l’Inca ou des centres de référence. L’exigence peut être adaptée en fonction de situations particulières, telles que les maladies rares.
Si l’évaluation effectuée par l’ANSM permet de penser que le rapport entre le bénéfice présumé et les effets indésirables encourus est favorable, l’ANSM élabore un projet de RTU avec en annexe un protocole définissant les modalités de recueil des données et de suivi des patients, portant sur les données d’efficacité, de sécurité et les conditions réelles d’utilisation du produit.
Le suivi doit comporter au moins un critère permettant d’évaluer le bénéfice thérapeutique du médicament ; le recueil des effets indésirables ; des données relatives aux conditions réelles d’utilisation.
Aux termes de l’article R. 5121-76-1 du même code, une convention précise les modalités de suivi des patients et de recueil des informations. Elle indique le rôle de chacun des intervenants dans le cadre du dispositif de suivi mis en place et, notamment, de l’Agence nationale de sécurité du médicament et des produits de santé, des professionnels de santé ainsi que du titulaire de l’autorisation de mise sur le marché.
Surtout, la convention peut comporter l’engagement du titulaire de l’autorisation de mise sur le marché de la spécialité de déposer une demande de modification de cette autorisation dans un délai déterminé par l’agence. Cette possibilité est essentielle et devra être pleinement utilisée par l’ANSM.
Les articles R. 5121-76-4 à R. 5121-76-7 du même code fixent les modalités de création, de suivi et de communication des RTU.
L’ANSM envoie par lettre recommandée avec accusé de réception le projet de RTU et, le cas échéant, le projet de convention au laboratoire. Le laboratoire dispose d’un délai d’un mois (un mois supplémentaire s’il en fait la demande) à partir de la date de réception du projet pour retourner à l’ANSM la convention signée. À l’expiration de ce délai, le directeur général de l’ANSM signe la RTU et en tant que de besoin, la convention.
Les RTU ainsi que leurs mises à jour sont adressées au ministre chargé de la santé, à la HAS, à l’Uncam et au Comité économique des produits de santé ainsi qu’à l’Inca et aux centres de référence. Elles sont rendues publiques sur le site internet de l’ANSM et sont envoyées aux Ordres professionnels et sociétés savantes concernées qui le transmettent aux professionnels.
Les entreprises ont l’obligation de mettre en place et de financer la collecte des données relatives au suivi des patients tel que décrit dans le protocole (R. 5121-76-7). Elles devront s’engager dans certains cas à déposer une demande de modification de l’autorisation de mise sur le marché en conformité avec la pratique de prescription.
Enfin, l’article R. 5121-76-8 du même code dispose qu’en cas de suspicion de risque pour la santé publique ou en cas de manquement à l’obligation de suivi des patients et de recueil d’informations ou si le directeur général de l’ANSM estime que les critères définis par la loi ne sont plus remplis, il peut modifier, suspendre ou retirer la RTU. Sauf en cas d’urgence, la modification, la suspension ou le retrait de la recommandation ne peut intervenir qu’à l’expiration d’un délai d’un mois après réception, par le titulaire de l’AMM d’un courrier recommandé.
Pour mémoire, l’article 57 de la loi de financement de la sécurité sociale pour 2013 a introduit un nouveau paragraphe V à l’article L. 5121-12-1 du code de la santé publique, prévoyant que par dérogation aux dispositions dans lesquelles une prescription « hors AMM » est possible, et en présence d’alternative médicamenteuse appropriée, une spécialité pharmaceutique peut faire l’objet d’une RTU.
Cette dernière ne peut être établie que dans l’objectif, soit de remédier à un risque avéré pour la santé publique, soit d’éviter des dépenses ayant un impact significatif sur les finances de l’assurance maladie. Le décret qui devait en préciser l’application n’a pas encore été publié.
Selon le directeur de l’ANSM, M. Dominique Maraninchi, les premières RTU pourraient concerner le baclofène ainsi que des médicaments en pédiatrie.
Enfin, pour mémoire, l’article 31 de la loi, d’application directe, précise les obligations des entreprises en matière de surveillance des prescriptions « hors AMM ». Surtout, il confie au Comité économique des produits de santé (CEPS) un rôle de police du « hors AMM » : les conventions qu’il signe avec les entreprises pourront comporter l’obligation de limiter l’usage constaté en « hors AMM » de certains médicaments. Il pourra, le cas échéant, sanctionner l’entreprise qui ne ferait aucun effort particulier d’information en direction des professionnels de santé en cas de prescription « hors AMM » injustifiée.
Selon le président du CEPS, auditionné par la mission, il est envisagé de mettre en place une solution pour relayer l’information auprès des prescripteurs. Ainsi, en cas de prescription fréquente d’un produit en « hors AMM », les visiteurs médicaux travaillant pour les exploitants de produits de santé devraient en informer les professionnels, afin de les dissuader de prescrire la spécialité hors des indications prévues par son autorisation de mise sur le marché.
IV.- LA PHARMACOVIGILANCE ET LE SUIVI POST-AMM DES PRODUITS DE SANTÉ
L’affaire du Mediator a révélé de nombreuses failles dans notre système de pharmacovigilance, qu’il était impératif de combler. Pour cela, les chapitres Ier, III et VI de la loi prévoient la réévaluation systématique des médicaments par la nouvelle agence, la transposition des dispositions communautaires relatives à la pharmacovigilance, le renforcement du système de notification des effets indésirables et la protection des lanceurs d’alerte, ainsi que la création d’un groupement d’intérêt public dédié aux études de santé publique.
A. LA RÉÉVALUATION SYSTÉMATIQUE DES MÉDICAMENTS APRÈS LEUR MISE SUR LE MARCHÉ
● La prise en compte de la valeur thérapeutique ajoutée des produits de santé dans leur évaluation
La question de la prise en compte de la valeur ajoutée thérapeutique dans l’évaluation du médicament a été plusieurs fois abordée lors de la discussion de la loi du 29 décembre 2011. Un médicament ne doit pas être seulement « un peu mieux que rien » ; il doit procurer un réel bénéfice au patient.
Cependant, les marges de manœuvre sont ici très étroites. En effet, la directive européenne n° 2001/83/CE du Conseil dispose que « d’une manière générale, les essais cliniques doivent être effectués sous forme d’essais contrôlés si possible, randomisés et le cas échéant par opposition à un placebo et par opposition à un médicament dont la valeur thérapeutique est déjà communément connue ; toute autre manière de procéder doit être justifiée. » Cette disposition ne permet pas d’imposer systématiquement la réalisation d’essais versus comparateur. De plus, dans certains cas, le dossier de demande d’autorisation de mise sur le marché versé ne permet pas d’évaluer le rapport bénéfices/risques du médicament étudié versus les alternatives pertinentes.
Il y a là un combat à mener au niveau européen, pour convaincre nos partenaires de la nécessité de prendre en compte le bénéfice d’un médicament par rapport à des médicaments comparables.
Dans le même esprit, l’article 14 prévoit que l’amélioration du service médical rendu, qui détermine les conditions de remboursement d’un médicament, doit s’effectuer non pas en comparaison avec un placebo, mais avec les traitements existants. Aux termes de l’article L. 162-17 du code de la sécurité sociale, la demande d’inscription d’un médicament sur la liste des produits remboursés est subordonnée à la réalisation d’essais cliniques contre des stratégies thérapeutiques, lorsqu’elles existent.
Les conditions d’application de cet article devaient être définies par décret en Conseil d’État. Ce texte n’a cependant pas encore été publié.
● Les nouveaux pouvoirs de l’ANSM en matière de contrôle post-AMM des produits de santé
L’intégralité des articles confiant de nouveaux pouvoirs à l’ANSM en matière de contrôle et d’évaluation des produits de santé après leur mise sur le marché sont d’application directe.
La nouvelle agence doit d’abord pouvoir fixer, dès le stade de l’autorisation de mise sur le marché, des lignes directrices en matière de santé publique, que devront respecter les entreprises. C’est ce que prévoit explicitement l’article 9 de la loi, avec la possibilité pour elle de demander, dès le stade de l’autorisation de mise sur le marché, des études de sécurité ou d’efficacité. La non communication de ces études dans les délais requis pourra faire l’objet de sanctions administratives.
L’évaluation ne s’arrête pas à l’autorisation de mise sur le marché. C’est pourquoi l’article 11 de la loi prévoit la possibilité pour la nouvelle agence de suspendre ou modifier l’autorisation de mise sur le marché d’un médicament si celui-ci est nocif ou présente une balance négative entre les bénéfices et les risques. Il s’agit ici de renforcer le principe de précaution trop longtemps oublié en la matière et renverser la charge de la preuve en faveur du patient.
Pour que cette évaluation en continue soit possible, il faut mobiliser les entreprises. Pour cela, l’article 12 prévoit que le titulaire de l’autorisation de mise sur le marché communique immédiatement à la nouvelle agence toute interdiction ou restriction imposée par l’autorité compétente de tout pays dans lequel le médicament est mis sur le marché et toute autre information nouvelle qui pourrait influencer l’évaluation des bénéfices et des risques de ce médicament. Chacun se rappellera que le Mediator avait été retiré de la vente en Italie et en Espagne, pour des raisons dites commerciales.
Afin de pouvoir évaluer en continu le rapport entre les bénéfices et les risques liés au médicament, la nouvelle agence pourra à tout moment demander au titulaire de l’autorisation de mise sur le marché de transmettre des données démontrant que ce rapport reste favorable.
Parallèlement, il doit être possible, pour des raisons de santé publique et lorsque le médicament est nocif ou présente une balance négative des bénéfices et des risques, d’interdire sa prescription et sa délivrance. C’est ce que prévoit l’article 22 de la loi.
L’ANSM a lancé un programme d’évaluation dans le cadre des dispositions de l’article L. 5311-1 du code de la santé publique qui prévoient notamment que l’ANSM procède à l’évaluation des bénéfices et des risques liés à l’utilisation des produits à finalité sanitaire destinés à l’homme et des produits à finalité cosmétique, surveille le risque lié à ces produits et effectue des réévaluations des bénéfices et des risques.
L’ordre d’examen au sein de chaque classe de la classification anatomique, thérapeutique et chimique, a été déterminé en trois phases :
– le tri selon un score quantitatif : dans un premier temps, la liste a été ordonnée sur un score quantitatif établi en fonction de l’ancienneté de la commercialisation, l’existence d’un SMR faible ou insuffisant, des ventes particulièrement faibles ou au contraire particulièrement importantes ;
– la hiérarchisation selon des critères qualitatifs : ont été pris en compte des critères d’appréciation qualitatifs tels que l’existence de signaux de pharmacovigilance, l’appartenance à une classe thérapeutique connue pour présenter un risque particulier, l’utilisation dans une population à risques telle que les enfants ou les personnes âgées, mais également le bénéfice thérapeutique des médicaments dans leur contexte thérapeutique actuel, ou encore la nécessité d’une mise à jour substantielle de l’information de leur RCP. Cette démarche a été réalisée par consultation des équipes internes de l’Afssaps, du Département de Pharmacovigilance et des Unités PTC.
– dans une dernière phase, les caractéristiques spécifiques de certains produits ont conduit à quelques ajustements de l’ordre de la liste.
Selon le rapport annuel de l’agence de 2013, le programme de réévaluation systématique du rapport bénéfices/risques des médicaments concerne les autorisations de mise sur le marché nationales délivrées jusqu’au 31 décembre 2005. Il s’ajoute aux réévaluations effectuées dans le cadre des renouvellements quinquennaux ainsi que celles concernant les produits ayant fait l’objet d’un signal de sécurité.
Ce programme systématique a débuté, dans le cadre d’une initiative interne de l’agence, fin 2011 et comporte deux phases.
La première est la révision de l’autorisation de mise sur le marché (AMM). Il s’agit d’une réévaluation en interne avec les données disponibles en terme d’efficacité (données AMM, essais en cours, SMR, ASMR, alternatives, mésusage, hors AMM) et de risque (rapport périodique de sécurité, avis de la commission de pharmacovigilance, enquête/suivi de pharmacovigilance, inspection, chiffres de ventes et données d’exposition, détection automatisée, risque de report, cas marquants). La révision peut aboutir à un maintien de l’AMM en l’état, à une prise de mesure sans réévaluation complète (enquête ou suivi, demande de modification de l’information, information auprès des professionnels, plan de gestion de risque, proposition de modification du remboursement, modification des conditions de prescription et de délivrance, listage, mise sous surveillance renforcée) ou bien à une décision d’informer le laboratoire du lancement d’une réévaluation du rapport bénéfices/risques.
La deuxième phase est la réévaluation complète du rapport bénéfices/risques. Il s’agit d’une réévaluation s’appuyant sur les travaux de la révision en ajoutant l’ensemble des données ainsi que la synthèse fournies par le laboratoire. Cette réévaluation se fait dans un cadre national ou européen selon la commercialisation ou non du produit dans d’autres pays européens. Elle peut conduire, le cas échéant, à une suspension, un retrait ou une modification des indications du produit.
Dans le cadre du programme de révision systématique, le passage à cette seconde phase n’est pas automatique, mais dépend des conclusions de la révision.
En revanche, en cas d’instruction suite à un signal de sécurité, la substance concernée est directement examinée dans le cadre d’une réévaluation complète.
Parmi l’ensemble des AMM nationales délivrées jusqu’au 31 décembre 2005 ont été sélectionnées en priorité celles ayant un profil considéré à risque, c’est-à-dire les produits par voie systémique, avec prescription médicale obligatoire et disponibles en ville. Cette sélection a abouti à 678 substances actives qui ont été priorisées selon un score quantitatif (basé sur la date d’AMM, les chiffres de ventes donnant les données d’exposition et le SMR) et un critère qualitatif (basé sur les signaux de pharmacovigilance, la classe thérapeutique, le contexte thérapeutique et les caractéristiques de la population exposée). Ainsi 161 de ces substances ont été placées en priorité élevée, 158 en priorité faible et 359 non prioritaires.
B. LA CONSOLIDATION DU SYSTÈME DE PHARMACOVIGILANCE
● La transposition de la directive communautaire de 2010 relative à la pharmacovigilance
L’article 28 transpose la directive européenne 2010/84/UE du 15 décembre 2010 en matière de pharmacovigilance. Cette réforme passe par trois axes : l’élargissement du cercle des notificateurs et leur protection, ainsi que l’amélioration de l’efficacité des remontées d’informations à l’ANSM ; la responsabilité des entreprises pharmaceutiques ; le rôle accru de l’ANSM.
Tout d’abord, il est procédé à un élargissement du cercle des notificateurs. Ainsi, les médecins, chirurgiens-dentistes, sages-femmes, pharmaciens auront l’obligation de déclarer tout effet indésirable suspecté d’être dû à un produit de santé. Tous les autres professionnels de santé, mais également les associations de patients, pourront également notifier les effets indésirables.
Il ne doit pas s’agir ici seulement des effets manifestement graves et inattendus, mais de tous les effets indésirables liés à la prise d’un médicament. C’est un point fondamental.
Afin que cette disposition ne reste pas lettre morte, compte tenu de la surcharge de travail des professionnels de santé, il convient de valoriser au mieux et simplifier la notification des effets indésirables.
Par un amendement adopté à l’Assemblée nationale, l’obligation d’assurer un retour d’information aux notificateurs a été introduite dans la loi. Il s’agissait d’une préconisation importante des Assises du médicament.
Par ailleurs, a été mise en place un statut protecteur des donneurs d’alerte. Cette revendication était portée depuis quelques années par certains professionnels et les associations de patients.
Le deuxième axe de cette réforme est la meilleure implication des entreprises. Celles-ci doivent notamment veiller au bon usage de leurs spécialités, c’est-à-dire au respect des indications de l’AMM, et prendre toutes les mesures d’information vis-à-vis des professionnels lorsque des prescriptions sont non conformes à l’AMM. Elles doivent également déclarer à l’Agence ces prescriptions hors AMM, ainsi que tout arrêt de commercialisation de leurs produits dans les autres pays (y compris dans les pays tiers) ainsi que toute restriction ou interdiction prise par une autre autorité compétente. L’ensemble de ces dispositions devrait ainsi permettre après exploitation de ces données une meilleure police sanitaire des produits.
La loi du 29 décembre 2011 pose enfin le rôle central de la nouvelle agence dans la mise en œuvre du système de pharmacovigilance. Elle sera chargée de :
– procéder à l’évaluation scientifique de toutes les informations ;
– examiner les options permettant de prévenir les risques ou les réduire et, au besoin prendre des mesures appropriées ;
– définir les orientations de la pharmacovigilance, animer et coordonner les actions des différents intervenants ;
– veiller au respect des procédures de surveillance ;
– participer aux activités de l’Union européenne dans ce domaine.
Il s’agit d’un véritable défi pour la nouvelle agence, doublé d’un relatif changement de culture qui doit s’accompagner de moyens suffisants. En effet, elle doit être capable de recueillir l’information et de la traiter correctement pour transformer des signalements en alerte.
Les Centres régionaux de pharmacovigilance (CRPV) saisissent les cas d’effets indésirables qu’ils reçoivent des professionnels de santé et des patients, dans une base de données : la base nationale de pharmacovigilance (BNPV). L’Agence assure la gestion de cette base et effectue quotidiennement une revue des cas saisis, notamment lorsqu’ils sont graves.
Comme l’indique le graphique ci-dessous, en 2012, 31 434 cas initiaux d’effets indésirables ont donné lieu à 6 865 mises à jour. Au premier trimestre 2013, 9 437 cas initiaux ont donné lieu à 2 137 mises à jour, ce qui représente une augmentation de plus de 20 % du nombre de cas initiaux et mises à jour saisis par les CRPV par rapport à la même période en 2012.
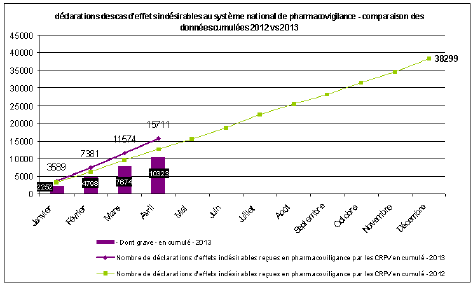
Concernant les erreurs médicamenteuses, 522 signalements ont été notifiés à l’Agence au 31 mai 2013, dont certains ont conduit, après évaluation, à des mesures afin de minimiser le risque identifié.
En parallèle, en 2013 deux réflexions générales avec des projets de recommandations ont été initiées portant sur la minimisation :
– des erreurs médicamenteuses liées aux dispositifs d’administration des solutions buvables (soit environ 400 spécialités) ;
– des erreurs d’administration par injection intrathécale de produits destinés à la voie intraveineuse (80 spécialités sont concernées).
● Le contrôle des dispositifs médicaux
Enfin, un des apports de la loi de 2011 réside dans l’amorce d’un encadrement de la mise sur le marché des dispositifs médicaux, dont le risque est souvent négligé au regard des médicaments.
L’article L. 165-1 du code de la sécurité sociale subordonne le remboursement des dispositifs médicaux par l’assurance maladie à leur inscription sur une liste établie après avis de la commission de la Haute Autorité de santé (HAS), l’inscription sur ladite liste étant soumise au respect de spécifications techniques, d’indications thérapeutiques ou diagnostiques et de conditions particulières de prescriptions et d’utilisation.
L’article 35 de la loi institue un contrôle de conformité effectué par l’ANSM des dispositifs médicaux aux spécifications techniques requises pour pouvoir bénéficier du remboursement par les caisses d’assurance maladie.
Il insère un nouvel article L. 165-1-2 dans le code de la sécurité sociale qui prévoit que pour les produits inscrits sur la liste, l’ANSM peut effectuer un contrôle du respect des spécifications techniques auxquelles l’inscription sur la liste est subordonnée. Si les produits sont saisis par l’agence, les fabricants ou leurs mandataires ou les distributeurs sont tenus de compenser la perte financière subie par l’acheteur du produit.
En cas de non-respect des spécifications techniques précédemment citées et après avoir mis l’entreprise en mesure de présenter ses observations, l’agence notifie au fabricant, à son mandataire ou au distributeur les manquements retenus à son encontre ainsi que les pénalités encourues. Une copie du courrier est adressée aux ministres chargés de la santé et de la sécurité sociale, au CEPS et au directeur général de l’Union nationale des caisses d’assurance maladie.
Après avoir recueilli les observations du fabricant ou de son mandataire ou du distributeur, l’agence peut lui infliger une pénalité financière dont le montant est fixé en fonction de la gravité du manquement constaté et qui ne peut être supérieur à 10 % du chiffre d’affaires hors taxes réalisé en France par le fabricant ou distributeur au titre du dernier exercice clos pour le produit concerné. La pénalité est alors recouvrée par les organismes d’assurance maladie et son produit est affecté aux régimes obligatoires de base d’assurance maladie.
Si le produit a été inscrit sur la liste des produits remboursables sans posséder les spécifications requise, les organismes nationaux d’assurance maladie ont la possibilité de recouvrer les remboursements indus.
Si le manquement retenu par l’agence a rendu nécessaire la dispensation d’actes de soins, de prestations ou de produits de santé à un assuré, le professionnel ou l’établissement de santé qui a connaissance de ce manquement et a accompli cette dispensation doit en informer l’organisme local d’assurance maladie.
Le décret n° 2012-1135 du 8 octobre 2012 précise le contrôle des spécifications techniques ainsi que la pénalité financière pouvant être infligée. Il crée à cet effet une section 11 au chapitre V du titre VI du livre 1er du code de la sécurité sociale.
Trois modalités de contrôle du respect des spécifications techniques sont prévues. Tout d’abord, celui-ci peut se faire dans le cadre d’un programme annuel défini par l’ANSM en concertation avec les ministres chargés de la santé et de la sécurité sociale, le CEPS, la HAS et les organisations représentatives des fabricants et des distributeurs. Ensuite, il peut avoir lieu sur demande du ministre chargé de la santé ou de la sécurité sociale. Enfin, il peut être mené à l’initiative de l’ANSM dans le cadre de ses missions de surveillance du marché et de matériovigilance ou sur signalement de la HAS.
La saisie des dispositifs objets du contrôle peut être inopinée et intervenir tant chez le fabricant ou le distributeur que sur le lieu de vente ou en établissement de santé. Dans ce cas, le fabricant ou distributeur doit compenser la perte financière provoquée.
L’ANSM est la seule autorité compétente pour prélever les dispositifs à contrôler, mais elle peut décider de déléguer le contrôle effectif à un autre organisme qu’elle désigne.
Le contrôle peut être complété par des vérifications sur place.
L’ANSM doit établir un rapport de contrôle et informer le fabricant ou le distributeur des éventuels manquements constatés. Le fabricant ou le distributeur dispose d’un délai d’un mois pour faire parvenir ses observations. L’ANSM doit ensuite les informer de ses conclusions définitives, des manquements retenus ainsi que de l’éventuelle pénalité financière appliquée. Si le CEPS envisage de prononcer une pénalité, il doit notifier sa décision au fabricant ou distributeur. L’entreprise dispose d’un délai d’un mois pour présenter ses observations et pour porter à la connaissance du CEPS les éléments de son chiffre d’affaires lui permettant de fixer le montant de la pénalité.
Enfin, le décret précise au nouvel article R. 165-5 que le manquement aux spécifications techniques est susceptible d’entrainer la fin du remboursement du dispositif médical par l’assurance maladie.
L’article 37 de la loi modifie les articles L. 165-11 à L. 165-13 du code de la sécurité sociale, afin de rendre obligatoire l’évaluation par la Haute autorité de santé des dispositifs médicaux relevant d’un financement dans les groupes homogènes de séjours (GHS) dans le but de contribuer à l’amélioration de la prise en charge des patients dans les établissements de santé.
Pour ce faire, les ministres de la santé et de la sécurité sociale, après avis de la Commission nationale d’évaluation des dispositifs médicaux et technologies de santé (CNEDIMTS) de la HAS, établissent une liste par arrêté des dispositifs médicaux évalués et pour lesquels un financement au titre des GHS est considéré comme justifié. Ainsi, seuls les dispositifs inscrits sur la liste pourront être achetés par les établissements de santé.
Les catégories homogènes de dispositifs qui relèvent de cette disposition comprennent les produits de santé qui doivent répondre, au regard de leur caractère invasif ou des risques qu’ils peuvent présenter pour la santé humaine, à au moins l’une de ces exigences : la validation de leur efficacité clinique ; la définition de spécifications techniques particulières ou l’appréciation de leur efficience au regard des alternatives thérapeutiques disponibles.
L’inscription sur la liste n’est pas définitive. Elle est prononcée pour une durée déterminée, renouvelable. L’inscription ou le renouvellement d’inscription peuvent être notamment assortis de conditions de prescription et d’utilisation et subordonnés à la réalisation par les fabricants ou leurs mandataires ou par les distributeurs d’études complémentaires.
Le décret n° 2012-1051 du 13 décembre 2012 prévoit les catégories homogènes de produits de santé soumises à évaluation et les modalités de leur inscription sur la liste permettant leur prise en charge par l’assurance maladie. Il créé une section 12 au chapitre V du titre VI du livre 1er du code de la sécurité sociale, codifié aux articles R. 165-49 et suivants.
Les catégories homogènes de produits de santé sont fixées par arrêté des ministres en charge de la santé et de la sécurité sociale en prenant en compte les indications fournies par l’ANSM, la HAS, les ARS ou les professionnels de santé exerçant dans les établissements de santé. Pour chaque catégorie homogène, les ministres doivent en outre préciser la durée d’utilisation de ces produits, dans une limite de 4 ans, et les modalités d’inscription.
L’inscription s’effectue soit par la description générique du produit, soit sous la forme du nom de marque ou du nom commercial. L’inscription de ces produits est prononcée pour une durée de 5 ans renouvelable et peut être subordonnée à des conditions de prescription et d’utilisation ou à la production par l’exploitant d’études post-inscription. La demande d’inscription est adressée à la Commission nationale d’évaluation des dispositifs médicaux et des technologies de santé qui rend un avis. L’inscription peut être demandée par les fabricants, les distributeurs mais également par les ministres en charge de la santé et de la sécurité sociale.
Les produits ne pourront être inscrits ou ne pourront pas voir leur inscription renouvelée s’ils ont enfreint les règles applicables à la publicité, si le service rendu est insuffisant, s’ils ne satisfont pas aux règles de mise sur le marché ou s’ils entrainent des dépenses injustifiées à la charge de l’assurance maladie.
La Commission dispose d’un délai de 180 jours pour publier la décision d’inscription ou de renouvellement d’inscription. En l’absence de publication de la décision et si le dossier de renouvellement comprend les éléments nécessaires à son évaluation, l’inscription est renouvelée tacitement.
Si le produit ne remplit plus les conditions requises pour être inscrit sur la liste permettant le remboursement par l’assurance maladie, la radiation est alors prononcée par les ministres. L’entreprise peut présenter ses observations dans un délai de 30 jours.
Les produits faisant l’objet d’une interdiction de mise sur le marché, d’utilisation, de prescription de délivrance ou d’administration sont radiés sans que l’entreprise ne puisse présenter ses observations ou être entendue par la Commission.
La Commission, à son initiative ou à la demande des ministres, peut décider de réévaluer le service attendu ou rendu des produits inscrits.
L’article 37 prévoit aussi deux manquements qui sont susceptibles d’entraîner une pénalité financière.
À l’article L. 165-12, il est précisé que les établissements de santé qui achètent ou utilisent des produits de santé appartenant aux catégories homogènes sans être inscrits sur la liste sont passibles d’une sanction financière. Cette sanction est prononcée par le directeur général de l’ARS à la suite d’un contrôle réalisé sur pièces et sur place et après que l’établissement a été mis en mesure de présenter ses observations. Le montant de cette sanction, fixé en fonction de la gravité du manquement constaté, ne peut excéder le coût total d’achat par l’établissement des produits considérés durant l’année précédant la constatation du manquement.
Si les études complémentaires post-inscription demandées ne sont pas effectuées dans les délais impartis par le fabricant, le mandataire ou le distributeur, le nouvel article L. 165-13 prévoit quant à lui que les ministres chargés de la santé et de la sécurité sociale peuvent prononcer, après que les intéressés ont été mis en mesure de présenter leurs observations, une pénalité financière à leur encontre, le montant de la pénalité ne pouvant être supérieur à 10 % du chiffre d’affaires hors taxes réalisé en France par le fabricant ou le mandataire ou par le distributeur, au titre du ou des produits considérés, durant les douze mois précédant la constatation du manquement. Le montant de la pénalité est fixé en fonction de la gravité du manquement constaté.
Le décret n° 2012-1095 du 28 septembre 2012 prévoit la mise en œuvre de la pénalité financière prévue à l’article L. 165-13, c’est-à-dire lorsque le fabricant ou le distributeur du produit n’a pas réalisé les études complémentaires qui peuvent être exigées par la Commission pour autoriser la prise en charge forfaitaire de certains dispositifs utilisés en établissement de santé.
Le nouvel article R. 165-45 précise que le manquement sera caractérisé dès lors que les études complémentaires n’ont pas été réalisées dans le délai requis ou que les études remises ne comportent pas les éléments attendus.
L’entreprise est tenue de déclarer aux ministres les éléments de son chiffre d’affaires permettant de fixer le montant de la pénalité. Lorsque la pénalité est prononcée, l’entreprise doit s’en acquitter dans un délai d’un mois.
Concernant la matériovigilance, portant sur les dispositifs médicaux, le rapport annuel de l’ANSM indique que la courbe des signalements d’incidents incluant ceux relatifs aux prothèses PIP révèle une très forte augmentation des déclarations d’incidents entre mars et août 2012 avec une forte augmentation du nombre de déclarations en mars et avril 2012 (+ 200% d’incidents) puis une diminution progressive de mai à août pour revenir « à la normale » à partir de septembre 2012. Cette augmentation soudaine est liée à l’affluence de déclarations d’explantations préventives de prothèses mammaires PIP sur cette période.
En revanche, la courbe des signalements d’incidents hors PIP montre que le nombre moyen de déclarations est sensiblement le même sur les premiers mois de 2013 que sur l’année 2012, bien que légèrement inférieur (12 % environ) au 1er trimestre 2013 par rapport au 1er trimestre 2012.
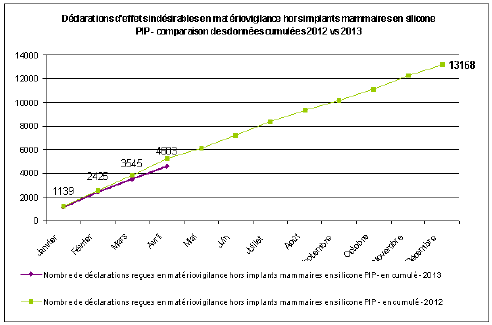
C. LE GROUPEMENT D’INTÉRÊT PUBLIC DESTINÉ À PRODUIRE DES ÉTUDES DE VIGILANCE ET D’ÉPIDÉMIOLOGIE
L’article 33 de la loi du 29 décembre 2011 insère un chapitre Ier ter, intitulé « Études en santé publique », au sein du titre II du livre Ier de la cinquième partie du code de la santé publique, afin de faciliter et d’encadrer la réalisation d’études pharmaco-épidémiologiques à partir des données de l’assurance maladie.
Ce chapitre est constitué d’un article L. 5121-27 qui prévoit la création d’un groupement d’intérêt public (GIP) pour autoriser l’accès au système national d’information inter régimes de l’assurance maladie (SNIIRAM) ou l’extraction de ses données afin de réaliser des études, notamment de pharmacovigilance et de pharmacoépidémiologie.
Pour pouvoir mener des études pharmaco-épidémiologiques et compléter ainsi les signaux issus de la pharmacovigilance traditionnelle, il importe d’accéder à des bases de données fiables. La principale est le système national d’information inter-régimes de l’assurance maladie qui possède trois atouts majeurs : une couverture nationale avec des données standardisées (codage), un numéro de patient unique et anonyme et une disponibilité rapide.
Le SNIIRAM est une immense base de données qui retrace les dépenses de l’ensemble des régimes d’assurance maladie par circonscription géographique, par nature de dépenses, par catégorie de professionnels responsables de ces dépenses et par professionnel ou établissements. Il comporte des éléments sur les pathologies dont souffrent les patients et les médicaments qu’ils ont consommés. Il couvre toute la population française. La seule limite du SNIIRAM est d’exclure les produits de santé non remboursables, et ceux remboursables qui ne sont pas présentés au remboursement.
L’exploitation de ces données est indispensable en vue de repérer les effets indésirables rares ou survenant à long terme qui n’apparaissent pas dans les essais cliniques, compte tenu du faible nombre de personnes qui y sont incluses et de leur brièveté.
La fonction du GIP institué par la loi est triple :
– l’encadrement de l’exploitation des données du SNIIRAM. L’autorisation serait accordée en fonction des finalités poursuivies par ces études et de la contribution qu’elles sont susceptibles d’apporter, par leur qualité scientifique et dans le respect de la protection des données personnelles, notamment du secret médical, à la santé publique ou à l’efficience des dépenses d’assurance maladie ;
– le lancement d’appels d’offres auprès d’équipes de recherches ;
– la réalisation directe d’études de pharmacovigilance et de pharmacoépidémiologie.
Le groupement réunira UNCAM, l’InVS, l’ANSM et la HAS, auxquels ils permettront d’avoir accès aux données de l’assurance maladie.
Un décret en Conseil d’État, pris après avis de la Commission nationale de l’informatique et des libertés (CNIL), doit intervenir pour déterminer un certain nombre de modalités d’application. Il n’a pas encore été publié.
S’il n’est pas question de remettre en cause l’utilité d’une meilleure exploitation des données de l’assurance maladie et la nécessité d’en encadrer l’usage, on peut s’interroger sur l’opportunité de créer une nouvelle entité.
En effet, un certain nombre d’acteurs du domaine de la santé, notamment le Collectif interassociatif sur la santé (Ciss) lors de son audition, ont estimé que le nouveau groupement revenait sur les compétences du GIP « Institut des données de santé » (GIP-IDS) déjà en charge de partager les données du SNIIRAM. Sa gouvernance est plus large que celle du GIP prévu par la loi du 29 décembre 2011, et inclut notamment les représentants de la société civile.
Installé fin mars 2007, le GIP-IDS, créé par la loi du 13 août 2004 réformant l’assurance maladie, doit assurer deux missions: permettre l’accès aux données de santé, notamment fournies par l’assurance maladie obligatoire et complémentaire, et mettre en place des échantillons statistiques représentatifs des assurés portant sur chacun des régimes.
Par un arrêté du 2 mars 2013, la convention constitutive de l’IDS a été prorogée jusqu’au 31 décembre 2013 au lieu d’être modifiée avec une restriction de son champ d’action au profit du futur GIP « Études de santé ».
La décision de créer un nouveau groupement est d’autant plus surprenante qu’a été lancée en décembre 2012 la modernisation de l’action publique qui prévoit notamment la rationalisation du nombre et du périmètre d’intervention des opérateurs de l’État. S’y ajoute une mission conjointe de l’Inspection générale des affaires sociales (IGAS) et de l’Inspection générale des finances (IGF) qui doit rendre prochainement un rapport sur les rôles et missions des agences sanitaires. Dans ce contexte, il serait peu opportun de créer un nouveau groupement alors même que les compétences de l’IDS pourraient être étendues.
Une mission sur les données de santé, confiée à M. Pierre-Louis Bras, inspecteur des affaires sociales, est en cours et devrait rendre ses conclusions d’ici la fin de l’été. Par ailleurs, le Premier ministre, Jean-Marc Ayrault, a annoncé l’organisation d’un débat spécifique sur l’ouverture des données de santé, lors de son discours de clôture de la sixième réunion du Conseil stratégique des industries de santé au ministère des affaires sociales et de la santé.
Dans cette perspective, vos rapporteurs, tout en estimant que la création d’un GIP « Études de santé » n’est pas nécessaire, plaident pour une ouverture mesurée et encadrée des données de santé. Seuls des besoins de santé publique et de sécurité sanitaire peuvent justifier l’accès des acteurs privés aux données de santé. Dans l’hypothèse où l’accès aux données de santé dans le cadre de la veille sanitaire serait confié à l’IDS, il conviendrait de préciser dans la loi à quelles conditions les acteurs non institutionnels pourront avoir accès aux données de santé.
D. LA PRESCRIPTION ET LA DÉLIVRANCE DES PRODUITS DE SANTÉ
● L’interdiction de la prescription, de la délivrance et retrait du marché d’un produit de santé par l’ANSM
L’article 22 insère un article L. 5121-14-2 dans le code de la santé publique.
Est conférée à l’ANSM la capacité d’interdire la délivrance et la prescription d’une spécialité pharmaceutique ou de la retirer du marché. Quelques-uns des motifs qui peuvent justifier cette décision reprennent les termes de la directive communautaire du 15 décembre 2010 relative à la pharmacovigilance. L’absence de contrôle sur la fabrication ou l’absence d’autorisation est également citée comme motif de retrait. Un décret en Conseil d’Etat doit déterminer les modalités d’application de cette mesure.
Par ailleurs, l’ANSM peut moduler l’interdiction qu’elle prononce en la limitant à certains lots de médicaments. De plus, dans des conditions définies par décret en Conseil d’Etat, le prolongement de traitements suivis par certains patients peut être autorisé avec un médicament interdit.
Le décret n° 2013-1244 du 8 janvier 2011 modifie l’article R. 5121-47 du code de la santé publique afin de prévoir que le directeur général de l’ANSM peut, par décision motivée indiquant les voies et les délais de recours, modifier d’office, suspendre, pour une période ne pouvant excéder un an, ou retirer une autorisation de mise sur le marché pour les motifs mentionnés à l’article L. 5121-9.
Toutefois, lorsque l’autorisation est suspendue, soit à titre conservatoire dans l’attente de l’issue d’une procédure d’arbitrage de l’Union européenne, soit conformément à une décision de la Commission européenne prise à l’issue de cette procédure, la suspension demeure en vigueur jusqu’à ce que la Commission européenne ordonne qu’il y soit mis fin.
L’autorisation de mise sur le marché est également modifiée d’office, suspendue ou retirée par le directeur général de l’agence :
– lorsqu’il apparaît que les renseignements fournis à l’occasion de la demande d’autorisation de mise sur le marché sont erronés ou n’ont pas été modifiés conformément à la demande de l’ANSM, que les conditions prévues à la présente section ne sont pas ou ne sont plus remplies ou que les contrôles n’ont pas été effectués ;
– lorsque l’étiquetage ou la notice du médicament ou du produit ne sont pas conformes aux prescriptions générales ou spécifiques qui s’y appliquent ;
– lorsque les obligations imposées en application des articles R. 5121-36-1 et R. 5121-43 ne sont pas exécutées, c’est-à-dire respectivement les études d’efficacité et de sécurité post-autorisation ou encore le système de pharmaco-vigilance, et les obligations relatives au système de gestion des risques ;
– lorsque l’autorisation doit être mise en conformité avec la décision prise par la Commission européenne à l’issue d’une procédure d’arbitrage de l’Union européenne ;
– lorsque l’autorisation doit être mise en conformité avec un accord ayant fait l’objet d’un consensus au sein du groupe de coordination mentionné à l’article 27 de la directive 2001/83/ CE du Parlement européen et du Conseil.
L’article R. 5121-48 du code de la santé publique, tel que modifié par le décret, prévoit que le directeur général de l’ANSM peut interdire la prescription et la délivrance d’une spécialité pharmaceutique et la retirer du marché pour les motifs mentionnés à l’article L. 5121-14-2.
La décision est motivée et, sauf en cas d’urgence, ne peut intervenir qu’après que le titulaire a été invité à présenter ses observations.
Pour une spécialité pharmaceutique dont la délivrance a été interdite, le directeur général de l’agence peut, dans des circonstances exceptionnelles et pour une période transitoire, autoriser la délivrance de la spécialité à des patients qui sont déjà traités, notamment lorsque l’une des conditions suivantes est remplie :
– il est nécessaire de mettre en œuvre une décroissance progressive du traitement afin de prévenir ou de limiter un effet rebond ou l’effet d’un syndrome de sevrage ;
– un délai est nécessaire pour adapter les recommandations de prise en charge de la pathologie concernée ;
– un délai est nécessaire pour que les professionnels de santé puissent définir la meilleure alternative thérapeutique possible pour leurs patients.
La durée de la période transitoire envisagée est indiquée dans le courrier par lequel le titulaire est invité à présenter ses observations conformément au premier alinéa.
Le décret précité crée également l’article R. 5121-49 du code de la santé publique, qui dispose que lorsque une action urgente est indispensable pour protéger la santé humaine ou l’environnement, le directeur général de l’ANSM peut suspendre l’utilisation d’une spécialité qui fait l’objet d’une autorisation de mise sur le marché délivrée, de sa propre initiative ou à la demande de la Commission européenne. La décision du directeur général de l’agence est motivée. Le titulaire de l’autorisation de mise sur le marché est préalablement informé de la décision et, si la situation le permet, il est préalablement invité à présenter ses observations. Lorsqu’il agit de sa propre initiative, le directeur général de l’agence informe la Commission européenne et l’Agence européenne des médicaments des raisons de cette mesure au plus tard le premier jour ouvrable suivant sa décision.
● L’expérimentation de la consultation du dossier pharmaceutique des patients dans les établissements de santé
Le dossier pharmaceutique a été créé par la loi du 30 janvier 2007 relative à l’organisation de certaines professions de santé. Il est alimenté lors de la dispensation de médicaments par les pharmaciens d’officine et les pharmacies à usage intérieur (PUI) des établissements de santé. Ce dossier permet de retracer tous les médicaments dispensés au patient les quatre mois précédents.
L’article 23 de la loi prévoit, à l’article L. 1111-23 du code de la santé publique, qu’à titre expérimental et pour une durée de trois ans, les médecins peuvent, dans certains établissements de santé et dans le cadre de la prise en charge des patients, consulter, avec leur autorisation, leur dossier pharmaceutique. Cette disposition a notamment pour but d’améliorer la continuité et la coordination des soins pour les patients hospitalisés et de diminuer le risque d’iatrogénie médicamenteuse.
Le décret du 9 janvier 2013 (5) fixe les conditions d’application de l’expérimentation et le type d’établissement éligible.
Les établissements de santé éligibles à l’expérimentation ont été désignés par un arrêté du 28 mai 2013 pris par la ministre de la santé, après avis du Conseil national de l’ordre des pharmaciens (CNOP), sur proposition des directeurs généraux des agences régionales de santé (ARS) et sur la base du cahier des charges annexé au décret. 55 établissements publics et privés situés dans 17 régions sont concernés par l’expérimentation (6).
Le champ d’application de l’expérimentation est limité aux seuls professionnels expressément cités par le décret. Ainsi en est-il des médecins anesthésistes-réanimateurs, des médecins exerçant dans les structures d’urgence, des médecins exerçant dans une unité de réanimation et des médecins exerçant dans les structures de médecine gériatrique
L’accès au dossier pharmaceutique ne sera possible qu’après information du patient sur l’expérimentation – une brochure doit être remise au patient au moment de l’utilisation de la carte vitale – et recueil de son consentement ou de celui de son représentant légal. En aucun cas, la participation du patient à l’expérimentation n’est obligatoire, celui-ci peut s’y opposer. Toutefois, le consentement ne sera pas requis si le patient présente une urgence vitale et que la connaissance des médicaments consommés peut s’avérer indispensable. Pour accéder au dossier, le professionnel de santé doit disposer de la carte vitale du bénéficiaire de l’assurance maladie ainsi que de sa carte professionnelle.
Prévue pour une durée de trois ans, jusqu’au 29 décembre 2014, l’expérimentation n’a pu être officiellement lancée qu’en mai 2013. Nous disposons donc de peu de recul pour en évaluer la portée. À l’issue de la période d’expérimentation, les établissements devront dresser un bilan au sein de leurs structures et les ARS devront évaluer le dispositif dans un rapport remis au ministre en charge de la santé.
En annexe du décret du 9 janvier 2013 figure le cahier des charges de l’expérimentation. Celui a pour objet de prévoir les conditions dans lesquelles seront menées les expérimentations de la consultation du dossier pharmaceutique en établissement de santé. Il fixe les conditions, prérequis et modalités de candidature des établissements à l’expérimentation et précise les conditions dans lesquelles l’évaluation du dispositif sera opérée. S’y ajoute l’instruction n° DGOS/PF2/2013/45 du 5 février 2013 relative à la mise en œuvre de l’expérimentation de la consultation du dossier pharmaceutique par les médecins exerçant dans certains établissements de santé, qui fixe le modèle des conventions signées entre l’établissement, le Conseil de l’ordre des pharmaciens et le ministère de la santé.
L’objectif de l’expérimentation est de tester l’apport d’une consultation du dossier pharmaceutique pour le corps médical. Il s’inscrit dans le contexte plus global d’amélioration de la qualité, de la sécurité et de la coordination des soins. Il répond en effet aux préoccupations du décloisonnement ville-hôpital, de l’amélioration de la sécurité de l’acte de prescription et de la sécurisation de la prise en charge médicamenteuse. L’expérimentation engendre un coût important, tant en équipement qu’en ressources humaines pour l’établissement de santé. Ces frais seront supportés en majorité par l’établissement de santé volontaire, à l’exception de certains frais pris en charge par l’ASIP santé (cartes CPS) et par le Conseil de l’ordre des pharmaciens (support d’information, déclarations à la CNIL).
Un accompagnement des établissements de santé à la mise en place du système a été prévu. L’instruction en date du 5 février 2013 précise les modalités de l’expérimentation. Les établissements ont eu jusqu’au 11 mars 2013 pour déposer leur candidature auprès de leur ARS qui ont disposé d’un délai de deux semaines pour examiner les candidatures et transmettre à la DGOS la liste des établissements éligibles.
Un comité de pilotage de l’expérimentation a été instauré au niveau national. Celui-ci est composé de la Direction générale de l’offre de soin (DGOS), du Conseil de l’ordre des pharmaciens (CNOP), de la délégation à la stratégie des systèmes d’information de santé (DSSIS), de la Haute Autorité de santé (HAS), de l’Agence technique d’information sur l’hospitalisation (ATIH) et du secrétariat général des ministères chargés des affaires sociales.
L’évaluation quantitative du dispositif sera réalisée par le Conseil de l’ordre des pharmaciens, qui doit mettre à disposition du comité de pilotage les informations recueillies sur la base de quatre indicateurs : le nombre de requêtes de consultation par service ; le nombre de requêtes de consultation par service ayant abouti à la consultation d’un dossier pharmaceutique ; le nombre de consultations par poste de travail connecté à l’intérieur de chaque service ; le nombre de consultations par jour et par service.
L’enquête qualitative comporte deux volets : d’une part, une évaluation réalisée par une équipe de recherche universitaire retenue par appel d’offre national, d’autre part, une enquête de satisfaction réalisée tous les six mois par le Conseil de l’ordre des pharmaciens qui permettra d’évaluer l’acceptation du dossier pharmaceutique par les professionnels de santé et les différents aspects techniques et organisationnels, ce volet de l’évaluation n’étant qu’une extension de l’enquête déjà mise en place auprès des officines reliées au dossier pharmaceutique.
La mise en œuvre de cette mesure soulève quelques difficultés. Tout d’abord, les professionnels de santé concernés par l’expérimentation devront être formés à l’utilisation du dossier pharmaceutique et les établissements équipés. Ensuite, l’expérimentation ne durera en réalité qu’un an et demi, voire moins, dans la mesure où les établissements viennent à peine d’être retenus et qu’un temps d’adaptation sera nécessaire pour équiper les établissements et former les personnels. Enfin, l’expérimentation, pour être probante, devra comporter un nombre suffisant de patients hospitalisés dans une des unités de soins concernées et acceptant la création de leur dossier pharmaceutique. La montée en charge du dispositif pourrait donc être plus lente que prévue. Il conviendra certainement de prolonger la durée de cette expérimentation.
● Les modalités d’organisation du système d’astreinte pour les grossistes-répartiteurs et d’encadrement des exportations parallèles
L’article 47 de la loi met en place un système d’astreinte pour les grossistes-répartiteurs, afin de lutter contre les ruptures d’approvisionnement.
Le nouvel article L. 5124-17-1 du code de la santé publique, tel qu’issu de la loi du 29 décembre 2011 dispose qu’un système d’astreinte est organisé pour répondre aux besoins urgents en médicaments en dehors des jours d’ouverture généralement pratiqués par les grossistes-répartiteurs sur leur territoire de répartition. Tous les grossistes-répartiteurs sont tenus de participer à ce système.
L’organisation du système d’astreinte est réglée par les organisations représentatives de la profession. À défaut d’accord ou si l’organisation retenue ne permet pas de satisfaire les besoins en santé publique, une décision du directeur général de l’agence régionale de santé territorialement compétente règle l’organisation dudit système.
En outre, l’article L. 5124-17-2 du code de la santé publique prévoit désormais que les grossistes-répartiteurs sont tenus de respecter sur leur territoire de répartition les obligations de service public déterminées par décret en Conseil d’État. Ils assurent l’approvisionnement continu du marché national de manière à couvrir les besoins des patients sur leur territoire de répartition.
Le décret n° 2012-1096 du 28 septembre 2012 relatif à l’approvisionnement en médicaments à usage humain est venu préciser ces dispositions.
L’article R. 5124-48-1 du code de la santé publique est complété afin de préciser que l’entreprise pharmaceutique exploitant des médicaments assure un approvisionnement approprié et continu de tous les établissements autorisés au titre d’une activité de grossiste-répartiteur de manière à couvrir les besoins des patients en France.
Le décret crée l’article R. 5124-49-1 qui définit la rupture d’approvisionnement comme l’incapacité pour une pharmacie d’officine ou une pharmacie à usage intérieur de dispenser un médicament à un patient dans un délai de 72 heures. Ce délai peut être réduit à l’initiative du pharmacien en fonction de la compatibilité avec la poursuite optimale du traitement du patient.
Le pharmacien tient à la disposition du directeur général de l’agence régionale de santé les éléments prouvant les ruptures d’approvisionnement.
Par ailleurs, il est prévu que lorsque l’exploitant anticipe une situation de rupture potentielle d’approvisionnement, il en informe l’Agence nationale de sécurité du médicament et des produits de santé en précisant les délais de survenue, les stocks disponibles, les modalités de disponibilité et les délais prévisionnels de remise à disposition et l’identification de spécialités, le cas échéant, pouvant se substituer à la spécialité pharmaceutique en défaut.
Il est précisé que les établissements pharmaceutiques exploitants doivent disposer de centres d’appel d’urgence permanents accessibles aux pharmaciens d’officine, aux pharmaciens de pharmacie à usage intérieur et aux pharmaciens responsables ou délégués des grossistes-répartiteurs. En cas de recours aux centres d’appel d’urgence, le pharmacien en informe l’agence régionale de santé. Les exploitants prennent toutes dispositions pour faire connaître les numéros d’appel auprès des professionnels de santé. L’exploitant assure la traçabilité des appels.
Ces centres doivent être organisés de manière à prendre en charge à tout moment les ruptures d’approvisionnement qui concernent les médicaments et à permettre la dispensation effective de la spécialité manquante. Cette prise en charge se fait en cas de rupture effective ou de manière anticipée lorsque la rupture est confirmée par le grossiste-répartiteur ou le dépositaire. L’exploitant informe trimestriellement l’agence régionale de santé dans le ressort de laquelle est le pharmacien des approvisionnements d’urgence en mentionnant chaque destinataire et les quantités fournies.
Un bilan trimestriel de ces approvisionnements d’urgence et des déclarations est réalisé par l’exploitant et adressé à l’agence, chronologiquement pour chaque médicament avec mention, le cas échéant, des quantités fournies et de leurs destinataires.
Il est prévu que l’Agence nationale de sécurité du médicament et des produits de santé informe les professionnels de santé des ruptures d’approvisionnement effectives ou anticipées et précise, s’il y a lieu, les recommandations éventuelles pour gérer cette pénurie.
Les grossistes-répartiteurs informent quant à eux l’exploitant de toute rupture d’approvisionnement dont ils n’ont pas été déjà informés par celui-ci ou par l’Agence nationale de sécurité du médicament et des produits de santé (article R. 5124-59-1 du code de la santé publique), de même que les pharmaciens d’officine ou exerçant dans les pharmacies à usage intérieur des établissements notamment par les centres d’appel d’urgence (articles R. 5125-46-1 et R. 5126-7-1 du même code).
Enfin, le décret renforce, à l’article R. 5124-59 du code de la santé publique, le régime de déclaration du territoire de répartition par les grossistes-répartiteurs. Le territoire déclaré par le grossiste-répartiteur doit être compatible avec ses obligations de service public (stocks, délais de livraison, astreintes). Le directeur général de l’ANSM pourra demander tout élément justifiant du respect de ces obligations et refuser tout ou partie de la modification demandée du territoire de répartition déclaré.
Le samedi à partir de 14 heures, ainsi que le dimanche et les jours fériés, les grossistes-répartiteurs devront livrer les médicaments dans les délais et au maximum dans les huit heures.
Par ailleurs, l’article 45 de la loi fait obligation aux distributeurs qui recourent à des exportations parallèles de déclarer leur activité, ce qui aurait pour effet de soustraire les quantités ainsi vendues ou revendues à l’étranger de l’assiette de la taxe annuelle auparavant perçue au profit de l’Afssaps (article L. 5127-17 du code de la santé publique) et de celle de la contribution sur le chiffre d’affaires des entreprises pharmaceutiques (article L. 245-6 du code de la sécurité sociale), qui sont assises sur les ventes en France. Parallèlement, une modification apportée à l’article L. 5123-1 du code de la santé publique permet aux entreprises pharmaceutiques de fixer librement leurs prix à l’exportation. Cette mesure devait être précisée par un décret.
Les premiers projets de texte prévenaient toute passerelle entre les activités de répartition et d’exportation. L’Autorité de la concurrence avait souhaité une version moins restrictive. Le texte donc été assoupli, les grossistes pouvant réaliser des exportations sur certains produits mais en informant les laboratoires exploitant sur les quantités exportées et leur destination. Dans la version définitive du décret précité, la notion d’exportation parallèle a été retirée, ce qu’approuvent vos rapporteurs, qui doutaient de l’efficacité de cette disposition pour lutter contre les ruptures d’approvisionnement.
V.- LA FORMATION ET L’INFORMATION DES PROFESSIONNELS ET DU PUBLIC
L’amélioration de la sécurité sanitaire ne dépend certes pas au premier chef des règles applicables à la promotion des produits de santé ou à l’information des professionnels. Examinées séparément, ces mesures, qu’il s’agisse de publicité, de visite médicale, d’information publique sur le médicament, de prescription en dénomination commune internationale ou de certification de logiciels, peuvent sembler secondaires aux yeux de certains acteurs du domaine de la santé. Pourtant, lorsqu’on les considère dans leur ensemble, il apparaît évident qu’elles contribuent à assainir le système de sécurité sanitaire et à l’orienter vers davantage d’objectivité et de prudence.
A. LA BASE DE DONNÉES PUBLIQUE SUR LES PRODUITS DE SANTÉ ET LEUR USAGE
L’article 8 de la loi du 29 décembre 2011 crée un nouvel article L. 161-40-1 du code de la sécurité sociale, qui prévoit que l’Agence nationale de sécurité du médicament et des produits de santé (ANSM), en liaison avec la Haute Autorité de santé (HAS) et l’Union nationale des caisses d’assurance maladie, sous l’égide du ministère chargé de la santé, met en œuvre une base de données administratives et scientifiques sur les traitements ainsi que sur le bon usage des produits de santé.
Cette base de données doit être consultable et téléchargeable gratuitement sur le site internet du ministère chargé de la santé.
Elle est destinée à servir de référence pour l’information des professionnels de santé, des usagers et des administrations compétentes en matière de produits de santé. Cette base de données doit répondre aux critères définis dans la charte de qualité des bases de données médicamenteuses destinées aux éditeurs de logiciels d’aide à la prescription certifiés. En effet, les deux outils d’information doivent être construits sur les mêmes bases.
Il est prévu qu’un décret fixe les conditions d’application de la base, et notamment les conditions dans lesquelles celle-ci est rendue gratuitement accessible au public. Ce décret n’a pas encore été pris, mais son projet a été communiqué à vos rapporteurs. Le futur contenu de la base de données est détaillé dans l’encadré ci-dessous. Vos rapporteurs estiment que la base de données doit à moyen terme intégrer des informations sur les produits non remboursés, ainsi que sur les dispositifs médicaux.
Contenu de la base de données sur les produits de santé
1° La dénomination de la spécialité en DCI ;
2° La composition qualitative et quantitative en substances actives ;
3° Le nom du titulaire de l’autorisation de mise sur le marché ;
4° La date de l’autorisation de mise sur le marché ou de l’enregistrement ou de l’autorisation d’importation parallèle ;
5°Les différentes présentations commercialisées, accompagnées de la date de la déclaration de leur commercialisation ;
6° Le résumé des caractéristiques du produit prévu à l’article R.5121-23 du code de la santé publique ;
7° La notice prévue à l’article R.5121-148 du code de la santé publique ;
8° Les conditions de prescription et de délivrance ;
9° Le code identifiant la spécialité et le code identifiant la présentation ;
10° Le type de procédure d’autorisation ;
11° L’appartenance à un groupe générique tel que défini à l’article L.5121-1 du code de la santé publique ;
12° Le motif et la date de l’avis de la commission de la Transparence ;
13° Les indications thérapeutiques du médicament ainsi que les niveaux de service médical rendu et d’amélioration du service médical rendu correspondants ;
14° Le lien vers les pages du site de la Haute autorité de santé présentant une synthèse de l’avis de la Commission de la Transparence ainsi que l’avis complet de la Commission de la Transparence ;
15° Le prix de vente au public, en euros toutes taxes comprises, de la spécialité, par présentation ;
16° Le taux de remboursement du médicament ;
17° Le cas échéant, la date d’arrêt de commercialisation de la spécialité ou de l’une de ses présentations ;
18° Le cas échéant, la date de suspension, de retrait ou d’abrogation de l’autorisation de mise sur le marché ou la date de suppression de l’enregistrement ou la date de fin de l’autorisation d’importation parallèle ;
19° Les informations de sécurité sanitaire ;
20° Le statut du médicament au regard de son autorisation de mise sur le marché et de sa commercialisation ;
21° L’appartenance à la liste des médicaments faisant l’objet d’une surveillance supplémentaire.
Le projet de décret communiqué à vos rapporteurs prévoit également que les données administratives et scientifiques sont mises à jour régulièrement. Les données relatives aux spécialités pharmaceutiques dont l’autorisation de mise sur le marché est retirée ou abrogée sont supprimées à compter d’un délai de deux ans après la date de la décision de l’ANSM ou de la Commission européenne portant retrait ou abrogation de l’autorisation.
Les données relatives aux spécialités dont l’autorisation d’importation parallèle a pris fin sont supprimées à compter d’un délai de deux ans après la date d’échéance de l’autorisation. Les données relatives aux spécialités dont l’enregistrement a été retiré sont supprimées à compter d’un délai de deux ans après la date de la décision de l’Agence nationale de sécurité du médicament et des produits de santé retirant l’enregistrement. Les spécialités dont toutes les présentations font l’objet d’un arrêt de commercialisation depuis plus de deux ans sont également supprimées.
B. LE CONTRÔLE DE LA PUBLICITÉ POUR LES MÉDICAMENTS ET LES DISPOSITIFS MÉDICAUX
● Le contrôle de l’information et de la publicité des médicaments à usage humain
L’article 29 de la loi met en place un régime plus strict que par le passé.
L’article L. 5122-3 du code de la santé publique pose une interdiction de principe de la publicité en cas de réévaluation du rapport bénéfices/risques à la suite d’un signalement de pharmacovigilance. L’exploitant du médicament doit alors en informer les professionnels de santé et l’information doit être conforme à celle délivrée par l’ANSM. La méconnaissance de cette disposition pourra entrainer la suspension ou le retrait du visa.
Les campagnes publicitaires à destination du public sont autorisées pour deux catégories de médicaments : les médicaments favorisant le sevrage nicotinique et les vaccins soumis à prescriptions médicales ou remboursables, mais ces derniers ne peuvent faire l’objet d’une campagne publicitaire non institutionnelle (cf. infra).
Le renforcement du contrôle de la publicité passe par la mise en place d’un contrôle a priori exercé par l’ANSM. En effet, l’article L. 5122-9 dispose que la publicité à destination des professionnels de santé pour un médicament est soumise à autorisation préalable de l’ANSM, nommé « visa de publicité ». Ce visa est valable pour une durée qui ne peut excéder celle de l’AMM. Enfin, l’article abroge le 5° de l’article L. 5122-16 qui donnait l’autorisation de faire une publicité de rappel qui faisait référence à une autre publicité classique.
Le décret n° 2012-741 du 9 mai 2012 est venu préciser ces dispositions.
L’article 2 du décret créé un article R. 5122-1-2 qui prévoit que si la réévaluation du rapport bénéfices/risques citée à l’article L. 5122-3 donne lieu à une modification de l’AMM ou de l’enregistrement imposant une modification des mentions figurant dans une publicité qui bénéficiait d’un visa, l’exploitant devra alors demander un nouveau visa qui pourra être déposé en dehors des périodes déterminées par l’agence. En l’absence de réponse du directeur général de l’ANSM dans les deux mois à compter de la date de réception de la demande, la demande sera réputée acceptée.
Pour les médicaments génériques, la publicité auprès du public et à destination des professionnels de santé devra obligatoirement préciser que la spécialité fait partie de la catégorie des génériques, le nom de la ou des spécialités de référence, leur dosage et leur forme pharmaceutique. Elle devra également faire apparaître la mention : « Médicament inscrit au répertoire des génériques. Lors de la substitution, consultez la liste des excipients à effet notoire figurant sur l’emballage ainsi que le répertoire des génériques pour prendre connaissance des mises en garde éventuelles y figurant. » Si la publicité est radiophonique, l’annonce devra mentionner la dénomination commune seulement si le médicament ne contient pas plus de deux principes actifs et, le cas échéant, que le médicament est un générique.
Le calendrier de dépôt des demandes de visas est fixé tous les ans par le directeur général de l’agence avant le 1e novembre. Quatre périodes de une à deux semaines devront être fixées pour le dépôt.
Le visa a une durée de validité de deux ans.
Lors du dépôt, un numéro interne de référencement est attribué et devra apparaître sur le support publicitaire, à l’exception des publicités radiophoniques.
La suspension du visa en cas d’urgence ne peut pas excéder une durée de trois mois.
À des fins publicitaire, la remise d’échantillons gratuits n’est possible que pendant les deux premières années de commercialisation effective du produit en France pour une spécialité bénéficiant d’un premier enregistrement ou d’une première AMM, ou, si la spécialité a déjà bénéficié d’un enregistrement ou d’une AMM, le dosage ou la forme pharmaceutique a été modifié et que la spécialité dispose d’une extension d’indication. Le décret limite la remise d’échantillons gratuits à quatre par an et par destinataire alors que l’ancien dispositif avait fixé la limite à dix par an et par destinataire.
Vos rapporteurs souhaitent attirer l’attention du Gouvernement sur une difficulté dans la mise en œuvre de cet article. Dans la période de dépôt des demandes d’autorisation, les entreprises peuvent déposer autant de dossiers qu’elles le souhaitent, elles devaient aussi redéposer ceux qui avaient fait l’objet d’une autorisation a posteriori accordée entre la promulgation de la loi et la sortie du décret. Certaines entreprises ont déposé jusqu’à 5 000 dossiers pour une demande de publicité. L’ANSM a des difficultés à traiter toutes les demandes dans le temps imparti (deux mois). Cette situation comporte le risque que certains dossiers bénéficient d’un accord tacite systématique, sans avoir été véritablement examinés, même si l’ANSM s’est efforcée de traiter certaines demandes en priorité. Une augmentation des droits pourrait avoir un effet dissuasif sur le nombre de dossiers déposés.
● Le contrôle de la publicité relative aux vaccins
En ce qui concerne les vaccins soumis à prescription médicale ou remboursables, ni le Gouvernement ni la commission n’ont en 2011 souhaité aller jusqu’à l’interdiction de toute campagne émanant des acteurs privés, au motif qu’une telle interdiction aurait pu avoir des effets négatifs en termes de prévention. En revanche, l’article L. 5122-6 du code de la santé publique, tel qu’issu de la loi du 29 décembre 2011, réserve la possibilité de campagnes publicitaires non institutionnelles aux vaccins soumis à prescription médicale ou remboursables mentionnés sur une liste établie par arrêté ministériel pris après avis du Haut Conseil de la santé publique.
Le contenu de ces campagnes publicitaires doit être assorti, de façon clairement identifiée, des mentions minimales obligatoires déterminées par cette instance. Ces mentions sont reproduites in extenso, doivent être facilement audibles et lisibles, selon le support du message publicitaire concerné.
Dans un avis du 25 mai 2012, le Haut Conseil de la santé publique a rappelé que dans un avis du 17 octobre 2008, il avait déjà déploré « que les firmes productrices de vaccin soient autorisées à faire des publicités télévisuelles et radiodiffusées pour le grand public ». Le Haut Conseil ne souhaite pas alimenter à ce jour la liste des vaccins qui pourraient faire l’objet de campagnes publicitaires non institutionnelles et propose de contribuer à la mise en place de procédures permettant des actions de communication indépendantes des firmes. Celles-ci nécessiteraient la consultation d’un panel de personnes représentant les différents acteurs de la vaccination, incluant les associations d’usagers et les professionnels de santé, ceci en fonction de critères définis par le Haut Conseil (7).
L’arrêté du 28 septembre 2012 fixant la liste des vaccins mentionnée à l’article L. 5122-6 du code de la santé publique a retenu les produits suivants :
– les vaccins contre la rougeole, les oreillons, la rubéole ;
– les vaccins contre la méningite C ;
– les vaccins contre la grippe saisonnière ;
– les vaccins contre la diphtérie, le tétanos, la poliomyélite, la coqueluche ;
– les vaccins contre la tuberculose ;
– les vaccins contre les infections à pneumocoques.
● L’alignement du régime de la publicité des dispositifs médicaux sur le régime prévu pour les médicaments
L’article 34 créé un chapitre III dans le titre 1er du livre II du code de la santé publique relatif à la publicité des dispositifs médicaux. Il vise à définir la publicité des dispositifs médicaux, à déterminer les cas dans lesquels elle est autorisée et à la soumettre au respect d’un certain nombre de règles.
Jusqu’à la loi du 29 décembre 2011, la publicité pour les dispositifs médicaux bénéficiait d’un encadrement limité aux directives européennes relatives à la concurrence déloyale (8) et à la publicité trompeuse et comparative (9) mais n’avait pas de régime propre. Le but de cet article a été d’harmoniser et de rendre plus lisibles les règles pouvant s’appliquer à la publicité des dispositifs médicaux.
L’article L. 5213- 1 définit la notion de publicité pour les dispositifs médicaux comme « toute forme d’information, y compris de démarchage, de prospection ou d’incitation qui vise à promouvoir la prescription, la délivrance, la vente ou l’utilisation de ces dispositifs, à l’exception de l’information dispensée dans le cadre de leurs fonctions par les pharmaciens gérant une pharmacie à usage intérieur ». Certaines informations ne rentrent pas dans le champ de cette définition. Ainsi, ne sont pas inclus :
– l’étiquetage et la notice d’instruction des dispositifs médicaux ;
– la correspondance accompagnée le cas échéant de tout document non publicitaire, nécessaire pour répondre à une question précise sur un dispositif médical particulier ;
– les informations relatives aux mises en garde, aux précautions d’emploi et aux effets indésirables relevés dans le cadre de la matériovigilance, ainsi que les catalogues de ventes et listes de prix s’il n’y figure aucune information sur le dispositif médical ;
– les informations relatives à la santé humaine s’il n’y a pas de référence même indirecte à un dispositif médical.
L’article L. 5213-2 précise les caractéristiques que la publicité doit revêtir : elle doit définir le produit de façon objective, ses performances et sa conformité aux exigences essentielles concernant la sécurité et la santé et doit favoriser son bon usage. La publicité ne peut être trompeuse ni présenter un risque pour la santé publique.
La publicité pour les dispositifs médicaux est néanmoins limitée aux dispositifs médicaux non remboursés par la sécurité sociale. La prise en charge, même partielle par les régimes obligatoires interdit la publicité pour le produit, sauf si le dispositif présente un faible risque pour la santé humaine, mais dans ce cas, la publicité ne peut pas faire mention de ce remboursement. La liste de ces dispositifs médicaux est fixée par arrêté des ministres chargés de la santé et de la sécurité sociale. L’arrêté fixant la liste de ces dispositifs médicaux a été publié le 21 décembre 2012.
Les dispositifs médicaux présentant un risque important pour la santé humaine sont soumis à autorisation préalable de l’ANSM pour pouvoir faire l’objet d’une publicité. L’autorisation est délivrée pour une durée de 5 ans renouvelable et peut être retirée ou suspendue par l’agence. L’arrêté fixant la liste de ces dispositifs médicaux a été pris le 24 septembre 2012. Il s’agit des dispositifs médicaux de classe I et IIa (faible degré de risque et degré moyen de risque).
L’ANSM peut mettre en demeure la personne concernée de retirer la publicité, de présenter ses observations et de régulariser la situation, au besoin, en assortissant cette mise en demeure d’une astreinte. L’agence peut interdire la publicité après avoir mis l’entreprise en demeure.
Les dispositions précédemment exposées ne s’appliquent pas aux produits mentionnés aux articles L. 5134-1 et L. 5134-2, à savoir, les moyens contraceptifs.
Est passible de deux ans d’emprisonnement et 30 000 euros d’amende le fait pour un fabricant de dispositif médical ou son mandataire, ainsi que toute personne qui se livre à la distribution ou à l’importation de dispositifs médicaux de réaliser ou diffuser :
– une publicité à caractère trompeur ou de nature à présenter un risque pour la santé humaine ;
– une publicité de dispositif médical soumise à autorisation préalable lorsque l’ANSM n’a pas délivré, a refusé de délivrer, a suspendu ou a retiré cette autorisation.
Des peines complémentaires sont également prévues. Les personnes physiques coupables de l’infraction encourent les peines complémentaires suivantes : diffusion de la décision de condamnation, communiqué informant le public, affichage de la décision prononcée, fermeture définitive ou pour une durée de 5 ans au plus des établissements ou de l’un ou de plusieurs des établissements de l’entreprise ayant servi à commettre les faits incriminés, l’interdiction de fabriquer, conditionner, d’importer et mettre sur le marché des dispositif médicaux pour une durée maximale de 5 ans. Les personnes morales déclarées responsables pénalement encourent quant à elles l’affichage de la décision prononcée ou diffusion de celle-ci et la fermeture définitive ou pour une durée de 5 ans au plus des établissements.
Le décret n° 2012-743 du 9 mai 2012 précise les règles applicables à la publicité pour les dispositifs médicaux en modifiant les articles R. 5213-1 et suivants du code de la santé publique.
Le décret fixe d’abord les conditions de la publicité pour les dispositifs médicaux :
– elle doit être conçue de telle sorte que le caractère publicitaire du message soit évident et que le produit soit clairement identifié comme un dispositif médical ;
– elle doit préciser la date à laquelle elle a été établie ou la date de la dernière modification et doit obligatoirement comporter une série de mentions comme, par exemple, la destination attribuée au dispositif médical par son fabricant, les informations indispensables pour un bon usage, une invitation expresse à lire attentivement les instructions figurant dans la notice.
En aucun cas, la publicité ne devra faire apparaître de mention précisant que le dispositif est pris en charge par l’assurance maladie. Le décret encadre très largement cette publicité en fixant une série de 15 éléments que la publicité ne pourra comporter. Par exemple, ne pourront apparaître aucune mention qui pourrait laisser penser que la consultation médicale ou l’intervention chirurgicale est superflue, que l’état de santé pourrait être amélioré par l’utilisation du dispositif médical ou encore qui pourrait conduire, par une description détaillée de symptômes, à un faux diagnostic.
La publicité à destination des professionnels de santé, devra, quant à elle, préciser la situation du dispositif médical au regard du remboursement par l’assurance maladie.
Si la publicité ne respecte pas les conditions posées par le décret, l’ANSM pourra mettre en demeure la personne de régulariser la situation dans un délai qui ne pourra être inférieur à un mois.
L’article R. 5213-5 fixe les conditions de l’autorisation préalable et, notamment le contenu de la demande d’autorisation. Le directeur général de l’agence doit notifier sa décision dans un délai de deux mois. En l’absence de notification de la décision dans ce délai, l’autorisation est réputée acquise. L’autorisation est délivrée pour une durée de 5 ans renouvelable. Les règles de suspension ou de retrait de l’autorisation sont identiques à celles fixées pour la publicité pour les médicaments à usage humain.
L’article 34 de la loi prévoit également un encadrement de la publicité pour les dispositifs médicaux de diagnostic in vitro. La publicité pour ces dispositifs médicaux doit suivre les mêmes règles que celles fixées pour les dispositifs médicaux.
Le décret n° 2012-744 du 9 mai 2012 fixe les règles applicables à la publicité pour les dispositifs médicaux de diagnostic in vitro. Les conditions et procédures applicables sont similaires à celles relatives aux dispositifs médicaux.
L’apport majeur de la loi est l’institution du contrôle a priori effectué par l’Agence. Le rapport annuel de l’ANSM précise que le contrôle a priori a été mis en place de manière effective le 1e juin 2012. A titre de comparaison, en 2012 et avant la mise en place du contrôle a priori, l’agence n’a interdit aucune publicité dans le cadre de l’évaluation a posteriori alors que 999 demandes d’autorisations de publicités à destination des professionnels de santé et 86 à destination du grand public ont été refusées par l’agence dans le cadre du contrôle a priori, ce qui montre que le contrôle est véritablement effectif et efficace.
C. L’OBLIGATION DE PRESCRIPTION EN DÉNOMINATION COMMUNE INTERNATIONALE
La prescription des médicaments en dénomination commune internationale, prévue par l’article 19, modifiant l’article L. 5121-1-2 du code de la santé publique, constitue une avancée majeure pour l’information des patients.
La dénomination commune internationale permet, en effet, d’identifier facilement les principes actifs d’un médicament, ainsi que les fausses innovations thérapeutiques. Ainsi, le principe actif du Mediator, le benfluorex, s’il avait été inscrit sur les ordonnances, aurait peut-être permis aux professionnels et aux patients de reconnaître le caractère anorexigène de ce médicament.
Cette mesure devrait également encourager la prescription de génériques et permettre d’éviter les contre-indications et les mauvaises associations de médicaments. C’est donc un outil d’une grande utilité pour les professionnels et pour les patients.
Il est toujours possible, en parallèle, de citer la dénomination de fantaisie, afin d’apporter toutes les garanties nécessaires à la bonne information du patient, tout en assurant la continuité avec le système antérieur et ne pas déstabiliser une partie de la population.
Cette obligation devra être respectée à compter du 1er janvier 2015, afin de donner aux médecins le temps nécessaire pour adapter leur pratique et s’équiper en logiciels d’aide à la prescription certifiés.
Les décrets d’application relatifs à la certification des logiciels d’aide à la prescription, et à la constitution d’une base de données publique sur les produits de santé ne sont pas parus. Or ils sont un préalable indispensable à la prescription en dénomination commune internationale. Vos rapporteurs insistent donc sur la nécessité d’une parution rapide de ces textes.
D. LES LOGICIELS D’AIDE À LA PRESCRIPTION ET À LA DISPENSATION
L’article 32 de la loi se fixe pour objectif de généraliser à court terme l’usage de logiciels certifiés d’aide à la prescription et à la dispensation. Cette certification doit être opérée par des organismes accrédités par le Comité français d’accréditation, et selon des référentiels établis par la Haute Autorité de santé (HAS) qui établit la procédure de certification des sites informatiques dédiés à la santé.
Ces logiciels doivent obligatoirement :
– intégrer les recommandations et avis médico-économiques identifiés par la Haute Autorité de santé ;
– permettre de prescrire directement en dénomination commune internationale ;
– afficher les prix des produits au moment de la prescription et le montant total de la prescription ;
– indiquer l’appartenance d’un produit au répertoire des génériques ;
– comporter une information relative à leur concepteur et à la nature de leur financement.
– participer à l’amélioration des pratiques de prescription médicamenteuse ;
– se conformer à des exigences minimales en termes de sécurité, de conformité et d’efficience de la prescription.
L’obligation de certification, qui devra être satisfaite au plus tard le 1er janvier 2015, repose sur les éditeurs des logiciels. Un décret en Conseil d’État devait être pris pour préciser les conditions d’application de l’article. Il n’a pas encore publié.
E. L’ENCADREMENT DE LA VISITE MÉDICALE
L’idée d’interdire purement et simplement la visite médicale a été écartée, dans la mesure où les visiteurs médicaux français ne sauraient être tenus pour seuls responsables des échecs qu’a connu notre système de sécurité sanitaire et où ils peuvent encore jouer un rôle positif en termes d’information technique sur les produits de santé.
Cela étant dit, il n’est pas contestable que la visite médicale soit appelée à évoluer en profondeur afin d’éviter les errements qu’a révélés le drame du Mediator.
L’article 30 de la loi prévoyait, à titre expérimental, et pour une durée de deux ans, que les visites médicales en faveur de certains produits de santé devraient être effectués de manière collective dans les établissements de santé.
Une convention conclue entre l’établissement de santé et l’entreprise pharmaceutique, dont les modalités devaient être fixées par arrêté du Ministre en charge de la santé pris après avis de la Haute Autorité de Santé (HAS) devait déterminer les conditions d’interventions des visiteurs au sein de l’établissement de santé.
Après de vives discussions au sein de l’Assemblée nationale et du Sénat, les parlementaires ont finalement décidé d’exclure de la liste des produits concernés par l’expérimentation les médicaments réservés à l’usage hospitalier, les médicaments à prescription hospitalière initiale ou non, et les dispositifs médicaux. Par conséquent, l’expérimentation porte principalement sur les médicaments prescrits à l’hôpital mais pouvant être délivrés en ville.
Le même article prévoyait qu’avant le 1e janvier 2013, le Gouvernement devait remettre un rapport au Parlement afin de dresser un bilan de l’expérimentation, après évaluation du dispositif par la HAS, pour mesurer l’opportunité de pérenniser le dispositif et de l’étendre aux professionnels de santé exerçant en ville.
Le Comité économique des produits de santé (CEPS) devait fixer des objectifs quantitatifs dans la pratique de la visite médicale, objectifs fixés par une nouvelle Charte de la visite médicale.
En cas de non-respect de ces objectifs par une entreprise, la loi donnait compétence au Comité économique des produits de santé pour prononcer une sanction financière à l’encontre de ladite entreprise ; la pénalité financière ne pouvant être supérieure à 10 % du chiffre d’affaires.
Un arrêté devait venir préciser les règles régissant la visite médicale ayant vocation à figurer dans le règlement intérieur de l’établissement, notamment :
– le nombre minimal de professionnels de santé participant à la rencontre avec le visiteur médical ;
– les modalités d’accès des visiteurs médicaux à l’établissement, les règles de référencement d’indentification et de circulation ainsi que les modalités de contrôle de l’accès aux structures à accès restreint ;
– les modalités d’organisation de la rencontre avec les professionnels de santé telles que la prise de rendez-vous et, le cas échéant, la détermination de locaux consacrés à la rencontre, l’accord éventuel du responsable de la structure interne ou autre ;
– la personne ou structure responsable du suivi de ces dispositions.
La convention devait être signée par le responsable légal de l’établissement après avis de la commission médicale d’établissement, de la commission des soins infirmiers, de rééducation et médicotechnique dans les hôpitaux, et après avis de la conférence médicale d’établissement dans les cliniques.
Les textes d’application de la mesure n’ont pas été publiés et la disposition n’a jamais été appliquée.
À l’occasion de l’examen du projet de loi de financement de la sécurité sociale pour 2013, le Gouvernement a voulu relancer en l’amendant le dispositif expérimental de visite médicale collective instauré par la loi du 29 décembre 2011. La mise en œuvre de l’expérimentation supposait en effet la conclusion de plusieurs conventions avec les industriels de santé, ce qui impliquait des formalités administratives trop lourdes. De plus, elle ne couvrait pas l’ensemble des médicaments.
L’article 58 de la loi de financement de la sécurité sociale pour 2013 a donc transformé l’expérimentation en disposition pérenne et simplifié sa mise en œuvre, le conventionnement avec les industriels étant supprimé au profit d’un encadrement de la mesure par le règlement intérieur de l’établissement. Pour mémoire, la généralisation de la visite médicale collective avait été limitée, par voie d’un amendement parlementaire, aux seuls médicaments pouvant être délivrés en ville, et ce, contre l’avis du Gouvernement. Une évaluation du dispositif devait être menée par la HAS et présentée par le Gouvernement au Parlement fin 2014.
Cependant, le Conseil constitutionnel, dans sa décision n° 2012-659 DC du 13 décembre 2012, a estimé que l’article 58 ne relevait pas de loi de financement de sécurité sociale et a censuré la disposition.
En conséquence, la mesure n’est pas appliquée.
Enfin, la Charte de la visite médicale, signée pour la première fois en 2004 (cf. encadré ci-dessous), est en cours de révision.
La Charte de la visite médicale
La charte de qualité destinée à encadrer la visite médicale est conclue entre, d’une part, le Comité économique des produits de santé et, d’autre part, « un ou plusieurs syndicats représentatifs des entreprises du médicament ». Selon l’article L. 162-17-8 du code de la sécurité sociale, cette charte « vise, notamment, à mieux encadrer les pratiques commerciales et promotionnelles qui pourraient nuire à la qualité des soins ».
Cette charte a été signée le 22 décembre 2004 entre le comité et le syndicat professionnel de l’industrie pharmaceutique, le LEEM. Elle prévoyait notamment le dispositif suivant :
« Le Comité économique des produits de santé arrête chaque année la liste des classes pharmaco-thérapeutiques, selon la nomenclature EPHMRA, pour lesquelles il estime qu’une réduction de la visite médicale est nécessaire. Le comité arrête sa décision après consultation de la Haute Autorité de santé, de l’Union nationale des caisses d’assurance maladie, des représentants des médecins dans le cadre du groupe de suivi de la charte de la visite médicale, qui font valoir à cette occasion leurs besoins d’information sur ces médicaments ainsi que du LEEM.
Ces classes sont désignées au vu de critères rendus publics intégrant le contenu de ces consultations notamment au regard du bon usage du médicament et des objectifs de santé publique ou de dépenses pour l’assurance maladie.
Après consultation du LEEM et des entreprises concernées, le [comité] fixe, pour chacune de ces classes, en excluant les médicaments appartenant à des groupes génériques et en tenant compte du lancement des produits nouveaux, un taux annuel d’évolution du nombre de contacts avec les médecins réalisés par les délégués médicaux. Les taux annuels d’évolution pour les deux années suivantes sont également fixés à titre indicatif.
En cas de non-respect, pour une classe, du taux fixé, le [comité] peut décider, conventionnellement ou à défaut par décision, une baisse, temporaire ou définitive, du prix des spécialités y figurant, dont l’importance est fonction notamment de l’écart entre l’évolution constatée et la décroissance fixée par le [comité]. »
Ce dispositif n’a toutefois pas pu recevoir d’application effective, le Conseil d’État (10) ayant jugé qu’aucune disposition législative ne donnait compétence aux signataires de la charte pour habiliter le comité à décider unilatéralement des mesures de réduction des visites médicales.
Des négociations sont en cours, associant le CEPS, la HAS et les industries pharmaceutiques, pour mettre au point une nouvelle charte au mois de novembre 2013. Les points suivants seraient en discussion :
– la définition d’un volume de visites médicales, qui pourrait être défini par classe thérapeutique ;
– le rôle des visiteurs médicaux dans le respect par les prescriptions des indications contenues dans l’autorisation de mise sur le marché du produit ;
– la mise en place d’une information scientifique et non promotionnelle ;
– l’encadrement et la transparence en matière d’avantages consentis par les visiteurs médicaux auprès des professionnels ;
– la qualité de la formation des visiteurs médicaux. La HAS demande à ce qu’un auditeur accompagne le VM sur le terrain afin de certifier son discours, si le médecin en est d’accord ;
– la rémunération des visiteurs médicaux, qui pourrait être moins quantitative et plus qualitative.
Vos rapporteurs estiment que la rédaction d’une Charte est une base essentielle qui permettra de fixer les grands principes déontologiques auxquels la visite médiale devra se soumettre. Cependant la Charte doit s’accompagner d’une disposition législative qui encadre la visite médicale, notamment dans les établissements de santé dont on connaît la place stratégique dans la prescription des médicaments.
Dans le but de renouveler les méthodes et l’esprit de la visite médicale, la solution proposée dans le projet de loi de financement de la sécurité sociale pour 2013 était équilibrée. Vos rapporteurs estiment qu’elle devrait être reprise dans le futur projet de loi de santé publique. Une telle mesure doit à terme s’accompagner d’un dispositif adapté aux soins de ville pour donner toute sa place à la visite médicale dans la juste prescription des produits de santé.
La Commission entend Mme Marisol Touraine, ministre des affaires sociales et de la santé, sur la mise en œuvre de la loi du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé au cours de sa séance du mardi 16 juillet 2013.
M. Christian Hutin, président. Nous recevons ce soir Mme Marisol Touraine, ministre des affaires sociales et de la santé, en vue de dresser le bilan de l’application de la loi du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé. Conformément aux dispositions de l’article 145-7 du Règlement, notre commission s’est saisie de l’application de ce texte, voté sous la précédente législature mais dont l’actualité demeure eu égard aux drames sanitaires que nous avons connus dans le domaine du médicament. Elle a désigné deux co-rapporteurs, l’un de la majorité, l’autre de l’opposition : Mme Catherine Lemorton, présidente de la commission, responsable du texte pour l’opposition lors de son précédent mandat, et M. Arnaud Robinet, qui en était le rapporteur. Nous examinerons demain en commission le rapport issu de leurs travaux, qui fait état des textes réglementaires publiés et des dispositions qui n’ont pas fait l’objet des textes d’application nécessaires.
Mme Marisol Touraine, ministre des affaires sociales et de la santé. La loi dite « de renforcement de la sécurité des produits de santé » du 29 décembre 2011, nécessite 37 décrets d’application. Si cette loi comporte des dispositions intéressantes, elle présente néanmoins des limites sur lesquelles il sera nécessaire de revenir dans notre débat.
Sur les 37 décrets nécessaires, 29 décrets ont déjà été pris, dont 19 depuis mai 2012, et 6 sont en train de l’être. Un autre décret, portant sur les données de santé, dépendra des conclusions d’une mission que j’ai confiée à Pierre-Louis Bras, inspecteur général des affaires sociales, dont les conclusions me seront remises au mois de septembre. Par ailleurs, 12 arrêtés et une circulaire étaient nécessaires : je les ai signés. J’ai également pris une ordonnance et trois autres le seront très prochainement.
Le travail d’évaluation de cette loi représente un enjeu tout à fait important en termes de sécurité sanitaire, c’est pourquoi je veux remercier et féliciter les deux co-rapporteurs qui s’étaient par ailleurs fortement impliqués dans l’examen de ce texte et résolument engagés en faveur de la sécurité sanitaire.
L’action que le Gouvernement mène pour appliquer la loi, telle qu’elle a été votée par le Parlement, s’articule autour de six priorités.
La première d’entre elles est le renforcement des instances chargées d’évaluer la sécurité, la qualité et l’efficacité des médicaments : c’est tout le sens de la création de l’Agence nationale de sécurité du médicament et des produits de santé (ANSM). Il ne s’agit pas là d’un simple changement de nom, pour cette structure qui a pris le relais de l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS), car cette agence s’est vue confier de nouvelles responsabilités pour mieux surveiller, pour mieux informer.
Cette agence ne fonctionne réellement, selon les nouveaux principes de la loi, que depuis six mois. Le renouvellement de son organisation et de sa gouvernance ont demandé un peu de temps. Désormais, et comme l’ont montré les crises sanitaires récentes, l’ANSM est opérationnelle et je tiens à saluer devant vous la qualité du travail qu’elle réalise au quotidien, même si nous pouvons réfléchir à la manière de toujours mieux garantir la sécurité de nos concitoyens.
La deuxième priorité est celle de la sécurisation des produits de santé. La loi a mis en place un renforcement du système de pharmacovigilance. En deux ans, plus de 110 révisions de médicaments ont été lancées en France et un programme de réévaluation des médicaments a été initié : la commercialisation de certains produits a ainsi été suspendue ou arrêtée. Parmi les 58 médicaments qui ont été réévalués, je citerai plus particulièrement Diane 35, qui nous a préoccupés cet hiver, ou, plus récemment encore, le tétrazépam (ou Myolastan).
La loi a également permis d’encadrer les prescriptions de médicaments en dehors des indications de leur autorisation de mise sur le marché (AMM). L’ANSM a ainsi reçu une trentaine de demandes de recommandation temporaire d’utilisation (RTU). La première RTU vient d’être annoncée par l’agence : elle concerne le Baclofène dans le traitement de la dépendance à l’alcool et devrait être mise en place à la rentrée.
Ce mouvement, je l’ai prolongé en renforçant encore la sécurisation des utilisations hors AMM pour des raisons économiques. J’ai ainsi soutenu, dans la loi de financement de la sécurité sociale pour 2013, le principe d’une nouvelle forme de RTU. Le décret est en cours de finalisation.
Ce sont des avancées majeures pour la sécurité des patients. Cependant, les crises récentes ont montré qu’il nous fallait aller plus loin. Je proposerai prochainement des mesures pour sécuriser encore les dispositifs médicaux, pour simplifier notre système de vigilance et pour promouvoir la notification.
La troisième priorité du Gouvernement est celle de la transparence et de l’indépendance de l’expertise.
Le crédit d’une étude ou d’une décision se mesure à l’aune de ces deux critères. S’assurer de leur effectivité, c’est là la condition de la confiance dans nos médicaments. Désormais, les responsables publics doivent publier une déclaration d’intérêts. C’est le cas, par exemple, des membres de mon cabinet et des dirigeants des agences sanitaires et des agences régionales de santé. Ces déclarations publiques d’intérêts sont disponibles sur Internet.
Dans le même temps, le décret qui instaure une charte de l’expertise dans le domaine de la santé et de la sécurité sanitaire, a été publié. Cette charte permettra de veiller aux principes d’impartialité, de transparence, de pluralité et d’indépendance de l’expertise.
Enfin, le décret dit « Sunshine Act », publié le 21 mai dernier après un important travail de concertation, permet désormais de clarifier les liens entre l’industrie et les professionnels de santé. Ce dispositif garantit la plus grande transparence possible dans l’état actuel de la loi. En effet, tout avantage d’une valeur supérieure ou égale à 10 euros sera rendu public. Ce seuil est le plus bas qui soit compatible avec la rédaction de la loi du 29 décembre 2011, comme l’a précisé le Conseil d’État. J’avais marqué ma préférence pour un seuil d’un euro, car j’estimais que tout avantage, aussi minime soit-il, méritait d’être rendu public. Mais le texte de la loi votée ne le permet pas et je le regrette. Par ailleurs, la publication des informations sera centralisée dans les meilleurs délais sur un site Internet unique. Dans l’attente de sa mise en place, ces informations seront publiées sur le site Internet des ordres professionnels concernés et sur ceux des entreprises, à partir du 1er octobre.
Dans cette bataille pour la transparence, il nous faudra encore franchir de nouvelles étapes : je pense notamment que le champ de la disposition pourrait être élargi, par exemple, aux organismes qui certifient les dispositifs médicaux. J’ai également à l’esprit le niveau du seuil visé à l’article 2.
La quatrième priorité est celle de l’accès à l’information, enjeu qui concerne aussi bien les professionnels de santé que les patients. Une large base de données sur le médicament est en train d’être créée. Une première version sera accessible à tous, dès le mois d’octobre. Elle permettra d’avoir un site unique, officiel, gratuit, exhaustif et indépendant. Cette version sera la première pierre d’un service public d’information en santé qui doit être lancé ensuite, au-delà du seul domaine du médicament.
La cinquième priorité, c’est de nous assurer du bon usage des médicaments.
Dans ce domaine, notre pays a progressé, même si nous restons dans le peloton de tête des grands consommateurs de médicaments.
Un mot sur la vente sur Internet des médicaments : comme l’imposaient la loi et les textes européens, nous l’avons autorisée, par l’ordonnance du 19 décembre 2012. Néanmoins, les médicaments ne sont pas des produits comme les autres. Il est du devoir des pouvoirs publics de garantir la sécurité de la chaîne d’approvisionnement, notamment pour combattre la contrefaçon. Il est aussi essentiel de ne pas encourager la surconsommation de médicaments. Voilà pourquoi le choix a été fait d’encadrer la vente sur Internet en mettant en place des garde-fous. Par exemple, les médicaments sous ordonnance n’y seront pas accessibles. Par ailleurs, seuls les sites Internet qui sont adossés à une pharmacie « physique » pourront vendre en ligne : les patients pourront ainsi se rendre dans une officine et échanger avec un pharmacien, si besoin. C’est également pourquoi le Gouvernement ne s’engagera pas dans l’ouverture partielle du monopole officinal, comme le suggère l’Autorité de la concurrence.
La dernière priorité vise à donner un souffle nouveau à la démocratie sanitaire. La participation des citoyens aux instances de santé est primordiale pour garantir la confiance. L’ANSM permet désormais à des représentants de patients et d’usagers d’intégrer ses instances de gouvernance, ainsi que ses groupes de travail. Je me réjouis de cette décision et je souhaite que ce mouvement se prolonge dans l’ensemble de nos instances de santé.
La réforme engagée en 2011 est une première étape importante, mais elle reste incomplète. Il nous revient ainsi de poursuivre les chantiers engagés, en centrant notre action sur le bon usage des médicaments, sur leur sécurisation, par la simplification de la pharmacovigilance et par la mise en place d’une surveillance de l’utilisation des médicaments. J’ai ainsi diligenté une mission, afin que soient étudiés les moyens d’analyser en continu les pratiques collectives de prescription. Enfin, le chantier de la sécurité des dispositifs médicaux doit être ouvert. Tous ces enjeux seront au cœur de la prochaine loi de santé publique : il nous appartient donc d’y travailler tous ensemble.
Mme Catherine Lemorton, rapporteure. Je voudrais souligner la situation singulière dans laquelle s’est retrouvé le nouveau Gouvernement en mai 2012 puisqu’il lui a incombé de mettre en musique la loi votée en décembre 2011, le précédent Gouvernement n’ayant eu que quatre mois devant lui pour travailler à son application.
J’aurai plusieurs questions précises.
Tout d’abord, sur la prévention des conflits d’intérêts : quand est prévue la mise en ligne sur un site Internet unique de l’ensemble des déclarations d’intérêts prévues par l’article 1er de la loi ? Envisage-t-on de centraliser aussi la publication des avantages consentis par les entreprises et d’en simplifier les modalités pour les plus petites ?
Ensuite, pourquoi ne pas avoir prévu la publication du montant de certaines rémunérations ou participations dans des entreprises par les experts soumis à déclarations publiques d’intérêts ? Pourquoi, de la même manière, ne pas avoir prévu la publication précise du contenu des conventions passées entre les laboratoires et les professionnels de santé ? Je pense notamment aux praticiens hospitaliers professeurs des universités (PUPH) qui utilisent la notoriété qu’ils ont acquise dans le cadre de leur activité à l’hôpital public pour la mettre au service de la promotion de certaines molécules en étant rémunérés par des laboratoires. Il y a eu des rapports mettant en lumière les montants touchés, dont un rapport de l’IGAS de janvier 2009. Sans mettre en cause la probité de ces professionnels, on peut néanmoins estimer que ces pratiques devraient s’inscrire dans un cadre transparent.
Les conventions conclues dans le cadre des relations commerciales qui ont pour objet l’achat de biens ou de services ne sont pas dans le champ de l’article 2 de la loi. Nous pensons qu’elles devraient y être intégrées.
M. Arnaud Robinet, rapporteur. La loi du 29 décembre 2011 a permis de changer les pratiques existantes pour plus de transparence, pour plus de sécurité et aussi pour éviter que les liens d’intérêt ne se transforment en conflits d’intérêt. La loi a permis de réaliser un grand nombre d’avancées, même si elle ne constitue qu’une première étape et que des ajustements seront sans doute nécessaires. A cet égard, la mission d’évaluation de la loi que nous avons menée avec Catherine Lemorton permettra sans doute d’avancer sur l’amélioration de ce texte.
Toujours sur les conflits d’intérêts, j’ai une question très précise sur le décret « Sunshine act » : pourquoi avoir fixé le seuil de déclaration des avantages accordés par les entreprises aux professionnels à 10 euros ? Vous l’avez évoqué mais je souhaiterais que vous nous précisiez le processus qui a abouti à cette décision, alors que les débats sur le texte avaient convergé sur un seuil à un euro.
S’agissant de l’ANSM et du pilotage du système sanitaire, Xavier Bertrand, avait annoncé, pour améliorer le pilotage de la politique du médicament, la création d’un comité stratégique de la politique des produits de santé et de la sécurité sanitaire réuni chaque semaine en comité opérationnel et en comité stratégique, tous les trimestres, sous la présidence du ministre. Cette pratique a-t-elle été mise en place ?
S’agissant de l’ANSM, l’augmentation de ses missions est un véritable défi et avec Gérard Bapt, nous sommes très conscients de ce que cela représente pour la nouvelle agence. Au-delà de cette augmentation, il y aussi un changement de culture qui touche tous les collaborateurs de l’agence. Or, il semble que les moyens humains attribués à l’Agence soient inférieurs aux chiffres annoncés par le ministre de l’époque. Quelles seront les principales orientations du contrat de performance signé avec l’ANSM et les moyens accordés pour remplir ses nouvelles missions ? Plus généralement comment pourrait-on réfléchir aux moyens d’alléger la charge de travail de l’ANSM, en simplifiant les procédures et en revoyant le partage des compétences entre les différentes agences ?
Mme Catherine Lemorton, rapporteure. Deux questions concernant la base de données publiques : les dispositifs médicaux et les médicaments non remboursés ont-ils vocation à y figurer, et tout un chacun pourra-t-il y accéder sur Internet de manière prioritaire par rapport aux informations communiquées par des sites commerciaux ?
Une question qui concerne également les compétences du ministère de la recherche : elle concerne la faiblesse de la formation initiale des professionnels de santé, qui présentent des carences importantes de contenu sur le médicament, en particulier pour toutes les professions qui relèvent du chapitre IV du code de la santé publique. S’agissant de la formation continue, une taxe a certes été mise en place, mais il n’y a pas de transparence sur les moyens dégagés par l’industrie pharmaceutique pour ce que l’on appelle désormais le développement professionnel continu des professionnels : on parle de 150 millions d’euros, mais cette estimation se situerait semble-t-il très en-deçà de la réalité.
S’agissant des campagnes de vaccination menées par les laboratoires pharmaceutiques, lors du débat sur le projet de loi, l’opposition de l’époque, dont j’étais, considérait qu’il s’agissait du domaine régalien. Je me réjouis donc de la parution d’une liste limitée à cinq vaccins pouvant faire l’objet de campagnes de publicité par l’industrie pharmaceutique, et notamment du fait que ne figure pas sur cette liste le vaccin contre le papillomavirus responsable du cancer de l’utérus, qui, à l’époque, avait fait l’objet d’une campagne de publicité qui avait fait grand bruit.
M. Arnaud Robinet, rapporteur. S’agissant de la formation des patients, je souhaiterais aborder la question d’un article qui avait été très débattu au sein même de la majorité de l’époque, concernant les visiteurs médicaux. Le texte a mis en place le principe d’une visite collective dans les hôpitaux. Une charte serait en cours de rédaction avec le CEPS pour encadrer la visite médicale : quelle sera sa valeur juridique ? Ne faut-il pas réformer les principes de la visite médicale comme cela avait d’ailleurs été prévu dans le cadre du projet de loi de financement de la sécurité sociale (PLFSS) pour 2013 ?
S’agissant ensuite de la nouvelle profession des attachés à la promotion du médicament, est-il envisageable d’encadrer celle-ci ainsi que les nouvelles méthodes qu’ils emploient au sein des officines ?
S’agissant de la question de l’encombrement thérapeutique et de la multiplication des médicaments, l’article 14 de la loi posait l’exigence de l’apport d’une valeur ajoutée d’un médicament pour sa mise sur le marché : le décret d’application de cet article n’a toujours pas été pris. Quand paraîtra-t-il ?
Sur la question de la pharmacovigilance, une simplification des procédures est-elle prévue, tant pour les centres régionaux de pharmacovigilance que pour les professionnels de santé, qui rencontrent souvent des difficultés pour faire remonter le signalement des effets indésirables auxquels ils sont confrontés ?
Mme Catherine Lemorton, rapporteure. Concernant la certification des logiciels d’aide à la prescription, le délai pour cette certification a été fixé à janvier 2015 : pensez-vous que ce délai est raisonnable ? Le processus est-il bien enclenché ?
S’agissant des données de santé, j’ai bien compris que nous attendions les résultats de la mission confiée à Pierre-Louis Bras, inspecteur général des affaires sociales, mais peut-être aurez-vous d’ores et déjà des éléments à nous communiquer.
Je restais pour ma part dans l’expectative concernant le GIP « Études de santé » prévu à l’article 33, dans la mesure où il me semblait qu’il était possible de perfectionner l’Institut des données de santé, qui avait l’avantage de regrouper de nombreux acteurs, y compris les patients. La question est donc la suivante : faut-il vraiment mettre en place une autre structure ?
La loi prévoit également que l’ANSM soit en mesure d’exiger des informations des entreprises et la réalisation d’études post-AMM, mais le décret fixant les sanctions que l’agence peut prononcer n’est toujours pas paru. Qu’en est-il ?
Sur le plan de la pharmacovigilance, j’ai avec moi deux notices de médicaments, l’un qui concerne un médicament administrable par voie orale, l’autre un médicament d’application locale. Pour ces deux médicaments, il est spécifié qu’en cas d’effet indésirable grave ou non prévu par cette notice, le patient est invité à en parler à son médecin ou à son pharmacien. Cela revient à limiter les types d’effets indésirables à déclarer, car pour ceux qui sont déjà spécifiés dans la notice, les patients ne sont pas incités à faire une démarche de déclaration. Or, certains effets indésirables rares, mais non graves, deviennent parfois fréquents lorsqu’un médicament est prescrit à plus large échelle. Je préconise que les notices des médicaments précisent la troisième voie qui est ouverte en matière de déclaration d’effets indésirables, autrement dit, la déclaration directe par le patient auprès du Centre régional de pharmacovigilance (CRPV). On pourra rétorquer que cela crée un risque d’engorgement des centres de pharmacovigilance, mais il existe aussi des risques d’effets en cascade en raison du nombre de prescriptions d’un médicament. Il faut donc bien distinguer entre effet indésirable grave et effet indésirable courant.
S’agissant des dispositifs médicaux, on connaît votre exigence, que vous portez au niveau européen. Il est en effet essentiel que le marquage des dispositifs médicaux ne soit pas de pacotille. On sait que dans certains pays, on peut choisir son certificateur ; cela n’est pas sérieux, et il convient de sécuriser encore ce dispositif.
Enfin, dernière question, très précise, concernant la pilule du lendemain : l’encadrement de la vente sur Internet est-il équivalent à certains médicaments soumis à prescription ? Autrement dit, le prix d’appel seul de la pilule est-il disponible sur Internet, à charge ensuite pour la personne d’aller l’acheter directement en pharmacie ? Ou, étant donné que la pilule du lendemain n’est pas soumise à prescription, l’achat peut-il s’effectuer directement par Internet, solution à laquelle je suis très opposée ?
Mme Marisol Touraine, ministre des affaires sociales et de la santé. Concernant tout d’abord la question de la prévention des conflits d’intérêts, l’enjeu est d’en finir avec l’ère du soupçon et de rétablir le lien de confiance entre les Français et les médicaments. En effet, cette confiance existe, mais elle est doublée de certaines inquiétudes et de certaines exigences. Il est donc important de supprimer l’idée qu’il existe de tels liens d’intérêts entre les laboratoires et les professionnels de santé : des mesures de transparence sont indispensables.
Le décret qui a été pris, dit du « Sunshine act » est issu d’un processus de réflexion. En effet, deux approches étaient possibles en la matière et qui ont d’ailleurs été en discussion au moment du vote de la loi. La première consiste à fixer un seuil au-delà duquel on considère que les avantages apportés ou les bénéfices consentis à un professionnel de santé doivent être déclarés ; en deçà de ce seuil, on considère qu’ils sont sans importance, comme par exemple, le fait d’offrir un café à un professionnel de santé. La seconde approche consiste à ne pas retenir de seuil, mais à adopter le principe de la transparence absolue, autrement dit, de dire qu’il n’y a aucune suspicion, que les relations entre les industriels et les professionnels de santé sont normales et n’ont pas à être interdites, mais qu’elles doivent juste être connues. C’est la première approche qui a été privilégiée par la loi, puisque celle-ci a retenu un seuil. Le Conseil d’État a préconisé de le fixer à 10 euros, considérant qu’en deçà, il n’y avait pas de seuil praticable. Il n’y a donc aucun fondement juridique dans la loi pour dire que c’est l’approche de la transparence complète qui a prévalu.
Concernant le pilotage du système sanitaire par l’ANSM, mon prédécesseur, M. Xavier Bertrand avait en effet mis en place un groupe de suivi de l’application de la loi, qui n’a tenu qu’une seule réunion en mars 2012. Ce comité de suivi n’était en réalité qu’une modalité du suivi de la loi, il n’était pas issu d’une prescription réglementaire. J’ai pour ma part opté pour un système de réunions informelles hebdomadaires, qui dressent un bilan régulier de la mise en œuvre de la loi. Ce que l’on peut dire aujourd’hui, c’est que les textes indispensables ont été pris ou sont en voie de l’être, autrement dit, sont en cours de finalisation. Le directeur général de l’ANSM est par ailleurs très régulièrement reçu par moi-même et mon cabinet.
Concernant le fonctionnement de l’ANSM, sa réorganisation est relativement récente et les progrès sont encore en cours, d’autant qu’elle a concerné près de 800 agents. L’Agence est bien prioritaire au regard des enjeux de la politique ministérielle. Un travail de simplification doit être mené, pour déboucher sur une meilleure articulation notamment avec la HAS, car même les professionnels de santé trouvent que l’articulation entre les deux instances n’est pas suffisamment claire.
Concernant la base de données prévue par l’article 8, elle a pour objet de renforcer l’information de nos concitoyens : elle doit procurer une information fiable et indépendante, mais n’a pas vocation à se substituer aux bases commerciales existantes. Elle sera publiée en octobre sur le site du ministère et on y trouvera des alertes de sécurité. Le principe qui a été retenu est celui d’une mise en ligne très rapide de cette base de données, qui a vocation à être améliorée en permanence, plutôt qu’une mise en ligne retardée pour une base de données plus complète. J’ajoute que l’information relative aux dispositifs médicaux devra en effet y figurer à terme.
Sur le thème des vaccins, je suis d’accord avec Catherine Lemorton et n’ai rien à ajouter.
Concernant la visite médicale, le président du CEPS doit aujourd’hui prendre en compte le nouvel objectif de la charte, qui fixera des objectifs annuels chiffrés d’évolution des pratiques. Un cycle de négociations a été engagé avec les industries du médicament. Une charte existe déjà depuis 2004 : un avenant doit donc intervenir sur ce point d’ici la fin de l’année. Indépendamment de cette révision de la charte, je pense en effet qu’il convient de réformer la visite médicale elle-même : l’article que nous avions présenté en projet de loi de financement de la sécurité sociale pour 2013 a été censuré comme cavalier social par le Conseil constitutionnel : il faudra y revenir.
S’agissant de l’accès aux données de santé, c’est bien entendu un sujet majeur. Nous devons aller vers un meilleur partage des données de santé dans l’intérêt de la sécurité sanitaire, et de la santé en général. Mais les règles doivent être précisément posées. Il faut éviter que tout cela soit d’un accès trop facile, car il y a un enjeu de confidentialité qui doit être respecté. J’attends donc bien sur ce point les conclusions de la mission confiée à Pierre-Louis Bras.
La certification des logiciels d’aide à la prescription doit être opérationnelle au plus tard le 1er janvier 2015 ; le processus se déroule de façon satisfaisante. Il n’y a pas de raison ni d’indices montrant que ces logiciels poseraient problème, en revanche, s’agissant d’une simple aide à la prescription, les problèmes éventuels peuvent venir d’une mauvaise maîtrise de ces outils par leurs usagers.
La présidente m’a interrogée sur les informations que l’ANSM peut demander aux laboratoires postérieurement à la délivrance de l’autorisation de mise sur le marché d’un médicament : le décret relatif à la pharmacovigilance qui introduit ces études « post-AMM » a été publié le 8 novembre 2012, l’Agence peut donc d’ores et déjà les demander. Elle aura le pouvoir d’infliger des sanctions qui seront fixées dans le cadre d’un décret en cours d’élaboration.
Concernant la notice des médicaments, dans le décret de novembre 2012 sur la pharmacovigilance, il est prévu que les notices des médicaments invitent expressément les patients à signaler eux-mêmes des effets indésirables. Il faudra toutefois simplifier le dispositif prévu pour ce faire.
J’ai demandé au niveau européen la mise en place d’une procédure d’autorisation de mise sur le marché pour les dispositifs médicaux les plus à risque, le renforcement de leur évaluation par les organismes notifiés ainsi que l’encadrement de ces organismes, et la mise en place des règles de transparence. En effet, en matière de dispositifs médicaux, certaines affaires récentes nous ont montré que la transparence de l’information était nécessaire.
M. Gérard Bapt. La loi dont nous suivons l’application vise à sécuriser l’usage des médicaments afin d’éviter que ne se reproduisent certains scandales sanitaires, mais aussi à restaurer la confiance de l’opinion publique en notre système de santé. La notion d’indépendance, sur laquelle la ministre a insisté, est très importante. La question des conflits d’intérêts est traitée très sérieusement par l’administration de la nouvelle Agence. En revanche, la transparence reste à assurer. La remise en question de la façon dont a été traité le règlement intérieur quant à la transparence sur les données d’études qui fondent les décisions montre que la mise en place de nouvelles structures ne suffit pas à changer les habitudes. Les salariés de l’Agence ont été traumatisés par la crise du Mediator, et les demandes de moyens supplémentaires du conseil d’administration méritent d’être entendues dans un contexte d’élargissement des missions de l’agence.
Concernant ce que l’on appelle le « Sunshine Act » à la française, c’est-à-dire la transparence sur les cadeaux, je comprends que le seuil de 10 euros apparaisse raisonnable, étant donné les contraintes posées par le Conseil d’État. Reste la question des leaders d’opinion qui, eux, ne sont soumis à aucune obligation de transparence lorsqu’ils passent des conventions avec l’industrie pharmaceutique. La présidente Catherine Lemorton et moi-même souhaitons proposer une amélioration législative sur ce point.
La transparence concerne aussi, comme vous l’avez souligné, l’accès aux données de santé, qui soulève deux questions distinctes : la possibilité pour les chercheurs de travailler sur des grands programmes de santé publique, et la surveillance en temps réel de la délivrance des médicaments. Sur ce sujet, la Caisse nationale d’assurance maladie est en conflit avec certaines grandes entreprises dont on retrouve le nom dans la présentation par la Caisse de l’évolution récente des prescriptions des contraceptifs oraux combinés. Je me suis exprimé au conseil d’administration de la Caisse en m’étonnant que certains médias aient déjà abandonné leur cheval de bataille contre les pilules de troisième et quatrième générations et soient passés à la promotion des œstradiols dits naturels.
De façon générale, la Haute Autorité de santé et l’ANSM devraient être particulièrement vigilantes sur les pratiques collectives de prescription déviantes.
Par ailleurs, j’ai été indigné, ce matin, en apprenant la façon dont, par des compromis secrets, le laboratoire Servier cherche à dissuader un certain nombre de victimes du Mediator d’aller au pénal. Cette possibilité de compromis entre le laboratoire et les victimes, qui avait été refusée à l’unanimité par les associations et par le Gouvernement de l’époque, ne peut pas être reproduite aujourd’hui dans des clauses secrètes. Il s’agit là d’un abus de faiblesse de la part de Servier. Le Gouvernement entend-il agir pour faire cesser ces pratiques ?
M. Elie Aboud. Je souhaiterais saluer le combat de la présidente qui a fait un travail exceptionnel sur l’amélioration de la sécurité du médicament. Je remercie aussi Madame la ministre de reconnaître que cette loi, votée par la précédente majorité, est intéressante. Vous dites aussi qu’elle a des limites, ce qui est vrai. Elle nécessitait plus de trente-cinq décrets, dont certains n’ont pas encore été publiés ; néanmoins, il faut avant tout laisser cette loi vivre et produire ses effets.
Concernant ce que vous appelez le premier pilier, l’ANSM : les relations avec les agences européennes sont-elles transparentes ? On a appris que certains praticiens français signent des conventions pour participer à des études réalisées dans d’autres pays européens, cela mérite une clarification.
S’agissant du deuxième pilier, la pharmacovigilance, on constate aujourd’hui que, dans les hôpitaux, les utilisations hors du cadre des autorisations de mise sur le marché sont de plus en plus fréquentes et échappent au contrôle attentif des agences. Par ailleurs, on parle des médicaments mais pas des prothèses : là aussi, il y a des conflits d’intérêts plus que visibles.
Le troisième pilier concerne les études cliniques et la transparence : je me félicite que désormais les études cliniques soient réparties sur tout le territoire et ne puissent plus être centralisées par certains patrons.
Concernant le cinquième pilier et la vente sur Internet, je m’inquiète sur ses conséquences en termes de surconsommation médicale.
Enfin, s’agissant de la formation, les visiteurs médicaux ont payé cher alors qu’ils n’étaient que la partie émergée de l’iceberg. Or, un tiers des généralistes se formaient grâce à ces visiteurs médicaux. Le système des visites regroupées dans les hôpitaux ne fonctionne pas bien. Cette question reste donc à approfondir.
M. Francis Vercamer. Le renforcement de la sécurité du médicament pose la question de la confiance de nos concitoyens dans notre système de santé. La France fait partie des pays les plus consommateurs de médicaments. Les patients sont à la fois surexposés et surinformés. La loi n’est pas encore parvenue à apporter une réponse d’ensemble à ces problèmes.
Pour assurer l’effectivité du contrôle des produits de santé, l’agence dispose-t-elle de moyens suffisants et en a-t-elle une visibilité pluriannuelle ? Nous avons besoin d’une agence qui anticipe les évènements. Comment entendez-vous développer et renforcer les évaluations post-autorisation de mise sur le marché ?
La confiance nait de la lisibilité. Le groupe centriste avait insisté sur la nécessité de réduire le nombre d’intervenants dans la régulation de notre système de santé. Ne pourrait-on pas opérer des regroupements entre l’ANSM et la Haute Autorité de santé ?
La qualité de l’information est aussi un élément déterminant de la confiance de nos concitoyens. À quelle date la base de données chère à la présidente Catherine Lemorton verra-t-elle le jour et comment cela va-t-il se passer pour ceux qui utilisent l’automédication : comment vont-ils pouvoir utiliser cette base de données ?
Le rôle du médecin dans la prescription est incontournable. Comment comptez-vous développer la formation continue ?
La baisse du prix des médicaments est-elle compatible avec l’amélioration de la sécurité ? C’est la chaîne de distribution qui est la plus impactée par ces économies.
Enfin, quel est le rôle de l’ANSM dans la contrefaçon, et comment collabore-t-elle avec les services des douanes ?
M. Jean-Louis Roumegas. Le groupe écologiste avait pris l’initiative d’un débat dans l’hémicycle sur la sécurité sanitaire du médicament le 26 février dernier ; vous connaissez donc notre vigilance sur les questions de santé publique, sur la précaution, l’indépendance de l’expertise et les conflits d’intérêts.
Au vu des mesures que vous aviez alors annoncées, il nous reviendra de mesurer le chemin parcouru lors de l’examen des textes budgétaires cet automne et lors du projet de loi de santé publique, tant attendu.
Vous avez souhaité que la politique de sécurité sanitaire du médicament repose sur un triptyque, je vous cite « vigilance, transparence, confiance ». Nous avons partagé vos constats : le système de pharmacovigilance est compliqué, peu lisible ; son organisation est morcelée ; les règles sont mal connues des professionnels de santé ; les alertes ne sont pas toujours prises en compte ; les prescripteurs sont insuffisamment impliqués dans leur rôle de vigie en matière de sécurité sanitaire.
Le dispositif d’information sur le médicament repose principalement sur le secteur privé. Les stratégies industrielles interdisent parfois aux patients l’accès à certains médicaments ou entraînent la délocalisation des fabrications.
Enfin vous avez relevé l’insuffisante association des patients à la pharmacovigilance, à l’encontre des principes de la démocratie sanitaire.
Nous saluons donc les décrets que vous venez d’annoncer et celui du 21 mai 2013 qui constitue un « Sunshine act » à la française.
Quels moyens de contrôle seront mis en œuvre, assortis de quelles sanctions ?
Il convient en outre de prendre les décrets relatifs à la commission nationale de déontologie pour l’expertise indépendante, prévus par la loi du 3 avril 2013 relative à l’indépendance de l’expertise en matière de santé et d’environnement et à la protection des lanceurs d’alerte, dont j’ai eu l’honneur d’être le rapporteur.
Je souhaiterais également vous entendre sur la mobilisation des patients contre le vaccin DT-Polio contenant des sels d’aluminium. Certains, Madame la Ministre, font la grève de la faim devant votre ministère depuis plus d’un mois : ils souhaitent seulement que vous les entendiez. Je ne comprends pas que vous ne les ayez pas reçus après trente jours. Lors de la campagne présidentielle, vous aviez pourtant exprimé des doutes sur les adjuvants à base de sels d’aluminium et, faisant état de nombreuses études scientifique, vous aviez déclaré que « les familles doivent avoir le droit de faire procéder à des vaccinations obligatoires par des vaccins sans sels d’aluminium, ce qui était le cas jusqu’en 2008 ». Je ne comprends donc pas la fin de non-recevoir que vous opposez à une revendication pourtant modérée : il s’agit seulement de demander le libre-choix pour les patients et non la suppression des sels d’aluminium. Vous invoquez l’attente d’un rapport sur le sujet ; pourtant il suffirait, sans attendre, de mettre à la disposition du public un vaccin qui était d’utilisation très large jusqu’en 2008 lorsque Sanofi a décidé de reprendre les vaccins DT-Polio. Le libre choix peut donc être accordé aux familles bien avant la publication de ce rapport qui se prononcera sur l’opportunité d’interdire les sels d’aluminium. Il me semble que nous ne pouvons pas laisser l’industrie pharmaceutique décider seule en la matière.
Mme Véronique Massonneau. Le but principal de l’ANSM est de surveiller et de prévenir les risques et les dysfonctionnements tels que ceux occasionnés par l’utilisation détournée, hors-AMM, du Mediator ou de la Diane 35 notamment.
Je m’interroge sur les caractéristiques communes des acheteurs des médicaments dont l’utilisation était détournée : il s’agissait selon les cas majoritairement ou exclusivement de femmes. J’ai déjà pu souligner que la problématique des essais cliniques sur des femmes se pose, alors que les personnes sur qui les essais sont pratiqués sont majoritairement des hommes, pour des raisons économiques et parfois physiologiques. C’est donc plus particulièrement pour les femmes que les conséquences de l’insuffisance des essais cliniques peuvent être dramatiques.
Comment l’ANSM peut-elle donc agir en matière de surveillance des essais cliniques et en ce qui concerne les différences physiologiques entre femmes et hommes. Est-il possible d’aller plus loin que la législation actuelle ? À l’échelle européenne, quels sont les résultats du Conseil européen du 28 juin dernier qui devait étudier la question des essais cliniques ?
M. Jean-Louis Touraine. Ma question s’inscrit dans le droit fil de l’intervention de Gérard Bapt. Quelle est la perspective d’un système de pharmacovigilance en temps réel ? Celui-ci est possible ? Il suppose un accès ouvert aux données de santé. La CNIL semble favorable à une telle ouverture. Quelle est la position du ministère et quelle serait celle de l’assurance maladie ? L’enjeu me semble important.
Mme Marisol Touraine, ministre des affaires sociales et de la santé. Sur les données de santé, en réponse aux questions de MM. Bapt et Vercamer, il convient de distinguer deux processus différents.
Un premier sujet concerne la mise à disposition de données dans un but d’intérêt collectif à des acteurs publics. C’est ce qui fait l’objet de la mission de Pierre-Louis Bras qui doit définir comment des acteurs publics pourront, de façon encadrée, utiliser des données qui sont aujourd’hui celles de la CNAM.
Vous avez, M. Bapt, évoqué le deuxième sujet qui renvoie à la pharmaco surveillance des prescriptions. Comment, à partir des données de l’assurance maladie, peut-on suivre les données collectives et en tirer un certain nombre d’enseignements en temps le plus réel possible ? J’avais mis ce point en avant lorsque nous nous sommes rendus compte de la prescription massive de Diane 35, en dehors de son autorisation de mise sur le marché, comme pilule contraceptive et non pas comme anti-acnéique. Ceci était apparu lorsqu’ont été rendues publiques les informations montrant que les pilules de troisième et de quatrième génération étaient massivement prescrites en première intention au lieu de l’être en deuxième intention, conformément aux recommandations. J’avais donc commandé une mission aux professeurs Bégaud et Costagliola : leur rapport me sera remis dans les prochains jours.
Sur la question des conventions, madame la Présidente, je me rallie à vos conclusions : de nouvelles dispositions législatives sont nécessaires pour que soient publiés le montant des conventions et des avantages consentis sur leur fondement. La loi prévoit la publication du principe et de la nature des conventions, mais pas leur montant. La définition législative est très précise : une nouvelle disposition législative est donc nécessaire.
Sur le Mediator, la clause litigieuse qui permettrait au laboratoire Servier de tenter d’obtenir un accord en amont avec certaines victimes a bien été signalée. Elle fait l’objet d’une saisine de la direction des affaires juridiques du ministère. Je serai très vigilante pour que la procédure spécifique mise en place à l’Office national d’indemnisation des accidents médicaux (ONIAM) afin d’indemniser les victimes du Mediator ne soit pas détournée par le laboratoire. Il ne faut pas que de nouveaux obstacles apparaissent sur la voie de l’indemnisation des victimes.
M. Aboud, concernant les essais cliniques, vous vous interrogiez sur les liens entre l’ANSM et l’agence européenne. Tout essai clinique conduit en France y est autorisé par l’ANSM. Tout essai clinique réalisé dans un pays européen est publié sur le site de l’agence européenne. Les mécanismes de transparence sont donc bien présents, aux deux niveaux. Je suis attentive à ce qu’il n’y ait pas de rupture entre ces deux niveaux de responsabilité.
Pour ce qui est de la visite médicale, vous avez parfaitement raison de souligner que la visite médicale collective à l’hôpital ne fonctionne pas. Dans le cadre de l’examen du dernier projet de loi de financement, j’avais donc, avec de nombreux parlementaires, souhaité l’adoption de mesures permettant de définir un autre processus ; mais cette disposition a été considérée comme un cavalier social par le Conseil constitutionnel. Il nous faudra donc trouver un autre vecteur pour mettre en œuvre des mesures nouvelles de sécurité sanitaire.
Concernant les dispositifs médicaux, je vous confirme que j’ai la volonté d’encadrer la mise sur le marché de dispositifs médicaux implantables. Il s’agit de bien les distinguer des autres dispositifs médicaux sans empêcher l’innovation médicale à laquelle nos concitoyens doivent accéder dans de bonnes conditions. J’ai engagé une démarche au niveau européen, isolée au départ et qui a rallié de nombreux soutiens. Je souhaite que les dispositifs médicaux soient à terme traités comme les médicaments sur les sites d’information mis en place par les autorités publiques.
M. Vercarmer, vous m’avez interrogé sur les moyens accordés à l’ANSM pour garantir son bon fonctionnement. Si les moyens de fonctionnement des autres opérateurs sont en diminution, ce n’est pas le cas pour cette agence. La réorganisation a été engagée : il convient également de continuer à rendre plus lisible les relations entre l’ANSM et la Haute autorité de santé.
Concernant les relations entre l’agence et les services des douanes: en application de la directive sur les médicaments falsifiés, une convention reliant l’agence et les douanes a été signée le 8 juillet dernier. Les liens sont donc renforcés dans l’objectif, si important à mes yeux, de lutte contre la contrefaçon.
M. Roumegas, vous m’avez demandé de préciser les sanctions appliquées en cas de non-respect des dispositions du Sunshine act : la non publication expose les entreprises à une amende de 45 000 euros ; les personnes physiques peuvent également faire l’objet de sanctions. Le contrôle relève de la direction générale de la concurrence, de la consommation et de la répression des fraudes : les sanctions seront mises en place à partir du 1er octobre prochain.
Mme Marisol Touraine, ministre des affaires sociales et de la santé. Un certain nombre de textes a déjà été pris, mais j’ai indiqué que nous devions aller plus loin, parce que nous nous trouvons dans un domaine dans lequel les progrès et les évolutions sont constants. La crise appelle certes la réaction, mais nous ne devons pas attendre les crises pour mettre en œuvre de nouveaux mécanismes. J’ai ainsi commandé des études qui nous permettront d’avancer concrètement sur la pharmaco-surveillance et la prescription collective.
S’agissant de la transparence, au moment de « l’affaire de la pilule », les autorités n’avaient pas pour habitude de diffuser des informations sur le nombre de prescriptions : il n’existait pas de réelle culture de la transparence. J’ai donc demandé à l’Agence nationale de sécurité du médicament de réaliser désormais un point presse mensuel donnant l’évolution des données. Le premier point presse a suscité d’importantes réactions et interrogations, mais il est désormais considéré comme un processus normal de partage de données, qui doivent être soumises au public et pouvoir être analysées.
Un chemin important a donc été parcouru, mais il reste des évolutions à engager, dont certaines doivent passer par le vote de lois et d’autres par la mise en place de dispositifs de politiques publiques ministérielles.
Sur les vaccins sans sels d’aluminium, une première grève de la faim a eu lieu il y a quelques mois, dont les protagonistes ont été reçus et ont exigé la mise en place d’un groupe de travail. Ils affirment aujourd’hui que ce groupe n’a pas été réuni, ce qui justifie à leurs yeux la reprise de leur grève. Or, ils ont été reçus à de multiples reprises par les services du ministère et par mon cabinet, et le groupe de travail a été constitué. D’ailleurs, les acteurs qui souhaitent évaluer la possibilité de remettre sur le marché des vaccins sans sels d’aluminium, ont demandé la réalisation d’études. Un comité de pilotage a donc été mis en place pour financer un projet de recherche spécifique, qui sera mené par le professeur Roman Ghirardi.
Les vaccins sans sels d’aluminium n’existent nulle part en Europe. Pourquoi ont-ils été retirés du marché ? Parce que des effets secondaires très graves ont été constatés dans un nombre limité de cas, ce qui a conduit à s’interroger sur leur maintien. Les autorités sanitaires européennes et françaises et l’Académie de médecine considèrent aujourd'hui qu’il n’y a pas d’éléments concluants sur l’existence d’un risque lié à l’adjuvant aluminique dans les vaccins. Je pense, cependant, qu’il faut poursuivre les recherches à ce sujet, et obtenir également des garanties sur le fait qu’un autre vaccin ne comporterait pas de risques, qui soient quantitativement ou qualitativement supérieurs. Les industriels devront, par ailleurs, solliciter une autorisation de mise sur le marché.
J’insiste sur la dimension européenne de cette réflexion aujourd'hui. Je ne veux pas laisser croire que la ministre de la santé a la capacité de décider seule de la mise sur le marché de ces vaccins : au contraire, ma responsabilité est de commander des études pour évaluer ces produits, puisque des doutes subsistent. Par principe, je suis favorable au choix des vaccins sans sels d’aluminium, mais les rapports dont nous disposons nous conduisent à nous interroger. Je n’oppose pas une fin de non-recevoir, contrairement à ce que vous prétendez : nous finançons de nouveaux projets de recherches.
Mme Massonneau m’a demandé pourquoi les essais cliniques étaient insuffisants chez les femmes. Il s’agit d’une vraie préoccupation mais, en même temps, cette situation de fait s’explique : s’il y a plus d’hommes dans les essais cliniques, c’est parce que l’on ne peut pas exclure, dans les essais, l’impact du médicament testé sur une grossesse. Or la plupart des essais cliniques se déploient sur plusieurs années et de nombreuses femmes se retirent en cours de protocole, lorsqu’elles attendent un enfant, ce qui coûte cher aux commanditaires. Il y a là un vrai enjeu : davantage de femmes doivent être représentées dans ces essais et nous devons réfléchir à la promotion du financement de projets incluant des femmes, car ceux-ci présentent des coûts supérieurs par rapport à ceux uniquement effectués sur des hommes.
M. Jean-Pierre Barbier. Pourriez-vous nous apporter un éclairage sur la question de la contraception d’urgence et de sa vente sur Internet, car je n’ai pas entendu votre réponse à cet égard ?
Mme Marisol Touraine, ministre des affaires sociales et de la santé. À l’origine, j’avais écarté la contraception d’urgence de la liste des médicaments qui pouvaient être vendus sur Internet, puis les produits concernés ont été réintégrés par le Conseil d’État, au motif qu’il s’agissait de produits non soumis à prescription.
La première liste que j’avais établie était beaucoup plus restrictive que celle qui a été finalement publiée. Mais nous sommes face à des textes européens qui ont été transposés en droit français. Toutefois, Internet ne me semble pas un moyen adapté à une situation d’urgence : un jour, c’est un jour. Or commander en ligne prend du temps. De deux choses l’une : soit vous recevez le produit chez vous, et vous vous trouvez hors délais, soit vous allez le chercher dans une pharmacie et, dans ce cas, vous bénéficiez d’un conseil médical de même nature que si vous vous rendiez directement dans une officine.
Nous sommes, de plus, face à une réalité qui existe en dehors d’Internet : une jeune femme peut aller de pharmacie en pharmacie et stocker chez elle des boîtes de contraception d’urgence. Je comprends votre interrogation et je la partage : nous serons donc vigilants à la mise en place de la vente de médicaments par Internet, du point de vue de la sécurité sanitaire, et si des problèmes étaient identifiés, nous étudierions comment y répondre.
La Commission examine le rapport de Mme Catherine Lemorton et M. Arnaud Robinet sur la mise en œuvre de la loi du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé au cours de sa séance du mercredi 17 juillet 2013.
M. Christian Hutin, président. Nous avons tout d’abord à examiner le rapport sur la mise en œuvre de la loi du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé présenté par Mme Catherine Lemorton et M. Arnaud Robinet. Ce texte a une particularité : adopté par l’ancienne majorité, il revient à la nouvelle majorité de le mettre en œuvre.
Lors de notre séance d’hier soir nous avons entendu Marisol Touraine, ministre des affaires sociales et de la santé, sur la mise en œuvre de cette loi et cette audition a constitué une excellente introduction à nos débats d’aujourd’hui.
Madame la Présidente, je vous donne la parole.
Mme Catherine Lemorton, rapporteure. C’est un plaisir en effet d’avoir travaillé avec M. Arnaud Robinet sur le rapport d’application, comme lors de l’examen de la loi, qui avait été un exemple de co-production législative fructueuse, même si certains dispositifs manquaient, selon nous, au texte à l’époque. Nos travaux avaient duré près de trois mois et nous n’avions jamais perdu de vue le cœur de la réforme : l’intérêt et la sécurité du patient après le drame du Mediator.
Nous interviendrons conjointement, M. Arnaud Robinet et moi-même, pour vous présenter un premier bilan de l’application de la loi relative à la sécurité du médicament et des produits de santé du 29 décembre 2011.
Pour réaliser ce rapport, nous avons mené de larges auditions, au cours desquelles nous avons reçu les représentants des professionnels du secteur et de ses usagers, les administrations, l’ancien ministre de la santé dont la loi porte aujourd’hui le nom, et madame Marisol Touraine qui nous a hier soir éclairés sur l’état d’application de la loi.
Je rappelle que le précédent Gouvernement avait eu quatre mois pour publier les premiers textes d’application de la loi. Il est donc revenu à la nouvelle ministre des affaires sociales et de la santé d’en reprendre le cours après les élections de mai et juin 2012.
Ce rapport porte principalement sur l’application réglementaire de la loi. L’exercice, certes formel, a la vertu d’accélérer l’élaboration des textes et leur signature par le pouvoir exécutif. Cependant, plus d’un an après la promulgation de la loi, il est d’ores et déjà possible d’examiner les conditions d’application et la pertinence de certaines dispositions ; les lacunes à combler ; le chemin qu’il reste à parcourir pour atteindre l’objectif premier de la loi : prévenir les drames sanitaires et restaurer la confiance des français dans leur système sanitaire. En effet nos compatriotes ont une attitude pour le moins paradoxale : grands consommateurs de médicaments, ils se défient, dans le même temps, de leur système sanitaire.
Pour mémoire, la loi de 2011 nécessitait la publication de 29 décrets, 21 ont déjà été publiés, dont 8 par l’ancien Gouvernement, 13 par le nouveau. 7 textes sont encore en attente de publication.
Le premier axe de la loi portait sur la prévention des conflits d’intérêts et la transparence sur les liens entre entreprises pharmaceutiques et professionnels au sens large, au-delà des médecins. Seules l’indépendance et l’impartialité fondent le crédit de l’expertise sanitaire. C’est pourquoi nous devons accorder à cette question une attention particulière.
L’article 1er prévoit la rédaction d’une charte de l’expertise sanitaire, qui ne constitue pas en soi une avancée majeure. Il généralise surtout la déclaration d’intérêts détaillée et rendue publique pour les professionnels des autorités et organismes sanitaires.
Si les autorités concernées ont bien mis en place un système de déclaration publique d’intérêts, le choix de prévoir deux modalités de déclaration ne facilite pas le traitement des données. De plus le site Internet regroupant les déclarations n’est pas encore créé, la ministre l’ayant annoncé pour 2014. Il est indispensable qu’il soit mis en place rapidement. D’autres points méritent toute notre attention.
Ainsi, les montants des rémunérations perçues ou des participations financières de certains professionnels dans des entreprises ne sont pas accessibles au public. Nous proposons de modifier la loi pour que ces informations deviennent accessibles. De même, il nous faudrait réfléchir plus sérieusement aux liens entre les étudiants et les entreprises pharmaceutiques. Il n’est pas normal qu’ils soient encore si étroits et nous pourrions envisager de modifier la loi pour mieux les encadrer et les limiter. Pourquoi ? On le sait, plus tôt les liens sont établis, plus les étudiants seront soumis à l’influence prégnante des industries pharmaceutique. S’y ajoute la création de filières d’études par l’industrie pharmaceutique. Je ne suis pas opposée à ces liens, mais il convient d’établir toute la transparence nécessaire sur leur nature.
M. Arnaud Robinet, rapporteur. L’article 2 de la loi prévoyait la mise en œuvre d’un Sunshine Act à la française : toute les conventions passées entre entreprises pharmaceutiques et professionnels de santé, éditeurs de logiciels d’aide à la prescription ou associations d’usager, doivent être rendues publiques, de même que les avantages accordés par les entreprises, au-delà d’un certain seuil.
Nous avions clairement évoqué au cours des débats un seuil symbolique de un euro. Le Conseil d’Etat a jugé que ce montant était insuffisant pour juridiquement constituer un seuil. Le décret du 29 mai 2013 l’a fixé à 10 euros. Nous pensons que les petits cadeaux influencent considérablement la prescription : si nous voulons changer les cultures, nous devons mettre en place un système véritablement dissuasif. Nous proposons donc de modifier la loi afin de supprimer la référence à un seuil minimal de publication des avantages. Ceci pour être en parfait accord avec l’esprit de la loi dite « Bertrand ».
Par ailleurs, la rédaction de l’article 2 ne permet pas la publication de l’objet exact des conventions passées entre les entreprises et les professionnels, ni leur contenu. Les conventions conclues dans le cadre des relations commerciales qui ont pour objet l’achat de biens ou de services ne sont pas non plus dans le champ. Sur ces deux points, la loi doit changer.
Le deuxième axe de la loi portait sur la gouvernance du système sanitaire, plus particulièrement la création d’une nouvelle agence clairement identifiée comme le contrôleur des produits de santé. L’Agence nationale de sécurité du médicament et des produits de santé (ANSM) a remplacé l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) en mai 2012. Il faut saluer l’immense travail de reconfiguration de l’Agence effectué par la direction et l’ensemble de son personnel et que les parlementaires membres du conseil d’administration, dont je suis, ont pu constater, notamment en termes de prévention des conflits d’intérêts. Il faut noter aussi que l’augmentation de ses missions est un véritable défi pour l’ANSM, doublé d’un changement de culture.
Il nous faut veiller à ce que le contrat de performance signé entre l’État et l’ANSM lui accorde les moyens nécessaires pour remplir ses nouvelles missions. Le financement de la nouvelle agence par le budget de l’État, acté par le précédent Gouvernement, ne doit en aucun cas se traduire par une réduction de son budget au détriment de la sécurité sanitaire.
Dans le cadre général d’une rationalisation de l’architecture des agences sanitaires, il serait souhaitable de trouver des voies de simplification des procédures administratives et de resserrement des compétences de l’ANSM pour qu’elle puisse se concentrer sur ses missions fondamentales.
Enfin, point de détail qui a son importance, il convient de publier dans les meilleurs délais le texte prévoyant les pouvoirs de sanction de l’ANSM en cas de non-respect par les entreprises de leurs obligations en matière de pharmacovigilance et de surveillance post-AMM.
Mme Catherine Lemorton, rapporteure. Cette dernière remarque m’amène à la pharmacovigilance qui est évidemment un axe fondamental de la loi de 2011. Notre système de veille sanitaire et de pharmacovigilance présentait de graves défaillances, souvent par méconnaissance du système. Un changement de méthodes, de culture s’imposait. Que ce soit la crise du Médiator ou celle des prothèses PIP, les deux ont mis en évidence l’impérieuse nécessité de mieux garantir la qualité et la sécurité des produits de santé.
Parmi les apports de la loi, on peut souligner la transposition de la directive communautaire de 2010, la réévaluation par la nouvelle agence des médicaments tout au long de leur vie. Nous avons donc inscrit dans la loi la nécessité d’évaluer les médicaments tous les cinq ans. Citons aussi le renforcement du système de notification des effets indésirables et la protection des lanceurs d’alerte. Il faut enfin noter l’amorce de contrôle des dispositifs médicaux.
L’application de cette partie du texte appelle les remarques suivantes.
Tout d’abord, concernant la mise sur le marché et la réévaluation des médicaments tout au long de leur vie, nous avons introduit à l’article 14 le critère d’une véritable valeur ajoutée du produit dans son évaluation, car l’encombrement thérapeutique et la multiplication des médicaments sur le marché ne sont pas synonymes de meilleur traitement pour les patients. Nous attendons la publication du décret d’application de cet article, ainsi que des précisions sur son articulation avec l’évaluation médico économique des médicaments. J’en profite pour mentionner le fait que l’articulation entre les décisions relatives au service médical rendu d’un médicament et son remboursement devrait être améliorée et les délais réduits. Je pense ici aux médicaments anti-Alzheimer, dont la dégradation du service médical rendu n’a entraîné une baisse de prix que trop tardivement.
Il faut aussi mettre en place un régime plus strict de mise sur le marché et de contrôle des dispositifs médicaux, plus particulièrement des dispositifs médicaux implantables. La ministre a annoncé hier son intention de plaider auprès des institutions européennes pour la création d’une autorisation de mise sur le marché pour les dispositifs médicaux à risque. Nous soutenons pleinement cette démarche.
Deuxième point : l’accès aux données de santé, condition sine qua non d’une pharmacovigilance efficace. Il convient tout d’abord de publier l’arrêté nécessaire à l’accès direct de l’ANSM aux données de l’assurance maladie pour permettre le lancement effectif de son programme de travail.
Concernant le GIP « Études de santé », prévu par l’article 33 de la loi, pour produire des études de pharmaco-épidémiologie, il faut étudier la possibilité d’étendre les prérogatives de l’Institut des données de santé (IDS), dont la gouvernance est plus large et inclut des membres de la société civile. Ce GIP est peut être redondant par rapport à l’IDS. La ministre nous a déclaré hier attendre les conclusions de la mission de M. Bras pour prendre une décision. En tout état de cause, seuls des besoins de santé publique et de sécurité sanitaire, et une meilleure prise en charge des patients peuvent justifier l’accès des acteurs privés aux données de l’assurance maladie, accès qu’il faut encadrer avec la plus grande prudence.
Dernière remarque concernant la pharmacovigilance. Certes les textes comptent, mais c’est la pratique qui doit aussi changer. A ce titre, il faut simplifier les modalités de déclaration des effets indésirables, en rappelant que tous les effets, y compris ceux visés par la notice, doivent être signalés, et surtout garantir un véritable retour d’information aux notificateurs. Il faut également améliorer la lisibilité des notices et y mentionner les voies et moyens de notification des effets indésirables autres que la déclaration aux médecins et pharmaciens. Enfin, il convient de renforcer la communication entre les centres régionaux de pharmacovigilance et l’ANSM.
M. Arnaud Robinet, rapporteur. Le meilleur usage des médicaments passe par la formation et l’information des professionnels et des patients. Qu’il s’agisse de publicité, de visite médicale, d’information publique sur le médicament, de prescription en dénomination commune internationale ou de certification de logiciels, toutes ces mesures contribuent à orienter notre système sanitaire vers davantage d’objectivité et de prudence.
Leur mise en œuvre appelle les remarques suivantes.
Tout d’abord, nous attendons encore plusieurs textes d’application. C’est le cas du décret relatif à la certification des logiciels d’aide à la prescription et à la délivrance. Je rappelle qu’elle sera obligatoire à compter du 1er janvier 2015. Dans la mesure où la prescription en dénomination internationale y est liée, nous insistons pour que ce texte sorte au plus vite.
C’est aussi le cas de la base de données publique créée par l’article 8, qui devrait être finalisée à la rentrée, et permettra à tous nos concitoyens, patients ou professionnels de santé de s’y retrouver dans les produits de santé. Elle devra être la plus complète possible, indiquant le prix au remboursement, les recommandations de la Haute Autorité de santé (HAS) et de l’ANSM ainsi que le service médical rendu. Nous insistons pour que les dispositifs médicaux y soient intégrés. Il faudra aussi assurer le référencement de cette base pour qu’elle devienne la principale source d’information sur les produits de santé.
Par ailleurs, la visite médicale attend toujours d’être transformée d’instrument de promotion en moyen d’information. C’est un sujet qui avait fait débat en 2011. Nous proposons de mettre en œuvre la visite médicale collective à l’hôpital telle que prévue par le projet de loi de financement de la sécurité sociale pour 2013, et d’engager une vraie réflexion sur l’information des médecins libéraux, qui en manquent parfois cruellement. Il conviendra aussi de mener une réflexion sur la profession d’attaché de promotion du médicament.
En matière de publicité pour les produits de santé, dont le régime d’autorisation est aujourd’hui plus strict, nous avons noté des difficultés de mise en œuvre : l’ANSM doit traiter un nombre exponentiel de dossiers déposés par les entreprises. Peut-être faudrait-il augmenter les droits liés au dépôt d’une demande et définir des priorités dans le traitement des dossiers, pour éviter que certains dossiers ne bénéficient d’un accord tacite sans avoir été véritablement examinés.
Quant à la publicité non institutionnelle relative aux vaccins, nous approuvons l’arrêté du 28 septembre 2012 qui en a limité la liste.
Enfin, lors de la discussion de la loi, nous avions insisté sur la faiblesse de la formation initiale et continue de certains professionnels aux problématiques des produits de santé. Nous veillerons donc à ce que le développement professionnel continu des professionnels inclue des modules relatifs aux médicaments ou à la pharmacovigilance.
Mme Catherine Lemorton, rapporteure. Pour finir, j’aimerais insister sur un sujet dont l’importance est majeure : les actions de groupe, mesure reportée et renvoyée à des textes futurs, quelle que soit la majorité. Nous avions déposé un amendement pour introduire une telle disposition dans la loi de 2011 avec mon collègue Gérard Bapt, le Gouvernement de l’époque l’avait rejeté. Le mois dernier, la ministre des affaires sociales et de la santé a annoncé que les actions de groupe concernant le domaine de la santé seraient une des mesures de la loi de santé publique que nous examinerons en 2014. Nous veillerons à ce que cette promesse soit respectée et à ce que le texte soit le plus équilibré possible, au service des patients.
M. Christian Hutin, président. La loi du 29 décembre 2011 était intervenue dans des circonstances exceptionnelles. Je me réjouis de constater que les deux rapporteurs ont travaillé dans un esprit républicain et sont parvenus, à l’heure de dresser un bilan de l’application de cette loi, à une forme de consensus.
Mme Bernadette Laclais. Le rapport que nous examinons aujourd’hui intervient à l’issue d’un long processus dans lequel le rôle de notre collègue Gérard Bapt doit aussi être salué. Le rapport traduit l’expertise qu’ont développée nos deux rapporteurs, et pour un lecteur moins aguerri, l’importance prise par la notion de transparence frappe tout particulièrement. Elle s’impose désormais dans le choix des experts et inspire aussi bien la notion de lien d’intérêts que celle de conflit d’intérêts.
Le décret dit Sunshine Act, créant une obligation de publication des liens entre les entreprises de produits de santé et de cosmétiques, et les professionnels de santé, constitue par ailleurs une avancée notable.
S’agissant de l’ANSM, de nouvelles missions lui ont été confiées, de nouvelles sanctions ont été créées et doivent être mises en œuvre : cela soulève la question des moyens financiers et humains qui doivent être dévolus à l’agence. Je tiens d’ailleurs à saluer l’engagement du personnel, qui a souffert de certaines polémiques. Il a fait preuve d’une remarquable capacité d’adaptation et de réforme.
En ce qui concerne la pharmacovigilance, gardons-nous d’une vision réductrice centrée sur le médicament, et intéressons-nous aux dispositifs médicaux.
Dans le domaine de la formation et de l’information, la base de données publique sur le médicament, qui doit se déployer à partir du mois d’octobre, devra faire l’objet d’une évaluation appropriée.
Enfin, s’agissant des visites médicales, des progrès ont été accomplis, mais il faut veiller à organiser une concertation avec les professionnels, qui se sont émus auprès des élus des réformes envisagées.
M. Jean-Pierre Door. La loi de 2011 est intervenue à la suite du cataclysme de l’affaire du Mediator. Elle a été précédée par les Assises du médicament, mais aussi par des rapports parlementaires dont la contribution au débat a été précieuse. L’immense majorité des préconisations du rapport d’information sur le Mediator et la pharmacovigilance, dont j’étais le rapporteur et qui était présidée par M. Gérard Bapt, a été reprise dans la loi de 2011.
Le Sunshine Act a constitué une avancée fondamentale. Mais la déclaration des liens d’intérêts marque également un progrès qu’il convient de souligner : plus ces déclarations seront nombreuses, plus elles réduiront les conflits d’intérêts. S’agissant du recrutement des experts, il convient d’être attentif à ce qu’ils ne soient pas complètement coupés du « terrain », des services hospitaliers et des laboratoires de recherche. J’aimerais en outre savoir ce qu’il advient des déclarations adressées aux ordres professionnels.
S’agissant de l’ANSM, notons qu’à chaque drame est créée une nouvelle agence, sous un nouveau nom. La faiblesse du lien entre l’Agence européenne, l’EMA (European Medicine Agency), et l’Agence française de sécurité sanitaire des produits de santé avait été relevée comme un dysfonctionnement ayant concouru au drame du Mediator ; l’EMA avait en effet été saisie dès 1995 des problèmes causés par le médicament. Qu’en est-il des relations entre l’ANSM et l’EMA ?
S’agissant des autorisations de mise sur le marché (AMM), elles doivent faire l’objet d’une réévaluation tous les cinq ans. Mais qu’en est-il des médicaments prescrits en hors-AMM ? Ils sont parfois utiles, et des exceptions doivent pouvoir être aménagées, à condition de faire l’objet d’une évaluation. De plus, le suivi des produits de santé en post-AMM est indispensable : il doit être conduit de manière plus sérieuse.
En ce qui concerne le groupement d’intérêt public (GIP) de pharmaco-épidémiologie, le décret qui devait consacrer sa création n’a-t-il pas souffert d’un certain retard dans sa publication ?
Les visiteurs médicaux se sont sentis légitimement concernés par la réforme des règles encadrant les visites médicales. La charte de 2004 doit être révisée, et les professionnels ont besoin de visibilité, notamment sur les évolutions susceptibles d’être occasionnées par le développement de la formation et de l’information numériques. Une directive européenne a interdit la délivrance d’échantillons gratuits ; ces échantillons n’étaient peut-être pas inutiles.
Il convient d’aborder la question du renforcement des centres régionaux de pharmacovigilance. Le sujet a-t-il été abordé au cours de l’audition de la ministre des affaires sociales et de la santé ?
Il importe également de veiller à la traçabilité des médicaments en vente sur Internet.
Enfin, s’agissant de l’action de groupe, opposition et majorité ont débattu, avancé puis fait marche arrière. Le futur projet de loi sur la santé publique doit nous permettre d’aborder de nouveau cette question.
Mme Véronique Louwagie. Il me semble que les rapporteurs ont exprimé une inquiétude quant aux moyens de l’ANSM. Avez-vous des éléments étayant cette inquiétude ? Une évaluation des besoins de l’Agence a-t-elle été réalisée ?
S’agissant de la base de données publique sur le médicament, les pays scandinaves font figure de précurseurs. Qu’en est-il des autres pays européens ? L’Union européenne a-t-elle pris des initiatives en ce sens ?
M. Jean-Pierre Barbier. Le médicament n’est pas un produit comme un autre. Je voudrais revenir sur la transparence qui entoure l’usage des médicaments. Elle est nécessaire, mais sans l’expertise des professionnels de la santé, je redoute les effets qu’elle peut entrainer. Si la base de données comporte une information sur les indications des médicaments, chacun ne risque-t-il pas de s’improviser prescripteur ? De même, si elle contient des informations sur les contre-indications, ne risque-t-on pas d’alimenter l’inquiétude des patients, qui pourraient renoncer à leur traitement ? Il en va de même des informations sur les effets secondaires.
Mme Catherine Lemorton, rapporteure. Monsieur Door, les mesures d’application relatives au GIP n’ont pas été prises : dès juillet 2012 j’avais demandé à la ministre d’expertiser le sujet et de voir si l’on ne pourrait pas faire au mieux avec l’existant. La mission conduite par M. Pierre-Louis Bras sur les données de santé va donc nous aider à identifier la meilleure piste.
Concernant le nombre de visiteurs médicaux, la tendance naturelle est à la baisse, indépendamment des mesures législatives ou réglementaires qui pourraient être prises. J’y vois trois raisons : l’arrivée des génériques rend inutile la visite médicale pour les médicaments princeps ; l’industrie pharmaceutique est aujourd’hui à la recherche de médicaments de niche, notamment en cancérologie, dont le ciblage rend inutile une information large au moyen de visites médicales ; enfin, les logiciels d’aide à la prescription vont être certifiés : le médecin pourra mieux gérer son temps d’information au moyen d’Internet ou d’une application plutôt que par la succession de visites à son cabinet.
L’industrie pharmaceutique a fait état d’un « effort » réalisé depuis 2005 avec le passage de 25 000 à 17 000 visiteurs médicaux : mais les syndicats de visiteurs médicaux confirment que cette baisse provient essentiellement d’un changement de stratégie industrielle.
Concernant le renforcement des centres régionaux de pharmacovigilance (CRPV), ceux-ci peuvent certes manquer de praticiens mais pas de petites mains : ils utilisent beaucoup les internes et externes en pharmacie et en médecine pour saisir les données relatives aux effets secondaires dont ils ont connaissance. Les tensions en matière d’effectif ne sont donc pas insurmontables, pour le moment du moins.
La vente des médicaments sur Internet procède d’une directive européenne de juin 2011 qui devait être transposée avant le 31 décembre 2012. Dans le but d’assurer la sécurité des patients, la Ministre a souhaité restreindre au maximum la liste des médicaments vendus sur Internet, en assumant le risque d’être désavouée et par les instances européennes et par les autorités de la concurrence. La Ministre a donc retenu la liste, définie sous le gouvernement précédent, des 450 médicaments d’accès libre dans les officines. Mais ce périmètre est restreint par rapport au critère retenu par la directive, qui concerne l’ensemble des médicaments vendus sans prescription médicale.
L’Union européenne a donc exigé la vente sur Internet de tous les médicaments non soumis à prescription médicale. C’est la raison pour laquelle, par exemple, la pilule du lendemain, exclue par la Ministre de la liste rendue publique en décembre dernier, figure désormais parmi les médicaments en vente sur Internet.
Je m’accorde avec mes collèges Gérard Bapt et Bernadette Laclais sur l’intérêt des actions de groupe en matière de médicament. Le médicament n’est pas, en effet, un produit comme les autres. Il s’agit d’une consommation subie : il est prescrit par un « sachant », le médecin, à un « non-sachant », le patient. Si l’on constate des effets secondaires graves imputables au médicament, l’action de groupe a toute sa pertinence.
Mme Louwagie m’a interrogé sur les bases de données publiques en matière de médicament dans les autres pays. Les pays scandinaves les ont en effet développées. Mais elles sont moins utiles chez nos partenaires européens dans la mesure où ils ne connaissent pas notre addiction au médicament. Celle-ci provient notamment d’un manque de prévention : nous avons créé un système de soin au lieu de bâtir un système de santé.
Les pilules contraceptives de troisième et de quatrième génération fournissent un exemple parlant : lorsque la France a saisi les autorités européennes pour évaluer les effets de ces pilules en cas de prescription en première intention, elle a constaté que partout ailleurs, la prescription de première intention restait circonscrite, dans la prescription collective, aux contraceptifs oraux-combinés de première ou de deuxième génération. La dérive avait donc été propre à la France.
Les modalités de décision sur l’innovation thérapeutique y sont souvent différentes : en Italie ou en Angleterre, les professionnels de santé travaillent de façon collégiale au niveau d’un territoire, d’un quartier ou d’un cabinet pour arbitrer entre la prescription du médicament de dernière innovation ou le maintien de l’existant, au demeurant souvent suffisant pour la plupart des patients.
De telles différences de comportements expliquent que les bases de données publiques sont moins attendues par nos voisins.
Concernant les informations en ligne évoquées par Jean-Pierre Barbier, je préfère une base de données publique validée par les autorités sanitaires au tout venant des informations médicales actuellement disponibles. Ceci constituera un véritable progrès. Mais rien ne vaut le contact humain avec le professionnel de santé. Je m’accorde avec vous sur le fait que ces sites devront notifier que l’interlocuteur de référence reste le praticien de santé.
M. Arnaud Robinet, rapporteur. Concernant les moyens de l’ANSM, nous avons demandé une veille sur le contrat de performance signé entre l’État et l’Agence. La mise en route de l’Agence a été difficile aussi bien pour la direction que pour le personnel. Un an et demi d’efforts et de travail ont permis à la direction et aux salariés de maintenir un dialogue de qualité et de veiller au respect du contrat avec l’État.
Il y a aujourd’hui quelques difficultés en termes d’équivalents temps plein au regard des missions assignées à l’Agence. Le contrôle a priori de la publicité représente par exemple une charge de travail considérable.
En outre les redéploiements de postes ont été nombreux, à la fois pour faire face aux nouvelles missions et pour diffuser la nouvelle « culture de l’Agence » : les mouvements internes visent ainsi à éviter que des agents qui se seraient maintenus dans les mêmes postes ne reconduisent les pratiques héritées de la précédente structure.
Après les difficultés initiales, la situation s’améliore donc, mais il convient de rester vigilant.
Concernant les Centres régionaux de pharmacovigilance (CRPV), on avait annoncé un financement supplémentaire. Il y a bel et bien eu des moyens supplémentaires, à l’exemple du CRPV de Champagne-Ardenne, mais il me semble qu’il convient de passer à une nouvelle étape par la voie de conventions avec l’ANSM, avec les agences régionales de santé (ARS) et parfois avec les centres hospitalo-universitaires (CHU), puisque ces centres ont également une mission d’enseignement.
Lors de l’examen du projet de loi en 2011, le débat a été largement centré sur la question de la transparence des liens d’intérêts afin d’éviter qu’ils ne se transforment en conflits d’intérêts. C’est pourtant l’article du projet de loi relatif aux visiteurs médicaux qui a mobilisé le plus fortement notre énergie. Nous avions alors auditionné les représentants des visiteurs médicaux qui redoutaient que la loi ne fragilise leur profession. Or, je rejoins ma collègue Catherine Lemorton : l’industrie pharmaceutique n’a pas attendu la loi de 2011 pour réduire considérablement les effectifs de visiteurs médicaux. Nombre d’entre eux s’orientent aujourd’hui vers d’autres professions.
Une nouvelle charte de la visite médicale va être mise en place par le Comité économique des produits de santé (CEPS) avec la HAS et les industries pharmaceutiques : elle devrait voir le jour en novembre 2013. Elle devrait encadrer le volume des visites médicales, qui pourrait être défini par classe thérapeutique. Elle va aborder la contribution des visiteurs médicaux au respect par les prescriptions des indications contenues dans l’autorisation de mise sur le marché du produit. Elle prévoira la mise en place d’une information scientifique et non promotionnelle : ceci rendra peut-être nécessaire de mieux définir les contours de la nouvelle profession des attachés à la promotion du médicament qui ne sont pas des visiteurs médicaux mais dont l’activité devra être regardée attentivement.
De même, la charte prévoira l’encadrement et la transparence en matière d’avantages consentis par les visiteurs médicaux auprès des professionnels : c’est un des axes majeurs de la loi sur le médicament.
Enfin, la charte doit définir la qualité de la formation des visiteurs médicaux et la rémunération des visiteurs médicaux, qui pourrait être moins quantitative et plus qualitative.
Il s’agit donc aujourd’hui d’une charte déontologique : il me semble que le législateur devra accompagner cette charte afin que la loi confère un caractère obligatoire à certaines de ses dispositions pour encadrer la visite médicale.
Enfin, en matière de transparence, j’ai récemment pu constater personnellement que le périmètre du Sunshine Act est encore incomplet. Devant préparer une intervention en vue d’un congrès pour les pharmaciens hospitaliers, j’ai dû rencontrer des représentants d’une entreprise pharmaceutique : le prix du petit-déjeuner auquel ils m’invitaient dépassant dix euros, ils m’ont remis le formulaire de déclaration à remplir en tant que professionnel de santé, puisque je suis pharmacologue et hospitalo-universitaire. Or je ne suis ni médecin ni pharmacien et n’appartiens à aucun ordre professionnel : je n’entre donc pas dans le champ du Sunshine Act bien que les missions que j’exerce au sein d’un CHU sont comparables à celles des médecins et des pharmaciens. Les scientifiques, non soumis à un ordre professionnel, lorsqu’ils occupent de telles fonctions, notamment dans la biologie, devraient être soumis aux dispositions du Sunshine Act. Il reste donc encore des choses à améliorer pour parfaire ce mécanisme.
*
La commission autorise, à l’unanimité, en application de l’article de 145-7 du Règlement, le dépôt du rapport sur la mise en œuvre de la loi du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé en vue de sa publication.
ANNEXE 1
LISTE DES PERSONNES AUDITIONNÉES
(par ordre chronologique)
Ø Table ronde réunissant :
● Fédération chimie énergie (FCE-CFDT) – M. Jean-François Chavance, délégué fédéral « pharmacie »
● Fédération nationale de la pharmacie, du cuir et de l’habillement (FNP-FO) –M. Jean-Claude Adam, secrétaire général adjoint
● Fédération nationale des industries chimiques (FNIC-CGT) –Mme Isabelle André, M. Dominique Boulay et Mme Marmar Kabir-Ahmadi, membres de la commission paritaire nationale
● Syndicat énergie chimie de l’Ile-de-France (SECIF-CFDT) – Mme Fabienne Malfroy, secrétaire générale adjointe branche pharmacie
● Union nationale des syndicats autonomes chimie pharmacie pétrole (CPP-UNSA) – M. Alain Maspataud, trésorier national, et M. Yann Maillard, visiteur médical élu au comité d’entreprise
● Fédération de la chimie CFE-CGC – Mme Isabelle Freret, secrétaire générale adjointe
● Fédération chimie mines textiles énergie CFTC – M. Nicolas Perducat, animateur adjoint de la branche Pharmacie, membre actif du Comité professionnel national de la visite médicale (organisme paritaire) et délégué hospitalier en activité.
Ø Agence nationale de sécurité du médicament et des produits de santé (ANSM) – M. Dominique Maraninchi, directeur général, M. François Hébert, directeur général adjoint en charge des opérations, et Mme Carole Le Saulnier, directrice en charge des affaires juridiques et réglementaires
Ø M. Édouard Couty, conseiller maître honoraire à la Cour des comptes, président de la Fédération hospitalière de France région Rhône-Alpes, et président du Haut conseil des professions paramédicales (HCPP)
Ø M. Lionel Benaiche, magistrat, secrétaire général du Service central de prévention de la corruption (SCPC), ancien responsable de la cellule de veille déontologique de l’AFSSAPS
Ø Conseil national de l’Ordre des pharmaciens – M. Xavier Desmas, membre du conseil national et président de la Commission de l’exercice professionnel
Ø Conseil national de l’Ordre des médecins – M. François Rousselot, président de la commission des relations médecins-industrie
Ø Collectif interassociatif sur la santé (CISS) – Mme Claude Rambaud, présidente
Ø Caisse nationale d’assurance maladie des travailleurs salariés (CNAMTS) – M. Frédéric Van Roekeghem, directeur général, et M. Hubert Allemand, médecin conseil
Ø Société française de pharmacologie et de thérapeutique – Mme Marie-Christine Pérault-Pochat, présidente
Ø Direction générale de la santé (DGS) – M. Jean-Yves Grall, directeur général, Mme Catherine Choma, sous-directrice de la politique des produits de santé et qualité des pratiques et des soins, et M. Frédéric Séval, chef de la Division droits des usagers, affaires juridiques et éthiques
Ø Haute autorité de santé (HAS) – Pr Jean-Luc Harousseau, président du collège, Mme Christine Vincent, chef de la mission juridique
Ø Fédération française des industries de santé (FEFIS) – M. Christian Lajoux, président
Ø Collectif Europe et médicament – Mme Laure Lechertier et M. Pierre Chirac
Ø Formindep – Mme Anne Chailleu, membre du conseil d’administration
Ø Comité économique des produits de santé (CEPS) – M. Dominique Giorgi, président
Ø Institut des données de santé (IDS) – M. Christian Babusiaux, président, et M. Richard de Cottignies, directeur
Ø Syndicat national de l’industrie des technologies médicales (SNITEM) –M. Éric Leroy, délégué général, et M. François-Régis Moulines, directeur Affaires publiques et communication
Ø Syndicat des entreprises du médicament (LEEM) – M. Philippe Lamoureux, directeur général, et Mme Muriel Carroll, directeur des affaires publiques
Ø Chambre syndicale de la répartition pharmaceutique (CSRP) –M. Emmanuel Déchin, délégué général
Ø M. Xavier Bertrand, ancien ministre de la santé et des solidarités, député de l’Aisne
ANNEXE 2
TEXTES RÉGLEMENTAIRES ET AUTRES MESURES D’APPLICATION
Echéancier de mise en application de la loi n° 2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé
Date de dernière mise à jour de l’échéancier : 14/06/2013
Articles |
Base légale |
Objet |
Décrets et arrêtés (décisions et circulaires) |
Chapitre Ier, article 1, I, 3° |
Article L. 1451-1, code de la santé publique |
Commissions et conseils, cabinets ministériels - Déclaration d’intérêts lors de leur prise de fonctions pour les agents des autorités et des organismes, dont les missions ou la nature des fonctions le justifient |
Décret n° 2012-745 du 9/05/2012 + Arrêté du 05/07/2012 (document type) + Arrêté du 02/08/2012 (instances concernées) |
Chapitre Ier, article 1, I, 6° |
Article L. 1451-3, code de la santé publique |
Modèle et contenu de la déclaration d’intérêts, conditions dans lesquelles elle est rendue publique ainsi que ses modalités de dépôt, d’actualisation et de conservation. | |
Chapitre Ier, article 1, I, 6° |
Article L. 1451-4, code de la santé publique |
Contrôle de la véracité des informations délivrées dans la déclaration d’intérêts par la commission éthique mise en place au sein de chaque agence |
Non appliqué |
Chapitre Ier, article 1, I, 8° |
Article L. 1452-2, code de la santé publique |
Approbation de la charte de l’expertise sanitaire |
Décret n° 2013-413 du 21/05/2013 |
Chapitre II, article 2, I |
Article L. 1453-1, code de la santé publique, II |
Avantages consentis par les entreprises : seuil des avantages en nature ou en espèces |
Décret n° 2013-414 du 21/05/2013 + Circulaire du 29 mai 2013 n° DGS/Pf2/2013/224" |
Chapitre II, article 2, I |
Article L. 1453-1, code de la santé publique, III |
Avantages consentis par les entreprises : nature des informations à rendre publiques (objet, date des conventions, délais et modalités de publication et d’actualisation de ces informations) | |
Article 5, V |
Article L. 5312-4-1, code de la santé publique |
Amendes administratives qui peuvent être assorties d’astreintes journalières |
Cf. ordonnance prévue par l’article 39 |
Article 5, VII |
Article L. 5421-8, code de la santé publique, 2° |
Signalement d’un effet indésirable pour toute personne exploitant un médicament ou produit mentionnés à l’article L. 5121, à l’Agence nationale de sécurité du médicament et des produits de santé. |
Décret n° 2012-1244 du 08/11/2012 |
Article 7, III |
Article L. 5322-1, code de la santé publique |
Sujets sur lesquels délibère le conseil d’administration de l’Agence nationale de sécurité du médicament et des produits de santé |
Décret n° 2012-597 du 27/04/2012 + Décret du 01/05/2012 |
Article 7, IV |
Article L. 5324-1, code de la santé publique |
Publicité de l’ordre du jour, des comptes rendus, assortis des détails et explications des votes, y compris les opinions minoritaires |
Décret n° 2012-745 du 09/05/2012 |
Article 8 |
Article L. 161-40-1, code de la sécurité sociale |
Gratuité et accessibilité de la base de données administratives et scientifiques sur les traitements ainsi que sur le bon usage des produits de santé |
En cours, publication prévue en septembre 2013 |
Chapitre 1er, article 9, II |
Article L. 5121-8-1, code de la santé publique |
Études que l’Agence nationale de sécurité du médicament et des produits de santé peut exiger du titulaire de l’autorisation |
Décret n° 2012-1244 du 08/11/2012 + Arrêté du 8 novembre 2012 |
Chapitre 1er, article 10 |
Article L. 1121-15, code de la santé publique |
inscription sur la liste définie des essais cliniques préalables à la délivrance de l’autorisation de mise sur le marché est obligatoire. |
Arrêté du 12 juin 2012 |
Chapitre 1er, article 11 |
Article L. 5121-9, code de la santé publique |
Suspension, retrait ou modification de l’autorisation de mise sur le marché délivrée par l’Agence française de sécurité sanitaire des produits de santé |
Décret n° 2012-1244 du 08/11/2012 + Arrêté du 8 novembre 2012 |
Chapitre 1er, article 14 |
Article L. 162-17, code de la sécurité sociale |
Réalisation d’essais cliniques contre des stratégies thérapeutiques |
En cours; publication prévue en septembre 2013 |
Chapitre II, article 17 |
Articles L. 5125-1-1 et L. 5125-1-1-1, code de la santé publique |
Préparations à l’officine |
En cours; publication prévue en septembre 2013 |
Chapitre II, article 18, I |
Article L. 5121-12-1, I, code de la santé publique |
Recommandations temporaires d’utilisation établie par l’Agence nationale de sécurité du médicament et des produits de santé |
Décret n° 2012-742 du 09/05/2012 |
Chapitre II, article 21 |
Article L. 162-17-4-1, I, code de la sécurité sociale |
Règles et délais de procédure, modes de calcul de la pénalité financière prononcée par le comité économique des produits de santé |
Décret n° 2012-1095 du 28/09/2012 |
Chapitre III, article 22 |
Article L. 5121-14-2, I, code de la santé publique |
Interdiction de la prescription, de la délivrance et retrait du marché d’une spécialité pharmaceutique par l’Agence nationale de sécurité du médicament et des produits de santé |
Décret n° 2012-1244 du 08/11/2012 |
Chapitre III, article 22 |
Article L. 5121-14-2, II, code de la santé publique |
Délivrance d’une spécialité pharmaceutique interdite ou qui a été retirée du marché, à des patients qui sont déjà traités avec elle | |
Chapitre III, article 23, II |
Extension du DP au PUI |
Décret n° 2012-1131 du 05/10/2012 | |
Chapitre III, article 23, III |
Modalités de désignation des établissements objets de l’expérimentation relative à la consultation du dossier pharmaceutique par les médecins avec autorisation des patients |
Décret n° 2013-31 du 09/01/2013 | |
Chapitre III, article 25 |
Article L. 6326-1, code de la santé publique |
Délivrance des médicaments, des dispositifs médicaux et approvisionnement des centres médicaux du service de santé des armées et leurs équipes mobiles |
En cours; publication prévue courant 2013. |
Chapitre IV, article 26 |
En cours; publication prévue courant 2013. |
ATU |
Décret n° 2013-66 du 18/01/2013 |
Chapitre V, article 27 |
Article L. 162-17-2-1, code de la santé publique |
Prise en charge dérogatoire de produits de santé de certains produits de santé, hors du périmètre de remboursement de droit commun |
Décret n° 2012-740 du 09/05/2012 |
Chapitre VI, article 28, I |
Article L. 5121-26 |
Règles applicables à la pharmacovigilance |
Décret n° 2012-1244 du 08/11/2012 + Arrêté du 8 novembre 2012 |
Chapitre VII, article 29, IV |
Article L. 5122-6, code de la santé publique |
Article L. 5122-6, code de la santé publique |
Arrêté du 28/09/2012 (liste des vaccins) + Arrêté du 28/09/2012 (mentions minimales des campagnes pub) |
Chapitre VII, article 29, II et V |
Articles L. 5122-3 et L. 5122-9, code de la santé publique |
Articles L. 5122-3 et L. 5122-9, code de la santé publique |
Décret n°2012-741 du 09/05/2012 |
Chapitre VII, article 30, II, 2° |
Article L. 162-17-8, code de la sécurité sociale |
Règles et délais de procédure, modes de calcul de la pénalité financière pour non respect d’une décision du Comité économique des produits de santé |
Décret n° 2012-1095 du 28/09/2012 |
Chapitre VIII, article 32 |
Article L. 161-38, code de la sécurité sociale, IV |
Certification des logiciels d’aide à la prescription médicale ayant respecté un ensemble de règles de bonne pratique |
En cours; Décret en cours de finalisation |
Chapitre IX, article 33 |
Article L. 5121-28, code de la santé publique |
Réalisation d’études de vigilance et d’épidémiologie - GIP- |
En cours, mission sur les données de santé de Pierre-louis Bras |
Article 34, I |
Articles L. 5213-7 et L. 5213-4, code de la santé publique |
Publicité pour les dispositifs médicaux de diagnostic in vitro. |
Décret n° 2012-743 du 9/05/2012 + Arrêté du 24 septembre 2012 |
Article 34, II |
Articles L. 5223-5 et L. 5223-3, code de la santé publique |
Publicité pour les dispositifs médicaux de diagnostic in vitro. |
Décret n° 2012-744 du 9/05/2012 + Arrêté du 24 septembre 2012 |
Article 34, |
Article L. 5213-3 |
Publicité des DM à faible risque pour la santé humaine |
Arrêté du 21/12/2012 |
Article 35 |
Article L. 165-1-2, code de la sécurité sociale, V |
Contrôles du respect des spécifications fonctionnelles et pénalité financière des DM |
Décret n° 2012-1135 du 8/10/2012 |
Article 36, I |
Article L. 162-1-20, code de la sécurité sociale |
Information sur le droit de se faire assister du conseil de son choix pendant les vérifications ou l’enquête administrative |
Décret n° 2012-1033 du 7/09/2012 |
Article 37 |
Article L. 165-11, code de la sécurité sociale, V |
Modalités de détermination des catégories homogènes de produits de santé concernées |
Décret n°2012-1051 du 13/09/12 |
Article 37 |
Article L. 165-13, code de la sécurité sociale |
Règles, délais de procédure et modes de calcul de la pénalité financière due en cas d’absence de réalisation dans les délais requis, par le fabricant ou le mandataire ou par le distributeur d’un dispositif médical, des études complémentaires demandées |
Décret n° 2012-1095 du 28/09/2012 |
Article 44 |
Article L. 5134-1, code de la santé publique |
Délivrance et administration de contraceptifs locaux ou hormonaux par certains infirmiers |
Décret n°2012-910 du 24 juillet 2012 |
Article 47 |
Article L. 5124-17-1, code de la santé publique |
Modalités d’organisation du système d’astreinte pour les grossistes-répartiteurs |
Décret n° 2012-1096 du 28/09/2012 |
Article 47 |
Article L. 5124-17-2, code de la santé publique |
Obligations de service public à respecter par les grossistes-répartiteurs sur leur territoire de répartition |
Ordonnances : loi n°2011-2012 du 29 décembre 2011 relative au renforcement de la sécurité sanitaire du médicament et des produits de santé | |||
Article de la loi |
Terme de l’habilitation |
Objet |
Ordonnances |
Article 38 |
dans un délai de douze mois à compter de la promulgation de la loi |
Transposition de la directive 2011/62/UE du Parlement européen et du Conseil, du 8 juin 2011, modifiant la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain et mesures nécessaires à leur extension et à leur adaptation aux îles Wallis et Futuna et, en tant qu’elles relèvent des compétences de l’Etat, à la Nouvelle-Calédonie et à la Polynésie française. |
Ordonnance n° 2012-1427 du 19 décembre 2012 relative au renforcement de la sécurité de la chaîne d’approvisionnement des médicaments, à l’encadrement de la vente de médicaments sur internet et à la lutte contre la falsification de médicaments + Décret n°2012-1562 du 31 décembre 2012 + Arrêté du 20 juin 2013 (bonnes pratiques de vente sur internet) Projet de loi de ratification déposé devant le parlement en mars 2013 Projet de loi DDADUE en cours d’examen au CE |
Article 39, 1° |
délai de vingt-quatre mois à compter de la promulgation de la présente loi |
Harmoniser et mettre en cohérence les dispositions relatives aux sanctions pénales et aux sanctions administratives dans le domaine des produits mentionnés à l’article L. 5311-1 du code de la santé publique avec les dispositions de la présente loi instituant de telles sanctions |
En cours, Septembre 2013 |
Article 39, 2° |
délai de vingt-quatre mois à compter de la promulgation de la présente loi |
Adapter les prérogatives des agents et des autorités chargés de constater les manquements punis par ces sanctions et de mettre celles-ci en œuvre |
En cours, Septembre 2013 |
Article 40 |
délai de douze mois à compter de la promulgation de la présente loi, |
Mesures relevant du domaine de la loi nécessaires à l’extension et à l’adaptation des dispositions de la présente loi aux îles Wallis et Futuna et, en tant qu’elles relèvent des compétences de l’Etat, à la Nouvelle-Calédonie et à la Polynésie française. |
|
Rapports | |||
Article de la loi |
Base légale |
Objet |
À déposer avant le |
Article 67 de la loi du 9 décembre 2004 |
Rapport sur l’application de la loi six mois après sa publication |
30/06/2012 | |
Article 3 |
Rapport sur le financement des associations d’usagers du système de santé et leurs besoins, au plus tard le 30 juin 2012. |
30/06/2012 | |
Article 15 |
Rapport d’activité mentionnant les modalités et principes selon lesquels les commissions spécialisées mettent en œuvre les critères d’évaluation des produits de santé en vue de leur prise en charge par l’assurance maladie. |
Fait, Annuel | |
Article 24 |
Rapport dressant le bilan de l’expérimentation, impact sur les dépenses et du bon usage des produits concernés |
Dans le cadre de l’examen du projet de loi de financement de la sécurité sociale pour 2014 | |
Article 30 |
Rapport dressant le bilan de l’expérimentation sur l’information par démarchage ou la prospection pour les produits de santé réalisé à partir d’une évaluation conduite par la Haute Autorité de santé |
01/01/2013 | |
Article 33 |
Rapport d’activité décrivant le résultat des études menées et formulant des recommandations |
Annuel | |
Article 41, IX |
Rapport formulant des propositions en matière de réparation des dommages quand le risque lié à un médicament se réalise. |
01/01/2013 | |
Article 41, X |
Rapport dressant le bilan des règles applicables à la sécurité des dispositifs médicaux et présentant les mesures susceptibles de l’améliorer - ANSM |
30/06/2012 | |
ANNEXE 3
RÉSUMÉ DES REMARQUES ET PRÉCONISATIONS DU RAPPORT
● La lutte contre les conflits d’intérêts et la transparence des avantages consentis par les entreprises
– modifier l’article L. 1451-1 du code de la santé publique et par suite l’article R. 1451-1 du même code afin d’assurer la communication, dans les déclarations publiques d’intérêts, du montant des rémunérations perçues par les professionnels visés par cet article ou de leurs participations financières ;
– modifier l’article L. 1453-2 du code de la santé publique afin de préciser que le contenu précis des conventions signées entre les entreprises et les professionnels du secteur doit être publié ;
– mettre en ligne dans les meilleurs délais un site internet unique regroupant l’ensemble des déclarations publiques d’intérêts et envisager une publication regroupée et harmonisée des conventions signées entre entreprises et professionnels de santé ;
– modifier l’article L. 1453-2 du code de la santé publique afin de supprimer la référence à un seuil pour la publication des avantages accordés par les entreprises aux professionnels du secteur ;
– intégrer au champ des conventions rendues publiques celles qui sont conclues entre les entreprises et les professionnels du secteur dans le cadre de relations commerciales qui ont pour objet l’achat de biens ou de services ;
– préciser par voie législative la définition de l’expertise et les notions de liens et de conflits d’intérêt figurant dans la charte de l’expertise sanitaire ;
– s’assurer que les experts qui présentent une situation de conflits d’intérêt ne sont pas associés à la délibération et à la prise de décision ;
● La gouvernance des produits de santé
– veiller à ce que le contrat de performance signé entre l’État et l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) lui accorde les moyens nécessaires pour remplir ses nouvelles missions. Le financement de la nouvelle agence par l’État, acté par le précédent Gouvernement, ne doit pas se traduire par une réduction de son budget au détriment de la sécurité sanitaire ;
– dans le cadre général d’une éventuelle reconfiguration de l’architecture des agences sanitaires, trouver des voies de simplification des procédures administratives et de resserrement des compétences de l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) afin qu’elle puisse de se concentrer sur ses missions fondamentales.
● L’évaluation des produits de santé après leur mise sur le marché et la pharmacovigilance
– publier dans les meilleurs délais le décret d’application de l’article 14 de la loi, qui prévoit l’obligation d’essais comparatifs contre des stratégies thérapeutiques existantes pour soumettre le médicament au remboursement, et articuler cette disposition avec la mise en place de l’évaluation médico-économique des produits de santé ; travailler à cette occasion à une meilleure articulation des avis de la commission de la transparence de la Haute autorité de santé et des décisions relatives au remboursement des produits de santé ;
– en lieu et place du GIP « Études de santé » prévu par l’article 33 de la loi pour encadrer l’accès aux données de santé et produire des études de pharmaco-épidémiologie, étendre les prérogatives de l’Institut des données de santé. Encadrer avec la plus grande prudence l’accès des acteurs privés aux données de l’assurance maladie que seuls des besoins de santé publique et de sécurité sanitaire peuvent justifier ;
– publier l’arrêté nécessaire à l’accès direct de l’Agence nationale de sécurité du médicament et des produits de santé aux données de santé (ANSM) afin de permettre le lancement effectif de son programme de travail ;
– publier dans les meilleurs délais le texte prévoyant les pouvoirs de sanction de l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) en cas de non-respect par les entreprises de leurs obligations en matière de pharmacovigilance et d’études post-autorisation de mise sur le marché ;
– faciliter et simplifier les modalités de déclaration des effets indésirables, en rappelant que tous les effets, y compris ceux visés par la notice, doivent être signalés, et garantir un véritable retour d’information aux notificateurs ;
– simplifier et améliorer la lisibilité des notices et y mentionner les voies et moyens de notification des effets indésirables, autres que le signalement aux pharmaciens et médecins ;
– mettre en place au niveau européen un régime plus strict de mise sur le marché et de contrôle des dispositifs médicaux, plus particulièrement des dispositifs médicaux implantables ;
– plaider pour l’application au niveau européen de règles plus strictes en termes de transparence, d’évaluation des produits de santé et d’indépendance des agences ;
– inscrire dans la loi de santé publique les actions de groupe dans le domaine de la santé, ce qui suppose une concertation avec les acteurs concernés et une réflexion préalable sur les notions de produit défectueux et d’imputabilité des effets indésirables et incidents.
● La formation et l’information des professionnels et du public
– mettre en ligne avant le 1er octobre 2013 la base de données publique sur le médicament à l’usage des professionnels et des particuliers. Cette base de données devra comporter des informations sur les médicaments remboursés et non remboursés, ainsi qu’à terme, sur les dispositifs médicaux ; il conviendra également d’assurer son référencement par les moteurs de recherche, afin de privilégier l’accès des patients aux informations publiques sur le médicament ;
– publier dans les meilleurs délais le décret d’application de l’article 32 de la loi relatif à la certification des logiciels d’aide à la prescription ;
– renforcer la place des enseignements relatifs aux produits de santé et la sensibilisation au problème de la pharmacovigilance dans la formation initiale et continue des médecins ;
– se limiter à la liste des vaccins pouvant faire l’objet d’une publicité non institutionnelle prévue par le décret du 28 septembre 2012 ;
– mettre en œuvre la visite médicale collective à l’hôpital, telle que prévue par le projet de loi de financement de la sécurité sociale pour 2013, et engager une réflexion sur l’amélioration de l’information des médecins exerçant en libéral ;
– envisager une augmentation des droits acquittés par les entreprises au titre du dépôt d’une demande d’autorisation de publicité pour un produit de santé auprès de l’ANSM et définir des priorités dans le traitement des dossiers, afin d’éviter que, face à l’augmentation exponentielle des demandes, certains dossiers ne bénéficient d’un accord tacite sans avoir été véritablement examinés.
1 () Les comités de protection des personnes, l’Office national de l’indemnisation des accidents médicaux, l’Établissement français du sang, l’Agence française de sécurité sanitaire, de l’alimentation, de l’environnement et du travail, l’Institut national du cancer, l’Institut national de prévention et d’éducation pour la santé, l’Agence de la biomédecine, l’Établissement de préparation et de réponse aux urgences sanitaires, la Haute Autorité de santé, le Comité économique des produits de santé, l’Agence française de sécurité sanitaire des produits de santé et l’Autorité de sûreté nucléaire.
2 () Norme établie en mai 2003 par l’Association française de normalisation.
3 () Séance du mardi 27 septembre 2011.
4 () CJUE, grande chambre, 9 novembre 2010, affaires jointes C-92/09 et C-93/09 Volker und Markus Schecke GbR et Hartmut Eifert.
5 () Décret n° 2013-31 du 9 janvier 2013 fixant les conditions de l’expérimentation relative à la consultation du dossier pharmaceutique par les médecins exerçant dans certains établissements de santé.
6 () - Alsace : centre hospitalier universitaire (CHU) de Strasbourg
- Aquitaine: CHU de Bordeaux, centre hospitalier (CH) de la Haute-Gironde à Blaye, CH Charles-Perrens à Bordeaux, CH de Mont-de-Marsan, CH de Dax, Association de gestion de l’institut Hélio Marin, CH d’Agen, polyclinique d’Aguilera à Biarritz, polyclinique Côte Basque Sud à Saint-Jean de Luz, SAS Capio Bayonne, CH de Pau
- Basse-Normandie : CH de Bayeux
- Franche-Comté : CHU de Besançon
- Guadeloupe : clinique Les Eaux claires (groupe Kapa Santé)
- Haute-Normandie : clinique du Cèdre à Bois-Guillaume
- Île-de-France : Assistance publique-hôpitaux de Paris (AP-HP), Service de santé des armées, CH Victor-Dupouy à Argenteuil
- Languedoc-Roussillon: CHU de Nîmes, polyclinique Pasteur à Pézenas, polyclinique Saint-Jean à Montpellier, UGECAM Languedoc-Roussillon Midi-Pyrénées, CHU de Montpellier
- Limousin : Association clinique de la Croix blanche, CHU de Limoges, CH Jacques-Boutard à Saint-Yrieix
- Lorraine : CH de Luneville, polyclinique Louis-Pasteur à Essey-lès-Nancy, CH de Verdun, CH de Remiremont
- Midi-Pyrénées : CH du pays d’Olmes à Lavelanet, CH du Val d’Ariège à Foix-Pamiers, CHU de Toulouse, Médipôle Garonne, CH Saint-Jacques à Saint-Céré, CH de Lourdes, CH de Bagnères-de-Bigorre, Hôpitaux de Lannemezan, CH du pays d’Autan à Castres-Mazamet, CH de Montauban
- Nord-Pas-de-Calais : CH de Valenciennes, CH d’Armentières, CH de Bailleul, CH d’Hazebrouck, CH de Béthune, CH de Calais, CH de Boulogne-sur-Mer
- Pays-de-la-Loire : CHU de Nantes
- Picardie : CHU d’Amiens
- Poitou-Charentes : CH de Niort
- Provence-Alpes-Côte d’Azur : CHU de Nice, Institut Paoli-Calmettes, CH Marie-Josée Treffot à Hyères
- Rhône-Alpes : CHU de Grenoble
7 () Rapport annuel 2012 du Haut Conseil de la santé publique.
8 () Directive 2005/29/CE du Parlement européen et du Conseil du 11 mai 2005 relative aux pratiques commerciales déloyales des entreprises vis-à-vis des consommateurs dans le marché intérieur.
9 () Directive 2006/114/CE du 12 décembre 2006 relative à la publicité trompeuse et comparative.
10 () CE, 8 octobre 2008, n°299043, Société Laboratoire GloxoSmithKline et Société Pfizer
© Assemblée nationale