

N° 1997
______
ASSEMBLÉE NATIONALE
CONSTITUTION DU 4 OCTOBRE 1958
TREIZIÈME LÉGISLATURE
Enregistré à la Présidence de l’Assemblée nationale le 28 octobre 2009.
RAPPORT D’INFORMATION
DÉPOSÉ
PAR LA COMMISSION DES AFFAIRES EUROPÉENNES(1)
sur le « paquet » médicaments
(E 4184, E 4185, E 4186, E 4187 et E 4188) ,
ET PRÉSENTÉ
PAR Mme Valérie ROSSO-DEBORD,
Députée
——
La Commission des affaires européennes est composée de : M. Pierre Lequiller, président ; MM. Michel Herbillon, Jérôme Lambert, Thierry Mariani, Didier Quentin, vice-présidents ; M. Jacques Desallangre, Mme Marietta Karamanli, MM. Francis Vercamer, Gérard Voisin secrétaires ; M. Alfred Almont, Mme Monique Boulestin, MM. Pierre Bourguignon, Yves Bur, François Calvet, Christophe Caresche, Philippe Cochet, Bernard Deflesselles, Lucien Degauchy, Michel Delebarre, Michel Diefenbacher, Jean Dionis du Séjour, Marc Dolez, Daniel Fasquelle, Pierre Forgues, Mme Arlette Franco, MM. Jean-Claude Fruteau, Jean Gaubert, Hervé Gaymard, Guy Geoffroy, Mmes Annick Girardin, Anne Grommerch, Elisabeth Guigou, Danièle Hoffman-Rispal, MM. Régis Juanico, Marc Laffineur, Robert Lecou, Michel Lefait, Lionnel Luca, Philippe Armand Martin, Jean-Claude Mignon, Jacques Myard, Michel Piron, Franck Riester, Mmes Chantal Robin-Rodrigo, Valérie Rosso-Debord, Odile Saugues, MM. André Schneider, Philippe Tourtelier.
SOMMAIRE
___
Pages
INTRODUCTION 7
PREMIERE PARTIE : UN SECTEUR ÉCONOMIQUE SPÉCIFIQUE ET UN ENJEU DE SANTÉ PUBLIQUE IMPORTANT 11
I. UNE ACTIVITÉ STRATÉGIQUE EN COURS DE MUTATION 11
A. DE GRANDES ENTREPRISES EUROPÉENNES DE POINTE DANS UNE ACTIVITÉ OÙ DOMINENT LES SOCIÉTÉS AMÉRICAINES 11
B. UNE FRANCE AU PREMIER PLAN AU SEIN DE L’UNION EUROPÉENNE 12
1. Premier marché de l’Union européenne 12
2. Premier producteur de l’Union européenne 12
3. Une entreprise de taille mondiale, Sanofi-Aventis, et de nombreux laboratoires de plus petite taille 13
C. DES ENJEUX D’AVENIR ESSENTIELS ET IDENTIFIÉS PAR LA COMMISSION EUROPÉENNE TANT SUR LE PLAN HUMAIN ET SANITAIRE QUE SUR LE PLAN ÉCONOMIQUE 15
II. UN CADRE JURIDIQUE EUROPÉEN EXIGEANT ET FONDÉ SUR LA COEXISTENCE DE DEUX RÉGIMES : L’UN COMMUNAUTAIRE, L’AUTRE NATIONAL 19
A. DES RÈGLES COMMUNAUTAIRES ANCIENNES DONT LA TENEUR S’EST RENFORCÉE AVEC LE MARCHÉ UNIQUE 19
B. LA COEXISTENCE ACTUELLE DE COMPÉTENCES COMMUNAUTAIRES ET DE COMPÉTENCES NATIONALES ET AINSI DE DEUX PROCÉDURES D’AUTORISATION DE MISE SUR LE MARCHÉ, L’UNE NATIONALE, L’AUTRE COMMUNAUTAIRE 20
DEUXIEME PARTIE : LES PROPOSITIONS DU PAQUET PHARMACEUTIQUE 23
I. L’INFORMATION DES PATIENTS SUR LES MÉDICAMENTS SOUMIS À PRESCRIPTION MÉDICALE : DES PROPOSITIONS INACCEPTABLES EN L’ÉTAT, POUR UN SUJET QUI APPARAÎT DEVOIR ÊTRE TRAITÉ SUR DE TOUT AUTRES BASES 23
A. LE DROIT COMMUNAUTAIRE ACTUEL : UNE TRÈS LARGE INTERDICTION DE LA PUBLICITÉ ET UN STRICT ENCADREMENT DES INFORMATIONS DÉLIVRÉES AUX PATIENTS ET AUX PROFESSIONNELS DE SANTÉ PAR LES LABORATOIRES PHARMACEUTIQUES 23
B. LA PROPOSITION DE LA COMMISSION EUROPÉENNE : AUTORISER, SANS LA DÉFINIR, L’INFORMATION DU PATIENT SUR LES MÉDICAMENTS DÉLIVRÉS SUR ORDONNANCE, TOUT EN MAINTENANT LES RÈGLES ACTUELLES D’INTERDICTION DE LA PUBLICITÉ 25
C. UN DISPOSITIF QUI SE HEURTE EN L’ÉTAT À L’HOSTILITÉ DE LA PRESQUE TOTALITÉ DES ETATS MEMBRES POUR UN SUJET QUI NE POURRAIT ÊTRE REPRIS QUE SUR D’AUTRES BASES 27
1. Un sujet sensible et identifié comme tel depuis longtemps 27
2. L’hostilité ou les réserves d’une très large majorité d’Etats membres 29
3. Des professionnels nuancés et des patients réservés 29
4. La nécessité d’aborder le sujet, en tout état de cause, sur de tout autres bases 30
a) Une absence de définition de l’information et un manque de clarté préjudiciables 30
b) La mise en jeu d’importants équilibres de fond 30
c) Des textes non acceptables en l’état et la nécessité de prévoir d’autres points de départ 31
II. LE DÉVELOPPEMENT DE LA PHARMACOVIGILANCE : DES PROPOSITIONS ACCEPTABLES, SOUS RÉSERVE DE CLARIFICATIONS 33
A. LES RÈGLES ACTUELLES EN MATIÈRE DE PHARMACOVIGILANCE 33
1. Les enjeux du suivi des effets indésirables des médicaments 33
2. Un domaine déjà réglé par le droit communautaire 33
B. LES PROPOSITIONS DE LA COMMISSION EUROPÉENNE : UN NOUVEAU COMITÉ AU SEIN DE L’AGENCE EUROPÉENNE DES MÉDICAMENTS, UN OBJECTIF DE CLARIFICATION DES RÔLES AVEC UN RENFORCEMENT DU NIVEAU COMMUNAUTAIRE, COMME DES INSTRUMENTS DE SUIVI DU MÉDICAMENT 36
1. Une conception et une définition plus larges de l’effet indésirable 37
2. Un réaménagement des instruments de suivi post-AMM des médicaments 37
3. Le renforcement du niveau communautaire et une clarification des rôles 39
a) La base de données EudraVigilance 39
b) La création d’un portail Web européen sur la sécurité des médicaments et la coordination des portails nationaux 40
c) L’institutionnalisation d’un Comité consultatif pour l’évaluation des risques en matière de pharmacovigilance au sein de l’Agence européenne des médicaments et l’accroissement des mission du groupe de coordination des Etats membres pour l’amélioration du suivi post-AMM 41
d) La clarification de la procédure communautaire pour les risques de sécurité grave 42
e) L’adjonction de redevances pour le financement de la pharmacovigilance 42
C. DES AMÉLIORATIONS DE FOND À PRÉVOIR POUR CLARIFIER LES RÔLES ET PERMETTRE LE BON FONCTIONNEMENT DU NIVEAU COMMUNAUTAIRE 43
III. LA LUTTE CONTRE LES MÉDICAMENTS FALSIFIÉS : UN OBJECTIF CONSENSUEL ET PARTAGÉ, MAIS DES MODALITÉS À PRÉCISER ET AMÉLIORER 47
A. LA PROPOSITION DE LA COMMISSION EUROPÉENNE : UN RENFORCEMENT DE LA SÉCURITÉ DE LA CHAÎNE DE FABRICATION ET DE DISTRIBUTION DU MÉDICAMENT 47
a) Une approche large de santé publique, reposant sur la notion de médicament falsifié, et complémentaire de celle du Conseil de l’Europe 50
b) Des mesures pour sécuriser la chaîne de fabrication et de distribution des médicaments 50
c) La prise en compte des flux avec les pays tiers 53
3. Des compléments à prévoir 53
TRAVAUX DE LA COMMISSION 57
PROPOSITION DE RESOLUTION 59
ANNEXE : LISTE DES PERSONNES RENCONTREES 63
Mesdames, Messieurs,
Le médicament est l’un des grands résultats du progrès. Sa diffusion dans les pays développés, et donc en Europe, est intervenue au cours des Trente glorieuses. D’abord, le développement scientifique et la révolution thérapeutique qui a débuté dès l’entre-deux-guerres, ont assuré sa disponibilité. Les vaccinations se sont généralisées. Les antibiotiques, les traitements contre le cholestérol, les neuroleptiques et les antalgiques, entre autres, sont devenus d’usage courant. La mise en place des régimes de sécurité sociale a permis aux patients de contourner l’ancienne contrainte du coût des produits et le recours au médecin comme au pharmacien a été banalisé.
Cette vague d’innovations se poursuit encore. Le médicament connaît des développements aussi permanents que sensibles. De nouveaux produits n’ont cessé d’apparaître, notamment avec la chimiothérapie en cancérologie. Les maladies qui concernent des publics peu nombreux, les maladies rares ou orphelines, peuvent même parfois être soignées. Toutefois, les progrès à accomplir en la matière restent encore considérables.
D’abord, il reste encore un très grand nombre de pathologies sans traitement ou bien pour lesquelles les traitements actuels peuvent encore faire d’immenses progrès. Tel est le cas du paludisme, du SIDA ou du cancer, par exemple.
Ensuite, certains acquis ou éléments considérés comme tels ne le sont pas. La résistance microbienne pose le défi de la découverte de nouvelles substances, notamment d’antibiotiques.
En outre, de nouvelles pathologies apparaissent. Si parmi elles, le SRAS ou le virus Ebola ont pu être maîtrisés, le virus du SIDA, évoqué dès 1982, n’a pu être ni éradiqué ni cantonné.
De plus, les domaines des thérapies innovantes, la thérapie génique, la thérapie cellulaire et la thérapie tissulaire restent encore à explorer et à exploiter, de même que les nanothérapies expérimentées cette année chez la souris pour certains cancers.
Par ailleurs, la disponibilité des médicaments constitue un autre défi. Celle-ci doit être suffisante pour que le nombre de patients à traiter le soit effectivement. L’identification des menaces de pandémie grippale dès la première moitié de la décennie, d’abord avec le virus H5N1, puis avec le virus de la grippe A H1N1 a permis de mesurer l’importance de cette dimension industrielle et territoriale. L’Europe comme la France doivent être en mesure de produire vaccins et médicaments efficaces dans les meilleurs délais.
Enfin, l’amélioration de la sécurité du médicament se doit d’être continue. Le médicament sûr n’est pas un acquis. D’une part, tout médicament repose sur l’analyse d’un rapport bénéfice/risque. Il n’est pas toujours aisé de réduire le second terme au minimum souhaitable. La lutte contre les effets indésirables est une nécessité permanente. D’autre part, la mondialisation, dans ceux de ses aspects qui sont négatifs, a accru les possibilités de falsification des médicaments, et celles-ci ne laissent pas d’être utilisées d’une manière croissante par les réseaux de la criminalité pharmaceutique.
Dans ce contexte, on mesure l’importance de deux des trois orientations du «paquet médicaments » ou « paquet pharmaceutique » présenté par la Commission européenne, avec cinq textes(2), soit deux propositions de règlement et trois propositions de directive. Ces deux sujets incontestables sont, d’une part, l’amélioration de la pharmacovigilance et, d’autre part, la lutte contre les médicaments falsifiés, et parmi eux contre les médicaments contrefaits.
La troisième de ces orientations, l’information du patient, relève d’une tout autre démarche, celle du développement d’une forme nouvelle de communication entre la publicité, largement interdite en Europe en la matière, et les informations au patient délivrées dans le cadre de la notice.
Son insertion dans un ensemble par ailleurs cohérent, justifie pleinement en l’espèce la critique de certains à l’encontre de la démarche du « paquet » communautaire, auquel on reproche de noyer les problèmes dans des ensembles trop divers et trop vastes pour permettre de les régler de manière adaptée dans des conditions correctes.
Toutefois, ce n’est pas cet élément de forme, mais, au contraire, d’importantes réserves et critiques de fond qui ont conduit la rapporteure a considérer que la proposition de règlement et la proposition de directive relatives à l’information des patients n’étaient pas acceptables en l’état, et à émettre, à l’opposé, une opinion favorable aux autres textes, ceux sur la pharmacovigilance et celui sur la lutte contre la falsification, sous réserve naturellement que les clarifications et aménagements qui leur sont nécessaires leur soient apportés.
Avant d’évoquer les motifs de ces prises de position, il convient cependant de rappeler tant les principaux enjeux du médicament pour l’Europe que le cadre juridique communautaire qui lui est applicable.
PREMIERE PARTIE :
UN SECTEUR ÉCONOMIQUE SPÉCIFIQUE ET UN ENJEU DE SANTÉ PUBLIQUE IMPORTANT
I. UNE ACTIVITÉ STRATÉGIQUE EN COURS DE MUTATION
A. De grandes entreprises européennes de pointe dans une activité où dominent les sociétés américaines
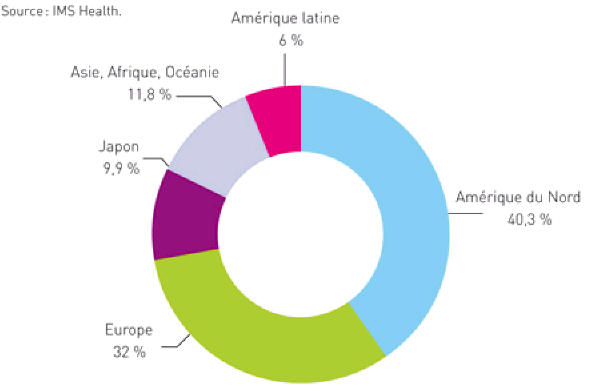
Sur le plan mondial, le marché du médicament est estimé à 773 milliards de dollars en 2008(3). Le marché nord-américain en représente plus de 40 %, selon les données d’HMS Health, pour 2008, dont plus de 37,5 % pour les seuls Etats-Unis.
Le marché européen est le deuxième marché, à raison de 32 % en 2008, devant le Japon. Les autres marchés sont de moindre ampleur à raison de 6 % pour l’Amérique latine et de moins de 12 % pour l’Asie hors Japon. Le graphique suivant récapitule ces éléments.
Répartition du marché mondial du médicament
Ce rôle majeur des Etats-Unis est en partie le fruit de l’histoire – les Etats-Unis ont été les premiers à exploiter et à commercialiser une large part des médicaments issus des découvertes de l’entre-deux-guerres, notamment la pénicilline et les antibiotiques, après la Seconde guerre mondiale. Elle est aussi le résultat d’un incomparable effort de recherche à l’origine de performances industrielles certaines. Parmi les dix premières entreprises mondiales, cinq sont d’origine américaine, dont les deux premières d’entre elles, à savoir Pfizer et Johnson & Johnson, les autres étant Abott, Merck et BMS (Bristol Myers Squibb).
Les autres principaux laboratoires sont suisses (Roche et Novartis), anglais avec GlaxoSmithKline et AstraZeneca, issu de la fusion en 1999 de Zeneca (Royaume-uni) et de l’entreprise suédoise Astra, et français avec Sanofi-Aventis.
Pour l’avenir, les éléments d’un maintien de ce rôle clef des Etats-Unis sont en place. Selon les éléments communiqués par Les entreprises du médicament (LEEM), l’avance américaine, acquise dès les années 1980 avec la création de petites sociétés innovantes proches des universités de San Francisco et Los Angeles, reste patente à raison de 195 000 personnes employées par le secteur, soit une moyenne de 130 personnes par entreprise, contre 81 947 en Europe, soit 47 employés par entreprise.
La France est le premier marché du médicament au sein de l’Union européenne, à raison de 5,5 % du marché mondial en 2008, devançant ainsi l’Allemagne (5,3 %), l’Italie (3,3 %), ainsi que l’Espagne et le Royaume-Uni (2,9 % chacun).
C’est un marché assez peu concentré puisque le premier fournisseur de médicaments, Sanofi-Aventis, n’en représente que 15 %. Les parts de marché des entreprises suivantes n’atteignent pas 7 % : Pfizer, GlaxoSmithKline, AstraZeneca et Bristol-Myers Squibb.
La France est également le premier producteur européen de médicaments, à raison de 35 milliards d’euros en 2007, devançant nettement l’Allemagne et le Royaume-Uni (25 milliards d’euros chacun), puis l’Italie, la Suisse et, enfin, l’Irlande, producteur négligeable au début des années 1990, et qui a bénéficié des implantations des grandes firmes étrangères, notamment américaines.
Le graphique suivant illustre ces éléments.
Les principaux pays européens producteurs de médicaments

3. Une entreprise de taille mondiale, Sanofi-Aventis, et de nombreux laboratoires de plus petite taille
En 2008, selon le LEEM, 326 entreprises industrielles étaient recensées dans le secteur, mais une seule de grande taille, Sanofi-Aventis, avec un chiffre d’affaires de l’ordre de 27 milliards d’euros en 2008 et 100.000 collaborateurs dans le monde.
Avec les quatre entreprises qui suivent, de moindre taille, cette entreprise a constitué une structure ad hoc, le G5.
Celles-ci sont Servier (3,7 milliards d’euros de chiffre d’affaires et 20 000 collaborateurs), Pierre Fabre (1,8 milliard d’euros de chiffre d’affaires en 2008, dont la moitié à l’exportation, et 9 800 collaborateurs pour le groupe, toutes activités groupées), IPSEN (971 millions d’euros de chiffre d’affaires en 2008) et LFB, spécialisé dans les protéines thérapeutiques et les dérivés du plasma (352 millions de chiffre d’affaires en 2008, et 1 531 collaborateurs).
Les autres laboratoires sont de plus petites tailles.
Le secteur du médicament est pour la France un secteur fortement exportateur à raison de 21 milliards d’euros en 2008, soit 45 % de son activité.
Les autres pays d’Europe représentent la majeure partie de son marché extérieur, à raison de presque 60 %, dont les trois quarts pour l’Union européenne, devant les Amériques (16 %) et l’Afrique (9 %), comme l’indique le graphique suivant.
Répartition des exportations françaises de médicaments

Plus précisément, les principaux clients de la France sont les Etats-Unis (2,35 milliards d’euros en 2008), suivis par la Belgique (1,93 milliard d’euros) et l’Allemagne (1,78 milliard d’euros), puis l’Italie, le Royaume-Uni et l’Espagne.
Pour l’avenir, le maintien de cette primauté de la France au sein de l’Union européenne n’est pas nécessairement acquis.
En matière de médicament d’avenir, de biotechnologies, c’est en effet le Royaume-Uni qui détient la première position, notamment grâce aux synergies entre les entreprises et les universités telles que Cambridge, sur le modèle américain.
Selon les données de BioCentury, le Royaume-Uni comprenait en 2008 presque trois fois plus d’entreprises de biotechnologies cotées que l’Allemagne, numéro deux en Europe, et quatre fois plus que la France, comme l’indique le graphique suivant.
Le secteur de biotechnologies dans quelques pays européens

C. Des enjeux d’avenir essentiels et identifiés par la Commission européenne tant sur le plan humain et sanitaire que sur le plan économique
Sur le plan économique, l’activité pharmaceutique repose sur un modèle spécifique.
En effet, les coûts sont essentiellement des coûts de recherche, de conception et de développement. Une fois celui-ci conçu, la fabrication proprement dite du médicament est une opération moins onéreuse. C’est une logique de rendement croissant : les unités supplémentaires de produits vendus sont très rentables, une fois les coûts de recherche amortis.
Du point de vue européen, et également américain ou japonais, le secteur du médicament doit actuellement surmonter trois défis majeurs. Ceux-ci sont bien identifiés, notamment par le rapport établi par le cabinet Ernst & Young intitulé Beyond Borders : Global biotechnolgy report (2008).
Le premier défi est celui de l’arrivée à échéance d’un certain nombre de brevets, ce qui modifie les conditions d’exploitation des firmes traditionnelles, en accroissant la concurrence des producteurs de génériques.
Le deuxième est celui de la mondialisation, avec ses deux aspects. D’un côté, la taille des marchés s’accroît, même si les conditions économiques font que les conditions de commercialisation ne sont pas les mêmes sur ces nouveaux marchés. D’un autre côté, la concurrence devient plus aigue. Les pays émergents sont en mesure de produire à des prix très compétitifs, notamment des génériques. L’Inde est ainsi qualifiée de « pharmacie des pays pauvres », avec des entreprises solides telles que CIPLA et Ranbaxy. Les plus en pointe d’entre eux disposent même de la capacité à concevoir et développer des nouveaux produits. La politique d’éducation a porté ses fruits et les compétences sont sur place. Le maintien d’activités pharmaceutiques sur le continent européen fait donc face à deux risques : celui de la concurrence des producteurs des pays émergents ; celui de la délocalisation vers ces mêmes pays des activités de laboratoires européens ou américains.
Le troisième défi est celui des médicaments du futur et des nouvelles technologies, notamment des biotechnologies. Comme l’indique le rapport précité, il comprend une importante part de risque, celle liée à l’innovation. C’est ce qui explique l’intérêt marqué des grandes firmes pour les petites entreprises innovantes et les actuelles politiques de rachat de celles qui sont les plus prometteuses. Il en résulte des modifications de fond dans la gestion des entreprises, avec une préférence pour l’organisation en réseau, de manière à maintenir et cultiver la spécificité des entreprises innovantes après leur intégration dans un ensemble plus vaste.
Enfin, les médicaments du futur ne sont pas uniquement un défi technologique, mais également un défi médical avec le développement futur de la médecine personnalisée, fondée sur une adaptation du traitement à la personne et dans certains cas la thérapie génique.
Dans sa communication du 10 décembre dernier, intitulée « Des médicaments sûrs, innovants et accessibles: une vision nouvelle du secteur pharmaceutique » (document COM (2008) 666 final), la Commission européenne propose une stratégie pour assurer le maintien d’une activité pharmaceutique performante sur le territoire européen.
Il s’agit pour elle fort justement d’un impératif incontournable. C’est un enjeu non seulement économique mais également de santé publique.
C’est la disponibilité des médicaments sur le territoire de l’Union européenne qui est le seul garant de la capacité à y soigner les patients.
Les actuelles inégalités entre les pays face à la pandémie de grippe A de type H1N1 rappellent que le médicament, le vaccin en l’occurrence, comme l’alimentation ou l’énergie, est un secteur où l’indépendance de l’approvisionnement est essentielle.
Très récemment, le manque de disponibilité de l’enzyme permettant de lutter contre la leucodystrophie, maladie rare qui touche 160 cas en France, révélée à l’occasion des déboires de l’association ELA qui avait fait appel au parrainage de MM. François Pinault, Frank Riboud et Zinedine Zidane, a rappelé que les goulets d’étranglement ne concernaient pas les seuls pays en voie de développement.
Sur le plan économique également, le secteur pharmaceutique est un secteur clef pour l’avenir de l’Europe.
C’est, en effet, un secteur qui prend pleinement part à la stratégie de Lisbonne, laquelle vise, grâce à la recherche et à la technologie, à rattraper l’écart entre l’Europe et les secteurs de pointe de l’économie américaine. L’Union européenne dispose actuellement d’atouts essentiels : une base de recherche forte, un système éducatif éprouvé, une main-d’oeuvre qualifiée et une industrie européenne bien établie avec de nombreuses entreprises innovantes.
Les objectifs de santé publique de la politique du médicament ne sont pas nécessairement antinomiques, sur le fond, avec ces objectifs économiques.
Même si par définition, la conception et la mise sur le marché d’un médicament reposent sur un rapport bénéfice/risque et sont donc toujours susceptibles de receler une certaine part de risque, une part d’effets secondaires indésirables, l’idéal est que ces derniers soient le plus réduit possible.
Un médicament sûr est toujours un médicament plus compétitif qu’un médicament qui l’est moins, pour une même indication thérapeutique.
C’est dans le cadre d’un tel équilibre entre enjeux industriels et enjeux de santé publique que l’on peut ainsi concevoir le bien fondé de la démarche de la Commission européenne qui a associé à sa communication du 12 décembre, les propositions de règlement et propositions de directive relatives à la pharmacovigilance et à la lutte contre les médicaments falsifiés.
II. UN CADRE JURIDIQUE EUROPÉEN EXIGEANT ET FONDÉ SUR LA COEXISTENCE DE DEUX RÉGIMES : L’UN COMMUNAUTAIRE, L’AUTRE NATIONAL
L’Europe du médicament s’est construite depuis le milieu des années 1960, par étapes.
Le premier texte, la directive 65/65/CEE du 26 janvier 1965, concernant le rapprochement des dispositions législatives, réglementaires et administratives, relatives aux spécialités pharmaceutiques, est intervenu après l’affaire de la thalidomide. Il a prévu l’obligation d’une autorisation préalable avant la mise sur le marché des médicaments, l’autorisation de mise sur le marché (AMM), et en a fixé les principales conditions de délivrance, notamment la production de résultats d’essais physico-chimiques, biologiques ou microbiologiques, pharmacologiques et toxicologiques ainsi que cliniques. Cet effort d’harmonisation a eu un objectif non seulement sanitaire, mais également économique, car il s’agissait de jeter les bases de la circulation des médicaments dans le marché commun.
En 1975, en raison de l’insuffisance de ces règles, une nouvelle étape a été franchie avec la directive 75/319/CE du 20 mai 1975 laquelle a notamment renforcé les obligations relatives au dossier de demande d’AMM, et la directive 75/318/CEE du 20 mai 1975, relative au rapprochement des législations des Etats membres concernant les normes et protocoles analytiques, toxico-pharmacologiques et cliniques en matière d’essais de spécialités pharmaceutiques. Un comité des spécialités pharmaceutiques a été créé, afin de faciliter l’adoption par les Etats membres d’une approche commune. Ont, en outre, été instituées des règles communes pour les importations provenant de pays tiers.
Initialement limité au seul médicament stricto sensu, le champ de l’harmonisation a ultérieurement été étendu par trois des directives du « paquet » de 1989 : la directive 89/342/CEE du 3 mai 1989 pour les médicaments immunologiques consistant en vaccins, toxines, sérums ou allergènes ; la directive 89/343/CEE du 3 mai 1989 pour les médicaments radiopharmaceutiques ; la directive 89/381/CEE du 14 juin 1989 pour les médicaments dérivés du sang ou du plasma humains. La quatrième directive de ce « paquet » a, quant à elle, prévu des mesures d’harmonisation des notices.
Dans la perspective du marché unique, une nouvelle étape importante a été franchie avec l’adoption, en 1992, d’un autre « paquet » regroupant plusieurs directives relatives à la distribution en gros des médicaments (directive 92/25/CEE), à leur classification en vue de leur délivrance (directive 92/26/CEE), l’étiquetage et la notice (directive 92/27/CEE), ainsi qu’à la publicité avec la directive 92/28/CEE, laquelle a notamment repris l’interdiction de publicité pour les médicaments soumis à prescription.
Une nouvelle étape a ensuite été franchie en 1993, avec les trois directives et le règlement qui organisent l’architecture actuelle du droit européen du médicament, avec la coexistence à partir de 1995 de deux procédures d’AMM : l’une nationale, avec reconnaissance mutuelle des AMM délivrées par chaque Etat membre ; l’autre dite centralisée, procédure d’AMM communautaire délivrée par l’Agence européenne d’évaluation des médicaments (EMEA), située à Londres.
B. La coexistence actuelle de compétences communautaires et de compétences nationales et ainsi de deux procédures d’autorisation de mise sur le marché, l’une nationale, l’autre communautaire
L’actuel droit européen du médicament provient pour l’essentiel des textes adoptés en 2001 puis en 2004, à l’issue de la procédure dite de « révision pharmaceutique ». Il est constitué de deux corps de règles.
Le premier est celui de la directive 2001/83/CE du 6 novembre 2001 instituant un code communautaire relatif aux médicaments à usage humain, qui comprend les dispositions propres aux AMM nationales et à leur reconnaissance mutuelle par les autres Etats membres.
Le second est le règlement (CE) n° 726/2004 établissant des procédures communautaires de mise sur le marché et relatif à l’Agence européenne des médicaments.
Ils organisent la coexistence entre les deux procédures d’autorisation de mise sur le marché d’un médicament, à savoir la procédure nationale ou décentralisée, en application de laquelle un laboratoire dépose un dossier de demande d’AMM auprès de l’autorité nationale compétente, et engage ensuite, s’il le souhaite, la procédure de reconnaissance de cette AMM par les autres Etats membres, et la procédure communautaire.
S’agissant de la France, la procédure nationale relève des compétences de l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS). Celle-ci délivre, en effet, les AMM après avis de la Commission nationale d’autorisation de mise sur le marché, laquelle procède à l’évaluation technique et scientifique des données soumises par le laboratoire pharmaceutique concerné. Tout dossier de demande d’AMM comporte, en effet, les résultats d’études pharmaceutiques et biologiques, d’études pharmacologiques et toxicologiques, ainsi que des études cliniques. Cette évaluation repose sur des critères de qualité, efficacité et sécurité d’emploi du médicament. L’autorisation de mise sur le marché est régulièrement réévaluée. Elle peut être modifiée, suspendue ou retirée par décision du directeur général de l’AFSSAPS après avis de la commission d’AMM.
Plus précisément, c’est dans la directive 2001/83/CE que sont prévues les principales règles qui s’imposent aux Etats membres pour les AMM nationales et la procédure de reconnaissance mutuelle, par un autre Etat membre, d’une AMM délivrée par un Etat membre. Ce texte impose également le retrait du marché pour les cas suivants notamment : le médicament est nocif ; l’effet thérapeutique fait défaut ; le rapport bénéfice/risque n’est pas favorable ; le médicament n’a pas la composition déclarée.
Il prévoit également l’obligation d’imposer des autorisations de fabrication de même que, pour les médicaments provenant des pays tiers, des autorisations d’importation, et précise également les règles relatives à l’étiquetage et à la notice, à la classification des médicaments, à leur distribution en gros (avec l’obligation de prévoir une autorisation d’exercer l’activité de grossiste en médicament), à la publicité, à la pharmacovigilance et à la surveillance du marché (obligation pour les autorités compétentes de prévoir des inspections régulières (inspection des établissements de fabrication et des laboratoires, prélèvement d’échantillons, examen de documents) et, si nécessaire, des inspections inopinées.
Pour ce qui concerne la procédure communautaire, le règlement précité (CE) n° 726/2004 fixe les condition relative à l’AMM centralisée, laquelle présente l’avantage de donner directement accès au marché européen, à l’ensemble des Etats membres, sans passer par la procédure de reconnaissance mutuelle.
Pour sa part, le règlement (CE) n° 726/2004 ne se limite pas à préciser les règles relatives à l’AMM communautaire, délivrée par la Communauté, à savoir par la Commission européenne, sur avis du comité des médicaments à usage humain placé auprès de l’Agence européenne du médicament. Les critères d’autorisation sont également la qualité, la sécurité et l’efficacité du médicament considéré, dans le cadre d’une évaluation du rapport bénéfice/risque.
L’application de la procédure communautaire, à l’exclusion de toute possibilité d’une AMM nationale, est obligatoire pour les médicaments suivants :
– les médicaments issus de la biotechnologie ;
– les médicaments de thérapie innovante ;
– les médicaments orphelins ;
– les médicaments qui contiennent une substance active entièrement nouvelle et dont l’indication thérapeutique est le traitement du SIDA, du cancer, d’une maladie neurodégénérative, du diabète, des maladies auto-immunes et d’autres dysfonctionnements immunitaires ainsi que des maladies virales.
Elle est facultative et donc alternative, selon le choix du fabricant, pour deux cas :
– les autres médicaments contenant une nouvelle substance active ;
– les médicaments représentant une innovation thérapeutique, scientifique ou technique ou bien un intérêt au niveau communautaire.
Dans l’ensemble, elle ne se substitue donc pas à la procédure nationale.
La directive 2001/83/CE et règlement (CE) n° 726/2004 organisent par ailleurs le cadre européen de surveillance de marchés et donc de la pharmacovigilance.
DEUXIEME PARTIE :
LES PROPOSITIONS DU PAQUET PHARMACEUTIQUE
I. L’INFORMATION DES PATIENTS SUR LES MÉDICAMENTS SOUMIS À PRESCRIPTION MÉDICALE : DES PROPOSITIONS INACCEPTABLES EN L’ÉTAT, POUR UN SUJET QUI APPARAÎT DEVOIR ÊTRE TRAITÉ SUR DE TOUT AUTRES BASES
A. Le droit communautaire actuel : une très large interdiction de la publicité et un strict encadrement des informations délivrées aux patients et aux professionnels de santé par les laboratoires pharmaceutiques
La directive précitée 2001/83/CE prévoit les règles en matière de publicité et d’information sur les médicaments.
Elle définit la publicité d’une manière large. La rédaction retenue vise, en effet, toute forme de démarchage, d’information, de prospection ou d’incitation qui vise à promouvoir la prescription, la délivrance, la vente ou la consommation de médicaments. Il s’agit ainsi de la publicité auprès du public et auprès des personnes habilitées à la prescription et à la délivrance (médecins et pharmaciens), de la visite de délégués médicaux, de la fourniture d’échantillons, ainsi que du parrainage de réunions promotionnelles et de congrès scientifiques auxquels assistent des personnes habilitées à prescrire ou à délivrer des médicaments.
Ne sont exclus de la notion de publicité que, d’une part, la notice et, d’autre part, l’étiquetage, ainsi que la correspondance pour répondre à une question précise et les informations relatives à la santé humaine ou à des maladies humaines sans référence à un médicament.
Ensuite, plusieurs règles sont prévues pour encadrer, et interdire dans la plupart des cas, la publicité. Elles reposent sur la distinction entre la publicité auprès du public, largement interdite, et celle auprès des personnels habilités à prescrire, les médecins d’une manière générale.
Quelques règles sont cependant communes : l’interdiction totale de publicité pour les médicaments qui ne bénéficient pas d’une autorisation de mise sur le marché ; l’obligation pour la publicité, lorsqu’elle est autorisée, de favoriser l’usage rationnel du médicament et de ne pas être trompeuse.
Pour ce qui concerne la publicité auprès du public, il faut distinguer plusieurs cas.
Celle-ci est interdite pour les médicaments uniquement délivrés sur prescription médicale et pour ceux qui contiennent des psychotropes ou des stupéfiants. On observera également que l’article 14 de la directive 89/552/CEE a interdit la publicité télévisée pour les médicaments et traitements médicaux soumis à prescription médicale.
En revanche, la publicité est autorisée pour les médicaments d’auto-médication, plus précisément ceux destinés, par leur composition ou leur objectif, à être utilisés sans intervention d’un médecin mais avec, au besoin, le conseil du pharmacien.
En outre, les Etats membres peuvent en tout état de cause interdire la publicité pour les médicaments remboursables.
Enfin, le cas des campagnes de vaccination faites par l’industrie et approuvées par les autorités compétentes des Etats membres n’est pas couvert par ces interdictions.
Selon la même inspiration, les Etats membres interdisent la distribution directe de médicaments au public à des fins promotionnelles.
Lorsqu’elle n’est pas interdite, la publicité auprès du public doit respecter des règles strictes :
– elle doit être clairement identifiable comme telle et comporter au moins les éléments suivants: le nom du médicament ; les informations indispensables à son bon usage ; une invitation à lire attentivement les instructions de son emploi ;
– elle doit respecter un nombre important de restrictions. Elle ne doit ainsi comprendre aucun élément incitant à se passer du médecin (interdiction d’inclure des éléments qui font apparaître la consultation médicale ou l’intervention chirurgicale comme superflues), ni comparant le médicament à d’autres traitements ou médicaments, ni suggérant que la bonne santé normale d’une personne peut être améliorée par l’utilisation du médicament ou affectée en cas de non-utilisation, ni s’adressant principalement ou exclusivement aux enfants, ni se référant à une recommandation émanant de scientifiques, de professionnels de la santé ou d’autres personnes pouvant, par leur notoriété, inciter à la consommation de médicaments, ni assimilant le médicament à une denrée alimentaire, à un produit cosmétique ou à un autre produit de consommation, ni suggérant que la sécurité ou l’efficacité du médicament est due au fait qu’il s’agit d’une substance naturelle, ni pouvant induire par une figuration, ou une description détaillée, à un faux autodiagnostic, ni se référant de manière abusive, effrayante ou trompeuse, à des attestations de guérison, ni utilisant de manière similaire des représentations visuelles des altérations du corps humain dues à des maladies ou à l’action d’un médicament dans le corps humain.
Pour ce qui concerne les professionnels de santé, la publicité auprès des personnes habilitées à prescrire ou à délivrer des médicaments (médecins, pharmaciens, etc.) est autorisée, mais doit suivre des règles précises.
Elle doit dans tous les cas mentionner les informations essentielles sur le médicament et sa classification en matière de délivrance.
Les Etats membres peuvent en outre exiger l’inclusion d’informations complémentaires sur le prix de vente ou le tarif indicatif, ainsi que sur les conditions de remboursement par la sécurité sociale.
Toute documentation sur le médicament contient en outre la date de dernière mise à jour et l’information qu’elle contient doit être exacte, actuelle, vérifiable et suffisamment complète.
Les échantillons gratuits, qui doivent comporter cette mention, ne peuvent en outre être remis qu’à titre exceptionnel aux prescripteurs.
La directive prévoit également plusieurs dispositions sur la formation des délégués médicaux de même que les obligations d’information et les règles qu’ils doivent respecter pendant leur travail : interdiction d’octroyer des primes ou des avantages significatifs comme technique de promotion des médicaments, limitation de l’hospitalité offerte lors de manifestations de promotion, restriction dans la distribution d’échantillons gratuits.
Enfin, les Etats membres doivent veiller à la mise en œuvre de moyens adéquats et efficaces pour contrôler la publicité à l’égard des médicaments.
B. La proposition de la Commission européenne : autoriser, sans la définir, l’information du patient sur les médicaments délivrés sur ordonnance, tout en maintenant les règles actuelles d’interdiction de la publicité
C’est en application de l’article 88 bis de la directive 2001/83/CE, introduit en 2004 lors de la révision pharmaceutique, que la Commission européenne a présenté au Parlement européen et au Conseil, après consultation des organisations de patients et de consommateurs, des organisations de médecins et de pharmaciens et des Etats membres, un rapport sur les pratiques actuelles en matière de communication d’information – notamment par Internet – et sur leurs risques et leurs avantages pour les patients, ainsi que les deux propositions de directive et de règlement concernées.
L’objectif de ces dernières est d’offrir aux sociétés pharmaceutiques la possibilité de communiquer au grand public des informations sur les médicaments soumis à prescription médicale.
Elles sont fondées sur les principes suivants.
D’abord, les titulaires des AMM seraient autorisés à délivrer directement des informations aux patients pour les médicaments soumis à prescription médicale, tout en maintenant l’interdiction de la publicité.
Néanmoins, de manière paradoxale, cette information n’est pas définie de manière directe et positive par le dispositif de la proposition de directive.
Elle l’est « en creux » puisque la proposition de directive précise que les informations en cause ne seront juridiquement pas de la publicité, et de manière indirecte, puisque elle donne des indications sur les types d’informations susceptibles d’être délivrées par le titulaire d’une AMM.
Le dispositif prévoit, en effet, les éléments suivants :
– une liste des types d’informations autorisés, à savoir, pour l’essentiel : les éléments du résumé des caractéristiques du produit, de l’étiquetage et de la notice, ainsi que de la version accessible au public du rapport d’évaluation du médicament ; des informations sur l’impact environnemental du médicament, sur le prix, ou des informations concrètes et les documents de référence relatifs, notamment, aux changements d’emballages ou aux mises en garde concernant les effets indésirables ; des informations portant sur des études scientifiques non interventionnelles (à savoir autres que cliniques) ou bien sur des mesures d’accompagnement de la prévention et du traitement de maladies ; des informations qui présentent le médicament dans le contexte de la condition (ou de la pathologie) à éviter ou à traiter(4) ;
– des « normes de qualité » : objectivité et impartialité ; adaptation aux besoins et aux attentes de patients ; nécessité de preuves pour les fonder ; nécessité d’une mise à jour ; fiabilité et absence de caractère trompeur ; nécessité d’être compréhensible par le public ; mention de la source des informations ; mention du fait que médicament n’est disponible que sur prescription ; mention du fait que les informations visent à soutenir et non à remplacer le contact entre le patient et les professionnels de santé et que le patient doit contacter un professionnel de santé s’il a besoin d’une clarification ;
– l’interdiction de la comparaison entre médicaments ;
– un code de conduite et des orientations établies par la Commission européenne sur l’information autorisée ;
– une liste limitative des canaux d’information autorisés. D’une part, le principe de l’interdiction de la radio et de la télévision est posé. D’autre part, seuls seraient autorisés, d’une part, les publications spécialisées dans le domaine de la santé, à l’exclusion des « gratuits », ainsi que les sites Internet consacrés aux médicament et les réponses écrites aux demandes d’information des particuliers ;
– des règles pour les sites Internet des firmes pharmaceutiques ;
– l’obligation pour les Etats membres d’instaurer un système de contrôle afin d’éviter les abus, de garantir le respect de ces dispositions susmentionnées en matière de contenu des informations, de normes de qualité et de canaux de diffusion. La Commission européenne propose néanmoins de laisser une grande latitude aux Etats membres.
Plus précisément, si elle pose le principe du contrôle de l’information avant sa diffusion, elle prévoit en pratique un contrôle a posteriori puisque le principe de ce contrôle a priori ne concerne ni les informations déjà approuvées par les autorités compétentes (cas notamment des extraits de la notice), ni les cas où un contrôle approprié, efficace et de niveau équivalent est assuré par un autre mécanisme. Parmi ces mécanismes équivalents, le contrôle par des organismes d’autoréglementation ou de « corégulation » est prévu.
C. Un dispositif qui se heurte en l’état à l’hostilité de la presque totalité des Etats membres pour un sujet qui ne pourrait être repris que sur d’autres bases
La proposition de directive et la proposition de règlement sur l’information des patients concernent l’un des sujets les plus délicats parmi ceux relatifs au médicament.
Il a, en effet, été l’un des plus controversés parmi ceux examinés dans le cadre de la « révision pharmaceutique » lancée en 2001 (document E 1902).
Dans une partie déjà consacrée à l’information des patients, mais visant en fait la publicité, la Commission européenne avait proposé « d’ouvrir une possibilité d’information auprès du public pour les classes de médicaments autorisés et prescrits dans le cadre des affections suivantes : SIDA, asthme et affections broncho-pulmonaires chroniques, diabète ».
Le Parlement européen s’y était opposé lors de la première lecture des textes du « paquet », en séance plénière, le 23 octobre 2002, comme l’avait rappelé la communication de M. Gilbert Chabroux, sénateur, du 5 mars 2003, sur la « révision pharmaceutique ».
S’agissant du Conseil, la France était pour sa part opposée à cette initiative.
En l’absence de texte communautaire, la question relève actuellement de la compétence de chaque Etat membre. La Suède dispose ainsi, selon les informations communiquées, de son propre dispositif.
S’agissant de la France, la loi n° 2009-879 du 21 juillet 2009 dite « hôpital, patients, santé et territoires », insère la notion d’éducation thérapeutique du patient aux articles L. 1161-1 et suivants du code de la santé publique.
Ce même article L. 1161-1 la définit ainsi : « l’éducation thérapeutique s’inscrit dans le parcours de soins du patient. Elle a pour objectif de rendre le patient plus autonome en facilitant son adhésion aux traitements prescrits et en améliorant sa qualité de vie. Elle n’est pas opposable au malade et ne peut conditionner le taux de remboursement de ses actes et des médicaments afférents à sa maladie. »
Il est également précisé que « tout contact direct entre un malade et son entourage et une entreprise se livrant à l’exploitation d’un médicament ou une personne responsable de la mise sur le marché d’un dispositif médical ou d’un dispositif médical de diagnostic in vitro est interdit».
Pour sa part, l’article L. 1161-2 précise les conditions de contrôle et de délivrance des programmes d’éducation thérapeutique : « Les programmes d’éducation thérapeutique du patient sont conformes à un cahier des charges national dont les modalités d’élaboration et le contenu sont définis par arrêté du ministre chargé de la santé. Ces programmes sont mis en œuvre au niveau local, après autorisation des agences régionales de santé. Ils sont proposés au malade par le médecin prescripteur et donnent lieu à l’élaboration d’un programme personnalisé. Ces programmes sont évalués par la Haute Autorité de santé. »
L’article L. 1161-3 prohibe l’intervention des seules firmes pharmaceutiques dans la définition des programmes et actions d’éducation thérapeutique : « Les programmes ou actions définis aux articles L. 1161-2 et L. 1161-3 ne peuvent être ni élaborés ni mis en œuvre par des entreprises se livrant à l’exploitation d’un médicament, des personnes responsables de la mise sur le marché d’un dispositif médical ou d’un dispositif médical de diagnostic in vitro ou des entreprises proposant des prestations en lien avec la santé. Toutefois, ces entreprises et ces personnes peuvent prendre part aux actions ou programmes mentionnés aux articles L. 1161-2 et L. 1161-3, notamment pour leur financement, dès lors que des professionnels de santé et des associations mentionnées à l’article L. 1114-1 élaborent et mettent en œuvre ces programmes ou actions. »
Bien que la question ait été, avec la fixation des prix et la mesure de l’efficacité relative, l’un des trois grands thèmes abordés dans le cadre du Forum pharmaceutique créé en 2005 pour donner suite au processus « G10 médicaments » qui avait rendu ses conclusions en 2002, elle ne fait pas encore l’objet d’un consensus entre les Etats. Au contraire, la grande majorité n’est pas favorable en l’état au texte proposé.
Les propositions de la Commission européenne en matière d’information du public ont fait l’objet, dans le cadre des travaux préparatoires au Conseil d’un accueil plus que réservé.
Pour des motifs et avec une ampleur variables, la plupart des Etats membres sont opposés à leur adoption, soit pour des raisons de principes, soit pour des raisons de fond en l’état dirimantes.
Selon les éléments communiqués, seul le Royaume-Uni a indiqué que le texte ne lui posait pas de difficulté majeure. Vingt Etats membres se sont en revanche manifestés comme hostile ou réservés. En effet, douze pays (Allemagne, Espagne, Danemark, Suède, Finlande, Portugal, Autriche, Pologne, Belgique, Luxembourg, Pays Bas, Lituanie) ont clairement demandé le retrait de ce texte alors que huit autres (Estonie, Italie, Hongrie, Slovénie, Royaume-Uni, Irlande, Grèce, ainsi que France), ont demandé à la présidence de donner la priorité aux deux autres textes du paquet pharmaceutique (pharmacovigilance et médicaments falsifiés).
La Présidence suédoise n’a pas inscrit ce texte à l’ordre du jour du groupe concerné.
Pour ce qui concerne les entreprises pharmaceutiques, l’organisme professionnel qui les représente, l’EFPIA, s’est prononcé pour l’information des patients.
Elle considère que le défaut actuel d’harmonisation communautaire conduit à des situations différentes selon les Etats membres, ce qui pose notamment problème à ses adhérents en raison de la diversité des règles à prendre en compte, et que l’intérêt des patients est de disposer d’une information de qualité.
Ce point de vue n’est pas unanime. En effet, s’appuyant sur le cas des Etats-Unis, certaines entreprises craignent d’avoir à faire face à une inflation des coûts, chacune d’entre elles étant tenue de prévoir et de financer des actions supplémentaires de communication, la concurrence excluant a priori la possibilité de se tenir à l’écart.
Pour ce qui concerne les patients, les associations qui les représentent sont réservées. Elles considèrent également qu’Internet pose problème, avec une information « libre » d’origine indéterminée et qui n’est pas maîtrisée, mais qui est abondamment consultée par certains patients et parfois invoquée auprès du médecin.
S’agissant de leur dispositif, l’une des principales objections que l’on peut adresser aux propositions de la Commission européenne est de ne pas différencier l’information de la publicité, faute de définition adéquate de la première.
Ce manque de clarté juridique est renforcé par le fait que les conclusions du Conseil Emploi, Politique sociale, Santé, Consommateur (EPSCO) de juin 2008, sous présidence slovène, n’ont été que partiellement reprises.
Il soulève trois problèmes dirimants :
- il crée une insécurité juridique préjudiciable ;
- il implique des procédures de contrôle particulièrement lourdes et coûteuses, pour des résultats qui plus est incertains, ce qui n’est pas admissible ;
- il affecte d’une manière risquée l’équilibre de la relation patient-médecin, point qui mérite un développement particulier.
Actuellement, l’usage et l’administration des médicaments délivrés sur prescription médicale, c’est-à-dire sur ordonnance, interviennent dans un environnement très rassurant pour le patient. C’est nécessaire. Le médicament n’est pas un produit de consommation : il est destiné à soigner.
Le médecin surtout qui prescrit, mais aussi le pharmacien qui délivre, sont soumis à des règles déontologiques précises, lui donnent l’information qu’il souhaite. Ils répondent à ses questions comme à ses doutes, dans le cadre d’un dialogue. En France en outre, le libre choix du médecin comme de celui du pharmacien font que la relation avec les professionnels de santé est avant tout fondée sur un sentiment de confiance, et donc sur la fonction de conseil.
Les informations dont peut par ailleurs disposer le patient sont celles de la notice, du « mode d’emploi », visées par l’autorité sanitaire compétente en matière de médicament.
Les professionnels de santé sont pour leur part destinataires d’une information qui leur est délivrée par les firmes, et qui par définition est destinée à des personnes compétentes et formées.
En proposant qu’un message puisse directement aller des firmes vers le patient, on affecte ces équilibres et la relation médecin patient.
Il convient donc d’être très prudent dans la démarche, et ne pas méconnaître l’importance des professionnels de santé.
Les propositions de la Commission européenne n’étant pas acceptables en l’état, pour les raisons qui viennent d’être indiquées, le Gouvernement a précisé avoir envisagé quels pourraient être de nouveau points pour engager sur des bases partagées les travaux préparatoires au Conseil.
Il s’agit des points suivants :
- la reprise des critères de qualité identifiés par le Conseil « EPSCO » de juin 2008, sous présidence slovène, et qui avaient été mentionnés dans les conclusions précitées : des informations sur les produits pharmaceutiques et sur les autres traitements qui soient de qualité, objectives, impartiales, fiables, complètes, compréhensibles, pertinentes, appropriées, orientées vers le patient et non publicitaires, conformément aux exigences légales, et qui encouragent une consommation rationnelle et appropriée des médicaments ;
- un définition permettant de différencier de manière claire l’information de la publicité ;
- la prise en compte des professionnels de santé, et non seulement des entreprises, comme acteurs de la diffusion de l’information ;
- des contrôles renforcés, compte tenu de l’importance des enjeux.
Ils sont tout à fait légitimes.
II. LE DÉVELOPPEMENT DE LA PHARMACOVIGILANCE : DES PROPOSITIONS ACCEPTABLES, SOUS RÉSERVE DE CLARIFICATIONS
La pharmacovigilance est une démarche aussi sensible qu’importante. C’est, en effet, la surveillance des médicaments, une fois qu’ils ont été mis sur le marché, c’est-à-dire après la délivrance de l’AMM, de manière à prévenir et à suivre leurs effets indésirables, que ceux-ci soient réels ou supposés.
L’enjeu est donc essentiel. Dans certains cas, les données issues de la pharmacovigilance peuvent conduire à une réévaluation du rapport bénéfice/risque, ainsi qu’à la confirmation ou à la modification, voire même à la suspension, au retrait ou au non renouvellement d’une AMM.
C’est en effet à la lumière de l’expérience et de la consommation importante de médicaments par un grand nombre de patients que l’on peut constater des exigences telles qu’un renforcement des contre-indications d’un médicament ou d’une indication amoindrie.
La pharmacovigilance fait partie des domaines déjà harmonisés au niveau communautaire. De même que pour l’AMM, cette harmonisation assure la coordination entre les deux niveaux, le niveau communautaire et le niveau national.
Les dispositions communautaires actuellement applicables ont été fixées à l’occasion de la « révision pharmaceutique » engagée en 2001 et achevée en 2004. Elles figurent au titre IX de la directive précitée 2001/83/CE instituant le code communautaire du médicament à usage humain.
Pour ce qui concerne les médicaments relevant de l’AMM nationale, les dispositions prévoient plusieurs obligations pour les Etats membres :
– mettre en place un système de pharmacovigilance pour le suivi des effets indésirables des médicaments dans des conditions normales d’utilisation, ainsi que sur les cas de mésusage et d’abus pouvant avoir une incidence sur le rapport bénéfice/risque. Ce système a pour mission le recueil des informations et leur évaluation scientifique ;
– prévoir des mesures pour la notification, le cas échéant obligatoire, des effets indésirables ou inattendus par les médecins et autres professionnels de santé ;
– garantir l’indépendance des activités de pharmacovigilance, grâce à un contrôle de la gestion des fonds destinés à leurs activités.
La directive prévoit en outre la possibilité, pour un État membre, de recommander la modification, la suspension ou le retrait de l’autorisation de mise sur le marché à la suite de l’évaluation des données de pharmacovigilance. En cas d’urgence, l’État membre concerné peut également suspendre l’autorisation de mise sur le marché d’un médicament, tout en respectant certaines obligations d’information.
Pour ce qui concerne les titulaires d’AMM, plusieurs obligations sont également prévues :
– avoir dans son personnel une personne qualifiée responsable de la pharmacovigilance ;
– enregistrer et notifier à l’autorité compétente, ainsi qu’à l’Etat membre de référence pour les médicaments relevant de la procédure nationale, toute présomption d’effet indésirable, qu’ils soient ou non transmis par des professionnels de santé. A défaut d’autre procédure, la notification à l’autorité compétente prend la forme d’un rapport périodique actualisé de sécurité, transmis à intervalles réguliers (mis à jour tous les six mois en général). Ces rapports font l’objet de lignes directrices établies par la Commission européenne. Par ailleurs, un délai de quinze jours est imposé pour toute présomption d’effet indésirable grave ayant été portée à son attention par un professionnel de la santé ;
– conserver les rapports détaillés de tous les effets indésirables supposés intervenus dans la Communauté ou dans les pays tiers.
Au niveau communautaire, la directive prévoit la mise en place par l’Agence européenne des médicaments d’un réseau informatique pour faciliter l’échange d’informations relatives à la pharmacovigilance. Il s’agit notamment de permettre la circulation de l’information pour transmettre les rapports périodiques aux autres Etats membres et informer les titulaires d’AMM de tous les incidents intervenus. Les Etats membres doivent également s’assurer que les notifications d’effets indésirables graves présumés sont portées aussitôt à l’attention de l’Agence comme du titulaire de l’AMM.
Pour sa part le règlement (CE) n° 726/2004, comprend un certain nombre de dispositions sur la dimension communautaire de la pharmacovigilance, avec :
– une reprise des obligations prévues pour les titulaires d’autorisations d’AMM nationales : présence d’une personne qualifiée parmi les membres du personnel ; notification des présomptions d’effets indésirables graves aux autorités des Etats membres et à l’Agence ; transmission des rapports périodiques actualisés semestriels, à défaut d’autre instrument ;
– la transmission de toute information pertinente à l’Agence européenne du médicament, par les titulaires d’AMM, et par les autorités compétentes des Etats membres ;
– des précisions sur le réseau informatique précité (base EudraVigilance).
Pour la France, c’est l’Agence française de sécurité sanitaire des produits de santé (AFSSAPS) qui est l’autorité compétente en matière de pharmacovigilance et qui est le premier élément du système national de pharmacovigilance.
Par ailleurs, la Commission nationale de pharmacovigilance, composée de six membres de droit et trente trois membres nommés, a pour missions d’évaluer les informations sur les médicaments et produits à usage humain, de proposer les enquêtes et travaux et de donner des avis au directeur général de l’AFSSAPS sur les mesures à prendre pour faire cesser les incidents et accidents liés à l’emploi des médicaments.
Sur le plan opérationnel, l’Unité de pharmacovigilance de l’AFSSAPS anime le réseau national de pharmacovigilance. Son rôle consiste notamment à évaluer les déclarations d’effets indésirables qui lui parviennent, informer les professionnels de santé des procédures et des recommandations établies, et coordonner l’activité des centres régionaux de pharmacovigilance.
A l’échelon territorial, ce sont, en effet, les trente et un Centres régionaux de pharmacovigilance (CRPV) qui sont chargés de surveiller, d’évaluer et de prévenir les risques médicamenteux potentiels ou avérés et de promouvoir le bon usage du médicament.
Ce sont également eux qui assurent le recueil des effets indésirables et leur transmission à l’AFSSAPS, ainsi qu’une mission d’expertise avec notamment la conduite d’enquêtes et d’évaluations.
Les professionnels de santé, les patients et les associations de patients, ainsi que les entreprises du médicament sont les autres acteurs du système national de pharmacovigilance.
Ce système s’intègre dans l’organisation européenne de la pharmacovigilance structurée autour de l’Agence européenne des médicaments.
Le recueil des déclarations est opéré au niveau des Etats membres et transmis au niveau communautaire, où les questions relèvent du groupe de travail européen de pharmacovigilance, placé auprès du Comité des médicaments à usage humain (CHMP, selon l’acronyme anglais) et composé des responsables des départements de pharmacovigilance de chacun des vingt sept Etats membres ainsi que d’un représentant de la commission européenne et du secrétariat de l’Agence. Selon l’AFSSAPS, il s’agit d’un véritable forum européen de discussion et d’échanges en pharmacovigilance qui peut être saisi à la demande du CHMP ou des Etats membres. Il est en liaison avec les instances de pays tiers, notamment la FDA américaine, et avec l’OMS.
Par ailleurs, on rappellera que la base de données européenne EudraVigilance précédemment mentionnée a, en pratique, pour objectif de développer les outils de traitement et la transmission électronique des observations de cas individuels et, en favorisant leur communication, de faciliter la coordination entre les autorités nationales concernées.
B. Les propositions de la Commission européenne : un nouveau comité au sein de l’Agence européenne des médicaments, un objectif de clarification des rôles avec un renforcement du niveau communautaire, comme des instruments de suivi du médicament
Les nouvelles dispositions sur la pharmacovigilance font l’objet de deux textes parallèles : la proposition de directive modifiant, en ce qui concerne la pharmacovigilance, la directive 2001/83/CE instituant un code communautaire relatif au médicament à usage humain (document E 4187), qui vise notamment les médicaments bénéficiant d’une AMM nationale ; la proposition de règlement modifiant, en ce qui concerne la pharmacovigilance des médicaments à usage humain et le rôle de l’Agence européenne du médicament, le règlement (CE) n° 726/2004 établissant des procédures communautaires pour l’autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant une Agence européenne des médicaments (document E 4186), concerne notamment les médicaments ayant une AMM communautaire et le renforcement des instances communautaires.
Ces deux textes sont le résultat de la procédure de révision initiée, dès 2004, par une étude sur le système communautaire de pharmacovigilance et qui a fait l’objet d’une consultation publique en 2007.
Quatre objectifs sont poursuivis : rationaliser et simplifier les procédures de décision et les règles actuelles pour éviter notamment les différences d’interprétation entre les Etats membres ; améliorer la transparence et la communication en la matière ; renforcer le dispositif applicable dans les firmes pharmaceutiques ; simplifier les procédures.
Ce sont autant d’objectifs partagés. On estime, en effet, que 5 % des hospitalisations sont dues aux effets indésirables des médicaments.
La Commission européenne propose d’abord une conception plus étendue de la pharmacovigilance avec deux modifications.
D’une part, la notion d’effet indésirable présumé serait introduite, en remplacement de celle d’effet indésirable inattendu, et définie, sous réserve de la nécessité de prévoir des coordinations techniques avec d’autres textes de moindre niveau pour éviter tout risque de confusion, comme « un effet indésirable pour lequel un lien de causalité entre l’événement et le médicament ne peut être exclu ».
D’autre part, la définition de l’effet indésirable serait élargie. Celle-ci ne concernerait plus seulement les « effets indésirables des médicaments dans des conditions normales d’utilisation », c’est-à-dire dans des conditions conformes au résumé des caractéristiques du produits (RCP) et ainsi à l’AMM mais, d’une manière générale, les risques pour les patients et pour la santé publique, ce qui se traduit de manière très concrète par l’adjonction des effets en cas d’utilisation hors AMM, y compris en cas de surdosage (abus), de mésusage ou d’erreur de médication.
Si la France est favorable à cette conception plus large, de nombreux Etats membres sont en revanche défavorables.
La Commission européenne propose également un réaménagement des instruments de suivi post-AMM des médicaments, décliné selon plusieurs modalités.
La première d’entre elles, conçue comme une simplification, est la création d’un « dossier permanent de système de pharmacovigilance », pour alléger en contrepartie le dossier de demande d’AMM. Celui-ci resterait dans l’entreprise et seuls les éléments essentiels en seraient présentés dans la demande d’AMM.
La deuxième modalité tient à l’aménagement du contenu des rapports périodiques actualisés de sécurité (PSUR, selon l’acronyme anglais).
Actuellement, les dispositions applicables précisent que ces rapports servent à la notification des effets indésirables et qu’ils sont accompagnés d’une évaluation scientifique du rapport bénéfice/risque du médicament. Les mesures proposées prévoient que ces rapports contiennent dorénavant un résumé des informations en rapport avec le bénéfice et les risques du médicament, une évaluation scientifique de ce rapport réalisée « sur la base de toutes les informations disponibles, y compris pour des essais cliniques réalisés pour des indications et des populations non autorisées » et toutes informations sur le volume des ventes de médicament. L’objet des rapports serait donc modifié de manière à les centrer sur le rapport bénéfice/risque du médicament, selon la Commission européenne. La proposition de directive prévoit en effet explicitement leur évaluation et le lien entre ces évaluations et la mise à jour des AMM (modification ou retrait, notamment). Par ailleurs, ces rapports ne seraient plus systématiques pour les risques considérés comme faibles, notamment les génériques, médicaments d’usage bien établi et les médicaments à base de plantes dûment enregistrés.
La troisième modalité vise à permettre aux autorités compétentes d’imposer un système de gestion des risques pour tout dossier de demande d’AMM. La Commission européenne précise qu’un tel système doit être proportionné aux risques avérés ou potentiels, ainsi qu’au besoin d’informations supplémentaires sur le médicament. On rappellera qu’un tel système vise à décrire, prévenir ou réduire au minimum les risques d’un médicament.
La quatrième modalité tient au renforcement du rôle des études de sécurité post-autorisation. Des dispositions et des principes directeurs sont prévus pour harmoniser et contrôler ces études non interventionnelles (c’est-à-dire qui ne sont pas des essais cliniques), et éviter qu’elles ne prennent un caractère publicitaire. Ces études sont envisagées pour trois hypothèses : leur réalisation sur une base volontaire par le laboratoire concerné ; leur réalisation obligatoire, en raison de leur mention dans une AMM conditionnelle ; leur réalisation obligatoire imposée par l’autorité nationale compétente après procédure contradictoire, « s’il existe des craintes quant aux risques posés par le médicament autorisé ». Dans ce cas, l’étude devient une condition de l’AMM.
La quatrième modalité tient en effet à l’aménagement des dispositions relatives aux AMM, avec la création de l’AMM conditionnelle distincte du cas des circonstances exceptionnelles, la délivrance de l’autorisation étant alors subordonnée à l’une ou plusieurs des conditions suivantes : des mesures garantissant l’utilisation sûre du médicament incluses dans le système de gestion des risques, la réalisation d’études de sécurité post-autorisation, le respect de dispositions plus rigoureuses en matière d’enregistrement ou de notification d’effets indésirables, des mesures autres garantissant une utilisation sûre et efficace du médicament.
La cinquième modalité concerne la mise en place au niveau communautaire, par l’Agence européenne des médicaments, d’une liste des médicaments faisant l’objet d’une surveillance approfondie.
Enfin, on peut observer une sixième modalité, destinée à améliorer la rapidité de l’information du patient, avec la création d’une nouvelle rubrique intitulée « informations essentielles » prévue pour être insérée dans le résumé des caractéristiques du produit comme dans la notice. Il s’agirait des éléments nécessaires pour une utilisation sûre et efficace du médicament. Serait également mentionnée la mise sous surveillance du médicament considéré.
Plusieurs modifications sont proposées de manière à clarifier les rôles et les procédures. Elles se traduisent également par un renforcement du niveau communautaire.
La Commission européenne propose de confirmer le rôle de la base de données européenne EudraVigilance, qui devrait recenser les effets indésirables sur tous les médicaments autorisés dans la Communauté, avec accès complet aux autorités compétentes des Etats membres et de la Commission européenne, et accès partiel aux titulaires d’AMM, pour leur permettre de s’acquitter de leurs obligations.
Ainsi, les règles de notification des effets indésirables de l’ensemble des médicaments disponibles en Europe, sont prévues pour être aménagées, avec, d’une part, une notification des professionnels de santé, qui les notifient aux autorités nationales ou aux titulaires des AMM et, d’autre part, une notification des laboratoires titulaires d’AMM, que celle-ci soit nationale ou centralisée, au niveau communautaire. Pour les titulaires d’AMM, l’obligation concerne tous les incidents, et non seulement ceux intervenus sur le territoire de la Communauté, avec une distinction selon la gravité de l’effet indésirable, et ainsi un délai de transmission de quinze jours pour les cas graves et de quatre vingt dix jours pour les cas non graves.
Des mesures sont par ailleurs proposées pour faciliter la notification par les patients et par les professionnels de santé, grâce à des formulaires en ligne élaborés par l’Agence, en collaboration avec les Etats membres.
L’une des innovations des propositions de la Commission européenne est en effet la notification des effets indésirables par les patients.
S’agissant de la France, la loi précitée n° 2009-879 du 21 juillet 2009 dite « hôpital, patients, santé et territoires », a déjà introduit une telle disposition dans le droit de la santé.
De manière complémentaire, la Commission européenne propose d’enrichir le contenu de la base, avec l’obligation pour l’Agence d’organiser une veille bibliographique des publications scientifiques. L’objectif est de recenser et d’y insérer les cas observés d’effets indésirables.
Au titre du renforcement de la transparence enfin, des dispositions sont prévues pour assurer un certain accès du public à la base EudraVigilance : mise à disposition des données sous une forme agrégée avec indication de la façon de les interpréter ; possibilité de demander à consulter des notifications d’effets indésirables.
b) La création d’un portail Web européen sur la sécurité des médicaments et la coordination des portails nationaux
Les propositions de la Commission européenne prévoient un renforcement et une coordination de la communication sur la sécurité des médicaments.
Trois éléments doivent être mentionnés :
– d’une part, la création d’un portail Internet européen sur la sécurité des médicaments, géré par l’Agence européenne du médicament. Il s’agit d’un outil distinct de la base de données Eudravigilance, car c’est une plate-forme de diffusion des avertissements relatifs à la sécurité des médicaments au niveau de l’Union européenne et un relais de portails nationaux, grâce à des liens avec eux. Ce portail mentionnerait notamment les éléments relatifs à la pharmacovigilance dans l’Union, ceux afférents au comité du médicament à usage humain (CHMP) et au comité consultatif pour les risques en matière de pharmacovigilance (CERP), notamment leur composition, la déclaration d’intérêts de leurs membres, un résumé de leur réunion ainsi que leurs évaluations, recommandations avis et décisions en matière de pharmacovigilance sur des médicaments précis ;
– d’autre part, l’obligation pour les Etats membres de mettre en place un portail Internet sur la sécurité des médicaments, de manière à assurer la mise en ligne des éléments suivants, au moins : les systèmes de gestion des risques relatifs aux médicaments autorisés ; la liste des médicaments faisant l’objet d’une surveillance approfondie au niveau communautaire ; des formulaires structurés pour la diffusion en ligne des effets indésirables ;
– enfin, un rôle de coordination de la communication au sein des Etats membres, reconnu à l’Agence européenne des médicaments.
c) L’institutionnalisation d’un Comité consultatif pour l’évaluation des risques en matière de pharmacovigilance au sein de l’Agence européenne des médicaments et l’accroissement des mission du groupe de coordination des Etats membres pour l’amélioration du suivi post-AMM
Les propositions de la Commission européenne visent également à clarifier les rôles et les responsabilités avec notamment une meilleure définition des rôles des différentes parties concernés : Etats membres, Agence européenne des médicaments et laboratoires titulaires des AMM.
La principale des mesures prévues vise à renforcer la visibilité de la pharmacovigilance au sein de l’Agence et le rôle de l’instance qui est chargée, grâce à la création du comité consultatif pour l’évaluation des risques en matière de pharmacovigilance (CERP en français ou PRAC en anglais). Comme on l’a vu, ces questions sont actuellement traitées par un groupe de travail placé auprès du Comité des médicaments à usage humain.
Ce comité serait composé de quinze membres nommés pour un mandat de trois ans renouvelable une fois, sur la base de leur compétence, à raison de dix membres titulaires (et dix suppléants) désignés par le conseil d’administration de l’Agence sur proposition des autorités compétentes nationales et de cinq membres titulaires (et cinq membres suppléants) désignés par la Commission européenne sur la base des résultats d’un appel public à manifestation d’intérêt, après consultation du Parlement européen.
Par ailleurs, les représentants des autorités compétentes nationales pourraient assister aux réunions sans voix délibérative, en s’abstenant d’influencer les décisions.
La CERP jouerait un rôle en matière de prise de décision par ses évaluations en matière de pharmacovigilance, grâce à ses rapports et ses recommandations soit au comité des médicaments à usage humain, soit au groupe de coordination des Etats membres.
A ce titre, il devrait intervenir sur les suites à donner tant aux rapports périodiques actualisés de sécurité, dont il établit des rapports d’évaluation, qu’aux « signes de risques nouveaux » détectés dans le cadre des systèmes de gestion de risques dont sont assortis certains médicaments. Est également prévue une surveillance des études de sécurité post-autorisation.
La seconde mesure de clarification des rôles concerne le groupe de coordination entre les Etats membres, dont le rôle est également précisé et étendu une compétence sur touts question relative à la pharmacovigilance en liaison avec son champ d’activité, à savoir la coordination pour les AMM décentralisées.
La cohérence de l’ensemble est assurée par le fait que le groupe de coordination serait assisté en matière pharmacovigilance par le CERP.
Les propositions de la Commission visent également à clarifier la procédure communautaire applicable en cas de risque de sécurité grave portant sur un médicament.
Il s’agit plus précisément des cas où un Etat membre, soit envisage de suspendre ou de retirer une AMM, d’interdire la délivrance d’un médicament, de refuser le renouvellement d’une AMM, soit est informé, par le titulaire d’une AMM, d’une interruption de livraison, d’un retrait ou d’un projet de retrait d’AMM en cas de risque grave pour la sécurité ou d’un constat de défaillances graves lors d’une inspection de pharmacovigilance.
Dans ce cas, l’Etat concerné peut prendre les mesures de suspension de l’AMM ou d’interdiction d’utilisation et doit en informer dans les vingt quatre heures l’Agence de sécurité des médicaments, ainsi que la Commission et les autres Etats membres.
Les principales phases de la procédure ainsi engagée sont alors les suivantes :
– la saisine du CERP, qui procède à l’instruction du dossier et peut organiser pour ses travaux des auditions publiques ;
– la diffusion dans les soixante jours d’une recommandation motivée, son contenu pouvant aller de l’absence de mesure préconisée au retrait de l’AMM, en passant notamment par des mesures de réduction du risque ;
– la saisine, si aucune AMM concernée n’est une AMM centralisée, du groupe de coordination, ou, dans l’hypothèse inverse, du comité des médicaments à usage humain ;
– lorsque le groupe de coordination est saisi, deux cas sont à distinguer : s’il y a consensus, les Etats membres doivent se conformer à son avis ; s’il n’y pas consensus, l’avis majoritaire est transmis à la Commission européenne, qui arrête sa décision avec intervention de la procédure de comitologie.
La Commission européenne propose d’élargir le financement des activités de pharmacovigilance, en autorisant la perception par les autorités compétentes des Etats membres, de redevances auprès des titulaires d’AMM.
Une telle mesure est vue avec suspicion par certaines associations de patients qui craignent une atteinte à l’indépendance de la surveillance post-AMM des médicaments.
Il peut être objecté que la disposition antérieure sur le contrôle permanent des autorités compétentes nationales afin de garantir leur indépendance est maintenue.
C. Des améliorations de fond à prévoir pour clarifier les rôles et permettre le bon fonctionnement du niveau communautaire
Dans l’ensemble, les objectifs de la Commission européenne étant partagés, les modifications à apporter à la proposition de règlement et à la proposition de directive concernées visent essentiellement à apporter au dispositif prévu des clarifications supplémentaires.
Il ne s’agit cependant pas de modifications de détail. Celles-ci conditionnent en effet l’adoption des deux textes concernés.
La première clarification à opérer concerne les autorités nationales. Les textes qui seront adoptés devront, en effet, garantir leur rôle et notamment leur expertise, et ainsi, s’agissant de la France, les compétences du réseau de centres régionaux, notamment. La pharmacovigilance est, en effet, un sujet pour lequel la proximité est essentielle.
De ce point de vue, il est notamment essentiel que les déclarations directes à la base de données EudraVigilance se fassent sans préjudice d’un maintien des déclaration aux Etats membres, de manière à ne pas risquer de déperdition d’information, tant qu’il n’existe pas d’alternative totalement fiable et n’ajoutant pas de délai préjudiciable.
La deuxième clarification à opérer concerne le niveau communautaire.
Elle a trait à plusieurs éléments.
Le premier d’entre eux concerne la composition du nouveau comité consultatif pour l’évaluation des risques en matière de pharmacovigilance, du CERP.
La Commission européenne a prévu une composition de quinze membres titulaires dont dix membres désignés sur proposition des Etats membres par l’Agence européenne des médicaments, et cinq par la Commission européenne, avec un même nombre de suppléants, comme on l’a vu.
Une telle composition n’est pas acceptable. D’une part, le groupe de travail auquel le comité succède comprend un membre par Etat membre, alors qu’il joue un rôle moindre. D’autre part, la pharmacovigilance exige une association étroite de tous les pays, car elle peut impliquer des actions de communication rapides et efficaces vers les patients résidant sur leur territoire.
Un renforcement de la composition du comité sur la base de la formule vingt-sept + cinq est donc nécessaire. On peut même aller au-delà dans la mesure où, par exemple, il serait légitime que les associations de patients soient représentées, par un membre par exemple.
Une telle modification, qui recueille l’assentiment des Etats membres, ne pose pas de difficulté.
Le deuxième élément concerne l’articulation du nouveau comité avec, d’une part, le comité des médicaments à usage humain (CHMP) de l’Agence européenne du médicament et, d’autre part, le groupe de coordination des Etats membres (CMD).
La décision appartenant soit au CHMP, soit au CMD, il convient de bien préciser les conditions d’une mise en œuvre harmonieuse de leurs rôles, de manière à éviter les doublons ainsi qu’à permettre le plein respect du principe des compétences de l’autonomie de chacun, ainsi que du principe de collégialité de la décision. Des aménagements sont ainsi à prévoir sur les délais de publicité des actes.
Si le collectif Europe et Médicaments souhaite que soit envisagée une séparation entre l’autorité qui autorise et celle qui intervient a posteriori, éventuellement pour modifier ou suspendre l’autorisation, une telle hypothèse apparaît délicate à envisager, à ce stade. Le risque de dysfonctionnement d’un système fondé sur ces hypothèses n’est pas négligeable, en raison d’une possibilité de « concurrence » entre les deux instances, comme d’un risque d’accaparement de la prise de décision effective par la seule instance de contrôle, puisqu’elle se prononcerait toujours en second. La crédibilité de l’ensemble pourrait ainsi être mise en cause. Ce serait particulièrement préjudiciable, s’agissant d’un domaine aussi sensible que la santé.
Le troisième élément concerne la base EudraVigilance. Son mode de fonctionnement actuel n’est pas en effet satisfaisant et, comme il l’a été indiqué à la rapporteure, le schéma proposé par la Commission européenne ne pourra fonctionner que si une importante mise à niveau est opérée, ne serait-ce que pour collecter des données exploitables par les autorités et les experts concernés.
Enfin, le quatrième élément concerne la question, délicate, des équilibres entre les exigences antérieures à l’octroi de l’AMM et celles ultérieures à sa délivrance.
Il convient, en effet, par des clarifications adaptées, de lever les craintes d’un développement des AMM prématurées. Le développement des études post-AMM, qui est la contrepartie d’un médicament plus sûr, est parfois perçu comme celle d’une réduction des exigences requises pour la délivrance des AMM.
Ce sujet relève d’ailleurs pour partie d’un autre corps de règles, celui défini par la directive 2001/20/CE du 4 avril 2001 sur la conduite des essais cliniques de médicaments à usage humain.
I. LA LUTTE CONTRE LES MÉDICAMENTS FALSIFIÉS : UN OBJECTIF CONSENSUEL ET PARTAGÉ, MAIS DES MODALITÉS À PRÉCISER ET AMÉLIORER
A. La proposition de la Commission européenne : un renforcement de la sécurité de la chaîne de fabrication et de distribution du médicament
Comme le rappelle l’Organisation mondiale de la santé (OMS) dans son rapport de 2000 sur les médicaments « contrefaits », « la contrefaçon des produits commerciaux est aussi vieille que le monde. »
Néanmoins, ce truisme interdit tout immobilisme, car si le phénomène n’est pas neuf, il connaît un développement sans précédent, lequel est facilité par la mondialisation et le développement des échanges commerciaux.
Des mesures nouvelles sont donc nécessaires pour le contrer et le réprimer.
C’est un enjeu de santé publique important. Les conséquences de la distribution de faux médicaments ne sont pas tolérables : des décès, des empoisonnements, des pathologies qui ne guérissent pas. En outre, les préjudices économiques et sociaux sont réels.
C’est le constat de l’OMS, qui a ainsi créé une structure spécifique, le Groupe spécial international de lutte contre la contrefaçon de produits à usage médical (IMPACT, selon l’acronyme anglais). C’est aussi celui fait au sein des institutions du Conseil de l’Europe, au sein desquelles la Pharmacopée représente une référence sur laquelle s’appuie d’ailleurs régulièrement l’Union européenne en matière de médicament.
Celui-ci a bien cerné le phénomène, qui dépasse largement la question de la contrefaçon stricto sensu, c’est-à-dire de violation des droits de propriété intellectuelle.
On constate en effet le développement du « commerce » de « médicaments » qui « ne présentent pas nécessairement la qualité à laquelle ils prétendent et peuvent porter des étiquettes indiquant une fausse identité et/ou provenance. Ils peuvent être importés, passer en contrebande, ou être fabriqués localement par de grands consortiums dans de grandes usines et des établissements dotés du matériel le plus moderne, ou par de petits agents dans des locaux de taille réduite et souvent mal équipés. »
Les exemples les plus couramment cités sont :
- les produits ne contenant aucun des principes actifs déclarés sur l’étiquetage ;
- les produits renfermant des principes actifs autres que ceux déclarés sur l’étiquetage ;
- les produits contenant la dose correcte des principes actifs déclarés, mais avec une provenance différente de celle qui est déclarée ;
- les produits renfermant les principes actifs déclarés, mais à des doses différentes de celles qui sont déclarées
– les produits où l’on retrouve des divergences au niveau de la nature et de la quantité des impuretés.
Comme pour toute activité illégale, on ne dispose pas de données chiffrées précises, mais uniquement d’estimations. Celles-ci vont de 5 % à 20 % du marché mondial.
Pour sa part, dans sa Recommandation n° 1794 (2007) sur la qualité des médicaments en Europe, l’Assemblée parlementaire du Conseil de l’Europe, dont l’importance des travaux est rappelée ci-après, retient une estimation 10 % de ce même marché mondial des médicaments et rappelle que l’OCDE a évalué les préjudices économiques correspondants à 500 milliards d’euros.
Cette dimension internationale a été rappelée par le Président Jacques Chirac, lors du lancement le 12 octobre dernier, à Cotonou, de la campagne internationale contre les médicaments falsifiés.
Le médicament falsifié n’affecte pas les pays de la même manière. Il touche moins les pays développés que les pays en développement.
Pour les premiers, selon les travaux précités du Conseil de l’Europe, le conditionnement est en général presque identique à celui d’un vrai médicament, et les circuits de fabrication comme de distribution sont systématiquement internationalisés de manière à éviter le plus possible la traçabilité, notamment parce que la fabrication des matières d’origine exige une industrie efficace dont peu de pays disposent. Des opérations de conditionnement sont parfois effectuées à bord de navires. Enfin, c’est un marché lucratif.
Pour les pays en développement, la falsification peut être plus grossière, car les médicaments se trouvent partout, dans les pharmacies, où la sécurité des approvisionnements n’est pas garantie, ainsi que dans les commerces et sur les marchés.
On sait également que ce phénomène contamine l’Union européenne en raison de certaines mutations, même si, comme dans les autres pays développés, le phénomène est moins répandu que dans le Tiers monde :
– le développement d’Internet, qui permet des achats « hors sol » dont l’origine n’est pas contrôlable, mais qui présentent notamment pour certains particuliers l’attrait de produits moins chers (l’original est peu ou pas remboursé) ou disponibles sans avoir à faire appel à une prescription médicale pour des raisons de « confidentialité ». Il n’est pas indifférent qu’une grande partie des annonces publicitaires « sauvages » sur Internet ou par e-mail concerne le Viagra ;
– la complexité de la chaîne de distribution, notamment avec le développement des échanges lié à la mondialisation ;
– le flou du statut des produits introduits dans l’Union européenne pour y transiter en principe, mais non pour y être mis sur le marché ;
– l’existence de réseaux et de chaînes de produits contrefaits, qui peuvent fabriquer et écouler un jour des marques de luxe, un autre jour des faux médicaments, un autre jour encore des stupéfiants ;
– la possibilité d’abuser la clientèle en raison de la difficulté à distinguer le vrai du faux.
On sait également que la France est a priori moins vulnérable pour trois raisons.
D’une part, le prix des médicaments est généralement assez bas. Celui des médicaments remboursables, qui représentent près des trois quarts du chiffre d’affaires du secteur, n’est pas libre, mais en principe fixé par accord ou convention entre le laboratoire et le Comité économique des produits de santé (CEPS) et, à défaut, par arrêté ministériel. Il n’y a donc pas d’incitation à se fournir sur des réseaux autres que celui des pharmacies.
D’autre part, le mécanisme de remboursement, avec non seulement l’assurance maladie, mais également les complémentaires, est favorable.
Enfin, la chaîne de distribution est bien contrôlée, avec un rôle clef des grossistes répartiteurs, cette activité étant soumise à autorisation.
Pour autant, notre pays ne peut pas pour autant se considérer comme indemne de tout risque.
a) Une approche large de santé publique, reposant sur la notion de médicament falsifié, et complémentaire de celle du Conseil de l’Europe
Pour la proposition de directive modifiant la directive 2001/83/CE en ce qui concerne la prévention de l’introduction dans la chaîne d’approvisionnement légale de médicaments falsifiés du point de vue de leur identité, de leur historique ou de leur source (E 4188), la Commission européenne a retenu une approche pragmatique et large du faux médicament.
Elle a, en effet, opté pour la notion de « médicament falsifié du point de vue de son identité, de son historique ou de sa source », et non celle de médicament « contrefait », laquelle ne recouvrirait que les seules violations des droits de propriété intellectuelle.
C’est l’approche que retient le groupe précité IMPACT de l’OMS.
La distinction est importante et tout à fait justifiée, ne serait-ce que parce que la falsification ne concerne pas seulement les médicaments protégés, les princeps, mais également les génériques.
On observera que la création d’un nouvel instrument communautaire est complémentaire des travaux du Conseil de l’Europe, dont il faut saluer l’importance et l’antériorité des initiatives en la matière.
En effet, s’appuyant sur l’expertise de la Direction européenne de la qualité du médicament et des soins de santé et sur la base politique de la Recommandation précitée n° 1794 (2007), dont le rapporteur était M. Bernard Marquet (Monaco), une nouvelle convention du Conseil de l'Europe est en cours de préparation, de manière à sécuriser la chaîne, renforcer les sanctions applicables et favoriser la coopération entre Etats.
Elle concernera, comme pour la Pharmacopée, un groupe de pays plus important que les seuls pays de l’Union européenne, et de l’EEE. Il y a quarante sept membres du Conseil de l’Europe. Il s’agit également d’une convention internationale classique, alors que les directives et règlements communautaires sont des instruments de mise en œuvre de règles communes dans le marché unique. Les deux démarches, celle du Conseil de l’Europe et celle de la Communauté, sont donc complémentaires.
Sur le fond, la Commission européenne propose un ensemble de mesures qui visent à sécuriser la chaîne d’approvisionnement en médicaments, à tous les maillons, de la fabrication du principe actif, qui est le composant essentiel du médicament, jusqu’à la distribution au patient.
Il s’agit d’assurer l’identification, la traçabilité et l’authenticité des produits.
La première d’entre elles vise à imposer des dispositifs de sécurité permettant d’établir l’identification, l’authenticité et la traçabilité des médicaments soumis à prescription, c’est-à-dire délivrés sur ordonnance. Les acteurs situés entre le fabricant et le dernier acteur (le pharmacien en général) ou l’utilisateur auraient en principe l’interdiction d’enlever, de recouvrir ou de modifier ces dispositifs de sécurité.
Ces dispositifs sont destinés à permettre aux distributeurs en gros, aux pharmaciens et aux personnes autorisées de vérifier l’authenticité du médicament, d’identifier les emballages individuels et de contrôler qu’il n’y a pas eu forçage. Ils représentent aussi une sécurité pour le patient qui peut constater que le produit n’a pas été ouvert.
Plusieurs conditions sont prévues pour permettre le reconditionnement et les éventuelles manipulations en cours de chaîne, et éviter ainsi d’interdire de fait le commerce parallèle :
– seuls les titulaires d’une autorisation de fabrication peuvent intervenir ;
– un nouveau dispositif de sécurité équivalent doit être apposé pour établir de manière interrompue l’identification, l’authenticité et la traçabilité du médicament ;
– le remplacement du dispositif de sécurité est soumis au contrôle de l’autorité compétente, avec ainsi une inviolabilité pour le reste de la chaîne.
Les modalités d’application de cette mesure sont renvoyées à un règlement de la Commission européenne, étant à ce stade uniquement prévu qu’elles devront notamment tenir compte du prix et du volume de vente du médicament et du risque de falsification, ainsi que de la gravité des conditions à traiter.
Les travaux ne sont pas encore suffisamment avancés pour connaître la forme que prendra ce dispositif : cachet ; marque permettant d’assurer la traçabilité ; puce électronique ou autre dispositif.
L’enjeu technique et économique est important.
Il l’est d’autant plus qu’il interfère avec les choix techniques déjà opérés par certains pays. S’agissant de la France, l’avis aux titulaires d’autorisation de mise sur le marché de médicaments à usage humain et aux pharmaciens responsables des établissements pharmaceutiques, publié au Journal officiel du 16 mars 2007, a informé les professionnels que l’AFSSAPS avait retenu le principe du changement du code identifiant de présentation (CIP) de sept à treize caractères du code à barres 39 vers l’EAN 128 (associé à un marquage Data Matrix ECC.200), dans le cadre des obligations de traçabilité des médicaments. Il s’agit de la mise en place de la norme Data Matrix.
En outre, le dispositif de sécurité peut soulever des questions plus vastes. Ainsi, la puce électronique suscite d’ores et déjà des réserves d’associations de patients, car elle pourrait suivre non seulement le médicament, mais également le patient, ce qui exigerait que des précautions soient alors prises pour éviter tout risque d’atteinte aux droits de la personne s’il s’avérait que la technique permette effectivement de « tracer » le patient.
Les autres mesures prévues par la Commission européenne visent également à renforcer l’étanchéité de la chaîne de distribution des médicaments avec :
– l’obligation pour tous les acteurs d’informer les autorités compétentes en cas de mise au jour de médicaments falsifiés ou de soupçons ;
– des mesures nouvelles pour les distributeurs en gros avec, d’une part, une liste centralisée des distributeurs en gros titulaires des autorisations d’exercer et établis dans les Etats membres, gérée par l’Agence européenne des médicaments, et l’obligation pour ces mêmes distributeurs en gros de s’assurer de la fiabilité de leurs partenaires commerciaux, à savoir : obligation de contrôler ou de faire contrôler par un tiers accrédité que leur fournisseur distributeur respecte les bonnes pratiques de distribution ; obligation de vérifier que le fabricant ou l’importateur détient une autorisation de fabrication ;
– la création d’obligations nouvelles pour les acteurs de la chaîne de distribution autre que les distributeurs en gros. L’objectif est de couvrir le négoce et le courtage, dont les opérations ne conduisent pas nécessairement à entrer en contact avec le médicament. Les professionnels concernés devraient ainsi notifier leur activité à l’Etat membre où ils sont établis, ainsi que veiller à ce que les médicaments concernés soient couverts par une AMM ;
– la sécurisation des substances actives avec l’enregistrement des importateurs et fabricants de substances actives implantés dans les Etats membres, l’obligation pour les fabricants de médicaments de s’assurer que leurs fournisseurs de principes actifs respectent les bonnes pratiques de fabrication des substances actives, ainsi qu’un renforcement des exigences relatives à la production de principes actifs, y compris lorsqu’ils sont destinés à l’exportation ;
– des règles plus strictes en matière d’inspection, avec notamment des lignes directrices pour les inspections des laboratoires producteurs de médicaments et des producteurs de substances actives ;
– la publicité des rapports d’inspection effectués par les autorités compétentes, avec publication sur la base de données EudraGMP de l’Agence européenne des médicaments ;
– et, enfin, l’obligation pour les Etats membres de prévoir des sanctions en cas d’infraction.
Pour prendre en compte la dimension internationale de la criminalité pharmaceutique, deux mesures sont prévues.
La première vise à éviter tout risque de détournement de trafic et ainsi prévoit l’obligation pour les Etats membres de veiller à l’interdiction d’importer des médicaments falsifiés, y compris lorsque ceux-ci ne sont pas destinés au marché européen mais à des pays tiers. Il s’agit d’interdire les opérations de transit.
La seconde vise à renforcer la garantie concernant les substances actives importées des pays tiers. Ceux-ci ne pourront être introduites sur le territoire communautaire que si elles sont fabriquées :
– soit dans un pays figurant sur la liste, établie par la Commission européenne, des pays qui disposent d’un cadre règlementaire du niveau de celui de l’Union européenne ;
– soit selon des normes de bonnes pratiques de fabrication au moins équivalentes à celles en vigueur dans l’Union européenne et accompagnées d’un certificat du pays tiers attestant de l’application de telles normes au moins équivalentes par l’établissement concerné.
Dans l’ensemble, la proposition de directive propose un cadre satisfaisant.
Les aménagements à y apporter ne concernent que quelques points, mais elles sont indispensables dans la mesure où le futur texte doit nécessairement être à la hauteur des enjeux. Pour l’essentiel, cinq d’entre eux doivent être mentionnés ici.
La première amélioration à prévoir vise à une sécurité juridique renforcée. Elle consiste à préciser le dispositif en y insérant, sur la base des travaux du groupe précité IMPACT de l’OMS, une définition du médicament falsifié.
La deuxième d’entre elles concerne un problème de fond. La proposition de la Commission européenne ne prévoit de dispositif de sécurité que pour les seuls médicaments soumis à prescription. C’est une position qui se conçoit, puisque ce sont les médicaments les plus importants.
Néanmoins, vis-à-vis du public, il apparaît difficile d’exclure les médicaments en vente libre, qui ne manqueront pas d’apparaître comme des produits de « deuxième zone ».
En outre, la liste des médicaments soumis à prescription est différente d’un pays à l’autre, puisqu’elle relève de la compétence des autorités nationales en application du principe de subsidiarité. Par conséquent, la mesure proposée risque en l’état de créer de grandes difficultés pratiques pour les médicaments soumis à prescription dans certains pays et pas dans d’autres.
Enfin, comme ce sont des produits en vente libre et dont le prix l’est aussi, les risques de contrefaçon et de falsification ne doivent pas non plus être négligés. Il convient donc d’étendre le champ des produits couverts par le futur dispositif.
La troisième amélioration à prévoir est d’ordre technique. Elle vise à inclure les excipients dans le champ de la proposition de directive, de manière à ce qu’ils offrent les mêmes garanties de qualité que les substances actives.
On rappellera que les excipients sont les substances sans activité thérapeutique entrant dans la composition du médicament ou utilisées pour sa fabrication. Il s’agit d’améliorer l’aspect ou le goût du médicament, ou d’assurer sa conservation, de faciliter la mise en forme et son administration. Un excipient peut aussi servir à acheminer la substance active vers son site d’action et à contrôler son absorption par l’organisme.
Le cas d’un sirop pédiatrique contre la toux dont l’excipient était frauduleux montre qu’il ne s’agit pas d’une hypothèse théorique.
Le quatrième aménagement est également d’ordre technique et consiste à bien distinguer le médicament falsifié de celui du défaut de fabrication du médicament. Dans le premier cas, l’intention du faux est délibérée. Il convient de sanctionner. Dans le second, il s’agit d’un incident d’ordre industriel qui n’est pas acceptable en matière de médicaments, mais qui se matérialise en principe par le rappel des lots concernés.
L’un des enjeux d’ailleurs du marquage précité Data Matrix, qui dès lors qu’il sera appliqué sur chaque boîte, permettra au pharmacien d’opérer, le cas échéant, un rappel directement auprès du patient.
Un cinquième aménagement viserait à assurer la coordination avec la future convention, précitée, du Conseil de l’Europe.
Par ailleurs, plusieurs précisions ou renforcement des mesures prévues sont également à prévoir notamment sur les compétences des autorités sanitaires, ainsi que l’équivalence entre les bonnes pratiques de fabrication en vigueur dans l’Union européenne et celles mises en œuvre dans les pays tiers.
Ces modifications laissent de côté l’une des interrogations que n’a pas manqué de susciter la proposition de la Commission européenne : alors qu’Internet est l’un des principaux vecteurs des médicaments falsifiés, celle-ci est en effet muette sur ce sujet et il convient de combler cette lacune.
Quelques éléments rendent, à ce stade, une telle initiative inopportune.
En premier lieu, la proposition de directive est neutre et vise à éviter que les médicaments falsifiés ne puissent pénétrer le réseau légal, indépendamment de sa forme.
Pour la France, les quelque onze grossistes répartiteurs ne sont pas les seuls interlocuteurs des 22 500 officines pharmaceutiques, qui selon les données du LEEM en septembre dernier, commandent directement au fabriquant, par voie électronique le cas échéant, un cinquième environ des médicaments qu’elles distribuent.
En deuxième lieu, il convient d’éviter toute initiative qui légaliserait au niveau communautaire les pharmacies sur Internet et imposerait de fait une solution aux Etats membres.
C’est un débat qui relève actuellement du débat national. C’est à ce niveau que la question doit être tranchée.
Actuellement, tous les Etats membres n’ont pas les mêmes règles en la matière.
La Belgique a, en effet, récemment autorisé la pharmacie sur Internet. Un arrêté royal du 21 janvier 2009 portant instructions pour les pharmaciens, permet aux pharmacies ouvertes au public et autorisées, à vendre par Internet des médicaments à usage humain non soumis à prescription et des dispositifs médicaux, uniquement.
Des conditions très strictes sont prévues et rappelées sur le site de l’Agence Fédérale des Médicaments et des Produits de Santé (AFMPS) : les produits sont délivrés « à partir de la pharmacie, sous l’entière responsabilité du pharmacien et en suivant les règles de bonnes pratiques officinales. Des dispositions sont également prévues pour garantir que l’offre en vente, la commande, l’empaquetage et la fourniture soient organisés de manière à respecter le droit à la protection de la vie privée du patient. Le site Internet doit aussi être conçu de manière à favoriser l’usage rationnel des médicaments et des dispositifs médicaux, et comporter ainsi obligatoirement un certain nombre d’informations visant à leur bon usage. »
Néanmoins, la question est très complexe, car elle a évidemment des implications transfrontalières. La consultation d’un site dont l’extension est « .be » montre que le public français est aussi visé.
En tout état de cause, si une initiative communautaire est nécessaire, elle doit intervenir dans le cadre d’un texte spécifique qui aborde bien l’ensemble de la question.
La Commission s’est réunie le mercredi 28 octobre 2009, sous la présidence de M. Didier Quentin, Vice-président, pour examiner le présent rapport d’information.
L’exposé du rapporteur a été suivi d’un débat.
M. Jean Gaubert. Les textes sur l’information des patients suscitent d’importantes réserves – c’est justifié. Il faut néanmoins tenir compte de ce que les patients, de plus en plus, se réfèrent à Internet pour chercher le nom des médicaments adaptés à leurs cas. C’est est un véritable problème qui n’a actuellement pas de solution. Les problèmes liés aux faux médicaments ne sont heureusement pas fréquents en France, alors qu’ils deviennent préoccupants et graves dans certains pays du monde. J’approuve donc la proposition de résolution.
M. Jean-Claude Mignon. Ce rapport est très intéressant mais il ne fait pas référence au travail de la Pharmacopée qui travaille sur ce thème très approfondi au sein du Conseil de l’Europe. Il faut également citer la Recommandation n°1794 de 2007 de l’Assemblée parlementaire du Conseil de l’Europe. Il paraît souhaitable que celle-ci soit mentionnée expressément dans le corps du rapport écrit.
M. Philippe Tourtelier. Je rejoins les propos de M. Gaubert et je souhaite insister sur les médicaments falsifiés qui font des ravages dans les pays pauvres. La France risque de n’en être pas à l’abri compte tenu du développement d’une population à faible pouvoir d’achat. Quelle est par ailleurs la définition d’un médicament falsifié ?
M. André Schneider. Ce rapport est très satisfaisant. Il est important de noter que le développement des génériques a induit une contrebande et une falsification des médicaments encore pire qu’avant dans les pays pauvres et, notamment, en Afrique. Ces pays ne recueillent même pas la plupart du temps les bénéfices de ces trafics dans la mesure où les vrais bénéficiaires sont quelquefois dans les pays riches. Que pensez-vous des perspectives dans ce domaine ?
Mme Valérie Rosso-Debord, rapporteure. Internet est un véritable problème et les faux médicaments représentent un marché très rentable et bien moins risqué que celui d’autres trafics. La France est assez à l’abri car, d’une part, la filière d’approvisionnement est claire et bien normée et, d’autre part, le remboursement annihile tout intérêt de se fournir en dehors des circuits normaux. Le problème posé par Internet ne pourrait être résolu que par une directive qui y serait consacrée. Une parade efficace résiderait dans des règles permettant au niveau médical d’assurer la sécurité de la distribution des médicaments.
Le rôle du Conseil de l’Europe et de la Pharmacopée est évoqué dans le corps du rapport écrit. Il est nécessaire que l’Union européenne, dont le périmètre est différent de celui du Conseil de l’Europe et qui a besoin de normes juridiques plus exigeantes en raison du marché unique, ait ses propres règles de pharmacovigilance.
M. André Schneider. Il faut que les réseaux de distribution des médicaments soient sûrs, on rencontre certains problèmes notamment pour les médicaments reconditionnés. Le système français, qui est satisfaisant, devrait être adapté au niveau de l’Union européenne.
M. Didier Quentin, Président. L’ancien Président de la République, M. Jacques Chirac a également engagé une action, au niveau international, dans la lutte contre les médicaments falsifiés. Je signale que des associations qui récupéraient des médicaments ont du arrêter cette activité.
Mme Valérie Rosso-Debord, rapporteure. C’est regrettable, mais c’est aussi inévitable compte tenu de l’exigence de sécurité. Les organisateurs des circuits de fraude tirent parti de toutes les opportunités.
A l’issue de ce débat, la Commission a adopté la proposition de résolution dont le texte figure ci-après.
L’Assemblée nationale,
Vu l’article 88-4 de la Constitution,
Vu la proposition de règlement du Parlement européen et du Conseil modifiant, en ce qui concerne la diffusion auprès du public d’informations relatives aux médicaments à usage humain soumis à prescription médicale, le règlement (CE) n° 726/2004 établissant des procédures communautaires pour l’autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant une Agence européenne des médicaments (COM [2008] 662 final/n° E 4184),
Vu la proposition de directive du Parlement européen et du Conseil modifiant, en ce qui concerne la diffusion auprès du public d’informations relatives aux médicaments soumis à prescription médicale, la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain (COM [2008] 663 final/n° E 4185),
Vu la proposition de règlement du Parlement européen et du Conseil modifiant, en ce qui concerne la pharmacovigilance des médicaments à usage humain, le règlement (CE) n° 726/2004 établissant des procédures communautaires pour l’autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant une Agence européenne des médicaments (COM [2008] 664 final/n° E 4186),
Vu la proposition de directive du Parlement européen et du Conseil modifiant, en ce qui concerne la pharmacovigilance, la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain (COM [2008] 665 final/n° E 4187),
Vu la proposition de directive du Parlement européen et du Conseil modifiant la directive 2001/83/CE en ce qui concerne la prévention de l’introduction dans la chaîne d’approvisionnement légale de médicaments falsifiés du point de vue de leur identité, de leur historique ou de leur source (COM [2008] 668 final/n° E 4188),
Vu également la communication de la Commission au Parlement européen, au Conseil, au Comité économique et social et au Comité des régions « Des médicaments sûrs, innovants et accessibles : une vision nouvelle du secteur pharmaceutique » (COM [2008] 666 final),
Considérant que l’intérêt des patients est de disposer de médicaments sûrs, adaptés à leurs pathologies et disponibles sur le territoire de l’Union européenne ;
Saluant ainsi la communication précitée pour un secteur pharmaceutique efficace et innovant, dès lors qu’elle prévoit les conditions scientifiques et industrielles nécessaires à l’accomplissement de cet objectif ;
1. Considère, s’agissant de l’information du public :
a) que la proposition de directive et la proposition de règlement précitées (documents E 4184 et E 4185) ne sont pas, en l’état, acceptables, notamment en ce qu’elles ne différencient pas l’information de la publicité ;
b) et que cette question ne pourra être à nouveau abordée à l’avenir que sur de tout autres bases, lesquelles devront notamment tenir compte des initiatives prises par les Etats membres en matière d’éducation thérapeutique, comme du rôle éclairé des personnels médicaux, notamment médecins et pharmaciens, dans ce domaine ;
2. Salue, pour ce qui concerne la pharmacovigilance, l’objectif d’un renforcement du suivi des médicaments après leur autorisation de mise sur le marché et les orientations des mesures proposées, notamment lorsqu’elles assurent une meilleure appréciation du rapport bénéfices/risques des médicaments et garantissent la transparence des décisions, mais estime néanmoins que :
a) le dispositif de la proposition de règlement et de la proposition de directive précitées (documents E 4186 et E 4187) doit être clarifié pour garantir tant la capacité d’expertise et de décision des autorités nationales que leur rôle d’interlocuteur de proximité dans le suivi, l’analyse et l’évaluation des effets indésirables ;
b) l’articulation du comité consultatif pour les risques en matière de pharmacovigilance avec le comité des médicaments à usage humain relevant de l’Agence européenne des médicaments, comme avec le groupe de coordination des Etats membres, doit être précisée et sa composition doit être modifiée, de manière à assurer la représentation d’experts nommément désignés par chaque Etat membre ;
c) le fonctionnement de la base de données EudraVigilance doit être amélioré pour que le niveau communautaire puisse effectivement jouer le rôle qui lui sera dévolu, sans préjudice d’un maintien de l’obligation de déclaration des effets indésirables à l’autorité compétente de l’Etat membre concerné ;
d) et que le respect des équilibres entre, d’une part, les essais, évaluations et études à opérer avant l’autorisation de mise sur le marché d’un médicament et, d’autre part, des études et instruments de suivi qui ont vocation à intervenir après sa délivrance, doit être garanti ;
3. Salue, enfin, la proposition de directive relative à la lutte contre les médicaments falsifiés (document E 4188) et souhaite son adoption dans les meilleurs délais, dès lors que les améliorations nécessaires à sa mise en œuvre efficace et la plus large possible lui auront été apportées, notamment une définition appropriée des médicaments falsifiés et l’intégration des excipients dans son champ d’application.
ANNEXE :
LISTE DES PERSONNES RENCONTREES
Ø Ministère de la santé et des sports
– M. Vincent Richez, conseiller diplomatique au cabinet de Mme Roselyne Bachelot-Narquin, ministre de la santé et des sports ;
– M. Vincent Houdry, conseiller technique ;
– M. Maxime Durier, attaché parlementaire ;
– Mme Danielle Golinelli, direction générale de la santé, adjointe à la sous-directrice,
sous-direction de la politique des pratiques et des produits de santé.
Ø Agence française de sécurité sanitaire des produits de santé (AFSSAPS)
– Mme Fabienne Bartoli, adjointe au directeur général.
Ø Collectif Europe et Médicament
– Mme Marie-Josée Augé-Caumon, Union syndicale des pharmaciens d’officine (USPO) ;
– M. Pierre Chirac, revue Prescrire ;
– Mme Florence Vandevelde, Internal Society of Drug Bulletin.
Ø Les entreprises du médicament (LEEM)
– M. Dominique Amory, administrateur et président de la Commission Europe, président de Lilly France ;
– Mme Aline Bessis, directrice des affaires publiques ;
– Mme Béatrice Kressman, directrice des affaires européennes et internationales ;
– Mme Chrystel Lanxade, conseiller juridique.
Ø Mutualité française
– Mme Isabelle Millet-Caurier, directrice des affaires publiques ;
– M. Vincent Figuereau,, responsable du département des relations extérieures ;
– Mme Laure Lechertier, responsable du département Politique du médicament.
1 () La composition de cette Commission figure au verso de la présente page.
2 () La proposition de règlement du Parlement européen et du Conseil modifiant, en ce qui concerne la diffusion auprès du public d’informations relatives aux médicaments à usage humain soumis à prescription médicale, le règlement (CE) n° 726/2004 établissant des procédures communautaires pour l’autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant une Agence européenne des médicaments (COM(2008) 662 - E 4184) ; la proposition de directive du Parlement européen et du Conseil modifiant, en ce qui concerne la diffusion auprès du public d’informations relatives aux médicaments soumis à prescription médicale, la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain (COM(2008) 663 - E 4185) ; la proposition de règlement du Parlement européen et du Conseil modifiant, en ce qui concerne la pharmacovigilance des médicaments à usage humain, le règlement (CE) n° 726/2004 établissant des procédures communautaires pour l’autorisation et la surveillance en ce qui concerne les médicaments à usage humain et à usage vétérinaire, et instituant une Agence européenne des médicaments (COM(2008) 664 - E 4186) ; la proposition de directive du Parlement européen et du Conseil modifiant, en ce qui concerne la pharmacovigilance, la directive 2001/83/CE instituant un code communautaire relatif aux médicaments à usage humain (COM(2008) 665 - E 4187) ; la proposition de directive du Parlement européen et du Conseil modifiant la directive 2001/83/CE en ce qui concerne la prévention de l’introduction dans la chaîne d’approvisionnement légale de médicaments falsifiés du point de vue de leur identité, de leur historique ou de leur source (COM(2008) 668 - E 4188).
3 () Les classes de médicaments qui en représentent la proportion la plus importante sont actuellement les hypolipémiants (4,7 %), contre le cholestérol, les antiulcéreux (4,4 %), les anticancéreux (3,8 %), les antipsychotiques (3,5 %), les antidépresseurs (3,1%), et les antiépileptiques (2,5 %). On observe également que les antiviraux HIV représentent 1,9 % de ce total.
4 () S’agissant des médicaments autorisés par AMM centralisées, la proposition de règlement prévoit un contrôle a priori de l’Agence européenne des médicaments, avec mécanisme d’approbation tacite en l’absence d’objection dans un délai de 60 jours, pour ces deux dernières catégories.