

N° 2497
______
ASSEMBLÉE NATIONALE
CONSTITUTION DU 4 OCTOBRE 1958
TREIZIÈME LÉGISLATURE
Enregistré à la Présidence de l’Assemblée nationale le 5 mai 2010.
RAPPORT D’INFORMATION
DÉPOSÉ
PAR LA COMMISSION DES AFFAIRES EUROPÉENNES(1)
sur la proposition de règlement sur les biocides
(E 4532),
ET PRÉSENTÉ
PAR MM. Jean GAUBERT et Robert LECOU,
Députés
——
La Commission des affaires européennes est composée de : M. Pierre Lequiller, président ; MM. Michel Herbillon, Jérôme Lambert, Thierry Mariani, Didier Quentin, vice-présidents ; M. Jacques Desallangre, Mme Marietta Karamanli, MM. Francis Vercamer, Gérard Voisin secrétaires ; M. Alfred Almont, Mme Monique Boulestin, MM. Pierre Bourguignon, Yves Bur, François Calvet, Christophe Caresche, Philippe Cochet, Bernard Deflesselles, Lucien Degauchy, Michel Delebarre, Michel Diefenbacher, Jean Dionis du Séjour, Marc Dolez, Daniel Fasquelle, Pierre Forgues, Jean-Claude Fruteau, Jean Gaubert, Hervé Gaymard, Guy Geoffroy, Mmes Annick Girardin, Anne Grommerch, Elisabeth Guigou, Danièle Hoffman-Rispal, MM. Régis Juanico, Marc Laffineur, Robert Lecou, Michel Lefait, Lionnel Luca, Philippe Armand Martin, Jean-Claude Mignon, Jacques Myard, Michel Piron, Franck Riester, Mmes Chantal Robin-Rodrigo, Valérie Rosso-Debord, Odile Saugues, MM. André Schneider, Philippe Tourtelier.
SOMMAIRE
___
Pages
INTRODUCTION 7
PREMIERE PARTIE : UN DROIT EUROPÉEN RÉCENT ET ENCORE AU DÉBUT DE SA MISE EN œUVRE 11
I. UNE PREMIÈRE DIRECTIVE EN 1998 11
A. LES BIOCIDES : UNE CATÉGORIE DIVERSE ET IMPORTANTE 11
1. Les biocides et leurs usages 11
2. Un segment de taille assez réduite de l’industrie chimique avec des entreprises de toutes dimensions, dont beaucoup de PME 13
3. Des produits potentiellement dangereux dont le risque doit être évalué avant leur mise sur le marché 15
B. UNE PREMIÈRE INTERVENTION EUROPÉENNE PLUS TARDIVE QUE DANS D’AUTRES SECTEURS ÉGALEMENT SOUMIS À AUTORISATION DE MISE SUR LE MARCHÉ 16
II. LA DIRECTIVE 98/8/CE : UN PREMIER ENCADREMENT AU NIVEAU EUROPÉEN, MAIS UNE APPLICATION ENCORE PARTIELLE EN RAISON DU DÉLAI, PLUS LONG QUE PRÉVU, D’ÉVALUATION DES SUBSTANCES ACTIVES 17
A. DES COMPÉTENCES RÉPARTIES ENTRE LE NIVEAU COMMUNAUTAIRE ET LE NIVEAU NATIONAL 17
1. Des listes européennes des substances actives 17
2. Des autorisations nationales de mise sur le marché de produits biocides, avec reconnaissance mutuelle entre Etats membres 18
3. Une obligation d’information de l’utilisateur 19
4. Des procédures d’évaluation harmonisées des substances actives et des produits 20
5. Le maintien à titre provisoire des dispositions nationales antérieures 21
6. La transposition par la France de la directive 98/8/CE 22
B. UN PREMIER BILAN 24
1. Le programme de travail de dix ans pour l’évaluation systématique des substances actives : un délai initial dépassé et des effets complexes à interpréter 24
a) Le déroulement prévu 24
b) Un calendrier originel qui n’a pas été tenu 24
c) Une réduction du nombre des substances actives 25
d) Des substances actives et des produits retirés du marché, soit pour des motifs sanitaires, soit également pour des raisons économiques 26
e) Une première autorisation de produit biocide, en France, en application des dispositions de la directive 98/8/CE 28
2. Des difficultés et des perspectives qui justifient une refonte du texte actuel 29
a) L’opportunité d’un nouveau texte 29
b) Les acquis de la directive 98/8/CE : une première avancée dans la protection de la santé, du consommateur et de l’environnement 29
c) Des défauts à corriger 29
(1) Des incertitudes pour les entreprises 29
(2) Le coût des dossiers : une contrainte forte, notamment pour les PME 30
(3) Des différences d’approche entre les Etats membres, notamment sur le niveau des redevances et les délais d’instruction des dossiers 31
DEUXIEME PARTIE : LA PROPOSITION DE RÈGLEMENT PRÉSENTÉE PAR LA COMMISSION EUROPÉENNE : DES AVANCÉES, MAIS UN DISPOSITIF NÉANMOINS PERFECTIBLE, NOTAMMENT POUR ÉLIMINER LES MENACES QU’IL FAIT PESER SUR LES PME 35
I. UN OBJECTIF DE SÉCURITÉ SANITAIRE PARTAGÉ ET DES AVANCÉES IMPORTANTES 35
A. LE REMPLACEMENT D’UNE DIRECTIVE PAR UN RÈGLEMENT POUR UNE APPLICATION HOMOGÈNE DANS LES DIFFÉRENTS ETATS MEMBRES 35
B. UN CHAMP D’APPLICATION ÉTENDU AUX PRODUITS ET ARTICLES TRAITÉS AVEC DES PRODUITS BIOCIDES, POUR ÉVITER TANT LE RISQUE DE DUMPING ENVIRONNEMENTAL ET SANITAIRE DES PAYS TIERS QU’UN ÉVENTUEL MOTIF DE DÉLOCALISATION DE L’INDUSTRIE CHIMIQUE HORS DE L’EUROPE 35
C. UN RÔLE DE COORDINATION POUR L’AGENCE EUROPÉENNE DES PRODUITS CHIMIQUES 36
D. LE DURCISSEMENT, POUR UNE PLUS GRANDE SÉCURITÉ SANITAIRE, DES EXIGENCES POUR L’INSCRIPTION COMMUNAUTAIRE DES SUBSTANCES ACTIVES 37
a) Des critères d’exclusion sanitaires 38
b) Des critères de substitution pour les substances actives présentant les risques les plus élevés 39
c) Une plus grande cohérence du dispositif d’inscription des substances actives avec notamment le principe d’une liste européenne unique en remplacement des trois listes actuelles 40
E. UN RÉAMÉNAGEMENT SUBSTANTIEL DU DISPOSITIF D’AUTORISATION DE MISE SUR LE MARCHÉ DES PRODUITS BIOCIDES, DANS LE RESPECT DU PRINCIPE DE LA COMPÉTENCE NATIONALE 40
1. Le maintien du principe de l’autorisation nationale 40
2. Une possibilité d’autorisation communautaire, avec accès direct au marché de chacun des Etats membres, pour les produits à faible risque et les produits innovants contenant de nouvelles substances actives 41
3. Une réforme des procédures de reconnaissance mutuelle des autorisations nationales 42
4. Un effort d’harmonisation et de clarification des trois éléments sur lesquels s’exerce la concurrence entre autorités sanitaires : le déroulement des procédures d’autorisation ; les délais d’instruction des dossiers ; les redevances versées aux autorités compétentes 43
a) Une clarification du déroulement des procédures 43
b) L’encadrement des délais 43
c) Une première harmonisation des redevances, avec une certaine prise en compte du cas spécifique des PME 44
d) Une certaine perspective d’allègement des dossiers avec l’amélioration des dérogations aux exigences en matière de données 45
e) La clarification et le renforcement des règles de protection des données, ainsi que la suppression des études redondantes sur les vertébrés 46
5. Une approche plus large de la notion de formulation cadre 47
6. Une gestion mieux coordonnée des décisions nationales, avec un registre communautaire des produits biocides 48
7. La confirmation des obligations en matières de classification, d’étiquetage et d’emballage des produits 48
8. Des dispositions sur l’accès du public à des informations par voie électronique 48
II. UN TEXTE ENCORE PERFECTIBLE, TANT DANS SES GRANDS ÉQUILIBRES, TROP DÉFAVORABLES AUX PME ET AU MAINTIEN DE LA DIVERSITÉ DES PRODUITS, QUE DANS SES DISPOSITIFS, QUI DOIVENT ENCORE ÊTRE COMPLÉTÉS ET CLARIFIÉS 49
A. DÉFINIR UN MEILLEUR ÉQUILIBRE ÉCONOMIQUE ET SANITAIRE 49
1. Des risques bien identifiés : la disparition d’un grand nombre de PME, de certains produits et par conséquent de la diversité chimique indispensable pour éviter l’apparition de résistances 49
2. Des éléments de réponse notamment institutionnels qui pourraient être insuffisants 50
3. Des pistes supplémentaires à explorer 51
B. PRÉVOIR DES CLARIFICATIONS ET DES COMPLÉMENTS 53
1. Etablir un haut niveau d’exigence sur les critères d’exclusion et les facultés de dérogation, avec l’ajout de facteurs environnementaux et de dispositions sur les nanomatériaux 53
2. Corriger la disposition sur le commerce parallèle 53
3. Mettre sur un pied d’égalité les différents producteurs de substances actives en évitant les comportements de type passager clandestin ou « free riders » 54
4. Envisager un renforcement du dispositif sur les produits à faible risque par un allongement des protections intellectuelles 55
TRAVAUX DE LA COMMISSION 57
ANNEXE : LISTE DES PERSONNES ENTENDUES PAR LES RAPPORTEURS ET REMERCIEMENTS 63
Mesdames, Messieurs,
La proposition de règlement concernant la mise sur le marché et l’utilisation des produits biocides a été présentée par la Commission européenne le 12 juin dernier. Elle a été transmise pour examen le 22 juin suivant à l'Assemblée nationale et au Sénat, en application de l'article 88-4 de la Constitution.
Depuis, elle a été examinée à l’occasion de plusieurs réunions du groupe Environnement, dans le cadre des travaux préparatoires au Conseil. Pour sa part, le Parlement européen a également commencé ses travaux. La commission de l’environnement, de la santé publique et de la sécurité alimentaire, saisie au fond, devrait bientôt se prononcer, le 3 juin prochain, sur les propositions de la rapporteure, Mme Christa Klass (PPE, Allemagne). Le vote en plénière est en l’état prévu pour le 6 juillet. Deux commissions sont par ailleurs saisies pour avis : la commission de l’industrie, de la recherche et de l’énergie, dont le rapporteur est M. Sajjad Karim (CRE, Royaume-Uni), et la commission du marché intérieur et de la protection des consommateurs, dont la rapporteure est Mme Amalia Sartori (PPE, Italie).
Ce délai d’examen, assez rapide pour une proposition de règlement dont le dispositif s’appliquera directement et de plein droit dans tous les Etats membres, sans qu’une transposition soit nécessaire, ne doit pas conduire à négliger l’importance de ce texte.
En effet, ses dispositions affecteront, sans que nous en ayons d’ailleurs toujours conscience, la vie quotidienne.
Une grande partie des produits biocides sont d’usage courant. Ce sont, en effet, l’ensemble des produits de désinfection, à usage domestique comme à usage professionnel, notamment l’eau de Javel, ainsi que les produits de protection contre les nuisibles (antimites, souricides, raticides, insecticides et acaricides, pour l’essentiel) et de protection des matériaux (protection du cuir, protection du bois etc.). Ils sont chaque jour, ou presque, utilisés ou présents dans les logements comme sur les lieux de travail.
A l’opposé, une autre partie est d’usage exceptionnel, mais néanmoins indispensable. Tel est le cas des produits de lutte contre les termites ou de ceux destinés à éradiquer la mérule, ce champignon qui détruit le bois dans certaines constructions de l’ouest de la France. D’autres produits encore ne sont connus que des professionnels, tels les produits de thanatopraxie.
Les produits biocides sont donc très nombreux et très divers. Leur nombre est actuellement de l’ordre de 25.000 en France.
Ces produits sont pour l’essentiel des produits « de niche ». Ils sont donc le fait de petits producteurs, de PME, plutôt que de grands groupes. Ceux-ci ne sont pas absents, mais sont davantage présents dans la fabrication des substances actives, qui sont les composants chimiques qui leur donnent leur efficacité. Les PME jouent le rôle de formulateurs, associant à une ou plusieurs substances actives des produits autres tels que, notamment, les excipients, adjuvants, colorants, parfums, et assurant leur conditionnement sous une forme utilisable (poudre, blocs, sachets, liquide à répandre, pulvérisation etc.).
C’est toute la difficulté d’une réglementation de ce secteur, particulièrement sensible à tout ce qui peut entraîner une augmentation de ses charges.
Sur le plan politique, les produits biocides exigent une mise en œuvre pragmatique du principe de précaution. Ce sont par définition des produits qui « tuent la vie » (telle est leur signification étymologique), mais on ne peut et on ne doit pourtant pas les interdire, car ce sont des produits indispensables.
L’ampleur des progrès réalisés depuis la première partie du XIXe siècle, dernière période où l’espèce humaine a vécu dans des conditions parfaitement « naturelles », suffit à s’en convaincre. Toute interdiction ou limitation trop stricte constituerait un facteur de régression.
Il faut donc gérer, en éliminant de manière pragmatique ce qui est trop dangereux, un double risque :
– d’une part, un risque sanitaire direct, celui que l’utilisation des produits biocides fait courir à l’homme, et aussi aux animaux et organismes autres que l’espèce ou des espèces visées. Un contrôle de la toxicité s’impose donc ;
– d’autre part, un risque environnemental. Un contrôle de l’écotoxicité s’avère indispensable pour éviter la contamination dommageable de l’eau, de l’air et la terre plus généralement de tous les éléments de l’environnement.
Une telle démarche est récente. Longtemps, dans la plupart des pays, la majorité des produits biocides n’a fait l’objet d’aucune réglementation, étant librement mis sur le marché dès lors qu’ils n’étaient composés d’aucun produit interdit. Ils n’étaient pas non plus mis en question, dès lors qu’ils faisaient partie du progrès et que les conditions de leur emploi étaient indiquées à l’utilisateur.
Néanmoins, le développement de leur usage, la prise de conscience des dangers diffus qu’ils pouvaient receler, la nature, sensible, de certaines de leurs utilisations, telles que la désinfection de l’eau potable ou celle des eaux de piscine, ont conduit les autorités nationales à prévoir des procédures d’homologation ou d’autorisation préalable.
Le même modèle que celui en vigueur pour les pesticides, et issu du médicament, a alors été suivi. Les biocides pouvant présenter les niveaux de risque les plus élevés ont fait l’objet d’une autorisation de mise sur le marché, d’une AMM.
Au niveau communautaire, c’est avec la réalisation du marché unique que s’est imposée l’exigence de prévoir une harmonisation. Celle-ci est intervenue assez tard, en 1998, avec la directive 98/8/CE du 16 février 1998 concernant la mise sur le marché des produits biocides.
Un dispositif à deux niveaux a été établi avec :
– une procédure d’homologation des substances actives au niveau communautaire, avec l’obligation de n’utiliser que les seules substances actives inscrites sur les listes européennes en annexe à la directive ;
– une procédure d’autorisation de mise sur le marché applicable à tous les produits biocides, au niveau national, avec une procédure de reconnaissance mutuelle entre les Etats membres.
La directive 98/8/CE n’a cependant fait l’objet que d’une application très partielle.
En effet, la condition principale de sa mise en œuvre, l’évaluation de toutes les substances actives présentes sur le marché lors de son entrée en vigueur, le 14 juin 2000, n’a pu être réalisée dans le délai initial. Le programme de travail décennal établi par la Commission européenne, en liaison avec les Etats membres, et leurs autorités sanitaires chargées de tâches concrètes d’évaluation, s’est avéré plus long que prévu. Il a donc dû être prolongé de 4 ans, jusqu’en 2014.
Néanmoins, dès 2008, conformément à la clause de rendez-vous du texte de 1998, la Commission européenne a pu dégager les premiers enseignements du dispositif de 1998 et proposer plusieurs modifications, dont la nature et l’ampleur l’ont conduite à proposer un texte entièrement nouveau.
Soumise en application de l’article 88-4 de la Constitution à l’examen de la commission des affaires européennes de l’Assemblée nationale, ce texte peut être dans l’ensemble approuvé, sous réserve que lui soient apportées plusieurs améliorations, tant pour mieux atteindre ses objectifs de sécurité sanitaire et environnementale, que pour éviter tout risque de déstructuration d’une filière qui comprend un grand nombre de PME.
PREMIERE PARTIE :
UN DROIT EUROPÉEN RÉCENT ET ENCORE AU DÉBUT DE SA MISE EN œUVRE
Les biocides sont destinés à éliminer ou à prévenir les organismes nuisibles. Ils sont actuellement définis au niveau européen comme les produits destinés à détruire, repousser ou rendre inoffensifs les organismes nuisibles, ou bien destinés soit à en prévenir l’action, soit à les combattre de tout autre manière, par des actions chimiques ou biologiques.
Il s’agit donc des désinfectants autres que ceux à usage médical, ainsi que de tous les produits destinés à lutter contre les champignons et les moisissures, et contre les autres nuisibles, notamment les rongeurs et les acariens. En pratique, ce sont les désinfectants de l’eau, de l’air, du sol, des surfaces ainsi que, notamment des lieux de travail. Certains d’entre eux sont simples, tels que l’eau de Javel, mais d’autres sont complexes, tels que les insecticides sélectifs. Ils sont parfois appelés pesticides non agricoles.
C’est une catégorie juridique large. Le produit biocide est défini par sa finalité. La seule mention d’une qualité biocide fait entrer un produit dans le champ des textes européens.
Il s’agit essentiellement, mais pas uniquement de produits chimiques. La mention des actions biologiques fait que des produits dits naturels, tels que les microorganismes, bactéries, champignons et virus susceptibles d’avoir une action biocide, sont également couverts par la réglementation.
Ce n’est pas illégitime puisque tous les mécanismes naturels ne sont pas nécessairement bénéfiques ni bénins. La nature présente elle aussi ses propres dangers. Il convient donc d’évaluer les mécanismes d’action des produits naturels, les moins connus, avant de les utiliser, même si les procédures doivent être adaptées.
La classification européenne dont les produits biocides font l’objet permet de percevoir l’étendue précise de la catégorie à raison de 23 types de produits différents se répartissant en quatre grands groupes.
Le premier de ces groupes comprend les désinfectants et les produits biocides généraux.
Il se décompose en plusieurs types de produits très différents, avec notamment les produits d’hygiène humaine qui ne relèvent pas d’autres catégories juridiques telles que les médicaments ou les dispositifs médicaux, ainsi que les produits d’hygiène publique, les désinfectants d’élevage, les désinfectants alimentaires et les désinfectants pour l’eau potable. C’est dans ce groupe que l’on trouve notamment les désinfectants pour eau de piscine et les produits pour l’entretien des conduits de climatisation.
Le deuxième groupe comprend les produits de protection. Il s’agit notamment des produits de protection du bois tels que les traitements contre les parasites (insectes, champignon, mérule etc.), les produits de protection des cuirs et textiles, notamment les antimites et les produits anti-moisissure.
Les troisième groupe comprend les antiparasitaires, avec plusieurs types de produits importants : les rodenticides contre les rongeurs (rats, souris, loirs et aussi taupes, bien que celles-ci ne soient pas biologiquement des rongeurs), qu’il s’agisse de poisons ou de gaz ; les avicides pour tuer, éloigner ou endormir les oiseaux (très peu sont autorisés) ; les molluscicides, contre les limaces et les escargots ; les piscicides contre les poissons et les insectes d’eau ; les insecticides et les acaricides (contre les acariens).
Le quatrième groupe concerne l’ensemble des autres biocides avec aussi bien les produits de protection pour les denrées, que les produits de taxidermie, les produits antisalissure et les produits de lutte contre certains vertébrés (ceux de lutte contre les écureuils relèvent de cette catégorie, bien que cet animal soit un rongeur).
Le tableau suivant récapitule ces éléments :
La classification européenne des produits biocides
GROUPE 1: Désinfectants et produits biocides généraux
Type de produits 1 : Produits biocides destinés à l’hygiène humaine
Type de produits 2 : Désinfectants utilisés dans le domaine privé et dans le domaine de la santé publique et autres produits biocides
Type de produits 3 : Produits biocides destinés à l’hygiène vétérinaire
Type de produits 4 : Désinfectants pour les surfaces en contact avec les denrées alimentaires et les aliments pour animaux
Type de produits 5 : Désinfectants pour eau de boisson
GROUPE 2 : Produits de protection
Type de produits 6 : Produits de protection utilisés à l’intérieur des conteneurs
Type de produits 7 : Produits de protection pour les pellicules
Type de produits 8 : Produits de protection du bois
Type de produits 9 : Produits de protection des fibres, du cuir, du caoutchouc et des matériaux polymérisés
Type de produits 10 : Protection des ouvrages de maçonnerie
Type de produits 11 : Protection des liquides utilisés dans les systèmes de refroidissement et de fabrication
Type de produits 12 : Produits anti-moisissures
Type de produits 13 : Produits de protection des fluides utilisés dans la transformation des métaux
GROUPE 3: Produits antiparasitaires
Type de produits 14 : Rodenticides
Type de produits 15 : Avicides
Type de produits 16 : Molluscicides
Type de produits 17 : Piscicides
Type de produits 18 : Insecticides, acaricides et produits utilisés pour lutter contre les autres arthropodes
Type de produits 19 : Répulsifs et appâts
GROUPE 4: Autres produits biocides
Type de produits 20 : Produits de protection pour les denrées alimentaires ou les aliments pour animaux
Type de produits 21 : Produits antisalissure
Type de produits 22 : Fluides utilisés pour l’embaumement et la taxidermie
Type de produits 23 : Lutte contre d’autres vertébrés
En définitive, les biocides sont une catégorie résiduelle et donc fourre-tout, identifiée par opposition à des catégories juridiquement identifiées plus tôt, telles que les dispositifs médicaux et les pesticides, notamment.
2. Un segment de taille assez réduite de l’industrie chimique avec des entreprises de toutes dimensions, dont beaucoup de PME
Les produits biocides représentent un secteur économique et un marché de faible ampleur.
Le marché mondial est estimé à 5 milliards de dollars, alors que celui des pesticides agricoles s’élève à quelque 40 milliards de dollars, selon l’Observatoire des résidus de pesticides.
L’Europe représente environ un quart du marché mondial des biocides.
Les acteurs sont de tailles très diverses avec :
- d’une part, les grandes entreprises du secteur chimique et leurs filiales spécialisées, notamment pour la production des substances actives qui donnent au produit biocide sa capacité d’action. Ces acteurs sont au nombre de quelques centaines ;
- d’autre part, des PME, présentes sur les marchés de « niches ». Plusieurs milliers de produits sont recensés.
On estime que les grandes entreprises représentent 25 % du marché européen des biocides.
Il faut relever que les différents acteurs n’ont pas le même rôle.
Les producteurs de substances actives sont fournisseurs des fabricants de produits biocides. Pour leur part, les producteurs de produits biocides utilisent soit des substances actives seules, soit un ensemble de composants en suivant une formule précise. Le cas le plus simple, d’un produit biocide utilisé seul, soit dans un cadre industriel, soit dans le domaine domestique, est celui de l’eau de Javel. L’un des cas les plus opposés est celui des peintures, auxquelles l’ajout de produits biocides assure une protection et une durabilité, pour lesquelles les coformulants sont nombreux. Les formulateurs réalisent des mélanges où les biocides, ainsi que d’autres additifs, s’ajoutent aux pigments colorants, aux solvants, aux liants et aux résines notamment. Les produits peuvent être présents même lorsque la peinture n’a pas une fonction biocide (par exemple, les peintures contre les insectes ont une fonction biocide).
La chaîne économique est complexe : directement fournis par les importateurs ou les fabricants de substances biocides ou de mélanges en comprenant, les formulateurs vendent soit directement à leur clientèle, professionnels ou particuliers, soit à des producteurs d’articles de détail, directement ou par l’intermédiaires de distributeurs.
Les biocides concernent donc un secteur sensible et stratégique de l’industrie européenne.
Pour sa part, l’industrie chimique de la France se situe au 5e rang mondial, avec 86 milliards d’euros de chiffre d’affaires, dont la moitié est exportée, 180 000 salariés et 859 entreprises, après les Etats-Unis, la Chine, le Japon et l’Allemagne. Elle est donc à la deuxième place, au niveau européen, se situant comme on vient de le voir après l’Allemagne, mais avant le Royaume-Uni et l’Italie.
Il faut enfin préciser que c’est un secteur d’activité attentif aux initiatives européennes, qui est encore en train de mettre en application, dans des conditions peu aisées, les dispositions du règlement REACH.
3. Des produits potentiellement dangereux dont le risque doit être évalué avant leur mise sur le marché
Par définition, les biocides ne sont pas des produits inoffensifs, pour la plupart d’entre eux, mais au contraire des produits toxiques. Dans une configuration idéale, ils ne le sont que vis-à-vis des espèces qu’ils visent à éviter ou à éradiquer.
Comme tout produit indispensable du même type, tel que les médicaments ou les phytosanitaires, ils relèvent d’une approche bénéfices/risques.
De manière plus détaillée, les cinq critères d’évaluation sont :
– l’efficacité du produit ;
– sa toxicité ;
– son écotoxicité, à savoir ses nuisances pour l’environnement (air, eau, sols et espèces non visées) ;
– les risques d’exposition lors de ses manipulations par l’homme ;
– les dangers résultant des propriétés physiques et chimiques (inflammabilité, explosivité, caractère corrosif etc.).
L’objectif est de disposer de produits qui soient le plus sûr possible et présentant des niveaux de risque maîtrisés.
Les produits biocides font l’objet d’une grande attention de la part de l’opinion et des associations de protection de l’environnement, ainsi que des associations de consommateurs, très sensibilisées aux risques domestiques. Le principe de précaution, qui veut en la matière que les risques soient évalués ex ante avant la mise sur le marché d’un produit, et non découverts ex post, doit donc jouer de manière adaptée, avec un régime d’autorisation préalable, d’autorisation de mise sur le marché (AMM), comme pour les pesticides ou pour les médicaments.
B. Une première intervention européenne plus tardive que dans d’autres secteurs également soumis à autorisation de mise sur le marché
La directive 98/8/CE concernant la mise sur le marché des produits biocides est le premier texte communautaire intervenu en la matière.
L’intervention du niveau européen est ainsi postérieure à ce qu’elle a été pour le médicament, pour lequel la première directive est intervenue en 1965, ainsi que pour les phytosanitaire, avec la directive 91/414/CEE adoptée dans la perspective de la réalisation du marché unique.
Les seules réglementations applicables avant 1998 aux produits biocides étaient celles définies au niveau national.
Sans être exhaustif, on peut indiquer qu’elles étaient partielles, éclatées et incomplètes avec, comme le montre l’exemple français, la coexistence de plusieurs régimes :
– la production et de la vente libre, régime applicable de plein droit en l’absence d’autres règles prévues ;
– les normes définies au niveau européen, régime notamment applicable aux désinfectants et produits biocides généraux non soumis à une procédure plus exigeante d’autorisation de mise sur le marché ;
– la liste nationale des produits prohibés, régime notamment applicable à l’emploi de gaz toxiques ;
– la liste positive des substances actives utilisables, couplée avec une autorisation par produit, selon le régime relatif aux produits de désinfection des eaux de piscines publiques ;
– la certification, comme c’est le cas pour les produits de protection du bois et les produits anti-termites pour les sols et les murs ;
– l’autorisation de mise sur le marché (AMM), prévue notamment pour les désinfectants utilisés dans l’élevage.
Au total, sur les vingt-trois types de produits biocides identifiés, huit seulement avaient été couverts en France par une procédure d’AMM.
II. LA DIRECTIVE 98/8/CE : UN PREMIER ENCADREMENT AU NIVEAU EUROPÉEN, MAIS UNE APPLICATION ENCORE PARTIELLE EN RAISON DU DÉLAI, PLUS LONG QUE PRÉVU, D’ÉVALUATION DES SUBSTANCES ACTIVES
La directive 98/8/CE a prévu un mécanisme d’homologation des substances actives : seules peuvent être utilisées les substances actives inscrites sur l’une des trois listes « positives » annexées à son dispositif. L’inscription initiale se fait pour une durée maximale de dix ans. Elle est ensuite renouvelable dans les mêmes conditions. La décision est prise par la Commission européenne, sur avis du comité permanent des produits biocides, et dans le cadre de la procédure de comitologie.
Ces trois listes sont les suivantes :
– la première d’entre elles est la liste de droit commun des substances actives homologuées (annexe I de la directive) ;
– la deuxième est la liste des substances dites à faible risque (annexe I A de la directive),
– la dernière est la liste des substances de base(2) (annexe I B de la directive).
L’inscription d’une substance active sur l’une de ces listes ne se fait qu’après évaluation. Un dossier en ce sens est déposé auprès d’un Etat membre. Les critères d’évaluation sont ceux précédemment évoqués : efficacité, toxicité, écotoxicité et dangers propres à la substance active.
La substance active ne donne pas lieu à une évaluation générale, mais à une évaluation précise en vue d’un type de produit biocide. L’évaluation porte donc plutôt sur un couple substance active/type de produit biocide.
2. Des autorisations nationales de mise sur le marché de produits biocides, avec reconnaissance mutuelle entre Etats membres
Pour ce qui les concerne, les produits biocides proprement dits doivent faire, préalablement à leur commercialisation, l’objet d’une autorisation de mise sur le marché (AMM).
Les AMM sont délivrées par les autorités compétentes des Etats membres (chaque Etat membre a l’obligation de désigner une telle autorité compétente). Elles le sont pour une période initiale maximale de dix ans. L’autorisation est ensuite reconductible dans les mêmes conditions. Une AMM peut également être révisée, modifiée ou annulée, lorsque les circonstances le commandent.
Les critères de délivrance de l’AMM sont notamment, comme pour les substances actives, l’efficacité, la toxicité et l’écotoxicité.
De manière plus précise, il est nécessaire que le produit biocide :
– ne comprenne pas de substance active autres que celles autorisées ;
– soit suffisamment efficace ;
– n’ait aucun effet inacceptable sur les organismes cibles ;
– n’ait aucun effet inacceptable sur la santé humaine ou animale, ou encore sur les eaux, eaux de surface ou eaux souterraines ;
– n’ait pas non plus d’effet inacceptable sur l’environnement.
En outre, des contraintes d’utilisation sont fixées : les propriétés physiques et chimiques du produit doivent être jugées acceptables pour assurer son utilisation, son stockage et son transport dans des conditions adéquates.
La directive prévoit également un mécanisme de reconnaissance mutuelle par les autres Etats membres d’une AMM délivrée par un Etat membre. La reconnaissance n’est pas automatique. Un Etat membre peut refuser dans certains cas la reconnaissance mutuelle. Dans ce cas, la décision appartient à la Commission européenne après saisine du comité permanent pour les biocides.
En outre, pour trois catégories de produits, la reconnaissance mutuelle peut être refusée (avicides, piscicides et produits de lutte contre les autres vertébrés).
La directive 98/8/CE prévoit également un cas intermédiaire : un Etat membre peut demander l’adaptation aux circonstances locales lorsque l’espèce cible n’est pas présente en quantités nocives sur son territoire, lorsqu’une tolérance ou une résistance inacceptable a été démontrée ou lorsqu’une autorisation inchangée aurait des conséquences inacceptables pour l’homme ou pour l’environnement.
Cette disposition est distincte de la clause de sauvegarde qui permet à un Etat membre de limiter ou bien d’interdire provisoirement l’utilisation ou la vente d’un produit biocide s’il a une raison d’estimer que celui-ci présente des risques inacceptables pour la santé humaine ou animale, ou pour l’environnement. Il est alors tenu d’en informer sans délai la Commission européenne et les autres Etats membres, en précisant quels en sont les motifs.
Une clause dérogatoire permet enfin à un Etat membre d’autoriser la mise sur le marché d’un produit qui ne respecte pas ses prescriptions, de manière temporaire, pour une durée de 120 jours (quatre mois), en vue d’un usage limité et contrôlé, la mise sur le marché de produits biocides, si un danger imprévu le rend nécessaire et si celui-ci ne peut être contrôlé par d’autres moyens.
En outre, dans certaines conditions, un Etat membre peut également autoriser, pour une durée maximale de trois ans, la mise sur marché d’un produit biocide contenant une nouvelle substance active non inscrite.
La délivrance des AMM fait l’objet d’une procédure d’information mutuelle : chaque trimestre, tous les Etat membres doivent informer les autres États membres et la Commission européenne de tous les produits biocides enregistrés et autorisés sur son territoire ainsi que de tous les produits dont l’autorisation a été refusée, modifiée, renouvelée ou annulée.
La directive 98/8/CE prévoit un certain nombre de dispositions que les Etats membres doivent mettre en œuvre pour la bonne information de l’utilisateur de produits biocides.
Ceux-ci doivent ainsi veiller à leur classification, leur étiquetage, leur emballage(3) selon des règles précises. La mise sur le marché des produits biocides peut également être subordonnée à l’emploi de la langue nationale ou, le cas échéant, des langues nationales.
Par ailleurs, depuis 2003, en outre, chaque Etat membre doit remettre tous les trois ans à la Commission européenne un rapport sur les éventuels empoisonnements dus à des produits biocides.
Les procédures d’évaluation sont fixées dans les annexes à la directive 98/8/CE non seulement pour les substances actives, qui font l’objet d’une harmonisation communautaire, mais également pour les produits biocides eux-mêmes.
En outre, il faut rappeler que la réalisation concrète des évaluations et des essais fait l’objet d’une harmonisation communautaire et même, au-delà, d’une harmonisation internationale.
En effet, les principes relatifs aux bonnes pratiques de laboratoire (BPL) ont fait l’objet d’une recommandation de l’OCDE dès l’année 1981. Ils ont ensuite été réexaminés et modifiés en 1997.
S’agissant de l’Union européenne, les principes de BPL sont fixés par la directive 2004/10/CE concernant le rapprochement des dispositions législatives, réglementaires et administratives relatives à l’application des principes de BPL et au contrôle de leur application pour les essais sur les substances chimiques.
Son dispositif prévoit notamment que lors de la remise des résultats des essais, les laboratoires doivent certifier que les essais ont été effectués conformément aux principes de BPL.
En pratique, comme l’ont indiqué les responsables de l’AFSSET aux rapporteurs, le dossier de demande d’inscription d’une substance active comprend différentes parties relatives :
– à l’évaluation du danger avec un exposé de ses caractéristiques physico-chimiques (identification, analyse, impuretés…), de son efficacité, de sa toxicité, de son écotoxicité et des dangers liés à ses propriétés physiques et chimiques (tels que l’inflammabilité ou le caractère explosif) ;
– mais aussi à l’évaluation des risques liés à la présence de substance active pour les usages pour lesquels l’inscription de la substance est demandée : quels risques pour l’homme, notamment en milieu professionnel, lors de l’utilisation du produit biocide ? Quels risques pour l’environnement, à savoir pour le « compartiment » aquatique, le « compartiment » atmosphérique, le « compartiment » terrestre, ainsi que pour la chaîne alimentaire ?
En outre, doivent être étudiés les risques liés aux propriétés physiques et chimiques du produit et les mesures de protection à prévoir, notamment lors de son conditionnement et de son transport, ainsi que lors de son utilisation.
Pour les produits biocides, la démarche est la même mais prend en outre en compte l’évaluation de l’exposition au produit lors de son utilisation et de son application, ainsi qu’après son application.
En particulier, il convient de connaître la toxicité d’une substance ou d’un produit lors de son inhalation, de son absorption orale ou de son contact avec les muqueuses (contact cutanée et contact avec les muqueuses les plus sensibles telles que la cornée).
L’évaluation des risques se fait donc en quatre phases :
– l’identification du danger intrinsèque à la substance ;
– l’évaluation de ce danger ;
– l’évaluation de l’exposition à ce danger ;
– la caractérisation de ce danger.
La directive 98/8/CE n’a pas été prévue pour faire l’objet d’une application immédiate.
En effet, la mise en œuvre de son dispositif est conditionnée à l’évaluation préalable des substances actives, avant leur inscription sur les listes communautaires.
Un programme de travail pour l’évaluation systématique de toutes les substances actives utilisées dans les produits présents sur le marché au 14 mai 2000 a été établi. Sa durée de réalisation a été estimée à dix ans, avec une échéance initiale au 14 mai 2010.
Par conséquent, les dispositions de la directive ont été applicables aux seuls produits nouveaux. Pour les produits biocides présents sur le marché à la date 14 mai 2000, des dispositions transitoires ont permis aux Etats membres des continuer à appliquer leurs dispositions ou leurs pratiques nationales antérieures.
A été ainsi explicitement prévue la possibilité de continuer d’utiliser les substances actives dans l’attente de leur éventuelle inscription ou interdiction.
Comme précisé ci-après, la réalisation du programme de travail s’est avérée plus longue que prévu et l’échéance initiale du 14 mai 2010 a été reportée de quatre années, au 14 mai 2014.
La directive 98/8/CE a été transposée en droit français par l’ordonnance du 11 avril 2001, et les dispositions correspondantes codifiées aux articles L.522-1 à L.522-18 du Code de l’environnement, ainsi que par le décret no 2004-187 du 26 février 2004 relatif à la mise sur le marché des produits biocides, dont les dispositions ont ensuite été codifiées par le décret no 2007-1467 du 12 octobre 2007 dans la partie règlementaire du code de l’environnement.
On observera que les sanctions pénales prévues sont lourdes. L’article L. 522-16 du code de l’environnement mentionne des peines de deux ans d’emprisonnement et 75.000 euros d’amende en cas de mise sur le marché ou de vente d’une substance active non inscrite ou bien d’un produit biocide ne bénéficiant pas d’une AMM, ainsi que de six mois d’emprisonnement et 7.500 euros d’amende en cas d’utilisation d’un produit biocide non autorisé.
C’est le ministère de l’écologie et du développement durable (MEDD) est qui est l’autorité compétente chargée de la mise en œuvre au niveau national des dispositions sur les produits biocides.
Le décret précité no 2004-187 du 26 février 2004 a confié à l’Agence française de sécurité sanitaire de l’environnement et du travail (AFSSET) la mission de coordonner, au niveau national, l'évaluation des dangers, des risques et de l'efficacité des substances actives et des produits biocides dont les dossiers sont soumis à la France.
Dans le cadre de cette coordination, l'AFSSET est amenée à formuler des avis adressés au MEDD, sur la recevabilité des dossiers ainsi que sur les conditions d'utilisation qui seront définies dans l'AMM.
L’Agence s’appuie notamment sur l’expertise extérieure d’organismes évaluateurs notamment l’Agence française de sécurité sanitaire des aliments (AFSSA) et l’Agence française de sécurité sanitaire des produits de santé (AFSSaPS), l’Ifremer et le FCBA (Institut technologique forêt, bois, construction et ameublement) ou encore l'Ecole nationale vétérinaire de Lyon (ENVL) pour les produits rodenticides.
Au 1er juillet prochain, ce dispositif sera adapté pour tenir compte de la fusion entre l’AFSSA et l’AFSSET, en une Agence nationale chargée de la sécurité sanitaire de l’alimentation, de l’environnement et du travail, ce qui correspond à la création d’un grand pôle d’expertise scientifique à Maisons-Alfort.
Pour ce qui concerne leur contenu, les dispositions nationales applicables aux produits biocides prévoient :
– les conditions de la mise en œuvre de l’obligation d’étiquetage, ainsi que l’obligation de fournir les fiches de données de sécurité ;
– la déclaration des produits au ministère de l’écologie et du développement durable.
Cette déclaration, qui vient compléter les dispositions antérieures maintenues au titre de la période transitoire, a pour objet de connaître les produits biocides utilisés en France ainsi que de veiller au respect des règles nouvelles au fur et à mesure de leur application.
Pour ce qui concerne par comparaisons, certains autres Etats membres, la situation est similaire à celle de la France, avec des variantes.
Au Royaume-Uni, la matière est régie par le Biocides Products Regulation de 2001 et l’autorité compétente au niveau national est le Health and Safety Executive (HSE), et plus spécifiquement, en son sein, le Chemicals Regulation Directorate (CDR).
En Roumanie, la transposition date de 2005. L’autorité compétente est le ministère de la santé, et en son sein, les missions sont exercées par la Commission nationale pour les biocides.
En Allemagne, les biocides sont de la compétence de la commission d’autorisation de l’Institut fédéral pour la sécurité au travail et la médecine du travail (Bundesanstalt für Arbeitsschutz und Arbeitsmedizin - BAuA). Tout produit actuellement présent sur le marché doit être notifié.
1. Le programme de travail de dix ans pour l’évaluation systématique des substances actives : un délai initial dépassé et des effets complexes à interpréter
Le programme de travail pour l’examen systématique des risques de toutes les substances actives sur le marché à la date de 14 mai 2000, en tant que substances actives d'un produit biocide, a été mis en œuvre en deux phases, selon les dispositions du règlement (CE) no 1896/2000 de la Commission européenne.
La première étape a eu pour objet l’identification des substances actives, c'est-à-dire leur recensement : les entreprises concernées ont dû ainsi identifier les produits qu’elles fabriquaient et les notifier, si elle souhaitaient continuer à les exploiter, en accompagnant leurs demandes d’un dossier d’évaluation.
A son issue, la deuxième phase, dédiée à l’évaluation, a été organisée par le règlement (CE) no 2032/2003 de la Commission européenne dressant l’inventaire des substances notifiées, établissant le calendrier correspondant et précisant également la liste des Etats membres rapporteurs désignés pour les deux premières listes prioritaires (produits de protection du bois et produits rodenticides – ceux utilisés contre les rongeurs).
Ce texte a été modifié en 2005 et en 2006 puis remplacé par le règlement (CE) no 1451/2007.
La réalisation du programme d’évaluation s’est avérée plus longue que les dix années originellement prévues.
Le calendrier initial avait été établi sur la base d’un délai de deux ans pour la procédure d’évaluation et d’autorisation d’une substance active précise. Il s’agissait d’un délai complet allant du dépôt du dossier correspondant par l’entreprise ou les entreprises intéressées jusqu’à l’intervention de la décision d’inscription communautaire.
Dans son rapport au Conseil et au Parlement européen du 8 octobre 2008 (document COM (2008) 620 final), la Commission européenne a indiqué que ce délai s’était avéré impossible à tenir.
Un délai d’évaluation minimum de trois ans a été constaté, pour une substance active, et le délai moyen a été estimé à quatre à cinq ans.
En conséquence, la directive 2009/170/CE du 26 octobre 2010 a reporté de quatre ans l’échéance antérieure et l’a fixée au 14 mai 2014. Il a également prolongé la période transitoire jusqu’à cette même date.
En outre, le texte prévoit, si nécessaire, une nouvelle prolongation de deux ans de la période transitoire, si nécessaire.
Cette question des délais d’évaluation des substances actives a fait l’objet de plusieurs remarques des interlocuteurs des rapporteurs.
Les entreprises sont attentives à leur brièveté. Elles indiquent d’ailleurs que c’est l’un des éléments sur lequel la concurrence s’exerce entre autorités sanitaires.
Pour ce qui concerne l’AFSSET, celle-ci invoque le nombre et l’importance des difficultés qui peuvent surgir lors de l’examen d’un dossier. Parmi celles évoquées, on peut retenir les suivantes : l’absence de validité de certaines études, par exemple lorsqu’elle sont trop anciennes ou insuffisamment documentées ; l’absence de disponibilité de certaines études ; les difficultés d’extrapoler certains résultats, notamment pour l’extrapolation temporelle des résultats sur une durée plus importante que celle du test ou encore sur un usage plus fréquent du produit ; la difficulté d’extrapoler à l’homme des résultats établis sur les animaux ; la complexité d’évaluer l’exposition.
Cette question des délais est de nouveau abordée au (4) du c) du 2 ci-après.
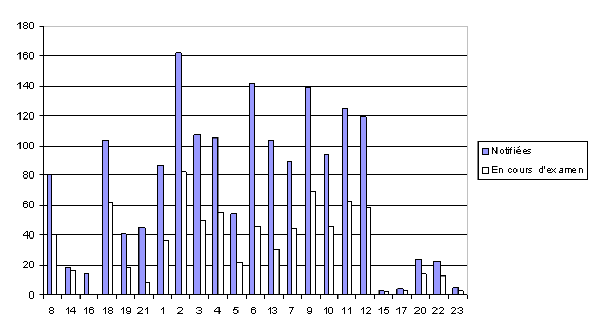
Initialement, on a estimé à environ 2.000 le nombre des substances actives incorporées dans les produits biocides, plus précisément des couples substance active-utilisation biocide.
A l’issue de la première phase, celle du recensement et du dépôt des dossiers par les entreprises, 964 substances actives ont été identifiées comme présentes sur le marché avant le 14 mai 2000.
Sur ce total, 416 ont été notifiées en vue de leur utilisation sur un type ou plusieurs types de produits biocides, au 1er septembre 2006. 548 substances actives n’avaient donc pas été défendues.
Ensuite, des retraits sont intervenus et le nombre des substances actives soutenues a été de 364.
Le graphique suivant donne par type de produits le nombre des combinaisons substance active/type de produits initialement notifiées et finalement défendues.
Combinaisons substance active/type de produit initialement notifiées et finalement défendues par le secteur
La première inscription d’une substance active est intervenue en 2006.
Depuis, vingt-neuf autres substances actives ont été inscrites à l’annexe 1 de la directive 98/8/CE, et une (le gaz carbonique) à l’annexe 1 A sur les substances à faible risque, soit trente substances au total.
Il s’agit de dix-sept substances utilisées dans les produits de protection du bois, de dix rodonticides, ainsi que trois insecticides et acaricides.
d) Des substances actives et des produits retirés du marché, soit pour des motifs sanitaires, soit également pour des raisons économiques
Le principal effet de la directive 98/8/CE a été de réduire le nombre des substances actives utilisables.
En effet, une substance active ne peut continuer à être exploitée que si elle a été notifiée, et si elle est ensuite soutenue. A défaut, l’incorporation de la substance active dans les produits biocides doit cesser.
L’absence de notification ou de soutien de plusieurs centaines de substances a eu pour effet, comme on vient de le voir, de réduire le nombre des substances actives disponibles.
Cette réduction peut être attribuée à trois facteurs.
Le premier de ces facteurs est d’ordre industriel et technique : la substance n’était en fait plus utilisées dans les produits biocides.
Le deuxième, plus important, est d’ordre sanitaire : la substance présente des caractéristiques toxicologiques ou écotoxicologiques particulièrement défavorables.
Le troisième est d’ordre économique. Les coûts d’évaluation, et plus globalement les coûts de constitution du dossier qui sont à la charge des industriels, sont trop élevés eu égard à la taille du marché.
En l’absence de données plus précises, il n’est pas possible d’envisager de préciser la part respective de chacun de ces trois facteurs.
En revanche, on peut constater que leurs effets concrets sont fort différents.
Si le retrait du marché de substances actives particulièrement toxiques est bénéfique, le retrait pour des motifs économiques est regrettable à deux points de vue.
D’une part, il entraîne une réduction de l’offre et de la diversité des produits. Il en résulte donc une tendance à l’augmentation des prix et des difficultés de lutte contre les espèces cibles, lesquelles s’accoutument à un produit donné, ce qui implique de changer périodiquement de traitement selon le principe de la rotation.
D’autre part, il présente le risque de laisser l’utilisateur final sans solution de remplacement pour lutter contre certains organismes nuisibles.
Ce risque, la Commission européenne ne l’a d’ailleurs pas exclu.
Pour la France, ce cas de figure a déjà été rencontré pour la lutte anti-vectorielle, celle contre les moustiques porteurs de maladie.
Plus précisément, selon les informations communiquées, il a fallu recourir au mécanisme de l’usage essentiel pour le Téméphos, substance de la famille des organophosphorés que les sociétés exploitantes avaient décidé de ne pas soutenir pour l’inscription à la liste des substances actives biocides autorisées au niveau communautaire. En conséquence, n’ayant pas fait l’objet d’une notification, le téméphos aurait dû être retiré du marché français le 1er septembre 2006, conformément à l’article 4 du règlement CE no 1451/2007.
Cependant, dans le cadre de l’article 5 de ce même règlement qui prévoit la possibilité pour les Etats membres de demander une prolongation d’autorisation de mise sur le marché pour certaines substances actives dans le cadre d’un usage essentiel, la France a présenté en mai 2006 des informations démontrant la nécessité de prévoir la plus large gamme possible de larvicides disponibles pour lutter contre les moustiques vecteurs de maladies graves touchant les populations (paludisme, dengue, chikungunya…), et a demandé le maintien sur le marché du Téméphos pour un usage en lutte anti-vectorielle. Cette demande de dérogation a été accordée uniquement à des fins de lutte anti-vectorielle dans les départements d’Outre-mer. La substance est utilisée uniquement lorsque les traitements avec les autres substances disponibles ne sont pas efficaces. Chaque année, un rapport est transmis à la Commission européenne sur les utilisations du produit, et sur l'avancée des travaux engagés pour trouver des substituts sur le long terme.
Par ailleurs, le recours à une autorisation provisoire de 120 jours est intervenu pour le Malathion, cette substance insecticide contre les moustiques adultes n'étant finalement plus soutenue dans le cadre du programme de travail européen et son utilisation allant être interdite au 21 février 2009. En application de l'article 15 de la directive 98/8/CE, la France a eu recours à une autorisation provisoire octroyée pour 3 mois à la fin du mois de février 2009 pour utiliser du Malathion en Guyane, laquelle était confrontée à une épidémie de dengue depuis la première semaine de janvier 2009. Ces dispositions dérogatoires ont été prises par l'arrêté du 27 février 2009. Seul le Malathion était disponible sur place et l'autorisation provisoire à permis de pouvoir continuer à utiliser ce produit le temps que le substitut soit disponible. A l'issue de ces trois mois, la Commission européenne a permis à la France de pouvoir renouveler cette autorisation provisoire (décision 2009/521/CE), ce qu’elle n’a pas fait car un substitut est arrivé sur place entre-temps.
Par ailleurs, on peut observer que la soude caustique, qui est un produit biocide autrefois utilisé dans les élevages, mais qui ne l’est plus en raison de ses difficultés de manipulation, n’est pas soutenue. Or, c’est un produit qui est utilisé dans la lutte contre la fièvre aphteuse, notamment pour désinfecter les véhicules qui partent des élevages contaminés.
e) Une première autorisation de produit biocide, en France, en application des dispositions de la directive 98/8/CE
Un premier produit biocide a été autorisé, en France d’ailleurs, dans le cadre du nouveau régime de la directive 98/8/CE, en octobre 2009. Il concerne l’élimination des petits rongeurs à l’aide de pièges à gaz carbonique, qui est une substance à faible risque. C’est un appareillage destiné aux usages professionnels.
La directive 98/8/CE n’a fait l’objet que d’une application partielle. L’établissement d’un bilan et la révision de son dispositif à un tel stade peuvent donc sembler relever d’une approche paradoxale. Néanmoins, comme l’a fait la Commission européenne dans le cadre du rapport précité de 2008, établi selon la clause d’évaluation qui figure dans le corps de la directive, des propositions d’aménagement peuvent d’ores et déjà être formulées.
Leur contenu est d’ailleurs corroboré par les éléments recueillis par les rapporteurs au cours des auditions préparatoires au présent rapport.
b) Les acquis de la directive 98/8/CE : une première avancée dans la protection de la santé, du consommateur et de l’environnement
La directive 98/8/CE a permis quelques avancées en matière de protection de la santé, de protection des utilisateurs de produits, ainsi que de protection de l’environnement.
D’une part, les recensements auxquels elle a donné lieu ont été l’occasion d’avoir pour la première fois une vision globale des substances actives et des produits utilisés. D’autre part, le secteur a cessé de se développer « au fil de l’eau », dans des conditions de sécurité reposant sur des règles générales et non sur une approche adaptée. C’est d’autant plus légitime que, dans l’esprit d’un grand nombre de consommateurs, les produits biocides sont des produits qui ne sont pas librement produits et mis en vente, mais qui sont contrôlés. Enfin, comme on l’a vu, la démarche systématique d’identification et d’évaluation des substances actives a entraîné l’élimination de certaines substances dont la nocivité est la raison de fond de l’absence de soutien.
La Commission européenne cite dans son rapport le cas de la strychnine, autrefois utilisée comme taupicide, de composés d’arsenic et du tributylétain, identifiés comme dangereux pour la conchyliculture et donc interdit en France pour les peintures pour coques de bateau dès les années 1980, et dorénavant prohibés en Europe.
Lorsqu’elles n’ont pas été soutenues, les substances actives ont été interdites, comme on l’a vu précédemment, de même que les produits biocides utilisant ces substances actives.
Des substituts ont dû être utilisés.
Certains professionnels ont néanmoins indiqué qu’ils avaient été contraints de recourir à ces substituts avant même qu’ils aient été évalués. C’est un élément d’incertitude, dans la mesure où en cas de non-inscription future du substitut dans la liste des substances actives autorisées, il leur faudra de nouveau prévoir une solution alternative avec les coûts que cela implique.
La procédure d’évaluation systématique tant des substances actives que des produits biocides implique l’obligation de produire un dossier d’évaluation.
Le dossier reste conséquent en raison de la quantité des informations à fournir, même si, plusieurs interlocuteurs l’ont indiqué, la numérisation a permis de réduire, pour l’essentiel, sa lourdeur matérielle.
La principale critique porte sur le coût de réalisation du dossier.
Ce coût a deux composantes principales : d’une part, il s’agit des frais engagés pour la réalisation des évaluations toxicologiques et écotoxicologiques ; d’autre part, il s’agit des redevances, question évoquée au point suivant.
En l’état, seules les substances actives, plutôt produites par les grandes entreprises, ont été concernées.
Néanmoins, la perspective de devoir prochainement déposer des dossiers de demande d’AMM inquiète les PME.
Si le montant des redevances est déjà élevé, et constitue un élément connu, tel n’est pas le cas du coût de constitution d’un dossier proprement dit, qui exige de faire appel à une expertise extérieure, ainsi que du coût des évaluations, c'est-à-dire des analyse scientifiques. Hors redevance, le coût d’un dossier peut atteindre plusieurs centaines de milliers d’euros.
S’agissant des redevances, celles-ci sont, pour la France, fixées par arrêté du 26 janvier 2007 modifiant l’arrêté du 24 juin 2004 fixant le montant de la rémunération due au titre de l’autorisation de mise sur le marché des substances et produits biocides.
Pour ce qui concerne les substances actives, le total est de :
-165.000 euros pour un nouveau dossier, c’est-à-dire pour la demande d’inscription en vue d’un produit biocide ;
- 85.000 euros pour une demande d’inscription supplémentaire pour un produit biocide autre que ceux pour lesquels la même substance est déjà inscrite ;
- 104.000 euros lorsque la substance a déjà été examinée dans le cadre d’une autre réglementation ;
- 132.000 euros lorsque la substance a déjà été examinée dans le cadre d’une autre réglementation, mais qu’un examen supplémentaire est nécessaire ;
- 185.000 euros si la substance exige des données supplémentaires.
Ces tarifs comprennent la somme de 11.000 euros perçue au titre de la recevabilité du dossier. Ils sont majorés de 15.000 euros s’il y a nécessité d’une évaluation du risque alimentaire, et de la même somme s’il y a nécessité d’une évaluation du risque hospitalier.
Pour leur part, les produits biocides font l’objet des redevances suivantes :
- 32.500 euros pour un nouveau dossier ;
- 16.000 euros si l’évaluation des dangers est déjà disponible ;
- 25.500 euros si l’évaluation des dangers écotoxocologiques peut être obtenue par calcul ;
- 37.000 euros si des données supplémentaires sont requises.
Ces tarifs comprennent la somme de 2.500 euros perçue au titre de la recevabilité.
Ils sont majorés de 7.500 euros en présence d’une substance préoccupante, ainsi que de 2.500 euros lorsque est exigée l’évaluation du risque alimentaire de même que quand celle du risque hospitalier est également nécessaire.
(3) Des différences d’approche entre les Etats membres, notamment sur le niveau des redevances et les délais d’instruction des dossiers
La mise en œuvre, même partielle, de la directive 98/8/CE a montré des différences d’approche entre les Etats membres.
Un premier exemple concerne les données à fournir pour les produits à faible risque, où les niveaux d’exigence des Etats membres ne seraient pas les mêmes.
Un deuxième exemple concerne les formulations cadres, à savoir les déclinaisons d’un même produit. La formulation cadre est définie comme un groupe de produits très proches et contenant les mêmes substances actives, destinés aux mêmes utilisateurs et aux mêmes utilisations, présentant les mêmes niveaux de risques et d’efficacité, mais avec soit une réduction de la quantité de substance active soit le remplacement de colorants, piments ou parfums. Avant même que des formulations cadres ne puissent être délivrées, certains Etats membres ont appliqué par anticipation le principe de formalités simplifiées lorsque le même produit était décliné selon différentes couleurs.
L’élément le plus flagrant concerne le niveau des redevances.
Le texte de la directive impose aux Etats membres de mettre en place un dispositif de redevance pour couvrir les coûts administratifs de procédure.
La Commission européenne a constaté que tant les mécanismes que les montants des redevances sont hétérogènes. Pour l’évaluation d’une substance active, le coût de la redevance varie de 50.000 euros à 350.000 euros. Les modalités de paiement varient aussi. Les graphiques suivants récapitulent ces écarts.
Redevances et fourchettes de redevances perçues par les
etats membres pour l’evaluation des substances actives

Redevances et fourchettes de redevances perçues par les etats membres pour l’autorisation des produits
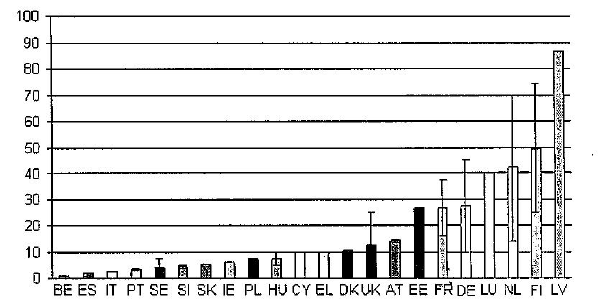
Globalement, les professionnels ont fait état de différences d’approche entre les Etats membres, avec des Etats attractifs, et des Etats qui le seraient moins. Le cas du Royaume-Uni a été avancé.
L’une des critiques les plus couramment adressées au dispositif de la directive 98/8/CE est la longueur des délais d’instruction.
Elle concerne pour l’instant uniquement les substances actives, qui sont les seules à avoir été évaluées.
Le cas « unique » du seul produit biocide autorisé en France dans le cadre de la nouvelle réglementation ne peut être considéré comme significatif, même s’il a été très rapide.
L’une des difficultés d’interpréter ces délais tient à l’importance des arrêts, ou « arrêts d’horloge ». Il s’agit d’une suspension des délais et des procédures d’évaluation le temps que des informations complémentaires soient communiquées. Ces arrêts ne sont pas nécessairement défavorables aux entreprises, qui bénéficient actuellement du régime provisoire.
En effet, selon les données communiquées aux rapporteurs par l’AFSSET, si la durée moyenne des arrêts de computation des délais est du même ordre de grandeur que l’examen des dossiers, comme l’indique le tableau suivant :
Durée moyenne d’examen des dossiers communiquée par l’AFSSET
Période du : |
30/11/2008 |
30/11/2007 |
30/11/2006 |
30/11/2005 | |
au : |
30/11/2009 |
30/11/2008 |
30/11/2007 |
30/11/2006 | |
Durée moyenne |
Nb de jours |
263,1 |
223,3 |
333,4 |
172,3 |
de la recevabilité |
Nb de mois |
8,8 |
7,4 |
11,1 |
5,7 |
Durée moyenne |
Nb de jours |
606,5 |
576,6 |
651,1 |
478,6 |
des arrêts |
Nb de mois |
20,2 |
19,2 |
21,7 |
16,0 |
Durée moyenne |
Nb de jours |
434,7 |
460,0 |
456,3 |
621,0 |
d’évaluation |
Nb de mois |
14,5 |
15,3 |
15,2 |
20,7 |
Source : AFSSET.
Néanmoins, on doit relever que l’examen de la recevabilité du dossier prend déjà plusieurs mois.
Au chapitre des éléments positifs néanmoins, il apparaît que l’AFSSET a réalisé des avancées significatives sur les durées d’évaluation, c'est-à-dire d’examen des dossiers au fond, avec un gain de six mois entre 2006 et 2009
Le bilan de la mise en œuvre de la directive 98/8/CE a permis de recenser également plusieurs lacunes d’ordre technique.
La plus importante d’entre elles concerne le champ d’application du texte. Celui-ci ne concerne que les seuls produits biocides utilisés en Europe, dans un cadre professionnel ou domestique, et non les articles traités avec ces produits. Il en résulte donc une inégalité de traitement avec les produits importés, et une possibilité de dumping environnemental à leur profit.
Le problème s’est clairement posé avec les produits de traitement du cuir.
En 2008, des articles (chaussures et canapés, notamment) traités avec du fumarate de diméthyle, interdit en Europe mais autorisé en Chine, ont provoqué vingt-huit cas d’allergie grave en France. La presse et l’audiovisuel s’en sont alors fait largement écho.
DEUXIEME PARTIE :
LA PROPOSITION DE RÈGLEMENT PRÉSENTÉE PAR LA COMMISSION EUROPÉENNE : DES AVANCÉES, MAIS UN DISPOSITIF NÉANMOINS PERFECTIBLE, NOTAMMENT POUR ÉLIMINER LES MENACES QU’IL FAIT PESER SUR LES PME
A. Le remplacement d’une directive par un règlement pour une application homogène dans les différents Etats membres
Pour remédier aux différences d’interprétation déjà constatées entre les Etats membres dans l’application de la directive 98/8/CE, la Commission européenne propose de remplacer l’actuelle directive par un règlement.
Contrairement à une directive, un règlement n’exige pas de texte de transposition, puisqu’il est d’application de plein droit dans tous les Etats membres. Il en résulte deux avantages.
D’une part, les risques de divergence entre les Etats membres dans l’application du dispositif européen sont réduits.
D’autre part, c’est plus simple pour les entreprises comme pour les distributeurs et les utilisateurs puisqu’une seule règlementation est applicable sur le territoire de l’Union européenne, et non plus vingt-sept législations.
B. Un champ d’application étendu aux produits et articles traités avec des produits biocides, pour éviter tant le risque de dumping environnemental et sanitaire des pays tiers qu’un éventuel motif de délocalisation de l’industrie chimique hors de l’Europe
Actuellement, le champ d’application de la directive 98/8/CE relève d’une conception assez classique : il ne vise que les seuls produits qu’il concerne directement, c'est-à-dire les produits biocides.
Cette approche logique mais stricte n’est pas suffisante, car si l’on se place d’un autre point de vue, celui des articles et produits qui font l’objet d’un traitement biocide, elle ne concerne que ceux qui sont fabriqués en Europe.
Elle laisse donc la possibilité en Europe d’importer librement ces mêmes produits et articles qui subissent hors d’Europe un traitement biocide à base de substances ou produits interdits dans les 27 Etats membres.
Une telle situation n’est pas acceptable de deux points de vue :
– le premier est d’ordre sanitaire : des produits ou articles sont importés alors qu’ils présentent des niveaux de risque trop élevés ;
– l’autre d’ordre économique et commercial : il y a distorsion de concurrence, et donc un risque supplémentaire de délocalisation, puisque les produits importés peuvent faire l’objet, dans les pays tiers, de traitements moins coûteux que les produits fabriqués à l’intérieur de l’Union, car les procédés moins nocifs sont en général innovants et donc plus coûteux.
Il faut donc se féliciter que la proposition telle que présentée par la Commission européenne prévoie que les articles traités par un produit biocide ne puissent être mis sur le marché, c'est-à-dire commercialisés dans le marché intérieur, que si ce produit est autorisé dans l’un des Etats membres de l’Union européenne.
Des aménagements doivent cependant être prévus sur certaines modalités d’application.
Tel est notamment le cas, pour des raisons pratiques, pour le marquage et l’étiquetage des produits vendus en vrac tels que les poteaux de bois.
La proposition de règlement confie un rôle nouveau, de coordination, à l’Agence européenne des produits chimiques (ECHA).
Créée dans le cadre du règlement REACH et implantée en Finlande, à Helsinki, l’ECHA est chargée des procédures d’enregistrement, d’évaluation, d’autorisation et de restriction à l’utilisation des substances chimiques.
De manière spécifique, la proposition de règlement prévoit, pour l’essentiel, que l’Agence sera chargée des missions suivantes :
– la réception des demandes d’inscription des substances actives et la désignation de l’autorité nationale chargée de la demande d’évaluation ;
– la gestion des éléments relatifs aux substances dont la substitution est envisagée ;
– la préparation des avis, soumis à la Commission européenne, d’inscription des substances actives ;
– la mise en œuvre de la procédure d’autorisation communautaire des produits biocides ;
– un rôle consultatif sur les questions scientifiques et techniques, en cas d’opposition d’un Etat membre à une demande de reconnaissance mutuelle comme en cas d’intention d’adaptation aux circonstances locale de l’AMM délivrée par un autre Etat membre ;
– la tenue de la base de données sur les substances actives et les produits biocides ;
– la gestion du registre de partage des données sur les produits biocides.
Il est également prévu de constituer au sein de l’Agence, pour préparer ses décisions en la matière, un comité des produits biocides, de manière à disposer d’une instance spécialisée, dont les membres sont désignés sur proposition des Etats membres. Les règles prévues par le règlement REACH sont applicables à cette instance. Elles imposent notamment aux Etats membres et à leurs autorités compétentes de mettre à leur disposition les éléments nécessaires à l’exercice de leurs fonctions.
Ce comité est distinct du comité permanent des produits biocides, instance de comitologie placée auprès de la Commission européenne.
D. Le durcissement, pour une plus grande sécurité sanitaire, des exigences pour l’inscription communautaire des substances actives
Pour l’essentiel, le texte de la directive 98/8/CE prévoit actuellement que l’inscription sur les listes communautaires de substances actives utilisables dans la fabrication des produits biocides repose sur deux catégories de critères.
La première catégorie regroupe ceux relatifs à l’efficacité et aux risques, selon la démarche d’évaluation appliquée au domaine sanitaire au sens large :
– les substances actives ne sont inscrites que si les produits qui en seront issus seront suffisamment efficaces ;
– ceux-ci ne doivent pas avoir d’effet inacceptable sur les organismes cibles (résistance inacceptable, résistance croisée, souffrances et douleurs inutiles chez les vertébrés) ;
– ils ne doivent pas non plus soit intrinsèquement, soit par l’effet de leurs résidus, avoir d’effet inacceptable sur la santé humaine ou animale, et ils ne doivent pas avoir un tel effet soit directement soit indirectement, par l’intermédiaire par exemple de l’eau potable, sur des aliments destinés à la consommation humaine ou animale ou de l’air ;
– ils ne doivent pas non plus avoir d’effet inacceptable sur l’environnement, notamment entraîner une contamination des eaux ;
– en outre, les propriétés physiques et chimiques du produit biocide qui sera issu de cette substance doivent permettre son utilisation, son stockage et son transport dans des conditions adéquates.
La deuxième catégorie regroupe des critères de refus d’inscription et destinés à promouvoir les substances qui présentent de moindres risques.
Ainsi, une substance active peut faire l’objet d’un refus d’inscription lorsque son évaluation montre qu’elle présente des risques préoccupants pour la santé et l’environnement dans des conditions normales d’utilisation et qu’il lui existe un substitut qui présente significativement moins de risques.
Par ailleurs, l’inscription dans liste des substances actives à faible risque, celle de l’annexe 1 A est soumise à condition. Ne peuvent y figurer les substances classées comme cancérigènes, mutagènes, toxiques pour la reproduction (CMR), ou bien sensibilisatrices ou susceptibles de bioaccumulation et ne se dégradant pas rapidement.
Le texte proposé par la Commission européenne renforce notablement les exigences en matière de substances actives, et repose sur une approche plus systématique avec une conception beaucoup plus claire des critères d’exclusion et l’introduction de la notion de substance candidate à la substitution, selon les principes du règlement REACH.
Le texte prévoit, en effet, un renforcement des critères d’exclusion avec le principe de la non inscription dans la liste des substances actives des substances cancérigènes, mutagènes ou toxiques pour la reproduction (CMR) de catégories 1 A et 1 B (celles dont le risque est avéré) ainsi que des perturbateurs endocriniens.
Les exceptions prévues concernent des cas précis :
– le risque maîtrisé : l’exposition des humains est négligeable dans des conditions normales d’utilisation du produit biocide, notamment lorsque le produit est utilisé dans des systèmes fermés et strictement contrôlés ;
– l’impératif sanitaire : la substance active est nécessaire pour lutter contre un grave problème de santé publique ;
– enfin, la nécessité et l’absence de solution alternative : la non-inscription de la substance active aurait des conséquences disproportionnées par rapport aux risques de son utilisation pour la santé humaine et l’environnement, parce qu’il n’existe pas de substance ou de technologie de substitution.
Conformément à la démarche engagée au niveau européen pour tous les produits chimiques avec le règlement REACH, le principe de substitution est posé pour les substances actives présentant les dangers les plus importants.
Sont en effet considérées comme substances comme candidates à la substitution, c'est-à-dire dont la substitution est envisagée :
– les substances plus toxiques que « la moyenne », à savoir celles dont la dose journalière admissible ou la dose aiguë de référence, ou encore pour lesquelles le niveau acceptable d’exposition de l’opérateur, sont inférieurs à ceux de la majorité des substances actives inscrites ;
– les substances répondant à deux des trois critères PBT : persistance bioaccumulation et toxicité ;
– les substances engendrant des préoccupations particulières, même avec des mesures très strictes de gestion des risques, à savoir :
. celles contenant un pourcentage important d’isomères non actifs ;
. les CMR de catégories 1A et 1B (CMR avérés) ;
. les perturbateurs endocriniens.
On observera que ces deux dernières catégories concernent des substances qui ne sont pas en principe inscriptibles, et ne peuvent donc être utilisées que dans le cadre des dérogations précitées à cette interdiction. Le principe de substitution représente donc pour elles un « deuxième filtre », une « seconde sécurité ».
Cette indentification a deux conséquences majeures :
– l’une sur la substance elle-même : le renouvellement de l’inscription des substances candidates à la substitution est prévu pour être décennal, et non illimité, comme pour les autres ;
– l’autre sur les produits biocides qui en sont issus, avec la création d’une faculté d’évaluation comparative : tout produit contenant une substance répondant aux critères de substitution précités peut faire l’objet d’une étude comparative lors de sa demande d’AMM et cette AMM peut être limitée (facultés d’usage réduites) ou refusée s’il existe un produit déjà autorisé ou une méthode non chimique présentant significativement moins de risques, ne présentant pas d’inconvénient économique majeur et si la diversité chimique est suffisante pour limiter les risques de résistance.
c) Une plus grande cohérence du dispositif d’inscription des substances actives avec notamment le principe d’une liste européenne unique en remplacement des trois listes actuelles
La cohérence du dispositif d’ensemble d’inscription des substances actives est renforcée avec, comme on l’a vu, un rôle central de l’Agence européenne des produits chimiques (ECHA), qui devient :
– le coordinateur de l’évaluation des substances actives dès l’entrée en vigueur du règlement, ce qui concerne la suite du programme d’évaluation systématique ;
– par la suite, pour les nouvelles substances actives, l’organisme recevant les demandes des industriels. L’évaluation est néanmoins prévue pour être confiée à un Etat membre, celui demandé par le pétitionnaire.
Par ailleurs, les trois listes actuelles des substances actives sont remplacées par une liste unique : le faible succès des demandes d’inscription de substances à faible risque conduit comme on l’a vu la Commission européenne à préconiser une autre approche pour promouvoir ce faible risque.
E. Un réaménagement substantiel du dispositif d’autorisation de mise sur le marché des produits biocides, dans le respect du principe de la compétence nationale
La Commission européenne n’a pas proposé de bouleverser l’équilibre actuel qui repose sur l’inscription au niveau européen des substances actives, et l’autorisation au niveau national des produits biocides, avec reconnaissance mutuelle des autorisations accordées par les Etats membres.
C’est une option à laquelle les Etats membres sont attachés, même si, à l’opposé, la logique du marché intérieur pourrait plaider en faveur d’une autorisation communautaire unique.
La proposition qu’une évolution limitée mais significative, avec la création d’une AMM communautaire pour deux catégories de produits, les produits à faible risque et les produits contenant une ou plusieurs substances actives, selon les modalités évoquées au 2 ci-après.
Plusieurs interlocuteurs des rapporteurs ont estimé qu’il conviendrait d’aller au-delà.
Quelques arguments semblent bien plaider néanmoins, à ce stade et en l’état, pour la prudence en la matière.
En premier lieu, l’importance du nombre des produits biocides (quelque 25.000 pour le seul marché français) fait qu’il n’est pas certain que le niveau communautaire dispose de la capacité de traitement des dossiers, d’autant que l’ECHA, déjà mobilisée par la mise en œuvre de la directive REACH, ne dispose pas de la capacité matérielle d’expertise nécessaire.
En deuxième lieu, le développement des AMM communautaires paraît devoir se heurter au goulet d’étranglement que constitue la comitologie, puisque leur délivrance n’intervient en effet qu’après avis du comité permanent des produits biocides.
En troisième lieu, sur le plan financier, le niveau de la redevance annoncé par la Commission européenne pour une AMM communautaire de produit biocide apparaît, à raison de 100.000 euros, supérieure au total d’une redevance à verser au titre d’une AMM nationale et de vingt-sept redevances au titre de la reconnaissance mutuelle, selon les éléments communiqués aux rapporteurs.
Enfin, en quatrième et dernier lieu, toute extension du champ de l’autorisation communautaire, déjà facultative, ne peut que nuire à l’objectif de faire de l’AMM communautaire un élément d’incitation au développement de produits innovants ou bien à faible risque.
2. Une possibilité d’autorisation communautaire, avec accès direct au marché de chacun des Etats membres, pour les produits à faible risque et les produits innovants contenant de nouvelles substances actives
Dans la promotion des produits biocides à faible risque, les dispositions de la directive 98/8/CE n’ont pas été pertinentes : la liste des substances actives pour inclusion dans les produits biocides à faible risque n’a pas été alimentée comme on aurait pu le souhaiter. Une seule substance a été inscrite : le dioxyde de carbone.
Sur ce constat d’échec, la Commission européenne propose un mécanisme plus ambitieux, fondé sur la possibilité d’une autorisation communautaire pour les produits biocides à faible risque. Il s’agit d’une possibilité et non d’une obligation pour les fabricants.
L’enjeu est de leur ouvrir directement le marché européen
Pour favoriser l’innovation, cette même procédure européenne est prévue pour les produits biocides comprenant une ou plusieurs nouvelles substances actives.
Les demandes seront adressées à l’Agence européenne des produits chimiques (ECHA) et l’évaluation confiée à l’autorité compétente d’un Etat membre choisi par elle.
Il est prévu qu’avant la décision d’autorisation, émise par la Commission européenne sur avis de l’ECHA et après avis du comité permanent des produits biocide, un Etat membre puisse demander d’adapter certaines conditions de l’autorisation aux circonstances locales.
La Commission européenne a proposé deux aménagements afin de simplifier, faciliter et accélérer la procédure de reconnaissance mutuelle des AMM nationales.
Le premier d’entre eux concerne la création d’une nouvelle procédure dite de reconnaissance mutuelle simultanée.
Il s’agit d’une procédure différente de l’actuelle, suivant laquelle le fabricant ou l’importateur s’adresse successivement aux Etats membres dans lesquels il souhaite commercialiser le produit concerné. Afin de clarifier le texte, cette actuelle procédure serait renommée reconnaissance mutuelle séquentielle.
La procédure de reconnaissance mutuelle simultanée repose sur le principe du dépôt d’une demande d’AMM pour un fabricant en même temps devant plusieurs Etats membres. Celui-ci choisit lui-même une autorité nationale de référence pour procéder à l’évaluation. La procédure prévoit une consultation des autres autorités nationales compétentes concernées, ainsi qu’une coordination entre elles, avec une approbation par chacune du rapport d’évaluation de même que du résumé des caractéristiques du produit. En cas de désaccord, la Commission européenne est saisie. C’est à elle que revient alors en dernier ressort la décision d’autorisation, de limitation d’autorisation ou de refus.
Par ailleurs, la procédure d’adaptation aux circonstances locales est maintenue dans le cadre des procédures de reconnaissance mutuelle. Pour la mettre en œuvre, il faut démontrer que l’espèce cible n’est pas présente en quantités nuisibles, ou bien une tolérance ou une résistance inacceptable de l’organisme cible au produit biocide, ou bien des circonstances d’utilisation (climat ou période de reproduction des espèces cibles notamment) différentes de celles existant dans l’Etat membre d’autorisation initiale et qui font qu’une autorisation nationale inchangée peut présenter des risques inacceptables pour l’environnement.
Le second grand aménagement concerne la création d’une demande de reconnaissance mutuelle par des organismes officiels ou scientifiques, ou des organisations professionnelles. Il s’agit de permettre aux organismes tiers de pallier les éventuelles difficultés de l’initiative privée.
Enfin, les dispositions relatives au refus de reconnaissance mutuelle sont clarifiées. Deux éléments doivent être soulignés. D’une part, la proposition de règlement expose clairement les étapes de la procédure pour le règlement des litiges entre Etats membres. D’autre part, le texte proposé reprend la disposition qui permet à un Etat membre de refuser la reconnaissance mutuelle pour un certain nombre de catégories de produits (les avicides, les piscicides et les produits utilisés contre d’autres vertébrés) au titre de la protection de la santé humaine ou de la santé animale, de la protection des végétaux ainsi que de la protection d’éléments du patrimoine national ayant une valeur artistique, historique ou archéologique ou de la protection de la propriété intellectuelle et commerciale. Selon les débats préparatoires au Conseil, cette liste pourrait être élargie.
4. Un effort d’harmonisation et de clarification des trois éléments sur lesquels s’exerce la concurrence entre autorités sanitaires : le déroulement des procédures d’autorisation ; les délais d’instruction des dossiers ; les redevances versées aux autorités compétentes
La proposition de règlement prévoit plusieurs améliorations sur le déroulement des procédures. Son dispositif est plus précis que celui de la directive 98/8/CE. Il offre davantage de sécurité aux opérateurs car il prévoit explicitement leur consultation aux différents stades de la procédure.
L’une des principales critiques des professionnels à l’encontre des textes actuellement en vigueur porte sur la longueur des délais d’autorisation.
C’est une question essentielle. L’argument est souvent invoqué pour expliquer le retard dans la réalisation du programme d’évaluation systématique des substances actives.
Néanmoins, s’il est préjudiciable en raison des incertitudes qu’elle engendre (une substance sera-t-elle autorisée ou interdite), le retard s’accompagne de la prolongation du régime national antérieur et n’est donc pas toujours dommageable.
Pour régler la question, la Commission européenne prévoit d’encadrer à l’avenir les procédures d’inscription des substances actives et d’AMM de produits biocides, comme de reconnaissance mutuelle des AMM nationales, dans des délais précis figurant au règlement.
Ainsi, la proposition prévoit pour les produits biocides un délai d’un mois pour la recevabilité du dossier et de douze mois pour l’évaluation, soit une durée totale de treize mois. Pour le renouvellement des dossiers, la proposition de règlement retient l’hypothèse d’un délai d’un mois pour la recevabilité et de six mois pour l’évaluation.
S’agissant de l’autorisation communautaire, la procédure est plus complexe et le délai total est de vingt-neuf mois.
Par rapport à la procédure nationale, les délais de base (un mois pour la recevabilité et douze mois pour l’évaluation) sont les mêmes.
Le supplément est dû aux formalités complémentaires : un mois pour la saisine par l’ECHA de l’autorité compétente chargée de l’évaluation ; neuf mois pour que l’ECHA rende son avis sur l’évaluation qui a été effectuée par cette dernière ; six mois pour l’intervention de la décision au niveau communautaire, qui incombe à la Commission européenne avec l’intervention du comité permanent des produits biocides.
Pour le renouvellement des autorisations communautaires, on obtient un délai total de vingt-trois mois.
Pour les procédures de reconnaissance mutuelle, les délais sont aussi fixés et l’on obtient dix-huit mois pour la reconnaissance simultanée et un temps équivalent, de dix-sept mois, pour la reconnaissance mutuelle séquentielle, selon les éléments fournis aux rapporteurs.
c) Une première harmonisation des redevances, avec une certaine prise en compte du cas spécifique des PME
Le texte de la directive 98/8/CE se limite à prévoir l’obligation de redevances correspondant autant que possible aux coûts des différentes procédures.
Il y a donc une obligation de principe d’une redevance, mais une grande liberté laissée aux Etats membres sur le niveau et les modalités de sa perception.
La proposition de directive prévoit une certaine harmonisation entre les redevances nationales.
Il faut observer que la création d’une faculté d’autorisation communautaire, avec intervention de l’ECHA, entraîne aussi la mise en place d’un système de redevance européenne, établi par la Commission européenne.
Pour ce qui concerne les redevances nationales, la proposition de directive s’en tient à la fixation des grands principes avec :
– une redevance réduite pour les PME ;
– la prise en compte des coûts complets de constitution du dossier : la structure des redevances devra prendre en compte le fait que les informations aient été soumises conjointement par plusieurs entreprises ou séparément ;
– la possibilité, dans des conditions à préciser, pour l’autorité compétente ou l’ECHA de ne pas percevoir la redevance ;
– le caractère annuel de la redevance pour les personnes qui mettent sur le marché des produits biocides ;
– l’adéquation entre les recettes des autorités concernées, parmi lesquelles les redevances, et leurs coûts de fonctionnement.
Des dispositions transitoires sont par ailleurs prévues pour faciliter le passage d’un dispositif à l’autre et garantir les droits acquis dans le système actuel.
L’objectif est donc d’avoir des pratiques identiques dans tous les Etats membres et d’encourager, avec la prise en compte du nombre de demandeurs, le recours à l’innovation.
Pour les PME, le texte prévoit textuellement une « redevance réduite ».
d) Une certaine perspective d’allègement des dossiers avec l’amélioration des dérogations aux exigences en matière de données
Actuellement, le dispositif de la directive 98/8/CE prévoit, sur le plan des principes, la faculté de déroger aux exigences en matière de fourniture de données par les demandeurs d’une AMM.
La Commission européenne a estimé que, faute de précision, cette faculté n’était pas utilisée et que les Etats membres avaient été bloqués par les craintes issues d’une totale incertitude.
Aussi propose-t-elle de bien préciser les motifs de dispense de fourniture des données requises (informations non nécessaires compte tenu de l’exposition associée aux utilisations proposées, informations non nécessaires sur le plan scientifique et informations impossibles à fournir sur le plan technique), et d’ouvrir la faculté au demandeur de demander des adaptations des exigences en matière de données. Des justifications appropriées doivent être fournies. Elles doivent être appréciées selon des critères qui seront définis dans le cadre des mesures d’exécution du règlement, par la Commission européenne, suivant la procédure de réglementation avec contrôle qui laisse un droit de regard aux Etats membres et au Parlement européen.
Dès lors qu’il s’agit uniquement d’éviter toute fourniture d’éléments non pertinents, ces dispositions sont opportunes. D’une manière générale, il convient d’éviter que le respect de la procédure ne prime inutilement, et de manière néfaste, sur le fond.
e) La clarification et le renforcement des règles de protection des données, ainsi que la suppression des études redondantes sur les vertébrés
Dans la constitution des dossiers de demandes d’évaluation, les données toxicologiques et écotoxicologiques ne constituent pas uniquement un enjeu scientifique, mais également un enjeu économique.
Ces données sont notamment collectées dans le cadre d’études financées par les entreprises, et dont le coût peut, selon les éléments communiqués aux rapporteurs par les professionnels, représenter plusieurs centaines de milliers d’euros.
La directive 98/8/CE a prévu une période de protection des informations détenues par les autorités compétentes de dix ans pour les substances actives et pour les produits biocides, de quinze ans pour les nouvelles substances actives. Une nouvelle période complémentaire de protection des données de cinq ans est ouverte à l’occasion du réexamen du dossier, notamment en vue de la prolongation de l’inscription ou de l’AMM au-delà des dix premières années.
La proposition de directive complète et parachève ce dispositif en prévoyant la protection des données transmises après l’inscription de la substance active, c'est-à-dire des données collectées dans la phase d’évaluation ou de l’utilisation du produit biocide dont elle est un constituant, ainsi que de certaines données communiquées à l’occasion des études nouvellement produites. Il s’agit de renforcer la protection de la propriété intellectuelle.
De manière opposée, la proposition de règlement prévoit de rendre obligatoire le partage des données pour éviter le renouvellement des études sur les vertébrés, les plus onéreuses, les plus lourdes et les plus contestées par certains courants de l’opinion.
C’est une évolution majeure puisque la directive 98/8/CE a exigé un accord préalable du premier demandeur pour que le deuxième demandeur ou, plus généralement, le demandeur ultérieur puisse invoquer des données déjà détenues par l’autorité compétente. Formellement, le demandeur ultérieur doit disposer d’une lettre d’accès, accord écrit lui permettant d’invoquer ces éléments.
La proposition de règlement va plus loin en prévoyant le partage obligatoire des données les plus sensibles, celles qui sont relatives aux essais sur les vertébrés. Elle prévoit aussi le partage des coûts, avec en cas de conflit, des modalités d’arbitrage.
Les dispositions correspondantes sont légitimes, dès lors qu’il s’agit d’éviter les expérimentations lourdes et redondantes. Néanmoins, elles doivent être les plus claires et les plus équitables qui soient, afin de limiter au maximum tout risque de contentieux.
Il faut saluer la démarche de la Commission européenne, qui prévoit de définir de manière précise le contenu de la lettre d’accès, qui conserve toute sa pertinence pour les données qui ne relèvent pas de la procédure du partage obligatoire.
Le mécanisme de la formulation cadre permet de délivrer des AMM à des familles de produits très voisins. Il s’agit d’éviter de soumettre à plusieurs autorisations des produits qui ne diffèrent les uns des autres que par des éléments non essentiels.
Actuellement, le dispositif de la directive 98/8/CE offre surtout la possibilité de changer ainsi de pigment et de teinture, c’est-à-dire de couleur, et de parfum.
La proposition de règlement de la Commission européenne propose des assouplissements, permettant des variations plus importantes par rapport au produit biocide de référence, dès lors que ces variations n’ont d’effet ni sur l’efficacité du produit ni sur le niveau de risque.
Cela permet d’envisager précisément une diminution du pourcentage de substance active présente dans le produit biocide de référence et/ou une modification de la composition en pourcentage d'une ou de plusieurs substances non actives, ou encore le remplacement d'une ou de plusieurs substances non actives par d'autres présentant un niveau de risque identique ou inférieur.
6. Une gestion mieux coordonnée des décisions nationales, avec un registre communautaire des produits biocides
Afin de disposer d’une vision d’ensemble sur les produits biocides, la proposition de directive prévoit un registre communautaire des produits biocides.
Celui-ci serait alimenté par les autorités compétentes de Etats membres et contiendrait le résumé des caractéristiques du produit biocide, le rapport récapitulant les conclusions de l'évaluation du produit biocide, ainsi que les motifs justifiant son autorisation ou le rejet de la demande d'autorisation de ce produit, de même que les décisions administratives prises par l'autorité compétente réceptrice au sujet de la demande.
7. La confirmation des obligations en matières de classification, d’étiquetage et d’emballage des produits
La proposition de règlement prévoit les dispositions relatives à la classification, à l’emballage et à l’étiquetage des produits biocides.
Celles-ci n’appellent pas d’observation particulière dès lors que la faculté pour les Etats membres d’exiger un étiquetage dans leur(s) langue(s) nationale(s) est bien prévue et que l’emballage doit éviter tout risque de méprise avec les denrées alimentaires.
De plus, les mentions telles que « produit biocide à faible risque », « non toxique » et « ne nuit pas à la santé » sont interdites, de manière à éviter tout risque d’emploi non conforme aux prescriptions d’usage.
La proposition de règlement prévoit des dispositions sur la transparence, avec la mise à la disposition du public, gratuitement, d’un certain nombre de données scientifiques et techniques, sur les sites Internet de l’ECHA, des autorités compétentes et de la Commission européenne.
Des restrictions à ce principe sont cependant prévues pour permettre aux entreprises de préserver leurs intérêts commerciaux. Celles-ci doivent préalablement exposer les motifs de cette demande de confidentialité.
II. UN TEXTE ENCORE PERFECTIBLE, TANT DANS SES GRANDS ÉQUILIBRES, TROP DÉFAVORABLES AUX PME ET AU MAINTIEN DE LA DIVERSITÉ DES PRODUITS, QUE DANS SES DISPOSITIFS, QUI DOIVENT ENCORE ÊTRE COMPLÉTÉS ET CLARIFIÉS
1. Des risques bien identifiés : la disparition d’un grand nombre de PME, de certains produits et par conséquent de la diversité chimique indispensable pour éviter l’apparition de résistances
La perspective d’une AMM obligatoire pour les produits biocides n’est pas contestable sur le plan sanitaire ni sur le plan environnemental. Elle relève en outre d’une orientation forte de certains segments de sociétés d’appliquer le principe, constitutionnel, de précaution de la manière la plus étendue possible.
Néanmoins, avant toute décision, le législateur doit se poser la question des conséquences concrètes des mesures qu’il prévoit d’adopter, et donc se poser la question de leur évaluation ex ante. C’est une autre déclinaison du même principe de prudence.
En l’espèce, les conclusions que l’on tirer de la visite d’entreprises sur le terrain ne sont guère rassurantes. L’exemple d’une PME modeste, travaillant avec quelque 200 revendeurs, ayant un chiffre d’affaires de moins de 2 millions d’euros, mettant sur le marché plus de 170 formules déclarées (principalement des produits de désinfection et des insecticides à usage professionnel, ces produits étant issus de plus de 30 substances actives) et employant environ 40 salariés, notamment, un ingénieur, est éclairant.
A raison du chiffre d’affaires moyen de l’ordre de 10.000 euros par produit, les coûts d’une AMM sont trop élevés.
Outre la redevance, même annuelle et même minorée, l’entreprise devra supporter le coût des dossiers : évaluations ; rémunération d’un cabinet extérieur pour la confection de chacun d’entre eux puisque l’entreprise ne peut affecter à temps plein, même temporairement, son ingénieur à cette tâche : il y a 170 dossiers à réaliser. Le coût d’un dossier est estimé à 150.000 euros. Ce qui est est jugé clairement prohibitif.
Placée dans l’impossibilité de poursuivre son activité dans les mêmes conditions, l’entreprise indique alors ne pouvoir survivre que soit en devenant un sous traitant, soit en devenant un simple distributeur pour des entreprises de grande taille, avec toutes les incertitudes que cela implique, soit encore en diminuant la gamme de ses produits.
Cette dernière hypothèse, qui n’est pas incompatible avec l’une des deux autres, soulève un problème majeur de santé publique.
La diversité des produits de désinfection à usage professionnel, notamment des élevages ou des locaux sanitaires (cabinets vétérinaires, laboratoires d’analyse, établissements hospitaliers) résulte non d’une diversification de l’offre, mais du choix des clients pour l’utilisation de produits variés. Il est, en effet, impératif pour elles de prévoir une certaine rotation, afin d’éviter l’apparition des résistances, selon les principes de base de l’hygiène publique.
La réduction du nombre des produits disponibles, dans leur déclinaison, fait peser une menace importante.
Dans l’hypothèse la plus extrême, le risque est celui d’en venir à la solution du « tout eau de Javel », qui se heurte à une difficulté sanitaire majeure, en raison l’inefficacité du produit dans certaines circonstances (tel est par exemple le cas en présence de salissure sur un pneu de camion) et, sur le plan matériel, à un obstacle essentiel en raison de son caractère corrosif, lequel en interdit l’usage pour certains matériaux.
Ces risques avérés, invoqués par les professionnels, trouvent-ils une justification possible dans l’amélioration de la sécurité des produits résultant d’une évaluation telle que celle prévue ?
Implicitement, la Commission européenne répond positivement. On peut néanmoins en douter, au moins dans un premier temps qui ne couvre pas le seul court terme.
Lorsque ce risque d’une réduction du nombre des producteurs et des produits est évoqué, plusieurs éléments de réponse sont invoqués.
Aucun n’est néanmoins tout à fait convaincant.
Le premier élément de réponse tient à ce que la proposition de directive prévoit deux mesures, précédemment évoquées, en faveur des PME. La première d’entre elles est un niveau réduit de redevance. La seconde consiste en l’élargissement des possibilités de dispense des données, ce qui permet d’alléger le dossier en aval.
Le deuxième élément de réponse est que les entreprises pourront opérer certaines rationalisations, c'est-à-dire un réduction du nombre de leurs produits. Comme on vient de le voir, cela revient à risquer perdre une partie, tout au moins, de la nécessaire diversité des produits.
Le troisième élément est de nature institutionnelle. Le Gouvernement envisage de mettre en place au sein la future AFSSAET, qui sera issue à partir du 1er juillet prochain de la fusion entre l’AFSSET et l’AFSSA, une structure spécialisée, un Help Desk, pour venir en aide aux PME. Les services déconcentrés de l’Etat seraient également mobilisés. C’est un élément utile pour faciliter les démarches des PME. Néanmoins il ne règle ni la question de la confection matérielle des dossiers, ni celle de leur coût.
Le dernier repose sur des aides à la structuration du secteur, de telle sorte que les entreprises concernées créent des structures leur permettant de faire face aux charges nouvelles de manière collective, grâce à des entités communes qui seraient chargées de leur prêter assistance dans la confection des dossiers et qui auraient vocation à mutualiser le plus possibles les coûts correspondants, notamment ceux des évaluations.
De telles mesures sont en tout état de cause nécessaires. Nul n’en disconviendra. Néanmoins, la question de savoir si elles seront suffisantes reste clairement posée.
Lors de son audition par la Commission des affaires européennes le 6 avril dernier, le ministre d’Etat, ministre de l’écologie, de l’énergie, du développement durable et de la mer, M. Jean-Louis Borloo, est d’ailleurs convenu de ces difficultés et de l’absence de solution, en l’état.
Pour donner aux PME l’environnement leur permettant de poursuivre leurs activités sans avoir à affronter de mutation insurmontable, les mesures envisageables sont de deux ordres : des mesures nationales, telles que le Help Desk précité, et des mesures européennes.
S’agissant des secondes, le cadre même de la proposition de règlement offre plusieurs orientations susceptibles d’en améliorer l’équilibre.
La première d’entre elles concerne les exigences de données à fournir à l’appui des demandes d’AMM.
D’une part, des allégements supplémentaires par rapport aux dispenses de données semblent devoir être envisagés dès lors que les produits concernés simples, et tel est le cas puisque qu’un grand nombre n’est composé que d’une seule substance active (et non d’un mélange de substances actives) et d’excipients. On ne peut non plus envisager de supprimer toute autorisation dans la mesure où même pour les produits les plus simples et constitués de composants courants, les conditions d’emploi et les risques d’exposition doivent être vérifiés : un désinfectant utilisé par pulvérisation ne présente pas les mêmes risques que lorsqu’il est utilisé sous une forme liquide.
D’autre part, il convient également d’envisager un dispositif plus précis sur la lettre d’accès aux données relatives aux substances actives. La proposition de règlement prévoit, en effet, l’obligation pour les fabricants de produits de disposer d’une lettre d’accès aux données déposées auprès des autorités compétentes par les fabricants de substances actives. Elle fixe le contenu de cette lettre d’une manière plus précise que ne le fait la directive 98/8/CE. Néanmoins, elle laisse l’essentiel, à savoir les conditions d’accès à ce document, à la libre négociation entre le producteur de substance active et ses clients.
On doit se demander s’il ne serait pas opportun de prévoir des conditions d’accès facilitées à ces données pour les PME.
En effet, l’objet de la lettre d’accès n’est pas tant de répercuter les coûts d’inscription des substances actives sur l’aval du secteur, ce qui relève davantage de la politique globale des prix de l’entreprise, que de s’assurer que toutes les substances actives utilisées par les producteurs auront bien été évaluées et sont bien celles produites par des industriels qui ont déposé un dossier, et non par des « passagers clandestins » ou « free riders ».
De ce point de vue, le texte initial de la proposition de règlement présentait une lacune en laissant cette possibilité de producteurs « free riders » de substances actives.
Dès lors que celle lacune est comblée, en rendant obligatoire pour tout fournisseur de substance active de faire évaluer sa source, ce risque n’existe plus et l’enjeu de la lettre d’accès s’en trouve considérablement réduit. Il perd sa dimension économique pour devenir une formalité uniquement destinée à lever tout obstacle du côté des règles de la propriété intellectuelle : l’accès à des données protégées ne peut, en effet, intervenir sans le consentement de leur propriétaire.
Une deuxième hypothèse de travail concerne les formulations cadres et à les assouplir le plus possible, sur des bases scientifiques, pour les produits ne comprenant qu’une seule substance active.
Une troisième piste éventuelle dans une même optique, consisterait à traiter de manière également allégée certains produits biocides d’origine naturelle. L’exemple est parfois donné des peaux d’orange, manipulées par chacun d’entre nous, et dont les extraits ont des propriétés biocides.
1. Etablir un haut niveau d’exigence sur les critères d’exclusion et les facultés de dérogation, avec l’ajout de facteurs environnementaux et de dispositions sur les nanomatériaux
Sur les critères d’exclusion des substances actives, la proposition de règlement prévoit comme on l’a vu une approche de sécurité sanitaire. Ne sont visés que les CMR de catégorie 1A et 1B et les perturbateurs endocriniens.
Le projet de rapport préparé par la rapporteure du Parlement européen sur la proposition de règlement, Mme Christa Klass (PPE, Allemagne), au nom de la Commission de l’environnement, de la santé publique et de la sécurité alimentaire, propose d’élargir cette approche en y ajoutant des critères environnementaux. Une telle amélioration est tout à fait pertinente.
Il s’agit ainsi d’ajouter à la liste des exclusions, les substances persistantes, toxiques et bioaccumulables (PTB), les substances très persistantes et très bioaccumulables (vPvB), ainsi que les polluants organiques persistants (POP).
Ce même projet de rapport propose également d’ajouter une disposition sur les nanomatériaux. La disposition proposée vise à ce que les produits biocides en contenant fassent l’objet d’évaluations spécifiques. Son adoption paraît particulièrement opportune.
L’AFSSET vient d’ailleurs de rappeler à propos des nanomatériaux, dans un avis du 24 mars, que le risque qu’ils présente ne peut pas être évalué, mais ne peut pas non plus être exclu. Il en résulte que le principe de précaution doit en la matière jouer.
La proposition de règlement contient une disposition sur le commerce parallèle.
Bien que la terminaison ne soit pas appropriée, car faisant historiquement référence au commerce interlope organisé par des commerçants anglais dans l’Amérique latine du XVIIIe siècle, une telle disposition répond à une exigence juridique forte qui a été précisée par la Cour de Justice dans deux domaines reposant sur la même logique d’AMM que les produits biocides : d’une part, les médicaments ; d’autre part, les produits phytosanitaires, notamment dans l’arrêt du 11 mars 1999 British Agrochemicals Association (affaire C-100/96).
La notion de commerce parallèle est fondée sur les dispositions du traité qui mettent en œuvre le principe de libre circulation des biens.
Elle permet d’importer dans un Etat membre des produits suffisamment similaires à ceux qui y sont déjà autorisés. Dans la plupart des cas, il s’agit du commerce exercé par des professionnels en dehors des circuits de distribution mis en place par un producteur ou un distributeur dans un Etat membre donné.
Comme il s’agit de contourner des monopoles de fait qui ont pu se constituer, notamment à l’abri des règles administratives, elle correspond à la logique du marché intérieur.
La disposition proposée par la Commission européenne prévoit ainsi :
– l’exigence d’une autorisation d’importation parallèle, pour s’assurer qu’il s’agit bien d’un produit essentiellement identique au produit déjà autorisé (lequel est appelé produit de référence) ;
– une définition précise du produit biocide pour lequel l’importation parallèle est demandée : des substances actives de même source (même fabricant/ même lieu de fabrication) ; substances non actives identiques ou similaires ; une identité ou équivalence des effets potentiels néfastes sur la santé humaine ou animale ou sur l’environnement ;
– la faculté pour l’autorité compétente de l’Etat membre d’importation d’obtenir les informations permettant de déterminer si le produit concerné est essentiellement identique au produit de référence.
Ce mécanisme suscite l’inquiétude des professionnels.
D’une part, ils craignent qu’une brèche soit ouverte au profit d’entreprises qui ne rempliraient pas les formalités correspondant à une AMM ou à une reconnaissance mutuelle et ne couvriraient pas les coûts correspondants, notamment en termes de redevance.
D’autre part, se pose la question des éventuels retraits ou modifications d’AMM qui interviendraient sur le produit de référence et ne seraient pas répercutés sur les produits similaires faisant l’objet d’une autorisation d’importation parallèle.
Ces inquiétudes doivent être levées par un texte parfaitement clair.
3. Mettre sur un pied d’égalité les différents producteurs de substances actives en évitant les comportements de type passager clandestin ou « free riders »
Sur les substances actives, les dispositions de la proposition de règlement présentent une lacune qu’il peut être utile de combler.
Comme précédemment évoqué, elles laissent en effet la faculté à des producteurs de substances actives qui n’ont pas participé au programme d’examen systématique, de les produire et de les commercialiser. C’est le risque de voir apparaître des passagers clandestins, ou selon la terminologie anglaise, des free riders.
Conformément à ce que propose la rapporteure du Parlement européen, Mme Christa Klass, il convient de combler cette lacune et d’imposer à tout fabricant de substance active de faire évaluer sa source avant mise sur le marché.
Il importe cependant de prévoir des modalités d’application claires, notamment afin qu’il y ait comparabilité des exigences et, également, des coûts.
En contrepartie, on peut modifier, comme on l’a vu, les dispositions relatives à l’obligation pour tout fabricant ou importateur d’un produit biocide de disposer d’un dossier ou d’une lettre d’accès sur les données relatives aux substances actives utilisées.
Compte tenu du nombre des produits, cette disposition, qui vise indirectement à éliminer tout risque de free rider en matière de substance active, n’apparaît pas praticable.
4. Envisager un renforcement du dispositif sur les produits à faible risque par un allongement des protections intellectuelles
La notion de produit à faible risque présente un indéniable avantage. Elle permet de promouvoir, sur des éléments tangibles, un environnement plus sain.
Il n’est toutefois pas aisé de prévoir un dispositif efficace à la hauteur de l’enjeu. L’échec de la mesure de la directive 98/8/CE sur les substances actives à faible risque le montre.
Pour sa part, la Commission européenne a proposé de définir les produits biocides à faible risque selon deux critères :
– d’une part, une absence complète de risque pour l’environnement (la concentration prévue dans l’environnement ne dépasse pas le dixième de la concentration prévue sans effet) comme pour l’homme (une dose sans effet nocif observée -NOAEL- plus de 1.000 fois supérieure à la concentration d’exposition) ;
– d’autre part, l’absence de substances actives à la nocivité particulière (PTB, vPvB, perturbateurs endocriniens, CMR, neurotoxiques, immunotoxiques, toxiques pour la reproduction, sensibilisante).
En outre, il est prévu que soient également qualifiés de produits biocides à faible risque les produits dont la substance active est telle qu’elle ne peut donner lieu qu’à une exposition négligeable dans des conditions normales d’utilisation et lorsqu’il est manipulé dans des conditions strictement contrôlées à toutes les autres étapes de son cycle de vie.
Elle a seulement prévu comme avantage celui de l’autorisation communautaire, facultative.
Il n’est pas certain que cet avantage soit suffisant. Aussi, s’il s’avère nécessaire d’envisager un renforcement des incitations au développement de ces produits, peut-on suggérer, comme prévu en matière de médicament innovant ou de médicament pédiatrique, un renforcement des règles de la protection intellectuelle, et de la protection des données.
La Commission s’est réunie le 5 mai 2010, sous la présidence de M. Pierre Lequiller, Président, pour examiner le présent rapport d’information.
L’exposé des rapporteurs a été suivi d’un débat.
« M. Lucien Degauchy. Les produits de traitement biologique comme le jus de pyrèthre ou le purin d’ortie sont très peu chers à produire et efficaces mais se heurtent aux intérêts des groupes agrochimiques. Certains ont été retirés du marché par la Commission européenne sans que l’on sache pourquoi. Il y aurait certainement des recherches à faire dans ce domaine.
M. Jean Gaubert, co-rapporteur. Notre rapport ne concerne pas les pesticides. En tout état de cause, les grands groupes industriels n’ont en effet pas intérêt au développement de ces produits simples. Ceux-ci doivent cependant faire l’objet d’une autorisation car le fait d’être naturels ne les empêche pas d’être potentiellement dangereux, comme la ciguë, par exemple. Ils peuvent l’être, notamment lorsqu’ils sont en combinaison avec d’autres.
M. Robert Lecou, co-rapporteur. La différence essentielle est qu’un pesticide est destiné à protéger les végétaux, en agriculture ou en jardinage, alors que les biocides sont des produits qui ont une autre finalité, même s’il peut s’agir à la base des mêmes substances actives. Ce n’est pas la même réglementation.
M. Jérôme Lambert. Le purin d’ortie n’a pas été interdit à l’utilisation, par contre sa commercialisation l’a été car c’est un produit instable aux effets non garantis.
M. Philippe Armand Martin. Les produits biocides ont une utilisation très variée et certains antiparasitaires sont également utilisés pour les grandes cultures. Pour les produits phytosanitaires, utilisés en agriculture, là aussi les procédures d’homologation sont beaucoup trop compliquées, notamment pour les produits qui, de toute évidence, ne sont pas dangereux.
M. Robert Lecou, co-rapporteur. L’autorisation de mise sur le marché n’est pas une spécificité française, c’est une obligation européenne. Pour ce qui est des biocides, notre souhait est qu’il y ait une réduction du coût de la démarche, pour que les PME ne soient pas menacées. Les biocides biologiques sont des produits naturels mais pouvant être dangereux et qui doivent pour cela être expertisés.
M. Jean Gaubert, co-rapporteur. En se dégradant, ces produits peuvent devenir dangereux. La commercialisation est difficile car elle entraîne des problèmes de responsabilité. Nous souhaitons que les produits soient soumis à des degrés d’homologation différents en fonction de leur nature, les produits « bio » devant aussi être homologués. »
La Commission a ensuite adopté les conclusions dont le texte figure ci-après.
CONCLUSIONS ADOPTEES PAR LA COMMISSION
La Commission des affaires européennes,
Vu l'article 88-4 de la Constitution,
Vu la proposition de Proposition de règlement du Parlement européen et du Conseil concernant la mise sur le marché et l'utilisation des produits biocides (COM [2009] 267 final/no E 4532),
Vu le rapport de la Commission au Conseil et au Parlement européen intitulé « Evaluation de la mise en œuvre de la directive 98/8/CE concernant la mise sur le marché des produits biocides (présenté conformément à l’article 18, paragraphe 5, de ladite directive) et rapport sur l’état d’avancement du programme de travail visé à l’article 16, paragraphe 2, de cette même directive » (COM [2008] 620 final),
Vu la directive 98/8/CE du Parlement européen et du Conseil du 16 février 1998 concernant la mise sur le marché de produits biocides,
Vu également le règlement (CE) no 1907/2006 du Parlement européen et du Conseil du 18 décembre 2006 concernant l'enregistrement, l'évaluation et l'autorisation des substances chimiques, ainsi que les restrictions applicables à ces substances (REACH), instituant une Agence européenne des produits chimiques, modifiant la directive 1999/45/CE et abrogeant le règlement (CEE) no 793/93 du Conseil et le règlement (CE) no 1488/94 de la Commission, ainsi que la directive 76/769/CEE du Conseil et les directives 91/155/CEE, 93/67/CEE, 93/105/CE et 2000/21/CE de la Commission,
Considérant que la nature des produits biocides comme celle de leurs substances actives exigent préalablement à leur mise sur le marché une évaluation des risques, notamment de leur toxicité et leur écotoxicité, selon une approche similaire à celle en vigueur pour les produits comparables, tels que les produits phytopharmaceutiques, et adaptée aux conditions de leur usage dans la vie courante, conformément au principe de précaution ;
Regrettant que le programme de travail pour l’examen des substances actives prévu par la directive 98/8/CE précitée n’ait pu être réalisé dans le délai de dix ans initialement prévu et que, par conséquent, l’actuel cadre communautaire applicable aux produits biocides n’ait été que partiellement appliqué ;
Constatant qu’un nouveau texte européen peut néanmoins être envisagé dès maintenant, pour parvenir à un dispositif plus efficace, car protégeant davantage la santé humaine et la santé animale, de même que l’environnement, garantissant une application plus homogène des règles communautaires dans l’ensemble des Etats membres et offrant aux entreprises des procédures plus simples, plus rapides, aux coûts maîtrisés et tenant compte, par des adaptations spécifiques leur évitant des obligations disproportionnées, de la présence d’un grand nombre de PME dans ce secteur d’activité de l’industrie chimique ;
Considérant également qu’un tel dispositif ne doit pas se limiter à fixer les règles relatives aux produits biocides utilisables en Europe, mais doit aussi s’appliquer aux produits importés des pays tiers et traités avec des biocides, pour mettre les professionnels européens à égalité avec ceux de ces même pays tiers et éviter tout risque de délocalisation d’activité hors de l’Union européenne et de dumping sanitaire ou environnemental ;
1. Se félicite que la proposition précitée prévoie une disposition spécifique précisant que les articles et matériaux traités commercialisés dans l’Union européenne ne peuvent l’être que s’ils ont été traités par des produits biocides autorisés dans l’un au moins de ses Etats membre ;
2. Approuve également cette proposition, en ce qu’elle :
a) vise à remplacer une directive par un règlement, afin de garantir, notamment grâce au rôle de coordination de l’Agence européenne des produits chimiques, une application homogène des règles européennes dans tous les Etats membres ;
b) renforce les exigences relatives à l’inscription des substances actives en introduisant notamment des critères d’exclusion du recours aux substances présentant certains dangers ainsi que, pour les substances autorisées, des critères de substitution pour éviter à terme l’usage de celles qui présentent les risques les plus importants ;
c) vise à promouvoir les produits à faible risque et les produits innovants, notamment par la faculté d’une autorisation de mise sur le marché (AMM) communautaire qui leur est spécifique ;
d) simplifie et clarifie la procédure de délivrance des AMM nationales de produits biocides, notamment en prévoyant le dialogue entre l’autorité compétente et le demandeur, en fixant des délais de procédure, en rendant obligatoire le partage des données provenant d’études sur les vertébrés et en précisant, pour éviter les demandes inutiles, les conditions de dérogations aux exigences en matière de données ;
e) améliore la procédure de reconnaissance mutuelle des AMM nationales en créant une procédure de reconnaissance mutuelle simultanée, à côté de l’actuelle procédure, dorénavant appelée reconnaissance mutuelle séquentielle ;
f) prévoie une certaine harmonisation du niveau des redevances, en introduisant le principe d’un montant réduit pour les PME ;
3. Estime néanmoins que son dispositif ne pourra être adopté que s’il prévoit des mécanismes d’application adaptés aux besoins spécifiques des PME, évitant de leur imposer des obligations auxquelles elles ne pourront pas faire face et hors de proportion avec les enjeux environnementaux et sanitaires concernés, ces mécanismes étant complétés par des mesures pratiques au niveau des autorités compétentes nationales et, le cas échéant, de l’ECHA ;
4. Considère également, à ce stade, qu’un certain nombre d’améliorations et de compléments doivent être apportés à cette proposition de règlement pour :
a) ajouter aux critères d’exclusion des substances actives une dimension environnementale, pour exclure les substances persistantes, bioaccumulables et toxiques (PBT), celles qui sont très persistantes et très bioaccumulables (vPvB) et les polluants organiques persistants (POP) ;
b) prendre en considération les problèmes spécifiques soulevés par les nanomatériaux ;
c) renforcer encore les exigences sanitaires, en prévoyant, s’agissant des substances actives candidates à la substitution, des durées d’autorisations plus réduites que celles de droit commun ;
d) placer à égalité tous les producteurs et importateurs de substances actives, notamment les actuels et les futurs, en rendant obligatoire pour les fabricants de substances actives l’évaluation de leur source, pour éviter les risques de comportements de « passager clandestin » (« free rider ») vis-à-vis des opérateurs qui ont soutenu des dossiers dans le cadre du programme de travail précité ;
e) promouvoir les produits à faible risque les plus innovants et exigeant les technologies les plus modernes par des mécanismes renforcés, éventuellement fondés sur des durées accrues de protection intellectuelle ;
f) clarifier, pour sa parfaite compréhension par les professionnels, la disposition sur le commerce parallèle, conformément aux principes dégagés par la Cour de justice.
ANNEXE :
LISTE DES PERSONNES ENTENDUES PAR LES RAPPORTEURS
ET REMERCIEMENTS
Les rapporteurs tiennent à témoigner leur gratitude à l’ensemble des personnes qu’ils ont rencontrées.
*
* *
Ø Ministère de l’écologie, de l’énergie, du développement durable et de la mer, en charge des technologies vertes et des négociations sur le climat :
– M. Pablo Libreros, conseiller technique au cabinet du ministre d’Etat ;
– Mme Catherine Mir, adjointe au chef de service de la prévention des nuisances et de la qualité de l'environnement, direction générale de la prévention des risques ;
– M. Guillaume Bailly, chef bureau des substances et produits chimiques, direction générale de la prévention des risques ;
– M. Ludovic Chatelin, chargé de mission « biocides », direction générale de la prévention des risques.
Ø Agence française de sécurité sanitaire de l’environnement et du travail (AFSSET) :
– M. Martin Guespereau, directeur général ;
– M. Philippe Juvin, chef du département Réglementation chimie européenne.
Ø Association française des industries de la détergence (AFISE) et Chambre syndicale des industries de désinsectisation :
– Mme Claude Perrin, déléguée générale.
Ø Chambre syndicale de l'eau de Javel et des produits connexes :
– Mme Dominique Auzou, secrétaire générale.
Ø Fédération des industries des peintures, encres, couleurs, colles et adhésifs (FIPEC) :
– M. Michel Le Tallec, délégué général ;
– Mme Claudie Mathieu, responsable des affaires techniques et règlementaires.
Ø Institut technologique Forêt, Cellulose, Bois-construction, Ameublement (FCBA) :
– M. Marc Jequel, chef du laboratoire de biologie ;
– Dr Eric Heisel, chargé de profession.
Ø UFC Que choisir ?:
– M. Christophe Le Guehennec, spécialiste « santé ».
Ø Union des entreprises pour la protection des jardins et espaces verts (UPJ) :
– M. François Rollin, vice-président « biocides » ;
– M. Jacques My, directeur général ;
– Mme Mikaëline Billeret, animatrice de la Commission « Biocides ».
Ø Union des industries chimiques (UIC) :
– M. Philippe Prudhon, directeur des affaires techniques ;
– Mme Marie-Hélène Leroy, responsable santé et sécurité au travail, classification et étiquetage des produits.
Ø En outre, M. Jean Gaubert a rencontré des responsables d'entreprises concernées.
1 () La composition de cette Commission figure au verso de la présente page.
2 () Il s’agit des substances principalement utilisées dans des produits autres que les pesticides mais qui sont chacune marginalement utilisées en tant que biocide soit directement, soit dans un produit formé par la substance et un simple diluant, et qui n'est pas directement commercialisée pour une utilisation biocide. Sont nommément visées le dioxyde de carbone (gaz carbonique), l’azote, l’éthanol (alcool), l’alcool isopropylique, l’acide acétique et le Kieselguhr (composé de microalgues qui secrètent un squelette de silice, lequel tue certains insectes par absorption).
3 () Les produits biocides doivent être classés, emballés et étiquetés selon les règles de la directive 1999/45/CE concernant la classification, l’emballage et l’étiquetage des préparations dangereuses, texte applicable jusqu’au 1er juin 2015. Pour éviter tout risque de confusion avec d’autres produits, notamment des aliments ou des boissons, sont également prévues des exigences complémentaires d’emballage et d’étiquetage.